 Lakshmi Subramanian
Lakshmi Subramanian Maria Elisa Calcagnotto
Maria Elisa Calcagnotto Mercedes F. Paredes
Mercedes F. Paredes- 1Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research, University of California, San Francisco, San Francisco, CA, United States
- 2Neurophysiology and Neurochemistry of Neuronal Excitability and Synaptic Plasticity Laboratory, Department of Biochemistry, ICBS, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 3Graduate Program in Biological Sciences: Biochemistry, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 4Graduate Program in Neuroscience, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 5Department of Neurology, University of California, San Francisco, San Francisco, CA, United States
- 6Neuroscience Graduate Division, University of California, San Francisco, San Francisco, CA, United States
Creating a functional cerebral cortex requires a series of complex and well-coordinated developmental steps. These steps have evolved across species with the emergence of cortical gyrification and coincided with more complex behaviors. The presence of diverse progenitor cells, a protracted timeline for neuronal migration and maturation, and diverse neuronal types are developmental features that have emerged in the gyrated cortex. These factors could explain how the human brain has expanded in size and complexity. However, their complex nature also renders new avenues of vulnerability by providing additional cell types that could contribute to disease and longer time windows that could impact the composition and organization of the cortical circuit. We aim to discuss the unique developmental steps observed in human corticogenesis and propose how disruption of these species-unique processes could lead to malformations of cortical development.
Introduction
Malformations of cortical development (MCD) are an important and complex collection of neurodevelopmental disorders that underlie over 40% of medically refractory childhood seizures (Kuzniecky, 1994, 1995) with over three-quarters patients with MCD developing a seizure disorder (Leventer et al., 1999). A standout feature of MCD is their association with a broad range of cognitive deficits including mild to severe intellectual disability and autism (Guerrini and Dobyns, 2014). The heterogeneity in the genetic and phenotypic presentations that underlie MCD have limited our ability to classify these disorders and coincide with challenges to predict and manage these diseases. However, the increasing identification of genetic mutations have offered clues to common molecular pathways and cellular processes that are disrupted in cortical malformations. Mouse and rat models, the most frequently used to investigate the etiologies of MCD, have substantiated the clinical relevance of MCD-associated genetic mutations but have been unable to fully recapitulate the gross phenotypes observed in the clinical condition (Wong and Roper, 2016). A deeper understanding of human cortical development is necessary to more effectively apply the mechanistic findings from animal models to the disease state.
With this perspective, MCD offer an opportunity to decipher normal cortical development in the human brain. Proper development and organization of the mammalian brain requires the precise regulation of progenitor proliferation, cell type specification, and migration coordinated with neuronal differentiation, migration and cortical organization (Kriegstein and Alvarez-Buylla, 2009). Errors, due to gene mutations or environmental changes, can arise anywhere in this carefully choreographed series of events and result in alterations to cortical organization and a pathological states that are characteristic of MCD, including altered brain size, aberrant neuronal organization or clustering, and abnormal gyrification. In this review, we explore the neuro-developmental sequence in the human brain (Figure 1) and discuss various MCD associated with each of these stages, highlighting areas where human neurodevelopment differs from processes observed in mouse and rat models.
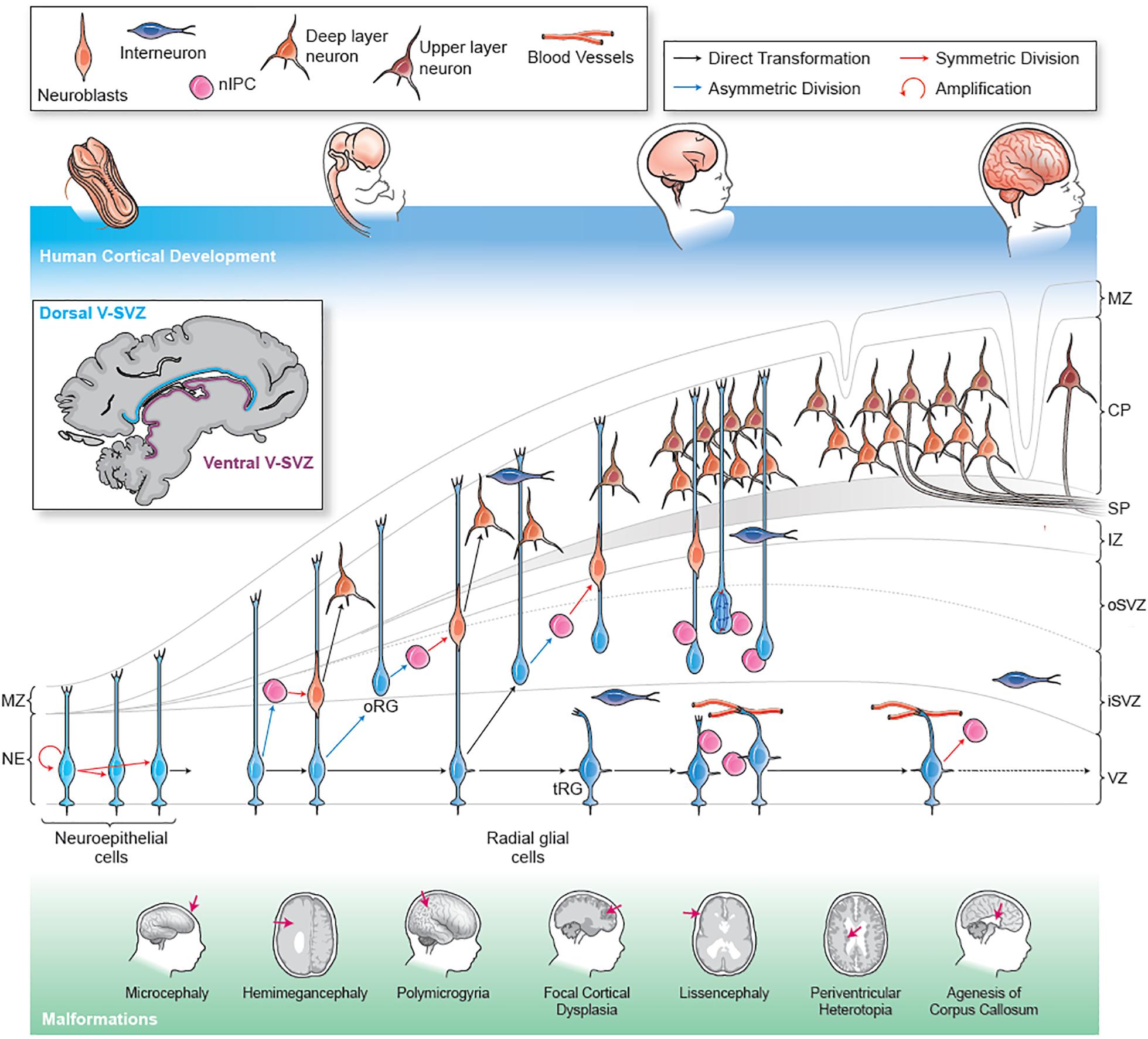
Figure 1. Human cortical development and stages of malformation. The human cerebral cortex forms early in the first trimester in the dorsal part of the telencephalon (forebrain). The human brain shows a rapid expansion in size and complexity during the 40 weeks of gestation as a result of extensive progenitor proliferation, migratory expansion and the generation of a complex connectivity pattern. During the first trimester, NE cells undergo symmetric division to expand the progenitor pool. NE cells elongate and convert into RG. By the end of the first trimester, RG are well established and can generate neurons (identified as migrating neuroblasts) directly through asymmetric division or indirectly by generation of IPCs. IPCs function as transient amplifying cells and can divide symmetrically one or more times to generate clones of neurons. Genetic mutations or environmental insults at this stage can cause microcephaly. In the second trimester, RG begin to give rise to RG-like cells that lack apical contact in the outer SVZ. These outer SVZ radial glia-like cells (oRG) are especially abundant in humans and other mammals with complex gyrencephalic cortices. oRG cells can generate neurons through IPCs and may contribute particularly to the generation of upper layer neurons. By the end of the second trimester, RG cells transform into truncated tRG. At this stage the RG scaffold is composed of the basal processes of the oRG cells. Proliferation errors or progenitor apoptosis in the second trimester can cause microcephaly or lissencephaly. Somatic mutations in mTOR pathway genes in NE, RG or oRG progenitors can result in FCD, HME or ME. Excitatory cortical pyramidal neurons are generated from RG and oRG progenitors via IPCs at the end of the first trimester. These neurons begin to migrate radially along the RG scaffold and until the middle of the third trimester. The pyramidal neurons maintain a radial organization as they migrate into and establish the cortical plate in an inside out manner, with the earliest generated neurons forming the deeper cortical layers while the youngest neurons contribute to the superficial layers. Errors in neuronal migration can result in heterotopias and lissencephaly. As they migrate, cortical pyramidal neurons begin to connect locally through transient connections in the subplate while they also begin to project axons that are myelinated by oligodendrocytes to form the cortical white matter. Errors in network connectivity can cause many forms of epilepsy, both de novo or secondary to other malformations along with ASD and schizophrenia. Errors of axonal projection lead to large scale connectivity defects like agenesis of corpus callosum. Toward the end of the second trimester, a combination of increased progenitor and neuronal numbers and rapidly expanding neuronal networks begins to generate physical stresses that contribute to the appearance of the main gyri. Over the course of the third trimester the secondary and tertiary gyrification of the cortex is established. Failure of gyrification may occur at any developmental stage leading to a range of malformations such as lissencephaly, polymicrogyria or pachygyria. Inhibitory interneurons migrate from ventrally located ganglionic eminences and appear in the cortex early in the second trimester. They migrate tangentially in the cortex along the marginal zone or in the subplate and SVZ and then move radially along the RG scaffold to integrate into the cortical circuits. Human interneurons continue to migrate into the cortex for a prolonged period through birth and early infancy. Failures of interneuron development, such as abnormal migration, arborization or maturation, can cause disinhibition within the cortical circuits resulting in epilepsy and cognitive dysfunction. Malformations of Cortical Development (MCD) (shown schematically at the bottom) arise at different stages along development. MZ, marginal zone; CP, cortical plate; IZ, intermediate zone, oSVZ, outer subventricular zone; iSVZ, inner sub-ventricular zone; VZ, ventricular zone; NE, neuroepithelium; RG, radial glia.
Developmental Stage 1: Progenitor Proliferation
The human cerebral cortex is a complex structure showing a remarkable increase in size when compared to other vertebrates. This increase can be attributed to an evolutionary increase in the numbers and types of progenitor cells that give rise to the various types of cortical neurons and glia. The human cerebral cortex displays a remarkable radial organization of its excitatory neurons that is a result of the carefully organized radial architecture established early in development (Rakic, 2009). Cortical excitatory neurons are generated from a parent population of neuro-epithelial (NE) cells that are the founder cells in the nervous system located in the ventricular zone (VZ). These NE cells are arranged in a pseudostratified epithelial organization with apical and basal contacts. Early on in development, NE cells proliferate symmetrically to generate more NE cells and expand the progenitor pool (Subramanian et al., 2017). This expansion of the NE progenitors has been hypothesized to be one of the key factors that contribute to an increased number of progenitor cells in the human brain (Rakic, 2009).
Around the beginning of neurogenesis, progenitor cells begin to show characteristic morphological, molecular and mitotic changes as NE cells transform into radial glial (RG) progenitors. Similar to NE cells, RG cells have contact with both the apical and basal surfaces, but their basal processes get progressively longer and form the radial scaffold that not only support the cortical architecture but also provide a framework for newly generated neurons to migrate along and establish the cortical plate, giving rise ultimately to the radial organization of the mature cortex. RG cells show a dramatic increase in the number of asymmetric divisions when compared to NE cells. These asymmetric divisions give rise to two different daughter cells, one of which is a self-renewed RG cell. The other daughter cell can be either a neuron, that migrates along the radial fiber of its sister cell to the cortical plate or more often a basal progenitor cell that no longer has apical contact with the ventricular surface. The basal progenitors are called intermediate progenitor cells (IPCs) and are predominantly located in the subventricular zone (SVZ). They undergo several rounds of proliferative divisions (Rakic, 2009) before generating differentiated neurons in a terminal division. At the end of neurogenesis, RG progenitors transform into translocating progenitor cells that lose contact with the apical surface and migrate through the cortex, eventually generating astrocytes. These translocating RG have been described extensively in multiple species including rat, ferrets, monkeys and humans (Schmechel and Rakic, 1979; Voigt, 1989; deAzevedo et al., 2003; Noctor et al., 2004).
A subtype of radial glia called the outer radial glial cells (oRGs) have been shown to generate neurons in humans, non-human primates and carnivores (Fietz et al., 2010; Hansen et al., 2010; Reillo et al., 2011; Kelava et al., 2012). Much smaller numbers of these cells have also been identified in mice (Shitamukai et al., 2011; Wang et al., 2011; Kalebic et al., 2019). Human oRG cells exhibit a characteristic mitotic behavior called “mitotic somatic translocation” (MST). The parent oRG cell moves rapidly along the basal process in the direction of the pial surface just prior to mitosis. This dramatic movement depends on the integrity of the basal process and contributes to the expansion of the oSVZ (Fietz et al., 2010; Ostrem et al., 2014; Kalebic et al., 2019). Recent studies have shown that the oRG cells become the predominant progenitor cell in the human cortex by mid-neurogenesis 17 gestational weeks (17 GW). At this stage, oRG cells also become the main contributor to the radial scaffold that supports the development of the cortical architecture as the vRG cells transform into truncated forms whose basal processes no longer reach the pial surface (Nowakowski et al., 2016). In humans, oRG cells are generated from ventricular radial glia (vRG) by a process that resembles epithelial-mesenchymal transitions (LaMonica et al., 2013; Pollen et al., 2015). vRG cells lose apical contact with the ventricular surface and translocate away into the SVZ to form an expanded progenitor rich, outer subventricular zone (oSVZ). Similar to vRG cells, oRG cells undergo multiple rounds of asymmetric division where they self-renew and generate daughter IPC cells.
Malformations of cortical development have been described that are associated with multiple progenitor cell types. In particular, changes to patterns of progenitor proliferation appear to be responsible for several developmental malformations. Progenitor proliferation consists of several events that are susceptible to errors leading to cortical malformations. These errors include but are not limited to changes in the proliferation rate, changes to symmetric or asymmetric division patterns, errors in mitosis resulting from changes to spindle orientation or centrosome maturation and distribution, errors in apical or basal attachment of progenitors affecting the position of the mitotic progenitors and increased progenitor apoptosis (Guarnieri et al., 2018; Pinson et al., 2019). Several of these errors may be the result of germline or somatic mutations in the patient, but there may also be environmental causes including viral infections in utero that predominantly affect progenitor cells.
Microcephaly
Primary microcephaly is a condition in which patients exhibit a marked decrease in the size of the head and the brain (>3 SDs over mean head size for the same age and gender). Although cortical organization is mostly preserved in the smaller brain, patients often have significant intellectual disability (Jayaraman et al., 2018). Microcephaly is predominantly associated with a decrease in progenitor numbers. This decrease can be due to decreased proliferation, changes in patterns of symmetric and asymmetric divisions and increased progenitor cell death.
Primary microcephaly is present at birth and can be caused by both genetic mutations and environmental insults like infections and toxins. Viral or parasitic infections such as Cytomegalovirus, Influenza, Herpes Simplex and Zika virus as well as parasitic infections like Toxoplasma gondii have all been linked to primary microcephaly (Devakumar et al., 2018). The microcephaly associated with the recent Zika virus epidemics have highlighted the role of progenitor cell proliferation in determining the size of the cerebral cortex in humans. Mouse studies, human in vitro models and studies on the developing human brain described widespread infection and consequent cell cycle arrest and apoptosis in infected NE, vRG and oRG cells (Li et al., 2016; Onorati et al., 2016; Retallack et al., 2016). Environmental toxins like alcohol in fetal alcohol syndrome have been shown to cause microcephaly through diverse effects on the developing neural tissue. Toxins and their metabolic by-products affect the survival and viability of progenitor and immature cells (Ehrhart et al., 2019). In addition, toxins can alter normal developmental signaling pathways including growth factor signaling, cause cytoskeletal disorganization in developing progenitor cells, alter the epigenetic landscape of developing neural tissues, initiate abnormal inflammatory responses, and alter patterns of programed cell death (Petrelli et al., 2019).
Abnormal cell biological changes in neural progenitor cells appear to be closely linked to primary microcephaly. The organization, maturation and distribution of the centrosome, and thus the organization of the spindle fibers appears to be especially vulnerable to these errors. The first gene mutations identified in patients with microcephaly, such as MCPH1, ASPM, CDK5RAP2, and CENPJ (Bond et al., 2002, 2005; Jackson et al., 2002; Zhong et al., 2006), had critical roles in the centrosome maturation and spindle organization and the list has been growing continually (Jayaraman et al., 2018). Centrosomes regulate the mitotic spindle and control both the ability of the progenitors to divide as well as the proper distribution of chromosomes across the two daughter cells. Errors in spindle organization can result in abnormal chromosomal numbers following cell division resulting in apoptosis of the daughter cells. Abnormal centrosome localization can also lead to abnormal orientation of the mitotic spindle, leading to a premature shift from proliferative symmetric divisions of ventricular progenitors to asymmetric divisions. Such a shift rapidly depletes the progenitor pool resulting in a smaller brain size. Mutations in the ASPM gene account for up to 40% of all autosomal recessive microcephaly (Pirozzi et al., 2018). Recent studies using genome editing approaches to knock out ASPM expression in the gyrencephalic ferret cortex (Johnson et al., 2018) have highlighted the role of this centrosomal protein in regulating the transition of apical vRG cells into basally located oRG cells. Such a change in progenitor sub-type results in fewer proliferative divisions. Further, loss of ASPM appears to disrupt the apical polarity complex that anchors vRG cells to the apical surface, leading to the delamination of the apical progenitors and an increase in the number of oRG cells. It is likely, therefore, that the effects of ASPM on the apical to basal transition in progenitors may be independent of or in addition to the effects of ASPM on the mitotic apparatus.
Mutations in the WDR62 gene are also frequently identified in patients with microcephaly. Mutations in human WDR62, however, result in a wide range of cortical malformations including microcephaly, pachygyria (unusually thick gyri) as well as callosal defects, lissencephaly and schizencephaly (Bilgüvar et al., 2010; Yu et al., 2010). Although gene knockout (KO) studies in mice have been very useful in understanding the role of WDR62 protein at the spindle pole as well as its interactions with ASPM (Jayaraman et al., 2016), other cortical phenotypes are not replicated in mice, which could be due to its absence of cortical folding. A recent report using gene edited human cerebral organoids has suggested a role for WDR62 in regulating progenitor proliferation as a result of delayed disassembly of the primary cilium, leading to cell cycle arrest and progenitor cell death (Zhang et al., 2019).
Other mutations associated with microcephaly appear to be involved in DNA regulation including DNA repair and chromatin organization (Jayaraman et al., 2018). These mutations appear to either affect cytokinesis of mitotic progenitors as a result of abnormal chromatin organization or increase progenitor apoptosis as a result of abnormal chromosome numbers.
mTORopathies and Progenitor Cells
There are also a spectrum of cortical malformations that are associated with abnormal cortical organization. These encompass a wide range of disorders involving local overgrowth or disorganization of specific cortical regions or cerebral hemispheres (Focal Cortical Dysplasias- FCD, Tuberous Sclerosis – TSC, partial or Hemimegalencephaly- HME) to enlargement of the entire cerebral cortex (Megalencephaly – ME) or the entire head (macrocephaly) (Pavone et al., 2017). While megalencephaly might contribute to an increase in head size, macrocephaly is more commonly associated with changes to the bone structure, vasculature or hydrocephalus. In contrast, FCD, TSC, HME and ME appear to be a series of related conditions resulting from specific changes to cortical architecture and size. In addition to abnormal cortical organization involving enlarged and aberrant neurons, disruptions of cortical layering, balloon cells as well as cortical tubers in TSC, patients with these conditions also suffer from intractable or medication-resistant epilepsy. In addition, some patients might also have some form of intellectual disability or autism spectrum disorders.
These malformations have been linked to mutations that hyper-activate the PI3K-AKT-mTOR signaling pathway. As a major signaling pathway regulating cell growth and proliferation, over-activation of the mTOR signaling pathway is thought to have an especially significant effect on progenitor cells, either their proliferation or their proper differentiation (Iffland and Crino, 2017). Sequencing studies from surgically resected patient lesions have identified mutations in multiple genes that are a part of or signal to the mTOR pathway AMPK, PI3K, AKT, PIK3CA, GATOR1 complex (DEPDC5-NPRL2-NPRL3), MTOR, TSC1, TSC2, PTEN, and STRAD (Iffland and Crino, 2017; Marsan and Baulac, 2018). While some of these mutations are germline mutations, multiple studies have demonstrated that in many cases the mTOR activating mutations are uniquely present within only a subset of cells associated with the lesion. This led to the hypothesis that the somatic mutation likely occurred in a single progenitor cell sometime in early neurodevelopment (Crino, 2011), with the more widespread malformations arising out of a mutagenic event in a progenitor cell at an earlier stage of development. Thus, the severity of the malformation may be directly linked to the stage of cortical development, with more severe malformations being the result of earlier mutations whereas mutations that occur later in development result in smaller malformations. Recent studies in mice have established that cortical malformations occur when the mTOR-activating mutation is present within the dorsal lineage (D’Gama et al., 2017). Studies using in utero electroporation approaches in rat and mouse models have been able to recapitulate the pathological and seizure phenotypes of mTOR-mediated cortical malformations by manipulating the molecular players in the mTOR signaling pathway. In utero electroporation approaches offer an elegant means of modeling the effects of somatic mutations arising in the fetal cortex in a focal subset of neural cells at varying developmental time-points. CRISPR-mediated gene deletion of DEPDC5 in rats (Hu et al., 2018) and activation of mTOR signaling in mice using constitutively active mTOR kinase or Rheb proteins (Lim et al., 2015; Nguyen et al., 2019) have been shown to cause abnormal neuronal morphology and migration defects in the developing cortex. Furthermore, the severity of the seizure phenotype appears to correlate well with the extent of the electroporation, providing evidence linking the severity of malformations with the timing of the mutagenic event.
Patients with mTOR-mediated cortical malformations also appear to have a combination of germline and somatic mutations within the mTOR pathway leading to the theory that the severity of the malformation may depend not just on the timing of the second somatic mutation but on a combination of the genes affected by the somatic mutation and the timing of the second hit (D’Gama et al., 2017; Ribierre et al., 2018). The two-hit hypothesis presents an intriguing model for human mTORopathies, particularly in light of recent evidence that mTOR signaling in human cortical progenitors is uniquely active only in the oRG cells (Nowakowski et al., 2017). oRG cells predominantly contribute to neurogenesis in humans between the middle and end of the second trimester (Hansen et al., 2010). This developmental time window may therefore be particularly vulnerable to the effects of mTOR-activating somatic mutations.
Cortical Folding and Progenitor Cells
The increase in the number of progenitor cells by proliferative expansion has been suggested to be a contributor to the gyrification, cortical infolding, of the human cortex. The progenitor-driven model of cortical folding was initially proposed following the development of folded brains in mice with excessive progenitor cells following constitutive activation of beta-catenin signaling (Chenn and Walsh, 2003). The more recent discovery of the expanded population of oRG cells in many gyrencephalic species (Martínez-Cerdeño et al., 2012) has led to the hypothesis that the presence of this expanded progenitor pool might be an important driver of cortical folding (Stahl et al., 2013; Wang et al., 2016; Borrell, 2018; Llinares-Benadero and Borrell, 2019). A recent study of the developing macaque cortex has put forth the idea that gyrification is a result of the expansion of the oSVZ progenitors but is driven by gliogenesis rather than neurogenesis (Rash et al., 2019). Several cortical malformations have been associated with abnormal gyrification including smooth brain (lissencephaly), excessive gyrification (polymicrogyria), and increased gyral thickness (pachygyria) but it is unclear at this time what the role of progenitor cells are in the generation of these malformations. Key gene mutations associated with gyrification defects including LIS1 and FLNA appear to regulate mitosis and early differentiation of progenitor cells in mouse models, affecting the orientation of spindle fibers, cell cycle length and cytokinesis (Vallee and Tsai, 2006; Fallet-Bianco et al., 2014; Moon et al., 2014; Sun and Hevner, 2014; Lian et al., 2019). Studies on human cerebral organoids generated from patients with Miller-Dieker syndrome (MDS) identified specific changes in the mitosis of oRG cells (Bershteyn et al., 2017). It remains to be seen, however, how changes in progenitor cell proliferation and migration relate to aberrant cortical folding patterns associated with gyral malformations.
Developmental Stage 2: Neuronal Migration
A fundamental property of the developing brain is that newborn neurons must leave their site of origin to migrate varying distances to their target regions. Within the cortex, they leave the V-SVZ and reach their appropriate location within the developing cortical plate (CP), the future six-layered cortex (Buchsbaum and Cappello, 2019). This process happens in a highly regulated pattern in the mammalian brain to correctly establish the distinct laminae of the cortex. The cortex is also one of the most complex parts of the brain across species not simply in size but in anatomical architecture and cellular organization. Errors in the movement and placement of incoming neurons, therefore, can have consequences in the final cortical network. These fall under the category of MCD and can manifest with a wide spectrum of phenotypes, including seizures and cognitive disability.
For excitatory/pyramidal cells, migration depends on the RG process of neural progenitors to serve as a scaffold between the progenitor niche and the CP (Noctor et al., 2001). Radial migration is the primary mode of excitatory neuron movement in the mammalian neocortex. Post-mitotic neurons leave the V-SVZ using locomotive behaviors to travel along the RG fiber to reach the CP. This longitudinal scaffold provided by vRG cells underlies the protomap or radial unit hypothesis for how the cerebral cortex is built (Rakic et al., 2009). The young neuronal progeny generated by positionally related progenitors are kept together by the physical restraints of the RG fibers. Thus the cortical surface can expand with individual neurons maintaining their spatial, and possibly molecular, identity within the developing cortical layers. Once arrived, they undergo somal translocation to position themselves within the correct lamina. Their migration is regulated by several key factors including gap junctions between RG and the migrating neuron (Elias and Kriegstein, 2008) and the guidance by the extracellular protein Reelin, expressed at the marginal zone by Cajal–Retzius cells (Hirota and Nakajima, 2017). These elements influence cytoskeletal dynamics and adhesion properties of the migratory neurons and a disruption, either genetic or environmental, leads to disorganized formation.
Inhibitory neurons (interneurons) are produced in the ventral/subpallial embryonic brain within the ganglionic eminences and undergo a long “tangential” migration to reach their appropriate layers in the CP (Marin et al., 2010). Their migration undergoes a more complex pattern characterized by saltatory motion where interneurons have abrupt changes in speeds and accentuated pauses (Bellion et al., 2005). These long-range movements observed by interneurons are guided by a variety of cues. Neuregulins influence ERBB4-expressing MGE-derived interneurons and CXCR4 and CXCR7 chemokine receptors mediate migration in response to stromal-cell-derived factor 1 (SDF1) present in the marginal zone and intermediate zones of the developing cortex (Tiveron and Cremer, 2008; Li et al., 2012). Interneurons eventually change to radial migration as they enter the CP. Dysregulation of these processes can lead to disorganized lamina and abnormal placement of neurons within the gray and white matter.
Lissencephalies
Lissencephaly, or “smooth brain,” is a set of conditions where the surface involutions (sulci and gyri) of the brain are missing or abnormal due to defects in neuronal migration. One report has implicated infection by cytomegalovirus, but most of our understanding of lissencephaly comes from the identification of associated genes that involve different aspects of cellular movement, including cytoskeletal integrity and extracellular matrix (ECM) interactions (Joseph et al., 2008; Mitchell, 2015). The first genes identified in patients with cortical malformations highlighted the importance of the cytoskeletal machinery. LIS1 and DCX mutations were identified in patients with lissencephaly (Reiner et al., 1993; des Portes et al., 1998; Fox et al., 1998; Gleeson et al., 1998); both genes encode microtubule-associated proteins. DCX is an X-linked gene and mutations in men result in complete lissencephaly while in females, the mutation is associated with ectopic neuronal layering, such as in subcortical band heterotopia or double cortex (Pilz, 1998). The product of LIS1 gene regulates transport along the microtubule motor protein, dynein, and the DCX protein, doublecortin, regulates microtubule stability and signaling during migration (Faulkner et al., 2000; Tanaka et al., 2004). Mutations in one of the seven tubulin isoforms, the proteins that polymerize into microtubules, are found in a broad spectrum of malformations (Bahi-Buisson et al., 2014; Bahi-Buisson and Cavallin, 2016). Tubulin-related malformations, or tubulinopathies, demonstrate the high overlap between different MCD and the intimate relationship between progenitor cell divisions and neuronal migration in normal cortical development. Tubulin is fundamental to the function of microtubules and the centrosome, thus defects can impact on both progenitor proliferation and neuron migration. Many tubulin mutations are associated with microcephaly, highlighting the importance of microtubules on the mechanics of cell division (Chakraborti et al., 2016). However, tubulinopathy phenotypes also include heterotopic cortical layering and abnormal gyration including microlissencephaly, classic lissencephaly (agyria), subcortical band heterotopia, and polymicrogyria-like cortical dysplasias (Jaglin and Chelly, 2009; Chakraborti et al., 2016).
The ECM is another arena where disrupted interactions between neural progenitors, migrating neurons, and supporting external macromolecules can lead to abnormal cortical layers and loss of gyration as seen in MCDs. ECM is a complex lattice of macromolecules including collagens, proteoglycans, and glycoproteins that occupies the extracellular space in tissue (Maeda, 2015). It serves many functions including as an adhesive substrate for cells and a reservoir for signaling molecules such as chemokines. The glycoprotein Reelin is the classic and most studied member of this group (for more detailed reviews please see Ishii et al., 2016; Lee and D’Arcangelo, 2016). Reelin mutations have been associated with lissencephaly with cerebellar hypoplasia, and the focus has been on expression by Cajal–Retzius cells at the meninges and the early embryonic role in regulating excitatory neuron migration for proper cortical layering. However, Reelin expression and members of the Reelin signaling pathway persists postnatally in the human brain (Abraham and Meyer, 2003; Deguchi et al., 2003). The function of Reelin at the end of gestation and in the early postnatal period is unknown. Reelin localization outside of the ECM and along dendrites suggests a role in synaptic remodeling and neuronal maturation (Roberts et al., 2005; Stranahan et al., 2013). Yet, Reelin receptors have been shown to function in neuronal migration in the postnatal rostral migratory stream (Andrade et al., 2007) and mouse cortical interneurons born at the end of gestation continue to respond to Reelin signaling in the brain, even at ages where Cajal–Retzius cells have disappeared (Hammond et al., 2006). Thus, Reelin could function later in cortical development to regulate neuronal migration, especially in the human brain where neuronal migration continues into infancy (Sanai et al., 2011; Paredes et al., 2016).
Other ECM components have been implicated in cortical malformations. Dystroglycans complexes serve as a physical link between the cytoskeleton and the ECM; their function is greatly modified by post translational changes such as glycosylation (Barresi, 2006). While “dystroglycanopathies” classically manifest as congenital muscular dystrophy, mutations in glycosyltransferase enzymes such as POMT1 and LARGE have also been shown to present with brain malformations such as cobblestone lissencephaly and polymicrogyria (Kano et al., 2002; Balci et al., 2005; Meilleur et al., 2014). A rare cortical malformation, bilateral frontoparietal polymicrogyria (BFPP), arises from mutations in the adhesion G-protein coupled receptor GPR56 (Piao, 2004). Collagen III is the ECM ligand for GPR56. Postmortem human and mouse model studies have shown that loss of GPR56 function leads to abnormal ECM organization within the meninges, resulting in pial heterotopias at the cortical surface and cobblestone lissencephaly (Li et al., 2008; Singer et al., 2013). The signaling pathways that are affected by these ECM-associated mutations is not known.
Heterotopias
Cortical malformations can also appear as neuronal clusterings, or heterotopias, in abnormal locations. These are commonly identified as periventricular heterotopias (PVH) arising from the ventricular wall or subcortical heterotopias within the cortical layers. Despite their focality, heterotopias have significant impact: the vast majority (over 80–90%) of patients with these localized collections have seizures (Srour et al., 2011; Barkovich et al., 2015). The phenotype becomes more severe if associated with more expansive heterotopia size or other types of cortical malformations (d’Orsi et al., 2004).
The most commonly identified gene in patients with PVH is FLNA (Fox et al., 1998; Sheen et al., 2005) that encodes for filamin A, an actin-binding cytoskeletal protein and serves as a scaffolding protein, binding as many as 45 different proteins (Lian and Sheen, 2015). Other cytoskeletal members linked to heterotopias in a genetic screen of individuals with PVH include TUBG1, KIF2A, and microtubule-associated protein 1B (MAP1B) (Poirier et al., 2013; Heinzen et al., 2018). FLNA mutant models in mice and rats have also shown defects in neural progenitor proliferation and abnormal RG scaffolding in addition to arrested neuronal migration from the V-SVZ (Nagano et al., 2004; Carabalona et al., 2012), highlighting the complex interplay of different developmental cell types in the brain. Further support of heterotopias involving more than neuronal migration was the association of PVH-microcephaly with mutations in the ARFGEF2 gene (Sheen et al., 2004). ADP-ribosylation factor guanine exchange factor 2 (ARFGEF2) directs vesicle trafficking and fusion and heterotopias from mutations in this gene are linked to a disrupted neuroependymal lining and abnormal cell-cell contact within the ventricular zone (Ferland et al., 2009). Progenitors and the neuroepithelium have also been highlighted in PVH by the presence of mutations in DCHS1 and FAT4, members of the protocadherin family; both these protocadherins are highly expressed in the ventricular zone of early fetal human brains compared to the intermediate zone and the developing cortical plate (Cappello et al., 2013).
Modeling these heterotopias in mouse and rat brains have highlighted differences between human and rodent brain development and suggest divergent regulatory processes at play in different species. Neither FLNA KO or the FAT4 KO mice develop heterotopias (Carabalona et al., 2012; Badouel et al., 2015). Furthermore, focal knockdown of either FAT4 or DCHS1 in embryonic mice led an increase in progenitor proliferation in addition to accumulation of cells in the mouse ventricular zone (Cappello et al., 2013). However, 3D modeling using human iPSC-derived organoids did replicate this hyperproliferation due to FAT4 or DCHS1 mutations (Klaus et al., 2019). Instead, these organoids revealed morphological and transcriptomic changes in mutant progenitor cells together with abnormal migratory behaviors including increased paused times. Heterogeneity was also observed in that despite all neurons bore the same mutation, only a subset had abnormal migration and formed clusters. Taken together, studies of heterotopias demonstrate the intimate link, both physical and molecular, between neural progenitors and migratory young neurons.
Interneuron Development and Malformations
The specific role of the GABAergic interneuron in MCD pathology and whether interneurons are directly disrupted or are secondarily affected by the abnormal development is unknown. Changes in interneuron distribution and number in MCDs have been observed in both the lissencephaly and FCD human cortex (Pancoast et al., 2005; Medici et al., 2016; Nakagawa et al., 2017), with parvalbumin-expressing subtype was the most affected interneuron. In patients with MDS (lissencephaly involving 17p13 deletion), the number of calretinin-expressing interneurons were abnormal in the fetal cortex but were no different at postnatal childhood ages when compared to the number in “control” brains; this suggested a specific effect on migration (Pancoast et al., 2005). Analysis of surgical resections from patients with FCD have also tried to shed light into how interneurons contribute to the seizure phenotype. Surgical tissue resected from FCD patients had a reduction in the frequency of spontaneous inhibitory currents onto pyramidal cells compared to currents in control (non-FCD) resected tissue (Calcagnotto et al., 2005); the change in inhibition was associated with abnormal interneuron distribution and altered GABA reuptake kinetics. A defining feature in FCD type II is the presence of morphologically aberrant cells, including cytomegalic neurons and balloon cells (Najm et al., 2018). The origins of these abnormal cells is unknown, but electrophysiological studies of cytomegalic neurons show that they have membrane properties that could render them as a seizure-generating (Wuarin et al., 1990; Tasker et al., 1992, 1996; Cepeda et al., 2006). Interestingly, in cases of severe FCD, cytomegalic neurons were found to be interneurons with more complex arborization and, unlike pyramidal cytomegalic neurons, had hyperexcitable properties, including the presence of spontaneous depolarization (André et al., 2007).
While all these studies suggest a mechanism for epileptogenesis in FCD patients, it remains unknown whether the changes were secondary to the emergence of the dysplasia. In a toxin-induced gyrencephalic model for cortical dysplasia, the MAM (methylmethoxymethanol)-exposed ferret, interneuron migration is disorganized and is associated with a disorganized distribution of Calbindin- and Parvalbumin-expressing interneuron subtypes (Poluch et al., 2008). This was hypothesized to be a non-intrinsic, or indirect, phenomenon as transplanted interneuron precursors cells from the MAM-treated ferret brain migrated normally in the normal (non-MAM-treated) cortex. One gene that directly ties abnormal interneuron development to MCD is the ARX (aristaless related homoebox) gene. ARX mutations have been associated with diverse symptoms including agenesis of the corpus callosum (ACC) and lissencephaly; XLAG syndrome includes severe cases associated with abnormal genitalia (Bonneau et al., 2002; Kato et al., 2004). Glutamatergic neurons do not express ARX though changes in ARX expression can indirectly affect their radial migration (Friocourt et al., 2008). The lissencephalic ARX brain is a 2–3 layered cortex with diminished neuronal populations and loss of interneurons (Forman et al., 2005; Okazaki et al., 2008; Marcorelles et al., 2010). ARX is a transcription factor that acts at several developmental stages including interneuron progenitor proliferation and neuronal migration (Friocourt et al., 2008; Friocourt and Parnavelas, 2011), though many questions remain about its exact function. The generation (Arshad et al., 2016), migration, and maturation of human cortical interneurons take place over a long timeline (Nicholas et al., 2013; Paredes et al., 2016; Close et al., 2017); therefore there are several points along their developmental trajectory where interneurons may directly or indirectly become dysfunctional and lead to abnormal circuit formation and connectivity.
Developmental Stage 3: Connectivity
As discussed, the telencephalon develops through a sequence of spatiotemporally coordinated events: cell proliferation, migration, differentiation, axonal growth which end with synaptogenesis and synaptic pruning. Altogether, this leads to the formation of functional neuronal connectivity (Rakic and Lombroso, 1998; Judas et al., 2003, 2005; Kostović and Jovanov-Milošević, 2006; Bystron et al., 2008; Raybaud, 2013; Raybaud et al., 2013). In mouse models, the organization and differentiation of the cortex, neuronal proliferation and migration are essentially complete at birth, and many neurons begin to be eliminated (Bystron et al., 2008; Stiles and Jernigan, 2010; Tau and Peterson, 2010; Raybaud and Widjaja, 2011), influencing the subsequent synaptic connectivity in the brain. Neuronal pruning reaches a peak at two postnatal weeks in mice. In contrast synaptogenesis and neuronal connectivity in the human cortex begin at 22 GW (Semple et al., 2013), but occur mainly after birth, particularly during the first two postnatal years (Huttenlocher, 1979; Herschkowitz et al., 1999). The timing of synaptogenesis in humans is region-dependent, reaching the maximum near postnatal age 3 months in the auditory cortex, 8–12 months in the visual cortex and 2–4 years of age in the prefrontal cortex (Huttenlocher et al., 1982; Lenroot and Giedd, 2006).
The proliferation and migration of neurons shape the coordinated network and connectivity of the developing neocortex. Interneurons migrate from the ganglionic eminences into the cortical plate to form local synapses with cortical pyramidal cells establishing microcircuits (Nadarajah and Parnavelas, 2002). The tangential migration of GABAergic interneurons in the cortex occurs in close association with the radial migration of glutamatergic pyramidal neurons (Bystron et al., 2008). Peak migratory activity of human cortical neurons is suggested to be at mid-gestation (weeks 20–22), though subpopulations of human interneurons continue to migrate through infancy, long after pyramidal neurons have stopped (Bystron et al., 2008; Raybaud et al., 2013). Furthermore, neurogenesis of interneurons occur at later stages of human fetal development (Letinic et al., 2002; Arshad et al., 2016). Late-developing and distinct lineages of GABAergic neurons in the human brain may add to the diversity of inhibitory neuron subtypes and ultimately impact the cortical circuitry that emerges.
The structural and functional development of the cerebral cortex is also regulated by electrical activity (Kirischuk et al., 2017) and connectivity is influenced by early neuronal activity in multiple ways. At embryonic stages, intermittent spontaneous activity is synchronized within small neuronal networks and become more complex during further development of the cerebral cortex, depending on maturation of network connectivity (Egorov and Draguhn, 2013; Yang et al., 2016). Spontaneous synchronous network activity is required to activate silent synapses by incorporating AMPA receptors into the postsynaptic membrane (Durand et al., 1996; Voigt et al., 2005), modeling the functional connectivity within the existing structural network. At this time, the neurotransmitter GABA has an excitatory effect on immature cells and is important in shaping connectivity (Ben-Ari, 2002; Dzhala et al., 2005; Batista-Brito and Fishell, 2009). GABAergic transmission by interneurons contributes to spontaneous network oscillations in the developing cortex through the synapse-driven coordinated activity patterns. In addition, neuronal spontaneous activity regulates GABA synthesis, affecting the inhibitory innervation patterns and the pruning process of redundant neuronal connections (Hata and Stryker, 1994; Chattopadhyaya et al., 2007; Kirischuk et al., 2017).
The connectivity process follows the radial gradient of the inside-out migration of cortical neurons from the deeper to the superficial cortical layers. By 18 GW, when the cortex is still smooth, radial (inside-out), but not tangential (horizontal), intracortical connections have formed (Noctor et al., 2001; Hadders-Algra, 2018). New connections subsequently induce an excessive tangential expansion of the superficial cortical layers (Huttenlocher and Dabholkar, 1997) which is associated with an increase in cortical compressive stress and initiation of cortical folding (Richman et al., 1975). The excessive tangential growth induced by the formation of intracortical horizontal connections is limited to the superficial cortical layers I to IV. As the deep layers and the white matter do not undergo tangential expansion, this process induces compressive stress, which has been hypothesized to lead to surface involution (Tallinen et al., 2014). The late migration of superficial neurons, the increased number of astrocytes, oligodendrocytes, and microglial cells, the intense neural connectivity formation and the laminar organization all contribute to cortical expansion and cortical folding. This process begins around 23 GW following the same tangential gradient as proliferation and connectivity (Huttenlocher et al., 1982; Moulton and Goriely, 2011; Raybaud et al., 2013; Budday et al., 2015b). Therefore, the connectivity-driven tangential growth, as a physics-based approach, mainly affects the superficial layers and induces the formation of gyri and sulci (Huttenlocher, 1979; Raybaud et al., 2013). Through term, secondary gyri formation extend concentrically around the primary sulci and after term, tertiary sulci develop together with short association fibers.
From 22 GW to the end of gestation, the connectivity and circuit organization in the human cortex continue to develop (Marin-Padilla, 1971). The synaptic density increases rapidly after birth, mainly within the early postnatal months and by 2 years of age reaches a level of about 50% higher than that is seen in adulthood (Herschkowitz et al., 1999). Synaptic elimination and remodeling in humans continue to adolescence, while in mice, the entire process appears to be completed by 3–4 weeks of age (Petanjek et al., 2011; Semple et al., 2013). Therefore, the human cortex remains relatively plastic and locally adapts its thickness and stress state as new neuronal connections form and dissolve thought the entire life (Budday et al., 2015a). For example, learning induces the formation of new connections, increases gray matter volume, and changes the brain surface morphology (White et al., 2010), while aging acts in the opposite way.
Neuronal connectivity in the cortex proceeds from the deeper to the superficial cortical layers. Thus, corticothalamic fibers from the deeper layer 6 and corticospinal from layer 5 first project single axons; thalamocortical fibers reach layer 4 and then neurons from layer 3 and 2 form long-association and commissural tracts, receiving multiple incoming fibers (Raybaud et al., 2013). Short-association tracts in the cortical gray matter and in the subcortical white matter results in a horizontal layering pattern of the neocortex (Marin-Padilla, 1970). The developing cortex contains the subplate neurons, the earliest generated neurons in the cerebral cortex of mammals (Luskin and Shatz, 1985; Perkins et al., 2008; Kanold, 2009). In humans, the subplate contains up to 50% of cortical neurons in the second trimester and it remains highly expanded during the first few years of life. The subplate neurons form one of the first functional cortical circuits (Kanold, 2009). They expand markedly during gestation, reaching peak numbers at about gestational week 28, guide efferent axons and establish transient connections with them until their cortical target cells are mature enough to become connected (Shu et al., 2003; Bi et al., 2018; Zhao et al., 2019). As these thalamocortical, corticocortical, interhemispheric commissural fibers and the intracortical connections gradually develop from 26 to 47 weeks, the migration path of the cortical cells traverses the white matter (Raybaud and Widjaja, 2011). Subplate neurons are thus uniquely positioned to establish the initial transient connections between these neurons (Bystron et al., 2008; Budday et al., 2015b).
The most prominent interhemispheric connective structure in the human brain is the corpus callosum (Luders et al., 2010). It begins to differentiate as a commissural plate around week 8, the axons appear around week 12, and adult callosal morphology is achieved around week 20 (Achiron and Achiron, 2001). The genesis of the corpus callosum, which connects the two cerebral hemispheres, depends on the functional integrity of callosal projection neurons and midline cell populations that produce various molecular cues such as semaphorins, netrins, fibroblast growth factors and slits that guide callosal axons to extend toward and cross the midline (Serafini et al., 1996; Bagri et al., 2002; Huffman et al., 2004; Andrews et al., 2006; Smith et al., 2006; Tole et al., 2006; Molyneaux et al., 2009; Niquille et al., 2009; Chinn et al., 2015). The anatomical midline structures that display a guidance activity for callosal axons include the glial wedge, the indusium griseum glia and the hippocampal commissure. The glial wedge, located in the medial wall of the lateral ventricle and composed of astrocytes (Bignami and Dahl, 1973), repels ipsilateral callosal axons toward the midline (Shu and Richards, 2001) and guides the axons toward contralateral cortex (Shu et al., 2003; Keeble et al., 2006). The indusium griseum glia are dorsal to the developing corpus callosum, express SLIT2 and guide commissural axons toward their site of midline crossing (Shu and Richards, 2001). The hippocampal commissure facilitates caudal callosal development, acting as a scaffold for the caudal corpus callosum (Paul et al., 2007).
Malformations and Cortical Connectivity
Subtle changes in neuronal layering and altered brain connectivity of specific circuits are a common finding in neurological and neuropsychiatric disorders, such as epilepsy, autism spectrum disorder and schizophrenia. Abnormal connectivity has been demonstrated in humans and in animal models by molecular anatomical and neuroimaging studies (Barkovich et al., 2012; Goodkind et al., 2015; Huang et al., 2016; Guarnieri et al., 2018). Recent functional connectivity studies using neuroimaging in humans have demonstrated long-range connectivity defects in patients with variable degrees of cortical malformations. These include patients with epilepsy associated with gray matter heterotopia (Shafi et al., 2015), polymicrogyria (Sethi et al., 2016), focal cortical dysplasia (Jeong et al., 2014; Hong et al., 2017; Rezayev et al., 2018), and tuberous sclerosis complex (Im et al., 2016), patients with schizophrenia (Adhikari et al., 2019; Zhao et al., 2019), autism spectrum disorders (Cooper et al., 2017; Bi et al., 2018) and ACC (Owen et al., 2013). The impaired neuronal connectivity and synaptic plasticity in these diseases have been associated with a decreased expression/function of neural ECM proteins, such as reelin, that might disrupt the axonal guidance cue gradients (Costa et al., 2004; Abdolmaleky et al., 2005; Dong et al., 2005; Berretta, 2012; Folsom and Fatemi, 2013; Jovanov Milošević et al., 2014). The abnormalities in location of particular neuronal populations, cell cues and inputs or presence of abnormal neurons may affect the subsequent developmental steps that control cortical synaptic connectivity (Easter et al., 1985; Redecker et al., 1998; Jacobs et al., 1999b) leading to aberrant interhemispheric, callosal, corticocortical and corticothalamic connectivity (Humphreys et al., 1991; Jacobs et al., 1999a).
Changes in structural connectivity and gyrus formation can also result in an imperfect tangential growth of the cortex and consequent white matter volume reduction. The white matter may become dysplastic together with the cortex, as a part of the cortical malformation, or secondary to the cortical abnormality, and may also become abnormal as a late result of an epileptogenesis process and/or behavioral abnormalities (Budday et al., 2015b). This distorted connectivity, with decreased volume of white matter in the corresponding portion of the hemisphere and the brainstem, has been described as the basis for the aberrant sulcation with no recognizable pattern seen in polymicrogyria (Raybaud and Widjaja, 2011). Microcephaly resulting from a deficient pool of neurons with a consequent lack of connectivity, is frequently associated with abnormalities of the corpus callosum and the level of reduction in white matter volume is correlated with the severity of the malformation (Raybaud and Widjaja, 2011). ACC is a complex condition in which the corpus callosum is partially or completely absent. ACC may or may not be associated with other MCD and can result from any of the following: defects in cellular proliferation and migration, axonal growth or in the midline structures (Jacobs et al., 1999a; Paul et al., 2007).
The synaptic connectivity in the brain is formed by distinct neuronal population interacting in a complex and yet organized spatiotemporal dynamic network. In a rat model for microgyria for example, the permanent loss of all connections coming via white matter, is partially compensated by an increased intracortical connectivity with no changes in the number of cortical neurons (Jacobs et al., 1999a). Such an area with preserved number of neurons and aberrant synaptic connectivity are the most probable sites to generate seizures (Chauvette et al., 2010; Timofeev et al., 2014; Timofeev, 2000). Brain oscillations emerge from this dynamic interaction between intrinsic cellular and network properties and correlate with distinct behavior state (Buzsáki and Draguhn, 2004). Since the circuits necessary for any abnormal oscillations are present in the neocortex, small shifts in the normal network and intrinsically bursting neurons involved in this circuit reorganization could result in either epileptiform activity or abnormal cortical oscillations that would affect the behavior (Chagnac-Amitai and Connors, 1989; Chagnac-Amitai et al., 1990). In the neocortex, recurrent excitatory connections are enhanced in focal cortical dysplasia and the aberrant synaptic connectivity produces a focal epileptogenic zone capable to generate epileptiform activities independent of connections with the malformation itself (Paz and Huguenard, 2015). Aberrant synaptic connectivity and displacement cortical interneurons are associated with abnormalities of gamma oscillations in patients with autism spectrum disorders (An et al., 2018; Hashemi et al., 2018). The neuronal disorganization and clusters of immature neurons in FCD I contribute to hyperexcitability and in the FCD II, in addition to increased excitation caused by immature neurons and reorganization of neuronal network, cytomegalic neurons (FCD II A) intensify the hyperexcitability and recruitment microcircuits in the cortex (Blümcke et al., 2011; Abdijadid et al., 2015). The presence of balloon cells (FCD IIB), claimed to not be involved in epileptogenesis, could play a role in modifying brain oscillation by interfering with neuronal connectivity. Abnormalities of high frequency oscillations and hyperexcitability were recorded in bottom part of the type II FCD cortical sulcus, independent of the presence of the balloon cells (Hu et al., 2019). Interestingly, in FCD I and II, the subcortical white matter neurons are excessive and are particularly frequent just beneath the depth of a sulcus or the base of a gyrus, the U-fiber layer (O’Halloran et al., 2017). The U-fiber system consists of subcortical arcuate fibers, following gyral contour and within gyral cores, originate from pyramidal neurons of layer 6 of cortex, acting as short- synaptic circuits that connect neighboring cortical regions or microcircuits (plexi) (Sarnat, 2018). The neuronal dispersion in the U-fibers compromises the short-range network connections, increasing the microcircuit (plexi) integration and consequently modifying the structure and function of local network contributing to the epileptogenesis.
Neuronal diversity and functional spatiotemporal dynamic in the network are key points to establish the normal connectivity in the brain. The cerebral cortex of mammals has a large diversity of cells operating in intricate circuits. This cellular diversity form complex circuits formed by synapses in distinct cellular compartment and time for encoding processing storage and sending information. Therefore, brain oscillations and behavior depends on the spatiotemporal dynamics of the network (Klausberger and Somogyi, 2008). Aberrant organization, plasticity of neuronal network recruiting distinct microcircuits of different location at specific time could alter synchronicity, leading to abnormal oscillations and consequent behavior, resulting in epilepsy and neuropsychiatric disorders associated with abnormalities of structural and functional connectivity.
Glial Populations
Neuronal connectivity involves not only the growth of neuronal dendrites and axons, but also the generation and expansion of astrocytes, oligodendrocytes and microglial cells, the formation of synapses, and the development of the vasculature system (Dulla et al., 2013). By 28 weeks of gestation in humans, when excitatory neuronal migration is mostly complete, the number of astrocytes, oligodendrocytes, and microglial cells increases, and myelination reaches its peak that corresponds to the shift from pre-oligodendrocytes to immature oligodendrocytes that produce myelin and induce white matter growth (Semple et al., 2013; Budday et al., 2015b). The white matter development and axonal outgrowth in mice take place only at postnatal day 1–3 which correspond to 23–32 weeks gestation in humans (Craig et al., 2003). Gray matter changes from a radial to a tangential organization during the third trimester. The degree of interconnection within the white matter, and consequent stiffness, remains low between weeks 22 and 38, until term (Raybaud, 2013; Raybaud et al., 2013), but increases after term, when myelination and the formation of astrocytic branches give rise to a highly connected microstructure. In mice, myelination begins during neurogenesis and occurs over a shorter timescale, peaking at approximately postnatal day 20 compared to adolescence in humans (Wiggins, 1986; Rockland and Defelipe, 2011); the increase in white matter stiffness from earlier myelination could in part explain, why mouse brains are less folded than mammalian brains.
Astrocytes with thousands of processes interact with all cell types of the CNS, and help drive nervous system development and sculpt its activity by guiding the migration of developing axons and neurons. Oligodendrocytes provide structural and metabolic support and axon myelination that facilitate nerve impulse conduction. The dynamic regulation of myelination may regulate the precise timing of information propagation and communication across functional circuits. Microglia control cell proliferation, differentiation and modify synapses. Toward the end of the synaptogenic period, weak and inappropriate synapses are eliminated by astrocytes and microglia, leaving neurons with their adult connectivity. The capillaries and neuro-glial-vascular coupling ensure oxygen and metabolic supply to neurons for proper connectivity development (Barres, 2008; Allen and Lyons, 2018). Thus glial dysfunction could contribute to the pathology of MCD via creating a local proinflammatory environment driven by abnormal gliovascular interaction (Kielbinski et al., 2016).
Conclusion
As our knowledge of human developmental neuroscience increases, we can better understand the heterogeneity and origins of MCD. Common themes that emerge from human studies, when compared to mouse analyses, are the expansion of unique progenitor populations that contribute to neurogenesis and the protracted developmental timeline over which progenitor proliferation and neuronal migration can occur. This has likely provided the substrate to allow for a larger cortex with complex connections, as in the human brain. But it also creates increased vulnerability for mistakes in the neurodevelopmental process at any point. Rodent models remain the foundation for deciphering the precise mechanistic pathways that are implicated in MCD by genetic studies. But a clearer comprehension of how these pathways diverge in the human brain will require the development of creative new approaches to study human neurobiology. Investigations directly on human cortical specimens are crucial to accurately study MCD. There is a need to improve access to tissue samples for research, both from post-mortem tissue specimens and surgical resections. Toward this goal, our scientific community must have ongoing communication with the public about the importance of human tissue-based research. Developing new models for human brain research, including patient-derived 2D and 3D culture systems, and identifying appropriate gyrencephalic animal models, will be fundamental to understanding the pathology of cortical malformations and establish ways to screen treatment approaches. Human cortical development is a long and intricate process but by the same token, it also has the potential for expanded windows of opportunity that may be utilized for therapeutic purposes. More studies are needed to understand this potential.
Author Contributions
All authors contributed to the ideas, preparation, and editing of this manuscript.
Funding
This work was supported by NINDS-K08 NS091537 (MP), NARSAD Young Investigator Award (LS and MP), The Roberta and Oscar Gregory Endowment in Stroke and Brain Research (MP), CURE Foundation Taking Flight Award (LS), Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq-465671/2014-4 (MC), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES (MC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Kenneth Xavier Probst and Noel Sirivansanti for figure preparation and Alice Paredes for editorial comments on the manuscript.
References
Abdijadid, S., Mathern, G. W., Levine, M. S., and Cepeda, C. (2015). Basic mechanisms of epileptogenesis in pediatric cortical dysplasia. CNS Neurosci. Ther. 21, 92–103. doi: 10.1111/cns.12345
Abdolmaleky, H. M., Cheng, K.-H., Russo, A., Smith, C. L., Faraone, S. V., Wilcox, M., et al. (2005). Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 134B, 60–66. doi: 10.1002/ajmg.b.30140
Abraham, H., and Meyer, G. (2003). Reelin-expressing neurons in the postnatal and adult human hippocampal formation. Hippocampus 13, 715–727. doi: 10.1002/hipo.10125
Achiron, R., and Achiron, A. (2001). Development of the human fetal corpus callosum: a high-resolution, cross-sectional sonographic study. Ultrasound Obstet. Gynecol. 18, 343–347. doi: 10.1046/j.0960-7692.2001.00512.x
Adhikari, B. M., Hong, L. E., Sampath, H., Chiappelli, J., Jahanshad, N., Thompson, P. M., et al. (2019). Functional network connectivity impairments and core cognitive deficits in schizophrenia. Hum. Brain Mapp. 40, 4593–4605. doi: 10.1002/hbm.24723
Allen, N. J., and Lyons, D. A. (2018). Glia as architects of central nervous system formation and function. Science 362, 181–185. doi: 10.1126/science.aat0473
An, K.-M., Ikeda, T., Yoshimura, Y., Hasegawa, C., Saito, D. N., Kumazaki, H., et al. (2018). Altered gamma oscillations during motor control in children with autism spectrum disorder. J. Neurosci. 38, 7878–7886. doi: 10.1523/JNEUROSCI.1229-18.2018
Andrade, N., Komnenovic, V., Blake, S. M., Jossin, Y., Howell, B., Goffinet, A., et al. (2007). ApoER2/VLDL receptor and Dab1 in the rostral migratory stream function in postnatal neuronal migration independently of Reelin. Proc. Natl. Acad. Sci. U.S.A. 104, 8508–8513. doi: 10.1073/pnas.0611391104
André, V. M., Wu, N., Yamazaki, I., Nguyen, S. T., Fisher, R. S., Vinters, H. V., et al. (2007). Cytomegalic interneurons: a new abnormal cell type in severe pediatric cortical dysplasia. J. Neuropathol. Exp. Neurol. 66, 491–504. doi: 10.1097/01.jnen.0000240473.50661.d8
Andrews, W., Liapi, A., Plachez, C., Camurri, L., Zhang, J., Mori, S., et al. (2006). Robo1 regulates the development of major axon tracts and interneuron migration in the forebrain. Development 133, 2243–2252. doi: 10.1242/dev.02379
Arshad, A., Vose, L. R., Vinukonda, G., Hu, F., Yoshikawa, K., Csiszar, A., et al. (2016). Extended production of cortical interneurons into the third trimester of human gestation. Cereb. Cortex 26, 2242–2256. doi: 10.1093/cercor/bhv074
Badouel, C., Zander, M. A., Liscio, N., Bagherie-Lachidan, M., Sopko, R., Coyaud, E., et al. (2015). Fat1 interacts with Fat4 to regulate neural tube closure, neural progenitor proliferation and apical constriction during mouse brain development. Development 142, 2781–2791. doi: 10.1242/dev.123539
Bagri, A., Marín, O., Plump, A. S., Mak, J., Pleasure, S. J., Rubenstein, J. L. R., et al. (2002). Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron 33, 233–248. doi: 10.1016/s0896-6273(02)00561-5
Bahi-Buisson, N., and Cavallin, M. (2016). “Tubulinopathies overview,” in GeneReviews, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington, Seattle).
Bahi-Buisson, N., Poirier, K., Fourniol, F., Saillour, Y., Valence, S., Lebrun, N., et al. (2014). The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 137, 1676–1700. doi: 10.1093/brain/awu082
Balci, B., Uyanik, G., Dincer, P., Gross, C., Willer, T., Talim, B., et al. (2005). An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker–Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul. Disord. 15, 271–275. doi: 10.1016/j.nmd.2005.01.013
Barkovich, A. J., Dobyns, W. B., and Guerrini, R. (2015). Malformations of cortical development and epilepsy. Cold Spring Harb. Perspect. Med. 5:a022392. doi: 10.1101/cshperspect.a022392
Barkovich, A. J., Guerrini, R., Kuzniecky, R. I., Jackson, G. D., and Dobyns, W. B. (2012). A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135, 1348–1369. doi: 10.1093/brain/aws019
Barres, B. A. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. doi: 10.1016/j.neuron.2008.10.013
Barresi, R. (2006). Dystroglycan: from biosynthesis to pathogenesis of human disease. J. Cell Sci. 119, 199–207. doi: 10.1242/jcs.02814
Batista-Brito, R., and Fishell, G. (2009). The developmental integration of cortical interneurons into a functional network. Curr. Top. Dev. Biol. 87, 81–118. doi: 10.1016/S0070-2153(09)01203-4
Bellion, A., Baudoin, J.-P., Alvarez, C., Bornens, M., and Métin, C. (2005). Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J. Neurosci. 25, 5691–5699. doi: 10.1523/JNEUROSCI.1030-05.2005
Ben-Ari, Y. (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Berretta, S. (2012). Extracellular matrix abnormalities in schizophrenia. Neuropharmacology 62, 1584–1597. doi: 10.1016/j.neuropharm.2011.08.010
Bershteyn, M., Nowakowski, T. J., Pollen, A. A., Di Lullo, E., Nene, A., Wynshaw-Boris, A., et al. (2017). Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 20, 435.e4–449.e4. doi: 10.1016/j.stem.2016.12.007
Bi, X.-A., Zhao, J., Xu, Q., Sun, Q., and Wang, Z. (2018). Abnormal functional connectivity of resting state network detection based on linear ICA analysis in autism spectrum disorder. Front. Physiol. 9:475. doi: 10.3389/fphys.2018.00475
Bignami, A., and Dahl, D. (1973). Differentiation of astrocytes in the cerebellar cortex and the pyramidal tracts of the newborn rat. An immunofluorescence study with antibodies to a protein specific to astrocytes. Brain Res. 49, 393–402. doi: 10.1016/0006-8993(73)90430-7
Bilgüvar, K., Oztürk, A. K., Louvi, A., Kwan, K. Y., Choi, M., Tatli, B., et al. (2010). Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210. doi: 10.1038/nature09327
Blümcke, I., Thom, M., Aronica, E., Armstrong, D. D., Vinters, H. V., Palmini, A., et al. (2011). The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia 52, 158–174. doi: 10.1111/j.1528-1167.2010.02777.x
Bond, J., Roberts, E., Mochida, G. H., Hampshire, D. J., Scott, S., Askham, J. M., et al. (2002). ASPM is a major determinant of cerebral cortical size. Nat. Genet. 32, 316–320. doi: 10.1038/ng995
Bond, J., Roberts, E., Springell, K., Lizarraga, S. B., Scott, S., Higgins, J., et al. (2005). A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 37, 353–355. doi: 10.1038/ng1539
Bonneau, D., Toutain, A., Laquerrière, A., Marret, S., Saugier-Veber, P., Barthez, M.-A., et al. (2002). X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann. Neurol. 51, 340–349. doi: 10.1002/ana.10119
Borrell, V. (2018). How cells fold the cerebral cortex. J. Neurosci. 38, 776–783. doi: 10.1523/JNEUROSCI.1106-17.2017
Buchsbaum, I. Y., and Cappello, S. (2019). Neuronal migration in the CNS during development and disease: insights from in vivo and in vitro models. Development 146:dev163766. doi: 10.1242/dev.163766
Budday, S., Steinmann, P., and Kuhl, E. (2015a). Physical biology of human brain development. Front. Cell. Neurosci. 9:257. doi: 10.3389/fncel.2015.00257
Budday, S., Steinmann, P., and Kuhl, E. (2015b). Secondary instabilities modulate cortical complexity in the mammalian brain. Philos. Mag. A 95, 3244–3256. doi: 10.1080/14786435.2015.1024184
Buzsáki, G., and Draguhn, A. (2004). Neuronal oscillations in cortical networks. Science 304, 1926–1929. doi: 10.1126/science.1099745
Bystron, I., Blakemore, C., and Rakic, P. (2008). Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 9, 110–122. doi: 10.1038/nrn2252
Calcagnotto, M. E., Paredes, M. F., Tihan, T., Barbaro, N. M., and Baraban, S. C. (2005). Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J. Neurosci. 25, 9649–9657. doi: 10.1523/JNEUROSCI.2687-05.2005
Cappello, S., Gray, M. J., Badouel, C., Lange, S., Einsiedler, M., Srour, M., et al. (2013). Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat. Genet. 45, 1300–1308. doi: 10.1038/ng.2765
Carabalona, A., Beguin, S., Pallesi-Pocachard, E., Buhler, E., Pellegrino, C., Arnaud, K., et al. (2012). A glial origin for periventricular nodular heterotopia caused by impaired expression of Filamin-A. Hum. Mol. Genet. 21, 1004–1017. doi: 10.1093/hmg/ddr531
Cepeda, C., André, V. M., Levine, M. S., Salamon, N., Miyata, H., Vinters, H. V., et al. (2006). Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav. 9, 219–235. doi: 10.1016/j.yebeh.2006.05.012
Chagnac-Amitai, Y., and Connors, B. W. (1989). Synchronized excitation and inhibition driven by intrinsically bursting neurons in neocortex. J. Neurophysiol. 62, 1149–1162. doi: 10.1152/jn.1989.62.5.1149
Chagnac-Amitai, Y., Luhmann, H. J., and Prince, D. A. (1990). Burst generating and regular spiking layer 5 pyramidal neurons of rat neocortex have different morphological features. J. Comp. Neurol. 296, 598–613. doi: 10.1002/cne.902960407
Chakraborti, S., Natarajan, K., Curiel, J., Janke, C., and Liu, J. (2016). The emerging role of the tubulin code: from the tubulin molecule to neuronal function and disease. Cytoskeleton 73, 521–550. doi: 10.1002/cm.21290
Chattopadhyaya, B., Di Cristo, G., Wu, C. Z., Knott, G., Kuhlman, S., Fu, Y., et al. (2007). GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron 54, 889–903. doi: 10.1016/j.neuron.2007.05.015
Chauvette, S., Volgushev, M., and Timofeev, I. (2010). Origin of active states in local neocortical networks during slow sleep oscillation. Cereb. Cortex 20, 2660–2674. doi: 10.1093/cercor/bhq009
Chenn, A., and Walsh, C. A. (2003). Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb. Cortex 13, 599–606. doi: 10.1093/cercor/13.6.599
Chinn, G. A., Hirokawa, K. E., Chuang, T. M., Urbina, C., Patel, F., Fong, J., et al. (2015). Agenesis of the corpus callosum due to defective glial wedge formation in Lhx2 mutant mice. Cereb. Cortex 25, 2707–2718. doi: 10.1093/cercor/bhu067
Close, J. L., Yao, Z., Levi, B. P., Miller, J. A., Bakken, T. E., Menon, V., et al. (2017). Single-cell profiling of an in vitro model of human interneuron development reveals temporal dynamics of cell type production and maturation. Neuron 93, 1035.e5–1048.e5. doi: 10.1016/j.neuron.2017.02.014
Cooper, R. A., Richter, F. R., Bays, P. M., Plaisted-Grant, K. C., Baron-Cohen, S., and Simons, J. S. (2017). Reduced hippocampal functional connectivity during episodic memory retrieval in autism. Cereb. Cortex 27, 888–902. doi: 10.1093/cercor/bhw417
Costa, E., Davis, J. M., Dong, E., Grayson, D. R., Guidotti, A., Tremolizzo, L., et al. (2004). A GABAergic cortical deficit dominates schizophrenia pathophysiology. Crit. Rev. Neurobiol. 16, 1–23.
Craig, A., Ling Luo, N., Beardsley, D. J., Wingate-Pearse, N., Walker, D. W., Hohimer, A. R., et al. (2003). Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp. Neurol. 181, 231–240. doi: 10.1016/s0014-4886(03)00032-3
Crino, P. B. (2011). mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 17, 734–742. doi: 10.1016/j.molmed.2011.07.008
deAzevedo, L. C., Fallet, C., Moura-Neto, V., Daumas-Duport, C., Hedin-Pereira, C., and Lent, R. (2003). Cortical radial glial cells in human fetuses: depth-correlated transformation into astrocytes. J. Neurobiol. 55, 288–298. doi: 10.1002/neu.10205
Deguchi, K., Inoue, K., Avila, W. E., Lopez-Terrada, D., Antalffy, B. A., Quattrocchi, C. C., et al. (2003). Reelin and disabled-1 expression in developing and mature human cortical neurons. J. Neuropathol. Exp. Neurol. 62, 676–684. doi: 10.1093/jnen/62.6.676
des Portes, V., Pinard, J. M., Billuart, P., Vinet, M. C., Koulakoff, A., Carrié, A., et al. (1998). A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92, 51–61. doi: 10.1016/s0092-8674(00)80898-3
Devakumar, D., Bamford, A., Ferreira, M. U., Broad, J., Rosch, R. E., Groce, N., et al. (2018). Infectious causes of microcephaly: epidemiology, pathogenesis, diagnosis, and management. Lancet Infect. Dis. 18, e1–e13. doi: 10.1016/s1473-3099(17)30398-5
D’Gama, A. M., Woodworth, M. B., Hossain, A. A., Bizzotto, S., Hatem, N. E., LaCoursiere, C. M., et al. (2017). Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 21:3754. doi: 10.1016/j.celrep.2017.11.106
Dong, E., Agis-Balboa, R. C., Simonini, M. V., Grayson, D. R., Costa, E., and Guidotti, A. (2005). Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 102, 12578–12583. doi: 10.1073/pnas.0505394102
d’Orsi, G., Tinuper, P., Bisulli, F., Zaniboni, A., Bernardi, B., Rubboli, G., et al. (2004). Clinical features and long term outcome of epilepsy in periventricular nodular heterotopia. Simple compared with plus forms. J. Neurol. Neurosurg. Psychiatry 75, 873–878. doi: 10.1136/jnnp.2003.024315
Dulla, C. G., Tani, H., Brill, J., Reimer, R. J., and Huguenard, J. R. (2013). Glutamate biosensor imaging reveals dysregulation of glutamatergic pathways in a model of developmental cortical malformation. Neurobiol. Dis. 49, 232–246. doi: 10.1016/j.nbd.2012.09.001
Durand, G. M., Kovalchuk, Y., and Konnerth, A. (1996). Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381, 71–75. doi: 10.1038/381071a0
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., et al. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213. doi: 10.1038/nm1301
Easter, S. S. Jr., Purves, D., Rakic, P., and Spitzer, N. C. (1985). The changing view of neural specificity. Science 230, 507–511. doi: 10.1126/science.4048944
Egorov, A. V., and Draguhn, A. (2013). Development of coherent neuronal activity patterns in mammalian cortical networks: common principles and local hetereogeneity. Mech. Dev. 130, 412–423. doi: 10.1016/j.mod.2012.09.006
Ehrhart, F., Roozen, S., Verbeek, J., Koek, G., Kok, G., van Kranen, H., et al. (2019). Review and gap analysis: molecular pathways leading to fetal alcohol spectrum disorders. Mol. Psychiatry 24, 10–17. doi: 10.1038/s41380-018-0095-4
Elias, L. A. B., and Kriegstein, A. R. (2008). Gap junctions: multifaceted regulators of embryonic cortical development. Trends Neurosci. 31, 243–250. doi: 10.1016/j.tins.2008.02.007
Fallet-Bianco, C., Laquerrière, A., Poirier, K., Razavi, F., Guimiot, F., Dias, P., et al. (2014). Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol. Commun. 2:69. doi: 10.1186/2051-5960-2-69
Faulkner, N. E., Dujardin, D. L., Tai, C. Y., Vaughan, K. T., O’Connell, C. B., Wang, Y., et al. (2000). A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2, 784–791. doi: 10.1038/35041020
Ferland, R. J., Batiz, L. F., Neal, J., Lian, G., Bundock, E., Lu, J., et al. (2009). Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum. Mol. Genet. 18, 497–516. doi: 10.1093/hmg/ddn377
Fietz, S. A., Kelava, I., Vogt, J., Wilsch-Bräuninger, M., Stenzel, D., Fish, J. L., et al. (2010). OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 13, 690–699. doi: 10.1038/nn.2553
Folsom, T. D., and Fatemi, S. H. (2013). The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 68, 122–135. doi: 10.1016/j.neuropharm.2012.08.015
Forman, M. S., Squier, W., Dobyns, W. B., and Golden, J. A. (2005). Genotypically defined lissencephalies show distinct pathologies. J. Neuropathol. Exp. Neurol. 64, 847–857. doi: 10.1097/01.jnen.0000182978.56612.41
Fox, J. W., Lamperti, E. D., Ekşioğlu, Y. Z., Hong, S. E., Feng, Y., Graham, D. A., et al. (1998). Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325. doi: 10.1016/s0896-6273(00)80651-0
Friocourt, G., Kanatani, S., Tabata, H., Yozu, M., Takahashi, T., Antypa, M., et al. (2008). Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J. Neurosci. 28, 5794–5805. doi: 10.1523/JNEUROSCI.1067-08.2008
Friocourt, G., and Parnavelas, J. G. (2011). Identification of Arx targets unveils new candidates for controlling cortical interneuron migration and differentiation. Front. Cell. Neurosci. 5:28. doi: 10.3389/fnbeh.2011.00028
Gleeson, J. G., Allen, K. M., Fox, J. W., Lamperti, E. D., Berkovic, S., Scheffer, I., et al. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72. doi: 10.1016/s0092-8674(00)80899-5
Goodkind, M., Eickhoff, S. B., Oathes, D. J., Jiang, Y., Chang, A., Jones-Hagata, L. B., et al. (2015). Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry 72, 305–315. doi: 10.1001/jamapsychiatry.14.2206
Guarnieri, F. C., de Chevigny, A., Falace, A., and Cardoso, C. (2018). Disorders of neurogenesis and cortical development. Dialogues Clin. Neurosci. 20, 255–266. doi: 10.31887/dcns.2018.20.4/ccardoso
Guerrini, R., and Dobyns, W. B. (2014). Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 13, 710–726. doi: 10.1016/s1474-4422(14)70040-7
Hadders-Algra, M. (2018). Early human motor development: from variation to the ability to vary and adapt. Neurosci. Biobehav. Rev. 90, 411–427. doi: 10.1016/j.JNEUROSCI.2018.05.009
Hammond, V., So, E., Gunnersen, J., Valcanis, H., Kalloniatis, M., and Tan, S.-S. (2006). Layer positioning of late-born cortical interneurons is dependent on Reelin but not p35 signaling. J. Neurosci. 26, 1646–1655. doi: 10.1523/JNEUROSCI.3651-05.2006
Hansen, D. V., Lui, J. H., Parker, P. R. L., and Kriegstein, A. R. (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561. doi: 10.1038/nature08845
Hashemi, E., Ariza, J., Rogers, H., Noctor, S. C., and Martínez-Cerdeño, V. (2018). The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb. Cortex 27, 1931–1943.
Hata, Y., and Stryker, M. P. (1994). Control of thalamocortical afferent rearrangement by postsynaptic activity in developing visual cortex. Science 265, 1732–1735. doi: 10.1126/science.8085163
Heinzen, E. L., O’Neill, A. C., Zhu, X., Allen, A. S., Bahlo, M., Chelly, J., et al. (2018). De novo and inherited private variants in MAP1B in periventricular nodular heterotopia. PLoS Genet. 14:e1007281. doi: 10.1371/journal.pgen.1007281
Herschkowitz, N., Kagan, J., and Zilles, K. (1999). Neurobiological bases of behavioral development in the second year. Neuropediatrics 30, 221–230. doi: 10.1055/s-2007-973495
Hirota, Y., and Nakajima, K. (2017). Control of neuronal migration and aggregation by reelin signaling in the developing cerebral cortex. Front. Cell Dev. Biol. 5:40. doi: 10.3389/fcell.2017.00040
Hong, S.-J., Bernhardt, B. C., Gill, R. S., Bernasconi, N., and Bernasconi, A. (2017). The spectrum of structural and functional network alterations in malformations of cortical development. Brain 140, 2133–2143. doi: 10.1093/brain/awx145
Hu, S., Knowlton, R. C., Watson, B. O., Glanowska, K. M., Murphy, G. G., Parent, J. M., et al. (2018). Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann. Neurol. 84, 140–146. doi: 10.1002/ana.25272
Hu, W.-H., Zhao, B.-T., Zhang, C., Wang, X., Sang, L., Shao, X.-Q., et al. (2019). Focal cortical dysplasia II-related seizures originate from the bottom of the dysplastic sulcus: a stereoelectroencephalography study. Clin. Neurophysiol. 130, 1596–1603. doi: 10.1016/j.clinph.2019.05.029
Huang, W.-C., Chen, Y., and Page, D. T. (2016). Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat. Commun. 7:13421. doi: 10.1038/ncomms13421
Huffman, K. J., Garel, S., and Rubenstein, J. L. R. (2004). Fgf8 regulates the development of intra-neocortical projections. J. Neurosci. 24, 8917–8923. doi: 10.1523/JNEUROSCI.2086-04.2004
Humphreys, P., Rosen, G. D., Press, D. M., Sherman, G. F., and Galaburda, A. M. (1991). Freezing lesions of the developing rat brain: a model for cerebrocortical microgyria. J. Neuropathol. Exp. Neurol. 50, 145–160. doi: 10.1097/00005072-199103000-00006
Huttenlocher, P. R. (1979). Synaptic density in human frontal cortex — Developmental changes and effects of aging. Brain Res. 163, 195–205. doi: 10.1016/0006-8993(79)90349-4
Huttenlocher, P. R., and Dabholkar, A. S. (1997). Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 387, 167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z
Huttenlocher, P. R., de Courten, C., Garey, L. J., and Van der Loos, H. (1982). Synaptogenesis in human visual cortex — evidence for synapse elimination during normal development. Neurosci. Lett. 33, 247–252. doi: 10.1016/0304-3940(82)90379-2
Iffland, P. H. II, and Crino, P. B. (2017). Focal cortical dysplasia: gene mutations, cell signaling, and therapeutic implications. Annu. Rev. Pathol. 12, 547–571. doi: 10.1146/annurev-pathol-052016-100138
Im, K., Ahtam, B., Haehn, D., Peters, J. M., Warfield, S. K., Sahin, M., et al. (2016). Altered structural brain networks in tuberous sclerosis complex. Cereb. Cortex 26, 2046–2058. doi: 10.1093/cercor/bhv026
Ishii, K., Kubo, K.-I., and Nakajima, K. (2016). Reelin and neuropsychiatric disorders. Front. Cell. Neurosci. 10:229. doi: 10.3389/fncel.2016.00229
Jackson, A. P., Eastwood, H., Bell, S. M., Adu, J., Toomes, C., Carr, I. M., et al. (2002). Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 71, 136–142. doi: 10.1086/341283
Jacobs, K. M., Hwang, B. J., and Prince, D. A. (1999a). Focal epileptogenesis in a rat model of polymicrogyria. J. Neurophysiol. 81, 159–173. doi: 10.1152/jn.1999.81.1.159
Jacobs, K. M., Kharazia, V. N., and Prince, D. A. (1999b). Mechanisms underlying epileptogenesis in cortical malformations. Epilepsy Res. 36, 165–188. doi: 10.1016/s0920-1211(99)00050-9
Jaglin, X. H., and Chelly, J. (2009). Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet. 25, 555–566. doi: 10.1016/j.tig.2009.10.003
Jayaraman, D., Bae, B.-I., and Walsh, C. A. (2018). The genetics of primary microcephaly. Annu. Rev. Genomics Hum. Genet. 19, 177–200. doi: 10.1146/annurev-genom-083117-021441
Jayaraman, D., Kodani, A., Gonzalez, D. M., Mancias, J. D., Mochida, G. H., Vagnoni, C., et al. (2016). Microcephaly proteins Wdr62 and Aspm define a mother centriole complex regulating centriole biogenesis, apical complex, and cell fate. Neuron 92, 813–828. doi: 10.1016/j.neuron.2016.09.056
Jeong, W., Jin, S.-H., Kim, M., Kim, J. S., and Chung, C. K. (2014). Abnormal functional brain network in epilepsy patients with focal cortical dysplasia. Epilepsy Res. 108, 1618–1626. doi: 10.1016/j.eplepsyres.2014.09.006
Johnson, M. B., Sun, X., Kodani, A., Borges-Monroy, R., Girskis, K. M., Ryu, S. C., et al. (2018). Aspm knockout ferret reveals an evolutionary mechanism governing cerebral cortical size. Nature 556, 370–375. doi: 10.1038/s41586-018-0035-0
Joseph, L. D., and Pushpalatha, Kuruvilla, S. (2008). Cytomegalovirus infection with lissencephaly. Indian J. Pathol. Microbiol. 51, 402–404.
Jovanov Milošević, N., Judaš, M., Aronica, E., and Kostovic, I. (2014). Neural ECM in laminar organization and connectivity development in healthy and diseased human brain. Prog. Brain Res. 214, 159–178. doi: 10.1016/B978-0-444-63486-3.00007-4
Judas, M., Rados, M., Jovanov-Milosevic, N., Hrabac, P., Stern-Padovan, R., and Kostovic, I. (2005). Structural, immunocytochemical, and mr imaging properties of periventricular crossroads of growing cortical pathways in preterm infants. AJNR Am. J. Neuroradiol. 26, 2671–2684.
Judas, M., Rasin, M. R., Kruslin, B., Kostović, K., Jukić, D., Petanjek, Z., et al. (2003). Dendritic overgrowth and alterations in laminar phenotypes of neocortical neurons in the newborn with semilobar holoprosencephaly. Brain Dev. 25, 32–39. doi: 10.1016/s0387-7604(02)00149-3
Kalebic, N., Gilardi, C., Stepien, B., Wilsch-Bräuninger, M., Long, K. R., Namba, T., et al. (2019). Neocortical expansion due to increased proliferation of basal progenitors is linked to changes in their morphology. Cell Stem Cell 24, 535.e9–550.e9. doi: 10.1016/j.stem.2019.02.017
Kano, H., Kobayashi, K., Herrmann, R., Tachikawa, M., Manya, H., Nishino, I., et al. (2002). Deficiency of α-dystroglycan in muscle–eye–brain disease. Biochem. Biophys. Res. Commun. 291, 1283–1286. doi: 10.1006/bbrc.2002.6608
Kanold, P. O. (2009). Subplate neurons: crucial regulators of cortical development and plasticity. Front. Neuroanat. 3:16. doi: 10.3389/neuro.05.016.2009
Kato, M., Das, S., Petras, K., Kitamura, K., Morohashi, K.-I., Abuelo, D. N., et al. (2004). Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 23, 147–159. doi: 10.1002/humu.10310
Keeble, T. R., Halford, M. M., Seaman, C., Kee, N., Macheda, M., Anderson, R. B., et al. (2006). The Wnt receptor Ryk is required for Wnt5a-mediated axon guidance on the contralateral side of the corpus callosum. J. Neurosci. 26, 5840–5848. doi: 10.1523/JNEUROSCI.1175-06.2006
Kelava, I., Reillo, I., Murayama, A. Y., Kalinka, A. T., Stenzel, D., Tomancak, P., et al. (2012). Abundant occurrence of basal radial glia in the subventricular zone of embryonic neocortex of a lissencephalic primate, the common marmoset Callithrix jacchus. Cereb. Cortex 22, 469–481. doi: 10.1093/cercor/bhr301
Kielbinski, M., Gzielo, K., and Soltys, Z. (2016). Review: roles for astrocytes in epilepsy: insights from malformations of cortical development. Neuropathol. Appl. Neurobiol. 42, 593–606. doi: 10.1111/nan.12331
Kirischuk, S., Sinning, A., Blanquie, O., Yang, J.-W., Luhmann, H. J., and Kilb, W. (2017). Modulation of neocortical development by early neuronal activity: physiology and pathophysiology. Front. Cell. Neurosci. 11:379. doi: 10.3389/fncel.2017.00379
Klaus, J., Kanton, S., Kyrousi, C., Ayo-Martin, A. C., Di Giaimo, R., Riesenberg, S., et al. (2019). Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat. Med. 25, 561–568. doi: 10.1038/s41591-019-0371-0
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. doi: 10.1126/science.1149381
Kostović, I., and Jovanov-Milošević, N. (2006). The development of cerebral connections during the first 20–45 weeks’ gestation. Semin. Fetal Neonatal Med. 11, 415–422. doi: 10.1016/j.siny.2006.07.001
Kriegstein, A., and Alvarez-Buylla, A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184. doi: 10.1146/annurev.neuro.051508.135600
Kuzniecky, R. I. (1994). Magnetic resonance imaging in developmental disorders of the cerebral cortex. Epilepsia 35, S44–S56. doi: 10.1111/j.1528-1157.1994.tb05988.x
Kuzniecky, R. I. (1995). MRI in cerebral developmental malformations and epilepsy. Magn. Reson. Imaging 13, 1137–1145. doi: 10.1016/0730-725x(95)02024-n
LaMonica, B. E., Lui, J. H., Hansen, D. V., and Kriegstein, A. R. (2013). Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat. Commun. 4:1665. doi: 10.1038/ncomms2647
Lee, G. H., and D’Arcangelo, G. (2016). New insights into reelin-mediated signaling pathways. Front. Cell. Neurosci. 10:122. doi: 10.3389/fncel.2016.00122
Lenroot, R. K., and Giedd, J. N. (2006). Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci. Biobehav. Rev. 30, 718–729. doi: 10.1016/j.neubiorev.2006.06.001
Letinic, K., Zoncu, R., and Rakic, P. (2002). Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649. doi: 10.1038/nature00779
Leventer, R. J., Phelan, E. M., Coleman, L. T., Kean, M. J., Jackson, G. D., and Harvey, A. S. (1999). Clinical and imaging features of cortical malformations in childhood. Neurology 53, 715–722.
Li, C., Xu, D., Ye, Q., Hong, S., Jiang, Y., Liu, X., et al. (2016). Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 120–126. doi: 10.1016/j.stem.2016.04.017
Li, H., Chou, S.-J., Hamasaki, T., Perez-Garcia, C. G., and O’Leary, D. D. M. (2012). Neuregulin repellent signaling via ErbB4 restricts GABAergic interneurons to migratory paths from ganglionic eminence to cortical destinations. Neural Dev. 7:10. doi: 10.1186/1749-8104-7-10
Li, S., Jin, Z., Koirala, S., Bu, L., Xu, L., Hynes, R. O., et al. (2008). GPR56 regulates pial basement membrane integrity and cortical lamination. J. Neurosci. 28, 5817–5826. doi: 10.1523/JNEUROSCI.0853-08.2008
Lian, G., and Sheen, V. L. (2015). Cytoskeletal proteins in cortical development and disease: actin associated proteins in periventricular heterotopia. Front. Cell. Neurosci. 9:99. doi: 10.3389/fncel.2015.00099
Lian, G., Wong, T., Lu, J., Hu, J., Zhang, J., and Sheen, V. (2019). Cytoskeletal associated filamin A and RhoA affect neural progenitor specification during mitosis. Cereb. Cortex 29, 1280–1290. doi: 10.1093/cercor/bhy033
Lim, J. S., Kim, W.-I., Kang, H.-C., Kim, S. H., Park, A. H., Park, E. K., et al. (2015). Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 21, 395–400. doi: 10.1038/nm.3824
Llinares-Benadero, C., and Borrell, V. (2019). Deconstructing cortical folding: genetic, cellular and mechanical determinants. Nat. Rev. Neurosci. 20, 161–176. doi: 10.1038/s41583-018-0112-2
Luders, E., Thompson, P. M., and Toga, A. W. (2010). The development of the corpus callosum in the healthy human brain. J. Neurosci. 30, 10985–10990. doi: 10.1523/JNEUROSCI.5122-09.2010
Luskin, M. B., and Shatz, C. J. (1985). Studies of the earliest generated cells of the cat’s visual cortex: cogeneration of subplate and marginal zones. J. Neurosci. 5, 1062–1075. doi: 10.1523/JNEUROSCI.05-04-01062.1985
Maeda, N. (2015). Proteoglycans and neuronal migration in the cerebral cortex during development and disease. Front. Neurosci. 9:98. doi: 10.3389/fnins.2015.00098
Marcorelles, P., Laquerrière, A., Adde-Michel, C., Marret, S., Saugier-Veber, P., Beldjord, C., et al. (2010). Evidence for tangential migration disturbances in human lissencephaly resulting from a defect in LIS1, DCX and ARX genes. Acta Neuropathol. 120, 503–515. doi: 10.1007/s00401-010-0692-z
Marin, O., Valiente, M., Ge, X., and Tsai, L.-H. (2010). Guiding neuronal cell migrations. Cold Spring Harb. Perspect. Biol. 2:a001834. doi: 10.1101/cshperspect.a001834
Marin-Padilla, M. (1970). Prenatal and early postnatal ontogenesis of the human motor cortex: a golgi study. I. The sequential development of the cortical layers. Brain Res. 23, 167–183. doi: 10.1016/0006-8993(70)90037-5
Marin-Padilla, M. (1971). Early prenatal ontogenesis of the cerebral cortex (neocortex) of the cat (Felis domestica). A Golgi study. I. The primordial neocortical organization. Z. Anat. Entwicklungsgesch. 134, 117–145. doi: 10.1007/bf00519296
Marsan, E., and Baulac, S. (2018). Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol. 44, 6–17. doi: 10.1111/nan.12463
Martínez-Cerdeño, V., Cunningham, C. L., Camacho, J., Antczak, J. L., Prakash, A. N., Cziep, M. E., et al. (2012). Comparative analysis of the subventricular zone in rat, ferret and macaque: evidence for an outer subventricular zone in rodents. PLoS One 7:e30178. doi: 10.1371/journal.pone.0030178
Medici, V., Rossini, L., Deleo, F., Tringali, G., Tassi, L., Cardinale, F., et al. (2016). Different parvalbumin and GABA expression in human epileptogenic focal cortical dysplasia. Epilepsia 57, 1109–1119. doi: 10.1111/epi.13405
Meilleur, K. G., Zukosky, K., Medne, L., Fequiere, P., Powell-Hamilton, N., Winder, T. L., et al. (2014). Clinical, pathologic, and mutational spectrum of dystroglycanopathy caused by LARGE mutations. J. Neuropathol. Exp. Neurol. 73, 425–441. doi: 10.1097/NEN.0000000000000065
Mitchell, K. J. (2015). “The genetic architecture of neurodevelopmental disorders,” in The Genetics of Neurodevelopmental Disorders, ed. K. J. Mitchell (Hoboken, NJ: John Wiley & Sons, Inc.), 1–28. doi: 10.1002/9781118524947.ch1
Molyneaux, B. J., Arlotta, P., Fame, R. M., MacDonald, J. L., MacQuarrie, K. L., and Macklis, J. D. (2009). Novel subtype-specific genes identify distinct subpopulations of callosal projection neurons. J. Neurosci. 29, 12343–12354. doi: 10.1523/JNEUROSCI.6108-08.2009
Moon, H. M., Youn, Y. H., Pemble, H., Yingling, J., Wittmann, T., and Wynshaw-Boris, A. (2014). LIS1 controls mitosis and mitotic spindle organization via the LIS1-NDEL1-dynein complex. Hum. Mol. Genet. 23, 449–466. doi: 10.1093/hmg/ddt436
Moulton, D. E., and Goriely, A. (2011). Circumferential buckling instability of a growing cylindrical tube. J. Mech. Phys. Solids 59, 525–537. doi: 10.1016/j.jmps.2011.01.005
Nadarajah, B., and Parnavelas, J. G. (2002). Modes of neuronal migration in the developing cerebral cortex. Nat. Rev. Neurosci. 3, 423–432. doi: 10.1038/nrn845
Nagano, T., Morikubo, S., and Sato, M. (2004). Filamin A and FILIP (Filamin A-Interacting Protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J. Neurosci. 24, 9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004
Najm, I. M., Sarnat, H. B., and Blümcke, I. (2018). Review: the international consensus classification of focal cortical dysplasia - a critical update 2018. Neuropathol. Appl. Neurobiol. 44, 18–31. doi: 10.1111/nan.12462
Nakagawa, J. M., Donkels, C., Fauser, S., Schulze-Bonhage, A., Prinz, M., Zentner, J., et al. (2017). Characterization of focal cortical dysplasia with balloon cells by layer-specific markers: evidence for differential vulnerability of interneurons. Epilepsia 58, 635–645. doi: 10.1111/epi.13690
Nguyen, L. H., Mahadeo, T., and Bordey, A. (2019). mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J. Neurosci. 39, 2762–2773. doi: 10.1523/JNEUROSCI.2260-18.2019
Nicholas, C. R., Chen, J., Tang, Y., Southwell, D. G., Chalmers, N., Vogt, D., et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. doi: 10.1016/j.stem.2013.04.005
Niquille, M., Garel, S., Mann, F., Hornung, J.-P., Otsmane, B., Chevalley, S., et al. (2009). Transient neuronal populations are required to guide callosal axons: a role for semaphorin 3C. PLoS Biol. 7:e1000230. doi: 10.1371/journal.pbio.1000230
Noctor, S. C., Flint, A. C., Weissman, T. A., Dammerman, R. S., and Kriegstein, A. R. (2001). Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720. doi: 10.1038/35055553
Noctor, S. C., Martínez-Cerdeño, V., Ivic, L., and Kriegstein, A. R. (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7, 136–144. doi: 10.1038/nn1172
Nowakowski, T. J., Bhaduri, A., Pollen, A. A., Alvarado, B., Mostajo-Radji, M. A., Di Lullo, E., et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. doi: 10.1126/science.aap8809
Nowakowski, T. J., Pollen, A. A., Sandoval-Espinosa, C., and Kriegstein, A. R. (2016). Transformation of the radial glia scaffold demarcates two stages of human cerebral cortex development. Neuron 91, 1219–1227. doi: 10.1016/j.neuron.2016.09.005
O’Halloran, R., Feldman, R., Marcuse, L., Fields, M., Delman, B., Frangou, S., et al. (2017). A method for u-fiber quantification from 7 T diffusion-weighted MRI data tested in patients with nonlesional focal epilepsy. NeuroReport 28, 457–461. doi: 10.1097/wnr.0000000000000788
Okazaki, S., Ohsawa, M., Kuki, I., Kawawaki, H., Koriyama, T., Ri, S., et al. (2008). Aristaless-related homeobox gene disruption leads to abnormal distribution of GABAergic interneurons in human neocortex: evidence based on a case of X-linked lissencephaly with abnormal genitalia (XLAG). Acta Neuropathol. 116, 453–462. doi: 10.1007/s00401-008-0382-2
Onorati, M., Li, Z., Liu, F., Sousa, A. M. M., Nakagawa, N., Li, M., et al. (2016). Zika Virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. 16, 2576–2592. doi: 10.1016/j.celrep.2016.08.038
Ostrem, B. E. L., Lui, J. H., Gertz, C. C., and Kriegstein, A. R. (2014). Control of outer radial glial stem cell mitosis in the human brain. Cell Rep. 8, 656–664. doi: 10.1016/j.celrep.2014.06.058
Owen, J. P., Li, Y.-O., Yang, F. G., Shetty, C., Bukshpun, P., Vora, S., et al. (2013). Resting-state networks and the functional connectome of the human brain in agenesis of the corpus callosum. Brain Connect. 3, 547–562. doi: 10.1089/brain.2013.0175
Pancoast, M., Dobyns, W., and Golden, J. A. (2005). Interneuron deficits in patients with the Miller-Dieker syndrome. Acta Neuropathol. 109, 400–404. doi: 10.1007/s00401-004-0979-z
Paredes, M. F., James, D., Gil-Perotin, S., Kim, H., Cotter, J. A., Ng, C., et al. (2016). Extensive migration of young neurons into the infant human frontal lobe. Science 354:aaf7073. doi: 10.1126/science.aaf7073
Paul, L. K., Brown, W. S., Adolphs, R., Michael Tyszka, J., Richards, L. J., Mukherjee, P., et al. (2007). Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat. Rev. Neurosci. 8, 287–299. doi: 10.1038/nrn2107
Pavone, P., Praticò, A. D., Rizzo, R., Corsello, G., Ruggieri, M., Parano, E., et al. (2017). A clinical review on megalencephaly: a large brain as a possible sign of cerebral impairment. Medicine 96:e6814. doi: 10.1097/MD.0000000000006814
Paz, J. T., and Huguenard, J. R. (2015). Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat. Neurosci. 18, 351–359. doi: 10.1038/nn.3950
Perkins, L., Hughes, E., Srinivasan, L., Allsop, J., Glover, A., Kumar, S., et al. (2008). Exploring cortical subplate evolution using magnetic resonance imaging of the fetal brain. Dev. Neurosci. 30, 211–220. doi: 10.1159/000109864
Petanjek, Z., Judaš, M., Šimic, G., Rasin, M. R., Uylings, H. B. M., Rakic, P., et al. (2011). Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 108, 13281–13286. doi: 10.1073/pnas.1105108108
Petrelli, B., Bendelac, L., Hicks, G. G., and Fainsod, A. (2019). Insights into retinoic acid deficiency and the induction of craniofacial malformations and microcephaly in fetal alcohol spectrum disorder. Genesis 57:e23278. doi: 10.1002/dvg.23278
Piao, X. (2004). G protein-coupled receptor-dependent development of human frontal cortex. Science 303, 2033–2036. doi: 10.1126/science.1092780
Pilz, D. (1998). LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum. Mol. Genet. 7, 2029–2037. doi: 10.1093/hmg/7.13.2029
Pinson, A., Namba, T., and Huttner, W. B. (2019). Malformations of human neocortex in development–their progenitor cell basis and experimental model systems. Front. Cell. Neurosci. 13:305. doi: 10.3389/fncel.2019.00305
Pirozzi, F., Nelson, B., and Mirzaa, G. (2018). From microcephaly to megalencephaly: determinants of brain size. Dialogues Clin. Neurosci. 20, 267–282.
Poirier, K., Lebrun, N., Broix, L., Tian, G., Saillour, Y., Boscheron, C., et al. (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 45, 639–647. doi: 10.1038/ng.2613
Pollen, A. A., Nowakowski, T. J., Chen, J., Retallack, H., Sandoval-Espinosa, C., Nicholas, C. R., et al. (2015). Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67. doi: 10.1016/j.cell.2015.09.004
Poluch, S., Jablonska, B., and Juliano, S. L. (2008). Alteration of interneuron migration in a ferret model of cortical dysplasia. Cereb. Cortex 18, 78–92. doi: 10.1093/cercor/bhm032
Rakic, P. (2009). Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735. doi: 10.1038/nrn2719
Rakic, P., Ayoub, A. E., Breunig, J. J., and Dominguez, M. H. (2009). Decision by division: making cortical maps. Trends Neurosci. 32, 291–301. doi: 10.1016/j.tins.2009.01.007
Rakic, P., and Lombroso, P. J. (1998). Development of the cerebral cortex: I. Forming the cortical structure. J. Am. Acad. Child Adolesc. Psychiatry 37, 116–117.
Rash, B. G., Duque, A., Morozov, Y. M., Arellano, J. I., Micali, N., and Rakic, P. (2019). Gliogenesis in the outer subventricular zone promotes enlargement and gyrification of the primate cerebrum. Proc. Natl. Acad. Sci. U.S.A. 116, 7089–7094. doi: 10.1073/pnas.1822169116
Raybaud, C. (2013). The premature brain: imaging, anatomy and uncertain outcome. Neuroradiology 55(Suppl. 2), 1–2. doi: 10.1007/s00234-013-1244-8
Raybaud, C., Ahmad, T., Rastegar, N., Shroff, M., and Al Nassar, M. (2013). The premature brain: developmental and lesional anatomy. Neuroradiology 55(Suppl. 2), 23–40. doi: 10.1007/s00234-013-1231-0
Raybaud, C., and Widjaja, E. (2011). Development and dysgenesis of the cerebral cortex: malformations of cortical development. Neuroimaging Clin. N. Am. 21, 483–543. doi: 10.1016/j.nic.2011.05.014
Redecker, C., Lutzenburg, M., Gressens, P., Evrard, P., Witte, O. W., and Hagemann, G. (1998). Excitability changes and glucose metabolism in experimentally induced focal cortical dysplasias. Cereb. Cortex 8, 623–634. doi: 10.1093/cercor/8.7.623
Reillo, I., de Juan Romero, C., García-Cabezas, M. Á., and Borrell, V. (2011). A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb. Cortex 21, 1674–1694. doi: 10.1093/cercor/bhq238
Reiner, O., Carrozzo, R., Shen, Y., Wehnert, M., Faustinella, F., Dobyns, W. B., et al. (1993). Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature 364, 717–721. doi: 10.1038/364717a0
Retallack, H., Di Lullo, E., Arias, C., Knopp, K. A., Laurie, M. T., Sandoval-Espinosa, C., et al. (2016). Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. U.S.A. 113, 14408–14413. doi: 10.1073/pnas.1618029113
Rezayev, A., Feldman, H. A., Levman, J., and Takahashi, E. (2018). Bilateral thalamocortical abnormalities in focal cortical dysplasia. Brain Res. 1694, 38–45. doi: 10.1016/j.brainres.2018.05.005
Ribierre, T., Deleuze, C., Bacq, A., Baldassari, S., Marsan, E., Chipaux, M., et al. (2018). Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest. 128, 2452–2458. doi: 10.1172/JCI99384
Richman, D. P., Stewart, R. M., Hutchinson, J. W., and Caviness, V. S. (1975). Mechanical model of brain convolutional development. Science 189, 18–21. doi: 10.1126/science.1135626
Roberts, R. C., Xu, L., Roche, J. K., and Kirkpatrick, B. (2005). Ultrastructural localization of reelin in the cortex in post-mortem human brain. J. Comp. Neurol. 482, 294–308. doi: 10.1002/cne.20408
Rockland, K. S., and Defelipe, J. (2011). Cortical white matter: beyond the pale. Front. Neuroanat. 5:67. doi: 10.3389/fnana.2011.00067
Sanai, N., Nguyen, T., Ihrie, R. A., Mirzadeh, Z., Tsai, H.-H., Wong, M., et al. (2011). Corridors of migrating neurons in the human brain and their decline during infancy. Nature 478, 382–386. doi: 10.1038/nature10487
Sarnat, H. B. (2018). The 2016 Bernard Sachs Lecture: timing in morphogenesis and genetic gradients during normal development and in malformations of the nervous system. Pediatr. Neurol. 83, 3–13. doi: 10.1016/j.pediatrneurol.2017.08.015
Schmechel, D. E., and Rakic, P. (1979). A Golgi study of radial glial cells in developing monkey telencephalon: morphogenesis and transformation into astrocytes. Anat. Embryol. 156, 115–152. doi: 10.1007/bf00300010
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M., and Noble-Haeusslein, L. J. (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106-107, 1–16. doi: 10.1016/j.pneurobio.2013.04.001
Serafini, T., Colamarino, S. A., Leonardo, E. D., Wang, H., Beddington, R., Skarnes, W. C., et al. (1996). Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87, 1001–1014. doi: 10.1016/s0092-8674(00)81795-x
Sethi, M., Pedersen, M., and Jackson, G. D. (2016). Polymicrogyric cortex may predispose to seizures via abnormal network topology: an fMRI connectomics study. Epilepsia 57, e64–e68. doi: 10.1111/epi.13304
Shafi, M. M., Vernet, M., Klooster, D., Chu, C. J., Boric, K., Barnard, M. E., et al. (2015). Physiological consequences of abnormal connectivity in a developmental epilepsy. Ann. Neurol. 77, 487–503. doi: 10.1002/ana.24343
Sheen, V. L., Ganesh, V. S., Topcu, M., Sebire, G., Bodell, A., Hill, R. S., et al. (2004). Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat. Genet. 36, 69–76. doi: 10.1038/ng1276
Sheen, V. L., Jansen, A., Chen, M. H., Parrini, E., Morgan, T., Ravenscroft, R., et al. (2005). Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 64, 254–262. doi: 10.1212/01.WNL.0000149512.79621.DF
Shitamukai, A., Konno, D., and Matsuzaki, F. (2011). Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J. Neurosci. 31, 3683–3695. doi: 10.1523/JNEUROSCI.4773-10.2011
Shu, T., and Richards, L. J. (2001). Cortical axon guidance by the glial wedge during the development of the corpus callosum. J. Neurosci. 21, 2749–2758. doi: 10.1523/JNEUROSCI.21-08-02749.2001
Shu, T., Sundaresan, V., McCarthy, M. M., and Richards, L. J. (2003). Slit2 guides both precrossing and postcrossing callosal axons at the midline in vivo. J. Neurosci. 23, 8176–8184. doi: 10.1523/JNEUROSCI.23-22-08176.2003
Singer, K., Luo, R., Jeong, S.-J., and Piao, X. (2013). GPR56 and the developing cerebral cortex: cells, matrix, and neuronal migration. Mol. Neurobiol. 47, 186–196. doi: 10.1007/s12035-012-8343-0
Smith, K. M., Ohkubo, Y., Maragnoli, M. E., Rasin, M.-R., Schwartz, M. L., Sestan, N., et al. (2006). Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat. Neurosci. 9, 787–797. doi: 10.1038/nn1705
Srour, M., Rioux, M.-F., Varga, C., Lortie, A., Major, P., Robitaille, Y., et al. (2011). The clinical spectrum of nodular heterotopias in children: report of 31 patients. Epilepsia 52, 728–737. doi: 10.1111/j.1528-1167.2010.02975.x
Stahl, R., Walcher, T., De Juan Romero, C., Pilz, G. A., Cappello, S., Irmler, M., et al. (2013). Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell 153, 535–549. doi: 10.1016/j.cell.2013.03.027
Stiles, J., and Jernigan, T. L. (2010). The basics of brain development. Neuropsychol. Rev. 20, 327–348. doi: 10.1007/s11065-010-9148-4
Stranahan, A. M., Erion, J. R., and Wosiski-Kuhn, M. (2013). Reelin signaling in development, maintenance, and plasticity of neural networks. Ageing Res. Rev. 12, 815–822. doi: 10.1016/j.arr.2013.01.005
Subramanian, L., Bershteyn, M., Paredes, M. F., and Kriegstein, A. R. (2017). Dynamic behaviour of human neuroepithelial cells in the developing forebrain. Nat. Commun. 8:14167. doi: 10.1038/ncomms14167
Sun, T., and Hevner, R. F. (2014). Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat. Rev. Neurosci. 15, 217–232. doi: 10.1038/nrn3707
Tallinen, T., Chung, J. Y., Biggins, J. S., and Mahadevan, L. (2014). Gyrification from constrained cortical expansion. Proc. Natl. Acad. Sci. U.S.A. 111, 12667–12672. doi: 10.1073/pnas.1406015111
Tanaka, T., Serneo, F. F., Higgins, C., Gambello, M. J., Wynshaw-Boris, A., and Gleeson, J. G. (2004). Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 165, 709–721. doi: 10.1083/jcb.200309025
Tasker, J. G., Hoffman, N. W., Kim, Y. I., Fisher, R. S., Peacock, W. J., and Dudek, F. E. (1996). Electrical properties of neocortical neurons in slices from children with intractable epilepsy. J. Neurophysiol. 75, 931–939. doi: 10.1152/jn.1996.75.2.931
Tasker, J. G., Peacock, W. J., and Dudek, F. E. (1992). Local synaptic circuits and epileptiform activity in slices of neocortex from children with intractable epilepsy. J. Neurophysiol. 67, 496–507. doi: 10.1152/jn.1992.67.3.496
Tau, G. Z., and Peterson, B. S. (2010). Normal development of brain circuits. Neuropsychopharmacology 35, 147–168. doi: 10.1038/npp.2009.115
Timofeev, I. (2000). Origin of slow cortical oscillations in deafferented cortical slabs. Cereb. Cortex 10, 1185–1199. doi: 10.1093/cercor/10.12.1185
Timofeev, I., Chauvette, S., and Soltani, S. (2014). Neocortical focus: experimental view. Int. Rev. Neurobiol. 114, 9–33. doi: 10.1016/B978-0-12-418693-4.00002-9
Tiveron, M.-C., and Cremer, H. (2008). CXCL12/CXCR4 signalling in neuronal cell migration. Curr. Opin. Neurobiol. 18, 237–244. doi: 10.1016/j.conb.2008.06.004
Tole, S., Gutin, G., Bhatnagar, L., Remedios, R., and Hébert, J. M. (2006). Development of midline cell types and commissural axon tracts requires Fgfr1 in the cerebrum. Dev. Biol. 289, 141–151. doi: 10.1016/j.ydbio.2005.10.020
Vallee, R. B., and Tsai, J.-W. (2006). The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes Dev. 20, 1384–1393. doi: 10.1101/gad.1417206
Voigt, T. (1989). Development of glial cells in the cerebral wall of ferrets: direct tracing of their transformation from radial glia into astrocytes. J. Comp. Neurol. 289, 74–88. doi: 10.1002/cne.902890106
Voigt, T., Opitz, T., and de Lima, A. D. (2005). Activation of early silent synapses by spontaneous synchronous network activity limits the range of neocortical connections. J. Neurosci. 25, 4605–4615. doi: 10.1523/JNEUROSCI.3803-04.2005
Wang, L., Hou, S., and Han, Y.-G. (2016). Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex. Nat. Neurosci. 19, 888–896. doi: 10.1038/nn.4307
Wang, X., Tsai, J.-W., LaMonica, B., and Kriegstein, A. R. (2011). A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 14, 555–561. doi: 10.1038/nn.2807
White, T., Su, S., Schmidt, M., Kao, C.-Y., and Sapiro, G. (2010). The development of gyrification in childhood and adolescence. Brain Cogn. 72, 36–45. doi: 10.1016/j.bandc.2009.10.009
Wong, M., and Roper, S. N. (2016). Genetic animal models of malformations of cortical development and epilepsy. J. Neurosci. Methods 260, 73–82. doi: 10.1016/j.jneumeth.2015.04.007
Wuarin, J.-P., Kim, Y. I., Cepeda, C., Tasker, J. G., Walsh, J. P., Peacock, W. J., et al. (1990). Synaptic transmission in human neocortex removed for treatment of intractable epilepsy in children. Ann. Neurol. 28, 503–511. doi: 10.1002/ana.410280406
Yang, J.-W., Reyes-Puerta, V., Kilb, W., and Luhmann, H. J. (2016). Spindle bursts in neonatal rat cerebral cortex. Neural Plast. 2016:3467832. doi: 10.1155/2016/3467832
Yu, T. W., Mochida, G. H., Tischfield, D. J., Sgaier, S. K., Flores-Sarnat, L., Sergi, C. M., et al. (2010). Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 42, 1015–1020. doi: 10.1038/ng.683
Zhang, W., Yang, S.-L., Yang, M., Herrlinger, S., Shao, Q., Collar, J. L., et al. (2019). Modeling microcephaly with cerebral organoids reveals a WDR62-CEP170-KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun. 10:2612. doi: 10.1038/s41467-019-10497-2
Zhao, W., Guo, S., Linli, Z., Yang, A. C., Lin, C.-P., and Tsai, S.-J. (2019). Functional, anatomical, and morphological networks highlight the role of basal ganglia–thalamus–cortex circuits in schizophrenia. Schizophr. Bull. sbz062. doi: 10.1093/schbul/sbz062
Keywords: human cortical development, MCD = malformation of cortical development, progenitors cells, neuronal migration, connectivity
Citation: Subramanian L, Calcagnotto ME and Paredes MF (2020) Cortical Malformations: Lessons in Human Brain Development. Front. Cell. Neurosci. 13:576. doi: 10.3389/fncel.2019.00576
Received: 10 August 2019; Accepted: 16 December 2019;
Published: 24 January 2020.
Edited by:
Laurent Nguyen, University of Liège, BelgiumReviewed by:
Veronica Martinez Cerdeño, University of California, Davis, United StatesEmma Louth, Aarhus University, Denmark
Copyright © 2020 Subramanian, Calcagnotto and Paredes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mercedes F. Paredes, bWVyY2VkZXMucGFyZWRlc0B1Y3NmLmVkdQ==