 Ziqian Cheng
Ziqian Cheng Xiaohan Zou
Xiaohan Zou Yang Jin1
Yang Jin1 Ranji Cui
Ranji Cui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 21 September 2018
Sec. Non-Neuronal Cells
Volume 12 - 2018 | https://doi.org/10.3389/fncel.2018.00325
This article is part of the Research Topic Role of Neural Molecules in the Neuroimmune Axis Regulation of Cognitive and Behavioral Activities View all 10 articles
Krüppel-like factor 4 (KLF4), a member of the family of zinc-finger transcription factors, is widely expressed in range of tissues that play multiple functions. Emerging evidence suggest KLF4’s critical regulatory effect on the neurophysiological and neuropathological processes of Alzheimer’s disease (AD), indicating that KLF4 might be a potential therapeutic target of neurodegenerative diseases. In this review, we will summarize relevant studies and illuminate the regulatory role of KLF4 in the neuroinflammation, neuronal apoptosis, axon regeneration and iron accumulation to clarify KLF4’s status in the pathogenesis of AD.
Kruppel-like factor 4 (KLF4) is a member of the family of zinc-finger transcription factor,which is expressed in various human tissues. It is well known as one of the four factors of the induction to pluripotent stem cells (iPSCs) (Ghaleb and Yang, 2017). KLF4 can regulate multiple important biological processes such as neuroinflammation, oxidative stress, proliferation, differentiation, and apoptosis (Kaushik et al., 2010; Mamonkin et al., 2013; Zhang et al., 2015; Miao et al., 2017; Xu et al., 2017). Amounts of previous studies focused on KLF4’s role in cancer development and progression (Karam et al., 2017; Yadav et al., 2018). KLF4 is a dual-function transcription factor, which can exert its role as an oncogene or a tumor suppressor gene depending on the cancer type or cancer stage (Evans and Liu, 2008). It can activate or inhibit transcription of genes involved in cell proliferation, differentiation and apoptosis (Ding et al., 2015). KLF4 can collaborate with other reprogramming factors to convert the somatic cells into iPSCs and inhibit the differentiation of stem cells (Takahashi and Yamanaka, 2006; van Schaijik et al., 2018). This provides therapeutic prospects for vascular diseases, immune diseases, anorexia and other diseases (Imbernon et al., 2014; Liu Y. et al., 2015; Murgai et al., 2017). Moreover, KLF4 can play a widely regulatory role in the central nervous system (CNS). Several studies indicate that KLF4 is linked to multiple neurological disorders, including Alzheimer’s disease (AD), epilepsy, Parkinson’s disease, hydrocephalus and schizophrenia (Qin et al., 2011; Xie et al., 2013; Han et al., 2015; Nishiguchi et al., 2015; Li L. et al., 2017).
AD is one of the most common chronic neurodegenerative diseases, which leads to cognitive and memory impairments, various mental symptoms and behavioral abnormality and progressive dementia is the most common clinical feature (Jiang et al., 2018). The current confirmed pathogenic factors of AD include the formation of senile plaques induced by abnormal amyloid-β (Aβ) deposition and the neurofibrillary tangles or dystrophic neuritis induced by tau accumulation (Querfurth and LaFerla, 2010; Shinohara et al., 2014). In addition, AD can be also affected by genetic factors. However, the elicit pathogenesis is still obscure. The most prevalent drugs for AD treatment include neurotransmitter enhancers, anti-Amyloid agents, neuroprotective peptides, and other drugs (Cacabelos, 2018). Notably, several studies have showed that KLF4 played a significant role in the pathogenesis of AD. In this review, we focus on the regulatory role of KLF4 in neuroinflammation, neuronal apoptosis, axonal regeneration, and iron accumulation to explain the association between KLF4 and the pathogenesis of AD, which might provide insights into the cellular and molecular mechanisms of neurodegenerative disorders.
KLF4 is a zinc finger-containing nuclear protein, isolated from NIH 3T3 library and located in the cell nucleus. It was first identified and characterized by Shields et al. (1996). The molecular mass of human KLF4 is 55kD and it is located on the chromosome 9q31. KLF4 covers a 6.3 kb gene segment and has five exons. Its cDNA coding region encodes a polypeptide consisting of 470 amino acid residues (Yet et al., 1998; Ghaleb and Yang, 2017). The carboxy terminus of KLF4 has a DNA binding structure region containing three Cys2His2 (C2H2) type zinc finger structures, which are formed by 81 highly conserved amino acids. It regulates transcription by high affinity with CACCC elements and GC-rich target gene DNA sequences (Shields and Yang, 1998; Pearson et al., 2008). Most of the DNA-binding sites of KLF4 are located within the zinc finger region, including N-terminal transcription activation domain for proteins interacting, C-terminal zinc finger structure for DNA binding and transcription inhibition zone (Bieker, 2001). KLF4 is involved in regulating the expression of many endogenous genes (Shields and Yang, 1998). There is a highly variable transcriptional regulatory domain at the amino terminus of KLF4. The amino acid residues located between the 91 and the 117 amino constitute a transcriptional activation domain, which is rich in proline and serine, while a transcriptional repression domain also exists. Therefore, KLF4 has two adverse effects: activating and inhibiting gene transcription (Yet et al., 1998; Wei et al., 2006).
During the embryonic development, KLF4 was higher expressed in the late stage of embryonic development. While in mature tissues and organs, KLF4 is mainly expressed in the gastrointestinal tract, oral cavity, skin epidermis, vascular endothelium and kidney, and is less expressed in the brain (Segre et al., 1999; Ghaleb et al., 2011; Liu et al., 2013; Chen et al., 2015; He et al., 2015; Bin et al., 2016). It is thought to play significant role in regulating cell proliferation and differentiation. Besides, KLF4 can also regulate cell cycle. KLF4 can activate P21 in a P53-dependent manner (Zhang et al., 2000). In addition, It was found that KLF4 (–/–) cells entered senescence phase earlier than KLF4 (+/+) cells, which can be explained by the less antioxidant gene expression and higher reactive oxygen species (ROS) level in KLF4 (–/–) cells. ROS can increase p53 and p21 expression and subsequently promote the DNA damage (Liu C. et al., 2015). It was found that PRMT5 can elevate the KLF4 expression in protein levels. PRMT5 was reported to increase the transcription of p21 and decrease the expression of bax via inhibiting KLF4 ubiquitylation (Hu et al., 2015). Furthermore, numerous studies have demonstrated that KLF4 is involved in regulation of apoptosis of neurons (Kong et al., 2016; Cui et al., 2017; Song et al., 2018). Physiological regulatory role of KLF4 that we have known are still little and further investigations are needed.
It is well established that AD is mainly characterized by memory and cognitive impairments and executive dysfunction (Goedert and Spillantini, 2006). Many studies have demonstrated that neuronal apoptosis and synaptic dysfunction are pathological basis of the decline of cognitive function (Caccamo et al., 2017; Guo et al., 2017; Yoon et al., 2018). The accumulated damage of Aβ deposition, oxidative stress and iron accumulation can lead to neuronal dysfunction and apoptosis of AD patients. Several studies have shown that KLF4’s regulatory role appears to be crucial in CNS. Considering that KLF4 was reported to regulate neuronal apoptosis, synaptic regeneration, oxidative stress and neuroinflammation, the relationship between KLF4 and the pathogenesis of AD might be a potential novel target for AD treatment.
Amounts of clinical studies have shown that Aβ can aggregate and is the main component of the extracellular deposits of the brain tissue of AD patients, which can impair the surrounding synapses and neurons, and lead to neuronal death. Abnormal secretion or excessive production of Aβ leads to pathological changes of AD, so Aβ deposition is the core link of AD (Rajmohan and Reddy, 2017). In addition, studies have shown that excessive Aβ deposition can stimulate glial cells to secrete ROS and other influencing factors, leading to oxidative stress. It was known that oxidative stress can stimulate the production of Aβ. Therefore, Aβ and oxidative stress can interact with each other and affect the progression of AD (Cheignon et al., 2018).
KLF4 was reported as a potential modulator and has a great effect on inflammation by mediating macrophages and endothelial cells (Figure 1) (Yoshida et al., 2014; Kapoor et al., 2015; Yang et al., 2018). In the CNS, excessive and chronic inflammatory reactions can cause damage of neuron and neurogliocyte. It was recently demonstrated that the KLF4 expression positively correlated with Aβ42-induced neuroinflammation. In microglial BV2 cells, oligomeric Aβ42 can increase KLF4 expression, which is mediated by activated P53 (Li L. et al., 2017). Under inflammatory conditions, such as Aβ accumulation, the release of pro-inflammatory cytokines may be stimulated in the generation of AD (Griffin and Barger, 2010). Neurotoxicity potency and pro-inflammatory potency of soluble Aβ42 oligomers is relatively higher than insoluble fiber deposit (Selkoe, 1991; Weinberg et al., 2018). Silence of KLF4 is able to restore Aβ42-mediated neuroinflammation, and overexpression of KLF4 can exacerbate Aβ42-mediated neuroinflammation (Li L. et al., 2017). Aβ accumulation induces activation of astrocytes and microglia (Rodríguez et al., 2016). Activated astrocytes can enhance the neuroinflammation by releasing pro-inflammatory factors such as IL-1, IL-6, and TNF-α (Rubio-Perez and Morillas-Ruiz, 2012; Doméné et al., 2016). The vicious cycle of inflammatory responses eventually leads to dysfunction and neuronal apoptosis.
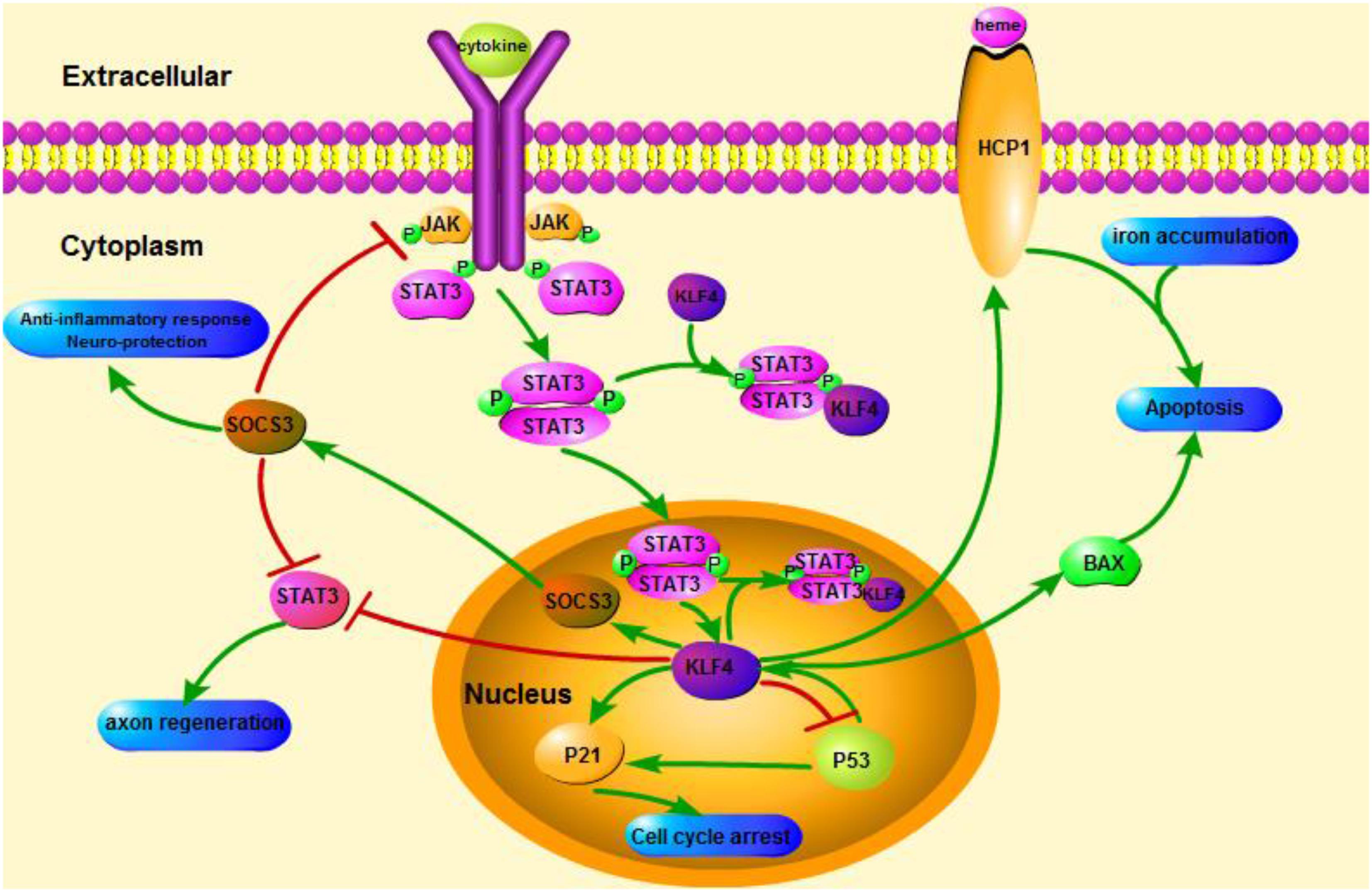
FIGURE 1. Schematicillustration of KLF4 related signaling pathways. This figure highlights the role of KLF4 in neuroprotection and axon regeneration. The arrows in the figure indicate activation or promotion, and the straight lines indicate related inhibition. KLF4, Kruppel-like factor 4; STAT3, Signal transducer and activator of transcription 3; JAK, Janus Kinase; SOCS3, Suppressor of cytokine signaling 3; HCP1, heme carrier protein 1; ERK5, mitogen-activated protein [MAP] kinase 5.
KLF4 plays a crucial role in regulating pro-inflammatory signals. In glial cells, gemfibrozil-induced KLF4 activation increases suppressor of cytokine signal 3 (SOCS3) via PI3-kinase-AKT pathway (Ghosh and Pahan, 2012). The SiRNA-mediated knockdown of KLF4 could attenuate the level of SOCS in astroglia and microglia of mice, which could subsequently affect the expression of inflammatory gene (Kaushik et al., 2010; Ghosh and Pahan, 2012). In addition, SOCS deletion can promote the survival of injured neurons and promote axon regeneration (Smith et al., 2009; Sun et al., 2011). And KLF4 positively regulates the production of IL-1β or other pro-inflammatory markers. It positively regulates cyclooxygenase-2 (Cox-2) and negatively regulates inducible nitric oxide synthase (iNOS) (Kaushik et al., 2013). In addition, KLF4 is an important regulatory factor for monocyte differentiation and a potential target for immune regulation (Alder et al., 2008). Therefore, KLF4 might promote neuroinflammation by regulating these negative regulators.
It is worth mentioning that in Parkinson’s disease model, KLF4 can promote MPP+-induced oxidative stress and neurotoxicity, and then increase neuronal apoptosis and delay the cell proliferation (Chen et al., 2013). Oxidative stress is an imbalance between peroxidation and antioxidation. Free radicals can cause changes in different macromolecules, leading to cell damage, cell aging and tissue damage (Parajuli et al., 2013; Nie et al., 2015). Oxidative stress can aggravate early inflammation and Aβ production and then aggravate AD (Cai et al., 2011). Therefore, KLF4 may be involved in oxidative stress in AD.
These findings imply KLF4 a key role in mediating neuroinflammation by activating the microglia and the consequently release of pro-inflammatory cytokines. It has potential to enhance neuroinflammation. So far, many studies on the pathogenesis of AD have focused on neuroinflammation. As a potential target for immune regulation, KLF4 can promote the inflammatory responses of microglia via affecting related negative regulators, which has a great effect on the development of AD.
Neurodegenerative changes include gradual loss of neurons and synapses in the representative brain regions, such as the cerebral cortex, hippocampus and other subcortical regions. The functional impairments of CNS induced by neuronal loss are permanent (Citron, 2010). Sustained oxidative stress can lead to neuronal apoptosis (Wu et al., 2010). A large number of studies have confirmed that AD is closely related to oxidative stress (Lee et al., 2012; Yui et al., 2015). It was found that chronic oxidative stress can enhance the expression of Phospholipase A2 group 3 (Pla2g3) in astrocytes and disrupt the balance of Aβ, and consequently lead to the development of AD (Yui et al., 2015).
Many studies have demonstrated that KLF4 plays an important role in inhibiting the development of oxidative stress (Shi et al., 2014; Liu C. et al., 2015). It was found that KLF4 can promote the cells apoptosis induced by H2O2, this action is likely to be caused by increased bax expression and decreased bcl-2 expression (Li et al., 2010). Quercetin could reduce KLF4 expression in human neuroblastoma SH-SY5Y cells, and increase the expression of bcl-2/bax ratio. Furthermore, Quercetin can moderate the apoptosis rate of SH-5YSY cell and reduce caspase-3 enzyme activity (Xi et al., 2012). A recent study investigated the neuroprotective effect of mitogen-activated protein (MAP) kinase 5 (ERK5) against oxidative stress. Activation of ERK5 can partially reduce H2O2-induced hippocampal neurons death and increase the NGF- and PC-induced neuroprotection (Su et al., 2014). Nils et al. used a mutant of MEK5 (MEK5D) to study the ERK5-activated transcription and functional responses in human endothelial cells, and identified KLF4 was a novel downstream ERK5 target (Ohnesorge et al., 2010). It was found that overexpression of KLF4 can suppress TNF-mediated inflammatory responses and reduce leukocyte adhesion and basal cell apoptosis. These results confirm that KLF4 has anti-inflammatory and anti-apoptotic properties (Ohnesorge et al., 2010). Subsequent experiments have demonstrated that the disappearance of cerebral cavernous malformation 1 (CCM1) in endothelial cells activates ERK5 via MEKK3-MEK5 signal pathway and increases KLF4 expression (Cuttano et al., 2016). ERK5 plays a mediating role in preconditioning (PC) and nerve growth factor (NGF) up-regulated the expression of KLF4 (Su et al., 2014). In addition, RNAi-mediated knocking-down of KLF4 can also reduce NGF- or PC-induced neuroprotection. Overexpression of KLF4 leads to higher bcl-2/bax ratio in H2O2-stressed cells (Su et al., 2014). Over-expressed KLF4 accelerates changes in bcl-2 and bax by combining with its corresponding promoter (Li et al., 2010). ERK5/KLF4 cascade may act as a pivot in various pathways which protect neurons from oxidative stress-induced death (Su et al., 2014).
Oxidative stress has been considered to be closely related to many degenerative diseases. KLF4 plays significant roles in maintaining genomic stability in oxidative stress. KLF4 and ERK5 act together to protect neurons from oxidative stress-induced apoptosis. Therefore,KLF4 may act as a therapeutic target to act against oxidative stress when it activated. It has been reported that statin drugs can activate ERK5, leading to the expression of KLF4 and its dependent genes (Ohnesorge et al., 2010), but the mechanism remains unclear, and KLF4 related upstream and downstream target genes are less studied in oxidative stress, there is a need for further study.
Early axon loss is a common feature of neurodegenerative diseases. Synaptic loss and transport impairment in AD can cause cognitive impairments (Holtzman et al., 2011; Coleman, 2013). The degree of declarative memory damage is related to the synaptic density in the hippocampus and cortex. Soluble Aβ oligomers reduce glutamate uptake and promote synaptic dysfunction, disrupting synaptic plasticity (Li et al., 2009). Therefore, it is particularly important to study how to repair the axons in the CNS. In retinal ganglion cells, axons have a strong ability to grow and regenerate during early development, but in the CNS of adult mammals, axons lose their regeneration capacity and the neurons may graduate to die or atrophy (Goldberg and Barres, 2000; Goldberg et al., 2002).
KLF4 plays an important role in inhibiting axon growth. In embryonic RGCs, overexpression of KLF4 can reduce the percentage of neurite elongation, the length of axons and dendrites, and the neurite branching. Besides, it was found that the overexpression of KLF4 can reduce long-term postnatal axon growth rates but failed to reduce short-term axon growth rates (Moore et al., 2009; Steketee et al., 2014). Later studies have found that the axon bundles of KLF4–cKO mice were thicker than control mice (Fang et al., 2016). In addition, removal of KLF4 expression during development can increase the reproductive potential of adult RGCs. In addition, KLF4 lacking the c-terminal DNA binding domain had no effect on the axon growth. There was no impact on the survival of cells after retinal ganglion cells were injured if the KLF4 was knocking-out (Moore et al., 2009).
KLF4 can also affect the axonal regeneration. A recent study reported that the decrease of KLF4 expression in adult retinal ganglion cells promoted axon regeneration through JAK-STAT3 pathway (Qin et al., 2013). KLF4 increased the phosphorylation of STAT3, and regulated the axon growth via JAK-STAT signaling (Qin and Zhang, 2012). Under the treatment of cytokines, members of STAT family of proteins are phosphorylated at the carboxy-terminal tyrosine and serine sites within the cell to form a stable dimer. This modification enhances transcription of cell-associated genes (Yuan et al., 2005). The interaction between KLF4 and STAT3 on cytokine-induced phosphorylation of tyrosin705 inhibits the expression of STAT3 by inhibiting the binding of STAT3 to DNA (Qin et al., 2013). KLF4 knockdown obviously improves axon’s regeneration in retinal ganglion cells after injury of optic nerve, and prevents the nerve from injury after mild brain injury. The actions are mediated by a decrease in p-p53 and an increase in pSTAT3 levels. KLF4 positively regulates neuronal apoptosis via the p53 and JAK-STAT3 pathways, and KLF4 negatively regulates axonal repair via the JAK-STAT3 pathway (Cui et al., 2017).
Therefore, we hypothesized that in AD, axonal regeneration can be accomplished by altering the expression of KLF4 or altering intracellular related signaling pathways, and controlling AD progression by reducing missing axons or reducing axonal dysfunction. However, how to use the KLF4 transcription factor in potential therapeutics still needs further exploration.
Iron is widely found in biological systems, iron-related metalloproteinases play a key role in transporting oxygen, transferring electrons, and catalyzing biochemical reactions (Aisen et al., 2001). However, any excess of iron beyond the normal physiological range can damage human health (Adlard and Bush, 2006). Studies have found that iron content in the hippocampus is negatively correlated with the performance of memory tests (Ding et al., 2009). Increased iron load in the brain accelerates the formation of Aβ plaques and hyperphosphorylated tau tangles, while also enhancing oxidative stress (Peters et al., 2015). Iron, which has a high degree of permeability, promotes nerve growth and cell-to-cell connections during brain development (Dallman and Spirito, 1977).
A recent study demonstrated that physiological stress caused activation of the KLF4-HCP1 signaling pathway and increased heme uptake (Li H. et al., 2017). Heme accounts for 95% of the functional iron in the human body. It is one of the main components of heme oxygenase (Hooda et al., 2014; Kurucz et al., 2018). Increasing the activity of oxygenase-1 can delay oxidation of the aging brain (Verdile et al., 2015; Serini and Calviello, 2016; Kurucz et al., 2018). This has a relief effect on AD. Physiological stress induces glucocorticoid level rise, glucocorticoid increases heme carrier protein 1 (HCP1) expression via KLF4, and then HCP1 promotes heme uptake (Li H. et al., 2017). Glucocorticoid and KLF4 regulate anti-inflammatory genes together, and cells with low glucocorticoid content cannot fully induce KLF4 expression (Sevilla et al., 2015). KLF4-induced increase in heme intake leads to iron accumulation in the brain. Iron promotes the release of ROS (Tronel et al., 2013). Iron element enhances brain oxidative stress in rats under psychological stress (Yu et al., 2011). Therefore, HCP1 may be regulated by KLF4 and glucocorticoid together. Increasing HCP1 enhances heme uptake, which leads directly to iron accumulation in the brain, exacerbates oxidation, increases apoptosis or dysfunction and worsens brain damage.
It is generally accepted that the memory and learning disability are the main symptoms of AD. A large number of clinical data have shown that Aβ plaque load and iron accumulation response to the development of learning and cognitive dysfunction in AD (van Bergen et al., 2018). Recently published data has suggested that, high-dose iron increases Aβ deposition and attenuates learning and memory in mice (Guo et al., 2013). Clinical studies have shown that iron-containing microglia is found in the hippocampus of AD patients under magnetic resonance imaging (Zeineh et al., 2015). Microglia acquires iron from transferring or non-transferring, extracellular and intracellular sources (McCarthy et al., 2018). Selective and sustained KLF4 expression can be induced in the nucleus and cytoplasm of ischemic hippocampal reactive astrocytes (Park et al., 2014). Studies have shown that KLF4 acts as a transcriptional repressor. It down-regulates the expression of ELK-3, and then ELK-3 inhibits the expression of HO-1 (Tsoyi et al., 2015). Heme oxygenase-1 (HO-1) is a stress protein that degrades heme into bilirubin, free iron, and carbon monoxide. Up-regulation of HO-1 in astrocytes can lead to abnormal iron deposition and mitochondrial dysfunction in the brain, leading to decreased cognitive ability (Schipper, 1999, 2004). Therefore, KLF4 may be involved in the process of iron accumulation in astrocytes, exacerbating oxidation in AD and aggravating brain damage.
KLF4 is commonly known to play a pivotal role in regulating cell proliferation, apoptosis, and differentiation. Previous studies have focused on the regulation of KLF4 in several important neurophysiological processes, including neuroinflammation, neuroprotection and synaptic regeneration. Recently, KLF4 has been found to play an important role in the pathogenesis of AD. In this article, we review the role of KLF4 in neuroprotection and neurogenesis in AD.
KLF4 is not only a regulator of regulation of cell proliferation and differentiation, but also a potential target for regulating immune responses. KLF4 may regulate negative inflammatory factors and promote inflammatory response, and have a great effect on the expression of astrocyte nuclear microglia. In addition, KLF4 and ERK5 can act together to exert neuroprotective actions. Furthermore, axon regeneration can be accomplished by altering the content of specific transcription factors, intracellular inhibitors, or altering intracellular signaling pathways. Knocking out KLF4 can enhance the axon regeneration and accelerate axon growth rate. Reduction of KLF4 expression promotes axon regeneration through the JAK-STAT3 pathway, and KLF4 promotes the JAK-STAT3 pathway to further axon regeneration. Therefore, KLF4 might be involved in the process of anti-inflammatory, anti-apoptosis, axon regeneration and iron accumulation in the CNS, which plays a pivotal role in the AD generation. These findings suggest that KLF4 represents a potential therapeutic target for AD. However, the deep cellular and molecular mechanisms of the effects of KLF4 on AD remain unclear and further investigations are needed.
ZQC, XHZ, and YJ wrote the manuscript. SHG and JYL modified the framework of the manuscript. BJL and RJC provided the critical revisions. All authors approved the final version of the manuscript for submission.
This work was supported by grants from the Natural Science Foundation of China (NSFC) (81871070, 31571126, 31471120) and Jilin Science and Technology Agency funding (Grant Nos. 20180519003JH, 20180414051GH, 20170414034GH and 20180414050GH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adlard, P. A., and Bush, A. I. (2006). Metals and Alzheimer’s disease. J. Alzheimers Dis. 10, 145–163. doi: 10.3233/JAD-2006-102-303
Aisen, P., Enns, C., and Wessling-Resnick, M. (2001). Chemistry and biology of eukaryotic iron metabolism. Int. J. Biochem. Cell Biol. 33, 940–959. doi: 10.1016/S1357-2725(01)00063-2
Alder, J. K., Georgantas, R. W., Hildreth, R. L., Kaplan, I. M., Morisot, S., Yu, X., et al. (2008). Kruppel-like factor 4 is essential for inflammatory monocyte differentiation in vivo. J. Immunol. 180, 5645–5652. doi: 10.4049/jimmunol.180.8.5645
Bieker, J. J. (2001). Krüppel-like factors: three fingers in many pies. J. Biol. Chem. 276, 34355–34358. doi: 10.1074/jbc.R100043200
Bin, L., Deng, L., Yang, H., Zhu, L., Wang, X., Edwards, M. G., et al. (2016). Forkhead box C1 regulates human primary keratinocyte terminal differentiation. PLoS One 11:e0167392. doi: 10.1371/journal.pone.0167392
Cacabelos, R. (2018). Have there been improvements in Alzheimer’s disease drug discovery over the past 5 years. Exp. Opin. Drug Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
Caccamo, A., Branca, C., Piras, I. S., Ferreira, E., Huentelman, M. J., Liang, W. S., et al. (2017). Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20, 1236–1246. doi: 10.1038/nn.4608
Cai, Z., Zhao, B., and Ratka, A. (2011). Oxidative stress and β-amyloid protein in Alzheimer’s disease. Neuromol. Med. 13, 223–250. doi: 10.1007/s12017-011-8155-9
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, J., Wang, X., Yi, X., Wang, Y., Liu, Q., and Ge, R. (2013). Induction of KLF4 contributes to the neurotoxicity of MPP+in M17 cells: a new implication in Parkinson’s disease. J. Mol. Neurosci. 51, 109–117. doi: 10.1007/s12031-013-9961-3
Chen, W. C., Lin, H. H., and Tang, M. J. (2015). Matrix-stiffness-regulated inverse expression of krüppel-like factor 5 and krüppel-like factor 4 in the pathogenesis of renal fibrosis. Am. J. Pathol. 185, 2468–2481. doi: 10.1016/j.ajpath.2015.05.019
Citron, M. (2010). Alzheimer’s disease: strategies for disease modification. Nat. Rev. Drug Discov. 9, 387–398. doi: 10.1038/nrd2896
Coleman, M. P. (2013). The challenges of axon survival: introduction to the special issue on axonal degeneration. Exp. Neurol. 246, 1–5. doi: 10.1016/j.expneurol.2013.06.007
Cui, D. M., Zeng, T., Ren, J., Wang, K., Jin, Y., Zhou, L., et al. (2017). KLF4 knockdown attenuates TBI-induced neuronal damage through p53 and JAK-STAT3 signaling. CNS Neurosci. Ther. 23, 106–118. doi: 10.1111/cns.12633
Cuttano, R., Rudini, N., Bravi, L., Corada, M., Giampietro, C., Papa, E., et al. (2016). KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 8, 6–24. doi: 10.15252/emmm.201505433
Dallman, P. R., and Spirito, R. A. (1977). Brain iron in the rat: extremely slow turnover in normal rats may explain long-lasting effects of early iron deficiency. J. Nutr. 107, 1075–1081. doi: 10.1093/jn/107.6.1075
Ding, B., Chen, K. M., Ling, H. W., Sun, F., Li, X., Wan, T., et al. (2009). Correlation of iron in the hippocampus with MMSE in patients with Alzheimer’s disease. J. Magn. Reson. Imaging 29, 793–798. doi: 10.1002/jmri.21730
Ding, B., Liu, P., Liu, W., Sun, P., and Wang, C. L. (2015). Emerging roles of Krüppel-like factor 4 in cancer and cancer stem cells. Asian Pac. J. Cancer Prev. 16, 3629–3633. doi: 10.7314/APJCP.2015.16.9.3629
Doméné, A., Cavanagh, C., Page, G., Bodard, S., Klein, C., Delarasse, C., et al. (2016). Expression of phenotypic astrocyte marker is increased in a transgenic mouse model of alzheimer’s disease versus age-matched controls: a presymptomatic stage study. Int. J. Alzheimers Dis. 2016:5696241. doi: 10.1155/2016/5696241
Evans, P. M., and Liu, C. (2008). Roles of Krüpel-like factor 4 in normal homeostasis, cancer and stem cells. Acta Biochim. Biophys. Sin. (Shanghai) 40, 554–564. doi: 10.1111/j.1745-7270.2008.00439.x
Fang, J., Shaw, P. X., Wang, Y., and Goldberg, J. L. (2016). Krüppel-like factor 4 (KLF4) is not required for retinal cell differentiation. eNeuro 3, ENEURO.117–ENEURO.115. doi: 10.1523/ENEURO.0117-15.2016
Ghaleb, A. M., McConnell, B. B., Kaestner, K. H., and Yang, V. W. (2011). Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the Krüppel-like factor 4 gene. Dev. Biol. 349, 310–320. doi: 10.1016/j.ydbio.2010.11.001
Ghaleb, A. M., and Yang, V. W. (2017). Krüppel-like factor 4 (KLF4): what we currently know. Gene 611, 27–37. doi: 10.1016/j.gene.2017.02.025
Ghosh, A., and Pahan, K. (2012). Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders. J. Biol. Chem. 287, 27189–27203. doi: 10.1074/jbc.M112.346932
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer’s disease. Science 314, 777–781. doi: 10.1126/science.1132814
Goldberg, J. L., and Barres, B. A. (2000). The relationship between neuronal survival and regeneration. Annu. Rev. Neurosci. 23, 579–612. doi: 10.1146/annurev.neuro.23.1.579
Goldberg, J. L., Klassen, M. P., Hua, Y., and Barres, B. A. (2002). Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 296, 1860–1864. doi: 10.1126/science.1068428
Griffin, W. S., and Barger, S. W. (2010). Neuroinflammatory cytokines-the common thread in alzheimer’s pathogenesis. US Neurol. 6, 19–27. doi: 10.17925/USN.2010.06.02.19
Guo, C., Wang, T., Zheng, W., Shan, Z. Y., Teng, W. P., and Wang, Z. Y. (2013). Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 562–575. doi: 10.1016/j.neurobiolaging.2012.05.009
Guo, J. W., Guan, P. P., Ding, W. Y., Wang, S. L., Huang, X. S., Wang, Z. Y., et al. (2017). Erythrocyte membrane-encapsulated celecoxib improves the cognitive decline of Alzheimer’s disease by concurrently inducing neurogenesis and reducing apoptosis in APP/PS1 transgenic mice. Biomaterials 145, 106–127. doi: 10.1016/j.biomaterials.2017.07.023
Han, F., Wang, W., Chen, B., Chen, C., Li, S., Lu, X., et al. (2015). Human induced pluripotent stem cell-derived neurons improve motor asymmetry in a 6-hydroxydopamine-induced rat model of Parkinson’s disease. Cytotherapy 17, 665–679. doi: 10.1016/j.jcyt.2015.02.001
He, M., Zheng, B., Zhang, Y., Zhang, X. H., Wang, C., Yang, Z., et al. (2015). KLF4 mediates the link between TGF-β1-induced gene transcription and H3 acetylation in vascular smooth muscle cells. FASEB J. 29, 4059–4070. doi: 10.1096/fj.15-272658
Holtzman, D. M., Morris, J. C., and Goate, A. M. (2011). Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3:77sr1. doi: 10.1126/scitranslmed.3002369
Hooda, J., Shah, A., and Zhang, L. (2014). Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients 6, 1080–1102. doi: 10.3390/nu6031080
Hu, D., Gur, M., Zhou, Z., Gamper, A., Hung, M. C., Fujita, N., et al. (2015). Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 6:8419. doi: 10.1038/ncomms9419
Imbernon, M., Sanchez-Rebordelo, E., Gallego, R., Gandara, M., Lear, P., Lopez, M., et al. (2014). Hypothalamic KLF4 mediates leptin’s effects on food intake via AgRP. Mol. Metab. 3, 441–451. doi: 10.1016/j.molmet.2014.04.001
Jiang, S., Nandy, P., Wang, W., Ma, X., Hsia, J., Wang, C., et al. (2018). Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol. Neurodegener. 13:5. doi: 10.1186/s13024-018-0238-8
Kapoor, N., Niu, J., Saad, Y., Kumar, S., Sirakova, T., Becerra, E., et al. (2015). Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J. Immunol. 194, 6011–6023. doi: 10.4049/jimmunol.1402797
Karam, J., Fadous-Khalifé, M. C., Tannous, R., Fakhreddine, S., Massoud, M., Hadchity, J., et al. (2017). Role of Krüppel-like factor 4 and heat shock protein 27 in cancer of the larynx. Mol. Clin. Oncol. 7, 808–814. doi: 10.3892/mco.2017.1412
Kaushik, D. K., Gupta, M., Das, S., and Basu, A. (2010). Krüppel-like factor 4, a novel transcription factor regulates microglial activation and subsequent neuroinflammation. J. Neuroinflamm. 7:68. doi: 10.1186/1742-2094-7-68
Kaushik, D. K., Thounaojam, M. C., Kumawat, K. L., Gupta, M., and Basu, A. (2013). Interleukin-1β orchestrates underlying inflammatory responses in microglia via Krüppel-like factor 4. J. Neurochem. 127, 233–244. doi: 10.1111/jnc.12382
Kong, B., Wu, P. C., Chen, L., Yang, T., Yuan, Y. Q., Kuang, Y. Q., et al. (2016). microRNA-7 protects against 1-Methyl-4-phenylpyridinium iodide-induced cell apoptosis in SH-SY5Y cells by directly targeting krüpple-like factor 4. DNA Cell Biol. 35, 217–225. doi: 10.1089/dna.2015.3097
Kurucz, A., Bombicz, M., Kiss, R., Priksz, D., Varga, B., Hortobágyi, T., et al. (2018). Heme oxygenase-1 activity as a correlate to exercise-mediated amelioration of cognitive decline and neuropathological alterations in an aging rat model of dementia. Biomed. Res. Int. 2018:7212861. doi: 10.1155/2018/7212861
Lee, H. P., Pancholi, N., Esposito, L., Previll, L. A., Wang, X., Zhu, X., et al. (2012). Early induction of oxidative stress in mouse model of Alzheimer disease with reduced mitochondrial superoxide dismutase activity. PLoS One 7:e28033. doi: 10.1371/journal.pone.0028033
Li, L., Zi, X., Hou, D., and Tu, Q. (2017). Krüppel-like factor 4 regulates amyloid-β (Aβ)-induced neuroinflammation in Alzheimer’s disease. Neurosci. Lett. 643, 131–137. doi: 10.1016/j.neulet.2017.02.017
Li, H., Zhang, C., Shen, H., Shen, Z., Wu, L., Mo, F., et al. (2017). Physiological stress-induced corticosterone increases heme uptake via KLF4-HCP1 signaling pathway in hippocampus neurons. Sci. Rep. 7:5745. doi: 10.1038/s41598-017-06058-6
Li, S., Hong, S., Shepardson, N. E., Walsh, D. M., Shankar, G. M., and Selkoe, D. (2009). Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. doi: 10.1016/j.neuron.2009.05.012
Li, Z., Zhao, J., Li, Q., Yang, W., Song, Q., Li, W., et al. (2010). KLF4 promotes hydrogen-peroxide-induced apoptosis of chronic myeloid leukemia cells involving the bcl-2/bax pathway. Cell Stress Chaper. 15, 905–912. doi: 10.1007/s12192-010-0199-5
Liu, H., Lin, H., Zhang, L., Sun, Q., Yuan, G., Zhang, L., et al. (2013). miR-145 and miR-143 regulate odontoblast differentiation through targeting Klf4 and Osx genes in a feedback loop. J. Biol. Chem. 288, 9261–9271. doi: 10.1074/jbc.M112.433730
Liu, Y., Shi, J., and Chen, X. (2015). Identification of novel targets for seasonal allergic rhinitis during and outside the pollen season by microarray analysis. Acta Otolaryngol. 135, 1330–1336. doi: 10.3109/00016489.2015.1067906
Liu, C., La Rosa, S., and Hagos, E. G. (2015). Oxidative DNA damage causes premature senescence in mouse embryonic fibroblasts deficient for Krüppel-like factor 4. Mol. Carcinog. 54, 889–899. doi: 10.1002/mc.22161
Mamonkin, M., Shen, Y., Lee, P. H., Puppi, M., Park, C. S., and Lacorazza, H. D. (2013). Differential roles of KLF4 in the development and differentiation of CD8 + T cells. Immunol. Lett. 156, 94–101. doi: 10.1016/j.imlet.2013.09.008
McCarthy, R. C., Sosa, J. C., Gardeck, A. M., Baez, A. S., Lee, C. H., and Wessling-Resnick, M. (2018). Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 293, 7853–7863. doi: 10.1074/jbc.RA118.001949
Miao, X., Wu, X., and Shi, W. (2017). MicroRNA-346 regulates neural stem cell proliferation and differentiation by targeting KLF4. Am. J. Transl. Res. 9, 5400–5410.
Moore, D. L., Blackmore, M. G., Hu, Y., Kaestner, K. H., Bixby, J. L., Lemmon, V. P., et al. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301. doi: 10.1126/science.1175737
Murgai, M., Ju, W., Eason, M., Kline, J., Beury, D. W., Kaczanowska, S., et al. (2017). KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. 23, 1176–1190. doi: 10.1038/nm.4400
Nie, X., Liang, L., Xi, H., Jiang, S., Jiang, J., Tang, C., et al. (2015). 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin induces premature senescence of astrocytes via WNT/β-catenin signaling and ROS production. J. Appl. Toxicol. 35, 851–860. doi: 10.1002/jat.3084
Nishiguchi, M., Kikuyama, H., Kanazawa, T., Tsutsumi, A., Kaneko, T., Uenishi, H., et al. (2015). Increases in iPS transcription factor (Oct4, Sox2, c-Myc, and Klf4) gene expression after modified electroconvulsive therapy. Psychiatry Investig 12, 532–537. doi: 10.4306/pi.2015.12.4.532
Ohnesorge, N., Viemann, D., Schmidt, N., Czymai, T., Spiering, D., Schmolke, M., et al. (2010). Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4). J. Biol. Chem. 285, 26199–26210. doi: 10.1074/jbc.M110.103127
Parajuli, B., Sonobe, Y., Horiuchi, H., Takeuchi, H., Mizuno, T., and Suzumura, A. (2013). Oligomeric amyloid β induces IL-1β processing via production of ROS: implication in Alzheimer’s disease. Cell Death Dis. 4, e975. doi: 10.1038/cddis.2013.503
Park, J. H., Riew, T. R., Shin, Y. J., Park, J. M., Cho, J. M., and Lee, M. Y. (2014). Induction of Krüppel-like factor 4 expression in reactive astrocytes following ischemic injury in vitro and in vivo. Histochem. Cell Biol. 141, 33–42. doi: 10.1007/s00418-013-1134-5
Pearson, R., Fleetwood, J., Eaton, S., Crossley, M., and Bao, S. (2008). Krüppel-like transcription factors: a functional family. Int. J. Biochem. Cell Biol. 40, 1996–2001. doi: 10.1016/j.biocel.2007.07.018
Peters, D. G., Connor, J. R., and Meadowcroft, M. D. (2015). The relationship between iron dyshomeostasis and amyloidogenesis in Alzheimer’s disease: two sides of the same coin. Neurobiol. Dis. 81, 49–65. doi: 10.1016/j.nbd.2015.08.007
Qin, S., Liu, M., Niu, W., and Zhang, C. L. (2011). Dysregulation of kruppel-like factor 4 during brain development leads to hydrocephalus in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 21117–21121. doi: 10.1073/pnas.1112351109
Qin, S., and Zhang, C. L. (2012). Role of Kruppel-like factor 4 in neurogenesis and radial neuronal migration in the developing cerebral cortex. Mol. Cell. Biol. 32, 4297–4305. doi: 10.1128/MCB.00838-12
Qin, S., Zou, Y., and Zhang, C. L. (2013). Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat. Commun. 4:2633. doi: 10.1038/ncomms3633
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Rajmohan, R., and Reddy, P. H. (2017). Amyloid-Beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J. Alzheimers Dis. 57, 975–999. doi: 10.3233/JAD-160612
Rodríguez, J. J., Butt, A. M., Gardenal, E., Parpura, V., and Verkhratsky, A. (2016). Complex and differential glial responses in Alzheimer’s disease and ageing. Curr. Alzheimer Res. 13, 343–358. doi: 10.2174/1567205013666160229112911
Rubio-Perez, J. M., and Morillas-Ruiz, J. M. (2012). A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012:756357. doi: 10.1100/2012/756357
Schipper, H. M. (1999). Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox. Res. 1, 57–70. doi: 10.1007/BF03033339
Schipper, H. M. (2004). Heme oxygenase-1: transducer of pathological brain iron sequestration under oxidative stress. Ann. N. Y. Acad. Sci. 1012, 84–93. doi: 10.1196/annals.1306.007
Segre, J. A., Bauer, C., and Fuchs, E. (1999). Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 22, 356–360. doi: 10.1038/11926
Selkoe, D. J. (1991). The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498. doi: 10.1016/0896-6273(91)90052-2
Serini, S., and Calviello, G. (2016). Reduction of oxidative/nitrosative stress in brain and its involvement in the neuroprotective effect of n-3 PUFA in Alzheimer’s disease. Curr. Alzheimer Res. 13, 123–134. doi: 10.2174/1567205012666150921101147
Sevilla, L. M., Latorre, V., Carceller, E., Boix, J., Vodák, D., Mills, I. G., et al. (2015). Glucocorticoid receptor and Klf4 co-regulate anti-inflammatory genes in keratinocytes. Mol. Cell. Endocrinol. 412, 281–289. doi: 10.1016/j.mce.2015.05.015
Shi, M., Cui, J., Du, J., Wei, D., Jia, Z., Zhang, J., et al. (2014). A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin. Cancer Res. 20, 4370–4380. doi: 10.1158/1078-0432.CCR-14-0186
Shields, J. M., Christy, R. J., and Yang, V. W. (1996). Identification and characterization of a gene encoding a gut-enriched Krüppel-like factor expressed during growth arrest. J. Biol. Chem. 271, 20009–20017. doi: 10.1074/jbc.271.33.20009
Shields, J. M., and Yang, V. W. (1998). Identification of the DNA sequence that interacts with the gut-enriched Krüppel-like factor. Nucleic Acids Res. 26, 796–802. doi: 10.1093/nar/26.3.796
Shinohara, M., Fujioka, S., Murray, M. E., Wojtas, A., Baker, M., Rovelet-Lecrux, A., et al. (2014). Regional distribution of synaptic markers and APP correlate with distinct clinicopathological features in sporadic and familial Alzheimer’s disease. Brain 137, 1533–1549. doi: 10.1093/brain/awu046
Smith, P. D., Sun, F., Park, K. K., Cai, B., Wang, C., Kuwako, K., et al. (2009). SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 64, 617–623. doi: 10.1016/j.neuron.2009.11.021
Song, Y., Liu, Y., and Chen, X. (2018). MiR-212 attenuates MPP+-induced neuronal damage by targeting KLF4 in SH-SY5Y Cells. Yonsei Med. J. 59, 416–424. doi: 10.3349/ymj.2018.59.3.416
Steketee, M. B., Oboudiyat, C., Daneman, R., Trakhtenberg, E., Lamoureux, P., Weinstein, J. E., et al. (2014). Regulation of intrinsic axon growth ability at retinal ganglion cell growth cones. Invest. Ophthalmol. Vis. Sci. 55, 4369–4377. doi: 10.1167/iovs.14-13882
Su, C., Sun, F., Cunningham, R. L., Rybalchenko, N., and Singh, M. (2014). ERK5/KLF4 signaling as a common mediator of the neuroprotective effects of both nerve growth factor and hydrogen peroxide preconditioning. Age (Dordr) 36:9685. doi: 10.1007/s11357-014-9685-5
Sun, F., Park, K. K., Belin, S., Wang, D., Lu, T., Chen, G., et al. (2011). Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 480, 372–375. doi: 10.1038/nature10594
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tronel, C., Rochefort, G. Y., Arlicot, N., Bodard, S., Chalon, S., and Antier, D. (2013). Oxidative stress is related to the deleterious effects of heme oxygenase-1 in an in vivo neuroinflammatory rat model. Oxid Med. Cell Longev. 2013:264935. doi: 10.1155/2013/264935
Tsoyi, K., Geldart, A. M., Christou, H., Liu, X., Chung, S. W., and Perrella, M. A. (2015). Elk-3 is a KLF4-regulated gene that modulates the phagocytosis of bacteria by macrophages. J. Leukoc. Biol. 97, 171–180. doi: 10.1189/jlb.4A0214-087R
van Bergen, J. M. G., Li, X., Quevenco, F. C., Gietl, A. F., Treyer, V., Meyer, R., et al. (2018). Simultaneous quantitative susceptibility mapping and Flutemetamol-PET suggests local correlation of iron and β-amyloid as an indicator of cognitive performance at high age. Neuroimage 174, 308–316. doi: 10.1016/j.neuroimage.2018.03.021
van Schaijik, B., Davis, P. F., Wickremesekera, A. C., Tan, S. T., and Itinteang, T. (2018). Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: a review. J. Clin. Pathol. 71, 88–91. doi: 10.1136/jclinpath-2017-204815
Verdile, G., Keane, K. N., Cruzat, V. F., Medic, S., Sabale, M., Rowles, J., et al. (2015). Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediat. Inflamm. 2015:105828. doi: 10.1155/2015/105828
Wei, D., Kanai, M., Huang, S., and Xie, K. (2006). Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis 27, 23–31. doi: 10.1093/carcin/bgi243
Weinberg, R. P., Koledova, V. V., Shin, H., Park, J. H., Tan, Y. A., Sinskey, A. J., et al. (2018). Oil palm phenolics inhibit the in vitro aggregation of β-amyloid peptide into oligomeric complexes. Int. J. Alzheimers Dis. 2018:7608038. doi: 10.1155/2018/7608038
Wu, Z., Zhao, Y., and Zhao, B. (2010). Superoxide anion, uncoupling proteins and Alzheimer’s disease. J. Clin. Biochem. Nutr. 46, 187–194. doi: 10.3164/jcbn.09-104-2
Xi, J., Zhang, B., Luo, F., Liu, J., and Yang, T. (2012). Quercetin protects neuroblastoma SH-SY5Y cells against oxidative stress by inhibiting expression of Krüppel-like factor 4. Neurosci. Lett. 527, 115–120. doi: 10.1016/j.neulet.2012.08.082
Xie, L. Q., Sun, H. P., Wang, T., Tang, H. L., Wang, P., Zhu, J. H., et al. (2013). Reprogramming of adult human neural stem cells into induced pluripotent stem cells. Chin. Med. J. 126, 1138–1143.
Xu, L., Sun, H., Zhang, M., Jiang, Y., Zhang, C., Zhou, J., et al. (2017). MicroRNA-145 protects follicular granulosa cells against oxidative stress-induced apoptosis by targeting Krüppel-like factor 4. Mol. Cell. Endocrinol. 452, 138–147. doi: 10.1016/j.mce.2017.05.030
Yadav, S. S., Nair, R. R., and Yadava, P. K. (2018). KLF4 signalling in carcinogenesis and epigenetic regulation of hTERT. Med. Hypotheses 115, 50–53. doi: 10.1016/j.mehy.2018.03.012
Yang, H., Xi, X., Zhao, B., Su, Z., and Wang, Z. (2018). KLF4 protects brain microvascular endothelial cells from ischemic stroke induced apoptosis by transcriptionally activating MALAT1. Biochem. Biophys. Res. Commun. 495, 2376–2382. doi: 10.1016/j.bbrc.2017.11.205
Yet, S. F., McA’Nulty, M. M., Folta, S. C., Yen, H. W., Yoshizumi, M., Hsieh, C. M., et al. (1998). Human EZF, a Krüppel-like zinc finger protein, is expressed in vascular endothelial cells and contains transcriptional activation and repression domains. J. Biol. Chem. 273, 1026–1031. doi: 10.1074/jbc.273.2.1026
Yoon, G., Shah, S. A., Ali, T., and Kim, M. O. (2018). The adiponectin homolog osmotin enhances neurite outgrowth and synaptic complexity via AdipoR1/NgR1 signaling in Alzheimer’s disease. Mol. Neurobiol. 55, 6673–6686. doi: 10.1007/s12035-017-0847-1
Yoshida, T., Yamashita, M., Horimai, C., and Hayashi, M. (2014). Deletion of Krüppel-like factor 4 in endothelial and hematopoietic cells enhances neointimal formation following vascular injury. J. Am. Heart. Assoc. 3:e000622. doi: 10.1161/JAHA.113.000622
Yu, S., Feng, Y., Shen, Z., and Li, M. (2011). Diet supplementation with iron augments brain oxidative stress status in a rat model of psychological stress. Nutrition 27, 1048–1052. doi: 10.1016/j.nut.2010.11.007
Yuan, Z. L., Guan, Y. J., Chatterjee, D., and Chin, Y. E. (2005). Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273. doi: 10.1126/science.1105166
Yui, D., Nishida, Y., Nishina, T., Mogushi, K., Tajiri, M., Ishibashi, S., et al. (2015). Enhanced phospholipase A2 group 3 expression by oxidative stress decreases the insulin-degrading enzyme. PLoS One 10:e0143518. doi: 10.1371/journal.pone.0143518
Zeineh, M. M., Chen, Y., Kitzler, H. H., Hammond, R., Vogel, H., and Rutt, B. K. (2015). Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging 36, 2483–2500. doi: 10.1016/j.neurobiolaging.2015.05.022
Zhang, D., Zhu, L., Li, C., Mu, J., Fu, Y., Zhu, Q., et al. (2015). Sialyltransferase7A, a Klf4-responsive gene, promotes cardiomyocyte apoptosis during myocardial infarction. Basic Res. Cardiol. 110:28. doi: 10.1007/s00395-015-0484-7
Keywords: Alzheimer’s disease, KLF4, stem cell, neuroinflammation, apoptosis
Citation: Cheng Z, Zou X, Jin Y, Gao S, Lv J, Li B and Cui R (2018) The Role of KLF4 in Alzheimer’s Disease. Front. Cell. Neurosci. 12:325. doi: 10.3389/fncel.2018.00325
Received: 02 June 2018; Accepted: 07 September 2018;
Published: 21 September 2018.
Edited by:
Chuang Wang, Ningbo University, ChinaReviewed by:
Sergio Machado, Salgado de Oliveira University, BrazilCopyright © 2018 Cheng, Zou, Jin, Gao, Lv, Li and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bingjin Li, bGliaW5namluQGpsdS5lZHUuY24= Ranji Cui, Y3VpcmFuamlAamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.