 Zsuzsanna Szepesi
Zsuzsanna Szepesi Oscar Manouchehrian
Oscar Manouchehrian Sara Bachiller
Sara Bachiller Tomas Deierborg
Tomas Deierborg
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 27 September 2018
Sec. Non-Neuronal Cells
Volume 12 - 2018 | https://doi.org/10.3389/fncel.2018.00323
This article is part of the Research TopicMicroglia in Neurodegenerative Disorders: in Sickness and in HealthView all 36 articles
Microglia are ramified cells that exhibit highly motile processes, which continuously survey the brain parenchyma and react to any insult to the CNS homeostasis. Although microglia have long been recognized as a crucial player in generating and maintaining inflammatory responses in the CNS, now it has become clear, that their function are much more diverse, particularly in the healthy brain. The innate immune response and phagocytosis represent only a little segment of microglia functional repertoire that also includes maintenance of biochemical homeostasis, neuronal circuit maturation during development and experience-dependent remodeling of neuronal circuits in the adult brain. Being equipped by numerous receptors and cell surface molecules microglia can perform bidirectional interactions with other cell types in the CNS. There is accumulating evidence showing that neurons inform microglia about their status and thus are capable of controlling microglial activation and motility while microglia also modulate neuronal activities. This review addresses the topic: how microglia communicate with other cell types in the brain, including fractalkine signaling, secreted soluble factors and extracellular vesicles. We summarize the current state of knowledge of physiological role and function of microglia during brain development and in the mature brain and further highlight microglial contribution to brain pathologies such as Alzheimer’s and Parkinson’s disease, brain ischemia, traumatic brain injury, brain tumor as well as neuropsychiatric diseases (depression, bipolar disorder, and schizophrenia).
Microglia are the resident immune cells of the central nervous system, which represent about 5–12% of total CNS cells in the healthy brain and the spinal cord. Microglia are derived from the myeloid precursors cells in the embryonic yolk sac and they travel to the area of the developing CNS during early embryogenesis (Schulz et al., 2012; Ginhoux et al., 2013; Kierdorf and Prinz, 2013; Ginhoux and Prinz, 2015; Wieghofer and Prinz, 2016; Hoeffel and Ginhoux, 2018). Microglial progenitors are already present around the neural tube at embryonic day 9 in mice, and from the fifth gestational week in humans. After neural entry, microglia progenitors migrate through the developing nerve tissue and undergo massive proliferation before they reach their final density (Ginhoux et al., 2010; Verney et al., 2010). Embryonic microglia develop into highly ramified mature microglia via regulatory factors such as Pu.1, IL-34, and CSF-1 (Schulz et al., 2012; Kierdorf et al., 2013). Microglia are broadly distributed throughout the CNS although quantity and location among species varies (Lawson et al., 1990; Mittelbronn et al., 2001). Despite their broad distribution, microglia represent a rather heterogeneous community with different subpopulation based on specific brain regions. Each microglia subpopulation develop unique features and they can be distinguished by capacities and functions (Gertig and Hanisch, 2014; Neiva et al., 2014; Doorn et al., 2015; Grabert et al., 2016; De Biase et al., 2017). Microglia are considered to be the resident macrophages in the CNS and are long-lived, self-renewing cells, autonomous from peripheral monocytes that normally do not enter the brain.
Phenotypically, ramified microglia have been described as ‘quiescent’ or remaining in a ‘resting’ state. However, these views radically changed after in vivo imaging studies have revealed the extraordinary active nature of microglia processes in the healthy brain (Davalos et al., 2005; Nimmerjahn et al., 2005). Microglia are constantly restless and their processes undergo continuous cycles of extension and withdrawal and de novo formation to scan their environment for disruptions in brain homeostasis thereby ‘resting’ microglia are able to acquire numerous phenotypes. Each microglia process seems to have a defined territory and they are able to scan their environment within several hours. Microglia process movement is systematically aimed at synapses to monitor and regulate neuronal activity, indicating the presence of specific signaling mechanisms that direct microglial processes to synapses (Li et al., 2012, 2013; Dissing-Olesen et al., 2014). When microglia detect ‘danger’ signals that compromise CNS homeostasis- for instance through pathogen recognition receptors- they rapidly change their appearance by shortening of cellular processes, enlargement of their soma and they transform into a reactive phenotype. Reactive microglia can further evolve into phagocytic or amoeboid microglia that completely lack cellular processes (Streit, 2000; Stence et al., 2001; Ransohoff and Perry, 2009; Graeber and Streit, 2010; Kettenmann et al., 2011). In response to CNS injury or infections, microglia are able to upregulate expression of many cell surface receptors including toll-like receptors (TLRs), phagocytic receptors (CR3, CR4), scavenger receptors (CD36, CD91) and release various complement factors (Tian et al., 2012). However, microglia activation does not refer to a single phenotype, and a continuum of microglia activation is rather considered. They are able to acquiring numerous phenotypes upon activation ranging from a phagocytic to an antigen presenting phenotype that mainly depends on the type of stimuli provided in their environment (Town et al., 2005; Schwartz et al., 2006; Boche et al., 2013). Once activated, microglia can be potent immune effector cells and initiate both innate and adaptive immune responses and produce a number of cytokines, chemokines and growth factors (Ransohoff and Perry, 2009; Loane and Kumar, 2016; von Bernhardi et al., 2016). Microglia accept a wide variety of inputs and they are also able to provide an appropriate response to a multitude of reactions. Therefore, microglia activation is considered to be a flexible and adaptive process rather than being stereotypic and granted (Schwartz et al., 2006; Hanisch and Kettenmann, 2007; Deczkowska et al., 2018). Importantly, microglia are also capable of morphological remodeling without any indication of an insult or neurodegeneration. Chronic stress, enhanced glutamatergic neurotransmission or light deprivation can lead to hyper-ramification of microglia and more frequent neuron–microglia contacts (Tremblay et al., 2010; Hinwood et al., 2012; Walker et al., 2013; Torres-Platas et al., 2014; Yirmiya et al., 2015). These observations suggest that there is information transmission between microglia and neurons and microglia are continuously informed about the actual activity or state of neurons in their vicinity.
Microglia are in close contact with neurons as well as oligodendrocytes and astrocytes. Astrocytes are the most abundant glial cells in the CNS and they show significant contribution to synapse formation, maintenance and elimination, thus regulating the overall architecture and activity of neuronal circuits. Astrocytes perform direct contacts with their neuronal pre- and postsynaptic counterparts and they also release soluble factors to modulate synaptic transmission of both excitatory and inhibitory synapses. This led to the concept of the ‘tripartite synapse,’ a synapse composed of two neurons and an astrocyte as an integrated functional unit (Perea et al., 2009; Nistico et al., 2017; Farhy-Tselnicker and Allen, 2018). The tripartite synapses are supplemented by microglia, which are specially attracted by synapse activity, location of crosstalk between neurons and glial cells. In the healthy brain, microglia exhibit an actively repressed ‘surveying’ phenotype that is dependent on a dynamic crosstalk between microglia and neurons (Biber et al., 2007; Kettenmann et al., 2011; Kierdorf and Prinz, 2017). It has been proposed that the removal of this neuronal derived inhibitory control represents a type of danger signal for microglia, indicating that neuronal function is impaired and leads to alterations of microglia morphology and function. The reciprocal neuron–microglia communication is mediated by numerous soluble factors, extracellular vesicles (EVs) as well as contact-dependent mechanisms, and is essential for adaptive neuroplasticity and learning (Posfai et al., 2018).
In the healthy brain microglia remain remarkably quiescent or ‘unactivated’ while undertaking surveillance roles. It is now recognized that this ‘unactivated’ state is under the control, at least in part, of neuronal factors, including CD200 and fractalkine (CX3CL1) as the most studied cell-to-cell proteins (Eyo and Wu, 2013) (Figures 1, 2). CD200 is a glycoprotein widely found on the cell membranes of neurons, astrocytes and oligodendrocytes in the CNS (Wright et al., 2001; Barclay et al., 2002; Koning et al., 2009). The target receptor of CD200, CD200R, is only found on macrophages and microglia (Hernangomez et al., 2012). When neuronal CD200 interacts with microglial CD200R, the microglia will be kept in its inactivated, resting state (Hoek et al., 2000; Biber et al., 2007) thus CD200 signaling plays a critical role in neuronal protection. Defaults in CD200 signaling have been observed in several neuroinflammatory conditions like in multiple sclerosis (MS), Alzheimer’s disease, or in the aging brain (Hernangomez et al., 2012; Varnum et al., 2015; Xie et al., 2017).
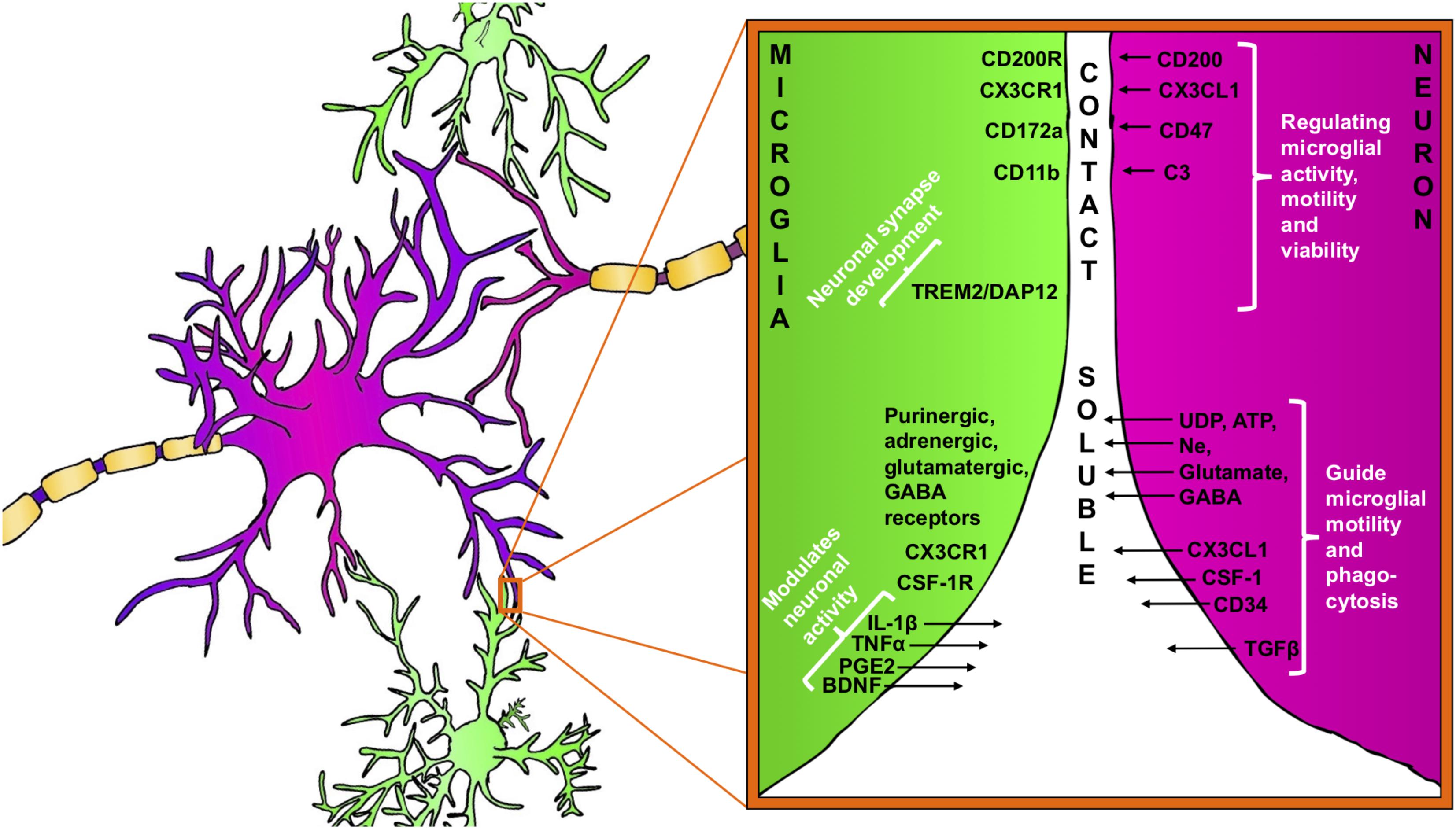
FIGURE 1. Bidirectional signaling between microglia (green) and neuron (purple). Neuron–microglia communication is mediated by receptor–ligand interactions as well as by various soluble factors. Microglia are equipped with a group of surface receptors, which trigger signals and regulate specific microglia function like phagocytosis, motility and viability. Many of the receptor ligands, such as CX3CR1, CD200R, and CD172a are released or expressed on the surface of neurons. Receptor–ligand interactions represent a classical contact dependent communication between microglia and neurons. Microglia and neurons reciprocally release soluble factors that can modulate cell functions and promote tissue homeostasis.
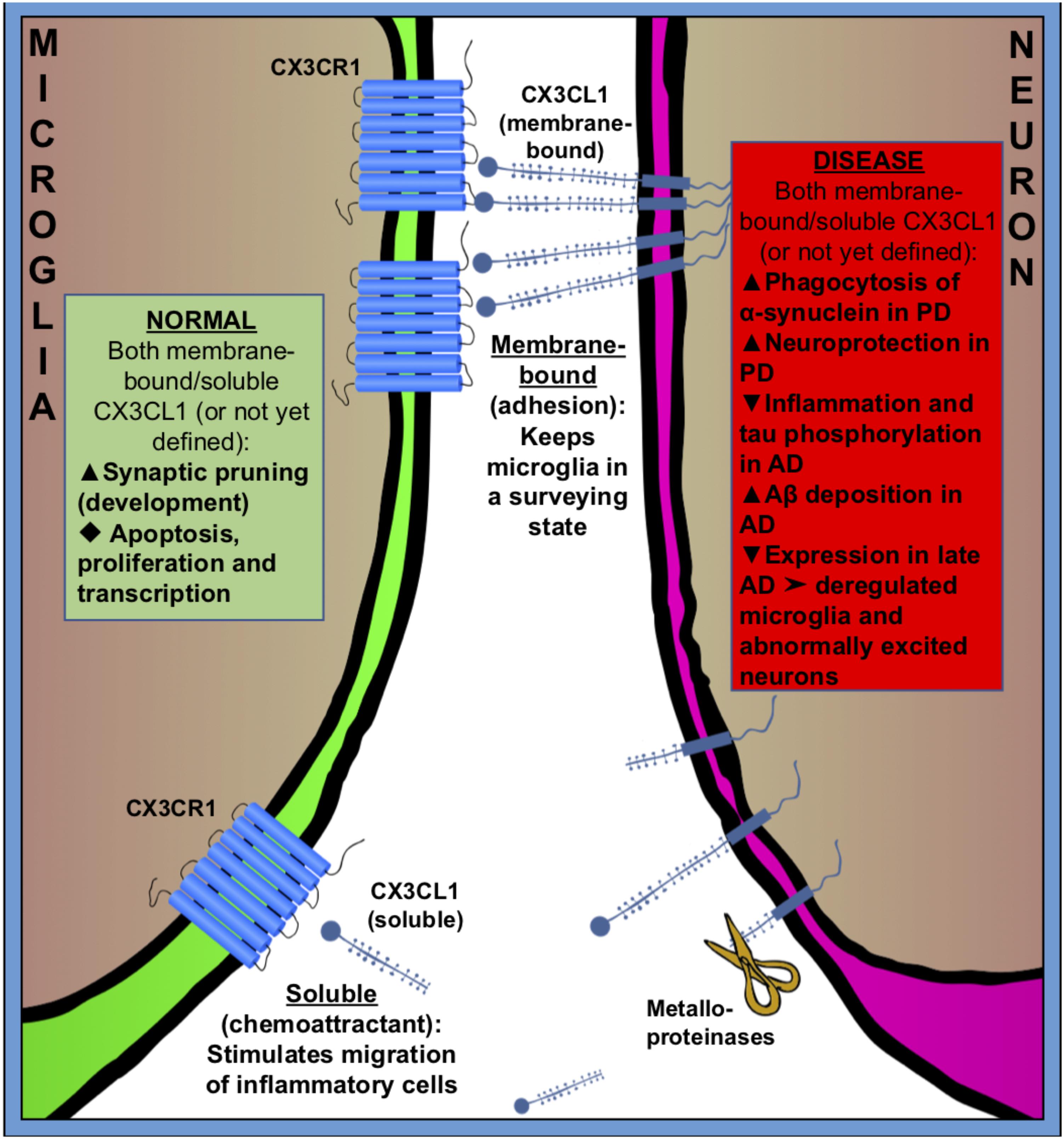
FIGURE 2. Fractalkine (CX3CR1–CX3CL1) signaling between microglia (green) and neuron (purple) in normal (green square) and pathologic (red square) states. CX3CL1 is either membrane-bound or cleaved by metalloproteinases to become soluble. Membrane bound CX3CL1 is an OFF-signal, that keeps microglia in a surveying state, and soluble CX3CL1 is believed to act as a chemoattractant, stimulating migration of inflammatory cells. Fractalkine signaling has many roles in both healthy and pathologic conditions. Black triangles indicate increase ( ), decrease (
), decrease ( ) or not specified (
) or not specified ( ).
).
The lack of CD200 in an experimental animal model for MS (EAE) revealed an accelerated progression of the MS symptoms, indicating that microglia attain a dysregulated activation in absence of CD200-signaling (Hoek et al., 2000).
Fractalkine, also known as CX3CL1, is a unique chemokine that is constitutively expressed in neurons (mainly in the prosencephalon) (Tarozzo et al., 2003). The fractalkine receptor, CX3CR1, is predominantly found on microglia and on neurons in less amount in the normal CNS (Harrison et al., 1998; Meucci et al., 2000; Hatori et al., 2002; Paolicelli et al., 2014) and CX3CL1–CX3CR1 signaling has been considered a neuronal ‘off signal’ that keeps microglia in a surveying phenotype (Nishiyori et al., 1998; Maciejewski-Lenoir et al., 1999; Biber et al., 2007). It should be noted that under chronic inflammatory conditions, glial cells also express CX3CL1 (Hughes et al., 2002).
CX3CL1 is found both as membrane-bound, to neuronal membranes, and cleaved (soluble) into the extracellular space (Figure 2). The cleavage of CX3CL1 in its heavily glycosylated chemokine domain is performed by metalloproteinases (MMPs) (Garton et al., 2001). CX3CL1 bound to the membrane is believed to be important for adhesion, while the soluble CX3CL1 acts a chemo-attractant, recruiting inflammatory cells (Bazan et al., 1997; Schall, 1997; Mizoue et al., 2001). The G-protein-linked receptor CX3CR1 triggers several intracellular secondary messengers, such as P13K, AKT, and NF-κB, and thus regulates apoptotic, proliferative, transcriptional and migratory functions in microglia (Al-Aoukaty et al., 1998; Chandrasekar et al., 2003). The fractalkine receptor is expressed by other cell types than microglia, such as monocytes, dendritic cells and natural killer cells, but because these cells don’t frequently cross the brain–blood barrier (BBB) in healthy brains, microglia are believed to be the main recipient of CX3CL1-signaling in the normal CNS (Imai et al., 1997; Harrison et al., 1998; Truman et al., 2008; Mizutani et al., 2012; Wolf et al., 2013).
Expression of fractalkine increases in the CNS during embryonic and postnatal maturation and promotes microglial recruitment to neuronal circuits that need rewiring during a period of activity-dependent remodeling. In return, microglia modulate neuronal function and survival via the release of trophic factors (Ueno et al., 2013). In the absence of fractalkine signaling, microglial colonization is impaired in the somatosensory, motor cortex and the hippocampus during the early postnatal development (Paolicelli and Gross, 2011; Hoshiko et al., 2012) (Figure 2).
Replacing the CX3XR1-gene with GFP, in the CX3CR1-GFP knock-in mouse line (Jung et al., 2000), has been proved to be a very useful tool to study microglial function and fractalkine signaling in the healthy brain. Experimental paradigms comparing CX3CR1-GFP homozygous mice (CX3CR1-/-) that entirely lack CX3CR1, and therefore of fractalkine signaling in the brain, with heterozygotes (CX3CR1-/+) proved to be extremely useful strategy to disclose the molecular players in neuron–microglia communication in a non-invasive way related to fractalkine signaling.
Besides fractalkine, numerous other chemokines are produced and released by neurons and glial cells (by both astrocytes and microglia) in the CNS. CCL2/MCP-1 is expressed in the neurons principally during pathological conditions as most studies have shown, although some observations support the notion of a constitutive neuronal CCL2 expression (Coughlan et al., 2000; Banisadr et al., 2005; Conductier et al., 2010). The receptor for CCL2 is found in microglia and its activation trigger chemotaxis of cultured microglia. An induction of neuronal CCL2 expression was described in response to various types of injury and degeneration such as ischemia, Alzheimer’s disease, MS, axonal injury, amyotrophic lateral sclerosis (ALS) or peripheral nerve injury (Barna et al., 1994; Che et al., 2001; Pang et al., 2001; Rancan et al., 2001; Schreiber et al., 2001; Baron et al., 2005; Bose and Cho, 2013; Perner et al., 2018). The chemokine CCL2 is also produced by glial cells (Barna et al., 1994; Hanisch, 2002; He et al., 2016) as perivascular astrocytes have been identified as the most common and predominant source of CCL2 in the CNS in various neuroinflammatory conditions (Andjelkovic et al., 2002; Guillemin et al., 2003). Another chemokine, CXCL12/SDF-1 (stromal cell-derived factor 1) and its receptor CXCR4 have been reported to be produced and expressed predominately by astrocytes and neurons. Astrocytic activation of CXCR4 can lead to the release of pro-inflammatory cytokines and prostaglandins, and it is able to trigger glutamate exocytosis from astrocytes thereby modulate neuronal activity. A unique chemokine, CCL21, is exclusively expressed on endangered neurons after injury and activate microglia through chemokine receptor CXCR3 (Biber et al., 2001; Dijkstra et al., 2004; van Weering et al., 2010).
Overall, chemokines are able to control neuron–microglia interactions through diverse intracellular signaling upon chemokine receptor activation and contribute to a variety of cellular functions such as modulation of neurotransmission, regulating cell survival and BBB permeability, exert neuroprotection, migration of neuronal progenitors, stem cells and axonal sprouting (Mackay, 2001; Mellado et al., 2001; Reaux-Le Goazigo et al., 2013; Zheng et al., 2017).
Furthermore, many other inhibitory receptors/molecules can be found on microglial surfaces that interact with ligands from neurons (both secreted and membrane-bound). An example is the integrin CD47, which communicates a “don’t eat me”-signal to microglial CD172a (van Beek et al., 2005; Biber et al., 2007) (Figure 1) by recruiting tyrosine-protein phosphatases SHP-1 and SHP-2, down-regulating phagocytosis as well as by increasing the synthesis of TGF-β (reviewed in Griffiths et al., 2007).
During the first week of postnatal life vast amounts of synapses are generated during an intensive period of synaptogenesis and this is also when microglia density reaches its maximum (Dalmau et al., 2003). Neuronal networks are refined by synaptic pruning, a selective and activity-driven process that eliminates redundant, developing synapses. Microglia make contacts with presynaptic axon terminals and postsynaptic dendritic spines that can be followed by a complement-dependent phagocytosis and elimination (Wakselman et al., 2008; Tremblay and Majewska, 2011; Cunningham et al., 2013; Jung and Chung, 2018).
Fractalkine signaling has been implicated in microglial pruning of dendritic spines during normal brain development (Figure 2). Paolicelli and Gross (2011) and Paolicelli et al. (2011) showed that a decreased microglial density in the hippocampus of CX3CR-/- mice was associated with a temporary boost in dendritic spine numbers on pyramidal neurons and an excess of immature synapses compared with CX3CR-/+ littermates. It is suggested that microglia lacking the CX3CR1 receptor fail to recognize synapses displaying fractalkine. Comparing homozygous CX3CL1-/- and heterozygous CX3CL1-/+ mice, microglial fractalkine signaling was shown to play a crucial role in the maturation of excitatory glutamatergic synapses and remodeling of neuronal circuits during postnatal development (Bertollini et al., 2006; Ragozzino et al., 2006; Hoshiko et al., 2012; Zhan et al., 2014) (Figure 2); although, the role of neuron–microglial crosstalk in this context is still not fully understood. In the developing barrel field of the somatosensory cortex, fractalkine (CX3CL1/CX3CR1) signaling has been also shown to regulate microglial recruitment to the location where developing thalamocortical synapses are concentrated (i.e., barrel centers). Hoshiko et al. (2012) showed that microglia entry into the barrel centers is temporally delayed in CX3CR1-/- mice compared with WTs at the developmental stage, when fractalkine is overexpressed within the barrels. CX3CR1 deficiency also delays the functional maturation of glutamate receptors and results in a significantly lower proportion of postsynaptic AMPARs in CX3CR1-/- than in CX3CR1-/+ mice. Moreover, the developmental switch from GluN2B to GluN2A-containing NMDA receptors, which are known to occur during the early weeks of postnatal development of thalamocortical area, was delayed in CX3CR1-/- mice but this delay was only transient. These observations support to the notion that microglia influence synaptic maturation during development and fractalkine signaling deficiency induced a transient impairment in the maturation of glutamate receptor functional expression at thalamocortical synapses. However, the exact mechanism by which microglial cells influence synapse maturation is complex and it is not fully understood. Microglia release several signaling molecules known to modulate the functional expression of glutamate receptors. For instance, glia-derived TNF-α facilitates AMPA receptors trafficking and membrane insertion (Beattie et al., 2002; Stellwagen et al., 2005). Also, TNF-α specifically controls the glutamate release step of gliotransmission in the hippocampal dentate gyrus (Santello et al., 2011). Similarly, brain-derived neurotrophic factor (BDNF) released by microglia has been demonstrated to modulate spine density and the expression of AMPA and NMDA receptors in cortical neurons of adult mice (Parkhurst et al., 2013). Pascual et al. (2012) have showed in vitro that microglia are able to regulate synaptic neurotransmission by releasing ATP, which binds to P2Y1R located on astrocytes and enhances excitatory postsynaptic current. Also, microglia regulate synaptic functions and neuronal development of cultured neurons through the interactions of the microglia-released interleukin 10 (IL-10) with IL-10 receptors (IL-10R) expressed by neurons (Lim et al., 2013). Thereby, fractalkine-dependent recruitment of microglia within the barrel centers might induce the release of microglia-derived signaling factors necessary for alterations of the functional expression of glutamate receptors at thalamocortical synapses.
Moreover, a lack of CX3CL1-signaling, and therefore a weakened crosstalk between neurons and microglia, affects transmission efficiency in synapses of the adult brain. It was observed that CX3CR1-/- had fewer multi-synaptic boutons in the hippocampus CA1 region compared to wild-type littermates indicating a lasting impairment of synaptic connectivity (Zhan et al., 2014). Changes in long-term potentiation (LTP) have been found in CX3CR1-/- mice, and were accompanied with deficits in behavior related to learning and memory (Rogers et al., 2011). The Rotarod test revealed that motor control in particular was negatively affected in these mice, compared to control animals. However, the CX3CR1-/- mice also appear to have deficits in associative learning, measured by fear-conditioning tests, and in hippocampal-dependent memory formation – as revealed with Morris water maze. These findings indicate that fractalkine signaling plays a significant role in learning and memory, and the effects appear to be dependent on the activity of interleukin 1β (IL-1β). The expression of IL-1β was increased in both heterozygous and homozygous CX3CR1 knockouts, and the hippocampal infusion of the IL-1β antagonist, IL-1ra, reversed the behavioral deficits in the CX3CR1-/- mice, indicating that IL-1β, released from microglia, could mediate these effects (Rogers et al., 2011). The dose-dependent inhibitory effects of IL-1β on LTP in the hippocampus are supported by other studies and this inhibition is consistent with the high distribution of IL-1R1 on hippocampal neurons (Lynch, 2015). The inhibitory effects of IL-1β in hippocampus have been linked with stimulation of the stress-activated kinases, p38 and JNK (O’Donnell et al., 2000; Vereker et al., 2000). Interestingly, the anti-inflammatory IL-10, which could be also released by microglia, has been demonstrated to antagonize certain actions of IL-1β. IL-10 is able to abrogate the IL-1β -induced inhibition of glutamate release and LTP and its stimulatory effect on JNK signaling (Kelly et al., 2001).
Although fractalkine signaling has a significant role in synapse pruning, it is not the only signaling pathway involved in this complex process. Microglial complement 3 receptor (C3R) has been also determined participating in developmental synaptic pruning. In the healthy developing brain, C1q, the protein that initiates the classical complement pathway of the complement system, promotes activation of C3, which opsonizes subsets of synapses for elimination and promote microglial engulfment and phagocytosis of synaptic elements. This process is significantly downregulated in the mature brain (Stevens et al., 2007; Schafer et al., 2012; Bialas and Stevens, 2013). However, in an early phase of AD this normal developmental synaptic pruning pathway can be locally reactivated and it mediates synapse loss. Hong et al. (2016) demonstrated that in an AD-like mouse strain, C1q was increased and co-localized with synapses even before visible plaque deposition, and blocking C1q, C3 or the microglial receptor CR3, decreased microglial phagocytosis and early synapse loss. The possible relationships between fractalkine signaling and the classical complement pathway could further complicate the topic of developmental and pathological synaptic pruning and it demands more research. Microglia-synapse interactions and synapse removal has also been discovered in many brain pathologies. Synaptic dysfunction and loss, as well as microglial activation, are early events in neurodegenerative diseases such as AD and Huntington’s disease (Perry and O’Connor, 2010). Despite the extensive research of the topic, the question is remaining: are microglia initiating synapse defects and loss in neurodegenerative diseases or is their activation just a secondary phenomenon during pathogenesis?
Apart from the fractalkine and complement signaling, a role for microglial DAP12 (DNAX-activation protein 12) has also been reported in the development of functional neuronal synapses. Intriguingly, in genetically deficient DAP12 mice, developmental apoptosis of neurons was decreased (Wakselman et al., 2008) but synaptic plasticity was enhanced (Roumier et al., 2004). Interestingly, DAP12 function has also been linked to TREM2 (triggering receptor expressed in myeloid cells 2) both are expressed on microglia in the brain and form a receptor-adaptor complex. TREM2/DAP12 signaling is known to regulate microglial phagocytosis and connected to numerous other intracellular signaling pathways implicated in regulation of synaptic plasticity (Kiialainen et al., 2005; Hsieh et al., 2009) (Figure 1).
Overall, the proper microglia functions and their interactions with synaptic elements are considered to be instrumental for appropriate neuronal development and also support the homeostasis of neuronal networks in adults.
Neurons are able to release further immune-related soluble factors that bind to cognate receptors on microglia and promote specific microglia phenotype (Figure 1). These comprise neurotrophins, neuropeptides, neurotransmitters, anti-inflammatory cytokines and chemokines (Biber et al., 2007; Kerschensteiner et al., 2009).
For instance, the multifunctional cytokine, transforming growth factor beta (TGF-β), which is expressed by both neurons and glial cells, has been recognized as a vital regulator for microglia differentiation that promotes a unique transcription profile and surface structure of adult microglia (Butovsky et al., 2014). Also, TGF-β is a potent regulator of cytotoxicity and neuroinflammation in the nervous system (John et al., 2003; Saud et al., 2005). Its importance is to down-regulate microglial responses as showed by increased microglial activity and neuronal loss in the brains of TGF-β-deficient mice (Brionne et al., 2003). Smad pathway has been considered as one of the main signal transduction pathway activated by TGF-β receptors, which is responsible for the regulatory and neuroprotective effects of TGF-β. TGF-β-Smad signaling is involved in the induction of the quiescent phenotype of microglia within the CNS (Abutbul et al., 2012). Binding of TGF-β and its receptor induces ligand dependent assembly of a heteromeric receptor complex, receptor-kinase activation and subsequent phosphorylation and activation of SMAD proteins. SMADs are transcriptional regulators that accumulate in the nucleus and directly regulate gene transcription to evoke cell-type-specific and context-dependent transcriptional programs (Schmierer and Hill, 2007). Besides Smad proteins, there are additional signaling pathways activated by TGF-β, including ERK, p38 and PI3K those can mediate synergistic or antagonistic effects thereby, the responses of activated microglia are the end result of interactions of different signaling pathways (Derynck and Zhang, 2003; Lee et al., 2007).
Furthermore, CD45, a leukocyte common antigen, is constitutively expressed at moderate levels on microglia. Its activation leads to inhibition of microglia activity via negative regulation of the Src/p44/42 MAPK cascade (Tan et al., 2000). The endogenous ligand of CD45 is CD22 has been found at neuronal membranes and neurons secrete CD22 following neuronal injury in order to inhibit microglial proinflammatory cytokine production (Mott et al., 2004).
Electrically active neurons can suppress the interferon-gamma (IFNγ)-induced increase in the expression of pro-inflammatory MHC class II molecules on microglia in the intact CNS (Vass and Lassmann, 1990) and in cultured hippocampal slices (Neumann et al., 1996). The neurotransmitter glutamate, which is released during synaptic activity, is involved in the regulation of microglial cells via glutamate receptors, including metabotropic receptors (Fazio et al., 2018). Glutamate does not necessarily act directly on microglial cells. Instead neuronal released trophic factors such as neurotrophin-3 (NT-3), BDNF and nerve growth factor (NGF) were identified as activity-dependent regulators of microglial MHC class II expression and induction of pro-inflammatory molecules. Neuron-derived glutamate stimulates BDNF and NGF production or release while the regulation of NT3 production by synaptic activity appears to be indirect. Neurotrophins bind to two different classes of receptors: the tyrosine kinase receptors (trkA, trkB, or trkC) or the p75 neurotrophin receptor (also known as the low-affinity NGF receptor). Neumann et al. (1998) and Neumann (2001) have showed that NGF and, to a lower extent, NT3 but not BDNF acted directly on isolated microglia through the p75 neurotrophin receptor as MHC class II inducibility could be enhanced by neutralizing locally released neurotrophins (NGF, BDNF, and NT3) or blocking the p75 neurotrophin receptor. Thus, neurotrophins secreted by electrically active neurons are able to control the antigen-presenting potential of microglia, which is mediated partly via the p75 neurotrophin receptor.
Several neurotransmitters have modulatory effects on microglial activity and proliferation that are related to local neurochemical environment and could differ across various brain regions (McCluskey and Lampson, 2001). As mentioned, glutamate can exert an inhibitory regulation of microglial cells via metabotropic glutamate receptors (mGluRs; Fazio et al., 2018). Microglia also express GABA-B receptors which activation strongly decreases the LPS-induced secretion of certain but not all inflammatory cytokines (Kuhn et al., 2004). Glycine, which is the other inhibitory neurotransmitter, also attenuates the production of inflammatory cytokines and the phagocytic activity of brain macrophages (Zeilhofer, 2008). Similarly, noradrenalin reduces the LPS-stimulated release of NO, IL-6, and TNF-α. Dopamine has also been reported to modulate the activation, proliferation, and cytokine release in immune cells (Sarkar et al., 2010). Both D1 and D2 dopamine receptors mediate anti-inflammatory effects and inhibit neuroinflammation and attenuate brain injury after intracerebral hemorrhage in mice (Zhang et al., 2015; Wang et al., 2018).
In response to any disturbance of their microenvironment, microglia are able to respond rapidly and these responses are mediated, in part, by neuron-released nucleotides such as ATP and UDP (Figure 1). The role of the nucleotides for microglial activation and proliferation has been established, particularly during the early phase of brain injury (Haynes et al., 2006; Ohsawa et al., 2012; Ulmann et al., 2013). The chemotactic properties of ATP are mediated by microglial P2Y12 purinergic receptors and the absence of P2Y12 leads to impaired microglia process motility during injury. Davalos et al. (2005) confirmed the relevance of ATP-induced microglial chemotaxis in vivo in the mouse cortex. The authors showed that laser-induced injury to brain tissue resulted in robust microglial branch extension toward the site of injury and the process of the chemotaxis was abolished by an ATP/ADP degrading enzyme (Davalos et al., 2005). ATP-induced microglial chemotaxis was then confirmed in acute mouse brain slices in the mouse spinal cord (Chen et al., 2010; Dibaj et al., 2010) and retina (Fontainhas et al., 2011) as well as in other animal models. Another purinergic receptor, P2Y6, is also expressed by microglia and UDP signaling through P2Y6 receptors triggered microglial phagocytosis following hippocampal excitotoxicity (Koizumi et al., 2007). Extensive and prolonged neuronal release of glutamate directly leads to neuronal death but also serves as activation signal for microglia through a variety of microglial glutamate receptors like AMPA, kainate, and mGluRs (Biber et al., 1999; Hagino et al., 2004). It has been demonstrated that the activation of microglia mGluR2 triggers TNF-α-induced neurotoxicity through activation of TNF receptor-1 and facilitating the activation of caspase-3. Microglia also released FasL, which further potentiated TNF-α neurotoxicity after mGlu2 stimulation (Taylor et al., 2005).
In return, microglia influence and modulate neuronal function by the release of soluble factors, including cytokines, prostaglandins and neurotrophic factors, which bind to neuronal receptors. IL-1β, prostaglandins (PGE2), BDNF and tumor necrosis factor-alpha (TNF-α) are often released by microglia in response to variations in neuron-derived signals.
Brain-derived neurotrophic factor plays and important role in neuronal survival and differentiation and as a neuromodulator, directly involved in the control of neuronal activity and synaptic plasticity (Santos et al., 2010) (Figure 1). The neuromodulatory effect of BDNF has been recognized both on glutamatergic and GABAergic synapses in the CNS (Gottmann et al., 2009). Apart from neuron and astroglia, microglia also release BDNF as it was first shown in microglia cultures (Elkabes et al., 1996) and then confirmed in different regions of the CNS during the course of various neurological disorders such as traumatic injury, Parkinson’s disease, MS, and neuropathic pain (Batchelor et al., 1999; Knott et al., 2002; Stadelmann et al., 2002; Trang et al., 2011; Song et al., 2016). The synthesis and release of BDNF in microglia appear to be tightly associated with the activation of ATP sensitive purinergic receptors, such as P2X4R. The activation of P2X4R leads a significant intracellular Ca2+ flow and the downstream activation of signaling pathways like p38 MAP kinase, which controls the synthesis and release of BDNF (Trang et al., 2009). Then, microglia-derived BDNF rapidly downregulates K+-Cl- co-transporter KCC2 expression in neuronal membranes through tyrosine kinase B receptor (TrkB) which, disrupting Cl- homeostasis and the strength of GABAA- and glycine receptor-mediated inhibition thereby leads to an altered neuronal network activity (Ferrini and De Koninck, 2013). Although the low levels of microglia-derived cytokines are demonstrated to support homeostatic neuroplasticity, these signaling pathways could be further augmented during inflammatory conditions and mediate neurotoxicity (Stellwagen et al., 2005; Parkhurst et al., 2013; Pribiag and Stellwagen, 2014; Stellwagen and Lewitus, 2014).
Extracellular vesicles are recently discovered way of communication between cells in the CNS that are providing new insights into the brain physiology and pathophysiology of several diseases (Basso and Bonetto, 2016; Rufino-Ramos et al., 2017; Paolicelli et al., 2018; Trotta et al., 2018). Indeed, we found recently that inflammatory stimuli to microglia leads to distinct populations of released EVs, both in terms of size and protein content (Yang et al., 2018a).
In one of the earliest study on EVs in the CNS, membrane exovesicles were identified as vehicles for spreading morphogens through epithelia during Drosophila melanogaster development (Greco et al., 2001). Since then the influence of EVs in tissue development has also been observed in the developing mouse brain and undifferentiated neuronal culture (Marzesco et al., 2005; Faure et al., 2006; Marzesco, 2013).
The family of EVs contains different types of vesicles that are distinguishable by their size, biological origin and function. The EV family comprises exosomes (40–120 nm) that are released from multivesicular endosomes, microvesicles (100–1,000 nm) that are budding from the plasma membrane and apoptotic bodies (800–5,000 nm) that are released by cells during apoptosis (Cocucci et al., 2009; Gyorgy et al., 2011; Colombo et al., 2014; Cocucci and Meldolesi, 2015; Dozio and Sanchez, 2017).
All brain cells, including neurons (Faure et al., 2006; Lachenal et al., 2011) astrocytes (Dickens et al., 2017) microglia (Hooper et al., 2012; Prada et al., 2013; Glebov et al., 2015) and oligodendrocytes (Kramer-Albers et al., 2007; Fitzner et al., 2011; Fruhbeis et al., 2013) secrete EVs. EVs contain different bioactive compounds including cell surface receptors, mitochondrial and cytosolic proteins, metabolic enzymes and genetic materials such as microRNAs and mRNAs (Abels and Breakefield, 2016; Dozio and Sanchez, 2017; Prada et al., 2018). Additionally, EVs could carry pathological markers, such as α-synuclein, tau, amyloid beta (Aβ) (Rajendran et al., 2006; Ngolab et al., 2017; Valdinocci et al., 2017) and pathogenic prion proteins (Schneider and Simons, 2013; Vilette et al., 2018) as well as huntingtin (Zhang et al., 2016; Deng et al., 2017) that implicate exchange of EVs in pathological conditions. EVs are able to influence the behavior of recipient cells in multiple ways: they may transfer receptors and/or bioactive lipids between cells; they can modulate functional target cells by delivering intracellular proteins or transferring mRNA; and may act as signaling complexes through the stimulation of target cells (Basso and Bonetto, 2016). In the CNS, there is an extensive cross talk between neurons and microglia and alongside the actual cell-to-cell contact and the cellular release of soluble factors, microglia and neurons can communicate by bidirectional release of EVs, which permits an exchange of a wide range of biomolecules across long distances (Garzetti et al., 2014; Lai et al., 2014; Rajendran et al., 2014; Zaborowski et al., 2015; Budnik et al., 2016; Kramer-Albers and Hill, 2016).
Microvesicles released by microglia are known to differ in shape and size (100 nm – 1 mm) and they are able to modulate the activity of neighboring microglial population and/or neurons in the surroundings. For instance, microglia-derived EVs can increase glutamate release at the presynaptic sites of neuronal synapses thereby enhances excitatory synaptic transmission. The stimulation of synaptic activity occurs via enhanced neuronal sphingolipid metabolism (Antonucci et al., 2012). Gabrielli et al. (2015) demonstrated that microglial EVs are also able to modulate synaptic transmission through the modulation of the endocannabinoid system. Endocannabinoids are secreted through microglial EVs that have on their surface N-arachidonoylethanolamine (AEA), which can stimulate type-1 cannabinoid receptors (CB1), and inhibit presynaptic transmission of GABAergic neurons.
Microvesicles shedding from microglia is an intriguing topic but little is yet known and therefore warrants further investigations. Extracellular ATP is a key stimulant for vesicles shedding from microglia via the P2X7 ATP receptor (P2X7R) that activates the p38 MAPK cascade through src kinase-mediated phosphorylation. Phosphorylated p38 triggers microvesicle shedding and IL-1β release from glia cells via a process that requires activation of acid sphingomyelinases (Bianco et al., 2005, 2009). Additionally, IL-1β-loaded microvesicles released by microglia via P2X7-p38 pathway can enhance the sensitivity of mechanical allodynia and thermal hyperalgesia induced by nerve injury (Li et al., 2017). Microglial P2X7R is also responsible for the release of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) into the extracellular space, which could be involved in the regulation of neuroinflammation and/or neuritogenesis in the brain (Takenouchi et al., 2015). Others and we have applied a proteomic approach to characterize the proteome of EVs released by microglia. Several proteins involved in protein translation, transcription, cell adhesion/extracellular matrix organization, autophagy-lysosomal pathway and cellular metabolism, that may influence the response of target cells to EVs were identified (Drago et al., 2017; Yang et al., 2018a). Interestingly, upon LPS stimulation proteins related to RNA processing and protein translation were upregulated in the EVs (Yang et al., 2018a). Furthermore, glial microvesicles can contain purine nucleoside phosphorylase, a crucial enzyme in purine metabolism which converts ribonucleosides into purine bases and it can be released into the extracellular space through P2X7R activation, indicating that glial cells may support neuronal activity by maintaining the homeostasis of the purinergic system (Pena-Altamira et al., 2018). Serotonin can also stimulate exosome release both in primary microglia cultures and BV2 cell lines (Glebov et al., 2015). Additionally, stimulation with lipopolysaccharide induces microvesicles release from microglia that can carry proinflammatory mediators (Jablonski et al., 2016; Kumar et al., 2017). We found LPS stimulation of microglia specifically upregulated TNF, and to a lower extent IL-6, in EVs released (Yang et al., 2018a).
Accumulated research confirms the role of microglia-released EVs in neurodegenerative conditions. For instance, shedding of microglia-derived microvesicles was demonstrated after traumatic brain injury (TBI), ischemic stroke, spinal cord injury and in neuropathic pain (Kumar et al., 2017; Huang et al., 2018; Mondello et al., 2018; Osier et al., 2018). Microvesicle from the animals with TBI are loaded with pro-inflammatory mediators (IL-1β and microRNA-155) and can activate additional glial cells that may contribute to progressive neuroinflammatory response in the injured brain (Kumar et al., 2017). ALS is a fatal disease characterized by progressive degeneration of motor neurons and by the formation of inclusions consisting of SOD1 and TDP-43 in motor neurons. Recently, exosomes from astrocytes have been noted that are able to transfer misfolded SOD1 to spinal neurons and subsequently cause selective motor neuron death (Basso et al., 2013). In AD, toxic Aβ and hyperphosphorylated tau can be spread between cells by exosomes and therefore they have been known for contributing to apoptosis and neuronal loss. Rajendran et al. (2006), have shown (in N2a and Hela cells) that Aβ is present in multivesicular bodies (MVBs), and it is released into the extracellular space in exosomes upon MVBs fusion with the plasma membrane. Extracellular Aβ also activates microglia, and activated microglia-shed microvesicles into their environment. α-Synuclein exposure to murine microglial cell line BV2 increased the secretion of EVs enriched in TNF-α and MHCII molecules and promotes neuronal apoptosis (Chang et al., 2013). Furthermore, Joshi et al. (2014) have demonstrated that microglia-shed microvesicles promote the extracellular formation of highly toxic soluble form of Aβ thereby induces neurotoxicity. They have also found that microglia generate neurotoxic species following Aβ internalization, which are delivered to neurons possibly on the external membrane of microvesicles, which leads to neuronal damage.
On the other hand, microglia are able to clear Aβ by phagocytosis of the EVs loaded with Aβ. Yuyama et al. (2015) and Yuyama and Igarashi (2016, 2017) have demonstrated that neuronal exosomes can sequester Aβ through its abundant glycosphingolipids and then Aβ could be taken up and digested by microglia. The administration of neuronal exosomes into the brain of APP transgenic mice results in a decreased amyloid deposition and neuronal exosomes can effectively ameliorates AD pathology (Yuyama et al., 2015; Yuyama and Igarashi, 2016, 2017). Tamboli et al. (2010) have described another possible way by which microglia-shed exosomes could digest extracellular Aβ and thereby promote amyloid clearance. The insulin degrading enzyme (IDE) that is known to be effectively degrade extracellular amyloid deposits was found to be associated and released by exosomes shed by microglia (Tamboli et al., 2010). Microglial exosomes are also involved in the spread of tau pathology as was shown by numerous studies (Saman et al., 2012; Medina and Avila, 2014; Asai et al., 2015; Wang et al., 2017; DeLeo and Ikezu, 2018; Guix et al., 2018). Thus, in the context of neurodegeneration, microglia-derived EVs could play complex and also controversial roles. Microglial EVs have been demonstrated to spread toxic and mutant proteins, while other studies indicated their positive impact on protein aggregates clearance and regulation of neuronal viability. In line with this, various studies demonstrated that microglia activation in early stages of AD pathogenesis could be neuroprotective. However, in the late stage of the disease, microglial MVs have been found responsible for the transportation and distribution of soluble toxic proteins like Aβ and α-synuclein peptides, and to promote the spread of the disease (Benilova et al., 2012; Tofaris, 2017; Croese and Furlan, 2018; Zhang S. et al., 2018).
It has become evident that microglial cells are involved in essentially all brain diseases ranging from AD and PD, TBI, brain ischemia and psychiatric diseases such as schizophrenia (Wohleb, 2016; Joe et al., 2018; Perea et al., 2018; Stephenson et al., 2018). The outcome of many pathologies of the CNS appear to rely heavily on the activity of microglia, including their release of cytokines, chemokines and growth factors (Ransohoff and Cardona, 2010). Importantly, microglial functions are largely dependent on the type of activation stimuli, time after stimulation and local factors during pathological conditions. Microglia responses are not inevitably neurotoxic and various neuroprotective effects of activated microglia have been observed in vivo (Turrin and Rivest, 2006; Szalay et al., 2016). In general, rapid and acute activation of microglia is associated with inflammatory changes but these are designed to combat the immediate insult and ultimately return the tissue homeostasis. This acute reaction could therefore be considered to be neuroprotective in the longer perspective, unless the acute response is augmented and prolonged. Persistent microglial activation with the associated increase in expression of inflammatory cytokines and chemokines, accompanied by recruitment of peripheral cells into the brain, is typically characterized as detrimental chronic neuroinflammation (Lynch, 2009; Lynch et al., 2010). This prolonged and chronic microglia activation is considered to be neurotoxic and can impair neuronal activity. This ambivalent role of microglia in neurodegenerative diseases has been extensively reviewed in the literature, searching for a consensus amidst conflicting data and summating whether the activity of microglia is helpful or not in pathologies of the brain (Hu et al., 2012; Jiang et al., 2014; Loane and Kumar, 2016; Szalay et al., 2016; Du et al., 2017).
Neuroinflammation occurs not only in the brain, but also in the retina and optic nerve – which are outgrowths from the diencephalon and thus considered parts of the CNS (London et al., 2013). In fact, retinal homeostasis is dependent on many of the same bidirectional signaling pathways between brain microglia and neurons, such as fractalkine signaling (Karlstetter et al., 2015). The retina and the brain are affected in several neurodegenerative diseases and because they are similar, they respond similarly to any disturbances of tissue homeostasis and share common pathogenic mechanisms (London et al., 2013). For instance the extracellular deposits of beta-amyloid and intraneuronal accumulation of hyperphosphorylated tau protein reported in AD are also found in the retina and optic nerve. Similarly, PD involves the degeneration of dopaminergic cells in the retina (Ramirez et al., 2017). Excellent reviews are available covering the topic of microglia involvement in retinal physiology and pathology (Wang J.W. et al., 2016; Akassoglou et al., 2017; Jin et al., 2017; Rathnasamy et al., 2018).
Alzheimer’s disease is a progressive neurodegenerative disorder characterized by the plaque-forming accumulation of amyloid-β (Aβ) and the deposition of neurofibrillary tangles (NFTs, composed of hyper-phosphorylated tau protein) within the brain parenchyma. Beta-amyloidosis is a result from an imbalance production versus clearance of Aβ peptides. Microglia are able to engulf and phagocyte the extracellular Aβ via stimulation of triggering-receptor-expressed on myeloid cells 2 (TREM2). TREM2 is a cell surface protein that is selectively and highly expressed by microglia and it is linked to an anti-inflammatory phenotype (Colonna and Facchetti, 2003; Mecca et al., 2018). TREM2 interacts with the adaptor protein DNAX-activating protein of 12-kDa (DAP12), which initiate intracellular signal transduction pathways that regulate microglial phagocytosis and activation. TREM2 is critical for microglial phagocytosis of for example, cellular debris in order to maintain tissue homeostasis (Neumann and Takahashi, 2007). In AD condition, TREM2-deficient microglia demonstrated a reduced uptake of Aβ-lipoprotein complexes in vitro (Yeh et al., 2016) and a less effective Aβ internalization in vivo (Wang et al., 2015; Wang Y. et al., 2016; Yuan et al., 2016). Microglia also have an ability to build a protective barrier around amyloid deposits, that is composed of tightly packed and thereby less toxic amyloid fibrils (Condello et al., 2015).
On the other hand, microglia have significant role in complement-mediated synapse loss in AD (Hong et al., 2016; Fonseca et al., 2017; Jiang and Bhaskar, 2017). In line with this, disabled microglial proliferation and microglial depletion protects against synapse loss in amyloidosis mouse models but does not necessarily have effect on the formation and maintenance of β-amyloid plaques (Grathwohl et al., 2009; Olmos-Alonso et al., 2016; Spangenberg et al., 2016). Complement activation also appears to exacerbate tau pathology and induces NFTs in AD mouse models via unclear mechanisms (Britschgi and Wyss-Coray, 2007; Fonseca et al., 2009). Asai et al. (2015) have showed that microglia are able to spread tau pathology across the brain by microglial uptake and exosomal release of tau in a mouse model of tauopathy. In addition to synapse elimination and aggravation of tau pathology, activated microglia release proinflammatory mediators in response to extracellular protein aggregates and thereby causing harm to neurons (Heneka et al., 2013; Shi et al., 2017).
Microglial fractalkine signaling plays variable roles in different stages of AD pathogenesis in association with neuroinflammation, neurotoxicity, and synaptic plasticity (Chen et al., 2016; Zhang L. et al., 2018). The neuronal soluble CX3CL1 is likely to keep the microglial phenotype in a rather neuroprotective state by acting on CX3CR1 in microglia, since the disruption of CX3CL1–CX3CR1 signaling leads to dysregulate microglial responses and neuronal damage (Cardona et al., 2006; Febinger et al., 2015) (Figure 2). Indeed, hAPP-CX3CR1-/- mice as well as hTau-CX3CR1-/- mice showed increased expression of inflammatory factors, enhanced tau phosphorylation, and exacerbated neuronal dysfunction and cognitive deficits (Bhaskar et al., 2010; Cho et al., 2011) (Figure 2). However, others have demonstrated that both APP-PS1/CX3CR1-/- and CRND8/CX3CR1-/- mice showed reduction in Aβ deposition with increased number of microglia (Lee et al., 2010; Liu et al., 2010). Moreover, in some cases the suppression of CX3CL1–CX3CR1 alleviated Aβ-induced neurotoxicity and memory deficiency (Wu et al., 2013; Dworzak et al., 2015). Additionally, the level of plasma soluble CX3CL1 was found to be significantly greater in the patients with mild to moderate AD than in the patients with severe AD, and the level of CX3CL1 is inversely correlated to AD severity (Kim et al., 2008), suggesting an early neuroinflammatory role in AD pathogenesis. Thus, CX3CL1/CX3CR1 signaling may play a beneficial role in controlling AD progression by inhibiting the inflammation and tau phosphorylation but at a cost of the increased Aβ deposition. One possible hypothesis is that, in early AD the intra-neuronal Aβ accumulation causes a mild decrease in neuron–microglia crosstalk via CX3CL1–CX3CR1 signaling that leads to an enhanced microglial phagocytosis of Aβ while resulting in tau hyper-phosphorylation. Later as AD is progressing, the communication between neurons and microglia is further exacerbated and the CX3CL1–CX3CR1 signaling is severely downgraded that gives rise to deregulated microglia and abnormally excited neuron, which leads to neuron damage and loss. Indeed, we have found remarkable early activation of inflammatory pathways in microglia at 6 weeks of age, before plaque deposition, in the 5xFAD mouse model of AD using a proteomic approach (Boza-Serrano et al., 2018).
Parkinson’s disease is characterized by the selective loss of dopaminergic neurons in substantia nigra. The pathological hallmarks are the formation of Lewy-bodies and intraneuronal protein inclusions mainly consist of α-synuclein. Microglial activation and neuroinflammation clearly contribute to neurodegeneration seen in PD, although the exact cellular mechanisms are not known yet. Neuron–microglia communication via fractalkine signaling provides an effective endogenous mechanism to moderate microglia activation and suppress the release of pro-inflammatory factors during the course of the disease (Grimmig et al., 2016). Maintaining or enhancing CX3CL1-CX3CR1 communication has been proved to be neuroprotective in multiple rodent models of PD (Pabon et al., 2011; Morganti et al., 2012; Nash et al., 2015). However, the action of fractalkine is intricate and regulated on multiple levels; for example, the CX3CR1-/- mice have been demonstrated an enhanced Aβ phagocytosis and at the same time a decline in α-synuclein degradation (Thome et al., 2015) (Figure 2). It remains to be determined if this action is mediated by membrane bound fractalkine or the cleaved, soluble form.
Injuries to the CNS, including TBI, are the leading causes of death and severe disability for people under 40 years of age in the developed world (Donat et al., 2017). The clinical consequences of TBIs are very diverse including immediate death, complete recovery and permanent cognitive, emotional and physical impairments. TBIs are major risk factors for dementia (Donat et al., 2017; Saber et al., 2017) and chronic traumatic encephalitis (CTE) (Omalu, 2014). The primary injury of TBIs, caused by trauma to the head which for example might stretch, compress or tear blood vessels and axons, is not the only reason why patients die or are disabled (Prins et al., 2013). A lot of the damage is also caused by the secondary events such as edema, metabolic and blood flow disturbances, free radical formation, glutamate excitotoxicity, blood–brain barrier breakage, as well as neuroinflammation, i.e., activation of microglia and subsequent recruitment of peripheral leukocytes (Prins et al., 2013; Donat et al., 2017; Saber et al., 2017). Microglial activation in this context is multifactorial, e.g., primed by pro-inflammatory cytokines (Donat et al., 2017) or triggered through purinergic receptors by ATP released from necrotic neurons (Davalos et al., 2005; Fang et al., 2009), and could persist for years if not resolved (Braun et al., 2017). The methods for investigating the role of microglial activity following TBI, have been utilizing the same mouse models as have been discussed earlier in this review. Subjecting CX3CR1GFP/GFP mice to experimental TBI have enabled studies of the fractalkine axis between microglia and neurons in a context of TBI (Febinger et al., 2015; Erturk et al., 2016; Zanier et al., 2016). Similar methods have also been applied for exploring the role of other signaling pathways in TBI, such as TREM2/DAP12, etc. (Saber et al., 2017). However, while these models could be beneficial in understanding the role of specific molecular pathways, it remains difficult to specify their cellular source, which could be microglial or peripheral macrophages (Sewell et al., 2004).
However, microglial signaling in neurodegeneration caused by TBI remains controversial (Bennett and Brody, 2014). A study by Bennett and Brody (2014) showed that in a mouse model of repetitive closed skull TBI (rcTBI) the partial depletion of Cd11b+ microglia did not affect axon degeneration at 7 or 14 days post injury (Bennett and Brody, 2014). Another study observed a big therapeutic effect of deleting or inhibiting complement system proteins C3 and C5 after traumatic brain cryoinjury – which was not attributed directly to the activity of microglia – but instead to a lesser invasion of neutrophils (Sewell et al., 2004).
So far, four peer reviewed studies have investigated the effects of the absence or partial absence of fractalkine signaling after experimental TBI, with some conflicting results (Febinger et al., 2015; Erturk et al., 2016; Zanier et al., 2016; Makinde et al., 2017). In one study, CX3CR1GFP/GFP mice demonstrated a decrease of pro-inflammatory cytokines after the controlled cortical impact (CCI) model of TBI (Febinger et al., 2015). However, in another investigation with CX3CR1-/- mice subjected to CCI, the trends in cytokine expression were not as clear (Zanier et al., 2016), although in both studies, early after insult, the mice lacking CX3CR1 performed the neuroscore test better than WT controls (Febinger et al., 2015; Zanier et al., 2016). This effect was reversed at a later stage (Febinger et al., 2015; Zanier et al., 2016), indicating that fractalkine signaling might exert negative influence at an early stage, but protective at later stages after TBI. It should be noted that, while fractalkine signaling is important in microglia–neuron bidirectional communication, CX3CR1 is also expressed in peripheral macrophages, and could contribute to their infiltration into the CNS (Erturk et al., 2016). A third study examined the effects of CCI in mice missing one allele of CX3CR1 and found that the female mice, but not the male mice, showed significantly better symptoms post-injury, including less neurodegeneration, leukocyte infiltration and cognitive deficits (Erturk et al., 2016). Here it was also shown that WT mice revealed neurodegeneration one year after insult, even in areas far from the lesion, indicating that targeting chronic fractalkine signaling is a promising, albeit sex-specific, way of combating detrimental events after TBI (Erturk et al., 2016). A fourth study subjected CX3CR1 null mice to CCI and observed that neutrophil infiltration was significantly reduced (Makinde et al., 2017).
The receptor adaptor complex TREM2/DAP12, important in synapse formation (Filipello et al., 2018), is also involved in microglial activation and phagocytosis (Mecca et al., 2018). A massive upregulation of TREM2 has been demonstrated in a mouse model of TBI and the absence of TREM2 was shown to improve hippocampal survival and cognition, lower disinhibitory behavior, as well as to decrease the immune cell activation throughout the brain, except in proximity to the lesion (Saber et al., 2017). However, the TREM2-dependent immune cell activity, believed to be negative in the TBI setting, was not primarily associated with microglia, but rather with peripherally derived macrophages (Saber et al., 2017).
TBI is associated with a wide range of deleterious events for cerebral vasculature and tissues that cause immune reactions and leukocyte infiltration in the brain (Saber et al., 2017; Monson et al., 2018). The activity of macrophages appears to be a potential therapeutic target in different TBI settings in adult, but not young, rodents (Febinger et al., 2015; Erturk et al., 2016; Hanlon et al., 2016; Zanier et al., 2016; Chhor et al., 2017; Saber et al., 2017). However, it appears that the peripherally derived macrophages, rather than macrophages derived from the resident microglial population, are the primary targets for attenuating detrimental effects after TBI (Bennett and Brody, 2014; Morganti et al., 2015; Saber et al., 2017). More research is required to elucidate the specific role of microglia and microglial pathways in the very multi-facetted context of TBI.
Brain ischemia, or stroke, is one of the leading cause to death and disability in adults (American Heart Association, 2017). Brain ischemia is typically characterized as focal or global brain ischemia. The most common type of brain ischemia is stroke (focal ischemia), which to 85% is related to an occlusion of a cerebral artery or to less extent (15%) rupture of an cerebral artery and few treatment options is currently available and then related to clot removal or to resolve to clot in the immediate phase (Siegel et al., 2017). In global ischemia, the entire brain is ischemic, where most common condition leading to global brain ischemia is cardiac arrest with successful resuscitation (Joundi et al., 2016). In this section, we will focus on focal ischemia, which is characterized by an ischemic core, which in the acute phase is surrounded by a penumbra region, with impaired function, and injured tissue that is amenable for tissue protection. Neuroinflammation and microglia reactions are involved in all stages of the ischemic cascade: from the acute event leading to the first neuronal cell death to later stages of parenchymal processing including phagocytosis of cell debris and tissue remodeling. In this section, we will focus on the direct interaction between microglia–neuron, i.e., CD200 and fractalkine. Other recent reviews cover the broader aspect of inflammation in stroke and ischemia pathogenesis (Dirnagl, 2012; Benakis et al., 2014; Chamorro et al., 2016). As mentioned, several messengers are involved in the communication between microglia and neurons, for example cytokines and purines. The glycoprotein CD200 is mainly expressed by neurons and its receptor CD200R is expressed on myeloid cells including microglia. This interaction is involved in maintaining microglial cells in a quiescent homeostatic stage. Lower expression of CD200 has been related to proinflammatory activation of microglia and increased influx of inflammatory cells into the brain (Denieffe et al., 2013). Autocrine release of CD200 from microglia is also suggested to keep the microglia in an alternative/non-inflammatory activation state, involving IL-4 signaling (Yi et al., 2012). The endocannabinoid anandamide (AEA) which can be released by microglia in neuroinflammatory conditions and have an anti-inflammatory effect in stroke (Capettini et al., 2012), is also reported as a mechanism for upregulation of CD200R on microglia (Hernangomez et al., 2012). Additionally, peroxisome proliferator-activated receptor gamma (PPAR-γ), which is a transcription factor controlling the inflammatory response, has been reported to regulate CD200 and CD200R1 gene expression, which could be related to the neuroprotective action of PPAR-γ agonists (Dentesano et al., 2014). Recently, Yang et al. (2018b) investigated the acute effect, up to 48 h after ischemia, of CD200 following permanent focal ischemia in mice. They found CD200 expression on neurons, but not on microglia, and a negative correlation with neuronal death in cortical tissue. Injection of recombinant CD200 intracerebroventricular, performed right after pMCAO induction, reduced microglial activation and expression of cytokine TNF, IL-1β, and IL-10 (Yang et al., 2018b). To be able to use the beneficial effect of CD200 signaling in ischemic stroke, Kong et al. (2018) used human mesenchymal stem cells with high CD200 expression from human placenta which they transplanted intracerebrally 24 h after transient middle cerebral artery occlusion (tMCAO). Interestingly, they report reduced microglia activation in the infarct boundary area, smaller infarct and improved behavior. Silencing of CD200 in the mesenchymal stem cells also reduced the inflammatory response in BV2 microglia when applying a co-culture system (Kong et al., 2018). Using a similar rat stroke model, Matsumoto et al. (2015) investigated if the CD200 could be used to distinguish M1 or M2 macrophage/microglia in the infarct at 7 days after ischemia, but were not able to make this distinction in M1/M2 based on CD200 expression. However, using rat macrophages Hayakawa et al. (2016b) used CD200 stimulation induced alternative activation (M2) via CREB-C/EBP-beta signaling.
Focal white matter ischemia, where a small ischemic lesion can generate large neurological deficits, has also been studied in the context of CD200 signaling using endothelin injections in mice (Hayakawa et al., 2016a). Hayakawa et al. (2016a) used CD200-Fc treatment to target CD200R and found a reduction in the macrophage phagocytosis of oligodendrocyte progenitors, which was related to reduced TLR4 expression in macrophages. Indeed, they could even detect an enhancement of remyelination following CD200-Fc treatment (Hayakawa et al., 2016a). Microglia studies of CD200-deficient mice reveal increased classical activation, which could be related to increased infiltration of T cells and macrophages (Denieffe et al., 2013). Using CD200-deficient mice, or overexpression of CD200, using conditional systems in various experimental stroke models with long-term recovery would be interesting to further elucidate its function in neuroinflammation and potential as a therapeutic target.
CD200 signaling has been discussed for decades as an important mechanism in neuroinflammatory conditions (Neumann, 2001), but the jury is still out on whether CD200 could be important target to use in ischemic brain injury and neurodegenerative diseases.
Crosstalk between microglia and neuron via fractalkine/CXCR1 has also been studied in experimental stroke models and been a target for neuroprotection. CXCR1 knockout mice subjected to focal brain ischemia was reported by Denes et al. (2008) to surprisingly have reduced IL-1B and TNF production together with reduced infarct size, better recovery and ameliorated neuronal cell death.
Soriano et al. (2002) used the CX3CL1 deficient mice and confirmed the results with the tMCAO model, showing reduced infarct size and reduced mortality when CX3CL1/CX3CR1 signaling was absent.
In stark contrast to these results, when CX3CL1 was administered to naive mice (wt) in combination with pMCAO a reduction in infarct size was reported. In this study, Cipriani et al. (2011) found that the adenosine receptor 1 (A1R) was implicate in this neuroprotection and using A1R antagonist or A1R-/- mice abolished the positive effect of CX3CL1 (Cipriani et al., 2011). However, when they administering CX3CL1 to the CX3CL1 deficient mice they found aggravated the brain ischemia, possibly related to the constitutive lack of CX3CL1 leading to a maladaptation of the CX3CL1/CX3CR1 signaling axis (Cipriani et al., 2011). Clinical data though suggest a potential protective role of CX3CL1, where patients with better clinical outcome after 6 months had higher levels of CX3CL1 in blood plasma (Donohue et al., 2012).
In a recent rat study, Liu et al. (2017) used a pMCAO model to study the effect of CXCR1 inhibitor (day 3–14 systemic administration) and found increased expression of BDNF and NGF, neuroprotection and improved neurology up to 14 days after ischemia.
In view of the dispersed data related to CX3CL1/CX3CR1 and ischemic stroke it is difficult to judge the neuroprotective role of CX3CL1/CX3CR1 signaling and the future possibility to manipulate this pathway pharmaceutically to reduce the sequel of stroke.
Prolonged microglia activation has also been observed in the environment of malignant brain tumors. In contrast to what is known in neurodegenerative conditions, tumor-infiltrated microglia exhibit immunologically suppressed phenotypes and they have been demonstrated rather promoting tumor proliferation and progression than exert an anti-tumor activity (Bettinger et al., 2002; Yang et al., 2010; Zhai et al., 2011; Schiffer et al., 2017). The pathological hallmarks of malignant brain tumors involve rapid tumor proliferation, diffuse brain invasion, tumor-induced brain edema and neuronal cell death. Gliomas are the most common and aggressive primary brain tumors that are composed of neoplastic and non-neoplastic cells. The majority of the non-neoplastic cells are tumor-associated macrophages along with fibroblast and endothelial cells (Morantz et al., 1979; Rossi et al., 1987; Simmons et al., 2011). Histopathologic studies of glioblastomas have revealed high number of microglia and macrophages (with peripheral origin) in and around of glioma tissue. This is due to the local release of numerous factors by tumor cells which mediate microglia chemoattraction including chemokines, ligands of complement receptors, ATP and neurotransmitters. Monocyte chemoattractant protein-1 (MCP-1, also known CCL2) was among the first chemoattractant factors, which has been identified to contribute tumor proliferation and progression (Platten et al., 2003; Vakilian et al., 2017). The chemokine stromal-derived factor-1 (SDF-1, also known as CXCL12) has been described as another potent microglia and macrophage recruiting molecules (Wang et al., 2012; Jiang et al., 2013). The growth factor glial cell–derived neurotrophic factor (GDNF), is secreted in mouse and human gliomas, which serves as a strong chemoattractant for microglia (Ku et al., 2013). Colony stimulating factor-1 (CSF-1) is released by glioma cells and acts as a microglial chemoattractant. Blockade of CSF-1R signaling using RNA interference or pharmacological inhibitors have been shown to significantly reduce the number of tumor-associated microglia and glioblastoma invasion (Coniglio et al., 2012). Additionally, CSF-1 is also able to convert microglia into a pro-tumorigenic anti-inflammatory phenotype (Pyonteck et al., 2013). Furthermore, granulocyte- macrophage colony-stimulating factor (GM-CSF) can also serve as a chemoattractant for microglia, as GM-CSF knockdown reduces microglia-dependent invasion in organotypic brain slices and attenuates the gliomas growth in vivo (Sielska et al., 2013). Although the tumor-associated microglia are high in numbers, they exert suppressed functions that involves reduced phagocytic activity and defective antigen presentation for cytotoxic and helper T cells activation. Therefore, microglia associated with malignant gliomas appear incapable of inducing an effective anti-tumor T cell response (Wu et al., 2010; Hambardzumyan et al., 2016). Additionally, glioma cells produce numerous anti-inflammatory cytokines (e.g., IL-10, IL-6, TGF-β2, PGE2) that can revert activated microglia to an anti-inflammatory phenotype (Hishii et al., 1995; Pyonteck et al., 2013). For instance, TGF-β2 released by glioma cells inhibits proliferation and secretion of proinflammatory cytokines by microglia and lymphocytes (Suzumura et al., 1993).
In return, microglia secrete tumor proliferation promoting factors including epidermal growth factor (EGF) (Coniglio et al., 2012) and vascular endothelial growth factor (VEGF), TGF-β, arginase-1 (ARG1), and IL-10 (Gabrusiewicz et al., 2011; Li and Graeber, 2012). Additionally, TGF-β released predominantly from microglia are able to enhance tumor growth and invasion as the downregulation the TGF-β type II receptor expression with shRNAs abolished TGF-β-induced glioblastoma invasiveness and migratory responses in vitro (Wesolowska et al., 2008). Furthermore, microglia can help increase the spread of tumors by releasing of extracellular matrix degrading enzymes such as MMP-2, MMP-9, and MT1-MMP, into the tumor environment which support tissue remodeling and angiogenesis (Belien et al., 1999; Markovic et al., 2005; Konnecke and Bechmann, 2013).
Neurodegenerative actions of malignant gliomas resemble mechanisms also found in neurodegenerative diseases (e.g., AD, PD, ALS) and brain tumors can affect neuronal survival directly or by microglia-mediated factors. Tumor environments include elevated level of extracellular ATP, which can recruit microglia and macrophages into tumor regions and induce cellular release of inflammatory mediators initiating and sustaining tumor development. ATP is able to activate P2X7 purinergic receptors (P2X7R) expressed on glioma and immune responsive cells (microglia/macrophages). A critical point of ATP signaling in tumors is the prolonged duration in effect due to the inefficient hydrolysis of ATP. The high concentration of extracellular ATP is toxic for neurons and contributes to neuronal cell death (McLarnon, 2017). Additionally, the prolonged activation of microglial P2X7R leads to excessive inflammation by the release of inflammatory factors (IL-1β and TNF-α) and the activation of caspase activity in apoptosis, which can put neuronal survival at risk (Ferrari et al., 1997, 1999). Furthermore, gliomas have been shown to seize neuronal glutamate signaling for their own growth advantage (Savaskan et al., 2015). The cystine/glutamate antiporter xCT is expressed in various malignant tumors including brain tumors (Kim et al., 2001). The protein complex transports Na+-independent glutamate out of cells in exchange for cysteine thereby, releases high amounts of glutamate in the extracellular microenvironment. Extracellular glutamate represents a potent signaling molecule and neurotransmitter in the bran tissue and triggers membrane depolarization. However, excessive glutamate release and hence glutamate receptor activation can lead to excitatory neuronal cell death. In brain tumors, the cystine/glutamate antiporter expression is elevated that is consequently causes an increased extracellular glutamate levels in the peritumoral zone. The high extracellular glutamate concentration results in tumor-associated seizures, brain swelling and neuronal damage (Ye and Sontheimer, 1999; Savaskan et al., 2008; Buckingham et al., 2011). In line with this, other experimental and clinical studies have demonstrated that glioma cells secrete high levels of the neurotransmitter glutamate, resulting in neuronal damage and antagonizing ionotropic glutamate receptors alleviate neuronal degeneration in the tumor vicinity and lessen glioma growth in vivo (Behrens et al., 2000; Marcus et al., 2010).
As previously mentioned, microglia are one of the key mediators of neuroplasticity, acting in the remodeling of synaptic processes and circuitry formation during normal physiological conditions. Therefore, the bidirectional communication between neurons and microglia may be critical for preserving the homeostatic environment in the central nervous system and defects in microglia-neuronal activities have been suggested as potential contributors to neurodevelopmental alterations, resulting in psychiatric disorders such as schizophrenia, bipolar disorder (BD), and depression (Yirmiya et al., 2015; Laskaris et al., 2016).
Schizophrenia is a severe neurodevelopmental disorder characterized by psychosis, apathy and withdrawal, and cognitive impairment, which cause abnormal social behavior and self-care (Mueser and McGurk, 2004; Blank and Prinz, 2013). This disease affects ∼1% of the entire population (Yamamuro et al., 2015), emerges between 16 and 30 years and persists throughout the patient’s life (Mueser and McGurk, 2004). The origin of this disease is still unknown, but some evidences suggest that schizophrenia can arise from an interaction between neurodevelopmental processes, such as prenatal viral infections (Allswede and Cannon, 2018), and environmental factors (Davalieva et al., 2016). Brain structural irregularities have been reported in schizophrenic patients, such as the loss of gray matter in prefrontal, temporal and subcortical structures (Ellison-Wright and Bullmore, 2009; Fornito et al., 2009), the presence of white matter tracts connecting these areas (Bora et al., 2011; Zalesky et al., 2011), as well as an enlargement of ventricle (Olabi et al., 2011). Additionally, this disease presents multiple biochemical irregularities in the dopamine, serotonin, acetylcholine, glutamate, and GABA systems (Mueser and McGurk, 2004), as well as changes in the immune system (Nawa et al., 2000).
In the last few years, microglial cells have appeared as important players in the development of schizophrenia (Brown, 2011). Owing to their participation in inflammatory processes as well as in the modification of neuronal networks (Tremblay et al., 2010; Chew et al., 2013; Zhan et al., 2014b), microglial cells have been proposed as a possible mechanism that participate in the structural brain changes that appear in schizophrenia (Munn, 2000; Monji et al., 2009). Postmortem studies have shown an increased density of microglial cells in the brain of patients with schizophrenia specifically in frontal and temporal, but not in cingulate cortex (Garey, 2010). In addition, positron emission tomography (PET) imaging studies have shown that microglia are activated in these patients (van Berckel et al., 2008; Hercher et al., 2014). These microglia features have been also related with differences in behavior in animal studies, where specifically, schizophrenic rodent models present deficits in pre-pulse inhibition and working memory impairments (Ribeiro et al., 2013).
During the development of the nervous system, the communication between microglia and neuron is fundamental in the synaptic formation and maturation (Penzes et al., 2011) and persistent microglial activation has been used to explain how this process might cause neuronal degeneration and synaptic dysfunction (Monji et al., 2009). CX3CR1 is expressed on microglia during embryogenesis and throughout murine lifespan (Ginhoux et al., 2010; Paolicelli et al., 2011) and it has been proposed as a key mediator of neuron–microglia interactions. In fact, the CX3CR1-knockout mice model presents a reduction of microglia which resulted in deficits of synaptic pruning and a decreased functional brain connectivity (Zhan et al., 2014), as well as behavioral impairments measured by fear conditioning, Morris water maze and motor learning deficits (Rogers et al., 2011). Furthermore, meta-analysis using human blood and brain samples showed a significantly down-regulation of CX3CR1 in schizophrenic patients (Bergon et al., 2015), specifically individuals with Ala55Thr variant of this receptor present more susceptibility to develop this neurological disorder (Ishizuka et al., 2017). Apart from CX3CR1 receptors, secreted Neuregulin 1 (NRG1) can bind to NRG1 receptor on microglia and signal via a type of EGF receptor, ErbB2/3, leading to proliferation and activation of microglia (Calvo et al., 2010). Signaling deficits in NRG1/ErbB3 has been identified in schizophrenia patients (Corfas et al., 2004). Single nucleotide polymorphisms in NRG1; have been associated with psychosis and enlarged lateral ventricles and white matter disruption in schizophrenia (Bousman et al., 2018). Another important mediator between microglia and neuron communication is BDNF, where microglia is one of the central source of BDNF (Ferrini and De Koninck, 2013). Alterations in this gene have been pointed out as a responsible protein for the etiology of schizophrenia (Zhang et al., 2008). Moreover, a single polymorphism (Val66Met) in the BDNF gene has been related to the age of onset of this disorder and the manifestations that persist after durable antipsychotic treatment (Numata et al., 2006).
Bipolar disorder is a brain disorder that provokes intense emotion, changes in sleep patterns and activity levels and uncommon behaviors that may go along with mood episodes including symptoms of both manic and depression (Belmaker, 2004). This disorder affects approximately 1% of the population (Newman et al., 2002). The etiology of this psychiatric disorder is still unknown; however, as schizophrenia, the interaction between genes and environment have been related to the pathogenesis of BD (Belmaker, 2004). BD patients showed brain white matter abnormalities (Ganzola and Duchesne, 2017), as well as a decrease in hippocampal (Cao et al., 2016), corpus callosum (Lavagnino et al., 2015), and frontal cortical volumes, specifically in patients with manic (Abé et al., 2015).
Neuroinflammation has been suggested as a possible mechanism involved in mood disorders, such as BD (Perugi et al., 2015; Réus et al., 2015), and alterations in glial markers have been found in postmortem frontal cortex in BD subjects (Rao et al., 2010). Moreover, using PET imaging studies with the microglial tracer ([(11)C]-(R)-PK11195), Haarman et al. (2014) showed an intensification of neuroinflammation in the hippocampus of BD patients. After an insult, microglia can produce proinflammatory cytokines in response to activation via damage-associated molecular patterns (DAMPs) (Heneka et al., 2014). Indeed, numerous meta-analysis in patients with BD showed a significantly increase in peripheral cytokines, such as IL-6 or TNF-α (Goldsmith et al., 2016). Also, serum analysis of BD patients during acute manic/depressive episodes have shown an increased in DAMPs levels (Stertz et al., 2015).
As we have already mentioned, due to their participation in neuroinflammation and remodeling of synapses, microglial cells may contribute in the development of neuropsychiatric disorders, such as BD. Actually, postmortem studies showed a decreased in glial cells in the prefrontal cortex, especially in patients with familial history of mood disorders (Ongur et al., 1998). In particular, it has been suggested that the microglial function is altered in BD patients (Réus et al., 2015). The communication between microglial cells in the brain is mediated by purinergic signaling that includes the P2X purinergic receptor 7 for ATP (P2RX7) (for review, see Fields and Burnstock, 2006). In fact, a single nucleotide polymorphism in the gene that encodes for P2RX7 receptor has been associated with a highest susceptibility for BD pathology (Barden et al., 2006). Likewise, the role of fraktalkines mediated neuron–microglia interaction has been reported in BD (for review, see Pinto et al., 2018). Actually, CX3CR1 expression has been found reduced in monocytes of BD patients (Padmos et al., 2008).
Depression is a neuropsychiatric disorder characterized by psychophysiological alterations in affective symptoms (sadness, irritability, low mood, desperation, apathy, anhedonia) that decrease of interest in all daily activities and events (Belmaker and Agam, 2008). According to the World Health Organization, depression affects more than 300 million people worldwide. Conventionally, depressive symptoms have been attributed to an imbalance in the hypothalamic-pituitary-adrenal axis (Sousa et al., 2008) as well as a deregulation in the neuromodulation of neurotransmitters, specially by serotonin (Krishnan and Nestler, 2008). Brain abnormalities have also been found in depressed patients. Thus, postmortem brain studies showed reduced lobar volumes accompanied by a decreased number of synaptic contacts (Lampe et al., 2003; Kang et al., 2012). Moreover, it has been described a loss and a decrease in the size of GABAergic neurons, in occipital, prefrontal and limbic areas (Cotter et al., 2002; Rajkowska et al., 2007; Maciag et al., 2010).
Beyond the classical impairments in the HPA and/or in the neuromodulation by neurotransmitters, depression is also associated with alterations of microglia activity and inflammation (Yirmiya et al., 2015). Rodent models of depression showed significant variations in microglial function and morphology, specifically in brain areas sensitive to chronic stress leading depressive-like behavior, such as hippocampus, prefrontal cortex, amygdala and nucleus accumbens (Tynan et al., 2010; Kreisel et al., 2014). In depressive patients, PET imaging studies revealed a higher level of neuroinflammation in prefrontal cortex, anterior cingulate cortex and insula (Setiawan et al., 2015). Likewise, the communication between microglial cells also may participate in mood disorders. To note, in rodent models, excessive P2X7 receptor activation enhance depressive-like behavior (for review, see Stokes et al., 2015) and in humans, a single nucleotide polymorphism in this gene is associated with the develop of major depressive disorder (Lucae et al., 2006). Besides the alterations in the morphology and function, failures in the microglia–neuron communication may be involved in depression. Some authors showed that the manipulation of this interaction in the CX3CR1 pathway alters the stress reaction and depressive-like behavior (Corona et al., 2010; Milior et al., 2016). Furthermore, the CX3CR1-deficient mouse model showed resistance to stress-induced depressive-like behavior and changes in microglia morphology, suggesting that the hyper-ramification in microglia is controlled by neuron–microglia interaction (Hellwig et al., 2016). Another important mediator between microglia and neurons is BDNF, which has also been linked to depression (Castren, 2014; Kishi et al., 2017). Thus, in a rat model of chronic stress-induced depression, the infusion of this neurotrophin partially reverse the depressive-like behavior (Ye et al., 2011).
The dynamics of microglia–neuron communication in the healthy brain has attracted great attention in the field of neurobiology/neuroimmunology during the past decades. The bidirectional communication between the two cell-types involves several immunomodulatory factors and signaling pathways including purinergic, neurotransmitter, chemokine, and complement signaling. Many observations indicate that microglia modulates neuronal activity and prune synaptic elements such as dendritic spines in the healthy brain and during neural development. In return, neurons contribute to immune modulation through secreted and membrane associated molecules control microglial phagocytosis, motility and activation. Elucidating the functional significance of the bidirectional microglial–neuronal communication in the healthy brain is important in comprehending how the defects in physiological microglia function could contribute to or even trigger diseases. In brain injuries such as TBI and brain ischemia, microglia–neuron communication is very likely important in both the acute phase, in terms of altered neuroinflammation and cell death, as well as in the chronic phase including the rewiring of neuronal circuits. In neurodegenerative and neuropsychiatric disorders, such as AD, PD, BD depression and schizophrenia, the chronic activation of microglia is likely affecting the pathogenesis and aberrant neuronal signaling. Thus, a deeper understanding of microglia–neuron communication would be important in future therapies for diseases of the brain.
ZS, OM, SB, and TD have all designed and drafted sections of the manuscript, as well as significantly contributed to revisions and editing of the final version. OM has also prepared the figures.
SparbankFärs and Frosta Foundation to the list of contributors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge funding support from the Strategic Research Area MultiPark at Lund University, Lund, Sweden, the Swedish Research Council grant no. 2012-2229, the Basal Ganglia Disorders Linnaeus Consortium (BAGADILICO), and the Swedish Alzheimer Foundation, A.E. Berger Foundation, Olle Engkvist Byggmästare Foundation, Swedish Brain Foundation, Crafoord Foundation, Swedish Dementia Association, G&J Kock Foundation, Swedish National Stroke Foundation, Swedish Parkinson Foundation, Stohnes Foundation, and the Royal Physiographic Society.
Abé, C., Carl-Johan, E., Carl, S., Predrag, P., Martin, I., and Mikael, L. (2015). Manic episodes are related to changes in frontal cortex: a longitudinal neuroimaging study of bipolar disorder 1. Brain 138, 3440–3448. doi: 10.1093/brain/awv266
Abels, E. R., and Breakefield, X. O. (2016). Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol. Neurobiol. 36, 301–312. doi: 10.1007/s10571-016-0366-z
Abutbul, S., Shapiro, J., Szaingurten-Solodkin, I., Levy, N., Carmy, Y., Baron, R., et al. (2012). TGF-beta signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia 60, 1160–1171. doi: 10.1002/glia.22343
Akassoglou, K., Merlini, M., Rafalski, V. A., Real, R., Liang, L., Jin, Y., et al. (2017). In vivo imaging of CNS injury and disease. J. Neurosci. 37, 10808–10816. doi: 10.1523/JNEUROSCI.1826-17.2017
Al-Aoukaty, A., Rolstad, B., Giaid, A., and Maghazachi, A. A. (1998). MIP-3alpha, MIP-3beta and fractalkine induce the locomotion and the mobilization of intracellular calcium, and activate the heterotrimeric G proteins in human natural killer cells. Immunology 95, 618–624. doi: 10.1046/j.1365-2567.1998.00603.x
Allswede, D. M., and Cannon, T. D. (2018). Prenatal inflammation and risk for schizophrenia: a role for immune proteins in neurodevelopment. Dev. Psychopathol. 30, 1157–1178. doi: 10.1017/S0954579418000317
American Heart Association (2017). Correction to: Heart disease, and stroke statistics—2017 update: A report from the american heart association. Circulation 135:e646. doi: 10.1161/CIR.0000000000000491
Andjelkovic, A. V., Song, L., Dzenko, K. A., Cong, H., and Pachter, J. S. (2002). Functional expression of CCR2 by human fetal astrocytes. J. Neurosci. Res. 70, 219–231. doi: 10.1002/jnr.10372
Antonucci, F., Turola, E., Riganti, L., Caleo, M., Gabrielli, M., Perrotta, C., et al. (2012). Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 31, 1231–1240. doi: 10.1038/emboj.2011.489
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Banisadr, G., Gosselin, R. D., Mechighel, P., Rostene, W., Kitabgi, P., and Melik Parsadaniantz, S. (2005). Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J. Comp. Neurol. 492, 178–192. doi: 10.1002/cne.20729
Barclay, A. N., Wright, G. J., Brooke, G., and Brown, M. H. (2002). CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 23, 285–290. doi: 10.1016/S1471-4906(02)02223-8
Barden, N., Harvey, M., Gagne, B., Shink, E., Tremblay, M., Raymond, C., et al. (2006). Analysis of single nucleotide polymorphisms in genes in the chromosome 12Q24.31 region points to P2RX7 as a susceptibility gene to bipolar affective disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 374–382.
Barna, B. P., Pettay, J., Barnett, G. H., Zhou, P., Iwasaki, K., and Estes, M. L. (1994). Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J. Neuroimmunol. 50, 101–107. doi: 10.1016/0165-5728(94)90220-8
Baron, P., Bussini, S., Cardin, V., Corbo, M., Conti, G., Galimberti, D., et al. (2005). Production of monocyte chemoattractant protein-1 in amyotrophic lateral sclerosis. Muscle Nerve 32, 541–544. doi: 10.1002/mus.20376
Basso, M., and Bonetto, V. (2016). Extracellular vesicles and a novel form of communication in the brain. Front. Neurosci. 10:127. doi: 10.3389/fnins.2016.00127
Basso, M., Pozzi, S., Tortarolo, M., Fiordaliso, F., Bisighini, C., Pasetto, L., et al. (2013). Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 288, 15699–15711. doi: 10.1074/jbc.M112.425066
Batchelor, P. E., Liberatore, G. T., Wong, J. Y., Porritt, M. J., Frerichs, F., Donnan, G. A., et al. (1999). Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J. Neurosci. 19, 1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999
Bazan, J. F., Bacon, K. B., Hardiman, G., Wang, W., Soo, K., Rossi, D., et al. (1997). A new class of membrane-bound chemokine with a CX3C motif. Nature 385, 640–644. doi: 10.1038/385640a0
Beattie, E. C., Stellwagen, D., Morishita, W., Bresnahan, J. C., Ha, B. K., Von Zastrow, M., et al. (2002). Control of synaptic strength by glial TNFalpha. Science 295, 2282–2285. doi: 10.1126/science.1067859
Behrens, P. F., Langemann, H., Strohschein, R., Draeger, J., and Hennig, J. (2000). Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J. Neurooncol. 47, 11–22. doi: 10.1023/A:1006426917654
Belien, A. T., Paganetti, P. A., and Schwab, M. E. (1999). Membrane-type 1 matrix metalloprotease (MT1-MMP) enables invasive migration of glioma cells in central nervous system white matter. J. Cell Biol. 144, 373–384. doi: 10.1083/jcb.144.2.373
Belmaker, R. H., and Agam, G. (2008). Mechanisms of disease: major depressive disorder. N. Engl. J. Med. 358, 55–68. doi: 10.1056/NEJMra073096
Benakis, C., Garcia-Bonilla, L., Iadecola, C., and Anrather, J. (2014). The role of microglia and myeloid immune cells in acute cerebral ischemia. Front. Cell Neurosci. 8:461. doi: 10.3389/fncel.2014.00461
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Bennett, R. E., and Brody, D. L. (2014). Acute reduction of microglia does not alter axonal injury in a mouse model of repetitive concussive traumatic brain injury. J. Neurotrauma 31, 1647–1663. doi: 10.1089/neu.2013.3320
Bergon, A., Belzeaux, R., Comte, M., Pelletier, F., Herve, M., Gardiner, E. J., et al. (2015). CX3CR1 is dysregulated in blood and brain from schizophrenia patients. Schizophr. Res. 168, 434–443. doi: 10.1016/j.schres.2015.08.010
Bertollini, C., Ragozzino, D., Gross, C., Limatola, C., and Eusebi, F. (2006). Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology 51, 816–821. doi: 10.1016/j.neuropharm.2006.05.027
Bettinger, I., Thanos, S., and Paulus, W. (2002). Microglia promote glioma migration. Acta Neuropathol. 103, 351–355. doi: 10.1007/s00401-001-0472-x
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Bialas, A. R., and Stevens, B. (2013). TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 16, 1773–1782. doi: 10.1038/nn.3560
Bianco, F., Perrotta, C., Novellino, L., Francolini, M., Riganti, L., Menna, E., et al. (2009). Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 28, 1043–1054. doi: 10.1038/emboj.2009.45
Bianco, F., Pravettoni, E., Colombo, A., Schenk, U., Moller, T., Matteoli, M., et al. (2005). Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J. Immunol. 174, 7268–7277. doi: 10.4049/jimmunol.174.11.7268
Biber, K., Laurie, D. J., Berthele, A., Sommer, B., Tolle, T. R., Gebicke-Harter, P. J., et al. (1999). Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J. Neurochem. 72, 1671–1680. doi: 10.1046/j.1471-4159.1999.721671.x
Biber, K., Neumann, H., Inoue, K., and Boddeke, H. W. (2007). Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 30, 596–602. doi: 10.1016/j.tins.2007.08.007
Biber, K., Sauter, A., Brouwer, N., Copray, S. C., and Boddeke, H. W. (2001). Ischemia-induced neuronal expression of the microglia attracting chemokine secondary lymphoid-tissue chemokine (SLC). Glia 34, 121–133. doi: 10.1002/glia.1047
Blank, T., and Prinz, M. (2013). Microglia as modulators of cognition and neuropsychiatric disorders. Glia 61, 62–70. doi: 10.1002/glia.22372
Boche, D., Perry, V. H., and Nicoll, J. A. (2013). Review: activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 39, 3–18. doi: 10.1111/nan.12011
Bora, E., Fornito, A., Radua, J., Walterfang, M., Seal, M., Wood, S. J., et al. (2011). Neuroanatomical abnormalities in schizophrenia: a multimodal voxelwise meta-analysis and meta-regression analysis. Schizophr. Res. 127, 46–57. doi: 10.1016/j.schres.2010.12.020
Bose, S., and Cho, J. (2013). Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch. Pharm. Res. 36, 1039–1050. doi: 10.1007/s12272-013-0161-z
Bousman, C. A., Cropley, V., Klauser, P., Hess, J. L., Pereira, A., Idrizi, R., et al. (2018). Neuregulin-1 (NRG1) polymorphisms linked with psychosis transition are associated with enlarged lateral ventricles and white matter disruption in schizophrenia. Psychol. Med. 48, 801–809. doi: 10.1017/S0033291717002173
Boza-Serrano, A., Yang, Y., Paulus, A., and Deierborg, T. (2018). Innate immune alterations are elicited in microglial cells before plaque deposition in the Alzheimer’s disease mouse model 5xFAD. Sci. Rep. 8:1550. doi: 10.1038/s41598-018-19699-y
Braun, M., Vaibhav, K., Saad, N. M., Fatima, S., Vender, J. R., Baban, B., et al. (2017). White matter damage after traumatic brain injury: a role for damage associated molecular patterns. Biochim. Biophys. Acta 1863, 2614–2626. doi: 10.1016/j.bbadis.2017.05.020
Brionne, T. C., Tesseur, I., Masliah, E., and Wyss-Coray, T. (2003). Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 40, 1133–1145. doi: 10.1016/S0896-6273(03)00766-9
Britschgi, M., and Wyss-Coray, T. (2007). Systemic and acquired immune responses in Alzheimer’s disease. Int. Rev. Neurobiol. 82, 205–233. doi: 10.1016/S0074-7742(07)82011-3
Brown, A. S. (2011). The environment and susceptibility to schizophrenia. Progr. Neurobiol. 93, 23–58. doi: 10.1016/j.pneurobio.2010.09.003
Buckingham, S. C., Campbell, S. L., Haas, B. R., Montana, V., Robel, S., Ogunrinu, T., et al. (2011). Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 17, 1269–1274. doi: 10.1038/nm.2453
Budnik, V., Ruiz-Canada, C., and Wendler, F. (2016). Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 17, 160–172. doi: 10.1038/nrn.2015.29
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Calvo, M., Zhu, N., Tsantoulas, C., Ma, Z., Grist, J., Loeb, J. A., et al. (2010). Neuregulin-ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J. Neurosci. 30, 5437–5450. doi: 10.1523/JNEUROSCI.5169-09.2010
Cao, B., Bauer, I. E., Sharma, A. N., Mwangi, B., Frazier, T., Lavagnino, L., et al. (2016). Reduced hippocampus volume and memory performance in bipolar disorder patients carrying the BDNF val66met met allele. J. Affect. Disord. 198, 198–205. doi: 10.1016/j.jad.2016.03.044
Capettini, L. S., Savergnini, S. Q., da Silva, R. F., Stergiopulos, N., Santos, R. A., Mach, F., et al. (2012). Update on the role of cannabinoid receptors after ischemic stroke. Mediat. Inflamm. 2012:824093. doi: 10.1155/2012/824093
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924. doi: 10.1038/nn1715
Castren, E. (2014). Neurotrophins and psychiatric disorders. Handb. Exp. Pharmacol. 220, 461–479. doi: 10.1007/978-3-642-45106-5_17
Chamorro, A., Dirnagl, U., Urra, X., and Planas, A. M. (2016). Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 15, 869–881. doi: 10.1016/S1474-4422(16)00114-9
Chandrasekar, B., Mummidi, S., Perla, R. P., Bysani, S., Dulin, N. O., Liu, F., et al. (2003). Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem. J. 373, 547–558. doi: 10.1042/bj20030207
Chang, C., Lang, H., Geng, N., Wang, J., Li, N., and Wang, X. (2013). Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD. Neurosci. Lett. 548, 190–195. doi: 10.1016/j.neulet.2013.06.009
Che, X., Ye, W., Panga, L., Wu, D. C., and Yang, G. Y. (2001). Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 902, 171–177. doi: 10.1016/S0006-8993(01)02328-9
Chen, P., Zhao, W., Guo, Y., Xu, J., and Yin, M. (2016). CX3CL1/CX3CR1 in Alzheimer’s disease: a target for neuroprotection. Biomed. Res. Int. 2016:8090918. doi: 10.1155/2016/8090918
Chen, T., Koga, K., Li, X. Y., and Zhuo, M. (2010). Spinal microglial motility is independent of neuronal activity and plasticity in adult mice. Mol. Pain 6:19. doi: 10.1186/1744-8069-6-19
Chew, L. J., Fusar-Poli, P., and Schmitz, T. (2013). Oligodendroglial alterations and the role of microglia in white matter injury: relevance to schizophrenia. Dev. Neurosci. 35, 102–129. doi: 10.1159/000346157
Chhor, V., Moretti, R., Le Charpentier, T., Sigaut, S., Lebon, S., Schwendimann, L., et al. (2017). Role of microglia in a mouse model of paediatric traumatic brain injury. Brain Behav. Immun. 63, 197–209. doi: 10.1016/j.bbi.2016.11.001
Cho, S. H., Sun, B., Zhou, Y., Kauppinen, T. M., Halabisky, B., Wes, P., et al. (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 286, 32713–32722. doi: 10.1074/jbc.M111.254268
Cipriani, R., Villa, P., Chece, G., Lauro, C., Paladini, A., Micotti, E., et al. (2011). CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 31, 16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011
Cocucci, E., and Meldolesi, J. (2015). Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 25, 364–372. doi: 10.1016/j.tcb.2015.01.004
Cocucci, E., Racchetti, G., and Meldolesi, J. (2009). Shedding microvesicles: artefacts no more. Trends Cell Biol. 19, 43–51. doi: 10.1016/j.tcb.2008.11.003
Colombo, M., Raposo, G., and Thery, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell. Dev. Biol. 30, 255–289. doi: 10.1146/annurev-cellbio-101512-122326
Colonna, M., and Facchetti, F. (2003). TREM-1 (triggering receptor expressed on myeloid cells): a new player in acute inflammatory responses. J. Infect. Dis. 187(Suppl. 2), S397–S401. doi: 10.1086/374754
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 6:6176. doi: 10.1038/ncomms7176
Conductier, G., Blondeau, N., Guyon, A., Nahon, J. L., and Rovere, C. (2010). The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 224, 93–100. doi: 10.1016/j.jneuroim.2010.05.010
Coniglio, S. J., Eugenin, E., Dobrenis, K., Stanley, E. R., West, B. L., Symons, M. H., et al. (2012). Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 18, 519–527. doi: 10.2119/molmed.2011.00217
Corfas, G., Roy, K., and Buxbaum, J. D. (2004). Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat. Neurosci. 7, 575–580. doi: 10.1038/nn1258
Corona, A. W., Huang, Y., O’Connor, J. C., Dantzer, R., Kelley, K. W., Popovich, P. G., et al. (2010). Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J. Neuroinflamm. 7:93. doi: 10.1186/1742-2094-7-93
Cotter, D., Mackay, D., Chana, G., Beasley, C., Landau, S., and Everall, I. P. (2002). Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 12, 386–394. doi: 10.1093/cercor/12.4.386
Coughlan, C. M., McManus, C. M., Sharron, M., Gao, Z., Murphy, D., Jaffer, S., et al. (2000). Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience 97, 591–600. doi: 10.1016/S0306-4522(00)00024-5
Croese, T., and Furlan, R. (2018). Extracellular vesicles in neurodegenerative diseases. Mol. Aspects Med. 60, 52–61. doi: 10.1016/j.mam.2017.11.006
Cunningham, C. L., Martinez-Cerdeno, V., and Noctor, S. C. (2013). Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 33, 4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013
Dalmau, I., Vela, J. M., Gonzalez, B., Finsen, B., and Castellano, B. (2003). Dynamics of microglia in the developing rat brain. J. Comp. Neurol. 458, 144–157. doi: 10.1002/cne.10572
Davalieva, K., Maleva Kostovska, I., and Dwork, A. J. (2016). Proteomics research in schizophrenia. Front. Cell Neurosci. 10:18. doi: 10.3389/fncel.2016.00018
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
De Biase, L. M., Schuebel, K. E., Fusfeld, Z. H., Jair, K., Hawes, I. A., Cimbro, R., et al. (2017). Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron 95:e6. doi: 10.1016/j.neuron.2017.06.020
Deczkowska, A., Amit, I., and Schwartz, M. (2018). Microglial immune checkpoint mechanisms. Nat. Neurosci. 21, 779–786. doi: 10.1038/s41593-018-0145-x
DeLeo, A. M., and Ikezu, T. (2018). Extracellular vesicle biology in alzheimer’s disease and related tauopathy. J. Neuroimmune Pharmacol. 13, 292–308. doi: 10.1007/s11481-017-9768-z
Denes, A., Ferenczi, S., Halasz, J., Kornyei, Z., and Kovacs, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721. doi: 10.1038/jcbfm.2008.64
Deng, J., Koutras, C., Donnelier, J., Alshehri, M., Fotouhi, M., Girard, M., et al. (2017). Neurons export extracellular vesicles enriched in cysteine string protein and misfolded protein cargo. Sci. Rep. 7:956. doi: 10.1038/s41598-017-01115-6
Denieffe, S., Kelly, R. J., McDonald, C., Lyons, A., and Lynch, M. A. (2013). Classical activation of microglia in CD200-deficient mice is a consequence of blood brain barrier permeability and infiltration of peripheral cells. Brain Behav. Immun. 34, 86–97. doi: 10.1016/j.bbi.2013.07.174
Dentesano, G., Serratosa, J., Tusell, J. M., Ramon, P., Valente, T., Saura, J., et al. (2014). CD200R1 and CD200 expression are regulated by PPAR-gamma in activated glial cells. Glia 62, 982–998. doi: 10.1002/glia.22656
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi: 10.1038/nature02006
Dibaj, P., Nadrigny, F., Steffens, H., Scheller, A., Hirrlinger, J., Schomburg, E. D., et al. (2010). NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia 58, 1133–1144. doi: 10.1002/glia.20993
Dickens, A. M., Romo Tovar, L. B., Yoo, S. W., Trout, A. L., Bae, M., Kanmogne, M., et al. (2017). Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal. 10:eaai7696.
Dijkstra, I. M., Hulshof, S., van der Valk, P., Boddeke, H. W., and Biber, K. (2004). Cutting edge: activity of human adult microglia in response to CC chemokine ligand 21. J. Immunol. 172, 2744–2747. doi: 10.4049/jimmunol.172.5.2744
Dirnagl, U. (2012). Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann. N. Y. Acad. Sci. 1268, 21–25. doi: 10.1111/j.1749-6632.2012.06691.x
Dissing-Olesen, L., LeDue, J. M., Rungta, R. L., Hefendehl, J. K., Choi, H. B., and MacVicar, B. A. (2014). Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. 34, 10511–10527. doi: 10.1523/JNEUROSCI.0405-14.2014
Donat, C. K., Scott, G., Gentleman, S. M., and Sastre, M. (2017). Microglial activation in traumatic brain injury. Front. Aging Neurosci. 9:208. doi: 10.3389/fnagi.2017.00208
Donohue, M. M., Cain, K., Zierath, D., Shibata, D., Tanzi, P. M., and Becker, K. J. (2012). Higher plasma fractalkine is associated with better 6-month outcome from ischemic stroke. Stroke 43, 2300–2306. doi: 10.1161/STROKEAHA.112.657411
Doorn, K. J., Breve, J. J., Drukarch, B., Boddeke, H. W., Huitinga, I., Lucassen, P. J., et al. (2015). Brain region-specific gene expression profiles in freshly isolated rat microglia. Front. Cell Neurosci. 9:84. doi: 10.3389/fncel.2015.00084
Dozio, V., and Sanchez, J. C. (2017). Characterisation of extracellular vesicle-subsets derived from brain endothelial cells and analysis of their protein cargo modulation after TNF exposure. J. Extracell. Vesicles 6:1302705. doi: 10.1080/20013078.2017.1302705
Drago, F., Lombardi, M., Prada, I., Gabrielli, M., Joshi, P., Cojoc, D., et al. (2017). ATP modifies the proteome of extracellular vesicles released by microglia and influences their action on astrocytes. Front. Pharmacol. 8:910. doi: 10.3389/fphar.2017.00910
Du, L., Zhang, Y., Chen, Y., Zhu, J., Yang, Y., and Zhang, H. L. (2017). Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. 54, 7567–7584. doi: 10.1007/s12035-016-0245-0
Dworzak, J., Renvoise, B., Habchi, J., Yates, E. V., Combadiere, C., Knowles, T. P., et al. (2015). Neuronal Cx3cr1 deficiency protects against amyloid beta-induced neurotoxicity. PLoS One 10:e0127730. doi: 10.1371/journal.pone.0127730
Elkabes, S., DiCicco-Bloom, E. M., and Black, I. B. (1996). Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J. Neurosci. 16, 2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996
Ellison-Wright, I., and Bullmore, E. T. (2009). Meta-analysis of diffusion tensor imaging studies in schizophrenia. Schizophr. Res. 108, 3–10. doi: 10.1016/j.schres.2008.11.021
Erturk, A., Mentz, S., Stout, E. E., Hedehus, M., Dominguez, S. L., Neumaier, L., et al. (2016). Interfering with the chronic immune response rescues chronic degeneration after traumatic brain injury. J. Neurosci. 36, 9962–9975. doi: 10.1523/JNEUROSCI.1898-15.2016
Eyo, U. B., and Wu, L. J. (2013). Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. 2013:456857. doi: 10.1155/2013/456857
Fang, K. M., Yang, C. S., Sun, S. H., and Tzeng, S. F. (2009). Microglial phagocytosis attenuated by short-term exposure to exogenous ATP through P2X receptor action. J. Neurochem. 111, 1225–1237. doi: 10.1111/j.1471-4159.2009.06409.x
Farhy-Tselnicker, I., and Allen, N. J. (2018). Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev. 13:7. doi: 10.1186/s13064-018-0104-y
Faure, J., Lachenal, G., Court, M., Hirrlinger, J., Chatellard-Causse, C., Blot, B., et al. (2006). Exosomes are released by cultured cortical neurones. Mol. Cell Neurosci. 31, 642–648. doi: 10.1016/j.mcn.2005.12.003
Fazio, F., Ulivieri, M., Volpi, C., Gargaro, M., and Fallarino, F. (2018). Targeting metabotropic glutamate receptors for the treatment of neuroinflammation. Curr. Opin. Pharmacol. 38, 16–23. doi: 10.1016/j.coph.2018.01.010
Febinger, H. Y., Thomasy, H. E., Pavlova, M. N., Ringgold, K. M., Barf, P. R., George, A. M., et al. (2015). Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J. Neuroinflamm. 12:154. doi: 10.1186/s12974-015-0386-5
Ferrari, D., Chiozzi, P., Falzoni, S., Hanau, S., and Di Virgilio, F. (1997). Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J. Exp. Med. 185, 579–582. doi: 10.1084/jem.185.3.579
Ferrari, D., Los, M., Bauer, M. K., Vandenabeele, P., Wesselborg, S., and Schulze-Osthoff, K. (1999). P2Z purinoreceptor ligation induces activation of caspases with distinct roles in apoptotic and necrotic alterations of cell death. FEBS Lett. 447, 71–75. doi: 10.1016/S0014-5793(99)00270-7
Ferrini, F., and De Koninck, Y. (2013). Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013:429815. doi: 10.1155/2013/429815
Fields, R. D., and Burnstock, G. (2006). Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 7, 423–436. doi: 10.1038/nrn1928
Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48:e8. doi: 10.1016/j.immuni.2018.04.016
Fitzner, D., Schnaars, M., van Rossum, D., Krishnamoorthy, G., Dibaj, P., Bakhti, M., et al. (2011). Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell. Sci. 124, 447–458. doi: 10.1242/jcs.074088
Fonseca, M. I., Ager, R. R., Chu, S. H., Yazan, O., Sanderson, S. D., LaFerla, F. M., et al. (2009). Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 183, 1375–1383. doi: 10.4049/jimmunol.0901005
Fonseca, M. I., Chu, S. H., Hernandez, M. X., Fang, M. J., Modarresi, L., Selvan, P., et al. (2017). Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflamm. 14:48. doi: 10.1186/s12974-017-0814-9
Fontainhas, A. M., Wang, M., Liang, K. J., Chen, S., Mettu, P., Damani, M., et al. (2011). Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One 6:e15973. doi: 10.1371/journal.pone.0015973
Fornito, A., Yucel, M., Patti, J., Wood, S. J., and Pantelis, C. (2009). Mapping grey matter reductions in schizophrenia: an anatomical likelihood estimation analysis of voxel-based morphometry studies. Schizophr. Res. 108, 104–113. doi: 10.1016/j.schres.2008.12.011
Fruhbeis, C., Frohlich, D., Kuo, W. P., Amphornrat, J., Thilemann, S., Saab, A. S., et al. (2013). Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 11:e1001604. doi: 10.1371/journal.pbio.1001604
Gabrielli, M., Battista, N., Riganti, L., Prada, I., Antonucci, F., Cantone, L., et al. (2015). Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 16, 213–220. doi: 10.15252/embr.201439668
Gabrusiewicz, K., Ellert-Miklaszewska, A., Lipko, M., Sielska, M., Frankowska, M., and Kaminska, B. (2011). Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS One 6:e23902. doi: 10.1371/journal.pone.0023902
Ganzola, R., and Duchesne, S. (2017). Voxel-based morphometry meta-analysis of gray and white matter finds significant areas of differences in bipolar patients from healthy controls. Bipolar. Disord. 19, 74–83. doi: 10.1111/bdi.12488
Garey, L. (2010). When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J. Anat. 217, 324–333. doi: 10.1111/j.1469-7580.2010.01231.x
Garton, K. J., Gough, P. J., Blobel, C. P., Murphy, G., Greaves, D. R., Dempsey, P. J., et al. (2001). Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 276, 37993–38001.
Garzetti, L., Menon, R., Finardi, A., Bergami, A., Sica, A., Martino, G., et al. (2014). Activated macrophages release microvesicles containing polarized M1 or M2 mRNAs. J. Leukoc. Biol. 95, 817–825. doi: 10.1189/jlb.0913485
Gertig, U., and Hanisch, U. K. (2014). Microglial diversity by responses and responders. Front. Cell Neurosci. 8:101. doi: 10.3389/fncel.2014.00101
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Ginhoux, F., Lim, S., Hoeffel, G., Low, D., and Huber, T. (2013). Origin and differentiation of microglia. Front. Cell Neurosci. 7:45. doi: 10.3389/fncel.2013.00045
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7:a020537. doi: 10.1101/cshperspect.a020537
Glebov, K., Lochner, M., Jabs, R., Lau, T., Merkel, O., Schloss, P., et al. (2015). Serotonin stimulates secretion of exosomes from microglia cells. Glia 63, 626–634. doi: 10.1002/glia.22772
Goldsmith, D. R., Rapaport, M. H., and Miller, B. J. (2016). A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 21, 1696–1709. doi: 10.1038/mp.2016.3
Gottmann, K., Mittmann, T., and Lessmann, V. (2009). BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp. Brain Res. 199, 203–234. doi: 10.1007/s00221-009-1994-z
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Baillie, J. K., Stevens, M. P., et al. (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. doi: 10.1038/nn.4222
Graeber, M. B., and Streit, W. J. (2010). Microglia: biology and pathology. Acta Neuropathol. 119, 89–105. doi: 10.1007/s00401-009-0622-0
Grathwohl, S. A., Kalin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., et al. (2009). Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363. doi: 10.1038/nn.2432
Greco, V., Hannus, M., and Eaton, S. (2001). Argosomes: a potential vehicle for the spread of morphogens through epithelia. Cell 106, 633–645. doi: 10.1016/S0092-8674(01)00484-6
Griffiths, M., Neal, J. W., and Gasque, P. (2007). Innate immunity and protective neuroinflammation: new emphasis on the role of neuroimmune regulatory proteins. Int. Rev. Neurobiol. 82, 29–55. doi: 10.1016/S0074-7742(07)82002-2
Grimmig, B., Morganti, J., Nash, K., and Bickford, P. C. (2016). Immunomodulators as therapeutic agents in mitigating the progression of Parkinson’s disease. Brain Sci. 6:E41. doi: 10.3390/brainsci6040041
Guillemin, G. J., Croitoru-Lamoury, J., Dormont, D., Armati, P. J., and Brew, B. J. (2003). Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. Glia 41, 371–381. doi: 10.1002/glia.10175
Guix, F. X., Corbett, G. T., Cha, D. J., Mustapic, M., Liu, W., Mengel, D., et al. (2018). Detection of aggregation-competent tau in neuron-derived extracellular vesicles. Int. J. Mol. Sci. 19:E663. doi: 10.3390/ijms19030663
Gyorgy, B., Szabo, T. G., Pasztoi, M., Pal, Z., Misjak, P., Aradi, B., et al. (2011). Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol. Life Sci. 68, 2667–2688. doi: 10.1007/s00018-011-0689-3
Haarman, B. C., Riemersma-Van, der Lek, R. F., de Groot, Ruhe, J. C. H. G., Klein, H. C., et al. (2014). Neuroinflammation in bipolar disorder – A [(11)C]-(R)-PK11195 positron emission tomography study. Brain Behav. Immun. 40, 219–225. doi: 10.1016/j.bbi.2014.03.016
Hagino, Y., Kariura, Y., Manago, Y., Amano, T., Wang, B., Sekiguchi, M., et al. (2004). Heterogeneity and potentiation of AMPA type of glutamate receptors in rat cultured microglia. Glia 47, 68–77. doi: 10.1002/glia.20034
Hambardzumyan, D., Gutmann, D. H., and Kettenmann, H. (2016). The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–27. doi: 10.1038/nn.4185
Hanisch, U. K. (2002). Microglia as a source and target of cytokines. Glia 40, 140–155. doi: 10.1002/glia.10161
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hanlon, L. A., Huh, J. W., and Raghupathi, R. (2016). Minocycline transiently reduces microglia/macrophage activation but exacerbates cognitive deficits following repetitive traumatic brain injury in the neonatal rat. J. Neuropathol. Exp. Neurol. 75, 214–226. doi: 10.1093/jnen/nlv021
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U.S.A. 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Hatori, K., Nagai, A., Heisel, R., Ryu, J. K., and Kim, S. U. (2002). Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 69, 418–426. doi: 10.1002/jnr.10304
Hayakawa, K., Pham, L. D., Seo, J. H., Miyamoto, N., Maki, T., Terasaki, Y., et al. (2016a). CD200 restrains macrophage attack on oligodendrocyte precursors via toll-like receptor 4 downregulation. J. Cereb. Blood Flow Metab. 36, 781–793. doi: 10.1177/0271678X15606148
Hayakawa, K., Wang, X., and Lo, E. H. (2016b). CD200 increases alternatively activated macrophages through cAMP-response element binding protein – C/EBP-beta signaling. J. Neurochem. 136, 900–906. doi: 10.1111/jnc.13492
Haynes, S. E., Hollopeter, G., Yang, G., Kurpius, D., Dailey, M. E., Gan, W. B., et al. (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519. doi: 10.1038/nn1805
He, M., Dong, H., Huang, Y., Lu, S., Zhang, S., Qian, Y., et al. (2016). Astrocyte-derived CCL2 is associated with M1 activation and recruitment of cultured microglial cells. Cell Physiol. Biochem. 38, 859–870. doi: 10.1159/000443040
Hellwig, S., Brioschi, S., Dieni, S., Frings, L., Masuch, A., Blank, T., et al. (2016). Altered microglia morphology and higher resilience to stress-induced depression-like behavior in CX3CR1-deficient mice. Brain Behav. Immun. 55, 126–137. doi: 10.1016/j.bbi.2015.11.008
Heneka, M. T., Kummer, M. P., and Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. doi: 10.1038/nri3705
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hercher, C., Chopra, V., and Beasley, C. L. (2014). Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J. Psychiatry Neurosci. 39, 376–385. doi: 10.1503/jpn.130277
Hernangomez, M., Mestre, L., Correa, F. G., Loria, F., Mecha, M., Inigo, P. M., et al. (2012). CD200-CD200R1 interaction contributes to neuroprotective effects of anandamide on experimentally induced inflammation. Glia 60, 1437–1450. doi: 10.1002/glia.22366
Hinwood, M., Morandini, J., Day, T. A., and Walker, F. R. (2012). Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb. Cortex 22, 1442–1454. doi: 10.1093/cercor/bhr229
Hishii, M., Nitta, T., Ishida, H., Ebato, M., Kurosu, A., Yagita, H., et al. (1995). Human glioma-derived interleukin-10 inhibits antitumor immune responses in vitro. Neurosurgery 37, 1160–1166; discussion 66–67. doi: 10.1227/00006123-199512000-00016
Hoeffel, G., and Ginhoux, F. (2018). Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol. 330, 5–15. doi: 10.1016/j.cellimm.2018.01.001
Hoek, R. M., Ruuls, S. R., Murphy, C. A., Wright, G. J., Goddard, R., Zurawski, S. M., et al. (2000). Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290, 1768–1771. doi: 10.1126/science.290.5497.1768
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hooper, C., Sainz-Fuertes, R., Lynham, S., Hye, A., Killick, R., Warley, A., et al. (2012). Wnt3a induces exosome secretion from primary cultured rat microglia. BMC Neurosci. 13:144. doi: 10.1186/1471-2202-13-144
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N., and Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
Hsieh, C. L., Koike, M., Spusta, S. C., Niemi, E. C., Yenari, M., Nakamura, M. C., et al. (2009). A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x
Hu, X., Li, P., Guo, Y., Wang, H., Leak, R. K., Chen, S., et al. (2012). Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070. doi: 10.1161/STROKEAHA.112.659656
Huang, S., Ge, X., Yu, J., Han, Z., Yin, Z., Li, Y., et al. (2018). Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. 32, 512–528. doi: 10.1096/fj.201700673R
Hughes, P. M., Botham, M. S., Frentzel, S., Mir, A., and Perry, V. H. (2002). Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia 37, 314–327. doi: 10.1002/glia.10037
Imai, T., Hieshima, K., Haskell, C., Baba, M., Nagira, M., Nishimura, M., et al. (1997). Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91, 521–530. doi: 10.1016/S0092-8674(00)80438-9
Ishizuka, K., Fujita, Y., Kawabata, T., Kimura, H., Iwayama, Y., Inada, T., et al. (2017). Rare genetic variants in CX3CR1 and their contribution to the increased risk of schizophrenia and autism spectrum disorders. Transl. Psychiatry 7:e1184. doi: 10.1038/tp.2017.173
Jablonski, K. A., Gaudet, A. D., Amici, S. A., Popovich, P. G., and Guerau-de-Arellano, M. (2016). Control of the inflammatory macrophage transcriptional signature by miR-155. PLoS One 11:e0159724. doi: 10.1371/journal.pone.0159724
Jiang, S., and Bhaskar, K. (2017). Dynamics of the complement, cytokine, and chemokine systems in the regulation of synaptic function and dysfunction relevant to Alzheimer’s disease. J. Alzheimers Dis. 57, 1123–1135. doi: 10.3233/JAD-161123
Jiang, Z., Jiang, J. X., and Zhang, G. X. (2014). Macrophages: a double-edged sword in experimental autoimmune encephalomyelitis. Immunol. Lett. 160, 17–22. doi: 10.1016/j.imlet.2014.03.006
Jiang, Z., Zhou, W., Guan, S., Wang, J., and Liang, Y. (2013). Contribution of SDF-1alpha/CXCR4 signaling to brain development and glioma progression. Neurosignals 21, 240–258. doi: 10.1159/000339091
Jin, N., Gao, L., Fan, X., and Xu, H. (2017). Friend or Foe? Resident microglia vs bone marrow-derived microglia and their roles in the retinal degeneration. Mol. Neurobiol. 54, 4094–4112. doi: 10.1007/s12035-016-9960-9
Joe, E. H., Choi, D. J., An, J., Eun, J. H., Jou, I., and Park, S. (2018). Astrocytes, microglia, and Parkinson’s disease. Exp. Neurobiol. 27, 77–87. doi: 10.5607/en.2018.27.2.77
John, G. R., Lee, S. C., and Brosnan, C. F. (2003). Cytokines: powerful regulators of glial cell activation. Neuroscientist 9, 10–22. doi: 10.1177/1073858402239587
Joshi, P., Turola, E., Ruiz, A., Bergami, A., Libera, D. D., Benussi, L., et al. (2014). Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 21, 582–593. doi: 10.1038/cdd.2013.180
Joundi, R. A., Rabinstein, A. A., Nikneshan, D., Tu, J. V., Fang, J., Holloway, R., et al. (2016). Cardiac arrest in acute ischemic stroke: incidence, predisposing factors, and clinical outcomes. J. Stroke Cerebrovasc. Dis. 25, 1644–1652. doi: 10.1016/j.jstrokecerebrovasdis.2016.03.010
Jung, S., Aliberti, J., Graemmel, P., Sunshine, M. J., Kreutzberg, G. W., Sher, A., et al. (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell Biol. 20, 4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000
Jung, Y. J., and Chung, W. S. (2018). Phagocytic roles of glial cells in healthy and diseased brains. Biomol. Ther. (Seoul) 26, 350–357. doi: 10.4062/biomolther.2017.133
Kang, H. J., Voleti, B., Hajszan, T., Rajkowska, G., Stockmeier, C. A., Licznerski, P., et al. (2012). Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 18, 1413–1417. doi: 10.1038/nm.2886
Karlstetter, M., Scholz, R., Rutar, M., Wong, W. T., Provis, J. M., and Langmann, T. (2015). Retinal microglia: just bystander or target for therapy? Prog. Retin. Eye Res. 45, 30–57. doi: 10.1016/j.preteyeres.2014.11.004
Kelly, A., Lynch, A., Vereker, E., Nolan, Y., Queenan, P., Whittaker, E., et al. (2001). The anti-inflammatory cytokine, interleukin (IL)-10, blocks the inhibitory effect of IL-1 beta on long term potentiation, a role for JNK. J. Biol. Chem. 276, 45564–45572. doi: 10.1074/jbc.M108757200
Kerschensteiner, M., Meinl, E., and Hohlfeld, R. (2009). Neuro-immune crosstalk in CNS diseases. Neuroscience 158, 1122–1132. doi: 10.1016/j.neuroscience.2008.09.009
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kierdorf, K., Erny, D., Goldmann, T., Sander, V., Schulz, C., Perdiguero, E. G., et al. (2013). Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 16, 273–280. doi: 10.1038/nn.3318
Kierdorf, K., and Prinz, M. (2013). Factors regulating microglia activation. Front. Cell Neurosci. 7:44. doi: 10.3389/fncel.2013.00044
Kierdorf, K., and Prinz, M. (2017). Microglia in steady state. J. Clin. Invest 127, 3201–3209. doi: 10.1172/JCI90602
Kiialainen, A., Hovanes, K., Paloneva, J., Kopra, O., and Peltonen, L. (2005). Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol. Dis. 18, 314–322. doi: 10.1016/j.nbd.2004.09.007
Kim, J. Y., Kanai, Y., Chairoungdua, A., Cha, S. H., Matsuo, H., Kim, D. K., et al. (2001). Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim. Biophys. Acta 1512, 335–344. doi: 10.1016/S0005-2736(01)00338-8
Kim, T. S., Lim, H. K., Lee, J. Y., Kim, D. J., Park, S., Lee, C., et al. (2008). Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 436, 196–200. doi: 10.1016/j.neulet.2008.03.019
Kishi, T., Yoshimura, R., Ikuta, T., and Iwata, N. (2017). Brain-derived neurotrophic factor and major depressive disorder: evidence from meta-analyses. Front. Psychiatry 8:308. doi: 10.3389/fpsyt.2017.00308
Knott, C., Stern, G., Kingsbury, A., Welcher, A. A., and Wilkin, G. P. (2002). Elevated glial brain-derived neurotrophic factor in Parkinson’s diseased nigra. Parkinsonism Relat. Disord. 8, 329–341. doi: 10.1016/S1353-8020(02)00008-1
Koizumi, S., Shigemoto-Mogami, Y., Nasu-Tada, K., Shinozaki, Y., Ohsawa, K., Tsuda, M., et al. (2007). UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095. doi: 10.1038/nature05704
Kong, T., Park, J. M., Jang, J. H., Kim, C. Y., Bae, S. H., Choi, Y., et al. (2018). Immunomodulatory effect of CD200-positive human placenta-derived stem cells in the early phase of stroke. Exp. Mol. Med. 50:e425. doi: 10.1038/emm.2017.233
Koning, N., Swaab, D. F., Hoek, R. M., and Huitinga, I. (2009). Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. J. Neuropathol. Exp. Neurol. 68, 159–167. doi: 10.1097/NEN.0b013e3181964113
Konnecke, H., and Bechmann, I. (2013). The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas. Clin. Dev. Immunol. 2013:914104. doi: 10.1155/2013/914104
Kramer-Albers, E. M., Bretz, N., Tenzer, S., Winterstein, C., Mobius, W., Berger, H., et al. (2007). Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: trophic support for axons? Proteomics Clin. Appl. 1, 1446–1461. doi: 10.1002/prca.200700522
Kramer-Albers, E. M., and Hill, A. F. (2016). Extracellular vesicles: interneural shuttles of complex messages. Curr. Opin. Neurobiol. 39, 101–107. doi: 10.1016/j.conb.2016.04.016
Kreisel, T., Frank, M. G., Licht, T., Reshef, R., Ben-Menachem-Zidon, O., Baratta, M. V., et al. (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 19, 699–709. doi: 10.1038/mp.2013.155
Krishnan, V., and Nestler, E. J. (2008). The molecular neurobiology of depression. Nature 455, 894–902. doi: 10.1038/nature07455
Ku, M. C., Wolf, S. A., Respondek, D., Matyash, V., Pohlmann, A., Waiczies, S., et al. (2013). GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol. 125, 609–620. doi: 10.1007/s00401-013-1079-8
Kuhn, S. A., van Landeghem, F. K., Zacharias, R., Farber, K., Rappert, A., Pavlovic, S., et al. (2004). Microglia express GABA(B) receptors to modulate interleukin release. Mol. Cell Neurosci. 25, 312–322. doi: 10.1016/j.mcn.2003.10.023
Kumar, A., Stoica, B. A., Loane, D. J., Yang, M., Abulwerdi, G., Khan, N., et al. (2017). Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflamm. 14:47. doi: 10.1186/s12974-017-0819-4
Lachenal, G., Pernet-Gallay, K., Chivet, M., Hemming, F. J., Belly, A., Bodon, G., et al. (2011). Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell Neurosci. 46, 409–418. doi: 10.1016/j.mcn.2010.11.004
Lai, C. P., Tannous, B. A., and Breakefield, X. O. (2014). Noninvasive in vivo monitoring of extracellular vesicles. Methods Mol. Biol. 1098, 249–258. doi: 10.1007/978-1-62703-718-1_19
Lampe, I. K., Hulshoff Pol, H. E., Janssen, J., Schnack, H. G., Kahn, R. S., and Heeren, T. J. (2003). Association of depression duration with reduction of global cerebral gray matter volume in female patients with recurrent major depressive disorder. Am. J. Psychiatry 160, 2052–2054. doi: 10.1176/appi.ajp.160.11.2052
Laskaris, L. E., Di Biase, M. A., Everall, I., Chana, G., Christopoulos, A., Skafidas, E., et al. (2016). Microglial activation and progressive brain changes in schizophrenia. Br. J. Pharmacol. 173, 666–680. doi: 10.1111/bph.13364
Lavagnino, L., Cao, B., Mwangi, B., Wu, M. J., Sanches, M., Zunta-Soares, G. B., et al. (2015). Changes in the corpus callosum in women with late-stage bipolar disorder. Acta Psychiatr. Scand. 131, 458–464. doi: 10.1111/acps.12397
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-W
Lee, M. K., Pardoux, C., Hall, M. C., Lee, P. S., Warburton, D., Qing, J., et al. (2007). TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967. doi: 10.1038/sj.emboj.7601818
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 177, 2549–2562. doi: 10.2353/ajpath.2010.100265
Li, J., Li, X., Jiang, X., Yang, M., Yang, R., Burnstock, G., et al. (2017). Microvesicles shed from microglia activated by the P2X7-p38 pathway are involved in neuropathic pain induced by spinal nerve ligation in rats. Purinergic. Signal. 13, 13–26. doi: 10.1007/s11302-016-9537-0
Li, T., Pang, S., Yu, Y., Wu, X., Guo, J., and Zhang, S. (2013). Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain 136, 3578–3588. doi: 10.1093/brain/awt287
Li, W., and Graeber, M. B. (2012). The molecular profile of microglia under the influence of glioma. Neuro Oncol. 14, 958–978. doi: 10.1093/neuonc/nos116
Li, Y., Du, X. F., Liu, C. S., Wen, Z. L., and Du, J. L. (2012). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell 23, 1189–1202. doi: 10.1016/j.devcel.2012.10.027
Lim, S. H., Park, E., You, B., Jung, Y., Park, A. R., Park, S. G., et al. (2013). Neuronal synapse formation induced by microglia and interleukin 10. PLoS One 8:e81218. doi: 10.1371/journal.pone.0081218
Liu, Y. Z., Wang, C., Wang, Q., Lin, Y. Z., Ge, Y. S., Li, D. M., et al. (2017). Role of fractalkine/CX3CR1 signaling pathway in the recovery of neurological function after early ischemic stroke in a rat model. Life Sci. 184, 87–94. doi: 10.1016/j.lfs.2017.06.012
Liu, Z., Condello, C., Schain, A., Harb, R., and Grutzendler, J. (2010). CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J. Neurosci. 30, 17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010
Loane, D. J., and Kumar, A. (2016). Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp. Neurol. 275(Pt 3), 316–327. doi: 10.1016/j.expneurol.2015.08.018
London, A., Benhar, I., and Schwartz, M. (2013). The retina as a window to the brain-from eye research to CNS disorders. Nat. Rev. Neurol. 9, 44–53. doi: 10.1038/nrneurol.2012.227
Lucae, S., Salyakina, D., Barden, N., Harvey, M., Gagne, B., Labbe, M., et al. (2006). P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum. Mol. Genet. 15, 2438–2445. doi: 10.1093/hmg/ddl166
Lynch, A. M., Murphy, K. J., Deighan, B. F., O’Reilly, J. A., Gun’ko, Y. K., Cowley, T. R., et al. (2010). The impact of glial activation in the aging brain. Aging Dis. 1, 262–278.
Lynch, M. A. (2009). The multifaceted profile of activated microglia. Mol. Neurobiol. 40, 139–156. doi: 10.1007/s12035-009-8077-9
Lynch, M. A. (2015). Neuroinflammatory changes negatively impact on LTP: a focus on IL-1beta. Brain Res. 1621, 197–204. doi: 10.1016/j.brainres.2014.08.040
Maciag, D., Hughes, J., O’Dwyer, G., Pride, Y., Stockmeier, C. A., Sanacora, G., et al. (2010). Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biol. Psychiatry 67, 465–470. doi: 10.1016/j.biopsych.2009.10.027
Maciejewski-Lenoir, D., Chen, S., Feng, L., Maki, R., and Bacon, K. B. (1999). Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J. Immunol. 163, 1628–1635.
Mackay, C. R. (2001). Chemokines: immunology’s high impact factors. Nat. Immunol. 2, 95–101. doi: 10.1038/84298
Makinde, H. M., Cuda, C. M., Just, T. B., Perlman, H. R., and Schwulst, S. J. (2017). Nonclassical monocytes mediate secondary injury, neurocognitive outcome, and neutrophil infiltration after traumatic brain injury. J. Immunol. 199, 3583–3591. doi: 10.4049/jimmunol.1700896
Marcus, H. J., Carpenter, K. L., Price, S. J., and Hutchinson, P. J. (2010). In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J. Neurooncol. 97, 11–23. doi: 10.1007/s11060-009-9990-5
Markovic, D. S., Glass, R., Synowitz, M., Rooijen, N., and Kettenmann, H. (2005). Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 64, 754–762. doi: 10.1097/01.jnen.0000178445.33972.a9
Marzesco, A. M. (2013). Prominin-1-containing membrane vesicles: origins, formation, and utility. Adv. Exp. Med. Biol. 777, 41–54. doi: 10.1007/978-1-4614-5894-4_3
Marzesco, A. M., Janich, P., Wilsch-Brauninger, M., Dubreuil, V., Langenfeld, K., Corbeil, D., et al. (2005). Release of extracellular membrane particles carrying the stem cell marker prominin-1 (CD133) from neural progenitors and other epithelial cells. J. Cell Sci. 118, 2849–2858. doi: 10.1242/jcs.02439
Matsumoto, S., Tanaka, J., Yano, H., Takahashi, H., Sugimoto, K., Ohue, S., et al. (2015). CD200+ and CD200- macrophages accumulated in ischemic lesions of rat brain: the two populations cannot be classified as either M1 or M2 macrophages. J. Neuroimmunol. 282, 7–20. doi: 10.1016/j.jneuroim.2015.03.013
McCluskey, L. P., and Lampson, L. A. (2001). Local immune regulation in the central nervous system by substance P vs. glutamate. J. Neuroimmunol. 116, 136–146. doi: 10.1016/S0165-5728(01)00295-8
McLarnon, J. G. (2017). Roles of purinergic P2X7 receptor in glioma and microglia in brain tumors. Cancer Lett. 402, 93–99. doi: 10.1016/j.canlet.2017.05.004
Mecca, C., Giambanco, I., Donato, R., and Arcuri, C. (2018). Microglia and aging: the role of the TREM2-DAP12 and CX3CL1-CX3CR1 axes. Int. J. Mol. Sci. 19:E318. doi: 10.3390/ijms19010318
Medina, M., and Avila, J. (2014). The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell Neurosci. 8:113. doi: 10.3389/fncel.2014.00113
Mellado, M., Rodriguez-Frade, J. M., Vila-Coro, A. J., Fernandez, S., Martin, de Ana, A., et al. (2001). Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 20, 2497–2507. doi: 10.1093/emboj/20.10.2497
Meucci, O., Fatatis, A., Simen, A. A., and Miller, R. J. (2000). Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U.S.A. 97, 8075–8080. doi: 10.1073/pnas.090017497
Milior, G., Lecours, C., Samson, L., Bisht, K., Poggini, S., Pagani, F., et al. (2016). Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav. Immun. 55, 114–125. doi: 10.1016/j.bbi.2015.07.024
Mittelbronn, M., Dietz, K., Schluesener, H. J., and Meyermann, R. (2001). Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 101, 249–255.
Mizoue, L. S., Sullivan, S. K., King, D. S., Kledal, T. N., Schwartz, T. W., Bacon, K. B., et al. (2001). Molecular determinants of receptor binding and signaling by the CX3C chemokine fractalkine. J. Biol. Chem. 276, 33906–33914. doi: 10.1074/jbc.M101348200
Mizutani, M., Pino, P. A., Saederup, N., Charo, I. F., Ransohoff, R. M., and Cardona, A. E. (2012). The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J. Immunol. 188, 29–36. doi: 10.4049/jimmunol.1100421
Mondello, S., Thelin, E. P., Shaw, G., Salzet, M., Visalli, C., Cizkova, D., et al. (2018). Extracellular vesicles: pathogenetic, diagnostic and therapeutic value in traumatic brain injury. Exp. Rev. Proteom. 15, 451–461. doi: 10.1080/14789450.2018.1464914
Monji, A., Kato, T., and Kanba, S. (2009). Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin. Neurosci. 63, 257–265. doi: 10.1111/j.1440-1819.2009.01945.x
Monson, K. L., Converse, M. I., and Manley, G. T. (2018). Cerebral blood vessel damage in traumatic brain injury. Clin. Biomech. (Bristol, Avon). [Epub ahead of print].
Morantz, R. A., Wood, G. W., Foster, M., Clark, M., and Gollahon, K. (1979). Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. J. Neurosurg. 50, 305–311. doi: 10.3171/jns.1979.50.3.0305
Morganti, J. M., Jopson, T. D., Liu, S., Riparip, L. K., Guandique, C. K., Gupta, N., et al. (2015). CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci. 35, 748–760. doi: 10.1523/JNEUROSCI.2405-14.2015
Morganti, J. M., Nash, K. R., Grimmig, B. A., Ranjit, S., Small, B., Bickford, P. C., et al. (2012). The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 32, 14592–14601. doi: 10.1523/JNEUROSCI.0539-12.2012
Mott, R. T., Ait-Ghezala, G., Town, T., Mori, T., Vendrame, M., Zeng, J., et al. (2004). Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 46, 369–379. doi: 10.1002/glia.20009
Mueser, K. T., and McGurk, S. R. (2004). Schizophrenia. Lancet 363, 2063–2072. doi: 10.1016/S0140-6736(04)16458-1
Munn, N. A. (2000). Microglia dysfunction in schizophrenia: an integrative theory. Med. Hypotheses 54, 198–202. doi: 10.1054/mehy.1999.0018
Nash, K. R., Moran, P., Finneran, D. J., Hudson, C., Robinson, J., Morgan, D., et al. (2015). Fractalkine over expression suppresses alpha-synuclein-mediated neurodegeneration. Mol. Ther. 23, 17–23. doi: 10.1038/mt.2014.175
Nawa, H., Takahashi, M., and Patterson, P. H. (2000). Cytokine and growth factor involvement in schizophrenia–support for the developmental model. Mol. Psychiatry 5, 594–603. doi: 10.1038/sj.mp.4000730
Neiva, I., Malva, J. O., and Valero, J. (2014). Can we talk about microglia without neurons? A discussion of microglial cell autonomous properties in culture. Front. Cell Neurosci. 8:202. doi: 10.3389/fncel.2014.00202
Neumann, H. (2001). Control of glial immune function by neurons. Glia 36, 191–199. doi: 10.1002/glia.1108
Neumann, H., Boucraut, J., Hahnel, C., Misgeld, T., and Wekerle, H. (1996). Neuronal control of MHC class II inducibility in rat astrocytes and microglia. Eur. J. Neurosci. 8, 2582–2590. doi: 10.1111/j.1460-9568.1996.tb01552.x
Neumann, H., Misgeld, T., Matsumuro, K., and Wekerle, H. (1998). Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc. Natl. Acad. Sci. U.S.A. 95, 5779–5784. doi: 10.1073/pnas.95.10.5779
Neumann, H., and Takahashi, K. (2007). Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 184, 92–99. doi: 10.1016/j.jneuroim.2006.11.032
Newman, C. F., Robert, L. L., Aaron, B. T., Noreen, A., Reilly, H., and Laszlo, G. (eds). (2002). “Bipolar disorder: diagnostic, epidemiologic, etiologic, and prognostic issues,” in Bipolar Disorder: A Cognitive Therapy Approach, (Washington, DC: American Psychological Association).
Ngolab, J., Trinh, I., Rockenstein, E., Mante, M., Florio, J., Trejo, M., et al. (2017). Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol. Commun. 5:46. doi: 10.1186/s40478-017-0445-5
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Nishiyori, A., Minami, M., Ohtani, Y., Takami, S., Yamamoto, J., Kawaguchi, N., et al. (1998). Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 429, 167–172.
Nistico, R., Salter, E., Nicolas, C., Feligioni, M., Mango, D., Bortolotto, Z. A., et al. (2017). Synaptoimmunology – Roles in health and disease. Mol. Brain 10:26. doi: 10.1186/s13041-017-0308-9
Numata, S., Ueno, S., Iga, J., Yamauchi, K., Hongwei, S., Ohta, K., et al. (2006). Brain-derived neurotrophic factor (BDNF) Val66Met polymorphism in schizophrenia is associated with age at onset and symptoms. Neurosci. Lett. 401, 1–5. doi: 10.1016/j.neulet.2006.02.054
O’Donnell, E., Vereker, E., and Lynch, M. A. (2000). Age-related impairment in LTP is accompanied by enhanced activity of stress-activated protein kinases: analysis of underlying mechanisms. Eur. J. Neurosci. 12, 345–352. doi: 10.1046/j.1460-9568.2000.00900.x
Ohsawa, K., Sanagi, T., Nakamura, Y., Suzuki, E., Inoue, K., and Kohsaka, S. (2012). Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. J. Neurochem. 121, 217–227. doi: 10.1111/j.1471-4159.2012.07693.x
Olabi, B., Ellison-Wright, I., McIntosh, A. M., Wood, S. J., Bullmore, E. T., and Lawrie, S. M. (2011). Are there progressive brain changes in schizophrenia? A meta-analysis of structural magnetic resonance imaging studies. Biol. Psychiatry 70, 88–96. doi: 10.1016/j.biopsych.2011.01.032
Olmos-Alonso, A., Schetters, S. T., Sri, S., Askew, K., Mancuso, R., Vargas-Caballero, M., et al. (2016). Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 139, 891–907. doi: 10.1093/brain/awv379
Omalu, B. (2014). Chronic traumatic encephalopathy. Prog Neurol. Surg. 28, 38–49. doi: 10.1159/000358761
Ongur, D., Drevets, W. C., and Price, J. L. (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U.S.A. 95, 13290–13295. doi: 10.1073/pnas.95.22.13290
Osier, N., Motamedi, V., Edwards, K., Puccio, A., R. Diaz-Arrastia, Kenney, K., and Gill, J. (2018). Exosomes in acquired neurological disorders: new insights into pathophysiology and treatment. Mol. Neurobiol. [Epub ahead of print]. doi: 10.1007/s12035-018-1054-4
Pabon, M. M., Bachstetter, A. D., Hudson, C. E., Gemma, C., and Bickford, P. C. (2011). CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflamm. 8:9. doi: 10.1186/1742-2094-8-9
Padmos, R. C., Hillegers, M. H., Knijff, E. M., Vonk, R., Bouvy, A., Staal, F. J., et al. (2008). A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch. Gen. Psychiatry 65, 395–407. doi: 10.1001/archpsyc.65.4.395
Pang, L., Ye, W., Che, X. M., Roessler, B. J., Betz, A. L., and Yang, G. Y. (2001). Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke 32, 544–552. doi: 10.1161/01.STR.32.2.544
Paolicelli, R. C., Bergamini, G., and Rajendran, L. (2018). Cell-to-cell communication by extracellular vesicles: focus on microglia. Neuroscience [Epub ahead of print]. doi: 10.1016/j.neuroscience.2018.04.003
Paolicelli, R. C., Bisht, K., and Tremblay, M. E. (2014). Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell Neurosci. 8:129. doi: 10.3389/fncel.2014.00129
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Paolicelli, R. C., and Gross, C. T. (2011). Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol. 7, 77–83. doi: 10.1017/S1740925X12000105
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A., and Bessis, A. (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 109, E197–E205. doi: 10.1073/pnas.1111098109
Pena-Altamira, L. E., Polazzi, E., Giuliani, P., Beraudi, A., Massenzio, F., Mengoni, I., et al. (2018). Release of soluble and vesicular purine nucleoside phosphorylase from rat astrocytes and microglia induced by pro-inflammatory stimulation with extracellular ATP via P2X7 receptors. Neurochem. Int. 115, 37–49. doi: 10.1016/j.neuint.2017.10.010
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431. doi: 10.1016/j.tins.2009.05.001
Perea, J. R., Llorens-Martin, M., Avila, J., and Bolos, M. (2018). The role of microglia in the spread of tau: relevance for tauopathies. Front. Cell Neurosci. 12:172. doi: 10.3389/fncel.2018.00172
Perner, C., Perner, F., Stubendorff, B., Forster, M., Witte, O. W., Heidel, F. H., et al. (2018). Dysregulation of chemokine receptor expression and function in leukocytes from ALS patients. J. Neuroinflamm. 15:99. doi: 10.1186/s12974-018-1135-3
Perry, V. H., and O’Connor, V. (2010). The role of microglia in synaptic stripping and synaptic degeneration: a revised perspective. ASN Neuro 2:e00047. doi: 10.1042/AN20100024
Perugi, G., Quaranta, G., Belletti, S., Casalini, F., Mosti, N., Toni, C., et al. (2015). General medical conditions in 347 bipolar disorder patients: clinical correlates of metabolic and autoimmune-allergic diseases. J. Affect. Disord. 170, 95–103. doi: 10.1016/j.jad.2014.08.052
Pinto, J. V., Passos, I. C., Librenza-Garcia, D., Marcon, G., Schneider, M. A., Conte, J. H., et al. (2018). Neuron-glia interaction as a possible pathophysiological mechanism of bipolar disorder. Curr. Neuropharmacol. 16, 519–532.
Platten, M., Kretz, A., Naumann, U., Aulwurm, S., Egashira, K., Isenmann, S., et al. (2003). Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 54, 388–392. doi: 10.1002/ana.10679
Posfai, B., Cserep, C., Orsolits, B., and Denes, A. (2018). New insights into microglia-neuron interactions: a neuron’s perspective. Neuroscience [Epub ahead of print]. doi: 10.1016/j.neuroscience.2018.04.046
Prada, I., Furlan, R., Matteoli, M., and Verderio, C. (2013). Classical and unconventional pathways of vesicular release in microglia. Glia 61, 1003–1017. doi: 10.1002/glia.22497
Prada, I., Gabrielli, M., Turola, E., Iorio, A., D’Arrigo, G., Parolisi, R., et al. (2018). Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 135, 529–550. doi: 10.1007/s00401-017-1803-x
Pribiag, H., and Stellwagen, D. (2014). Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology 78, 13–22. doi: 10.1016/j.neuropharm.2013.06.008
Prins, M., Greco, T., Alexander, D., and Giza, C. C. (2013). The pathophysiology of traumatic brain injury at a glance. Dis. Model Mech. 6, 1307–1315. doi: 10.1242/dmm.011585
Pyonteck, S. M., Akkari, L., Schuhmacher, A. J., Bowman, R. L., Sevenich, L., Quail, D. F., et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272. doi: 10.1038/nm.3337
Ragozzino, D., Di Angelantonio, S., Trettel, F., Bertollini, C., Maggi, L., Gross, C., et al. (2006). Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J. Neurosci. 26, 10488–10498. doi: 10.1523/JNEUROSCI.3192-06.2006
Rajendran, L., Bali, J., Barr, M. M., Court, F. A., Kramer-Albers, E. M., Picou, F., et al. (2014). Emerging roles of extracellular vesicles in the nervous system. J. Neurosci. 34, 15482–15489. doi: 10.1523/JNEUROSCI.3258-14.2014
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Rajkowska, G., O’Dwyer, G., Teleki, Z., Stockmeier, C. A., and Miguel-Hidalgo, J. J. (2007). GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology 32, 471–482. doi: 10.1038/sj.npp.1301234
Ramirez, A. I., de Hoz, R., Salobrar-Garcia, E., Salazar, J. J., Rojas, B., Ajoy, D., et al. (2017). The role of microglia in retinal neurodegeneration: alzheimer’s disease, parkinson, and glaucoma. Front. Aging Neurosci. 9:214. doi: 10.3389/fnagi.2017.00214
Rancan, M., Otto, V. I., Hans, V. H., Gerlach, I., Jork, R., Trentz, O., et al. (2001). Upregulation of ICAM-1 and MCP-1 but not of MIP-2 and sensorimotor deficit in response to traumatic axonal injury in rats. J. Neurosci. Res. 63, 438–446. doi: 10.1002/1097-4547(20010301)63:5<438::AID-JNR1039>3.0.CO;2-P
Ransohoff, R. M., and Cardona, A. E. (2010). The myeloid cells of the central nervous system parenchyma. Nature 468, 253–262. doi: 10.1038/nature09615
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Rao, J. S., Harry, G. J., Rapoport, S. I., and Kim, H. W. (2010). Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol. Psychiatry 15, 384–392. doi: 10.1038/mp.2009.47
Rathnasamy, G., Foulds, W. S., Ling, E. A., and Kaur, C. (2018). Retinal microglia – A key player in healthy and diseased retina. Prog. Neurobiol. [Epub ahead of print]. doi: 10.1016/j.pneurobio.2018.05.006
Reaux-Le Goazigo, A., Van Steenwinckel, J., Rostene, W., and Melik Parsadaniantz, S. (2013). Current status of chemokines in the adult CNS. Prog. Neurobiol. 104, 67–92. doi: 10.1016/j.pneurobio.2013.02.001
Réus, G. Z., Fries, G. R., Stertz, L., Badawy, M., Passos, I. C., Barichello, T., et al. (2015). The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 300, 141–154. doi: 10.1016/j.neuroscience.2015.05.018
Ribeiro, B. M. M., do Carmo, M. R. S., Freire, R. S., Rocha, N. F. M., Borella, V. C. M., de Menezes, A. T., et al. (2013). Evidences for a progressive microglial activation and increase in iNOS expression in rats submitted to a neurodevelopmental model of schizophrenia: reversal by clozapine. Schizophr. Res. 151, 12–19. doi: 10.1016/j.schres.2013.10.040
Rogers, J. T., Morganti, J. M., Bachstetter, A. D., Hudson, C. E., Peters, M. M., Grimmig, B. A., et al. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011
Rossi, M. L., Hughes, J. T., Esiri, M. M., Coakham, H. B., and Brownell, D. B. (1987). Immunohistological study of mononuclear cell infiltrate in malignant gliomas. Acta Neuropathol. 74, 269–277. doi: 10.1007/BF00688191
Roumier, A., Bechade, C., Poncer, J. C., Smalla, K. H., Tomasello, E., Vivier, E., et al. (2004). Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J. Neurosci. 24, 11421–11428. doi: 10.1523/JNEUROSCI.2251-04.2004
Rufino-Ramos, D., Albuquerque, P. R., Carmona, V., Perfeito, R., Nobre, R. J., Pereira, et al. (2017). Extracellular vesicles: novel promising delivery systems for therapy of brain diseases. J. Control Release 262, 247–258. doi: 10.1016/j.jconrel.2017.07.001
Saber, M., Kokiko-Cochran, O., Puntambekar, S. S., Lathia, J. D., and Lamb, B. T. (2017). Triggering receptor expressed on myeloid cells 2 deficiency alters acute macrophage distribution and improves recovery after traumatic brain injury. J. Neurotr. 34, 423–435. doi: 10.1089/neu.2016.4401
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Santello, M., Bezzi, P., and Volterra, A. (2011). TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69, 988–1001. doi: 10.1016/j.neuron.2011.02.003
Santos, A. R., Comprido, D., and Duarte, C. B. (2010). Regulation of local translation at the synapse by BDNF. Prog. Neurobiol. 92, 505–516. doi: 10.1016/j.pneurobio.2010.08.004
Sarkar, C., Basu, B., Chakroborty, D., Dasgupta, P. S., and Basu, S. (2010). The immunoregulatory role of dopamine: an update. Brain Behav. Immun. 24, 525–528. doi: 10.1016/j.bbi.2009.10.015
Saud, K., Herrera-Molina, R., and Von Bernhardi, R. (2005). Pro- and anti-inflammatory cytokines regulate the ERK pathway: implication of the timing for the activation of microglial cells. Neurotox Res. 8, 277–287. doi: 10.1007/BF03033981
Savaskan, N. E., Fan, Z., Broggini, T., Buchfelder, M., and Eyupoglu, I. Y. (2015). Neurodegeneration and the brain tumor microenvironment. [corrected]. Curr. Neuropharmacol. 13, 258–265. doi: 10.2174/1570159X13666150122224158
Savaskan, N. E., Heckel, A., Hahnen, E., Engelhorn, T., Doerfler, A., Ganslandt, O., et al. (2008). Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 14, 629–632. doi: 10.1038/nm1772
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schall, T. (1997). Fractalkine–a strange attractor in the chemokine landscape. Immunol. Today 18:147. doi: 10.1016/S0167-5699(97)84655-5
Schiffer, D., Mellai, M., Bovio, E., and Annovazzi, L. (2017). The neuropathological basis to the functional role of microglia/macrophages in gliomas. Neurol. Sci. 38, 1571–1577. doi: 10.1007/s10072-017-3002-x
Schmierer, B., and Hill, C. S. (2007). TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982. doi: 10.1038/nrm2297
Schneider, A., and Simons, M. (2013). Exosomes: vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 352, 33–47. doi: 10.1007/s00441-012-1428-2
Schreiber, R. C., Krivacic, K., Kirby, B., Vaccariello, S. A., Wei, T., Ransohoff, R. M., et al. (2001). Monocyte chemoattractant protein (MCP)-1 is rapidly expressed by sympathetic ganglion neurons following axonal injury. Neuroreport 12, 601–606. doi: 10.1097/00001756-200103050-00034
Schulz, C., Gomez Perdiguero, E., Chorro, L., Szabo-Rogers, H., Cagnard, N., Kierdorf, K., et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. doi: 10.1126/science.1219179
Schwartz, M., Butovsky, O., Bruck, W., and Hanisch, U. K. (2006). Microglial phenotype: is the commitment reversible? Trends Neurosci. 29, 68–74.
Setiawan, E., Wilson, A. A., Mizrahi, R., Rusjan, P. M., Miler, L., Rajkowska, G., et al. (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72, 268–275. doi: 10.1001/jamapsychiatry.2014.2427
Sewell, D. L., Nacewicz, B., Liu, F., Macvilay, S., Erdei, A., Lambris, J. D., et al. (2004). Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J. Neuroimmunol. 155, 55–63. doi: 10.1016/j.jneuroim.2004.06.003
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Siegel, J., Pizzi, M. A., Brent Peel, J., Alejos, D., Mbabuike, N., Brown, B. L., et al. (2017). Update on neurocritical care of stroke. Curr. Cardiol. Rep. 19:67. doi: 10.1007/s11886-017-0881-7
Sielska, M., Przanowski, P., Wylot, B., Gabrusiewicz, K., Maleszewska, M., Kijewska, M., et al. (2013). Distinct roles of CSF family cytokines in macrophage infiltration and activation in glioma progression and injury response. J. Pathol. 230, 310–321. doi: 10.1002/path.4192
Simmons, G. W., Pong, W. W., Emnett, R. J., White, C. R., Gianino, S. M., Rodriguez, F. J., et al. (2011). Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J. Neuropathol. Exp. Neurol. 70, 51–62. doi: 10.1097/NEN.0b013e3182032d37
Song, S., Kong, X., Acosta, S., Sava, V., Borlongan, C., and Sanchez-Ramos, J. (2016). Granulocyte-colony stimulating factor promotes brain repair following traumatic brain injury by recruitment of microglia and increasing neurotrophic factor expression. Restor. Neurol. Neurosci. 34, 415–431. doi: 10.3233/RNN-150607
Soriano, S. G., Amaravadi, L. S., Wang, Y. F., Zhou, H., Yu, G. X., Tonra, J. R., et al. (2002). Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 125, 59–65. doi: 10.1016/S0165-5728(02)00033-4
Sousa, N., Cerqueira, J. J., and Almeida, O. F. (2008). Corticosteroid receptors and neuroplasticity. Brain Res. Rev. 57, 561–570. doi: 10.1016/j.brainresrev.2007.06.007
Spangenberg, E. E., Lee, R. J., Najafi, A. R., Rice, R. A., Elmore, M. R., Blurton-Jones, M., et al. (2016). Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 139, 1265–1281. doi: 10.1093/brain/aww016
Stadelmann, C., Kerschensteiner, M., Misgeld, T., Bruck, W., Hohlfeld, R., and Lassmann, H. (2002). BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain 125, 75–85.
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Stellwagen, D., and Lewitus, G. M. (2014). The complexity of homeostasis at the synapse. Neuropharmacology 78, 1–2. doi: 10.1016/j.neuropharm.2013.10.019
Stence, N., Waite, M., and Dailey, M. E. (2001). Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33, 256–266. doi: 10.1002/1098-1136(200103)33:3<256::AID-GLIA1024>3.0.CO;2-J
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219. doi: 10.1111/imm.12922
Stertz, L., Fries, G. R., Rosa, A. R., Kauer-Sant’anna, M., Ferrari, P., Paz, A. V., et al. (2015). Damage-associated molecular patterns and immune activation in bipolar disorder. Acta Psychiatr. Scand. 132, 211–217. doi: 10.1111/acps.12417
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Stokes, L., Spencer, S. J., and Jenkins, T. A. (2015). Understanding the role of P2X7 in affective disorders-are glial cells the major players? Front. Cell Neurosci. 9:258. doi: 10.3389/fncel.2015.00258
Streit, W. J. (2000). Microglial response to brain injury: a brief synopsis. Toxicol. Pathol. 28, 28–30. doi: 10.1177/019262330002800104
Suzumura, A., Sawada, M., Yamamoto, H., and Marunouchi, T. (1993). Transforming growth factor-beta suppresses activation and proliferation of microglia in vitro. J. Immunol. 151, 2150–2158.
Szalay, G., Martinecz, B., Lenart, N., Kornyei, Z., Orsolits, B., Judak, L., et al. (2016). Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 7:11499. doi: 10.1038/ncomms11499
Takenouchi, T., Tsukimoto, M., Iwamaru, Y., Sugama, S., Sekiyama, K., Sato, M., et al. (2015). Extracellular ATP induces unconventional release of glyceraldehyde-3-phosphate dehydrogenase from microglial cells. Immunol. Lett. 167, 116–124. doi: 10.1016/j.imlet.2015.08.002
Tamboli, I. Y., Barth, E., Christian, L., Siepmann, M., Kumar, S., Singh, S., et al. (2010). Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 285, 37405–37414. doi: 10.1074/jbc.M110.149468
Tan, J., Town, T., and Mullan, M. (2000). CD45 inhibits CD40L-induced microglial activation via negative regulation of the Src/p44/42 MAPK pathway. J. Biol. Chem. 275, 37224–37231. doi: 10.1074/jbc.M002006200
Tarozzo, G., Bortolazzi, S., Crochemore, C., Chen, S. C., Lira, A. S., Abrams, J. S., et al. (2003). Fractalkine protein localization and gene expression in mouse brain. J. Neurosci. Res. 73, 81–88. doi: 10.1002/jnr.10645
Taylor, D. L., Jones, F., Kubota, E. S., and Pocock, J. M. (2005). Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J. Neurosci. 25, 2952–2964. doi: 10.1523/JNEUROSCI.4456-04.2005
Thome, A. D., Standaert, D. G., and Harms, A. S. (2015). Fractalkine signaling regulates the inflammatory response in an alpha-synuclein model of parkinson disease. PLoS One 10:e0140566. doi: 10.1371/journal.pone.0140566
Tian, L., Ma, L., Kaarela, T., and Li, Z. (2012). Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflamm. 9:155. doi: 10.1186/1742-2094-9-155
Tofaris, G. K. (2017). A critical assessment of exosomes in the pathogenesis and stratification of Parkinson’s Disease. J. Parkinsons Dis. 7, 569–576. doi: 10.3233/JPD-171176
Torres-Platas, S. G., Cruceanu, C., Chen, G. G., Turecki, G., and Mechawar, N. (2014). Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav. Immun. 42, 50–59. doi: 10.1016/j.bbi.2014.05.007
Town, T., Nikolic, V., and Tan, J. (2005). The microglial “activation” continuum: from innate to adaptive responses. J. Neuroinflamm. 2:24.
Trang, T., Beggs, S., and Salter, M. W. (2011). Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. 7, 99–108. doi: 10.1017/S1740925X12000087
Trang, T., Beggs, S., Wan, X., and Salter, M. W. (2009). P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J. Neurosci. 29, 3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009
Tremblay, M. E., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Tremblay, M. E., and Majewska, A. K. (2011). A role for microglia in synaptic plasticity? Commun. Integr. Biol. 4, 220–222. doi: 10.4161/cib.4.2.14506
Trotta, T., Panaro, M. A., Cianciulli, A., Mori, G., Di Benedetto, A., and Porro, C. (2018). Microglia-derived extracellular vesicles in Alzheimer’s Disease: a double-edged sword. Biochem. Pharmacol. 148, 184–192. doi: 10.1016/j.bcp.2017.12.020
Truman, L. A., Ford, C. A., Pasikowska, M., Pound, J. D., Wilkinson, S. J., Dumitriu, I. E., et al. (2008). CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036. doi: 10.1182/blood-2008-06-162404
Turrin, N. P., and Rivest, S. (2006). Tumor necrosis factor alpha but not interleukin 1 beta mediates neuroprotection in response to acute nitric oxide excitotoxicity. J. Neurosci. 26, 143–151. doi: 10.1523/JNEUROSCI.4032-05.2006
Tynan, R. J., Naicker, S., Hinwood, M., Nalivaiko, E., Buller, K. M., Pow, D. V., et al. (2010). Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav. Immun. 24, 1058–1068. doi: 10.1016/j.bbi.2010.02.001
Ueno, M., Fujita, Y., Tanaka, T., Nakamura, Y., Kikuta, J., Ishii, M., et al. (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16, 543–551. doi: 10.1038/nn.3358
Ulmann, L., Levavasseur, F., Avignone, E., Peyroutou, R., Hirbec, H., Audinat, E., et al. (2013). Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia 61, 1306–1319. doi: 10.1002/glia.22516
Vakilian, A., Khorramdelazad, H., Heidari, P., Sheikh Rezaei, Z., and Hassanshahi, G. (2017). CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int. 103, 1–7. doi: 10.1016/j.neuint.2016.12.013
Valdinocci, D., Radford, R. A., Siow, S. M., Chung, R. S., and Pountney, D. L. (2017). Potential modes of intercellular alpha-synuclein transmission. Int. J. Mol. Sci. 18:E469. doi: 10.3390/ijms18020469
van Beek, E. M., Cochrane, F., Barclay, A. N., and van den Berg, T. K. (2005). Signal regulatory proteins in the immune system. J. Immunol. 175, 7781–7787. doi: 10.4049/jimmunol.175.12.7781
van Berckel, B. N., Bossong, M. G., Boellaard, R., Kloet, R., Schuitemaker, A., Caspers, E., et al. (2008). Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol. Psychiatry 64, 820–822. doi: 10.1016/j.biopsych.2008.04.025
van Weering, H. R., de Jong, A. P., de Haas, A. H., Biber, K. P., and Boddeke, H. W. (2010). CCL21-induced calcium transients and proliferation in primary mouse astrocytes: CXCR3-dependent and independent responses. Brain Behav. Immun. 24, 768–775. doi: 10.1016/j.bbi.2009.04.007
Varnum, M. M., Kiyota, T., Ingraham, K. L., Ikezu, S., and Ikezu, T. (2015). The anti-inflammatory glycoprotein, CD200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer’s disease. Neurobiol. Aging 36, 2995–3007. doi: 10.1016/j.neurobiolaging.2015.07.027
Vass, K., and Lassmann, H. (1990). Intrathecal application of interferon gamma. Progressive appearance of MHC antigens within the rat nervous system. Am. J. Pathol. 137, 789–800.
Vereker, E., O’Donnell, E., and Lynch, M. A. (2000). The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J. Neurosci. 20, 6811–6819. doi: 10.1523/JNEUROSCI.20-18-06811.2000
Verney, C., Monier, A., Fallet-Bianco, C., and Gressens, P. (2010). Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J. Anat. 217, 436–448. doi: 10.1111/j.1469-7580.2010.01245.x
Vilette, D., Courte, J., Peyrin, J. M., Coudert, L., Schaeffer, L., Andreoletti, O., et al. (2018). Cellular mechanisms responsible for cell-to-cell spreading of prions. Cell. Mol. Life Sci. [Epub ahead of print]. doi: 10.1007/s00018-018-2823-y
von Bernhardi, R., Heredia, F., Salgado, N., and Munoz, P. (2016). Microglia function in the normal brain. Adv. Exp. Med. Biol. 949, 67–92. doi: 10.1007/978-3-319-40764-7_4
Wakselman, S., Bechade, C., Roumier, A., Bernard, D., Triller, A., and Bessis, A. (2008). Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008
Walker, F. R., Nilsson, M., and Jones, K. (2013). Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr. Drug Targets 14, 1262–1276. doi: 10.2174/13894501113149990208
Wang, J. W., Chen, S. D., Zhang, X. L., and Jonas, J. B. (2016). Retinal microglia in glaucoma. J. Glaucoma 25, 459–465. doi: 10.1097/IJG.0000000000000200
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/jem.20151948
Wang, S. C., Hong, J. H., Hsueh, C., and Chiang, C. S. (2012). Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Invest. 92, 151–162. doi: 10.1038/labinvest.2011.128
Wang, T., Nowrangi, D., Yu, L., Lu, T., Tang, J., Han, B., et al. (2018). Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J. Neuroinflamm. 15:2. doi: 10.1186/s12974-017-1039-7
Wang, Y., Balaji, V., Kaniyappan, S., Kruger, L., Irsen, S., Tepper, K., et al. (2017). The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12:5. doi: 10.1186/s13024-016-0143-y
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wesolowska, A., Kwiatkowska, A., Slomnicki, L., Dembinski, M., Master, A., Sliwa, M., et al. (2008). Microglia-derived TGF-beta as an important regulator of glioblastoma invasion–an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 27, 918–930. doi: 10.1038/sj.onc.1210683
Wieghofer, P., and Prinz, M. (2016). Genetic manipulation of microglia during brain development and disease. Biochim. Biophys. Acta 1862, 299–309. doi: 10.1016/j.bbadis.2015.09.019
Wohleb, E. S. (2016). Neuron-microglia interactions in mental health disorders: “for better, and for worse”. Front. Immunol. 7:544.
Wolf, Y., Yona, S., Kim, K. W., and Jung, S. (2013). Microglia, seen from the CX3CR1 angle. Front. Cell Neurosci. 7:26. doi: 10.3389/fncel.2013.00026
Wright, G. J., Jones, M., Puklavec, M. J., Brown, M. H., and Barclay, A. N. (2001). The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology 102, 173–179. doi: 10.1046/j.1365-2567.2001.01163.x
Wu, A., Wei, J., Kong, L. Y., Wang, Y., Priebe, W., Qiao, W., et al. (2010). Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 12, 1113–1125. doi: 10.1093/neuonc/noq082
Wu, J., Bie, B., Yang, H., Xu, J. J., Brown, D. L., and Naguib, M. (2013). Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiol. Aging 34, 2843–2852. doi: 10.1016/j.neurobiolaging.2013.06.003
Xie, X., Luo, X., Liu, N., Li, X., Lou, F., Zheng, Y., et al. (2017). Monocytes, microglia, and CD200-CD200R1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J. Neurochem. 141, 222–235. doi: 10.1111/jnc.13972
Yamamuro, K., Kimoto, S., Rosen, K. M., Kishimoto, T., and Makinodan, M. (2015). Potential primary roles of glial cells in the mechanisms of psychiatric disorders. Front. Cell Neurosci. 9:154. doi: 10.3389/fncel.2015.00154
Yang, I., Han, S. J., Kaur, G., Crane, C., and Parsa, A. T. (2010). The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 17, 6–10. doi: 10.1016/j.jocn.2009.05.006
Yang, Y., Boza-Serrano, A., Dunning, C. J. R., Clausen, B. H., Lambertsen, K. L., and Deierborg, T. (2018a). Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflamm. 15:168. doi: 10.1186/s12974-018-1204-7
Yang, Y., Zhang, X. J., Zhang, C., Chen, R., Li, L., He, J., et al. (2018b). Loss of neuronal CD200 contributed to microglial activation after acute cerebral ischemia in mice. Neurosci. Lett. 678, 48–54. doi: 10.1016/j.neulet.2018.05.004
Ye, Y., Wang, G., Wang, H., and Wang, X. (2011). Brain-derived neurotrophic factor (BDNF) infusion restored astrocytic plasticity in the hippocampus of a rat model of depression. Neurosci. Lett. 503, 15–19. doi: 10.1016/j.neulet.2011.07.055
Ye, Z. C., and Sontheimer, H. (1999). Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 59, 4383–4391.
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yi, M. H., Zhang, E., Kang, J. W., Shin, Y. N., Byun, J. Y., Oh, S. H., et al. (2012). Expression of CD200 in alternative activation of microglia following an excitotoxic lesion in the mouse hippocampus. Brain Res. 1481, 90–96. doi: 10.1016/j.brainres.2012.08.053
Yirmiya, R., Rimmerman, N., and Reshef, R. (2015). Depression as a microglial disease. Trends Neurosci. 38, 637–658. doi: 10.1016/j.tins.2015.08.001
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/j.neuron.2016.05.003
Yuyama, K., and Igarashi, Y. (2016). Physiological and pathological roles of exosomes in the nervous system. Biomol. Concepts 7, 53–68. doi: 10.1515/bmc-2015-0033
Yuyama, K., and Igarashi, Y. (2017). Exosomes as carriers of alzheimer’s amyloid-ss. Front. Neurosci. 11:229. doi: 10.3389/fnins.2017.00229
Yuyama, K., Sun, H., Usuki, S., Sakai, S., Hanamatsu, H., Mioka, T., et al. (2015). A potential function for neuronal exosomes: sequestering intracerebral amyloid-beta peptide. FEBS Lett. 589, 84–88. doi: 10.1016/j.febslet.2014.11.027
Zaborowski, M. P., Balaj, L., Breakefield, X. O., and Lai, C. P. (2015). Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience 65, 783–797. doi: 10.1093/biosci/biv084
Zalesky, A., Fornito, A., Seal, M. L., Cocchi, L., Westin, C. F., Bullmore, E. T., et al. (2011). Disrupted axonal fiber connectivity in schizophrenia. Biol. Psychiatry 69, 80–89. doi: 10.1016/j.biopsych.2010.08.022
Zanier, E. R., Marchesi, F., Ortolano, F., Perego, C., Arabian, M., Zoerle, T., et al. (2016). Fractalkine receptor deficiency is associated with early protection but late worsening of outcome following brain trauma in mice. J. Neurotr. 33, 1060–1072. doi: 10.1089/neu.2015.4041
Zeilhofer, H. U. (2008). Loss of glycinergic and GABAergic inhibition in chronic pain–contributions of inflammation and microglia. Int. Immunopharmacol. 8, 182–187. doi: 10.1016/j.intimp.2007.07.009
Zhai, H., Heppner, F. L., and Tsirka, S. E. (2011). Microglia/macrophages promote glioma progression. Glia 59, 472–485. doi: 10.1002/glia.21117
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., et al. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406. doi: 10.1038/nn.3641
Zhang, L., Xu, J., Gao, J., Wu, Y., Yin, M., and Zhao, W. (2018). CD200-, CX3CL1-, and TREM2-mediated neuron-microglia interactions and their involvements in Alzheimer’s disease. Rev. Neurosci. [Epub ahead of print].
Zhang, S., Eitan, E., Wu, T. Y., and Mattson, M. P. (2018). Intercellular transfer of pathogenic alpha-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol. Aging 61, 52–65. doi: 10.1016/j.neurobiolaging.2017.09.016
Zhang, X., Abels, E. R., Redzic, J. S., Margulis, J., Finkbeiner, S., and Breakefield, X. O. (2016). Potential transfer of polyglutamine and CAG-Repeat RNA in extracellular vesicles in huntington’s disease: background and evaluation in cell culture. Cell Mol. Neurobiol. 36, 459–470. doi: 10.1007/s10571-016-0350-7
Zhang, X. Y., Zhou, D. F., Wu, G. Y., Cao, L. Y., Tan, Y. L., Haile, C. N., et al. (2008). BDNF levels and genotype are associated with antipsychotic-induced weight gain in patients with chronic schizophrenia. Neuropsychopharmacology 33, 2200–2205. doi: 10.1038/sj.npp.1301619
Zhang, Y., Chen, Y., Wu, J., Manaenko, A., Yang, P., Tang, J., et al. (2015). Activation of dopamine D2 receptor suppresses neuroinflammation through alphab-crystalline by inhibition of nf-kappab nuclear translocation in experimental ICH mice model. Stroke 46, 2637–2646. doi: 10.1161/STROKEAHA.115.009792
Keywords: fractalkine, extracellular vesicles, neuroinflammation, neurodegeneration, microglia, neuron, cytokines
Citation: Szepesi Z, Manouchehrian O, Bachiller S and Deierborg T (2018) Bidirectional Microglia–Neuron Communication in Health and Disease. Front. Cell. Neurosci. 12:323. doi: 10.3389/fncel.2018.00323
Received: 31 May 2018; Accepted: 06 September 2018;
Published: 27 September 2018.
Edited by:
Alessandro Tozzi, University of Perugia, ItalyReviewed by:
Alfonso Grimaldi, Istituto Italiano di Tecnologia (IIT), ItalyCopyright © 2018 Szepesi, Manouchehrian, Bachiller and Deierborg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomas Deierborg, dG9tYXMuZGVpZXJib3JnQG1lZC5sdS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.