 Marwa Elamin
Marwa Elamin David N. Ruskin
David N. Ruskin Susan A. Masino
Susan A. Masino Paola Sacchetti
Paola Sacchetti
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell. Neurosci. , 30 August 2018
Sec. Cellular Neurophysiology
Volume 12 - 2018 | https://doi.org/10.3389/fncel.2018.00263
This article is part of the Research Topic Cellular Mechanisms of Seizure Control through Cell Metabolism View all 9 articles
The ketogenic diet’s (KD) anti-seizure effects have long been documented. Recently, its therapeutic potential in multiple neurodegenerative and neurodevelopmental disorders has emerged. Yet experimental evidence for a fundamental mechanism underlying beneficial effects across numerous diseases remains lacking. We previously showed that feeding rats a KD produced an early (within 2 days) and persistent elevation of hippocampal nicotinamide adenine dinucleotide+ (NAD+), an essential metabolic coenzyme and signaling molecule. NAD+ is a marker of cellular health and a substrate for enzymes implicated in longevity and DNA damage repair such as sirtuins and poly-ADP ribose polymerase-1 (PARP-1). As a result, activation of NAD+-dependent enzymes’ downstream pathways could be the origin of KD’s broad beneficial effects. Here rats were fed ad libitum regular chow or KD for 2 days or 3 weeks and the levels of hippocampal sirtuins, PARP-1, and the oxidative DNA damage marker 8-hydroxy-2’-deoxyguanosine were quantified. We found a significant immediate and persistent increase in the collective activity of nuclear sirtuin enzymes, and a significant augmentation of Sirt1 mRNA at 2 days. Levels of PARP-1 and 8-hydroxy-2’-deoxyguanosine decreased after 2 days of treatment and further declined at 3 weeks. Our data show that a KD can rapidly modulate energy metabolism by acting on NAD+-dependent enzymes and their downstream pathways. Thus, therapy with a KD can potentially enhance brain health and increase overall healthspan via NAD+-related mechanisms that render cells more resilient against DNA damage and a host of metabolic, epileptic, neurodegenerative, or neurodevelopmental insults.
The ketogenic diet (KD) is a high-fat, low-carbohydrate, moderate protein therapy that shifts energy production away from glucose-based and toward ketone-based ATP production. It is effective in treating pharmacoresistant epilepsy (Neal et al., 2009; Sharma et al., 2013; Cervenka et al., 2017) and growing evidence supports its beneficial effects in diverse disorders (Yang and Cheng, 2010; Winter et al., 2017; Augustin et al., 2018) and in healthspan and lifespan (Newman et al., 2017; Roberts et al., 2017). As the fundamental mechanism(s) underlying its success remain unclear, molecular changes induced by the diet must be characterized in healthy cells.
Recently we proposed that the broad effectiveness of the KD is related to increased levels of the coenzyme nicotinamide adenine dinucleotide (NAD) (Elamin et al., 2017), a pivotal molecule for redox reactions and the backbone of ATP generation. Because fewer NAD molecules are reduced during ketone-based vs. glucose-based metabolism in brain, elevated levels of the oxidized form (NAD+) can be expected. Consistent with this prediction, we demonstrated that KD can induce rapid and sustained changes in the NAD+/NADH ratio in rat hippocampus - an energetically demanding brain region considered a seizure gate (Heinemann et al., 1992) - but not in the cerebral cortex (Elamin et al., 2017). Similar results have been found in aged mice with a ketone ester supplemented diet (Pawlosky et al., 2017).
Elevated NAD+ limits seizures and mediates lifespan extension (Lin and Guarente, 2003; Mills et al., 2016; Liu et al., 2017). Experimentally increased NAD+ levels enhance mitochondrial function, protect against oxidative stress damage and decrease cell death (Kussmaul and Hirst, 2006), diverse effects that could be linked to downstream pathways. NAD+ serves as a substrate for two enzyme groups, sirtuins and poly(ADP-ribose) polymerases (PARPs), that affect diverse cellular functions ranging from gene expression to DNA repair (Belenky et al., 2007).
The NAD+-dependent sirtuin enzymes are important regulators of metabolism, inflammation, and DNA repair (Michishita, 2005). Sirt1 is involved in the deacetylation of transcription factors, growth factors, anti-apoptotic, and anti-inflammatory proteins (Yang et al., 2006). Sirt1 is essential for normal cognition, preservation of memory and neuronal plasticity (Michan et al., 2010). Interestingly, Sirt1 has seizure-suppressing effects in animal models of epilepsy (Wang et al., 2016) and mediates benefits of caloric restriction, such as lifespan extension and promotion of cell survival (Lin et al., 2000; Cohen et al., 2004; Satoh et al., 2013). Sirt6 and Sirt7 participate directly in base-excision repair of DNA, thus decreasing age-associated DNA damage (Mostoslavsky et al., 2006; Vazquez et al., 2016).
PARP enzymes add polymers of ADP-ribose to nuclear proteins in an NAD+-dependent matter and are major consumers of NAD+. PARP-1 plays an important role in cell survival and DNA damage repair (Grube and Burkle, 1992; de Murcia and de Murcia, 1994). DNA oxidation by reactive oxygen species induces a steady-state level of DNA damage and is a by-product of normal cellular metabolism. PARP-1 is a molecular sensor for this type of DNA damage and both its activity and protein levels are affected directly by cellular levels of oxidative DNA damage (Dantzer et al., 2006; Bürkle and Virág, 2013; Shen et al., 2016). Although PARP enzymes play a vital role in DNA repair, their over-activation (and subsequent depletion of NAD+) has been linked to diverse neuropathological conditions (Morales et al., 2014; Martire et al., 2015).
Since the KD rapidly increases NAD+ levels (Elamin et al., 2017) and changes in NAD+ levels modulate the activity of the abovementioned enzymatic pathways, we hypothesized that consumption of a KD would lead to downstream beneficial changes in NAD+-dependent enzymes’ activity in rat hippocampus.
Sprague-Dawley male rats (age 11–14 week; 350–550 g; n = 22) were pair-housed at Trinity College, with water and food ad libitum. Animals received either a high-carbohydrate chow diet (Purina 5001, PharmaServ, Framingham, MA, United States) (CD), or a 6:1 (fat:protein+carbohydrates) KD (F3666, Bio-Serv, Frenchtown, NJ, United States) for 2 days (2d) or 3 weeks (3w); (n = 8 each). At the end of dietary treatment, animals were sacrificed, trunk blood was collected and the hippocampi were dissected. Plasma β-hydroxybutyrate was measured using Precision Xtra monitors (Abbott Laboratories; Abbott Park, Chicago, IL, United States). All experiments were in compliance with National Institutes of Health Guides and Trinity College Animal Care and Use Committee.
Analysis was done according to manufacturer’s instructions (Sigma-Aldrich, United States) as previously described (Elamin et al., 2017). Optical density of NAD+/NADH and NADH were obtained at 450 nm. NAD+ values were calculated by subtracting NADH values from total NAD values and normalized to protein concentrations.
Hippocampal tissues were homogenized in PBS lysis buffer [137 mM NaCl, 2.7 mM KCl, 10 mM Tris pH 7.4, 1 mM PMSF, 1:1000 Protease inhibitor cocktail (PIC)]. Pellets were suspended in Extraction lysis buffer (20 mM Tris pH 7.8, 125 mM NaCl, 5 mM MgCl2, 0.2 mM EDTA, 0.1% NP40, 12% glycerol, 200 mM PMSF, 200 mM DDT, 1:1000 PIC) and sonicated. After centrifugation, combined activity of nuclear sirtuin enzymes (Sirt1,6,7) was measured in hippocampal nuclear extracts following manufacturer’s instruction (Epigentek, United States).
Total hippocampal RNA (1 μg) was extracted (Qiagen, United States), reverse transcribed (Applied Biosystems, United States), and relative mRNA levels were detected by quantitative PCR (StepOnePlus, Thermo Fisher, United States). Predesigned rat Sirt1, Sirt6, Sirt7, and β-actin TaqMan Gene Expression probes (Assay IDs: Rn01428096_m1, Rn01408249_m1, Rn01471420_m1, and Rn00667869_m1, respectively) were used with TaqMan Gene Expression Master Mix (Thermo Fisher, United States). Analyses were performed using the standard curve method with Sirt transcripts normalized to β-actin as the endogenous control.
Hippocampal homogenates made in RIPA buffer were analyzed by western blot for PARP-1 (Cell Signaling) and β-actin (Neomarkers) proteins (1:1000 primary antibodies). Blots were revealed by Chemiluminescence (Thermo Fisher, United States) and quantified using ImageJ software (National Institutes of Health, Bethesda, MD, United States).
Purified DNA was obtained from hippocampal cells (DNeasy Blood and Tissue kit, Qiagen, United States) and levels of 8-OHdG (8-hydroxy-2′-deoxyguanosine) were quantified by competitive ELISA assay (StressMarq, United States) following manufacturer’s protocol.
One-way ANOVA and post hoc Tukey’s multiple comparisons test were performed for all experiments using GraphPad Prism software (GraphPad, United States). Data are expressed as mean ± SEM representing the average of two measurements per sample using the animal number indicated as subject number. ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001, non-significant (ns).
To re-establish previous observations on rapid and sustained KD-induced increases in NAD+ availability (Elamin et al., 2017), we treated a new cohort of rats with control diet or KD. In addition to replicating increased circulating ketones (mM: 0.76 ± 0.15 2d KD, 0.67 ± 0.23 3w KD, 0.05 ± 0.02 CD; p = 0.0189) and NAD+/NADH ratios in response to KD (Figure 1A), we established that the increase in ratio was solely due to an increase in the oxidized form of NAD (Figure 1B).
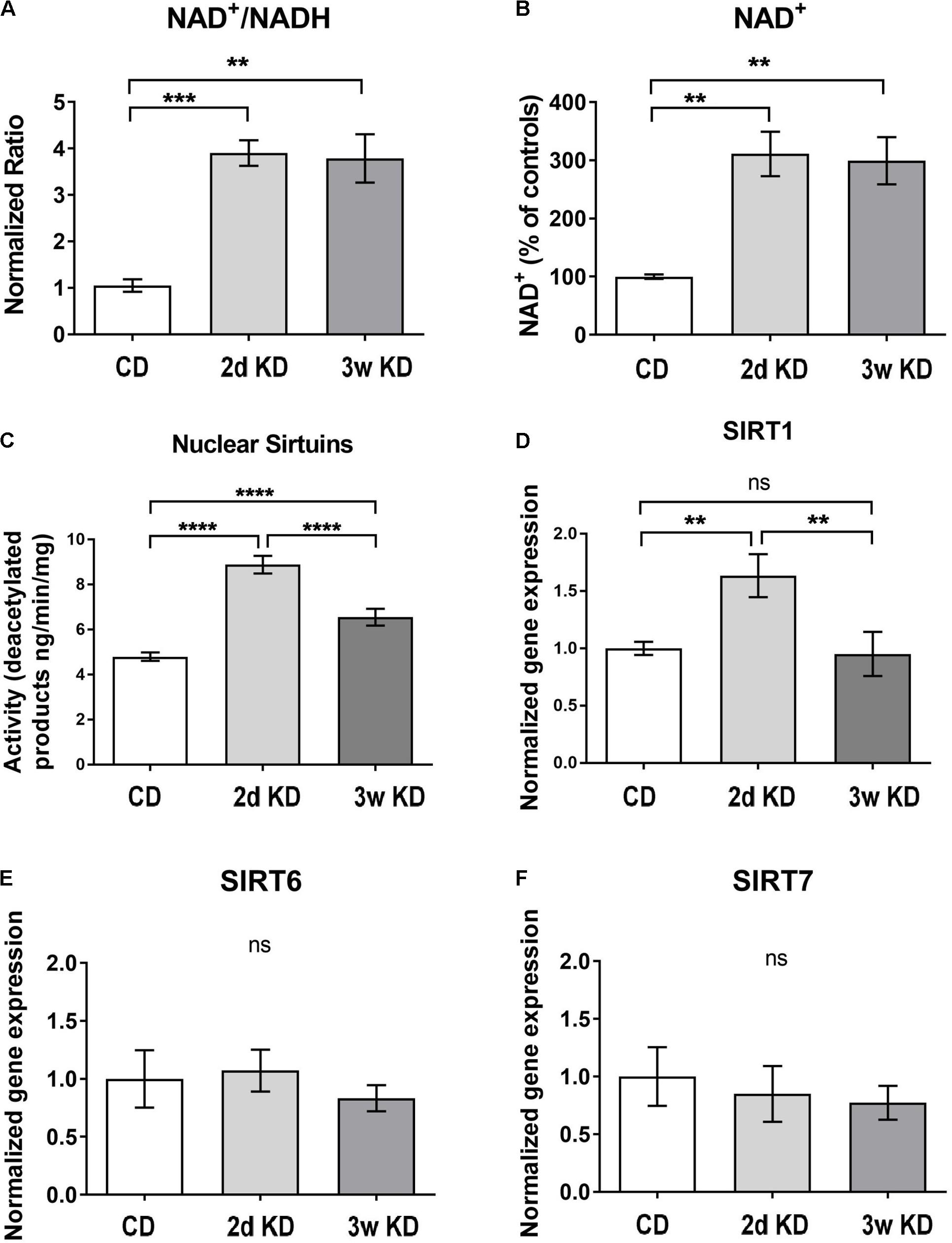
FIGURE 1. Hippocampal changes in NAD and sirtuins after ketogenic diet (KD) treatment. (A,B) NAD+/NADH ratios and NAD+ levels in the hippocampus after standard chow diet (CD; n = 5), 2 day (2d; n = 7), or 3 weeks KD treatment (3w; n = 7). (C) Collective deacetylation activity of Sirt1, Sirt6, and Sirt7 enzymes. A significant increase was observed at 2d and remained elevated at 3w. n = 6–8 animals (n = 2 per animal). (D–F) Real-time PCR analysis of Sirt1,–6,–7 gene expression in hippocampus. Sirt1 expression was increased only at 2d of treatment. CD, n = 6; 2d KD, n = 8; 3w KD, n = 8 (n = 2 per animal).
To address downstream effects sirtuin enzymes’ activity, and expression, which depend directly on the levels of the main substrate NAD+ (Landry et al., 2000; Chen et al., 2008), were examined and the collective deacetylation activity of nuclear sirtuin enzymes (Sirt1, Sirt6, Sirt7) was measured in hippocampal nuclear extracts. As shown in Figure 1C, a rapid, robust increase in sirtuins’ activity was detected at 2d of KD treatment compared to control, and remained elevated at 3w, albeit reduced. Changes in enzymatic activity could be reflective of alterations in gene expression of one or more of these enzymes. Analysis of nuclear sirtuins expression levels showed that Sirt1 mRNA was increased in animals fed a KD for 2d compared to control diet but normalized after 3w of treatment (Figure 1D). No significant mRNA changes were observed for Sirt6 and Sirt7 (Figures 1E,F).
NAD+ also serves as main substrate for PARP-1, an enzyme that functions as a DNA damage sensor and participates in DNA repair. We, therefore, examined potential changes in the hippocampal DNA damage response after KD treatment. A rapid and dramatic decline in PARP-1 protein level was detected after 2d, with further reduction upon longer KD exposure (Figure 2A). Although PARP-1 is a major consumer of NAD+, the activity and protein levels of this enzyme are modulated primarily by levels of oxidative DNA damage (Dantzer et al., 2006; Shen et al., 2016). Therefore DNA damage in hippocampal tissue was assessed further by quantification of 8-OHdG, a critical biomarker of oxidative stress. Dietary treatment with 2d KD decreased the levels of 8-OHdG, and a 3w treatment led to a further decrease in this biomarker (Figure 2B). PARP-1 protein levels strongly correlated with the changes in oxidative DNA damage here detected (Martinet et al., 2002; Huber et al., 2004). Together, observed decreases in PARP-1 and 8-OHdG levels suggest that consuming a KD decreases oxidative DNA damage.
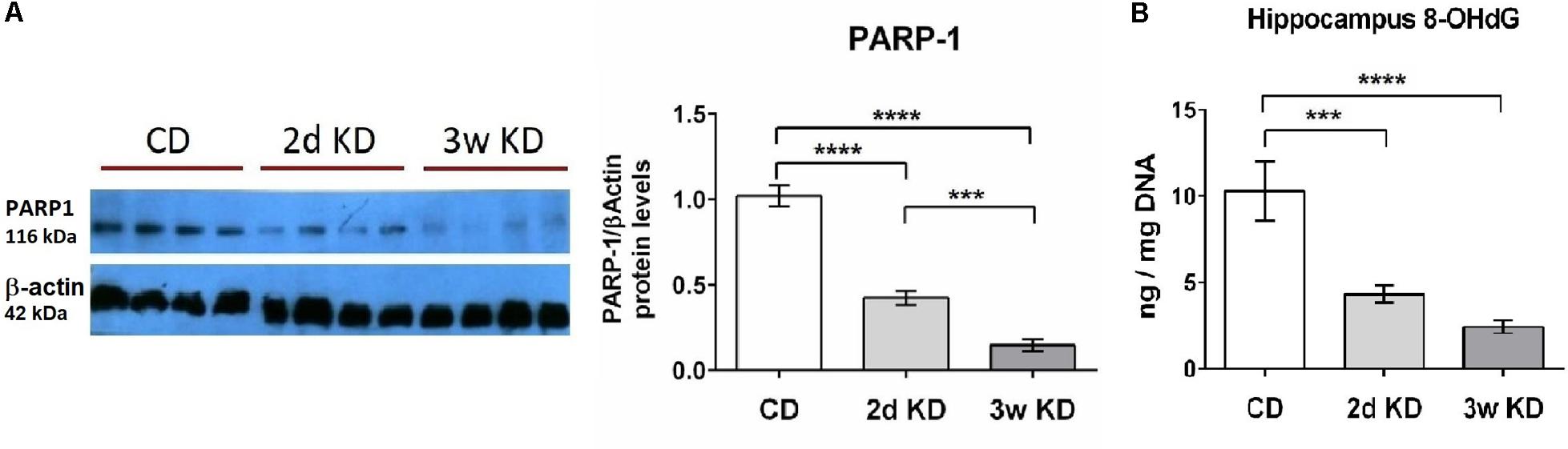
FIGURE 2. Effect of KD treatment on hippocampal PARP-1 levels and 8-OHdG levels. (A) Quantification of normalized PARP-1 protein levels in hippocampi obtained from animals fed standard chow (CD), or KD for 2d or 3w. Representative image of PARP-1 Western blot included. Each lane represents an individual animal. Blots were repeated 2–3 times. (B) Significant and progressive decrease in 8-OHdG levels was observed in hippocampi obtained from KD treated animals. CD, n = 6; 2d KD, n = 8; 3w KD, n = 8 (n = 2 for each animal).
Here we determined that KD increased NAD+, decreased levels of DNA damage and induced rapid changes in PARP-1 and sirtuin enzymes. This cohort of changes induced within 2 days of KD exposure could thus be protective for healthy cells against oxidative and metabolic damage and provide key mechanisms rendering KD beneficial across a range of neurological conditions.
The time-course of changes in Sirt1 activity and expression parallel the persistent increase in NAD+ levels (Elamin et al., 2017) and support the idea that elevated NAD+ increases activation of sirtuin enzymes. The ability of Sirt1 to affect the expression of genes implicated in a variety of functions ranging from neuroinflammation to proliferation and apoptosis (Cheng et al., 2003; Jeong et al., 2011) could explain the KD’s ability to impact diverse cellular pathways. Sirt1 activity reduces cell death and inflammation (Yeung et al., 2004; Kauppinen et al., 2013) and increases neuronal survival and life-span (Cohen et al., 2004; Kim et al., 2007; Khan et al., 2012). Therefore beneficial effects of ketogenic treatment in decreasing inflammation and combating neurodegenerative diseases could be attributed to these molecular mechanisms (Gasior et al., 2006; Ruskin et al., 2009; Kashiwaya et al., 2013).
Several genes controlling ketosis, fatty acid oxidation, and mitochondrial biogenesis are upregulated after long periods of KD treatment (Cullingford et al., 2002; Bough et al., 2006; Kennedy et al., 2007). Augmented sirtuin enzymatic activity without enhanced gene expression observed at 3 weeks may be due to elevated levels of the substrate NAD+. As Sirt1 was found to play a role in hepatocellular lipid metabolism (Hou et al., 2008), the transient upregulation of Sirt1 could be a cellular response to the shift in energy metabolism from glucose to fatty acids and disappear after adaptation occurs.
Previous work illustrated that ketogenic treatment reduced reactive oxygen species (Maalouf et al., 2007; Greco et al., 2016). Notably, Sirt1 can prevent neuronal cell death by reducing oxidative stress (Khan et al., 2012; Singh et al., 2017) which has been implicated in several neurodegenerative disorders (Andersen, 2004; Guo et al., 2013; Jiang et al., 2016; Islam, 2017). A consequence of oxidative stress and impaired mitochondrial function is DNA damage (Beal, 1995). This type of oxidative damage occurs as a by-product of normal cellular respiration, and can significantly contribute to the process of aging and neurodegeneration (Yakes and Van Houten, 1997). Moderate DNA damage triggers PARP-1 activation and DNA repair, resulting in reduced 8-OHdG levels (Hegedűs and Virág, 2014). Our quantified decrease in 8-OHdG and correlated PARP-1 changes suggest that ketogenic therapy can also rapidly and directly modulate DNA repair and oxidative stress. Therefore, potential direct effects, as well as elevation of Sirt activity and/or NAD+ inhibiting DNA damage (Hou et al., 2018) and decreasing reactive oxygen species (Lin and Guarente, 2003; Barzilai and Yamamoto, 2004; Kussmaul and Hirst, 2006) could contribute to decreased DNA damage. Interestingly, inhibition of PARP-1 can enhance mitochondrial metabolism and activate Sirt1 enzyme (Bai et al., 2011). Thus our data reinforce the existence of cross-talk between Sirt1 and PARP-1 (Cantó et al., 2013), possibly triggered by KD treatment, and modulated by NAD+ levels (Fouquerel and Sobol, 2014; Hegedűs and Virág, 2014; Mendelsohn and Larrick, 2017).
The rapid response of NAD+ and downstream effectors may relate to the anti-seizure response found in some patients after just a few days of KD treatment (Freeman and Vining, 1999; Cervenka et al., 2017). Moreover, elevation in PARP-1 activity, accompanied by NAD+ depletion and reduced Sirt1 activity, was shown to mediate neuronal death after seizure induction in animal models (Wang et al., 2013). Our data point to an opposite effect of KD on these three cellular measures, thus hinting at these effects as potential anti-seizure mechanisms. In addition, neuronal cell death and hippocampal 8-OHdG levels were increased following kainate-induced seizures in rats and prevented by administration of antioxidants (Liang et al., 2000). The KD’s ability to modulate oxidative DNA damage and reduce 8-OHdG levels in the hippocampus suggests that KD treatment may also be protective against seizure-induced neuronal death and DNA damage.
Here we quantified altered endogenous NAD+ levels and key downstream effectors as a direct result of consuming a KD and offered key potential mechanisms underlying anti-seizure and neuroprotective effects of ketogenic therapy. The changes found in disease-free animals in our current and previous studies (Elamin et al., 2017) indicate that the use of ketone bodies as an energy source can be associated with a healthier metabolic phenotype and an enhanced redox state even in the absence of disease or senescence. Furthermore, the NAD+ precursor nicotinamide ribose and PARP inhibitors are proposed to combat cancer and a host of inflammatory and neurodegenerative diseases (Basello and Scovassi, 2015; Lee et al., 2015; Ohmoto and Yachida, 2017; Vaur et al., 2017). We provide evidence that consuming a KD can mobilize similar mechanisms, and promote the metabolic resilience necessary to combat neurodegenerative and age-associated diseases.
ME, DR, SM, and PS conceived and designed the experiments, analyzed and interpreted the data, drafted the manuscript, and approved the final manuscript to be published. ME and DR acquired the data.
Supported by NIH (NS065957, NS066392, SM; AT008742, DR), Trinity College, Dorothy Goodwin Scholars Program (ME), and University of Hartford.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Andersen, J. K. (2004). Oxidative stress in neurodegeneration: cause or consequence? Nat. Rev. Neurosci. 10, S18–S25. doi: 10.1038/nrn1434
Augustin, K., Khabbush, A., Williams, S., Eaton, S., Orford, M., Cross, J. H., et al. (2018). Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet. Neurol. 17, 84–93. doi: 10.1016/S1474-4422(17)30408-8
Bai, P., Cantó, C., Oudart, H., Brunyánszki, A., Cen, Y., Thomas, C., et al. (2011). PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468. doi: 10.1016/j.cmet.2011.03.004
Barzilai, A., and Yamamoto, K. I. (2004). DNA damage responses to oxidative stress. DNA Repair 3, 1109–1115. doi: 10.1016/j.dnarep.2004.03.002
Basello, D. A., and Scovassi, A. I. (2015). Poly(ADP-ribosylation) and neurodegenerative disorders. Mitochondrion 24, 56–63. doi: 10.1016/j.mito.2015.07.005
Beal, M. F. (1995). Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 38, 357–366. doi: 10.1002/ana.410380304
Belenky, P., Bogan, K. L., and Brenner, C. (2007). NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19. doi: 10.1016/j.tibs.2006.11.006
Bough, K. J., Wetherington, J., Hassel, B., Pare, J. F., Gawryluk, J. W., Greene, J. G., et al. (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. doi: 10.1002/ana.20899
Bürkle, A., and Virág, L. (2013). Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 34, 1046–1065. doi: 10.1016/j.mam.2012.12.010
Cantó, C., Sauve, A. A., and Bai, P. (2013). Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 34, 1168–1201. doi: 10.1016/j.mam.2013.01.004
Cervenka, M. C., Hocker, S., Koenig, M., Bar, B., Henry-Barron, B., Kossoff, E. H., et al. (2017). Phase I/II multicenter ketogenic diet study for adult superrefractory status epilepticus. Neurology 88, 938–943. doi: 10.1212/WNL.0000000000003690
Chen, D., Bruno, J., Easlon, E., Lin, S. J., Cheng, H. L., Alt, F. W., et al. (2008). Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757. doi: 10.1101/gad.1650608
Cheng, C. M., Kelley, B., Wang, J., Strauss, D., Eagles, D. A., and Bondy, C. A. (2003). A ketogenic diet increases brain insulin-like growth factor receptor and glucose transporter gene expression. Endocrinology 144, 2676–2682. doi: 10.1210/en.2002-0057
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., et al. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392. doi: 10.1126/science.1099196
Cullingford, T. E., Eagles, D. A., and Sato, H. (2002). The ketogenic diet upregulates expression of the gene encoding the key ketogenic enzyme mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in rat brain. Epilepsy Res. 49, 99–107. doi: 10.1016/S0920-1211(02)00011-6
Dantzer, F., Amé, J., Schreiber, V., Nakamura, J., Ménissier-de Murcia, J., and de Murcia, G. (2006). Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 409, 493–510. doi: 10.1016/S0076-6879(05)09029-4
de Murcia, G., and de Murcia, J. M. (1994). Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem. Sci. 19, 172–173. doi: 10.1016/0968-0004(94)90280-1
Elamin, M., Ruskin, D. N., Masino, S. A., and Sacchetti, P. (2017). Ketone-based metabolic therapy: is increased NAD+ a primary mechanism? Front. Mol. Neurosci. 10:377. doi: 10.3389/fnmol.2017.00377
Fouquerel, E., and Sobol, R. W. (2014). ARTD1 (PARP1) activation and NAD(+) in DNA repair and cell death. DNA Repair 23, 27–32. doi: 10.1016/j.dnarep.2014.09.004
Freeman, J. M., and Vining, E. P. G. (1999). Seizures decrease rapidly after fasting: preliminary studies of the ketogenic diet. Arch. Pediatr. Adolesc. Med. 153, 946–949. doi: 10.1001/archpedi.153.9.946
Gasior, M., Rogawski, M. A., and Hartman, A. L. (2006). Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 17, 431–439. doi: 10.1097/00008877-200609000-00009
Greco, T., Glenn, T. C., Hovda, D. A., and Prins, M. L. (2016). Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 36, 1603–1613. doi: 10.1177/0271678X15610584
Grube, K., and Burkle, A. (1992). Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc. Natl. Acad. Sci. U.S.A. 89, 11759–11763. doi: 10.1073/pnas.89.24.11759
Guo, C., Sun, L., Chen, X., and Zhang, D. (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 8, 2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009
Hegedűs, C., and Virág, L. (2014). Inputs and outputs of poly(ADP-ribosyl)ation: relevance to oxidative stress. Redox Biol. 2, 978–982. doi: 10.1016/J.REDOX.2014.08.003
Heinemann, U., Beck, H., Dreier, J. P., Ficker, E., Stabel, J., and Zhang, C. L. (1992). The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res. Suppl. 7, 273–280.
Hou, X., Xu, S., Maitland-Toolan, K. A., Sato, K., Jiang, B., Ido, Y., et al. (2008). SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 283, 20015–20026. doi: 10.1074/jbc.M802187200
Hou, Y., Lautrup, S., Cordonnier, S., Wang, Y., Croteau, D. L., Zavala, E., et al. (2018). NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U.S.A. 115, E1876–E1885. doi: 10.1073/PNAS.1718819115
Huber, A., Bai, P., de Murcia, J. M., and de Murcia, G. (2004). PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair 3, 1103–1108. doi: 10.1016/j.dnarep.2004.06.002
Islam, M. T. (2017). Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 39, 73–82. doi: 10.1080/01616412.2016.1251711
Jeong, E. A., Jeon, B. T., Shin, H. J., Kim, N., Lee, D. H., Kim, H. J., et al. (2011). Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 232, 195–202. doi: 10.1016/j.expneurol.2011.09.001
Jiang, T., Sun, Q., and Chen, S. (2016). Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 147, 1–19. doi: 10.1016/j.pneurobio.2016.07.005
Kashiwaya, Y., Bergman, C., Lee, J.-H., Wan, R., King, M. T., Mughal, M. R., et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023
Kauppinen, A., Suuronen, T., Ojala, J., Kaarniranta, K., and Salminen, A. (2013). Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 25, 1939–1948. doi: 10.1016/J.CELLSIG.2013.06.007
Kennedy, A. R., Pissios, P., Otu, H., Xue, B., Asakura, K., Furukawa, N., et al. (2007). A high-fat, ketogenic diet induces a unique metabolic state in mice. AJP Endocrinol. Metab. 292, E1724–E1739. doi: 10.1152/ajpendo.00717.2006
Khan, R. S., Fonseca-Kelly, Z., Callinan, C., Zuo, L., Sachdeva, M. M., and Shindler, K. S. (2012). SIRT1 activating compounds reduce oxidative stress and prevent cell death in neuronal cells. Front. Cell. Neurosci. 6:63. doi: 10.3389/fncel.2012.00063
Kim, D., Nguyen, M. D., Dobbin, M. M., Fischer, A., Sananbenesi, F., Rodgers, J. T., et al. (2007). SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179. doi: 10.1038/sj.emboj.7601758
Kussmaul, L., and Hirst, J. (2006). The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7607–7612. doi: 10.1073/pnas.0510977103
Landry, J., Sutton, A., Tafrov, S. T., Heller, R. C., Stebbins, J., Pillus, L., et al. (2000). The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 5807–5811. doi: 10.1073/pnas.110148297
Lee, H. J., Hong, Y.-S., Jun, W., and Yang, S. J. (2015). Nicotinamide riboside ameliorates hepatic metaflammation by modulating NLRP3 inflammasome in a rodent model of type 2 diabetes. J. Med. Food 18, 1207–1213. doi: 10.1089/jmf.2015.3439
Liang, L. P., Ho, Y. S., and Patel, M. (2000). Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 101, 563–570. doi: 10.1016/S0306-4522(00)00397-3
Lin, S. J., Defossez, P. A., and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128. doi: 10.1126/science.289.5487.2126
Lin, S. J., and Guarente, L. (2003). Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 15, 241–246. doi: 10.1016/S0955-0674(03)00006-1
Liu, J., Yang, B., Zhou, P., Kong, Y., Hu, W., Zhu, G., et al. (2017). Nicotinamide adenine dinucleotide suppresses epileptogenesis at an early stage. Sci. Rep. 7:7321. doi: 10.1038/s41598-017-07343-0
Maalouf, M., Sullivan, P. G., Davis, L., Kim, D. Y., and Rho, J. M. (2007). Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. doi: 10.1016/j.neuroscience.2006.11.065
Martinet, W., Knaapen, M. W. M., De Meyer, G. R. Y., Herman, A. G., and Kockx, M. M. (2002). Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106, 927–932. doi: 10.1161/01.CIR.0000026393.47805.21
Martire, S., Mosca, L., and D’Erme, M. (2015). PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 14, 53–64. doi: 10.1016/j.mad.2015.04.001
Mendelsohn, A. R., and Larrick, J. W. (2017). The NAD+/PARP1/SIRT1 axis in aging. Rejuvenation Res. 20, 244–247. doi: 10.1089/rej.2017.1980
Michan, S., Li, Y., Chou, M. M.-H., Parrella, E., Ge, H., Long, J. M., et al. (2010). SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 30, 9695–9707. doi: 10.1523/JNEUROSCI.0027-10.2010
Michishita, E. (2005). Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635. doi: 10.1091/mbc.E05-01-0033
Mills, K. F., Yoshida, S., Stein, L. R., Grozio, A., Kubota, S., Sasaki, Y., et al. (2016). Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 24, 795–806. doi: 10.1016/J.CMET.2016.09.013
Morales, J., Li, L., Fattah, F. J., Dong, Y., Bey, E. A., Patel, M., et al. (2014). Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 24, 15–28. doi: 10.1615/CritRevEukaryotGeneExpr.2013006875
Mostoslavsky, R., Chua, K. F., Lombard, D. B., Pang, W. W., Fischer, M. R., Gellon, L., et al. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329. doi: 10.1016/j.cell.2005.11.044
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2009). A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 50, 1109–1117. doi: 10.1111/j.1528-1167.2008.01870.x
Newman, J. C., Covarrubias, A. J., Zhao, M., Yu, X., Gut, P., Ng, C.-P., et al. (2017). Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab 26, 547–557.e8. doi: 10.1016/j.cmet.2017.08.004
Ohmoto, A., and Yachida, S. (2017). Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 10, 5195–5208. doi: 10.2147/OTT.S139336
Pawlosky, R. J., Kemper, M. F., Kashiwaya, Y., King, M. T., Mattson, M. P., and Veech, R. L. (2017). Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 141, 195–207. doi: 10.1111/jnc.13958
Roberts, M. N., Wallace, M. A., Tomilov, A. A., Zhou, Z., Marcotte, G. R., Tran, D., et al. (2017). A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 26, 539–546.e5. doi: 10.1016/j.cmet.2017.08.005
Ruskin, D. N., Kawamura, M., and Masino, S. A. (2009). Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS One 4:e8349. doi: 10.1371/journal.pone.0008349
Satoh, A., Brace, C. S., Rensing, N., Cliften, P., Wozniak, D. F., Herzog, E. D., et al. (2013). Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430. doi: 10.1016/j.cmet.2013.07.013
Sharma, S., Sankhyan, N., Gulati, S., and Agarwala, A. (2013). Use of the modified Atkins diet for treatment of refractory childhood epilepsy: a randomized controlled trial. Epilepsia 54, 481–486. doi: 10.1111/epi.12069
Shen, Y., McMackin, M. Z., Shan, Y., Raetz, A., David, S., and Cortopassi, G. (2016). Frataxin deficiency promotes excess microglial DNA damage and inflammation that is rescued by PJ34. PLoS One 11:e0151026. doi: 10.1371/journal.pone.0151026
Singh, P., Hanson, P. S., and Morris, C. M. (2017). SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 18:46. doi: 10.1186/s12868-017-0364-1
Vaur, P., Brugg, B., Mericskay, M., Li, Z., Schmidt, M. S., Vivien, D., et al. (2017). Nicotinamide riboside, a form of Vitamin B3, protects against excitotoxicity-induced axonal degeneration. FASEB J. 31, 5440–5452. doi: 10.1096/fj.201700221RR
Vazquez, B. N., Thackray, J. K., Simonet, N. G., Kane-Goldsmith, N., Martinez-Redondo, P., Nguyen, T., et al. (2016). SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 35, 1488–1503. doi: 10.15252/embj.201593499
Wang, D., Li, Z., Zhang, Y., Wang, G., Wei, M., Hu, Y., et al. (2016). Targeting of microRNA-199a-5p protects against pilocarpine-induced status epilepticus and seizure damage via SIRT1-p53 cascade. Epilepsia 57, 706–716. doi: 10.1111/epi.13348
Wang, S., Yang, X., Lin, Y., Qiu, X., Li, H., Zhao, X., et al. (2013). Cellular NAD depletion and decline of SIRT1 activity play critical roles in PARP-1-mediated acute epileptic neuronal death in vitro. Brain Res. 1535, 14–23. doi: 10.1016/j.brainres.2013.08.038
Winter, S. F., Loebel, F., and Dietrich, J. (2017). Role of ketogenic metabolic therapy in malignant glioma: a systematic review. Crit. Rev. Oncol. Hematol. 112, 41–58. doi: 10.1016/j.critrevonc.2017.02.016
Yakes, F. M., and Van Houten, B. (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 94, 514–519. doi: 10.1073/pnas.94.2.514
Yang, T., Fu, M., Pestell, R., and Sauve, A. A. (2006). SIRT1 and endocrine signaling. Trends Endocrinol. Metab. 17, 186–191. doi: 10.1016/j.tem.2006.04.002
Yang, X., and Cheng, B. (2010). Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 42, 145–153. doi: 10.1007/s12031-010-9336-y
Keywords: ketone bodies, metabolism, hippocampus, longevity, oxidative stress, nicotinamide adenine dinucleotide, sirtuin, PARP-1
Citation: Elamin M, Ruskin DN, Masino SA and Sacchetti P (2018) Ketogenic Diet Modulates NAD+-Dependent Enzymes and Reduces DNA Damage in Hippocampus. Front. Cell. Neurosci. 12:263. doi: 10.3389/fncel.2018.00263
Received: 10 April 2018; Accepted: 31 July 2018;
Published: 30 August 2018.
Edited by:
Carl E. Stafstrom, Johns Hopkins Medicine, United StatesReviewed by:
Deep R. Sharma, SUNY Downstate Medical Center, United StatesCopyright © 2018 Elamin, Ruskin, Masino and Sacchetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Sacchetti, UHNhY2NoZXR0QGhhcnRmb3JkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.