 Manoela V. Fogaça
Manoela V. Fogaça Ronald S. Duman
Ronald S. Duman- Department of Psychiatry, Yale University School of Medicine, New Haven, CT, United States
Major depressive disorder (MDD) is a debilitating illness characterized by neuroanatomical and functional alterations in limbic structures, notably the prefrontal cortex (PFC), that can be precipitated by exposure to chronic stress. For decades, the monoaminergic deficit hypothesis of depression provided the conceptual framework to understand the pathophysiology of MDD. However, accumulating evidence suggests that MDD and chronic stress are associated with an imbalance of excitation–inhibition (E:I) within the PFC, generated by a deficit of inhibitory synaptic transmission onto principal glutamatergic neurons. MDD patients and chronically stressed animals show a reduction in GABA and GAD67 levels in the brain, decreased expression of GABAergic interneuron markers, and alterations in GABAA and GABAB receptor levels. Moreover, genetically modified animals with deletion of specific GABA receptors subunits or interneuron function show depressive-like behaviors. Here, we provide further evidence supporting the role of cortical GABAergic interneurons, mainly somatostatin- and parvalbumin-expressing cells, required for the optimal E:I balance in the PFC and discuss how the malfunction of these cells can result in depression-related behaviors. Finally, considering the relatively low efficacy of current available medications, we review new fast-acting pharmacological approaches that target the GABAergic system to treat MDD. We conclude that deficits in cortical inhibitory neurotransmission and interneuron function resulting from chronic stress exposure can compromise the integrity of neurocircuits and result in the development of MDD and other stress-related disorders. Drugs that can establish a new E:I balance in the PFC by targeting the glutamatergic and GABAergic systems show promising as fast-acting antidepressants and represent breakthrough strategies for the treatment of depression.
Introduction
Major depressive disorder (MDD) is a recurring neuropsychiatric illness that is among the leading contributors to social and economic burden, rm resilience to stress-induced affecting approximately one in five people in the United States (Kessler et al., 2005). The World Health Organization estimates that MDD will be the second leading cause of disability by 2020 (Reddy, 2010). Moreover, MDD induces a high level of personal suffering and suicidality, and also increases the risk of other comorbid medical conditions that can lead to further disability or death (Karch et al., 2009).
The current pharmacological treatment approaches set by national and international guidelines recommend the use of monoaminergic-based drugs, notably, serotonin reuptake inhibitors (SSRI), as first-line medications (Bauer et al., 2007). Although these drugs provide a significant therapeutic benefit, they still require weeks to months to induce an antidepressant response, and up to 33% of the patients are considered treatment resistant (i.e., fail to respond to two or more antidepressants). Also, the majority of patients experience recurrence after interrupting the treatment, and the adherence of patients to these medicines is relatively low, as they cause undesired side effects, including weight gain, sexual dysfunction, disruption of normal sleep patterns, memory deficits, and others. Moreover, most patients experience a worsening of symptoms in the first weeks of administration, which also contributes to the low treatment adherence (Trivedi et al., 2006).
Since the late 1980s, when the SSRIs were developed and the monoamine deficiency hypothesis of depression gained more support, there have been no considerable advances in pharmacological treatment for MDD. Considering the relatively low efficacy of monoaminergic drugs, there is an urgent need for development of novel medicaments that address current therapeutic limitations. Indeed, in recent years, a new class of fast-acting, efficacious antidepressants has emerged, showing immense promise in clinical and pre-clinical studies. Among them, ketamine, an NMDA receptor blocker, is the most studied due to its fast (within 2 h of administration) and sustained (up to 7 days) antidepressant effects (Berman et al., 2000; Zarate et al., 2006; Harmer et al., 2017). These glutamatergic-based drugs have shed light on yet unexplored avenues to explain the pathophysiology of depression, shifting efforts to the discovery and development of new classes of drugs. The accumulating evidence in the literature relating stress, GABA/glutamate deficits in the brain, and MDD, as well as the antidepressant efficacy of drugs that directly interact with these systems, have led to alternative hypotheses to explain the complex neurobiology of affective disorders that overcome inconsistences in monoaminergic theories.
In this review, we focus on the GABAergic deficit/imbalance hypothesis of MDD (Luscher et al., 2011; Fee et al., 2017), and elaborate this theory in the context of the glutamatergic hypothesis and the monoaminergic and neurotrophic deficits theories. We discuss the dysregulation of GABA neurotransmission and changes in specific GABAergic interneuron subtypes observed in MDD subjects and stress- or genetic-based animal models of depression. Although depression must be seen as a system-wide disorder, a broad range of GABA interneurons that orchestrate excitation–inhibition (E:I) balance in corticolimbic structures are located in the prefrontal cortex (PFC) and several studies point to this region as one of the primary brain regions involved in the pathophysiology of MDD. Indeed, multiple reports show a direct correlation between chronic stress and depression with decreased volume, synaptic atrophy/loss, and altered connectivity in the PFC (Duman et al., 2016). For this reason, we discuss in more detail evidence for cortical impairments in depression. Finally, based on recent findings, we will discuss how monoaminergic drugs can also modulate the GABAergic system and will explore novel non-monoaminergic fast-acting pharmacological approaches to treat MDD, including GABAA and GABAB receptor modulators (allosteric modulators, neurosteroids, agonists, and antagonists), NMDA receptors blockers (such as ketamine), and GABAergic interneurons-targeting neuropeptides.
GABAergic System in the PFC
GABA is the major inhibitory mediator of cortical interneurons in the brain that serves to modulate a wide range of local neurotransmitter systems, most notably, the glutamatergic excitatory counterpart. By targeting specific somatic domains of neighboring glutamatergic principal neurons, GABA interneurons control the E:I balance in the PFC as well as the excitatory output to projecting areas, such as the amygdala, bed nucleus of stria terminalis, and dorsal raphe nucleus. Due to this network orchestration of firing patterns, cortical GABAergic interneurons play an essential role in mediating complex emotional and cognitive processes in the brain. GABA is synthesized from glutamate by glutamate decarboxylase enzymes (GAD65 and GAD67) and stored in vesicles through the vesicular GABA transporter (VGAT1 and 2). The GABAergic signal is terminated by rapid uptake of GABA to glial cells and presynaptic neurons through plasma membrane GABA transporters (GAT1-4).
One-third of all synapses in the central nervous system (CNS) connects via GABA interneurons, which comprise 20–30% of neocortical neurons and can be classified accordingly to their diverse morphological, electrophysiological, and molecular characteristics (Markram et al., 2004; Rudy et al., 2011; DeFelipe et al., 2013; Tremblay et al., 2016). The most common nomenclature segregates interneurons accordingly to their expression profile of neurochemical markers. Three major non-overlapping interneuron groups in the neocortex include those that express the calcium-binding protein parvalbumin (PV), the neuropeptide somatostatin (SST), and the ionotropic serotonin receptor 3 (Rudy et al., 2011). These neurons can further co-express other markers, such as the neuropeptides cholecystokinin (CCK), vasoactive intestinal peptide (VIP), and neuropeptide Y, as well as other calcium-binding proteins, such as calbindin and calretinin (Rudy et al., 2011). Characterization of distinct subtypes of interneurons helps to identify vulnerable subpopulations that could be relevant to different neuropsychiatric disorders. Specifically, PV and SST interneurons have been extensively studied in stress-related disorders. The most abundant subtype, PV, correspond to 40% of cortical GABA interneurons, and have chandelier or, most commonly, basket cell morphology. PV basket interneurons mainly control firing synchronization and spike timing of neighboring excitatory neurons by providing somatic fast-spiking inhibition to pyramidal cells (Markram et al., 2004; Ferguson and Gao, 2018). On the other hand, 25–30% of cortical interneurons express SST, which consist mainly of Marinotti cells with low-threshold regular spiking properties and an independent high basal firing activity. SST cells make synapses on the dendritic tufts of pyramidal cells but can also inhibit local PV interneurons (Markram et al., 2004; Urban-Ciecko and Barth, 2016). These distinct properties and sub-localization confer to both SST and PV interneurons different roles in the cortical microcircuit: while SST cells control the spiking inputs to pyramidal neurons, PV interneurons regulate the spiking outputs from pyramidal neurons to projecting brain areas.
GABA interneurons express two subtypes of GABA receptors: GABAA and GABAB. The most prominent receptor, GABAA, is a ligand-gated Cl- ion channel (ionotropic) and has been extensively characterized as the target of many psychotropic agents, including benzodiazepines, ethanol, and barbiturates. These receptors are mostly located post-synaptically and control fast synaptic inhibition. GABAA receptors are tetrameric or pentameric in structure that are made up of multiple subunits (6α, 4β, and 3γ) in distinctive combinations that assemble together around a central chloride pore (Engin et al., 2018). GABAB receptors are Gi-coupled receptors (metabotropic) and composed of a heterodimer of two homologous subunits: GABAB1 and GABAB2; they are mainly located at pre-synaptic sites, functioning as autoreceptors and inhibiting GABA release, although they can also be found post-synaptically (Cryan and Kaupmann, 2005). Given the broad spectrum of neuronal activity controlled by GABA interneurons, it is increasingly clear that imbalance in the GABAergic system and hence in the E:I balance can contribute to the pathophysiology of several psychiatric disorders, including MDD.
Cortical Dysregulation of GABA Neurotransmission in Chronic Stress and Depression
Although the adaptive, innate stress response is essential for body homeostasis and survival, it is widely recognized that responses to sustained, chronic stress can become dysregulated and result in illness and abnormal behaviors. In the brain, chronic stress can produce changes in neurotransmitter function and appropriate neuroplasticity responses that could precipitate depression in humans and, therefore, has been extensively used as a rodent model for depression (Duman et al., 2016).
GABAergic neurons play an important role in the termination of stress response through regulation of the hypothalamus–pituitary–adrenal (HPA) axis, and disruption of this regulatory response contributes to the abnormal effects of chronic stress exposure. For example, chronic stress causes down-regulation of the transmembrane K-Cl cotransporter (KCC2), rendering GABA inputs ineffective to synaptic inhibition of the HPA axis (Hewitt et al., 2009). Moreover, deletion or mutation of the γ2 subunit of GABAA receptors (heterozygous γ2 knockout: γ2+/-) result in reduced GABAA receptor binding and consequent HPA axis hyperactivity, leading to anxiogenic and pro-depressive behaviors (Crestani et al., 1999; Chandra et al., 2005; Shen et al., 2010; Smith and Rudolph, 2012). A similar pro-depressive profile is found in α2 knockout mice (Vollenweider et al., 2011). Therefore, genetic modifications in GABAA receptors subunits have been increasingly used as animal models to study the influence of GABAergic system in the pathophysiology of anxiety and depression, as well as pharmacological approaches that have therapeutic potential.
Considering that GABA receptors are highly expressed and GABAergic interneurons are abundant in the PFC and exert an important GABAergic inhibitory control over HPA axis activity (Diorio et al., 1993; Akana et al., 2001; Radley et al., 2009) it is conceivable that the PFC GABAergic system plays an essential role in emotional processing that is vulnerable to stress. In this respect, acute psychological stress (threat-of-shock condition) decreased approximately 18% of PFC GABA levels relative to a “safe” condition in healthy subjects (Hasler et al., 2010). In rodents, repeated immobilization stress increased GAD activity and GABA turnover, and reduced GABA levels in the frontal cortex (Otero Losada, 1988), an effect that was also reported after a 3-week of chronic mild stress (CMS) exposure (Shalaby and Kamal, 2009). In a learned helplessness paradigm, a model of depression, rats that failed to learn the shuttlebox task showed a 25% reduction of GABAA receptors in cortical synaptoneurosomes (Drugan et al., 1989). Similar results were observed after other types of chronic stress, such as cold and isolation (Braestrup et al., 1979). A recent study reported that 9-weeks CMS exposure resulted in decreased cortical GABAA receptor function, decreased release probability at peri-somatic GABAergic synapses, and reduced postsynaptic GABAB receptor mediated inhibition in anhedonic rats, leading to higher excitability of pyramidal neurons (Czéh et al., 2018). Also, chronic unpredictable stress (CUS) or CMS exposure decreased innervation and function of GABAergic axons, and levels of GAD67, VGAT, and GAT3 in the PFC (Gilabert-Juan et al., 2013; Ma et al., 2016; Banasr et al., 2017). Besides chronic stress exposure of adult animals, there is also evidence that early life stress exposure impacts the GABAergic system later in life in the adult brain. Maternal separation stress and alteration of maternal care in the early (first weeks) postnatal period decreased expression of GABAA receptors in the frontal cortex and other limbic areas, as well as induced anxiety and depressive-like behaviors in adulthood (Caldji et al., 2000, 2003).
Collectively, these data provide support for the hypothesis that stress causes major changes in the GABAergic system in the PFC that could result in abnormal behavioral and synaptic responses, including dendritic reorganization of interneurons (Gilabert-Juan et al., 2013), as well as alterations of electrophysiological respones (Northoff and Sibille, 2014; McKlveen et al., 2016), that results in defective output from pyramidal neurons to other brain areas. However, even though numerous reports suggest that chronic stress decreases GABA levels and function, other studies have reported opposite effects. Chronic immobilization stress induced a small increase in GABAA receptor binding in the frontal cortex (Braestrup et al., 1979) and chronic social defeat stress increased GABAA-containing α5 subunit in the PFC and hippocampus of susceptible mice (Xiong et al., 2018). Likewise, chronic restraint stress (21 days) induced an increase in GABAA-α1 subunit mRNA expression in the mPFC but not α2, α3, α4, or γ2 (Gilabert-Juan et al., 2013). Moreover, McKlveen et al. (2016) found that CUS (14 days) increased the frequency of miniature inhibitory postsynaptic currents in the infralimbic area, as well as inhibitory appositions and terminals onto glutamatergic cells, suggesting a stress-induced enhancement of prefrontal inhibition. While difficult to reconcile, it is important to highlight that the results of stress studies may differ depending on the type and duration of the stressor, the GABA receptor subunit analyzed, and the specific subregions of the PFC studied.
In addition to these preclinical studies, there is accumulating evidence that dysfunction of the GABAergic system is associated with the pathophysiology of MDD and that normalization of GABA is associated with the remission of depressive symptoms (Godfrey et al., 2018). Pioneering studies showed that patients with depression have lower GABA levels in the plasma (Petty and Sherman, 1984) and the cerebrospinal fluid (CSF) (Gold et al., 1980; Gerner and Hare, 1981). Further studies extended this work through positron emission tomography (PET) imaging methods, which permits a direct and noninvasive quantification of GABA levels in the brain. These studies showed that GABA levels are reduced in unmedicated patients with MDD in several cortical areas, including the prefrontal (Hasler et al., 2007), occipital (Sanacora et al., 1999, 2004; Song et al., 2012), and anterior cingulate (ACC) cortices (Gabbay et al., 2012; Godfrey et al., 2018). Significant reduction in the ratio GABA/creatine + phosphocreatine was found in the ACC of female veterans with suicidal behavior (Prescot et al., 2018). Likewise, reduced GAD67 protein or gene expression were found in the dorsolateral PFC (dlPFC) and subgenual ACC (SgACC) of depressed patients (Karolewicz et al., 2010; Tripp et al., 2012), although other studies reported no significant effects (Sibille et al., 2011; Gilabert-Juan et al., 2012). Besides GABA levels, several studies reported decreased expression of GABAA receptors subunit genes in MDD cortices, including decreased α1, α3, α4, γ1, β2, and ρ1 (Merali et al., 2004; Sequeira et al., 2007; Klempan et al., 2009; Luscher et al., 2011). However, there were also reports of increased expression of certain subunits, including α5, γ2, β3, and δ in MDD subjects (Merali et al., 2004; Choudary et al., 2005; Sequeira et al., 2007; Klempan et al., 2009) suggesting that different GABAA receptor subunits may play distinct roles in the etiology of MDD.
Studies regarding the participation of GABAB receptors in the pathophysiology of MDD have received less attention and, therefore, the literature remains unclear. Although GABAB1 and GABAB2 subunits were reported to be decreased in the lateral cerebellum of MDD subjects (Fatemi et al., 2011), no evidence was found for altered GABAB receptor binding in the frontal cortex or hippocampus (Cross et al., 1988; Arranz et al., 1992). However, some variables should be considered as potential confounds in this study, as some of the MDD patients were taking antidepressants at the time of death, and in some cases there was a long post-mortem interval before tissue collection. Despite these limitations, it is notable that preclinical studies report that helpless rats showed decreased expression of GABAB receptors in the frontal cortex (Martin et al., 1989), and GABAB1 subunit knockout animals displayed antidepressant-like responses in the forced swim test (Mombereau et al., 2004). Taken together, the results demonstrate that modulation of GABAB receptors induces antidepressant effects (see the section “Conclusion and Future Directions”), and warrant additional studies with more cutting edge tools to further investigate the role of GABAB receptors in depression and treatment response.
GABA Interneuron-Related Deficits in Depression
Numerous studies suggest that the reduction in cortical GABA levels observed in MDD subjects and stressed rodents could not only result from decreased levels of the GABA synthetic enzymes GAD65/67, but could also result from a reduction in the density of specific GABA interneuron subpopulations (Table 1). MDD patients show a reduced volume of brain areas such as the PFC and hippocampus (MacQueen et al., 2008; Savitz and Drevets, 2009). Also, abnormalities in the GABAergic system in cortical areas can also robustly affect other brain regions. For example, low GABA levels in the ACC of MDD patients are associated with reduction in hippocampal volume (Abdallah et al., 2015). Reduced SST gene expression, mRNA, or protein levels were found in the CSF, SgACC, dlPFC, and amygdala of MDD subjects, and in the medial PFC (mPFC) and hippocampus of animals exposed to CUS (Rubinow et al., 1985; Rajkowska et al., 2007; Sibille et al., 2011; Tripp et al., 2011; Guilloux et al., 2012; Banasr et al., 2017). Interestingly, female MDD subjects show a more robust reduction in SST expression than males (Sibille et al., 2011; Tripp et al., 2011, 2012; Guilloux et al., 2012), suggesting that SST could be related to the twofold greater incidence of MDD in females (Kuehner, 2017). Although SST expression in MDD subjects and chronically stressed animals is decreased, Gilabert-Juan et al. (2013) reported dendritic hypertrophy of Martinotti cells (which includes SST-expressing interneurons) in the mPFC of mice exposed to chronic restraint stress, without changes in spine density.
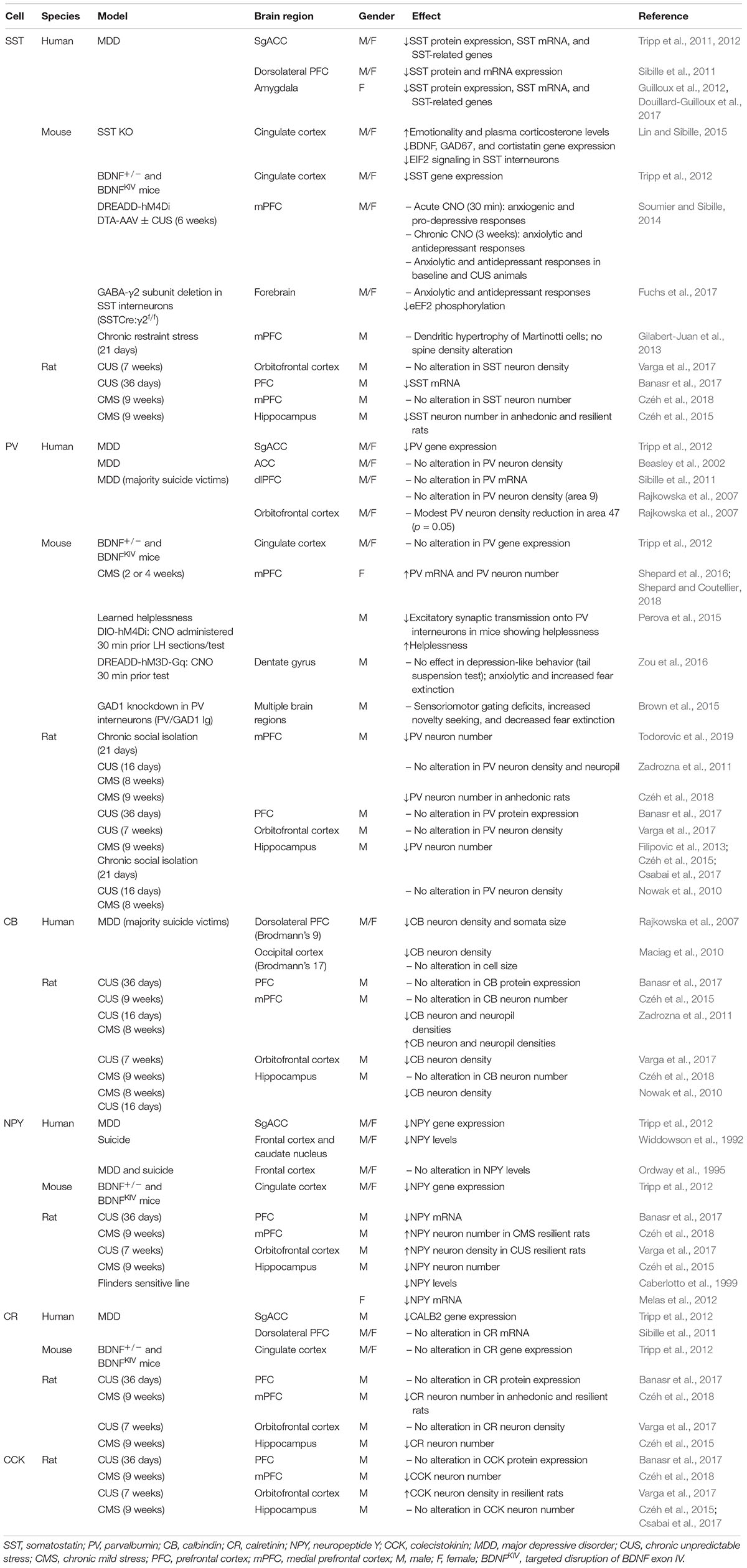
Table 1. Studies of GABAergic interneuron subtypes in MDD and animal models of depression.
The association between SST interneurons and the pathophysiology of MDD has been more directly supported through pharmacological and genetic manipulations in rodents. Mice lacking SST (SST-KO) exhibited increased anxiety- and depressive-like behaviors; elevated basal plasma corticosterone; and reduced BDNF, GAD67, and cortistatin genes expression (Lin and Sibille, 2015). Disinhibition of SST interneurons by deletion of GABAA-containing γ2 subunit in SST neurons (SSTCre:γ2f/f) resulted in enhanced inhibitory input to pyramidal cells in the hippocampus and cingulate cortex, and consequently produced anxiolytic- and antidepressant-like phenotypes (Fuchs et al., 2017). Interestingly, acute chemogenetic-induced inhibition of SST interneurons in the mPFC promoted anxiety and depressive-like responses, whereas chronic silencing or chemical ablation had the opposite effect (Soumier and Sibille, 2014). Furthermore, mice with constitutive, heterozygous deletion of the BDNF gene (BDNF+/-) or with targeted disruption of exon IV (BDNFKIV), causing a reduction or blockade of activity-dependent BDNF expression and depressive-like behaviors, showed reduced SST and NPY gene expression in the cingulate cortex (Tripp et al., 2012).
Somatostatin co-localizes with calbindin and NPY, and these neuropeptides have also been implicated in mood disorders. Reductions in calbindin and NPY markers were found in the frontal cortex of MDD patients (Widdowson et al., 1992; Rajkowska et al., 2007; Maciag et al., 2010; Tripp et al., 2011, 2012), as well as in the PFC and hippocampus of rats submitted to different rodent models of depression, including CUS, BDNF mutant mice, or the Flinders sensitive line of rat (Caberlotto et al., 1999; Nowak et al., 2010; Zadrozna et al., 2011; Melas et al., 2012; Tripp et al., 2012; Czéh et al., 2015, 2018; Banasr et al., 2017; Varga et al., 2017). However, other studies failed to detect statistical differences in chronic stress models (Czéh et al., 2015, 2018; Banasr et al., 2017). Additionally, NPY neuronal density was increased in the orbitofrontal cortex and mPFC (IL) of rats that were considered resilient to CUS behavioral effects, but unchanged in anhedonic animals (Varga et al., 2017; Czéh et al., 2018). In depressed patients, the levels of NPY in the CSF or plasma were inversely correlated with anxiety symptoms and with attempted suicide (Widerlov et al., 1988; Westrin et al., 1999). Other markers, such as CCK and calretinin, seem not to be consistently affected in stress models or MDD (Sibille et al., 2011; Zadrozna et al., 2011; Tripp et al., 2012; Banasr et al., 2017; Csabai et al., 2017), notwithstanding a study reporting decreased immunoreactivity of these peptides in the mPFC of rats submitted to CMS (Czéh et al., 2018).
Although several studies have failed to detect robust differences in the expression of PV in rodent models and MDD subjects (Beasley et al., 2002; Cotter et al., 2002; Rajkowska et al., 2007; Nowak et al., 2010; Sibille et al., 2011; Zadrozna et al., 2011; Banasr et al., 2017), there are reports that PV interneurons contribute to regulation of E:I within the PFC that influences emotional responses (Perova et al., 2015; Ferguson and Gao, 2018). One study has reported a decrease in PV gene expression in SgACC post-mortem tissues of depressed patients (Tripp et al., 2012), and a modest reduction in PV immunoreactivity was found in the orbitofrontal cortex (p = 0.05, Brodmann area 47) (Rajkowska et al., 2007). In rodents, CMS caused a reduction in PV neuron number in the mPFC of anhedonic rats, whereas SST density was unchanged (Czéh et al., 2018). A decrease in PV cell number was also found in rats submitted to chronic social isolation (Todorovic et al., 2019). By contrast, there was a report of increased PV expression in the PFC of female mice after 2 weeks of CUS (Shepard et al., 2016; Shepard and Coutellier, 2018). Also, mice that showed helplessness behavior in response to inescapable stress exposure showed a reduction in excitatory synaptic transmission onto PV interneurons in the mPFC, and selective chemogenetic inactivation of PV cells further increased helplessness responses (Perova et al., 2015).
However, other genetic approaches have reported complex behavioral changes. Knockdown of the Gad1 transcript specifically in PV interneurons (Pvalb/Gad1 Tg) produced a decrease in PV-induced GABAergic activity in multiple brain regions, leading to sensoriomotor gating deficits, increased novelty seeking, and decreased fear expression (Brown et al., 2015). Besides the PFC, PV cells located in the hippocampus also play an important role in the modulation of affective behaviors as well as memory. Acute activation of PV interneurons in the dentate gyrus of the hippocampus through DREADD-hM3D-Gq virus and CNO administration (30 min) did not affect depressive-like behavior in the tail suspension test, but produced anxiolytic-like responses and increased fear extinction (Zou et al., 2016). Also, CMS or chronic social isolation induced a decrease in PV-immunoreactive cells in the hippocampus, whereas CCK and calbindin expression remained unchanged (Filipovic et al., 2013; Czéh et al., 2015; Csabai et al., 2017). Thus, divergence in the responses observed among these studies in the literature highly are related to the brain region studied, the experimental protocol (timing, stress paradigms), and sex differences. Also, it is noteworthy that PV interneurons are under inhibitory control of other interneuron populations, such as SST, making the resultant responses even more complex.
Taken together, the studies mentioned highlight the complexity of segregating the GABAergic system into different subclasses of interneurons to study the pathophysiology of depression, but demonstrate the importance of understanding how these cells locally interact and integrate diverse neurocircuits that control affective behavioral responses.
The GABAergic System as a Therapeutic Target for the Treatment of MDD
Although extensive efforts have been conducted to develop new therapeutic interventions, the current pharmacological treatment approaches still recommend the use of SSRIs as first-line medications for the treatment of MDD. These drugs, along with other classic antidepressants, such as tricyclics and monoamine oxidase inhibitors, primarily facilitate monoaminergic systems, including 5-HT and norepinephrine. However, the emergence of fast-acting antidepressants, notably ketamine, provide evience for other neurotransmitter systems for the treatment, as well as pathophysiology of MDD. Evidence that normalization of GABA-mediated E:I imbalance in the PFC is a shared mechanism of action between different classes of antidepressants, providing further support for the involvement of GABAergic dysfunction in the etiology of MDD. In this section, we will review the literature showing how first-line monoaminergic antidepressants and rapid-acting agents can influence the GABAergic system. We will discuss how fast-acting antidepressants provide a new understanding of the pathophysiology of depression, leading to connections between the glutamatergic, GABAergic, and neurotrophic hypotheses of depression. Finally, we will discuss the antidepressant potential of agonists, antagonists, or allosteric modulators of GABAA and GABAB receptors, as well as neuropeptides that target specific subpopulations of GABA interneurons and cannabinoid agents.
Classic Monoaminergic Antidepressants: Effects Beyond Monoamines
The correlation between GABA deficits in the brain, stress, and MDD became more evident with investigations showing that SSRIs, electroconvulsive therapy, and transcranial magnetic stimulation normalize the reduction in cortical and plasmatic GABA levels, as well as in GAD67 expression in MDD subjects and rodents subjected to chronic stress (Sanacora et al., 1999, 2004; Bhagwagar et al., 2004; Goren et al., 2007; Kucukibrahimoglu et al., 2009; Karolewicz et al., 2010; Dubin et al., 2016). Besides decreased GABA levels, MDD patients and chronically stressed animals have reduced levels of allopregnanolone (brain and plasma), an endogenous neurosteroid that acts as a GABAA receptor positive allosteric modulator (discussed in more detail below). This deficit was reversed by chronic administration of SSRIs such as fluoxetine (Uzunov et al., 1996; Romeo et al., 1998; Uzunova et al., 1998, 2004; Strohle et al., 1999; Dong et al., 2001; Guidotti et al., 2001; Pinna et al., 2006, 2009); interestingly, in vitro evidence suggests that SSRIs can directly interact with the enzymes involved in neurosteroid synthesis (Griffin and Mellon, 1999). Chronic treatment with the classic monoaminergic antidepressant desipramine, but not fluoxetine, also normalized the elevated serum corticosterone levels and the pro-depressive behaviors of γ2+/- mice (Shen et al., 2010). In this same study, subchronic treatment with desipramine had no effect (Shen et al., 2010), suggesting that this drug acts over time to balance GABAergic inhibition deficits. In another study, chronic fluoxetine treatment induced pro-depressive and anxiogenic-like effects in γ2+/- mice (Benham et al., 2017), pointing to a requirement of GABAA-containing γ2 subunit in the antidepressant effect of SSRIs. Interestingly, studies reported that fluoxetine can act directly as an allosteric modulator of GABAA receptors (Robinson et al., 2003).
Indeed, direct interactions between the GABAergic and serotoninergic systems in the raphe nucleus and cortical regions have been reported (Celada et al., 2001; Puig et al., 2004; Santana et al., 2004; Llado-Pelfort et al., 2012). In the PFC, both pyramidal glutamatergic neurons and GABAergic interneurons, notably PV positive cells, express serotoninergic receptors (mainly 5HT1A and 5HT2A) (Santana et al., 2004; Celada et al., 2013). 5HT1A receptor agonists, such as 8-OH-DPAT, have a preferential action on GABA interneurons, resulting in pyramidal neuron disinhibition and enhancement of cell firing in PFC and targeted subcortical structures, such as the ventral tegmental area (Llado-Pelfort et al., 2012). On the other hand, the excitability of pyramidal neurons in the mPFC can be inhibited by activation of GABA interneurons through 5HT3 receptors (Puig et al., 2004). In this regard, multimodal drugs that are high affinity 5HT3 receptor antagonists, such as vortioxetine, show antidepressant efficacy in clinical studies and have been used as atypical antidepressants to treat MDD (Thase et al., 2016; Artigas et al., 2018). Moreover, the majority of serotoninergic cell bodies in the raphe nucleus express GABAB receptors, which control serotoninergic cell firing as well as the release of monoamines in other brain regions (Bowery et al., 1980; Abellan et al., 2000; Serrats et al., 2003). The antidepressant-like effects of the GABAB antagonist CGP56433A were abolished by prior treatment with a tryptophan hydroxylase inhibitor, which depletes serotonin levels (Slattery et al., 2005). Additionally, several different monoaminergic antidepressants increase GABAB receptor binding and function in the rat frontal cortex (Lloyd et al., 1985; Gray and Green, 1987).
Fast-Acting Glutamatergic Antidepressants: Is It All Glutamate?
In recent years, the mechanisms underlying the actions of ketamine have been extensively studied because of its rapid (within hours), sustained (up to 7 days), and efficacious effects (effective in patients considered treatment resistant) (Berman et al., 2000; Zarate et al., 2006). Related agents, including ketamine stereioisomers and metabolites, have also demonsrated rapid effects in rodent models. These drugs share the ability to influence, directly or indirectly the enhancement of glutamatergic signaling in the brain, promoting post-synaptic AMPA-mediated calcium influx that leads to BDNF release by pyramidal neurons (Lepack et al., 2014, 2016; Zhou et al., 2014). Extracellular BDNF, in turn, activates TrkB receptors in the membrane, resulting in stimulation of intracellular signaling cascades, including Akt, eukaryotic elongation factor 2 kinase (eEF2K), and mTORC1 that results in synaptic actions that contribute to antidepressant behavioral responses (Li et al., 2010; Autry et al., 2011; Duman et al., 2016).
The molecular and cellular mechanisms underlying the rapid enhancement of glutamatergic signaling in the PFC by ketamine have been of particular interest. One hypothesis is that ketamine first targets NMDA receptors specifically located in cortical interneurons, notably, SST and PV subtypes. Because these GABA inhibitory neurons are tonic firing they would be more sensitive to antagonist blockade as tonic activity would remove the Mg2+ block of the NMDA receptor allowing ketamine to enter the channel pore and block further activation of Ca2+ entry (Fee et al., 2017; Ghosal et al., 2017). Blockade of GABAergic interneuron firing would thereby decrease GABA release, resulting in disinhibition of excitatory pyramidal neurons and subsequently produce a glutamate burst that could drive activity dependent synaptic plasticity (Duman et al., 2016). An alternative hypothesis is that ketamine acts directly on pyramidal neurons to block resting state NMDA receptor activity driven by spontaneous glutamate release that produces synaptic changes via deactivation of eEF2K, resulting in increased synthesis of synaptic proteins (Autry et al., 2011). These two theories may not be mutually exclusive, although it is difficult to explain how NMDA receptors would be at resting levels in the presence of a known glutamate burst (Moghaddam et al., 1997). In either case, there is an increase in synaptic protein synthesis that underlies long-lasting changes (approximately 1 week) that correspond to the time course for the antidepressant behavioral actions of ketamine. It is also possible that more long-lasting ketamine metabolites contribute to the sustained actions of ketamine (Zanos et al., 2016; Fukumoto et al., 2019). The glutamate burst produced by ketamine appears to be contradictory with evidence of elevated glutamate levels in the brains of MDD subjects (Stone et al., 2012), although other studies have reported no significant differences (Valentine et al., 2011; Taylor et al., 2012). However, it is important to note that the ketamine-induced burst of glutamate is transient, lasting approximately 1 h, and then levels return to control (Moghaddam et al., 1997). Although transient, the glutamate burst results in activity-dependent synaptic changes that are long-lasting.
Although much of the current work has focused on glutamate synaptic changes in the actions of ketamine, there is also increasing evidence that GABA alterations contribute to the ketamine response, by reestablishing E:I balance in the PFC via homeostatic self-tuning adaptations. This local reorganization could influence microcircuits in target regions by reestablishing firing patterns, and thereby promoting antidepressant effects. This idea is supported by recent evidence that the fast antidepressant effects of ketamine are accompanied by a robust increase in GABA levels in the mPFC of MDD patients (Milak et al., 2016) and in the ACC of rats subjected to CUS (Perrine et al., 2014), although another study failed to detect differences in the occipital cortex (Valentine et al., 2011). One possibility for these discrepancies, in addition to the different cortical subregions analyzed, is the timepoint at which MRS data were collected. Whereas one study was conducted during ketamine infusion (Milak et al., 2016), the other was carried out after ketamine (Valentine et al., 2011); by the end of the infusion, it was shown in the former study that the increase in amino acid responses was no longer detectable (Milak et al., 2016).
Also, a SPECT study reports that S-ketamine administration leads to alterations of GABAA receptor binding in the dorsomedial PFC of healthy subjects (Heinzel et al., 2008). Likewise, studies in cultured murine neurons provide evidence that ketamine increases the activity of extrasynaptic GABAA receptors in the cortex and hippocampus (Wang et al., 2017). Combined administration of sub-effective doses of muscimol, a potent and selective agonist of GABAA receptors, and ketamine, produced antidepressant-like effects in female mice (Rosa et al., 2016). In this same study, the antidepressant effects of ketamine were blocked by the GABAB agonist baclofen, suggesting that the antidepressant actions of ketamine could involve activation of GABAA and blockade of GABAB receptors (Rosa et al., 2016). In support of glutamatergic and GABAergic interactions in ketamine responses is data showing that a single dose of the ketamine induced antidepressant-like effects and normalized the glutamatergic deficits, including reduced cell surface NMDA and AMPA receptor levels and impaired synaptic function in the hippocampus and mPFC of γ2+/- mice (Ren et al., 2016). Moreover, ketamine potentiated pre- and post-synaptic GABAergic synapses selectively in the ACC of these animals (Ren et al., 2016). In addition, we have found that a single dose of ketamine increases markers of GABA in the PFC, including increased levels of VGAT, GAD, and gephyrin (Ghosal et al., 2018, SfN abstract). Thus, although more studies are needed to clarify how ketamine modulates the GABAergic system, the current evidence indicates that ketamine enhances GABA levels/function in the brain as well as GABAA receptors activity.
In addition, it was reported that fast-acting agents, such as Ro-25-6981, a GluN2B-selective NMDA receptor antagonist, induce antidepressant effects by promoting GABAB receptor surface expression and increasing postsynaptic GABAB-mediated resting L-type calcium channel activity, resulting in an increased intracellular calcium, recruitment of BDNF/mTORC1 pathways, and protein synthesis (Workman et al., 2013, 2015). Accordingly, MDD and suicide patients have decreased levels of blood and brain BDNF levels and transcripts (Dwivedi et al., 2003; Shimizu et al., 2003; Kim et al., 2007; Guilloux et al., 2012; Banerjee et al., 2013); reduced BDNF levels in blood were absent in patients taking antidepressants (Shimizu et al., 2003). However, while monoaminergic antidepressants take weeks to modulate neurotrophic factor expression, the rapid elevation in BDNF “release” and signaling by ketamine is shared by other fast-acting agents, such as the non-selective muscarinic receptor antagonist scopolamine, the NMDA receptor modulator GLYX-13 (rapastinel), the ketamine metabolite (2R,6R)-Hydroxynorketamine [(2R,6R)-HNK], and the mGlu2/3 receptor antagonist LY341495 (Liu et al., 2012; Lepack et al., 2016; Ghosal et al., 2018; Kato et al., 2018) and may explain, at least in part, the fast versus slow response rates of these agents. Moreover, in vitro and in vivo studies suggest that BDNF induces antidepressant-like effects via increased phosphorylation of γ2 subunit, resulting in an increase of GABAA receptor accumulation and stability in the cell surface, and in an enhancement of synaptic inhibition efficacy in the hippocampus and PFC (Jovanovic et al., 2004; Vithlani et al., 2013). Thus, the upregulation of GABAB receptors induced by NMDA receptor blockade and consequent activation of BDNF/mTORC1 signaling, as well as the role of BDNF on GABAA receptors phosphorylation and enhancement of GABAergic mIPSC amplitude and frequency, could be a link associating the GABA/glutamate balance deficits to the neurotrophic theory of depression (Figure 1).
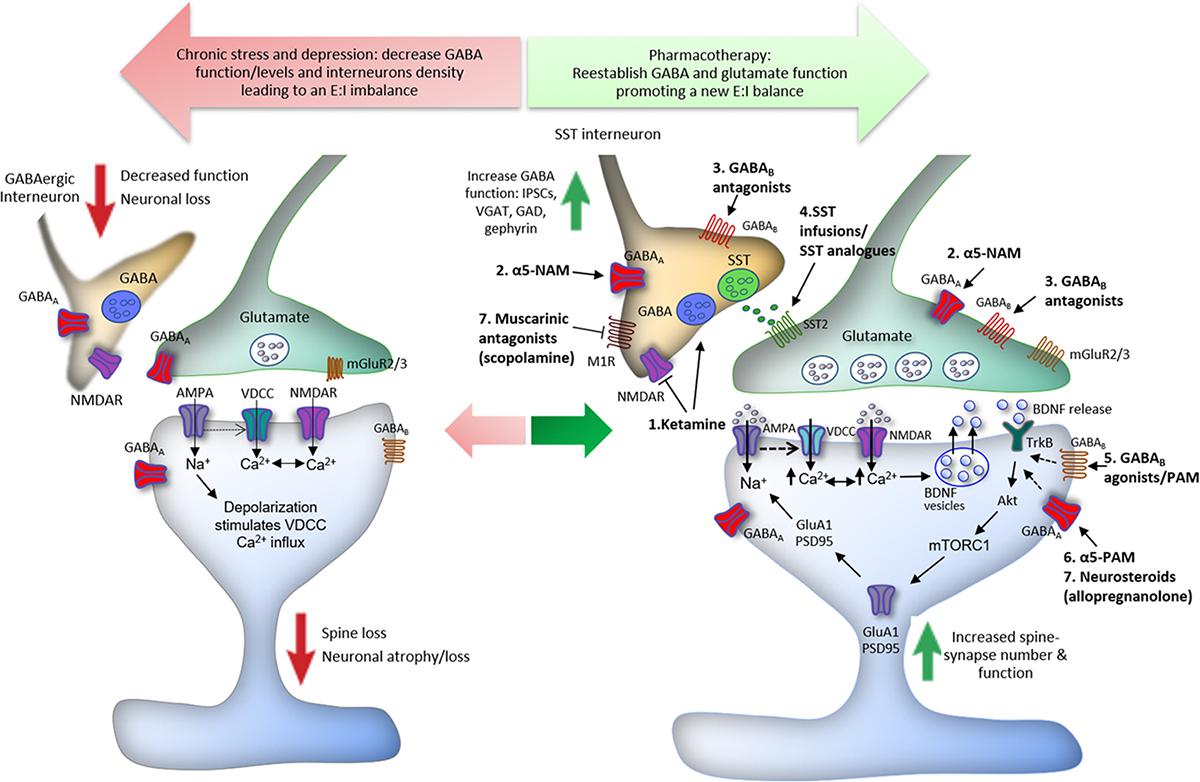
Figure 1. Proposed mechanisms underlying the action of ketamine and GABA-related drugs in the reestablishment of cortical excitatory–inhibitory (E:I) balance. Chronic stress induces spine loss and dendritic atrophy in pyramidal glutamatergic cells, and decreased GABAergic interneuron markers, leading to a reduction in the levels and function of GABA in the PFC. GABAergic dysfunction disturbs the optimal E:I balance in the brain and compromises the integrity of neurocircuits, contributing to the development of major depressive disorder (MDD) and other stress-related disorders. The E:I imbalance can be reversed by drugs via different GABA-related mechanisms. (1) Low doses of ketamine induce a glutamate burst in the PFC via blockade of NMDA receptors located in GABAergic interneurons; the tonic firing of these interneurons, notably parvalbumin (PV) and somatostatin (SST), is driven by NMDA receptors that are more sensitive to ketamine because of activity dependent of the Mg2+ block. This leads to disinhibition of pyramidal neurons causing activation of post-synaptic AMPA receptors; this in turn induces neuronal depolarization and activation of voltage-dependent Ca2+ channels (VDCCs). The enhancement of intracellular Ca2+ influx leads to BDNF release and stimulation of TrkB receptors, which activates mTORC1 signaling inducing protein synthesis required for the formation of new spines and synaptic plasticity. Ketamine also facilitates GABA-mediated effects, increasing IPSCs, VGAT, GAD, and gephyrin in the PFC, reversing the GABA deficits caused by chronic stress exposure. (2) Likewise, α5-GABAA negative allosteric modulators (α5-NAMs) and (3) GABAB receptors antagonists, probably located in GABAergic interneurons, enhance glutamatergic neurotransmission and produce ketamine-like effects. (4) Infusions of SST or SST analogs into limbic brain regions produce antidepressant-like effects through activation of SST2 receptors. (5) Finally, activation of post-synaptic GABAB receptors by agonists or positive allosteric modulators (PAMs), as well as activation of α5-GABAA by PAMs, and other GABAA subunits by neurosteroids, notably allopregnanolone (6), can also recruit BDNF expression and signaling that could contribute to antidepressant responses (dashed arrows).
GABA Ligands
GABAA-α2 Positive Allosteric Modulators
Deletion studies of α2-containing GABAA subunit demonstrate a role of these receptors in depressive behaviors and suggest that agonists or positive modulators could produce antidepressant effects. Benzodiazepines (BZD), GABAA positive allosteric modulators developed in the 1950s, are one of the most widely used thereapeutic agents for the treatment of psychiatric disorders, due to anxiolytic actions at the GABAA-α2 receptor and hypnotic effects at the GABAA-α1 (Mohler et al., 2002). However, the efficacy of classical BZD monotherapy for the treatment of MDD has not been consistently reported, in part due to methodological confounds (i.e., small sample size, variable duration of treatment, and cotreatment with antidepressant agents), as well as high comorbidity with anxiety disorders (Pehrson and Sanchez, 2015). However, the triazolobenzodiazepine alprazolam was shown to induce significant antidepressant effects similar to tricyclic drugs in several meta-analysis studies (Jonas and Cohon, 1993; Birkenhager et al., 1995; Petty et al., 1995; van Marwijk et al., 2012). This antidepressant potential has been attributed to its differential chemical structure formed by a triazol ring fused to the diazepine ring. Recently, selective agonists or positive modulators of GABAA-containing α2/α3 subunits, such as TPA023 and eszopiclone, have been developed and are proposed as potential antidepressants (Atack, 2011; Atack et al., 2011; Vollenweider et al., 2011). When co-administered with SSRIs, eszopiclone, a preferential α2/α3-GABAA positive modulator, induced a faster onset of efficacy and greater treatment response, suggesting a synergistic effect (Fava et al., 2006, 2011; Krystal et al., 2007). Although it is still unclear if increasing the activity of GABAA receptor is effective in relieving depression symptoms, given that some BZD seem to have greater antidepressant efficacy (i.e., alprazolam versus diazepam), a more thorough understanding of the role of different subunits/subtypes of GABAA receptors could result in the development of more selective and efficacious antidepressant drugs.
GABAA-α5 Negative Allosteric Modulators
Recently, a new class of fast-acting antidepressants that specifically target the GABAA-containing α5 subunit has emerged (Atack et al., 2009; Zanos et al., 2017; Xiong et al., 2018). These receptors were shown to be up-regulated in the cortex and hippocampus of depressed patients and stressed mice (Matsumoto et al., 2007; Xiong et al., 2018). Interestingly, preclinical studies have demonstrated that both positive and negative allosteric modulators of GABAA-α5 receptors produce rapid antidepressant-like effects or prevent the behavioral responses induced by chronic stress (Zanos et al., 2017; Xiong et al., 2018). This apparent discrepancy could be due to ketamine-like induction of a glutamate burst for negative modulators and GABAA receptor dependent effects of positive modulators. In one study, MRK-016, a negative allosteric modulator of GABAA-α5 receptors and partial inverse agonist of the BZD-binding site, produced a transient increase in electroencephalogram γ power, similar to ketamine. These effects of MRK-016 were abolished by NBQX, an AMPA-type glutamate receptor antagonist, suggesting a fast recruitment of the glutamatergic system and activity-dependent effects. Importantly, perhaps because of the restricted distribution of GABAA-α5 receptors in the brain (mainly in the cortex and hippocampus), MRK-016 did not induce the typical ketamine-like side effects indicative of psychotomimetic or cognitive impairment (Zanos et al., 2017). Unfortunately, further development of this compound has been discontinued because of low tolerability in elderly subjects (Atack et al., 2009; Atack, 2011; Rudolph and Knoflach, 2011). Another GABAA-α5 negative allosteric modulator, L-655,708, also restored alterations in hedonic behaviors induced by chronic stress and the excitatory synaptic strength in the CA1 region of the hippocampus (Fischell et al., 2015). A single dose of L-655,708 increased the expression of the GluA1 subunit of the AMPA receptor, suggesting that, similar to MRK-016, it produces an indirect potentiation of excitatory synapses. Other GABAA-α5 negative modulators have been tested as cognitive enhancers in clinical trials, but these agents have not been tested for effectiveness in depression (Rudolph and Knoflach, 2011).
Surprisingly, similar antidepressant-like effects were found after acute and chronic enhancement of α5-GABAA activity by a positive modulator; however, this response was restricted to female mice and absent in males, suggesting sex-effects (Piantadosi et al., 2016). Since the behavioral sex differences could not be explained by differential pharmacokinetic effects (i.e., different brain concentrations), it is possible that α5-GABAA positive modulators interact with steroid hormones to produce an antidepressant response. Indeed, GABAA receptor subunits, such as δ, are highly sensitive to neurosteroids (see next section) and are differentially modulated across the estrous cycle (Maggi and Perez, 1986; Maguire and Mody, 2007).
Neurosteroids
Endogenous neuroactive ligands synthetized from progesterone, deoxycorticosterone, or testosterone, referred to as neurosteroids interact with a number of targets, most notably GABAA receptors and act as positive or negative allosteric modulators. Numerous preclinical evidence demonstrate that neurosteroids modulate the HPA axis and adaptive responses to stress exposure (Crowley and Girdler, 2014), and exert anxiolytic or antidepressant effects in rodent models (Khisti et al., 2000; Guidotti et al., 2001; Rodriguez-Landa et al., 2007). Specifically, the progesterone-derived neurosteroids, allopregnanolone, a potent positive allosteric modulator of both synaptic and extrasynaptic GABAA receptors, were shown to rapidly modulate BDNF expression in the rat brain (Naert et al., 2007; Nin et al., 2011; Almeida et al., 2019), which could explain its fast onset for antidepressant responses. Allopregnanolone has been tested for the treatment of post-partum depression using a formulation developed by SAGE, referred to as brexanolone. The rationale for this study is based on the precipitous drop at the time of delivery of estrogen and progesterone, and consequently a drop in allopregnanolone resulting in a loss of this key positive allosteric modulator of GABAA receptors and a withdrawal like effect. Brexanolone has been delivered intravenously and tested in two Phase II and Phase III trials. Due to its very promising results, it was recently granted a FDA Breakthrough Therapy Designation for the treatment of post-partum depression, and it has also been tested in placebo-controlled Phase III trials for the treatment of MDD (Kanes S. et al., 2017; Kanes S.J. et al., 2017; Meltzer-Brody et al., 2018; Wilkinson and Sanacora, 2018). Another compound, SAGE-217, an improved allopregnanolone formula with higher oral bioavailability and longer half-life, which can be used for once daily oral administration, successfully completed a Phase II study for MDD and also received a FDA Breakthrough Therapy Designation (Sage Therapeutics, 2017). In addition, ganaxolone (Marinus Pharmaceuticals), a neuroactive steroid that acts as a GABAA positive allosteric modulator, was initially developed for the treatment of epilepsy and anxiety, and currently is under Phase II trials for post-partum depression (Wilkinson and Sanacora, 2018).
GABAB Receptors Ligands
The first prototypical GABAB receptor agonist, bacoflen, was synthetized in 1962 and it was an invaluable pharmacological tool that influenced studies that led to the characterization of GABAB receptors in the 1980s. Years later, with the development of the first GABAB receptor antagonists, phacoflen and saclofen, additional work has lead to the development of compounds that more specifically target GABAB receptors. Given that the GABAB receptor is a heterodimer of two subunits (GABAB1 and GABAB2), that GABAB1 has been reported to have several splice variants (mainly GABAB1A and GABAB1B), and that GABAB receptors are located both pre- and post-synaptically, pharmacological studies targeting these receptors report very challenging and complex results (Bowery et al., 1980, 1981; Kaupmann et al., 1997; Cryan and Kaupmann, 2005; Jacobson et al., 2018).
Preclinical studies suggest that GABAB agonists, positive allosteric modulators, and antagonists can produce antidepressant effects; unfortunately, there are very few clinical studies due to the lack of compounds adequate for human testing (Alexander, 2017). In rats, acute administration (i.p.) of baclofen or SKF97541, both GABAB receptors agonists, or CGP7930, a GABAB positive allosteric modulator, induced antidepressant-like effects in the forced swim test, whereas chronic administration increased the escape failures in the learned helplessness test (Nakagawa et al., 1999; Frankowska et al., 2007). However, other studies failed to find significant effects for agonists or positive allosteric modulators (Nakagawa et al., 1999; Slattery et al., 2005; Nowak et al., 2006). In humans, one study reported that bacoflen intensified depressive symptoms in MDD patients (Post et al., 1991). Studies of GABAB antagonists have yielded more consistent results, with a large range of studies showing antidepressant-like effects induced by several different compounds administered either acute- or chronically, such as CGP36742 (also known as SGS742), CGP51176, CGP51176A, CGP56433A, and SCH50911 (Bittiger et al., 1993; Nakagawa et al., 1999; Mombereau et al., 2004; Slattery et al., 2005; Nowak et al., 2006; Frankowska et al., 2007). Interestingly, CGP36742 decreased learned helplessness behavior in rats (Nakagawa et al., 1999) and increased BDNF and NGF release in the cortex and hippocampus (Heese et al., 2000), as well as increased extracellular glutamate and SST in the rat hippocampus (Nyitrai et al., 1999; Nyitrai et al., 2003). Notably, this is the first GABAB receptor antagonist that underwent clinical trials for cognition-enhancing activity and improved attention in patients with mild cognitive impairment (Froestl et al., 2004). Thus, considering that: (i) MDD patients in general have an upregulation of GABAB receptors; (ii) genetic deletion of GABAB receptors produce antidepressant-like effects; (iii) GABAB receptors are implicated in the antidepressant actions of fast agents such as ketamine (Workman et al., 2013, 2015; Rosa et al., 2016); and (iv) GABAB receptors antagonists offer a promising strategy for the development of novel fast-acting antidepressants, more studies and clinical trials are warranted to identify effective and safe agents.
Neuropeptides
Because of the postmortem evidence of selective alterations of GABA interneuron subytpes, it is interesting to speculate on approaches to target the function of specific subpopulations of interneurons based on expression of selective neuropeptides. Preclinical studies demonstrate promising pharmacological evidence for two neuropeptides, NPY and SST, to treat MDD. Intraperitoneal or direct intracerebral (lateral ventricle, hippocampus, amydgala, or septum) infusions of the SST peptide or small molecule SST agonists induce anxiolytic- and antidepressant-like effects in naïve and chronically stressed rodents, as well as exert inhibitory feedback on the HPA axis (Engin et al., 2008; Engin and Treit, 2009; Yeung et al., 2011; Prevot et al., 2017). There are five (1–5) SST Gi-protein-coupled receptors that are distributed on SST-expressing GABAergic interneurons, and are mainly coupled with induction of K+ conductance leading to neuronal hyperpolarization (Jiang et al., 2003; Meis et al., 2005). The development of selective SST compounds and genetic approaches using specific SST receptor subtypes knockout animals suggest that SST2 receptor, the most abundant subtype in the brain, is a key target receptor for the antidepressant effects of SST (Viollet et al., 2000; Engin and Treit, 2009; Prevot et al., 2017). However, the plasma half-life of SST is very short making it unsuitable for clinical trials (Pinter et al., 2006; Engin and Treit, 2009).
Two more stable analogs, octreotide and lanreotide, have been tested in clinical studies to treat a wide range of diseases, such as inflammation, tumor growth, and pain (De Jong et al., 1999; Hofland et al., 1992; Carlton et al., 2004; Pinter et al., 2006); although these drugs show a high affinity to SST2 receptors, they lack selectivity, and induce a broad spectrum of undesired effects in both periphery and CNS (Pawlikowski and Melen-Mucha, 2003). Drugs that act as selective SST2 receptor agonists, such as L-779,976, have never been tested in clinical trials. Thus, given that SST levels were reported to be lower in the brain of MDD patients and stressed rodents (Frye et al., 2003; Tripp et al., 2011), which can be normalized by monoaminergic drugs (Faron-Gorecka et al., 2016, 2018), and preclinical evidence that SST induces antidepressant-like effects, clinical studies testing the antidepressant potential of selective SST2 receptor analogs with longer half-life merit additional attention.
Early preclinical studies also provided evidence that central administration of NPY induces anxiolytic- and antidepressant-like effects (Heilig and Murison, 1987; Heilig et al., 1989, 1992; Pich et al., 1993; Broqua et al., 1995; Redrobe et al., 2002a,b), as well as promotes stress adaptation and resilience (Thorsell et al., 2000; Sajdyk et al., 2008; Yang et al., 2018). In the brain, at least four subtypes of Gi-coupled receptors for NPY were identified (Y1, Y2, Y4, and Y5) (Larhammar and Salaneck, 2004) and the antidepressant-like effects of NYP are suggested to be mediated by Y1R (Redrobe et al., 2002a; Karlsson et al., 2008). Interestingly, NPY levels were decreased in treatment-resistant MDD patients (Heilig et al., 2004) and increased after treatment with SSRIs, an effect that was inversely correlated to depression severity (Nikisch et al., 2005). NPY administration in humans also represents a challenge due to short half-life, as well as undesired effects. To overcome this problem, clinical studies have focused on the therapeutic potential of intranasal NPY administration (Lacroix and Mosimann, 1996; Lacroix et al., 1996; Hallschmid et al., 2004, 2003). While a recent randomized dose-ranging study found that intranasal NPY is effective for the treatment of posttraumatic stress disorder with reduced side effects (Sayed et al., 2018), the antidepressant efficacy of intranasal NPY in MDD patients has not been tested. Given that Y1 agonists or Y2 antagonists also show promise in preclinical studies as antidepressants (Redrobe et al., 2002b), further studies of selective drugs as well as intranasal administration of NPY in MDD patients are warranted.
Cannabinoid Agents
Endocannabinoids, such as anandamide and 2-arachidonoylglycerol, are pivotal endogenous neuromodulators that control GABA and glutamate release in the brain, mainly through actions on cannabinoid type 1 (CB1) and cannabinoid type 2 (CB2) receptors (although some endocannabinoids can also activate transient receptor potential vanilloid type 1 receptors) (Fogaça et al., 2012). CB1 and CB2 are Gi/o-coupled receptors mostly located pre-synaptically, and their activation results in hyperpolarization and reduction of neurotransmitter release (Szabo and Schlicker, 2005). In the neocortex, CB1 receptors are expressed by multiple interneuron subpopulations, mostly in CCK-, but are also found in SST-, calbindin-, and VIP-expressing cells, and at lower levels in glutamatergic neurons (Hill et al., 2007; Wedzony and Chocyk, 2009). Given that (i) CB1 receptors are highly expressed in cortical and limbic regions (Pettit et al., 1998; Wang et al., 2003), (ii) CB1 receptors are expressed in cortical interneurons and glutamatergic pyramidal cells, thereby modulating both GABA and glutamate release (Hill et al., 2007), and (iii) endocannabinoids act as retrograde messengers to mediate depolarization-induced suppression of E (DSE) and I (DSI) (Diana and Marty, 2004; Hill et al., 2007), it is not surprising that the endocannabinoid system plays an important role in orchestrating cortical E:I balance and controlling stress responses. Indeed, in the mPFC, endocannabinoids contribute to the termination of HPA activity during stress responses through inhibition of GABA release, increasing the outflow of principal interneurons to target regions (Hill et al., 2011).
Cannabinoid agents have shown promise for the treatment of anxiety disorders and depression (Poleszak et al., 2018; Stampanoni Bassi et al., 2018). The most studied compound for therapeutic use is cannabidiol (CBD), the major non-psychotomimetic substance from Cannabis sativa. Although CBD has a low affinity for CB1 and CB2 receptors, it enhances endocannabinoid neurotransmission by interfering with the function of fatty acid amide hydrolase (FAAH), the enzyme responsible for anandamide degradation (Bisogno et al., 2001; De Petrocellis et al., 2011; Fogaça et al., 2018). Also, CBD acts as an allosteric modulator of 5HT1A receptors and was recently shown to exert direct actions at GABAA receptors (Russo et al., 2005; Bakas et al., 2017). Accumulating clinical and pre-clinical evidence suggests that acute and chronic administration of CBD induces anxiolytic and antidepressant effects, as well as prevents the behavioral consequences of CUS (Schiavon et al., 2016; Campos et al., 2017; Crippa et al., 2018; Fogaça et al., 2018; Sales et al., 2018). Interestingly, the rapid molecular changes induced by CBD are similar to several glutamatergic and GABAergic rapid-acting drugs discussed so far, whereas the long-term effects resemble monoaminergic drugs. For example, a single injection of CBD promotes synaptogenesis in the mPFC and induces rapid and sustained antidepressant effects through increased mTORC1/BDNF signaling (Sales et al., 2018), and repeated administration of CBD prevents the decrease in neuronal remodeling/function and hippocampal neurogenesis induced by CUS (Campos et al., 2013; Fogaça et al., 2018). In spite of these advances in the mechanism of action of CBD and other cannabinoid agents, there are very few studies that have investigated the role of the GABAergic system. Thus, more causal studies should be performed to determine the subtype of interneuron populations that mediate the anxiolytic and antidepressant effects of cannabinoid drugs, as well as other GABA-related cellular and synaptic mechanisms that could be involved.
Conclusion and Future Directions
For decades, the monoaminergic deficit hypothesis of depression was the prevalent theoretical basis for studies of the mechanisms underlying the pathophysiology and treatment of depression. However, although increased extracellular monoamines underlies the acute actions of monoamingergic agents, altered monoamine levels alone in forebrain areas are insufficient to explain the molecular and cellular changes underlying the antidepressant actions of these agents. Moreover, there is little consensus evidence that depression results from a deficit of monoamines. Thus, research has focused on neurotransmitter systems and microcircuits that can explain both the efficacy of antidepressant drugs and the etiology of MDD. Given growing consensus that MDD patients have a decrease in GABA levels in the brain and the revolutionary discovery that NMDA receptors antagonists, such as ketamine, can produce rapid and sustained antidepressant responses, efforts have been made to link the deficits in amino acid neurotransmitter systems to the pathophysiology of depression. Notably, the GABA deficit and the imbalance of cortical E:I hypothesis of depression provide a broader understanding of depression, as it offers connections with other important conceptual frameworks, such as altered glutamate and neurotrophic factor deficit hypotheses. With recent advances and new approaches, researchers have renewed enthusiasm for the development of fast-acting antidepressants that target the GABAergic and glutamatergic systems and overcome current therapeutic limitations of monoaminergic drugs. Despite recent advances, significant challenges remain, including development of more selective GABA, NMDA, and neuropeptide receptor agonists, antagonists, and modulators, characterization of optimal doses and treatment schedules, and better design of clinical trials. Moreover, genetic, chemogenetic, and optogenetic approaches should be directed to elucidate the role of specific interneuron subtypes and mechanisms underlying the control of behaviors related to mood and emotion, as well as sex-specific differences involved in these processes, with a view to developing more selective and improved antidepressant treatments.
Author Contributions
MF designed and wrote the manuscript, revised the literature, and prepared the figure and table. RD revised, edited, and approved the manuscript, figure, and table, and contributed in writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdallah, C. G., Jackowski, A., Sato, J. R., Mao, X., Kang, G., Cheema, R., et al. (2015). Prefrontal cortical GABA abnormalities are associated with reduced hippocampal volume in major depressive disorder. Eur. Neuropsychopharmacol. 25, 1082–1090. doi: 10.1016/j.euroneuro.2015.04.025
Abellan, M. T., Adell, A., Honrubia, M. A., Mengod, G., and Artigas, F. (2000). GABAB-RI receptors in serotonergic neurons: effects of baclofen on 5-HT output in rat brain. Neuroreport 11, 941–945. doi: 10.1097/00001756-200004070-00009
Akana, S. F., Chu, A., Soriano, L., and Dallman, M. F. (2001). Corticosterone exerts site-specific and state-dependent effects in prefrontal cortex and amygdala on regulation of adrenocorticotropic hormone, insulin and fat depots. J. Neuroendocrinol. 13, 625–637. doi: 10.1046/j.1365-2826.2001.00676.x
Alexander, R. C. (2017). The potential efficacy of GABAB antagonists in depression. Curr. Opin. Pharmacol. 35, 101–104. doi: 10.1016/j.coph.2017.07.009
Almeida, F. B., Gomez, R., Barros, H. M. T., and Nin, M. S. (2019). Hemisphere-dependent changes in mRNA expression of GABAA receptor subunits and BDNF after intra-prefrontal cortex allopregnanolone infusion in rats. Neuroscience 397, 56–66. doi: 10.1016/j.neuroscience.2018.11.029
Arranz, B., Cowburn, R., Eriksson, A., Vestling, M., and Marcusson, J. (1992). Gamma-aminobutyric acid-B (GABAB) binding sites in postmortem suicide brains. Neuropsychobiology 26, 33–36. doi: 10.1159/000118893
Artigas, F., Bortolozzi, A., and Celada, P. (2018). Can we increase speed and efficacy of antidepressant treatments? Part I: general aspects and monoamine-based strategies. Eur. Neuropsychopharmacol. 28, 445–456. doi: 10.1016/j.euroneuro.2017.10.032
Atack, J. R. (2011). GABAA receptor subtype-selective modulators. I. alpha2/alpha3-selective agonists as non-sedating anxiolytics. Curr. Top. Med. Chem. 11, 1176–1202. doi: 10.2174/156802611795371350
Atack, J. R., Hallett, D. J., Tye, S., Wafford, K. A., Ryan, C., Sanabria-Bohorquez, S. M., et al. (2011). Preclinical and clinical pharmacology of TPA023B, a GABAA receptor α2/α3 subtype-selective partial agonist. J. Psychopharmacol. 25, 329–344. doi: 10.1177/0269881109354928
Atack, J. R., Maubach, K. A., Wafford, K. A., O’Connor, D., Rodrigues, A. D., Evans, D. C., et al. (2009). In vitro and in vivo properties of 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5-ylmethoxy)- pyrazolo[1,5-d]-[1,2,4]triazine (MRK-016), a GABAA receptor alpha5 subtype-selective inverse agonist. J. Pharmacol. Exp. Ther. 331, 470–484. doi: 10.1124/jpet.109.157636
Autry, A. E., Adachi, M., Nosyreva, E., Na, E. S., Los, M. F., Cheng, P. F., et al. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. doi: 10.1038/nature10130
Bakas, T., van Nieuwenhuijzen, P. S., Devenish, S. O., McGregor, I. S., Arnold, J. C., and Chebib, M. (2017). The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA receptors. Pharmacol. Res. 119, 358–370. doi: 10.1016/j.phrs.2017.02.022
Banasr, M., Lepack, A., Fee, C., Duric, V., Maldonado-Aviles, J., DiLeone, R., et al. (2017). Characterization of GABAergic marker expression in the chronic unpredictable stress model of depression. Chronic Stress 1. doi: 10.1177/2470547017720459
Banerjee, R., Ghosh, A. K., Ghosh, B., Bhattacharyya, S., and Mondal, A. C. (2013). Decreased mRNA and protein expression of BDNF, NGF, and their receptors in the hippocampus from suicide: an analysis in human postmortem brain. Clin. Med. Insights Pathol. 6, 1–11. doi: 10.4137/CMPath.S12530
Bauer, M., Bschor, T., Pfennig, A., Whybrow, P. C., Angst, J., Versiani, M., et al. (2007). World federation of societies of biological psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders in primary care. World J. Biol. Psychiatry 8, 67–104. doi: 10.1080/15622970701227829
Beasley, C. L., Zhang, Z. J., Patten, I., and Reynolds, G. P. (2002). Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol. Psychiatry 52, 708–715. doi: 10.1016/S0006-3223(02)01360-4
Benham, R. S., Hewage, N. B., Suckow, R. F., Engin, E., and Rudolph, U. (2017). Prodepressant- and anxiogenic-like effects of serotonin-selective, but not noradrenaline-selective, antidepressant agents in mice lacking alpha2-containing GABAA receptors. Behav. Brain Res. 332, 172–179. doi: 10.1016/j.bbr.2017.05.063
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., Charney, D. S., et al. (2000). Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354. doi: 10.1016/S0006-3223(99)00230-9
Bhagwagar, Z., Wylezinska, M., Taylor, M., Jezzard, P., Matthews, P. M., and Cowen, P. J. (2004). Increased brain GABA concentrations following acute administration of a selective serotonin reuptake inhibitor. Am. J. Psychiatry 161, 368–370. doi: 10.1176/appi.ajp.161.2.368
Birkenhager, T. K., Moleman, P., and Nolen, W. A. (1995). Benzodiazepines for depression? A review of the literature. Int. Clin. Psychopharmacol. 10, 181–195. doi: 10.1097/00004850-199510030-00008
Bisogno, T., Hanus, L., De Petrocellis, L., Tchilibon, S., Ponde, D. E., Brandi, I., et al. (2001). Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 134, 845–852. doi: 10.1038/sj.bjp.0704327
Bittiger, H., Froestl, W., Mickel, S., and Olpe, H. R. (1993). GABAB receptor antagonists: from synthesis to therapeutic applications. Trends Pharmacol. Sci. 14, 391–394. doi: 10.1016/0165-6147(93)90056-P
Bowery, N. G., Doble, A., Hill, D. R., Hudson, A. L., Shaw, J. S., Turnbull, M. J., et al. (1981). Bicuculline-insensitive GABA receptors on peripheral autonomic nerve terminals. Eur. J. Pharmacol. 71, 53–70. doi: 10.1016/0014-2999(81)90386-1
Bowery, N. G., Hill, D. R., Hudson, A. L., Doble, A., Middlemiss, D. N., Shaw, J., et al. (1980). (-)Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature 283, 92–94. doi: 10.1038/283092a0
Braestrup, C., Nielsen, M., Krogsgaard-Larsen, P., and Falch, E. (1979). Partial agonists for brain GABA/benzodiazepine receptor complex. Nature 280,331–333. doi: 10.1038/280331a0
Broqua, P., Wettstein, J. G., Rocher, M. N., Gauthier-Martin, B., and Junien, J. L. (1995). Behavioral effects of neuropeptide Y receptor agonists in the elevated plus-maze and fear-potentiated startle procedures. Behav. Pharmacol. 6, 215–222. doi: 10.1097/00008877-199504000-00001
Brown, J. A., Ramikie, T. S., Schmidt, M. J., Baldi, R., Garbett, K., Everheart, M. G., et al. (2015). Inhibition of parvalbumin-expressing interneurons results in complex behavioral changes. Mol. Psychiatry 20, 1499–1507. doi: 10.1038/mp.2014.192
Caberlotto, L., Jimenez, P., Overstreet, D. H., Hurd, Y. L., Mathe, A. A., and Fuxe, K. (1999). Alterations in neuropeptide Y levels and Y1 binding sites in the Flinders Sensitive Line rats, a genetic animal model of depression. Neurosci. Lett. 265, 191–194. doi: 10.1016/S0304-3940(99)00234-7
Caldji, C., Diorio, J., and Meaney, M. J. (2000). Variations in maternal care in infancy regulate the development of stress reactivity. Biol. Psychiatry 48, 1164–1174. doi: 10.1016/S0006-3223(00)01084-2
Caldji, C., Diorio, J., and Meaney, M. J. (2003). Variations in maternal care alter GABA(A) receptor subunit expression in brain regions associated with fear. Neuropsychopharmacology 28, 1950–1959. doi: 10.1038/sj.npp.1300237
Campos, A. C., Fogaca, M. V., Scarante, F. F., Joca, S. R. L., Sales, A. J., Gomes, F. V., et al. (2017). Plastic and neuroprotective mechanisms involved in the therapeutic effects of cannabidiol in psychiatric disorders. Front. Pharmacol. 8:269. doi: 10.3389/fphar.2017.00269
Campos, A. C., Ortega, Z., Palazuelos, J., Fogaca, M. V., Aguiar, D. C., Diaz-Alonso, J., et al. (2013). The anxiolytic effect of cannabidiol on chronically stressed mice depends on hippocampal neurogenesis: involvement of the endocannabinoid system. Int. J. Neuropsychopharmacol. 16, 1407–1419. doi: 10.1017/S1461145712001502
Carlton, S. M., Zhou, S., Du, J., Hargett, G. L., Ji, G., and Coggeshall, R. E. (2004). Somatostatin modulates the transient receptor potential vanilloid 1 (TRPV1) ion channel. Pain 110, 616–627. doi: 10.1016/j.pain.2004.04.042
Celada, P., Puig, M. V., and Artigas, F. (2013). Serotonin modulation of cortical neurons and networks. Front. Integr. Neurosci. 7:25. doi: 10.3389/fnint.2013.00025
Celada, P., Puig, M. V., Casanovas, J. M., Guillazo, G., and Artigas, F. (2001). Control of dorsal raphe serotonergic neurons by the medial prefrontal cortex: involvement of serotonin-1A, GABA(A), and glutamate receptors. J. Neurosci. 21, 9917–9929. doi: 10.1523/JNEUROSCI.21-24-09917.2001
Chandra, D., Korpi, E. R., Miralles, C. P., De Blas, A. L., and Homanics, G. E. (2005). GABAA receptor gamma 2 subunit knockdown mice have enhanced anxiety-like behavior but unaltered hypnotic response to benzodiazepines. BMC Neurosci. 6:30. doi: 10.1186/1471-2202-6-30
Choudary, P. V., Molnar, M., Evans, S. J., Tomita, H., Li, J. Z., Vawter, M. P., et al. (2005). Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. U.S.A. 102, 15653–15658. doi: 10.1073/pnas.0507901102
Cotter, D., Landau, S., Beasley, C., Stevenson, R., Chana, G., MacMillan, L., et al. (2002). The density and spatial distribution of GABAergic neurons, labelled using calcium binding proteins, in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia. Biol. Psychiatry 51, 377–386. doi: 10.1016/S0006-3223(01)01243-4
Crestani, F., Lorez, M., Baer, K., Essrich, C., Benke, D., Laurent, J. P., et al. (1999). Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat. Neurosci. 2, 833–839. doi: 10.1038/12207
Crippa, J. A., Guimaraes, F. S., Campos, A. C., and Zuardi, A. W. (2018). Translational investigation of the therapeutic potential of cannabidiol (CBD): toward a new age. Front. Immunol. 9:2009. doi: 10.3389/fimmu.2018.02009
Cross, J. A., Cheetham, S. C., Crompton, M. R., Katona, C. L., and Horton, R. W. (1988). Brain GABAB binding sites in depressed suicide victims. Psychiatry Res. 26, 119–129. doi: 10.1016/0165-1781(88)90066-2
Crowley, S. K., and Girdler, S. S. (2014). Neurosteroid, GABAergic and hypothalamic pituitary adrenal (HPA) axis regulation: what is the current state of knowledge in humans? Psychopharmacology 231, 3619–3634. doi: 10.1007/s00213-014-3572-8
Cryan, J. F., and Kaupmann, K. (2005). Don’t worry ’B’ happy!: a role for GABA(B) receptors in anxiety and depression. Trends Pharmacol. Sci. 26, 36–43. doi: 10.1016/j.tips.2004.11.004
Csabai, D., Seress, L., Varga, Z., Abraham, H., Miseta, A., Wiborg, O., et al. (2017). Electron microscopic analysis of hippocampal Axo-somatic synapses in a chronic stress model for depression. Hippocampus 27, 17–27. doi: 10.1002/hipo.22650
Czéh, B., Vardya, I., Varga, Z., Febbraro, F., Csabai, D., Martis, L. S., et al. (2018). Long-term stress disrupts the structural and functional integrity of GABAergic neuronal networks in the medial prefrontal cortex of rats. Front. Cell. Neurosci. 12:148. doi: 10.3389/fncel.2018.00148
Czéh, B., Varga, Z. K., Henningsen, K., Kovacs, G. L., Miseta, A., and Wiborg, O. (2015). Chronic stress reduces the number of GABAergic interneurons in the adult rat hippocampus, dorsal-ventral and region-specific differences. Hippocampus 25, 393–405. doi: 10.1002/hipo.22382
De Jong, M., Breeman, W. A., Bernard, H. F., Kooij, P. P., Slooter, G. D., Van Eijck, C. H., et al. (1999). Therapy of neuroendocrine tumors with radiolabeled somatostatin-analogues. Q. J. Nucl. Med. 43, 356–366.
De Petrocellis, L., Ligresti, A., Moriello, A. S., Allara, M., Bisogno, T., Petrosino, S., et al. (2011). Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 163, 1479–1494. doi: 10.1111/j.1476-5381.2010.01166.x
DeFelipe, J., Lopez-Cruz, P. L., Benavides-Piccione, R., Bielza, C., Larranaga, P., Anderson, S., et al. (2013). New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 14, 202–216. doi: 10.1038/nrn3444
Diana, M. A., and Marty, A. (2004). Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br. J. Pharmacol. 142, 9–19. doi: 10.1038/sj.bjp.0705726
Diorio, D., Viau, V., and Meaney, M. J. (1993). The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J. Neurosci. 13, 3839–3847. doi: 10.1523/JNEUROSCI.13-09-03839.1993
Dong, E., Matsumoto, K., Uzunova, V., Sugaya, I., Takahata, H., Nomura, H., et al. (2001). Brain 5alpha-dihydroprogesterone and allopregnanolone synthesis in a mouse model of protracted social isolation. Proc. Natl. Acad. Sci. U.S.A. 98, 2849–2854. doi: 10.1073/pnas.051628598
Douillard-Guilloux, G., Lewis, D., Seney, M. L., and Sibille, E. (2017). Decrease in somatostatin-positive cell density in the amygdala of females with major depression. Depress. Anxiety 34, 68–78. doi: 10.1002/da.22549
Drugan, R. C., Morrow, A. L., Weizman, R., Weizman, A., Deutsch, S. I., Crawley, J. N., et al. (1989). Stress-induced behavioral depression in the rat is associated with a decrease in GABA receptor-mediated chloride ion flux and brain benzodiazepine receptor occupancy. Brain Res. 487, 45–51. doi: 10.1016/0006-8993(89)90938-4
Dubin, M. J., Mao, X., Banerjee, S., Goodman, Z., Lapidus, K. A., Kang, G., et al. (2016). Elevated prefrontal cortex GABA in patients with major depressive disorder after TMS treatment measured with proton magnetic resonance spectroscopy. J. Psychiatry Neurosci. 41, E37–E45. doi: 10.1503/jpn.150223
Duman, R. S., Aghajanian, G. K., Sanacora, G., and Krystal, J. H. (2016). Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med. 22, 238–249. doi: 10.1038/nm.4050
Dwivedi, Y., Rizavi, H. S., Conley, R. R., Roberts, R. C., Tamminga, C. A., and Pandey, G. N. (2003). Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 60, 804–815. doi: 10.1001/archpsyc.60.8.804
Engin, E., Benham, R. S., and Rudolph, U. (2018). An emerging circuit pharmacology of GABAA receptors. Trends Pharmacol. Sci. 39, 710–732. doi: 10.1016/j.tips.2018.04.003
Engin, E., Stellbrink, J., Treit, D., and Dickson, C. T. (2008). Anxiolytic and antidepressant effects of intracerebroventricularly administered somatostatin: behavioral and neurophysiological evidence. Neuroscience 157, 666–676. doi: 10.1016/j.neuroscience.2008.09.037
Engin, E., and Treit, D. (2009). Anxiolytic and antidepressant actions of somatostatin: the role of sst2 and sst3 receptors. Psychopharmacology 206, 281–289. doi: 10.1007/s00213-009-1605-5
Faron-Gorecka, A., Kusmider, M., Kolasa, M., Zurawek, D., Szafran-Pilch, K., Gruca, P., et al. (2016). Chronic mild stress alters the somatostatin receptors in the rat brain. Psychopharmacology 233, 255–266. doi: 10.1007/s00213-015-4103-y
Faron-Gorecka, A., Kusmider, M., Solich, J., Kolasa, M., Pabian, P., Gruca, P., et al. (2018). Regulation of somatostatin receptor 2 in the context of antidepressant treatment response in chronic mild stress in rat. Psychopharmacology 235, 2137–2149. doi: 10.1007/s00213-018-4912-x
Fatemi, S. H., Folsom, T. D., and Thuras, P. D. (2011). Deficits in GABA(B) receptor system in schizophrenia and mood disorders: a postmortem study. Schizophr. Res. 128, 37–43. doi: 10.1016/j.schres.2010.12.025
Fava, M., McCall, W. V., Krystal, A., Wessel, T., Rubens, R., Caron, J., et al. (2006). Eszopiclone co-administered with fluoxetine in patients with insomnia coexisting with major depressive disorder. Biol. Psychiatry 59, 1052–1060. doi: 10.1016/j.biopsych.2006.01.016
Fava, M., Schaefer, K., Huang, H., Wilson, A., Iosifescu, D. V., Mischoulon, D., et al. (2011). A post hoc analysis of the effect of nightly administration of eszopiclone and a selective serotonin reuptake inhibitor in patients with insomnia and anxious depression. J. Clin. Psychiatry 72, 473–479. doi: 10.4088/JCP.09m05131gry
Fee, C., Banasr, M., and Sibille, E. (2017). Somatostatin-positive gamma-aminobutyric acid interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol. Psychiatry 82, 549–559. doi: 10.1016/j.biopsych.2017.05.024
Ferguson, B. R., and Gao, W. J. (2018). PV interneurons: critical regulators of E/I balance for prefrontal cortex-dependent behavior and psychiatric disorders. Front. Neural Circuits 12:37. doi: 10.3389/fncir.2018.00037
Filipovic, D., Zlatkovic, J., Gass, P., and Inta, D. (2013). The differential effects of acute vs. chronic stress and their combination on hippocampal parvalbumin and inducible heat shock protein 70 expression. Neuroscience 236, 47–54. doi: 10.1016/j.neuroscience.2013.01.033
Fischell, J., Van Dyke, A. M., Kvarta, M. D., LeGates, T. A., and Thompson, S. M. (2015). Rapid antidepressant action and restoration of excitatory synaptic strength after chronic stress by negative modulators of alpha5-containing GABAA receptors. Neuropsychopharmacology 40, 2499–2509. doi: 10.1038/npp.2015.112
Fogaça, M. V., Campos, A. C., Coelho, L. D., Duman, R. S., and Guimaraes, F. S. (2018). The anxiolytic effects of cannabidiol in chronically stressed mice are mediated by the endocannabinoid system: role of neurogenesis and dendritic remodeling. Neuropharmacology 135, 22–33. doi: 10.1016/j.neuropharm.2018.03.001
Fogaça, M. V., Lisboa, S. F., Aguiar, D. C., Moreira, F. A., Gomes, F. V., Casarotto, P. C., et al. (2012). Fine-tuning of defensive behaviors in the dorsal periaqueductal gray by atypical neurotransmitters. Braz. J. Med. Biol. Res. 45, 357–365. doi: 10.1590/S0100-879X2012007500029
Frankowska, M., Filip, M., and Przegalinski, E. (2007). Effects of GABAB receptor ligands in animal tests of depression and anxiety. Pharmacol. Rep. 59, 645–655.
Froestl, W., Gallagher, M., Jenkins, H., Madrid, A., Melcher, T., Teichman, S., et al. (2004). SGS742: the first GABAB receptor antagonist in clinical trials. Biochem. Pharmacol. 68, 1479–1487. doi: 10.1016/j.bcp.2004.07.030
Frye, M. A., Pazzaglia, P. J., George, M. S., Luckenbaugh, D. A., Vanderham, E., Davis, C. L., et al. (2003). Low CSF somatostatin associated with response to nimodipine in patents with affective illness. Biol. Psychiatry 53, 180–183. doi: 10.1016/S0006-3223(02)01343-4
Fuchs, T., Jefferson, S. J., Hooper, A., Yee, P. H., Maguire, J., and Luscher, B. (2017). Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state. Mol. Psychiatry 22, 920–930. doi: 10.1038/mp.2016.188
Fukumoto, K., Fogaca, M. V., Liu, R. J., Duman, C., Kato, T., Li, X. Y., et al. (2019). Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc. Natl. Acad. Sci. U.S.A. 116, 297–302. doi: 10.1073/pnas.1814709116
Gabbay, V., Mao, X., Klein, R. G., Ely, B. A., Babb, J. S., Panzer, A. M., et al. (2012). Anterior cingulate cortex gamma-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch. Gen. Psychiatry 69, 139–149. doi: 10.1001/archgenpsychiatry.2011.131
Gerner, R. H., and Hare, T. A. (1981). CSF GABA in normal subjects and patients with depression, schizophrenia, mania, and anorexia nervosa. Am. J. Psychiatry 138, 1098–1101. doi: 10.1176/ajp.138.8.1098
Ghosal, S., Bang, E., Yue, W., Hare, B. D., Lepack, A. E., Girgenti, M. J., et al. (2018). Activity-dependent brain-derived neurotrophic factor release is required for the rapid antidepressant actions of scopolamine. Biol. Psychiatry 83, 29–37. doi: 10.1016/j.biopsych.2017.06.017
Ghosal, S., Hare, B., and Duman, R. S. (2017). Prefrontal cortex GABAergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Curr. Opin. Behav. Sci. 14, 1–8. doi: 10.1016/j.cobeha.2016.09.012
Gilabert-Juan, J., Castillo-Gomez, E., Guirado, R., Molto, M. D., and Nacher, J. (2013). Chronic stress alters inhibitory networks in the medial prefrontal cortex of adult mice. Brain Struct. Funct. 218, 1591–1605. doi: 10.1007/s00429-012-0479-1
Gilabert-Juan, J., Varea, E., Guirado, R., Blasco-Ibanez, J. M., Crespo, C., and Nacher, J. (2012). Alterations in the expression of PSA-NCAM and synaptic proteins in the dorsolateral prefrontal cortex of psychiatric disorder patients. Neurosci. Lett. 530, 97–102. doi: 10.1016/j.neulet.2012.09.032
Godfrey, K. E. M., Gardner, A. C., Kwon, S., Chea, W., and Muthukumaraswamy, S. D. (2018). Differences in excitatory and inhibitory neurotransmitter levels between depressed patients and healthy controls: a systematic review and meta-analysis. J. Psychiatr. Res. 105, 33–44. doi: 10.1016/j.jpsychires.2018.08.015
Gold, B. I., Bowers, M. B. Jr., Roth, R. H., and Sweeney, D. W. (1980). GABA levels in CSF of patients with psychiatric disorders. Am. J. Psychiatry 137, 362–364. doi: 10.1176/ajp.137.3.362
Goren, M. Z., Kucukibrahimoglu, E., Berkman, K., and Terzioglu, B. (2007). Fluoxetine partly exerts its actions through GABA: a neurochemical evidence. Neurochem. Res. 32, 1559–1565. doi: 10.1007/s11064-007-9357-2
Gray, J. A., and Green, A. R. (1987). Increased GABAB receptor function in mouse frontal cortex after repeated administration of antidepressant drugs or electroconvulsive shocks. Br. J. Pharmacol. 92, 357–362. doi: 10.1111/j.1476-5381.1987.tb11331.x
Griffin, L. D., and Mellon, S. H. (1999). Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc. Natl. Acad. Sci. U.S.A. 96, 13512–13517. doi: 10.1073/pnas.96.23.13512
Guidotti, A., Dong, E., Matsumoto, K., Pinna, G., Rasmusson, A. M., and Costa, E. (2001). The socially-isolated mouse: a model to study the putative role of allopregnanolone and 5alpha-dihydroprogesterone in psychiatric disorders. Brain Res. Brain Res. Rev. 37, 110–115. doi: 10.1016/S0165-0173(01)00129-1
Guilloux, J. P., Douillard-Guilloux, G., Kota, R., Wang, X., Gardier, A. M., Martinowich, K., et al. (2012). Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol. Psychiatry 17, 1130–1142. doi: 10.1038/mp.2011.113
Hallschmid, M., Benedict, C., Born, J., Fehm, H. L., and Kern, W. (2004). Manipulating central nervous mechanisms of food intake and body weight regulation by intranasal administration of neuropeptides in man. Physiol. Behav. 83, 55–64. doi: 10.1016/S0031-9384(04)00349-X
Hallschmid, M., Gais, S., Meinert, S., and Born, J. (2003). NPY attenuates positive cortical DC-potential shift upon food intake in man. Psychoneuroendocrinology 28, 529–539. doi: 10.1016/S0306-4530(02)00038-0
Harmer, C. J., Duman, R. S., and Cowen, P. J. (2017). How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry 4, 409–418. doi: 10.1016/S2215-0366(17)30015-9
Hasler, G., van der Veen, J. W., Grillon, C., Drevets, W. C., and Shen, J. (2010). Effect of acute psychological stress on prefrontal GABA concentration determined by proton magnetic resonance spectroscopy. Am. J. Psychiatry 167, 1226–1231. doi: 10.1176/appi.ajp.2010.09070994
Hasler, G., van der Veen, J. W., Tumonis, T., Meyers, N., Shen, J., and Drevets, W. C. (2007). Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 64, 193–200. doi: 10.1001/archpsyc.64.2.193
Heese, K., Otten, U., Mathivet, P., Raiteri, M., Marescaux, C., and Bernasconi, R. (2000). GABA(B) receptor antagonists elevate both mRNA and protein levels of the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) but not neurotrophin-3 (NT-3) in brain and spinal cord of rats. Neuropharmacology 39, 449–462. doi: 10.1016/S0028-3908(99)00166-5
Heilig, M., McLeod, S., Koob, G. K., and Britton, K. T. (1992). Anxiolytic-like effect of neuropeptide Y (NPY), but not other peptides in an operant conflict test. Regul. Pept. 41, 61–69. doi: 10.1016/0167-0115(92)90514-U
Heilig, M., and Murison, R. (1987). Intracerebroventricular neuropeptide Y protects against stress-induced gastric erosion in the rat. Eur. J. Pharmacol. 137, 127–129. doi: 10.1016/0014-2999(87)90191-9
Heilig, M., Soderpalm, B., Engel, J. A., and Widerlov, E. (1989). Centrally administered neuropeptide Y (NPY) produces anxiolytic-like effects in animal anxiety models. Psychopharmacology 98, 524–529. doi: 10.1007/BF00441953
Heilig, M., Zachrisson, O., Thorsell, A., Ehnvall, A., Mottagui-Tabar, S., Sjögren, M., et al. (2004). Decreased cerebrospinal fluid neuropeptide Y (NPY) in patients with treatment refractory unipolar major depression: preliminary evidence for association with preproNPY gene polymorphism. J. Psychiatr. Res. 38, 113–121. doi: 10.1016/S0022-3956(03)00101-8
Heinzel, A., Steinke, R., Poeppel, T. D., Grosser, O., Bogerts, B., Otto, H., et al. (2008). S-ketamine and GABA-A-receptor interaction in humans: an exploratory study with I-123-iomazenil SPECT. Hum. Psychopharmacol. 23, 549–554. doi: 10.1002/hup.960
Hewitt, S. A., Wamsteeker, J. I., Kurz, E. U., and Bains, J. S. (2009). Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 12, 438–443. doi: 10.1038/nn.2274
Hill, E. L., Gallopin, T., Ferezou, I., Cauli, B., Rossier, J., Schweitzer, P., et al. (2007). Functional CB1 receptors are broadly expressed in neocortical GABAergic and glutamatergic neurons. J. Neurophysiol. 97, 2580–2589. doi: 10.1152/jn.00603.2006
Hill, M. N., McLaughlin, R. J., Pan, B., Fitzgerald, M. L., Roberts, C. J., Lee, T. T., et al. (2011). Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J. Neurosci. 31, 10506–10515. doi: 10.1523/JNEUROSCI.0496-11.2011
Hofland, L. J., van Koetsveld, P. M., Wouters, N., Waaijers, M., Reubi, J. C., and Lamberts, S. W. (1992). Dissociation of antiproliferative and antihormonal effects of the somatostatin analog octreotide on 7315b pituitary tumor cells. Endocrinology 131, 571–577.
Jacobson, L. H., Vlachou, S., Slattery, D. A., Li, X., and Cryan, J. F. (2018). The gamma-aminobutyric acid b receptor in depression and reward. Biol. Psychiatry 83, 963–976. doi: 10.1016/j.biopsych.2018.02.006
Jiang, N., Furue, H., Katafuchi, T., and Yoshimura, M. (2003). Somatostatin directly inhibits substantia gelatinosa neurons in adult rat spinal dorsal horn in vitro. Neurosci. Res. 47, 97–107. doi: 10.1016/S0168-0102(03)00183-4
Jonas, J. M., and Cohon, M. S. (1993). A comparison of the safety and efficacy of alprazolam versus other agents in the treatment of anxiety, panic, and depression: a review of the literature. J. Clin. Psychiatry 54(Suppl.), 25–45; discussion 46–28.
Jovanovic, J. N., Thomas, P., Kittler, J. T., Smart, T. G., and Moss, S. J. (2004). Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J. Neurosci. 24, 522–530. doi: 10.1523/JNEUROSCI.3606-03.2004
Kanes, S., Colquhoun, H., Gunduz-Bruce, H., Raines, S., Arnold, R., Schacterle, A., et al. (2017). Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet 390, 480–489. doi: 10.1016/S0140-6736(17)31264-3
Kanes, S. J., Colquhoun, H., Doherty, J., Raines, S., Hoffmann, E., Rubinow, D. R., et al. (2017). Open-label, proof-of-concept study of brexanolone in the treatment of severe postpartum depression. Hum. Psychopharmacol. 32:e2576. doi: 10.1002/hup.2576
Karch, D. L., Dahlberg, L. L., Patel, N., Davis, T. W., Logan, J. E., Hill, H. A., et al. (2009). Surveillance for violent deaths–national violent death reporting system, 16 States, 2006. MMWR Surveill. Summ. 58, 1–44.
Karlsson, R. M., Choe, J. S., Cameron, H. A., Thorsell, A., Crawley, J. N., Holmes, A., et al. (2008). The neuropeptide Y Y1 receptor subtype is necessary for the anxiolytic-like effects of neuropeptide Y, but not the antidepressant-like effects of fluoxetine, in mice. Psychopharmacology 195, 547–557. doi: 10.1007/s00213-007-0945-2
Karolewicz, B., Maciag, D., O’Dwyer, G., Stockmeier, C. A., Feyissa, A. M., and Rajkowska, G. (2010). Reduced level of glutamic acid decarboxylase-67 kDa in the prefrontal cortex in major depression. Int. J. Neuropsychopharmacol. 13, 411–420. doi: 10.1017/S1461145709990587
Kato, T., Fogaca, M. V., Deyama, S., Li, X. Y., Fukumoto, K., and Duman, R. S. (2018). BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol. Psychiatry 23, 2007–2017. doi: 10.1038/mp.2017.220
Kaupmann, K., Huggel, K., Heid, J., Flor, P. J., Bischoff, S., Mickel, S. J., et al. (1997). Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature 386, 239–246. doi: 10.1038/386239a0
Kessler, R. C., Chiu, W. T., Demler, O., Merikangas, K. R., and Walters, E. E. (2005). Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 62, 617–627. doi: 10.1001/archpsyc.62.6.617
Khisti, R. T., Chopde, C. T., and Jain, S. P. (2000). Antidepressant-like effect of the neurosteroid 3alpha-hydroxy-5alpha-pregnan-20-one in mice forced swim test. Pharmacol. Biochem. Behav. 67, 137–143. doi: 10.1016/S0091-3057(00)00300-2
Kim, Y. K., Lee, H. P., Won, S. D., Park, E. Y., Lee, H. Y., Lee, B. H., et al. (2007). Low plasma BDNF is associated with suicidal behavior in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 78–85.
Klempan, T. A., Sequeira, A., Canetti, L., Lalovic, A., Ernst, C., ffrench-Mullen, J., et al. (2009). Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol. Psychiatry 14, 175–189. doi: 10.1038/sj.mp.4002110
Krystal, A., Fava, M., Rubens, R., Wessel, T., Caron, J., Wilson, P., et al. (2007). Evaluation of eszopiclone discontinuation after cotherapy with fluoxetine for insomnia with coexisting depression. J. Clin. Sleep Med. 3, 48–55.
Kucukibrahimoglu, E., Saygin, M. Z., Caliskan, M., Kaplan, O. K., Unsal, C., and Goren, M. Z. (2009). The change in plasma GABA, glutamine and glutamate levels in fluoxetine- or S-citalopram-treated female patients with major depression. Eur. J. Clin. Pharmacol. 65, 571–577. doi: 10.1007/s00228-009-0650-7
Kuehner, C. (2017). Why is depression more common among women than among men? Lancet Psychiatry 4, 146–158. doi: 10.1016/S2215-0366(16)30263-2
Lacroix, J. S., and Mosimann, B. L. (1996). Attenuation of allergen-evoked nasal responses by local pretreatment with exogenous neuropeptide Y in atopic patients. J. Allergy Clin. Immunol. 98, 611–616. doi: 10.1016/S0091-6749(96)70095-7
Lacroix, J. S., Ricchetti, A. P., Morel, D., Mossimann, B., Waeber, B., and Grouzmann, E. (1996). Intranasal administration of neuropeptide Y in man: systemic absorption and functional effects. Br. J. Pharmacol. 118, 2079–2084. doi: 10.1111/j.1476-5381.1996.tb15647.x
Larhammar, D., and Salaneck, E. (2004). Molecular evolution of NPY receptor subtypes. Neuropeptides 38, 141–151. doi: 10.1016/j.npep.2004.06.002
Lepack, A. E., Bang, E., Lee, B., Dwyer, J. M., and Duman, R. S. (2016). Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 111, 242–252. doi: 10.1016/j.neuropharm.2016.09.011
Lepack, A. E., Fuchikami, M., Dwyer, J. M., Banasr, M., and Duman, R. S. (2014). BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 18:yu033. doi: 10.1093/ijnp/pyu033
Li, N., Lee, B., Liu, R. J., Banasr, M., Dwyer, J. M., Iwata, M., et al. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964. doi: 10.1126/science.1190287
Lin, L. C., and Sibille, E. (2015). Somatostatin, neuronal vulnerability and behavioral emotionality. Mol. Psychiatry 20, 377–387. doi: 10.1038/mp.2014.184
Liu, C. Y., Jiang, X. X., Zhu, Y. H., and Wei, D. N. (2012). Metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine produces antidepressant effects in rats: role of brain-derived neurotrophic factor. Neuroscience 223, 219–224. doi: 10.1016/j.neuroscience.2012.08.010
Llado-Pelfort, L., Santana, N., Ghisi, V., Artigas, F., and Celada, P. (2012). 5-HT1A receptor agonists enhance pyramidal cell firing in prefrontal cortex through a preferential action on GABA interneurons. Cereb. Cortex 22, 1487–1497. doi: 10.1093/cercor/bhr220
Lloyd, K. G., Thuret, F., and Pilc, A. (1985). Upregulation of gamma-aminobutyric acid (GABA) B binding sites in rat frontal cortex: a common action of repeated administration of different classes of antidepressants and electroshock. J. Pharmacol. Exp. Ther. 235, 191–199.
Luscher, B., Shen, Q., and Sahir, N. (2011). The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 16, 383–406. doi: 10.1038/mp.2010.120
Ma, K., Xu, A., Cui, S., Sun, M. R., Xue, Y. C., and Wang, J. H. (2016). Impaired GABA synthesis, uptake and release are associated with depression-like behaviors induced by chronic mild stress. Transl. Psychiatry 6:e910. doi: 10.1038/tp.2016.181
Maciag, D., Hughes, J., O’Dwyer, G., Pride, Y., Stockmeier, C. A., Sanacora, G., et al. (2010). Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biol. Psychiatry 67, 465–470. doi: 10.1016/j.biopsych.2009.10.027
MacQueen, G. M., Yucel, K., Taylor, V. H., Macdonald, K., and Joffe, R. (2008). Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol. Psychiatry 64, 880–883. doi: 10.1016/j.biopsych.2008.06.027
Maggi, A., and Perez, J. (1986). Estrogen-induced up-regulation of gamma-aminobutyric acid receptors in the CNS of rodents. J. Neurochem. 47,1793–1797. doi: 10.1111/j.1471-4159.1986.tb13090.x
Maguire, J., and Mody, I. (2007). Neurosteroid synthesis-mediated regulation of GABA(A) receptors: relevance to the ovarian cycle and stress. J. Neurosci. 27, 2155–2162. doi: 10.1523/JNEUROSCI.4945-06.2007
Markram, H., Toledo-Rodriguez, M., Wang, Y., Gupta, A., Silberberg, G., and Wu, C. (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807. doi: 10.1038/nrn1519
Martin, P., Pichat, P., Massol, J., Soubrie, P., Lloyd, K. G., and Puech, A. J. (1989). Decreased GABA B receptors in helpless rats: reversal by tricyclic antidepressants. Neuropsychobiology 22, 220–224. doi: 10.1159/000118620
Matsumoto, K., Puia, G., Dong, E., and Pinna, G. (2007). GABA(A) receptor neurotransmission dysfunction in a mouse model of social isolation-induced stress: possible insights into a non-serotonergic mechanism of action of SSRIs in mood and anxiety disorders. Stress 10, 3–12. doi: 10.1080/10253890701200997
McKlveen, J. M., Morano, R. L., Fitzgerald, M., Zoubovsky, S., Cassella, S. N., Scheimann, J. R., et al. (2016). Chronic stress increases prefrontal inhibition: a mechanism for stress-induced prefrontal dysfunction. Biol. Psychiatry 80, 754–764. doi: 10.1016/j.biopsych.2016.03.2101
Meis, S., Sosulina, L., Schulz, S., Hollt, V., and Pape, H. C. (2005). Mechanisms of somatostatin-evoked responses in neurons of the rat lateral amygdala. Eur. J. Neurosci. 21, 755–762. doi: 10.1111/j.1460-9568.2005.03922.x
Melas, P. A., Mannervik, M., Mathe, A. A., and Lavebratt, C. (2012). Neuropeptide Y: identification of a novel rat mRNA splice-variant that is downregulated in the hippocampus and the prefrontal cortex of a depression-like model. Peptides 35, 49–55. doi: 10.1016/j.peptides.2012.02.020
Meltzer-Brody, S., Colquhoun, H., Riesenberg, R., Epperson, C. N., Deligiannidis, K. M., Rubinow, D. R., et al. (2018). Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet 392, 1058–1070. doi: 10.1016/S0140-6736(18)31551-4
Merali, Z., Du, L., Hrdina, P., Palkovits, M., Faludi, G., Poulter, M. O., et al. (2004). Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J. Neurosci. 24, 1478–1485. doi: 10.1523/JNEUROSCI.4734-03.2004
Milak, M. S., Proper, C. J., Mulhern, S. T., Parter, A. L., Kegeles, L. S., Ogden, R. T., et al. (2016). A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol. Psychiatry 21, 320–327. doi: 10.1038/mp.2015.83
Moghaddam, B., Adams, B., Verma, A., and Daly, D. (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 17, 2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997
Mohler, H., Fritschy, J. M., and Rudolph, U. (2002). A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther. 300, 2–8. doi: 10.1124/jpet.300.1.2
Mombereau, C., Kaupmann, K., Froestl, W., Sansig, G., van der Putten, H., and Cryan, J. F. (2004). Genetic and pharmacological evidence of a role for GABA(B) receptors in the modulation of anxiety- and antidepressant-like behavior. Neuropsychopharmacology 29, 1050–1062. doi: 10.1038/sj.npp.1300413
Naert, G., Maurice, T., Tapia-Arancibia, L., and Givalois, L. (2007). Neuroactive steroids modulate HPA axis activity and cerebral brain-derived neurotrophic factor (BDNF) protein levels in adult male rats. Psychoneuroendocrinology 32, 1062–1078. doi: 10.1016/j.psyneuen.2007.09.002
Nakagawa, Y., Sasaki, A., and Takashima, T. (1999). The GABA(B) receptor antagonist CGP36742 improves learned helplessness in rats. Eur. J. Pharmacol. 381, 1–7. doi: 10.1016/S0014-2999(99)00567-1
Nikisch, G., Agren, H., Eap, C. B., Czernik, A., Baumann, P., and Mathe, A. A. (2005). Neuropeptide Y and corticotropin-releasing hormone in CSF mark response to antidepressive treatment with citalopram. Int. J. Neuropsychopharmacol. 8, 403–410. doi: 10.1017/S1461145705005158
Nin, M. S., Martinez, L. A., Pibiri, F., Nelson, M., and Pinna, G. (2011). Neurosteroids reduce social isolation-induced behavioral deficits: a proposed link with neurosteroid-mediated upregulation of BDNF expression. Front. Endocrinol. 2:73. doi: 10.3389/fendo.2011.00073
Northoff, G., and Sibille, E. (2014). Why are cortical GABA neurons relevant to internal focus in depression? A cross-level model linking cellular, biochemical and neural network findings. Mol. Psychiatry 19, 966–977. doi: 10.1038/mp.2014.68
Nowak, B., Zadrozna, M., Ossowska, G., Sowa-Kucma, M., Gruca, P., Papp, M., et al. (2010). Alterations in hippocampal calcium-binding neurons induced by stress models of depression: a preliminary assessment. Pharmacol. Rep. 62, 1204–1210. doi: 10.1016/S1734-1140(10)70383-2
Nowak, G., Partyka, A., Palucha, A., Szewczyk, B., Wieronska, J. M., Dybala, M., et al. (2006). Antidepressant-like activity of CGP 36742 and CGP 51176, selective GABAB receptor antagonists, in rodents. Br. J. Pharmacol. 149,581–590. doi: 10.1038/sj.bjp.0706845
Nyitrai, G., Kekesi, K. A., Emri, Z., Szarics, E., Juhasz, G., and Kardos, J. (2003). GABA(B) receptor antagonist CGP-36742 enhances somatostatin release in the rat hippocampus in vivo and in vitro. Eur. J. Pharmacol. 478, 111–119. doi: 10.1016/j.ejphar.2003.08.006
Nyitrai, G., Szarics, E., Kovacs, I., Kekesi, K. A., Juhasz, G., and Kardos, J. (1999). Effect of CGP 36742 on the extracellular level of neurotransmitter amino acids in the thalamus. Neurochem. Int. 34, 391–398. doi: 10.1016/S0197-0186(99)00042-X
Ordway, G. A., Stockmeier, C. A., Meltzer, H. Y., Overholser, J. C., Jaconetta, S., and Widdowson, P. S. (1995). Neuropeptide Y in frontal cortex is not altered in major depression. J. Neurochem. 65, 1646–1650. doi: 10.1046/j.1471-4159.1995.65041646.x
Otero Losada, M. E. (1988). Changes in central GABAergic function following acute and repeated stress. Br. J. Pharmacol. 93, 483–490. doi: 10.1111/j.1476-5381.1988.tb10302.x
Pawlikowski, M., and Melen-Mucha, G. (2003). Perspectives of new potential therapeutic applications of somatostatin analogs. Neuro Endocrinol. Lett. 24, 21–27.
Pehrson, A. L., and Sanchez, C. (2015). Altered gamma-aminobutyric acid neurotransmission in major depressive disorder: a critical review of the supporting evidence and the influence of serotonergic antidepressants. Drug Des. Devel. Ther. 9, 603–624. doi: 10.2147/DDDT.S62912
Perova, Z., Delevich, K., and Li, B. (2015). Depression of excitatory synapses onto parvalbumin interneurons in the medial prefrontal cortex in susceptibility to stress. J. Neurosci. 35, 3201–3206. doi: 10.1523/JNEUROSCI.2670-14.2015
Perrine, S. A., Ghoddoussi, F., Michaels, M. S., Sheikh, I. S., McKelvey, G., and Galloway, M. P. (2014). Ketamine reverses stress-induced depression-like behavior and increased GABA levels in the anterior cingulate: an 11.7 T 1H-MRS study in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 51, 9–15. doi: 10.1016/j.pnpbp.2013.11.003
Pettit, D. A., Harrison, M. P., Olson, J. M., Spencer, R. F., and Cabral, G. A. (1998). Immunohistochemical localization of the neural cannabinoid receptor in rat brain. J. Neurosci. Res. 51, 391–402. doi: 10.1002/(SICI)1097-4547(19980201)51:3<391::AID-JNR12>3.0.CO;2-A
Petty, F., and Sherman, A. D. (1984). Plasma GABA levels in psychiatric illness. J. Affect. Disord. 6, 131–138. doi: 10.1016/0165-0327(84)90018-1
Petty, F., Trivedi, M. H., Fulton, M., and Rush, A. J. (1995). Benzodiazepines as antidepressants: does GABA play a role in depression? Biol. Psychiatry 38, 578–591.
Piantadosi, S. C., French, B. J., Poe, M. M., Timic, T., Markovic, B. D., Pabba, M., et al. (2016). Sex-dependent anti-stress effect of an alpha5 subunit containing GABAA receptor positive allosteric modulator. Front. Pharmacol. 7:446. doi: 10.3389/fphar.2016.00446
Pich, E. M., Agnati, L. F., Zini, I., Marrama, P., and Carani, C. (1993). Neuropeptide Y produces anxiolytic effects in spontaneously hypertensive rats. Peptides 14, 909–912. doi: 10.1016/0196-9781(93)90065-O
Pinna, G., Costa, E., and Guidotti, A. (2006). Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake. Psychopharmacology 186, 362–372. doi: 10.1007/s00213-005-0213-2
Pinna, G., Costa, E., and Guidotti, A. (2009). SSRIs act as selective brain steroidogenic stimulants (SBSSs) at low doses that are inactive on 5-HT reuptake. Curr. Opin. Pharmacol. 9, 24–30. doi: 10.1016/j.coph.2008.12.006
Pinter, E., Helyes, Z., and Szolcsanyi, J. (2006). Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol. Ther. 112, 440–456. doi: 10.1016/j.pharmthera.2006.04.010
Poleszak, E., Wosko, S., Slawinska, K., Szopa, A., Wrobel, A., and Serefko, A. (2018). Cannabinoids in depressive disorders. Life Sci. 213, 18–24. doi: 10.1016/j.lfs.2018.09.058
Post, R. M., Ketter, T. A., Joffe, R. T., and Kramlinger, K. L. (1991). Lack of beneficial effects of l-baclofen in affective disorder. Int. Clin. Psychopharmacol. 6, 197–207. doi: 10.1097/00004850-199100640-00001
Prescot, A., Sheth, C., Legarreta, M., Renshaw, P. F., McGlade, E., and Yurgelun-Todd, D. (2018). Altered cortical gamma-amino butyric acid in female veterans with suicidal behavior: sex differences and clinical correlates. Chronic Stress 2. doi: 10.1177/2470547018768771
Prevot, T. D., Gastambide, F., Viollet, C., Henkous, N., Martel, G., Epelbaum, J., et al. (2017). Roles of hippocampal somatostatin receptor subtypes in stress response and emotionality. Neuropsychopharmacology 42, 1647–1656. doi: 10.1038/npp.2016.281
Puig, M. V., Santana, N., Celada, P., Mengod, G., and Artigas, F. (2004). In vivo excitation of GABA interneurons in the medial prefrontal cortex through 5-HT3 receptors. Cereb. Cortex 14, 1365–1375. doi: 10.1093/cercor/bhh097
Radley, J. J., Gosselink, K. L., and Sawchenko, P. E. (2009). A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J. Neurosci. 29, 7330–7340. doi: 10.1523/JNEUROSCI.5924-08.2009
Rajkowska, G., O’Dwyer, G., Teleki, Z., Stockmeier, C. A., and Miguel-Hidalgo, J. J. (2007). GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology 32, 471–482. doi: 10.1038/sj.npp.1301234
Reddy, M. S. (2010). Depression: the disorder and the burden. Indian J. Psychol. Med. 32, 1–2. doi: 10.4103/0253-7176.70510
Redrobe, J. P., Dumont, Y., Fournier, A., and Quirion, R. (2002a). The neuropeptide Y (NPY) Y1 receptor subtype mediates NPY-induced antidepressant-like activity in the mouse forced swimming test. Neuropsychopharmacology 26, 615–624.
Redrobe, J. P., Dumont, Y., and Quirion, R. (2002b). Neuropeptide Y (NPY) and depression: from animal studies to the human condition. Life Sci. 71, 2921–2937.
Ren, Z., Pribiag, H., Jefferson, S. J., Shorey, M., Fuchs, T., Stellwagen, D., et al. (2016). Bidirectional homeostatic regulation of a depression-related brain state by gamma-aminobutyric acidergic deficits and ketamine treatment. Biol. Psychiatry 80, 457–468. doi: 10.1016/j.biopsych.2016.02.009
Robinson, R. T., Drafts, B. C., and Fisher, J. L. (2003). Fluoxetine increases GABA(A) receptor activity through a novel modulatory site. J. Pharmacol. Exp. Ther. 304, 978–984. doi: 10.1124/jpet.102.044834
Rodriguez-Landa, J. F., Contreras, C. M., Bernal-Morales, B., Gutierrez-Garcia, A. G., and Saavedra, M. (2007). Allopregnanolone reduces immobility in the forced swimming test and increases the firing rate of lateral septal neurons through actions on the GABAA receptor in the rat. J. Psychopharmacol. 21, 76–84. doi: 10.1177/0269881106064203
Romeo, E., Strohle, A., Spalletta, G., di Michele, F., Hermann, B., Holsboer, F., et al. (1998). Effects of antidepressant treatment on neuroactive steroids in major depression. Am. J. Psychiatry 155, 910–913. doi: 10.1176/ajp.155.7.910
Rosa, P. B., Neis, V. B., Ribeiro, C. M., Moretti, M., and Rodrigues, A. L. (2016). Antidepressant-like effects of ascorbic acid and ketamine involve modulation of GABAA and GABAB receptors. Pharmacol. Rep. 68, 996–1001. doi: 10.1016/j.pharep.2016.05.010
Rubinow, D. R., Gold, P. W., Post, R. M., and Ballenger, J. C. (1985). CSF somatostatin in affective illness and normal volunteers. Prog. Neuropsychopharmacol. Biol. Psychiatry 9, 393–400.
Rudolph, U., and Knoflach, F. (2011). Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 10, 685–697. doi: 10.1038/nrd3502
Rudy, B., Fishell, G., Lee, S., and Hjerling-Leffler, J. (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61. doi: 10.1002/dneu.20853
Russo, E. B., Burnett, A., Hall, B., and Parker, K. K. (2005). Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res. 30, 1037–1043. doi: 10.1007/s11064-005-6978-1
Sage Therapeutics (2017). Sage Therapeutics Reports Positive Top-Line Results from Phase 2 Placebo-Controlled Trial of SAGE-217 in Major Depressive Disorder [Press Release]. Cambridge, MA: Sage Therapeutics.
Sajdyk, T. J., Johnson, P. L., Leitermann, R. J., Fitz, S. D., Dietrich, A., Morin, M., et al. (2008). Neuropeptide Y in the amygdala induces long-term resilience to stress-induced reductions in social responses but not hypothalamic-adrenal-pituitary axis activity or hyperthermia. J. Neurosci. 28, 893–903. doi: 10.1523/JNEUROSCI.0659-07.2008
Sales, A. J., Fogaca, M. V., Sartim, A. G., Pereira, V. S., Wegener, G., Guimaraes, F. S., et al. (2018). Cannabidiol induces rapid and sustained antidepressant-like effects through increased BDNF signaling and synaptogenesis in the prefrontal cortex. Mol. Neurobiol. doi: 10.1007/s12035-018-1143-4 [Epub ahead of print].
Sanacora, G., Gueorguieva, R., Epperson, C. N., Wu, Y. T., Appel, M., Rothman, D. L., et al. (2004). Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch. Gen. Psychiatry 61, 705–713. doi: 10.1001/archpsyc.61.7.705
Sanacora, G., Mason, G. F., Rothman, D. L., Behar, K. L., Hyder, F., Petroff, O. A., et al. (1999). Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 56, 1043–1047. doi: 10.1001/archpsyc.56.11.1043
Santana, N., Bortolozzi, A., Serrats, J., Mengod, G., and Artigas, F. (2004). Expression of serotonin1A and serotonin2A receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb. Cortex 14, 1100–1109. doi: 10.1093/cercor/bhh070
Savitz, J., and Drevets, W. C. (2009). Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci. Biobehav. Rev. 33, 699–771. doi: 10.1016/j.neubiorev.2009.01.004
Sayed, S., Van Dam, N. T., Horn, S. R., Kautz, M. M., Parides, M., Costi, S., et al. (2018). A randomized dose-ranging study of neuropeptide Y in patients with posttraumatic stress disorder. Int. J. Neuropsychopharmacol. 21, 3–11. doi: 10.1093/ijnp/pyx109
Schiavon, A. P., Bonato, J. M., Milani, H., Guimaraes, F. S., and Weffort de Oliveira, R. M. (2016). Influence of single and repeated cannabidiol administration on emotional behavior and markers of cell proliferation and neurogenesis in non-stressed mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 64, 27–34. doi: 10.1016/j.pnpbp.2015.06.017
Sequeira, A., Klempan, T., Canetti, L., ffrench-Mullen, J., Benkelfat, C., Rouleau, G. A., et al. (2007). Patterns of gene expression in the limbic system of suicides with and without major depression. Mol. Psychiatry 12, 640–655. doi: 10.1038/sj.mp.4001969
Serrats, J., Artigas, F., Mengod, G., and Cortes, R. (2003). GABAB receptor mRNA in the raphe nuclei: co-expression with serotonin transporter and glutamic acid decarboxylase. J. Neurochem. 84, 743–752. doi: 10.1046/j.1471-4159.2003.01557.x
Shalaby, A., and Kamal, S. (2009). Effect of Escitalopram on GABA level and anti-oxidant markers in prefrontal cortex and nucleus accumbens of chronic mild stress-exposed albino rats. Int. J. Physiol. Pathophysiol. Pharmacol. 1, 154–161.
Shen, Q., Lal, R., Luellen, B. A., Earnheart, J. C., Andrews, A. M., and Luscher, B. (2010). gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol. Psychiatry 68, 512–520. doi: 10.1016/j.biopsych.2010.04.024
Shepard, R., and Coutellier, L. (2018). Changes in the prefrontal glutamatergic and parvalbumin systems of mice exposed to unpredictable chronic stress. Mol. Neurobiol. 55, 2591–2602. doi: 10.1007/s12035-017-0528-0
Shepard, R., Page, C. E., and Coutellier, L. (2016). Sensitivity of the prefrontal GABAergic system to chronic stress in male and female mice: relevance for sex differences in stress-related disorders. Neuroscience 332, 1–12. doi: 10.1016/j.neuroscience.2016.06.038
Shimizu, E., Hashimoto, K., Okamura, N., Koike, K., Komatsu, N., Kumakiri, C., et al. (2003). Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol. Psychiatry 54, 70–75. doi: 10.1016/S0006-3223(03)00181-1
Sibille, E., Morris, H. M., Kota, R. S., and Lewis, D. A. (2011). GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int. J. Neuropsychopharmacol. 14, 721–734. doi: 10.1017/S1461145710001616
Slattery, D. A., Desrayaud, S., and Cryan, J. F. (2005). GABAB receptor antagonist-mediated antidepressant-like behavior is serotonin-dependent. J. Pharmacol. Exp. Ther. 312, 290–296. doi: 10.1124/jpet.104.073536
Smith, K. S., and Rudolph, U. (2012). Anxiety and depression: mouse genetics and pharmacological approaches to the role of GABA(A) receptor subtypes. Neuropharmacology 62, 54–62. doi: 10.1016/j.neuropharm.2011.07.026
Song, Z., Huang, P., Qiu, L., Wu, Q., Gong, Q., Zhang, B., et al. (2012). [Decreased occipital GABA concentrations in patients with first-episode major depressive disorder: a magnetic resonance spectroscopy study]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 29, 233–236.
Soumier, A., and Sibille, E. (2014). Opposing effects of acute versus chronic blockade of frontal cortex somatostatin-positive inhibitory neurons on behavioral emotionality in mice. Neuropsychopharmacology 39, 2252–2262. doi: 10.1038/npp.2014.76
Stampanoni Bassi, M., Gilio, L., Maffei, P., Dolcetti, E., Bruno, A., Buttari, F., et al. (2018). Exploiting the multifaceted effects of cannabinoids on mood to boost their therapeutic use against anxiety and depression. Front. Mol. Neurosci. 11:424. doi: 10.3389/fnmol.2018.00424
Stone, J. M., Dietrich, C., Edden, R., Mehta, M. A., De Simoni, S., Reed, L. J., et al. (2012). Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol. Psychiatry 17, 664–665. doi: 10.1038/mp.2011.171
Strohle, A., Romeo, E., Hermann, B., Pasini, A., Spalletta, G., di Michele, F., et al. (1999). Concentrations of 3 alpha-reduced neuroactive steroids and their precursors in plasma of patients with major depression and after clinical recovery. Biol. Psychiatry 45, 274–277. doi: 10.1016/S0006-3223(98)00328-X
Szabo, B., and Schlicker, E. (2005). Effects of cannabinoids on neurotransmission. Handb. Exp. Pharmacol. 168, 327–365. doi: 10.1007/3-540-26573-2_11
Taylor, M. J., Tiangga, E. R., Mhuircheartaigh, R. N., and Cowen, P. J. (2012). Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. J. Psychopharmacol. 26, 733–737. doi: 10.1177/0269881111405359
Thase, M. E., Mahableshwarkar, A. R., Dragheim, M., Loft, H., and Vieta, E. (2016). A meta-analysis of randomized, placebo-controlled trials of vortioxetine for the treatment of major depressive disorder in adults. Eur. Neuropsychopharmacol. 26, 979–993. doi: 10.1016/j.euroneuro.2016.03.007
Thorsell, A., Michalkiewicz, M., Dumont, Y., Quirion, R., Caberlotto, L., Rimondini, R., et al. (2000). Behavioral insensitivity to restraint stress, absent fear suppression of behavior and impaired spatial learning in transgenic rats with hippocampal neuropeptide Y overexpression. Proc. Natl. Acad. Sci. U.S.A. 97, 12852–12857. doi: 10.1073/pnas.220232997
Todorovic, N., Micic, B., Schwirtlich, M., Stevanovic, M., and Filipovic, D. (2019). Subregion-specific protective effects of fluoxetine and clozapine on parvalbumin expression in medial prefrontal cortex of chronically isolated rats. Neuroscience 396, 24–35. doi: 10.1016/j.neuroscience.2018.11.008
Tremblay, R., Lee, S., and Rudy, B. (2016). GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292. doi: 10.1016/j.neuron.2016.06.033
Tripp, A., Kota, R. S., Lewis, D. A., and Sibille, E. (2011). Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol. Dis. 42, 116–124. doi: 10.1016/j.nbd.2011.01.014
Tripp, A., Oh, H., Guilloux, J. P., Martinowich, K., Lewis, D. A., and Sibille, E. (2012). Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry 169, 1194–1202. doi: 10.1176/appi.ajp.2012.12020248
Trivedi, M. H., Rush, A. J., Wisniewski, S. R., Nierenberg, A. A., Warden, D., Ritz, L., et al. (2006). Evaluation of outcomes with citalopram for depression using measurement-based care in STAR∗D: implications for clinical practice. Am. J. Psychiatry 163, 28–40. doi: 10.1176/appi.ajp.163.1.28
Urban-Ciecko, J., and Barth, A. L. (2016). Somatostatin-expressing neurons in cortical networks. Nat. Rev. Neurosci. 17, 401–409. doi: 10.1038/nrn.2016.53
Uzunov, D. P., Cooper, T. B., Costa, E., and Guidotti, A. (1996). Fluoxetine-elicited changes in brain neurosteroid content measured by negative ion mass fragmentography. Proc. Natl. Acad. Sci. U.S.A. 93, 12599–12604. doi: 10.1073/pnas.93.22.12599
Uzunova, V., Sheline, Y., Davis, J. M., Rasmusson, A., Uzunov, D. P., Costa, E., et al. (1998). Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc. Natl. Acad. Sci. U.S.A. 95, 3239–3244. doi: 10.1073/pnas.95.6.3239
Uzunova, V., Wrynn, A. S., Kinnunen, A., Ceci, M., Kohler, C., and Uzunov, D. P. (2004). Chronic antidepressants reverse cerebrocortical allopregnanolone decline in the olfactory-bulbectomized rat. Eur. J. Pharmacol. 486, 31–34. doi: 10.1016/j.ejphar.2003.12.002
Valentine, G. W., Mason, G. F., Gomez, R., Fasula, M., Watzl, J., Pittman, B., et al. (2011). The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Res. 191, 122–127. doi: 10.1016/j.pscychresns.2010.10.009
van Marwijk, H., Allick, G., Wegman, F., Bax, A., and Riphagen, I. I. (2012). Alprazolam for depression. Cochrane Database Syst. Rev. 7:CD007139. doi: 10.1002/14651858.CD007139.pub2
Varga, Z., Csabai, D., Miseta, A., Wiborg, O., and Czeh, B. (2017). Chronic stress affects the number of GABAergic neurons in the orbitofrontal cortex of rats. Behav. Brain Res. 316, 104–114. doi: 10.1016/j.bbr.2016.08.030
Viollet, C., Vaillend, C., Videau, C., Bluet-Pajot, M. T., Ungerer, A., L’Heritier, A., et al. (2000). Involvement of sst2 somatostatin receptor in locomotor, exploratory activity and emotional reactivity in mice. Eur. J. Neurosci. 12, 3761–3770. doi: 10.1046/j.1460-9568.2000.00249.x
Vithlani, M., Hines, R. M., Zhong, P., Terunuma, M., Hines, D. J., Revilla-Sanchez, R., et al. (2013). The ability of BDNF to modify neurogenesis and depressive-like behaviors is dependent upon phosphorylation of tyrosine residues 365/367 in the GABA(A)-receptor gamma2 subunit. J. Neurosci. 33, 15567–15577. doi: 10.1523/JNEUROSCI.1845-13.2013
Vollenweider, I., Smith, K. S., Keist, R., and Rudolph, U. (2011). Antidepressant-like properties of alpha2-containing GABA(A) receptors. Behav. Brain Res. 217, 77–80. doi: 10.1016/j.bbr.2010.10.009
Wang, D. S., Penna, A., and Orser, B. A. (2017). Ketamine increases the function of gamma-aminobutyric acid type A receptors in hippocampal and cortical neurons. Anesthesiology 126, 666–677. doi: 10.1097/ALN.0000000000001483
Wang, X., Dow-Edwards, D., Keller, E., and Hurd, Y. L. (2003). Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience 118, 681–694. doi: 10.1016/S0306-4522(03)00020-4
Wedzony, K., and Chocyk, A. (2009). Cannabinoid CB1 receptors in rat medial prefrontal cortex are colocalized with calbindin- but not parvalbumin- and calretinin-positive GABA-ergic neurons. Pharmacol. Rep. 61, 1000–1007. doi: 10.1016/S1734-1140(09)70161-6
Westrin, A., Ekman, R., and Traskman-Bendz, L. (1999). Alterations of corticotropin releasing hormone (CRH) and neuropeptide Y (NPY) plasma levels in mood disorder patients with a recent suicide attempt. Eur. Neuropsychopharmacol. 9, 205–211. doi: 10.1016/S0924-977X(98)00026-1
Widdowson, P. S., Ordway, G. A., and Halaris, A. E. (1992). Reduced neuropeptide Y concentrations in suicide brain. J. Neurochem. 59, 73–80. doi: 10.1111/j.1471-4159.1992.tb08877.x
Widerlov, E., Lindstrom, L. H., Wahlestedt, C., and Ekman, R. (1988). Neuropeptide Y and peptide YY as possible cerebrospinal fluid markers for major depression and schizophrenia, respectively. J. Psychiatr. Res. 22, 69–79. doi: 10.1016/0022-3956(88)90030-1
Wilkinson, S. T., and Sanacora, G. (2018). A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Discov. Today doi: 10.1016/j.drudis.2018.11.007 [Epub ahead of print].
Workman, E. R., Haddick, P. C., Bush, K., Dilly, G. A., Niere, F., Zemelman, B. V., et al. (2015). Rapid antidepressants stimulate the decoupling of GABA(B) receptors from GIRK/Kir3 channels through increased protein stability of 14-3-3eta. Mol. Psychiatry 20, 298–310. doi: 10.1038/mp.2014.165
Workman, E. R., Niere, F., and Raab-Graham, K. F. (2013). mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology 73, 192–203. doi: 10.1016/j.neuropharm.2013.05.037
Xiong, Z., Zhang, K., Ishima, T., Ren, Q., Chang, L., Chen, J., et al. (2018). Comparison of rapid and long-lasting antidepressant effects of negative modulators of alpha5-containing GABAA receptors and (R)ketamine in a chronic social defeat stress model. Pharmacol. Biochem. Behav. 175, 139–145. doi: 10.1016/j.pbb.2018.10.005
Yang, Y., Babygirija, R., Zheng, J., Shi, B., Sun, W., Zheng, X., et al. (2018). Central neuropeptide Y plays an important role in mediating the adaptation mechanism against chronic stress in male rats. Endocrinology 159, 1525–1536. doi: 10.1210/en.2018-00045
Yeung, M., Engin, E., and Treit, D. (2011). Anxiolytic-like effects of somatostatin isoforms SST 14 and SST 28 in two animal models (Rattus norvegicus) after intra-amygdalar and intra-septal microinfusions. Psychopharmacology 216, 557–567. doi: 10.1007/s00213-011-2248-x
Zadrozna, M., Nowak, B., Lason-Tyburkiewicz, M., Wolak, M., Sowa-Kucma, M., Papp, M., et al. (2011). Different pattern of changes in calcium binding proteins immunoreactivity in the medial prefrontal cortex of rats exposed to stress models of depression. Pharmacol. Rep. 63, 1539–1546. doi: 10.1016/S1734-1140(11)70718-6
Zanos, P., Moaddel, R., Morris, P. J., Georgiou, P., Fischell, J., Elmer, G. I., et al. (2016). NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486. doi: 10.1038/nature17998
Zanos, P., Nelson, M. E., Highland, J. N., Krimmel, S. R., Georgiou, P., Gould, T. D., et al. (2017). A negative allosteric modulator for alpha5 subunit-containing GABA receptors exerts a rapid and persistent antidepressant-like action without the side effects of the NMDA receptor antagonist ketamine in mice. eNeuro 4:ENEURO.285-ENEURO.216. doi: 10.1523/ENEURO.0285-16.2017
Zarate, C. A. Jr., Singh, J. B., Carlson, P. J., Brutsche, N. E., Ameli, R., Luckenbaugh, D. A., et al. (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864. doi: 10.1001/archpsyc.63.8.856
Zhou, W., Wang, N., Yang, C., Li, X. M., Zhou, Z. Q., and Yang, J. J. (2014). Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry 29, 419–423. doi: 10.1016/j.eurpsy.2013.10.005
Keywords: depression, stress, prefrontal cortex, GABA, somatostatin, parvalbumin, ketamine
Citation: Fogaça MV and Duman RS (2019) Cortical GABAergic Dysfunction in Stress and Depression: New Insights for Therapeutic Interventions. Front. Cell. Neurosci. 13:87. doi: 10.3389/fncel.2019.00087
Received: 15 January 2019; Accepted: 20 February 2019;
Published: 12 March 2019.
Edited by:
Boldizsar Czeh, University of Pécs, HungaryReviewed by:
Juan Nacher, University of Valencia, SpainGrazyna Rajkowska, University of Mississippi Medical Center, United States
Copyright © 2019 Fogaça and Duman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronald S. Duman, cm9uYWxkLmR1bWFuQHlhbGUuZWR1