
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 17 December 2019
Sec. Cellular Neuropathology
Volume 13 - 2019 | https://doi.org/10.3389/fncel.2019.00550
This article is part of the Research Topic The Role of Cellular Senescence in Health and Disease View all 16 articles
 Hanqing Lin1,2
Hanqing Lin1,2 Hao Xiong1,2
Hao Xiong1,2 Zhongwu Su1,2Jiaqi Pang1,2Lan Lai1,2
Zhongwu Su1,2Jiaqi Pang1,2Lan Lai1,2 Huasong Zhang1,2Bingquan Jian1,2Weijian Zhang1,2
Huasong Zhang1,2Bingquan Jian1,2Weijian Zhang1,2 Yiqing Zheng1,2*
Yiqing Zheng1,2*Background: Mitochondrial dysfunction is considered to contribute to the development of age-related hearing loss (AHL). The regulation of mitochondrial function requires mitochondrial quality control, which includes mitophagy and dynamics. Dynamin-related Protein 1 (DRP-1) is believed to play a central role in this regulation. However, the underlying mechanism of DRP-1 in AHL remains unclear. Here, we examined whether the decline of DRP-1-dependent mitophagy contributes to the development of AHL.
Methods: We induced cellular and cochlear senescence using hydrogen peroxide (H2O2) and evaluated the level of senescence through senescence-associated β-galactosidase staining. We evaluated mitophagy levels via fluorescence imaging and Western Blotting of LC3II and P62. Mitochondrial function was assessed by ATP assay, mtDNA assay, and JC-1.
Results: We found that both the expression of DRP-1 and the mitophagy level decreased in senescent cells and aged mice. DRP-1 overexpression in HEI-OC1 cells initiated mitophagy and preserved mitochondrial function when exposed to H2O2, while cells with DRP-1 silencing displayed otherwise. Moreover, inhibition of DRP-1 by Mdivi-1 blocked mitophagy and exacerbated hearing loss in aged C57BL/6 mice.
Conclusion: These results indicated that DRP-1 initiated mitophagy, eliminated mitochondrial dysfunction, and may protect against oxidative stress-induced senescence. These results provide a potential therapeutic target for AHL.
Aging is a physiological phenomenon that occurs in all eukaryotes. Age-related hearing loss (AHL) is one of the most serious health-related social problems (Roth et al., 2011). It is characterized by an age-dependent decline of auditory function that is attributable to the loss and dysfunction of hair cells and spiral ganglion cells in the cochlea of the inner ear (Lin et al., 2013). There is agreement that cumulative mitochondrial dysfunction and Reactive Oxygen Species (ROS) exacerbate hearing loss. Still, the exact molecular mechanism of AHL remains unknown. A series of cellular activities are regulated in the cellular aging process. Among these, mitochondrial function is considered to be a central event in various diseases in all systems and at all ages. A recent study shows that mitochondrial dysfunction is one of the characteristics of the aging process (López-Otín et al., 2013), and it is thought to induce diseases such as Alzheimer’s (Lee et al., 2010) and AHL (Seo et al., 2017).
Cellular activities require mitochondria to provide energy, but meanwhile, adenosine triphosphate (ATP) synthesis oxidative stress is also produced, causing injury to cells and organelles. Excessive oxidative stress and damaged organelles can be eliminated and recycled by an essential process called autophagy (Mizushima et al., 2008). Specific mitochondrial autophagy is called mitophagy and allows dysfunctional mitochondria to go through a process of degradation. Mitophagy, along with mitochondrial biogenesis, maintains mitochondrial function.
Mitochondria constantly undergo fission and fusion to realize mitochondrial quality control. The activation of dynamin-related protein-1 (DRP-1) is required when mitochondria go through fission to create new mitochondria or to separate damaged mitochondria matrices so as to preserve normal mitochondrial function during high levels of cellular stress (Youle and van der Bliek, 2012). The main approach to eliminate injured mitochondria by autophagy is mitophagy. Disturbance of mitophagy may affect normal mitochondrial function and the physiological or pathological cellular condition. DRP-1 is a highly conserved GTPase that plays the core role in mitophagy by regulating the separation of mitochondria (Yoneda et al., 1994). When mitochondrial fission occurs, DRP-1 is recruited from the cytosol to the outer membrane, where it forms a spiral complex around the mitochondria (Ingerman et al., 2005). One situation is symmetrical replicative fission. This is where a healthy mitochondrion divides equally into two daughter mitochondria, which generates new mitochondria by mitochondrial biogenesis or by fusion (Gray et al., 1999). The other situation is where asymmetrical fission is initiated so that damaged mitochondria can be removed through mitophagy (Yang and Yang, 2013). Dysfunctional mitochondria display relatively depolarized membrane potential, which can be targeted for selective autophagy for further elimination.
In our study, we hypothesized that the expression and normal functions of DRP-1 and DRP-1-dependent mitophagy are disrupted in the aged cochlea with AHL. This disruption is considered harmful to the survival and function of cochlear hair cells. To determine whether DRP-1 and mitophagy play a role in aged cochlea with AHL, we examined DRP-1 and mitophagy and their roles in cellular senescence and AHL.
C57BL/6 mice were purchased from the Laboratory Animal Center, Sun Yat-sen University (Guangzhou, China). C57BL/6 mice were randomly divided into three groups: one “young control” group (1 month old), one “old control” group (12 months old), one “old Mdivi-1 IP” group (12 months old). Mdivi-1 was administrated intraperitoneally in the amount of 12 mg/kg every 3 days. All experiments were performed according to protocols approved by the Animal Research Center, Sun Yat-sen University.
Auditory brainstem response (ABR) tests were conducted using the Tucker-Davis Technology (TDT) System III (Alachua, FL, USA) as previously described (Pang et al., 2017). In brief, mice were anesthetized, and subdermal needle electrodes were inserted at the vertex (active) and below the left ear (reference) and the right ear (ground). ABR tests were measured at 8, 16, and 32 kHz. The average response to 1,024 stimuli was obtained through reduction of the sound intensity in 5 dB intervals near the threshold, which was defined as the lowest stimulus level where a positive wave was evident.
C57BL/6 mice were decapitated after the ABR tests. The temporal bones were dissected, and the cochleae were obtained and fixed with 4% paraformaldehyde at 4°C overnight and decalcified in 4% sodium ethylenediaminetetraacetic acid for 2 days. For protein preparation, cochlear basilar membranes were dissected from cochlea, snap-frozen in liquid nitrogen, and stored at −80°C.
Cochlear HEI-OC1 cells (kindly provided by F. Kalinec from the House Ear Institute, Los Angeles, CA, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, USA) and supplemented with 10% fetal bovine serum (Gibco) at 33°C under a 10% CO2 condition.
P2–3 C57BL/6 mice were purchased from Guangdong Medical Laboratory Animal Center (GDMLAC). The temporal bones were dissected, and the cochleae were obtained and then supplemented with 10% fetal bovine serum (Gibco) at 37°C under a 5% CO2 condition.
DRP-1 Silencer (Ruibo, Guangzhou, China), a commercial mixture of siRNA and ASO (Antisense oligonucleotides), was used to knock down the expression of DRP-1 in HEI-OC1 cells. Cell transfections were performed using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies, USA) following the manufacturer’s instructions. The HEI-OC1 cells were transfected with lentivirus-mediated green fluorescent protein (GFP)-LC3 to generate GFP-LC3-expressing cells. The lentivirus containing the GFP-LC3 fusion gene was purchased from Hanbio (Shanghai, China). HEI-OC1 cells were seeded into six-well dishes (1 × 105 per well) and infected with the recombinant lentivirus. After culturing in the presence of puromycin for 2 weeks, HEI-OC1 cells with GFP-LC3 were selected.
Western Blotting was conducted as previously described (Pang et al., 2017). Briefly, proteins were extracted from cells and cochlea tissues using radioimmunoprecipitation assay lysis buffer (Thermo plus, USA). Protein samples (20 μg) were resolved on a 10% SDS-polyacrylamide gel and transferred onto polyvinylidene fluoride membranes (Millipore, Burlington, MA, USA). After being blocked with 5% nonfat milk, the membranes were incubated with anti-DRP1 (1:1,000, Proteintech, Rosemont, IL, USA), anti-p-DRP1 (1:1,000, Abclonal, Woburn, MA, USA), anti-LC3I/II (1:1,000, CST, USA), anti-P62 (1:1,000, CST, USA), anti-P53 (1:1,000, Abclonal, Woburn, MA, USA), and anti-P21 (1:1,000, Abclonal, Woburn, MA, USA) at 4°C overnight, followed by incubation with secondary antibodies (1:3,000) at room temperature for 1 h. The immunoreactive bands were then detected by enhanced chemiluminescence (Millipore, Burlington, MA, USA). Band intensities were analyzed using ImageJ (NIH, USA). β-actin was applied as loading and internal control.
Cell viability was detected using CCK-8 kits (DOJINDO, Japan) according to the manufacturer’s protocols. Briefly, cells were plated into 96-well plate. At the indicated time after treatments, 10 μl of CCK-8 solution was added to the wells. Cells were then incubated at 37°C for 1 h. Finally, a microplate reader (Labsystems Dragon, Finland) was used to measure the absorbance at 450 nm.
For the population doubling rate assay, cells were counted, and 1 × 105 cells were plated in every 35 mm dish, pre-treated, replaced with a normal culture medium, and then incubated under permissive conditions. Cells were harvested and counted and re-planted every 72 h. The formula below was used to evaluate the population doubling rate.
(N1 = Number of cells counted, N2 = Number of cells plated)
Cellular senescence-associated β-galactosidase staining was conducted using a senescence β-galactosidase staining kit (Beyotime, Shanghai, China) following the manufacturer’s instruction. Briefly, cells were plated in 6-well plate and pre-treated. Cells were gently washed with PBS and fixed with fixing solution. After being washed with PBS three times, 1 ml of staining solution was added to each well, which was then sealed with parafilm and incubated at 37°C without CO2 overnight.
ATP assay was performed using an ATP Assay Kit (Beyotime, Shanghai, China) following the manufacturer’s instructions. Briefly, cells were homogenized with lysis buffer and centrifuged at 12,000 g for 5 min at 4°C. An ATP detection reagent was diluted with dilution buffer and added to 96-wells. Then, the samples were added into the wells and mixed with the detection solution. The chemiluminescence intensities of samples and standards were measured with a SpectraMax M5 microplate reader (Molecular Devices, San Jose, CA, USA). The levels of ATP were calculated based on the standard curve and normalized to the protein content.
Mitochondrial staining was conducted with the mitochondrial probe MitoTracker Red CMXRos (Yeasen, Shanghai, China) according to the manufacturer’s protocols. After being washed with PBS, the cells were counterstained with DAPI for 10 min and imaged with an Olympus BX63 microscope (Olympus, Japan).
Total genomic DNA was extracted from cells using a Universal Genomic DNA Extraction Kit (Takara) according to the manufacturer’s protocols. The mtDNA levels were quantified by qPCR on a Roche LightCycler 96 (Roche) using D-loop primers (forward: 5′-GGTTCTTACTTCAGGGCCATCA-3′, reverse: 5′-GATTAGACCCGTTACCATCGAGAT-3′). Nuclear gene beta2-microglobulin (B2M) primers (forward: 5′-ATGGGAAGCCGAACATACTG-3′, reverse: 5′-CAGTCTCAGTGGGGGTGAAT-3′) were used as a nuclear control.
All experiments were independently repeated at least three times. Data were presented as mean ± SD and were analyzed with SPSS and Graphpad Prism 5 software. Student’s t-test and one-way ANOVA were used for statistical analysis. Values with p < 0.05 were considered significant.
We first established cellular senescence by inducing oxidative stress. HEI-OC1 cells were briefly exposed to H2O2 (1 mM for 1 h), and we then further investigated the cellular molecular change between mitophagy and senescence. Our results revealed that cellular senescence was induced 24 h after H2O2 treatment at a rate of 54.4 ± 9.94% HEI-OC1 cells stained with β-gal staining (Figure 1A). In the meantime, there was 13.4 ± 2.25% of senescent β-gal-stained cells in the normal control HEI-OC1 cells (p < 0.0001, Figure 1B). We further assessed cellular senescence with cell viability, population doubling rate, and senescence-associated P53 and P21. Lower cell viability was detected in cells treated with H2O2, being 0.63 ± 0.03-fold lower than the control cells (p = 0.0006, Figure 1C). The population doubling rate was calculated to evaluate the aging pattern. Higher rates indicate a higher speed of cell growth. The population doubling rate dropped to 1.73 ± 0.27 compared to normal cells at 4.21 ± 0.08 (p = 0.0001, Figure 1D). Cellular senescence-associated P53 and P21 were further assessed by Western Blotting. H2O2 treatment of HEI-OC1 cells significantly elevated the expression of P53 and P21 (Figures 1E–G). These data demonstrated that H2O2 induced cellular senescence in HEI-OC1 cochlear cells.
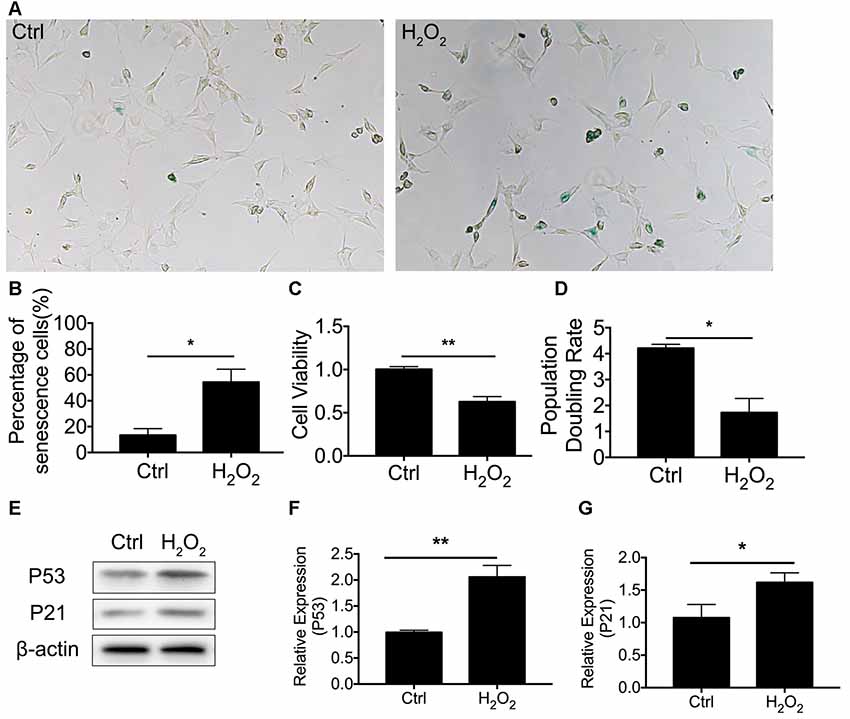
Figure 1. H2O2-induced cellular senescence in HEI-OC1 cells. (A) β-gal staining of senescent HEI-OC1 cells treated with H2O2. (B) Percentage of β-gal stained cells. (C) Cell viability of 1 mM H2O2 treated cells compared with control cells. (D) Population doubling rate in HEI-OC1 cells. (E–G) Representative Western Blot analysis using antibodies against P53 and P21 to assess cellular senescence. *p < 0.05, **p < 0.01.
To assess whether there was a molecular change between mitophagy and senescence in HEI-OC1 cells, we further examined blockage of the autophagy flux (Figure 2A). Western Blotting revealed that 1 mM of H2O2 treatment resulted in a decrease of LC3 II of 0.62 ± 0.08-fold relative to control and a 1.77 ± 0.18-fold relative increase of P62 (p < 0.05, Figures 2B,C). The Western Blotting results revealed the suppression of the autophagy function. To further determine mitophagy, we used transfected HEI-OC1 cells expressing GFP-LC3 and staining with the MitoTracker Red fluorescence probe. The yellow puncta displayed were considered as the merging of mitochondria and autophagosomes (Figure 2D). From each group, 20 random cells were counted. The percentage of mitophagosome with H2O2 treatment was 10 ± 1.16% compared to the control group, with 16.67 ± 0.88% (p = 0.0101, Figure 2E). The data indicated that mitophagy was suppressed in H2O2-induced senescent HEI-OC1 cells.
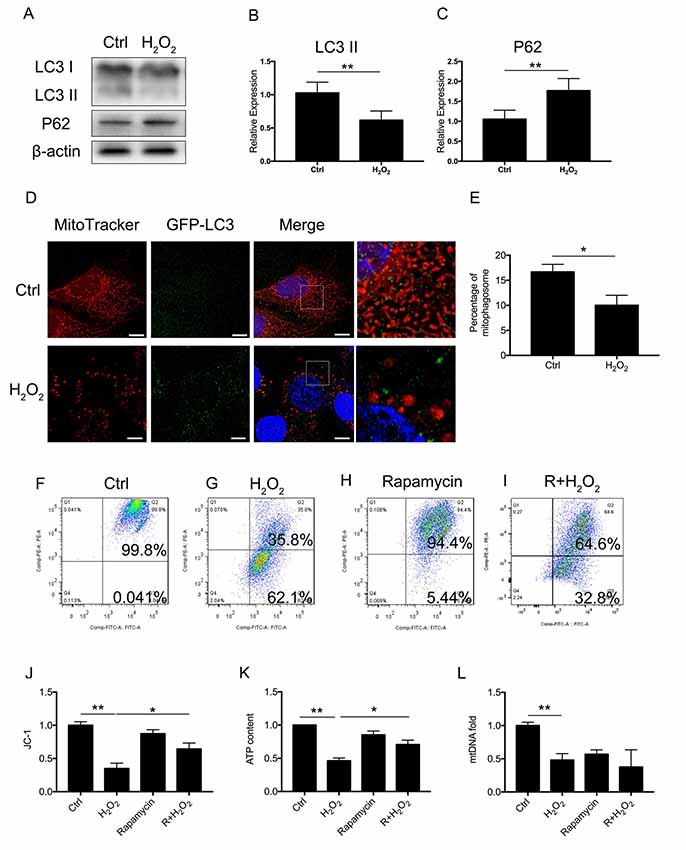
Figure 2. Mitophagy and mitochondrial function of HEI-OC1 cells. (A) Representative Western Blot analysis using antibodies against LC3 I/II and P62 to assess autophagy flux. (B,C) Relative expression of mitophagy proteins LC3 II and P62 between control cells and H2O2-treated cells. (D) Immunofluorescence image showing mitophagy of mitochondria and autophagosome in green fluorescent protein (GFP)-LC3-expressing HEI-OC1 cells. (E) Percentage of mitophagosome in GFP-LC3-expressing HEI-OC1 cells between control and H2O2 treatment. (F–I) Cellular mitochondrial membrane potential between control and H2O2 treatment according to JC-1 staining and measured by flow cytometry. (J) Mitochondrial membrane potential analysis. Rapamycin-initiated autophagy improved mitochondrial membrane potential under H2O2 treatment. (K) ATP content in cells treated with H2O2 or rapamycin. ATP content was largely preserved with rapamycin under H2O2 treatment. (L) Relative mtDNA fold change, representing relative mitochondria amount. No significant difference between H2O2 treatment and rapamycin treatment. *p < 0.05, **p < 0.01. Scale bar = 10 μm.
Next, we examined whether mitophagy is beneficial to mitochondrial function. We treated the cells with 0.5 μM rapamycin for 8 h to initiate autophagy and mitophagy. To evaluate mitochondrial dysfunction induced by oxidative stress and to determine if mitophagy would rescue mitochondrial function, we evaluated mitochondrial membrane potential by JC-1, ATP content, and mtDNA in HEI-OC1 cells treated with H2O2. The mitochondrial membrane potential of each group was evaluated with JC-1 stain by flow cytometry. Cells in the H2O2 treatment group displayed a lower percentage of mitochondria with normal potential at a rate of 39.91 ± 0.04% (p < 0.0001, Figures 2F,G,J). We found that treatment with rapamycin under oxidative stress increased the percentage of mitochondria with normal potential compared to H2O2 treatment alone (p = 0.0152, Figures 2H,I). Also, we examined the ATP content under H2O2 treatment and also with rapamycin. H2O2 decreased the ATP content of HEI-OC1 cells to ~0.46 ± 0.02-fold that of the control group, while, with pre-treatment with rapamycin, H2O2 only resulted in a decline of ATP content to ~0.71 ± 0.04 fold (p = 0.0058, Figure 2K). However, there is no significant difference between H2O2 and rapamycin treatment (p(H2O2, rapamycin) = 0.74, p(H2O2, R + H2O2) = 0.28, Figure 2L) in the amount of mtDNA. These results suggested that H2O2 suppressed autophagy and mitophagy and resulted in mitochondrial dysfunction.
DRP-1 plays the core role in mitophagy through regulation of mitochondrial fission. Mitochondrial fission separates injured mitochondria, and this is followed by mitophagy. With a link between declining levels of mitophagy and aging, we hypothesized that the accumulation of damaged mitochondria in senescent cells was due to suppressed mitochondrial fission and subsequent mitophagy. To investigate whether DRP-1 regulates mitophagy, we assessed mitophagy in DRP-1 overexpressing and silencing cells (Figures 3A,B). Autophagy flux and cellular senescence were assessed by Western Blotting of LC3II, P62, P53, and P21 (Figure 3C). Assessment via LC3 II and P62 revealed that autophagy flux was blocked in DRP-1 silencing cells but not in DRP-1 overexpressing cells (p < 0.01, Figures 3D,E). We further assessed cellular senescence in DRP-1 overexpressing and silencing cells under H2O2 treatment. We evaluated the expression of senescence-associated P53, P21 (Figures 3F,G), and β-Gal stain (Figure 3H). The elevation of P53 and P21 expression and an increased portion of β-Gal stained cells revealed that silencing DRP-1 suppressed resistance to H2O2-induced cellular senescence. We then evaluated mitochondrial function in DRP-1 overexpressing and silencing cells to further understand mitophagy levels. DRP-1 overexpressing or silencing in HEI-OC1 cells resulted in no difference in the percentage of mitochondrial membrane potential compared with control cells. However, when cells faced oxidative stress, DRP-1 overexpression rescued mitochondrial membrane potential, displaying 53.0 ± 0.05% of healthy mitochondria and DRP-1 silencing of 18.8 ± 0.05% (p < 0.05, Figures 3I,J). The ATP content in DRP-1 overexpressing cells decreased but remained at 0.71 ± 0.04-fold that of the control group. The mtDNA remained at 0.62 ± 0.04-fold that of the control in DRP-1 overexpressing cells treated with H2O2 (p < 0.05, Figures 3K,L). In contrast, cells silencing DRP-1 displayed a significant decrease in ATP content to 0.32 ± 0.07-fold that of the control but with mtDNA at 0.90 ± 0.03 that of the control (p < 0.05, Figures 3H,I). Together with the blockage of autophagy flux, we may assume that mitophagy in HEI-OC1 cells was also blocked. The data revealed that DRP-1 overexpression initiated mitophagy and that such cells retained most of the mitochondrial function under oxidative treatment, while silencing DRP-1 inhibited mitophagy. Cells retained most of the mitochondria but less ATP content, indicating that mitochondrial function was disturbed in DRP-1 silencing cells under oxidative stress.
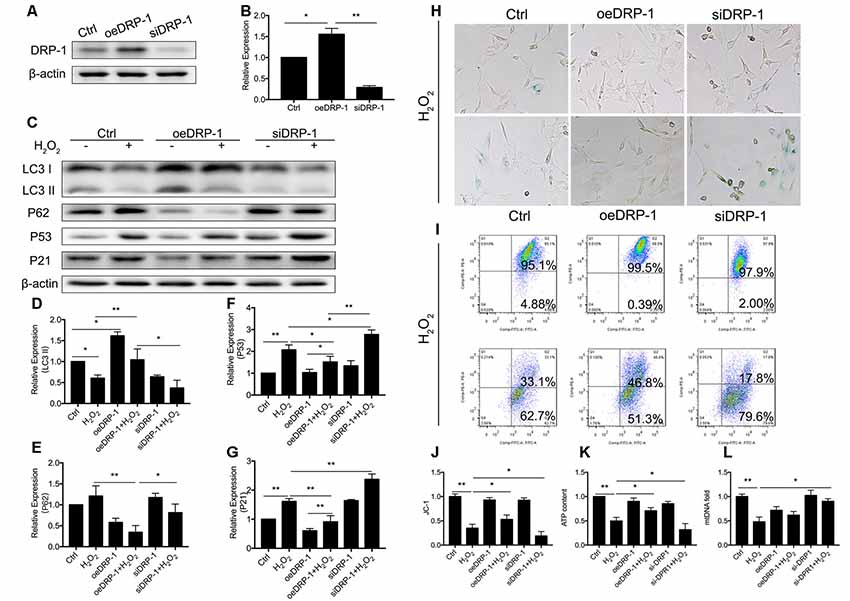
Figure 3. Regulation of Dynamin-related Protein 1 (DRP-1) in cellular senescence and mitochondrial function. (A) Representative Western Blot analysis using antibodies against DRP-1 to assess DRP-1 expression. (B) DRP-1 expression in overexpressing and silencing HEI-OC1 cells. Relative expression of DRP-1 in overexpressing and silencing HEI-OC1 cells compared to control. (C) Representative Western Blot analysis using antibodies against LC3 I/II, P62, P53, and P21 to assess the mitophagy level and cellular senescence in HEI-OC1 cells. (D,E) Relative expression of LC3 II and P62. H2O2 down-regulated mitophagy in all groups. The relative mitophagy was higher in DRP-1 overexpressing cells compared to the control. The mitophagy level was down-regulated in DRP-1 silencing cells. (F,G) Relative expression of P53 and P21. H2O2 induced cellular senescence in HEI-OC1 cells, which elevated the expression of P53 and P21. P53 and P21 expression were relatively higher in DRP-1 silencing cells than in DRP-1 overexpressing cells. (H) Senescent HEI-OC1 cells with DRP-1 overexpression and silencing, assessed by β-Gal stain. (I,J) Mitochondrial membrane potential, evaluated by FCM with JC-1 staining. (K) ATP content in cells overexpressing and silencing DRP-1 under H2O2 treatment. (L) The amount of mitochondria were evaluated by mtDNA measurement. *p < 0.05, **p < 0.01.
We observed the regulation of DRP-1 in mitochondrial functions and further investigated DRP-1 expression patterns in cochlear explants. Senescence was induced by 0.1 mM H2O2 treatment in the cochlear explants. We found a significant decrease of DRP-1 (0.66 ± 0.04-fold of control, p = 0.0017) and s616 phosphorylated DRP-1 (p-DRP1, 0.41 ± 0.08-fold of control, p = 0.0017, Figures 4A–E) in the cochlear explants. The mitophagy level of the cochlear explants was determined by the level of autophagy flux and mitochondrial function. Western Blotting showed the LC3 II and P62 protein levels to decrease (pLC3 II = 0.0164, pP62 = 0.0062), suggesting that autophagy flux was also blocked in H2O2-treated cochlear explants (p = 0.0188, Figures 4F,G). H2O2 also resulted in mitochondrial dysfunction in the cochlear explants by down-regulating the ATP content to 0.62 ± 0.08-fold (p = 0.0095) and mtDNA to 0.53 ± 0.09-fold (p = 0.0071) that of control (Figures 4H,I). These results indicated that H2O2 treatment induced DRP-1 down-regulation and the injury of mitophagy in cochlear explants.
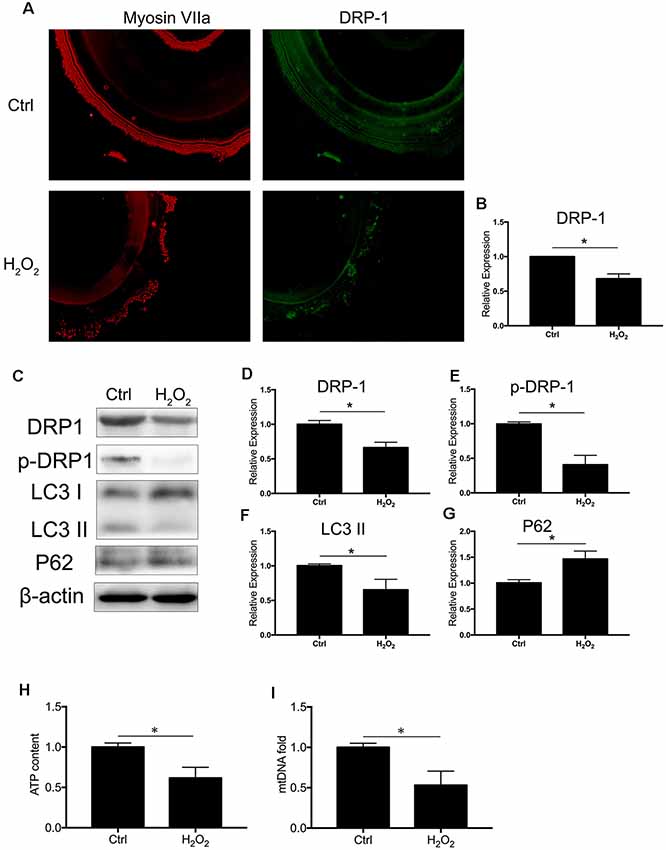
Figure 4. H2O2 inhibited DRP-1 and mitophagy in cochlea. (A) Representative image of immunofluorescence staining of DRP-1 in cochlear explant. (B) Down-regulated expression of DRP-1 in cochlea treated with H2O2. (C) Representative Western Blot analysis using antibodies against DRP-1, p-DRP-1, LC3 I/II, and P62 to assess the mitophagy level in cochlear explant. (D,E) Relative expressions of DRP-1 and p-DRP-1 in H2O2-treated cochlea. (F,G) Relative expression of LC3 II and P62 in H2O2-treated cochlea. (H,I) Mitochondrial function evaluated by ATP content and mtDNA. H2O2-treated cochlea revealed a low ATP content and mitochondrial amount. *p < 0.05. Scale bar = 300 μm.
Mitochondrial-division inhibitor Mdivi-1 inhibits mitochondrial fission by deactivation of DRP-1. To determine whether DRP-1 has an effect on cochlear senescence, we assessed the mitophagy function and senescent phenotype in the cochlear explants. We evaluated mitophagy in cochlear explants with pre-treatment with 50 μM of Mdivi-1 for 4 h before H2O2 treatment. Mdivi-1 pre-treatment de-activated DRP-1 and significantly down-regulated the mitophagy level under oxidative stress (Figures 5B,C). The LC3 II and P62 levels were evaluated to assess autophagy flux and mitophagy (Figures 5D,E). Western Blotting of protein extracts of H2O2-exposed cochlea pre-treated with Mdivi-1 revealed damaged mitophagy by down-regulation of LC3 II (p = 0.0243) and upregulation of P62 (p = 0.0022, Figures 5A–E). H2O2-induced cellular senescence was exacerbated by DRP-1 silencing. To further investigate the role of DRP-1 in aged cochlea, we assessed senescence-associated P53, P21 (Figures 5F,G), and β-gal stain in the cochlear explants. Mdivi-1 pre-treatment did not solely induce senescence in cochlea but exacerbated the senescent damage induced by H2O2 (Figure 5H). These results indicated that H2O2 inhibited mitophagy and could further induce senescence in cochlea.
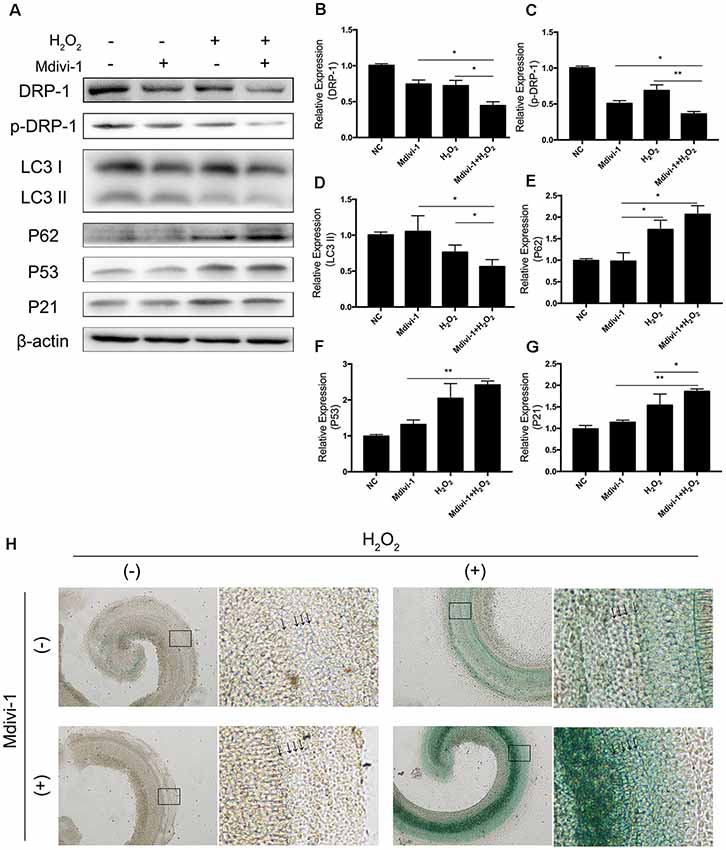
Figure 5. Mdivi-1 exacerbated senescence in H2O2-treated cochlea by inhibition of mitophagy. (A) Representative Western Blot analysis using antibodies against DRP-1, p-DRP-1, LC3 I/II, P62, P53, and P21 to assess mitophagy level and senescence in cochlear explants treated with H2O2 and DRP-1 inhibitor Mdivi-1. (B,C) Relative expression of DRP-1 and p-DPR-1. Mdivi-1 inhibited DRP-1 expression and phosphorylation. (D,E) Relative expression of LC3 II and P62. Mdivi-1 suppressed the mitophagy level in H2O2-treated cochlea. (F,G) Relative expression of P53 and P21. Mdivi-1 aggravated H2O2-induced senescence in cochlear explants. (H) Representative scanned images of the cochlear surface with β-gal staining of cochlea. With Mdivi-1, cochlea had a darker greenish color under H2O2 treatment. Black arrows indicate OHC and IHC. *p < 0.05, **p < 0.01.
C57BL/6 mice are known for their early-onset hearing loss and are often used as AHL models. To examine the hearing function of C57BL/6 mice, we performed ABR tests on aged and young C57BL/6 mice. We used 1-month-old mice as normal control, where hearing function had fully developed but no hearing loss had occurred. We evaluated the hearing threshold of the normal control group and a Mdivi-1 treatment group of 12 months old, by which time the C57BL/6 mice had developed hearing loss. The Mdivi-1 group was given Mdivi-1 via intraperitoneal administration beginning at the 8th month, before the C57BL/6 mice had developed hearing loss. The Mdivi-1 group developed greater threshold shifts at 8, 16, and 32 kHz than control mice and also exhibited greater hair cell loss (Figures 6A,B). We evaluated mitophagy using Western Blotting analysis, and the results revealed differences between aged mice and Mdivi-1-treated mice. There was a decrease of LC3 II (p = 0.013) and an increase of P62 (p = 0.0266, Figures 6C–E). Also, we found a decline of expression of DRP-1 and p-DRP-1 in aged cochlea (Figures 6F–H). However, there was no significant difference between aged mice and Mdivi-1-treated mice. The results indicated that mitophagy might be an effective way of hair cells survival under age stress and that inhibition of DRP-1 may block mitophagy and induce AHL.
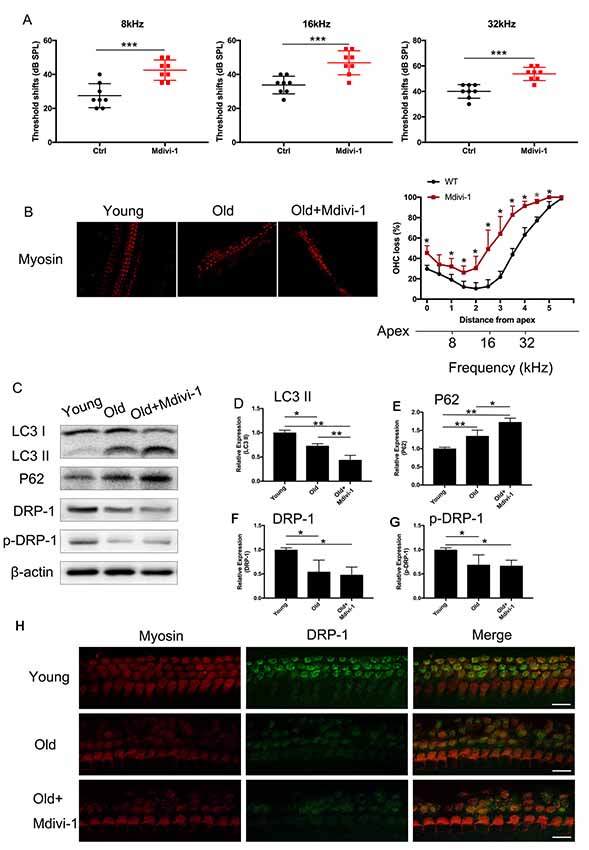
Figure 6. Administration of Mdivi-1 in C57BL/6 mice resulted in greater hearing loss and hair cell loss. (A) Auditory brainstem response (ABR) threshold shifts in C57BL/6 mice. One-month-old mice were considered as young control. There was a significant hearing threshold shift in 12-month-old Mdivi-1 IP mice compared to the control mice. Mdivi-1 IP administration resulted in greater shifts compared to in aged mice. (B) Hair cell count in the basilar membrane of young, old control, and Mdivi-1 IP mice. (C–H) Mitophagy level and DRP-1 expression assessment by LC3 II, P62, DRP-1, and p-DRP-1, comparing the cochlea of young, old control, and Mdivi-1 IP mice. (H) Representative image of immunofluorescence staining of DRP-1 in the cochlea of young, old, and old with Mdivi-1 administration mice. Significant down expression of DRP-1 in old-mouse cochlea compared to the young control mice. *p < 0.05, **p < 0.01, ***p < 0.001, scale bar = 100 μm.
Mitochondria form the cellular energy factory that synthesizes and provides ATP. However, dysfunctional mitochondria produce less ATP with more ROS and are more likely to induce apoptosis, necrosis, and senescence. Thus, there is a complicated and delicate mechanism of mitochondrial quality control to maintain mitochondrial function; this consists of mitochondrial fission and fusion, mitochondrial biogenesis, and mitophagy. Mitophagy is a process of selective cellular defense. Through this process, damaged mitochondrial matrices can be separated and eliminated to maintain mitochondrial function and homeostasis and alleviate cellular injury. The inhibition of mitophagy results in mitochondrial dysfunction and cellular senescence (García-Prat et al., 2016). On the other hand, an increase in mitophagy repairs cellular injury and delays the process of senescence (Dalle Pezze et al., 2014; Manzella et al., 2018).
Mitochondrial fission and fusion depend on a series of GTPases (van der Bliek et al., 2013). DRP-1 is a dynamic-related GTP-binding protein. It is key in controlling mitochondrial fission and is reported to initiate mitophagy (Lemasters, 2005). DRP-1 induces mitophagy to attenuate oxidative damage in other organs like the spinal cord and neurons. Oxidative stress damage also occurs and results in AHL. Here, we investigate whether DRP-1-induced mitophagy plays a role in the process of AHL.
We first established a cellular senescence model of HEI-OC1 cells and studied the expression of DRP-1 and mitophagy levels. We found a decline of DRP-1 expression and mitophagy level along with mitochondrial dysfunction. These results indicated that there was a correlation between DRP-1 and mitochondrial dysfunction in senescent HEI-OC1 cells. We hypothesized that mitochondrial dysfunction was induced by a disturbance in mitophagy. Our previous study revealed that rapamycin can preserve mitochondrial function by initiating autophagy (Pang et al., 2017). We found that in cellular senescence, the disturbed autophagy flux assessed by the LC3 II and P62 proteins confirmed our hypothesis. Cells treated with rapamycin displayed a normal level of ATP but a decreased level of mtDNA, indicating that rapamycin induced autophagy and preserved mitochondrial function. However, with oxidative stress, low ATP content and a lower autophagy level were found in senescent cells. The above results suggested that, in HEI-OC1 cells, oxidative stress suppressed autophagy, which might also suppress mitophagy and induce mitochondrial dysfunction. Other studies have demonstrated that DRP-1-induced mitophagy rescued mitochondrial function. These studies were related to oxidative stress in cardiac aging (Fernández et al., 2018), PD (Martinez et al., 2018), and early-stage AD (Wang et al., 2019). Our research confirmed that the initiation of mitophagy depends on DRP-1 and that DRP-1-induced mitophagy is key in the regulation of oxidative damage in hair cells.
We further investigated DRP-1 and mitochondrial function. We evaluated mitochondrial membrane potential, ATP content, and mtDNA in DRP-1 overexpressing and silencing HEI-OC1 cells. Mitochondrial membrane potential displayed no differences between DRP-1 overexpressing and silencing cells. However, in cells in which senescence had been induced by oxidative stress, we found more injured mitochondria with low membrane potential in DRP-1 silencing cells but relatively less in DRP-1 overexpressing cells. The tendency was the same in ATP content. We defined mitochondrial dysfunction as poor membrane potential and low ATP content. mtDNA, on the other hand, would represent the amount of mitochondria. A low amount of mitochondria, assessed by mtDNA, was found in H2O2 treatment and rapamycin treatment. In DRP-1 overexpressing cells, autophagy flux was initiated, while ATP content and membrane potential remained normal. However, mtDNA was mildly decreased in DRP-1 overexpressing cells, suggesting that fewer mitochondria would produce more ATP. In DRP-1 silencing cells treated with H2O2, the autophagy flux was disturbed, and mitochondrial dysfunction was displayed. We suggested that failure to eliminate dysfunctional mitochondria was the reason why mtDNA was at a normal level. Also, there were fewer merging fluorescent puncta of mitochondria (red) and lysosome (green). The expression of the autophagy-related proteins LC3 II and P62 decreased. The results may suggest that DRP-1 is the key to regulating mitophagy and mitochondrial function. As cellular senescence developed, the decline of DRP-1 expression resulted in decreased mitophagy levels and an accumulation of dysfunctional mitochondria. We further investigated DRP-1 expression and senescence in cochlear explants. In senescent cochlear explants, DRP-1 expression and mitophagy levels decreased along with impaired mitochondrial function. These results are similar to those from the cellular experiment. Therefore, we hypothesized that age-related decline in DRP-1 repressed mitophagy and there was an accumulation of impaired mitochondria, which caused cochlear senescence.
In aged C57BL/6 mice as AHL models, we identified the downregulation of DRP-1 expression and mitophagy by evaluating the protein expressions of DRP-1, LC3 II, and P62. DRP-1 and mitophagy were downregulated in aged mice and senescent cochlea. Impaired mitochondria accumulated during aging due to a gradual decline in mitophagy and due to mitochondrial dysfunction. Our previous study revealed that initiating autophagy with rapamycin could notably improve hearing conditions in aged mice compared to AHL mice. In our current study, we applied Mdivi-1, an DRP-1 inhibitor, to C57BL/6 mice. These mice developed even more severe hearing loss than those who went without Mdivi-1. All of the above implied that the inhibition of DRP-1 expression and function resulted in decreased levels of mitophagy and further AHL development. It is possible that there are still unknown mechanisms that alter mitochondrial function (resulting in senescence) that need further study. We propose that DRP-1 could be the center of mitochondrial quality control and could be a key factor in AHL.
In this research, we discovered the relationship between DRP-1 and hair cell senescence (Figure 7). We revealed the role of DRP-1 in AHL and examined the effects of inhibition of DRP-1 in hearing. However, there are still questions that remain to be answered in our study. One question regards mitochondrial morphology and mitochondrial function. According to some research, a giant mitochondrial network created through mitochondrial fusion can preserve damaged mitochondria to maintain their function. The dynamic morphology of mitochondria mismatches with mitochondrial function, making this unconvincing. Second, the exact mechanism that controls how oxidative stress suppresses DRP-1 expression and phosphorylation remains unknown. Research has implied that the phosphorylation of DRP-1 may affect the ROS-AMPK pathway (Li et al., 2017), which is one of the critical pathways in oxidative regulation and may be an essential target in the study of senescence and AHL. Third, our study would be more convincing if it were to show whether DRP-1 delays the development of AHL in transgenic mice. Lastly, we revealed that DRP-1 participated in the development of AHL and considered it as a critical part of AHL. We hope to uncover the mechanisms of senescence and AHL by studying DRP-1 and related to DRP-1 that could prevent, delay, or even cure AHL in the future.
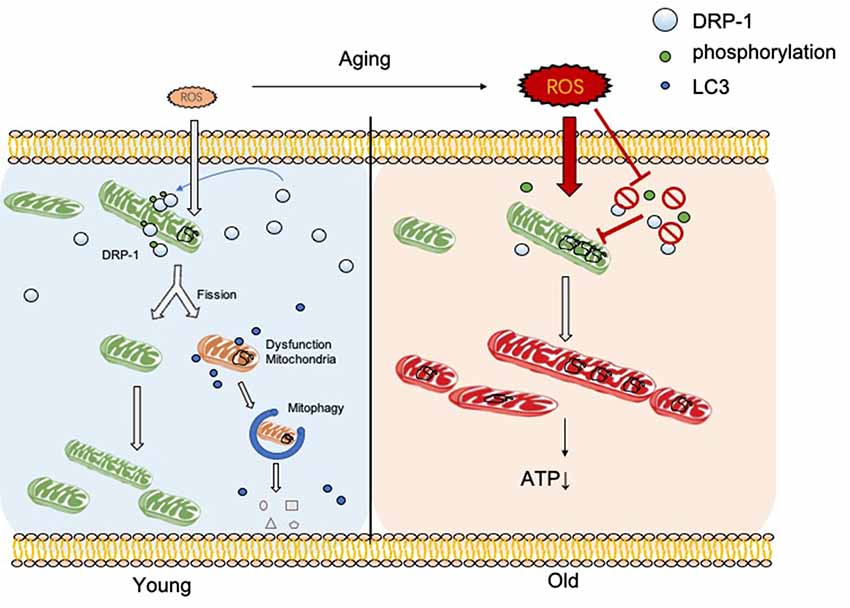
Figure 7. DRP-1 induces separation of damaged mitochondrial matrix before initiation of mitophagy in young cells. With aging, reactive oxygen species (ROS) and oxidative stress increase and inhibit DRP-1 function. Oxidatively damaged mitochondria accumulate and result in a dysfunctional ATP metabolism and thus cellular senescence.
All datasets generated for this study are included in the article.
The animal study was reviewed and approved by Sun Yat-sen University.
HL and HX: contributed equally to this work. HL: conception and design, designed and performed the experiments, data analysis and interpretation, and manuscript writing. YZ and HX: conception and design, manuscript writing, final approval of manuscript. ZS, JP, LL, HZ, BJ, and WZ: assembly of data and data analysis.
This work was supported by the National Natural Science Foundation of China (81570916, 81771018, 81873699 and 81900945) and the Yat-Sen Scholarship for Young Scientists.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Dalle Pezze, P., Nelson, G., Otten, E. G., Korolchuk, V. I., Kirkwood, T. B., von Zglinicki, T., et al. (2014). Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLoS Comput. Biol. 10:e1003728. doi: 10.1371/journal.pcbi.1003728
Fernández, Á. F., Sebti, S., Wei, Y., Zou, Z., Shi, M., McMillan, K. L., et al. (2018). Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140. doi: 10.1038/s41586-018-0162-7
García-Prat, L., Martínez-Vicente, M., Perdiguero, E., Ortet, L., Rodríguez-Ubreva, J., Rebollo, E., et al. (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. doi: 10.1038/nature16187
Gray, M. W., Burger, G., and Lang, B. F. (1999). Mitochondrial evolution. Science 283, 1476–1481. doi: 10.1126/science.283.5407.1476
Ingerman, E., Perkins, E. M., Marino, M., Mears, J. A., McCaffery, J. M., Hinshaw, J. E., et al. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027. doi: 10.1083/jcb.200506078
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Li, S., Sun, X., Xu, L., Sun, R., Ma, Z., Deng, X., et al. (2017). Baicalin attenuates in vivo and in vitro hyperglycemia-exacerbated ischemia/reperfusion injury by regulating mitochondrial function in a manner dependent on AMPK. Eur. J. Pharmacol. 815, 118–126. doi: 10.1016/j.ejphar.2017.07.041
Lin, F. R., Yaffe, K., Xia, J., Xue, Q. L., Harris, T. B., Purchase-Helzner, E., et al. (2013). Hearing loss and cognitive decline in older adults. JAMA Intern. Med. 173, 293–299. doi: 10.1001/jamainternmed.2013.1868
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Manzella, N., Santin, Y., Maggiorani, D., Martini, H., Douin-Echinard, V., Passos, J. F., et al. (2018). Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 17:e12811. doi: 10.1111/acel.12811
Martinez, J. H., Alaimo, A., Gorojod, R. M., Porte Alcon, S., Fuentes, F., Coluccio Leskow, F., et al. (2018). Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing α-synuclein. Mol. Cell. Neurosci. 88, 107–117. doi: 10.1016/j.mcn.2018.01.004
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Pang, J., Xiong, H., Lin, P., Lai, L., Yang, H., Liu, Y., et al. (2017). Activation of miR-34a impairs autophagic flux and promotes cochlear cell death via repressing ATG9A: implications for age-related hearing loss. Cell Death Dis. 8:e3079. doi: 10.1038/cddis.2017.462
Roth, T. N., Hanebuth, D., and Probst, R. (2011). Prevalence of age-related hearing loss in Europe: a review. Eur. Arch. Otorhinolaryngol. 268, 1101–1107. doi: 10.1007/s00405-011-1597-8
Seo, Y. J., Ju, H. M., Lee, S. H., Kwak, S. H., Kang, M. J., Yoon, J. H., et al. (2017). Damage of inner ear sensory hair cells via mitochondrial loss in a murine model of sleep apnea with chronic intermittent hypoxia. Sleep 40:zsx106. doi: 10.1093/sleep/zsx106
van der Bliek, A. M., Shen, Q., and Kawajiri, S. (2013). Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 5:a011072. doi: 10.1101/cshperspect.a011072
Wang, Z. T., Lu, M. H., Zhang, Y., Ji, W. L., Lei, L., Wang, W., et al. (2019). Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell 18:e12860. doi: 10.1111/acel.12860
Yang, J. Y., and Yang, W. Y. (2013). Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 4:2428. doi: 10.1038/ncomms3428
Yoneda, M., Miyatake, T., and Attardi, G. (1994). Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol. Cell. Biol. 14, 2699–2712. doi: 10.1128/mcb.14.4.2699
Keywords: presbycusis, oxidative stress, DRP-1, mitophagy, cochlea
Citation: Lin H, Xiong H, Su Z, Pang J, Lai L, Zhang H, Jian B, Zhang W and Zheng Y (2019) Inhibition of DRP-1-Dependent Mitophagy Promotes Cochlea Hair Cell Senescence and Exacerbates Age-Related Hearing Loss. Front. Cell. Neurosci. 13:550. doi: 10.3389/fncel.2019.00550
Received: 29 July 2019; Accepted: 28 November 2019;
Published: 17 December 2019.
Edited by:
Vassilis G. Gorgoulis, National and Kapodistrian University of Athens, GreeceReviewed by:
Daniel Munoz-Espin, University of Cambridge, United KingdomCopyright © 2019 Lin, Xiong, Su, Pang, Lai, Zhang, Jian, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiqing Zheng, emhlbmd5aXFAbWFpbC5zeXN1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.