
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 23 May 2018
Sec. Cellular Neuropathology
Volume 12 - 2018 | https://doi.org/10.3389/fncel.2018.00142
This article is part of the Research Topic Biology of Lipids in the Control of the Motor Function, in Health and Diseases View all 13 articles
 Xuan Yang1†
Xuan Yang1† Shurui Chen2†
Shurui Chen2† Zhenya Shao2
Zhenya Shao2 Yuanlong Li2
Yuanlong Li2 He Wu3
He Wu3 Xian Li4
Xian Li4 Liang Mao5Zipeng Zhou2
Liang Mao5Zipeng Zhou2 Liangjie Bai6
Liangjie Bai6 Xifan Mei2*Chang Liu3*
Xifan Mei2*Chang Liu3*Spinal cord injury (SCI) is a severe neurological trauma that involves complex pathological processes. Inflammatory response and oxidative stress are prevalent during the second injury and can influence the functional recovery of SCI. Specially, Apolipoprotein E (APOE) induces neuronal repair and nerve regeneration, and the deficiency of Apoe impairs spinal cord-blood-barrier and reduces functional recovery after SCI. However, the mechanism by which Apoe mediates signaling pathways of inflammatory response and oxidative stress in SCI remains largely elusive. This study was designed to investigate the signaling pathways that regulate Apoe deficiency-dependent inflammatory response and oxidative stress in the acute stage of SCI. In the present study, Apoe−/− mice retarded functional recovery and had a larger lesion size when compared to wild-type mice after SCI. Moreover, deficiency of Apoe induced an exaggerated inflammatory response by increasing expression of interleukin-6 (IL-6) and interleukin-1β (IL-1β), and increased oxidative stress by reducing expression of Nrf2 and HO-1. Furthermore, lack of Apoe promoted neuronal apoptosis and decreased neuronal numbers in the anterior horn of the spinal cord after SCI. Mechanistically, we found that the absence of Apoe increased inflammation and oxidative stress through activation of NF-κB after SCI. In contrast, an inhibitor of nuclear factor-κB (NF-κB; Pyrrolidine dithiocarbamate) alleviates these changes. Collectively, these results indicate that a critical role for activation of NF-κB in regulating Apoe-deficiency dependent inflammation and oxidative stress is detrimental to recovery after SCI.
Spinal cord injury (SCI) can lead to destructive and irreversible neurological deficits, including sensory, motor and autonomic impairments (Lang et al., 2015). The complex pathological process that occurs after SCI is biphasic, consisting of a primary injury followed by the spreading of secondary tissue damage (Bartus et al., 2016). The primary injury directly induces irreversible cell death at the initial traumatic site. In contrast, the secondary injury is characterized by inflammatory responses, oxidative damage, neuronal death and other molecular events, which can be alleviated (Oyinbo, 2011). Currently, treatment of SCI includes early interventions in the acute stage aimed at minimizing secondary complications and preventing the spread of damage beyond the primary injured site (Ramer et al., 2014). Numerous studies have also indicated that targeting this secondary damage induced by cytokines and signaling pathways could be a new therapeutic strategy to help improve neurological outcomes after SCI (Coll-Miró et al., 2016; Zheng et al., 2017). Because inflammation, oxidative stress and apoptosis play contributory roles in the pathogenesis of the secondary damage, it is indispensable and important to explore novel methods of inhibiting secondary injury as treatment strategies for patients with SCI.
Inflammatory and oxidative responses are two major components of the secondary injury and play fundamental roles in the pathogenesis of SCI (Silva et al., 2014; Anwar et al., 2016). NF-κB, a redox sensitive transcription factor, plays a vital regulatory role in inflammatory responses after SCI (Bethea et al., 1998; Rafati et al., 2008). Activation of NF-κB involved in the occurrence of SCI is necessary for expression of inflammatory factors, such as interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β; Lu et al., 2013). Nuclear factor erythroid-derived 2-related factor 2 (Nrf2) is accountable for anti-oxidative responses and anti-inflammatory properties in neurological disorders (Kanninen et al., 2015; Wang et al., 2017). After SCI, Nrf2 is up-regulation and activates a group of anti-oxidative and cytoprotective genes, such as heme oxygenase-1 (HO-1) and NADPH quinineoxidoreductase-1 (NQO1; Pomeshchik et al., 2014). Previous study shown that Nrf2 regulates the inflammation by limiting the up-regulation of NF-κB activity after SCI (Mao et al., 2010). Thus, inactivation of NF-κB and induction of the Nrf2 pathway in SCI might be an effective neuroprotective strategy.
Apolipoprotein E (APOE: protein, Apoe: gene), mainly synthesized in astrocytes of central nervous system (CNS), is involved in lipid metabolism and cholesterol transport for the promotion of neuronal repair (Bu, 2010; Mahley and Huang, 2012). It is well-established that APOE4 is the strongest genetic risk factor for early onset AD among its other three alleles (2, 3, and 4; Liu et al., 2017; Nuriel et al., 2017). Previous studies show APOE is up-regulated in vivo after neurological dysfunction, such as SCI (Seitz et al., 2003), traumatic brain injury (Iwata et al., 2005), chronic stroke (Kim et al., 2015), chronic constriction injury (Bellei et al., 2017). More specifically, Apoe possesses anti-inflammatory, anti-oxidative and anti-apoptosis properties (Laskowitz et al., 1997, 1998; Kitagawa et al., 2002; Hayashi et al., 2009; Li X.-Z. et al., 2017). For instance, APOE mimetic peptide significantly suppressed microglial activation in rats with SCI. Conversely, the excessive inflammation may be caused by the increased permeability of BSCB leading to the influx of inflammatory cells in peripheral blood in Apoe−/− mice after SCI (Wang et al., 2014; Cheng et al., 2018). Moreover, Apoe−/− mice showed that increased oxidative stress caused an exaggerated neuronal deposition and contributed to the neurodegenerative processes (Nuriel et al., 2017). Also, APOE plays a critical role in the clearance of apoptotic bodies during neuronal apoptosis (Elliott et al., 2007). Further studies revealed various signaling pathways were involved in the anti-inflammatory, anti-oxidative stress and anti-apoptosis role of APOE. For example, depletion of Apoe caused abnormal cerebrovascular and neuronal function by activating a proinflammatory CypA–NF-κB–MMP9 pathway (Bell et al., 2014). Also, extracellular APOE interacts with its receptor to activate mitogen-activated protein kinase (MAPK) signaling pathway to regulate inflammation and apoptosis (Pocivavsek and Rebeck, 2009; Gan et al., 2011; Safina et al., 2016).
Although recent studies shown APOE has a critical neuroprotective function after SCI, the molecular mechanism of Apoe interaction with NF-κB pathway in the alleviation of inflammatory response and oxidative stress of SCI remains unknown. Here, we demonstrated that deficiency of Apoe retarded the recovery of locomotor function and tissue repair via increasing the expression of inflammatory cytokines and oxidative response, as well as aggravating neural apoptosis of the spinal cord tissue in vivo. In addition, we further showed Apoe deficiency caused inflammation and oxidative stress could be alleviated through inhibition of NF-κB signaling pathway.
The present study was conducted using female Apoe−/− mice with a C57BL/6J background (strain B6/JNju-Apoeem1Cd82/Nju; stock number T001458), weighing 20–25 g, and 10–12 weeks. Apoe−/− mice were purchased from the Nanjing biomedical Research Institute of Nanjing University and have been described previously (Gallardo et al., 2008). Homozygous (ApoE−/−) mice were completely Knockout Apoe were verified by PCR. The age-matched, sex-matched littermates of Apoe−/− mice or WT controls were used for western blot (WB), immunofluorescence staining, as well as histological staining. All mice were housed in standard cages (five per cage) in a specific pathogen free (SPF) laboratory animal center of Jinzhou Medical University. They were given free access to food and water ad libitum, under a 12 h/12 h light/dark cycle. The Institutional Animal Care and Use Committee of Jinzhou Medical University approved all experimental protocols (permit number: SCXK (Liao) 2014-0004).
Mice were randomly allocated to the following four parts of the experiment using a completely random number table.
A total of 48 WT mice were randomly divided into eight experimental groups, including sham (Sham operation after 7 days), 12 h, 1 day, 3 days, 5 days, 7 days, 14 days and 28 days, to evaluate temporal changes of APOE protein levels after SCI.
WT (n = 6) and Apoe KO (n = 6) mice were used to evaluate the effect of Apoe deficiency on behavioral recovery, which was observed from before and after SCI days 1–28.
Sham (n = 6) and SCI (n = 6) group in WT and Apoe KO mice were sacrificed at 7 dpi for WB. The same numbers of WT and Apoe KO mice were sacrificed for histological analysis. These experimental mice were randomly assigned to four groups: the WT-Sham group, WT-SCI group, KO-Sham group and KO-SCI group.
WT (n = 24) and Apoe KO (n = 24) mice were randomly divided into four groups: WT-SCI and vehicle-treated group (WT-SCI-Veh), KO-SCI and vehicle-treated group (KO-SCI-Veh), WT-SCI and PDTC-treated (WT-SCI-PDTC), KO-SCI and PDTC-treated group (KO-SCI-PDTC). PDTC groups were intraperitoneal injection with an inhibitor of NF-κB, PDTC (P8765, Sigma-Aldrich, Darmstadt, Germany) 100 mg/kg/day as previously described (Jiménez-Garza et al., 2005; Bell et al., 2014). While the vehicle groups were injected with the equivalent volume of saline after 5 min SCI and then seven consecutive days.
All experimental interventions were carried out in accordance with the Guidelines for Animal Experiments provided by our institution. A mouse model of SCI was established using the modified weight-drop method (Ma et al., 2001). The mice were anesthetized with 10% chloral hydrate (0.33 mL/kg, intraperitoneal injection) until they were confirmed to be unconscious by toe-pinch. After immobilizing the vertebrae with a spinal stereotactic device, the skin and muscles were incised along the dorsal midline to expose the vertebral column, and laminectomy was performed at the T10 level. A 3 g-impactor device (diameter: 0.5 mm) was subsequently dropped from a height of 25 mm to the spinal cord to induce immediate hind limb paralysis. After SCI, the muscles and skin aseptically sutured in layers. Sham animals only received laminectomy. A manual bladder emptying was performed three times daily until the urinary emptying function was re-established in mice after SCI.
Mice were sacrificed at 7 days after SCI and injured spinal cord tissues (0.5 cm length from the injury epicenter) were rapidly extracted for WB. The spinal cord tissue was homogenized in RIPA lysis buffer containing PMSF buffer (P0013B, Beyotime, Beijing, China) for 30 min on ice. After centrifugation at 12,000 RMP (25 min, 4°C) to remove debris, the supernatant was quickly stored at −80°C. The BCA Protein Assay Kit (P0010, Beyotime, Beijing, China) was used to quantify protein levels. Protein (40 μg) was loaded at 10% or 12% SDS-PAGE. Following electrophoresis, the protein was transferred to PVDF membranes (0.22 μm, Merckmillipore, Darmstadt, Germany). The membranes were blocked with 5% dried skimmed milk in TBS with 0.1% Tween-20 for 2 h at RT, and then incubated at 4°C overnight with the following primary antibodies: mouse anti-GAPDH (1:10,000, HC301-01, Transgene, Beijing, China), rabbit anti-APOE (1:200, sc-13520, Santa Cruz Biotechnology, CA, USA), mouse anti-GFAP (1:1000, ab7260, Abcam, Cambridge, England), mouse anti-IL-6 (1:1000, ab9324, Abcam, Cambridge, England), rabbit anti-IL-1β (1:1000, ab9722, Abcam Cambridge, England), rabbit anti-Bcl-2 (1:1000, ab136285, Abcam, Cambridge, England), rabbit anti-Bax (1:1000, ab32503, Abcam, Cambridge, England), rabbit anti-Nrf2 (1:1000, ab31163, Abcam, Cambridge, England), rabbit anti-p-NF-κB-p65 (1:1000, ab86299, Abcam, Cambridge, England), mouse anti-HO-1 (1:1000, ab3248, Abcam, Cambridge, England), mouse anti-NQO-1(1:1000, ab2947, Abcam, Cambridge, England), mouse anti-Erk1(pT202/pY204) + Erk2(Pt185/Py187; 1:1000, 50011, Abcam, Cambridge, England), rabbit anti-ERK1 + ERK2 (1:1000, 184699, Abcam, Cambridge, England), rabbit anti-Phosphp-p38-MAPK(Thr180/Tyr182; 1:1000, 9211, Cell Signaling Technology, Danvers, MA, USA), rabbit anti-p38-MAPK (1:1000, 9212, Cell Signaling Technology, Danvers, MA, USA), and rabbit anti-JNK1 (pThr183/pThy185; 1:1000, NBP1-72242, Novus Biologicals, Littleton, CO, USA). The membranes were then incubated with the corresponding secondary antibodies for 2 h at RT. Subsequently, the membranes were visualized using a ChemiDoc-ItTMTS2 Imager (UVP, CA, USA). Finally, a blinded researcher analyzed the bands using the ImageJ2x software program (National Institute of Health, New York, NY, USA).
The mice were sacrificed with an overdose anesthetic and transcardially perfused with 0.9% saline, followed by 4% PFA in PBS at 7 days after injury. Spinal cord tissues containing areas rostral and caudal to the epicenter of injury (about 2 cm) were harvested and fixed in 4% PFA overnight at 4°C. Tissue segments then dehydrated in 30% sucrose-PFA until they sunk close to the bottom of the liquid. Finally, the spinal cord tissues (lesion epicenter about 2 cm) were cut with a cryostat microtome (CM3050S, Leica, Wetzlar, Germany) into 10 μm-thick longitudinal and 20 μm-thick transverse serial sections for immunofluorescence, Hematoxylin & Eosin (H&E) staining and Nissl staining.
Briefly, the spinal cord sections were incubated overnight at 4°C with primary antibodies. The following primary antibodies were used for immunofluorescence: rabbit anti-APOE (1:200, sc-13520, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-NeuN (1:500, 104224, Abcam, Cambridge, England), rabbit anti-GFAP antibody (1:200; DAKO, Glostrup, Denmark), mouse anti-GFAP (1:1000, ab7260, Abcam, Cambridge, England), mouse anti-CD68 (1:500, NBP2-33337, Novus, Plymouth, MN, USA) and rabbit anti-Cleaved-caspase-3 (1:200, 9664, Cell Signaling Technology, Danvers, MA, USA). On the following day, the sections were rinsed and incubated with appropriate secondary antibody, including goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor® 488 (1:400, A-11017, Invitrogen, Carlsbad, CA, USA) and goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor® 568 (1:400, A-11019, Invitrogen, Carlsbad, CA, USA) for 2 h at RT. Finally, the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 1:1000, 104139, Abcam, Cambridge, England) for 15 min at RT. All sections were mounted on Permount™ mounting medium before being screened using a fluorescence microscope (DMI4000B, Leica, Wetzlar, Germany).
Hind limb motor function was monitored by the Basso Mouse Scale (BMS) test at 1 day before SCI and then weekly for 4 weeks (Basso et al., 2006). Two raters who were blinded to the experimental groups collected and analyzed the data. Briefly, each mouse was observed for 4 min, and hind limb movement performance was assessed and corresponded to the nine-point scales. The final score of each animal was the mean values from both investigators who were blinded to the grouping and experimental design.
The spinal cord longitudinal sections were taken at 7 days after SCI and were immersed in hematoxylin solution for 3 min, following by rinsing distilled water quickly. Then the slides were differentiated in HCL/95% alcohol (1:50) solution for 10 s. After washing with distilled water for 5 min, the sections were stained with 0.5% eosin for 10 s. After dehydration used by gradient ethanol for 3 min (95%, 100%), and then transparency by xylene for 1 min. Next, the maximum area of the lesion epicenter in each section was examined with a light microscope (DMI4000B, Leica, Wetzlar, Germany).
In accordance with the manufacturer’s instructions of Nissl staining, the sections of spinal cord incubated in 0.1% Nissl staining solution (C0117, Beyotime, Beijing, China) for 3 min at 37°C, rinsed with distilled water, dehydrated in by 95% and 100% ethanol solutions for 2 min, respectively, and cleared in xylene for 5 min. The Nissl-stained images were captured on two sides of each section in the ventral horn at under an optical microscope (DMI4000B, Leica, Wetzlar, Germany).
For quantification of the lesion area, we performed as previously described (Cheng et al., 2016; Narang et al., 2017), the spinal cord segment of 0.5 cm contains the epicenter of the lesion. The lesion area was defined as the areas without normal tissue architecture, or tissue containing only cellular debris, which it showed a different optical density compared to the adjacent tissue. The border of the damage tissue was manually outlined with a dashed line. Four representative sections per animal (one of every 10 serial sections were stained), were traced three times manually outlined and analyzed by the ImageJ2x software program (National Institute of Health, New York, NY, USA). The extent of lesion expressed as percentage of lesion area per spinal cord on the same rostral-caudal axis was calculated in the sagittal plane. For quantification of the number of surviving neurons, transverse spinal cord sections were selected (four sections per animal) ranging from rostral (−300, −600 μm) to caudal (+300, +600 μm) around the epicenter. As the section thickness is 20 μm, it can be inferred that there are 15 sections in the 300 μm range. Neurons with a clear nucleolus and a Nissl bodies were counted by using previously published methods (Li et al., 1994; Li J. et al., 2017). According to Rexed’s lamina system of gray matter, the mean number of ventral motor neurons on two sides (lamina IX) was manually calculated through ImageJ2x software. For quantification of APOE-immunoreactive signal intensity, transverse spinal cord sections (four sections per animal) were selected ranging from rostral (−400, −700 μm) to caudal (+400, +700 μm) around the injury epicenter. An area of interest (ROI) was placed on the right and the left ventral horn of each section. Next, the APOE-immunoreactive optical density value was quantified by ImageJ2x software as described previously (Hong et al., 2017). For quantification of the NeuN/Cleaved-caspase-3 double-positive cells were performed as previously method (Bai et al., 2017), which were represented the positive of SCI-induced apoptotic cells. Four transverse sections were chosen from each animal in four position points, such as ±500 μm, ±800 μm. The optical density of double-labeled of Cleaved-caspase 3-positive neurons in the ventral motor regions was quantified using ImageJ2X. The double-staining of APOE and NeuN, GFAP and CD68 were selected ranging from rostral to caudal (±1000 μm) around the injury epicenter. All quantitative image analysis was conducted by two researchers blinded to the allocation.
Quantitative data are presented as mean ± SEM. Two experimental groups were analyzed by unpaired Student’s t-test. Comparisons of more than two groups were performed with a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. The BMS scores were analyzed by repeated measures two-way analyses of variance (ANOVAs), followed by Tukey’s post hoc test to compare the differences between each group. Statistical analysis was performed using GraphPad Prism software, version 7.00 (GraphPad Software Inc., La Jolla, CA, USA). Significance was considered at p < 0.05.
WBs and immunofluorescence were used to examine the dynamic changes of APOE protein expression in sham and injured spinal cords of WT mice. Compared with the sham group, the expression of APOE began to decline in the first 3 days, but then increased on day 5. The highest level of APOE expression occurred 7 days post-injury (dpi; WT-Sham vs. WT-SCI: 0.23 ± 0.05 vs. 0.63 ± 0.04, p < 0.001, Figures 1A,B), and subsequently returned to uninjured levels 14–28 dpi (Figures 1A,B). In addition, increased APOE expression was observed 7 dpi using immunofluorescence (WT-Sham vs. WT-SCI: 0.08 ± 0.01 vs. 0.12 ± 0.01, p < 0.01, Figures 1C,D), which was in agreement with the WB data. We next used Apoe KO mice to explore the role of endogenous APOE in the pathology of SCI. WB was used to confirm that deletion of Apoe caused deficient APOE expression. As shown in Figures 1E,F, APOE expression is lost in spinal cords of Apoe KO compared with WT mice (WT-Sham vs. Apoe KO: 0.34 ± 0.05 vs. 0.00 ± 0.00, p < 0.001, Figures 1E,F). The other two representative WBs images for APOE proteins assessed are presented on Supplementary Figure S3.
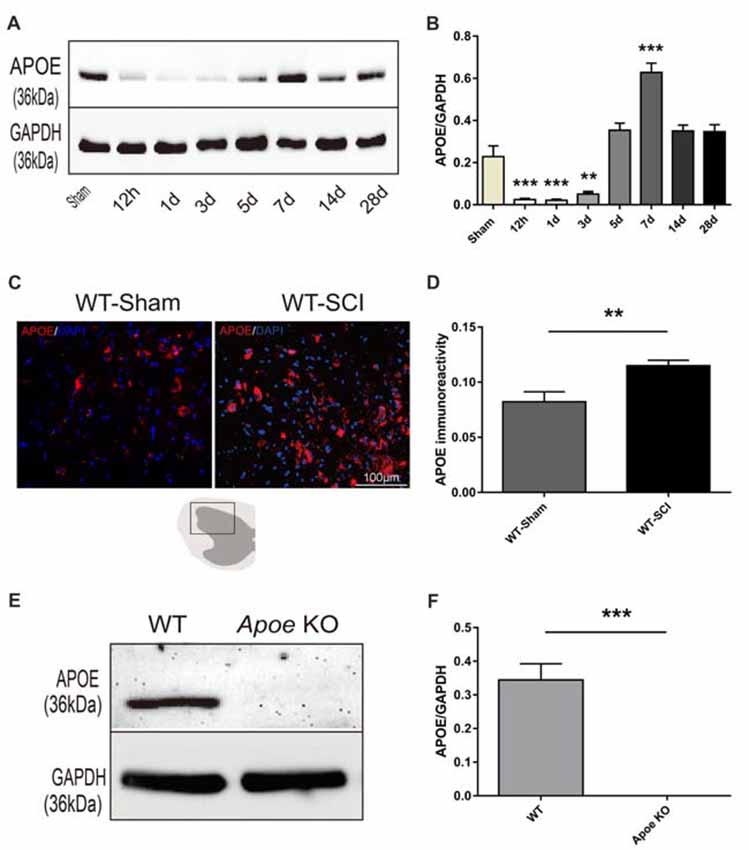
Figure 1. The temporal expression of Apolipoprotein E (APOE) protein after spinal cord injury (SCI) and identification of Apoe KO mice. (A) Representative western blots (WBs) of APOE and GAPDH expression in the lesion site after SCI. (B) Statistical analysis of WBs of APOE expression in WT mice after SCI. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). **p < 0.01, ***p < 0.001, Sham vs. 12 h, p = 0.0002, Sham vs. 1 day, p = 0.0001, Sham vs. 3 days, p = 0.0021, Sham vs. 7 days, p < 0.0001, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. (C) Representative immunofluorescent images of APOE at the lesion epicenter in Sham and SCI group 7 days post-injury. (D) Statistical analysis of APOE immunoreactivity after SCI. Four transverse sections were measured optical intensity at ± 400 μm, ±700 μm from rostral and caudal to the injury site per spinal cord. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). **p < 0.01, Sham vs. 7 days, p = 0.0047, Student’s t-test, Scale bars = 100 μm. (E) APOE expression in the spinal cord was detected by WBs from WT and Apoe KO mice. (F) Statistical analysis of APOE and GAPDH expression both in WT and Apoe KO mice. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). ***p < 0.001, WT vs. Apoe KO, Student’s t-test.
To determine the expression pattern and possible cellular source of APOE, immunofluorescent co-localization of APOE with neurons (NeuN), astrocytes (GFAP), and macrophages (CD68) in uninjured and contused WT mice at 7 dpi was conducted (Figures 2A–C). As previously demonstrated, APOE was mostly distributed in astrocytes of the CNS, also in some neurons and macrophages under normal situation. It was obviously increased in these cells after CNS injury. We demonstrated that APOE was detectable in normal spinal cord of WT mice. At 7 dpi, APOE expression increased in the injured spinal cord, with high-level co-localization in the cytoplasm of numerous NeuN-positive neurons, GFAP-positive astrocytes and CD68-positive macrophages.
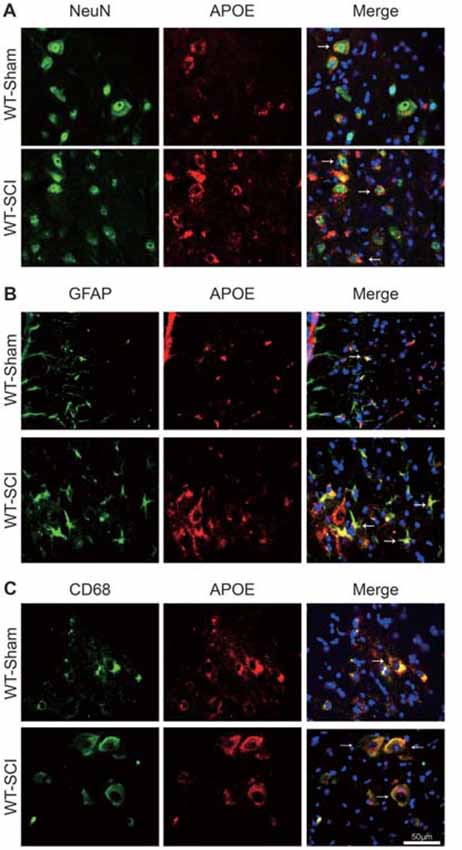
Figure 2. Expression of APOE protein in neurons, astrocytes, macrophages/microglia following SCI. (A–C) The cellular localization of APOE was observed by double immunofluorescent staining of APOE (red) with NeuN, GFAP, CD68 (green) and DAPI (blue), respectively. Four transverse sections were taken at ±1000 μm from rostral and caudal to the injury site per spinal cord (N = 6 per group, white arrows: APOE-positive neurons, astrocytes and macrophages, Scale bars = 100 μm).
Next, we evaluated the functional recovery of hind limb after SCI by BMS. There were not any differences in Apoe KO mice and WT mice before an injury, and then the movement function was abolished immediately on the first day after SCI. Already at 7 dpi, the BMS scores of Apoe KO mice were significantly reduced when compared to WT mice (WT-SCI vs. KO-SCI, 0.83 ± 0.21 vs. 0.42 ± 0.15, p < 0.05, Figure 3A), and this weakens in the locomotor performance of the Apoe KO mice for the subsequent 3 weeks period. Specifically, Apoe KO mice obtained an average BMS score of 2.25, which corresponds to a slight or extensive movement of the ankle following 28 dpi. On the contrary, an average BMS score of 2.92, WT mice showed extensive ankle movement and plantar paw placement without weight support (WT-SCI vs. KO-SCI, 2.92 ± 0.20 vs. 2.25 ± 0.17, p < 0.001, Figure 3A). These data clearly demonstrate that Apoe deficiency worsens the recovery of functional disabilities. Moreover, the lesion area evaluated by H&E staining in Apoe KO mice was greater than WT mice at 7 days after SCI (WT-SCI vs. KO-SCI, 0.53 ± 0.07 vs. 0.35 ± 0.03, p < 0.05, Figures 3B,C). Apoe KO mice exhibited less numbers of intact motor neurons and more damaged cells in the ventral horn of than in that of WT mice after 7 dpi (WT-SCI vs. KO-SCI, 9.11 ± 0.68 vs. 4.44 ± 0.47, p < 0.01, Figures 3D,E). Reactive astrogliosis are pathological markers of SCI. Glial scars can be used as a barrier to prevent regeneration after SCI. Therefore, we evaluated whether Apoe−/− mice exhibit aggravated astrogliosis. The level of GFAP protein was performed by WBs and GFAP immunofluorescence staining was measured to assess the degree of astrogliosis at 1 mm caudal and 1 mm rostral to the epicenter of the lesion. No significant differences in the Apoe KO mice and WT mice at 7 dpi (Supplementary Figure S1). These results suggest that Apoe deficiency impedes the neurological function and histological outcomes after SCI.
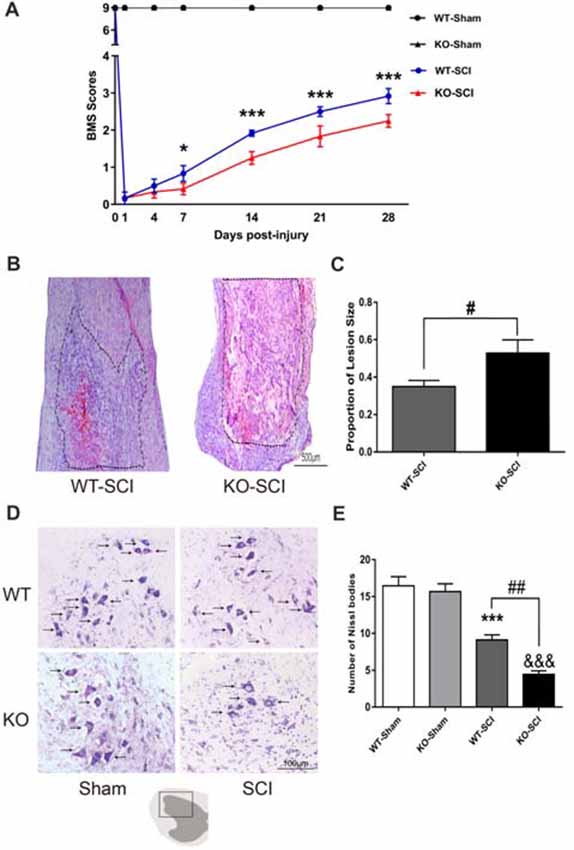
Figure 3. Apoe deficiency retards motor functional recovery and increases lesion area following SCI. (A) Statistical analysis of motor functional recovery of WT and Apoe KO mice before and after SCI using Basso Mouse Scale (BMS) scores. Data are expressed as mean ± SEM of seven independent experiments (n = 6 per group). *p < 0.05, ***p < 0.001, WT-SCI vs. KO-SCI, 7 days, p = 0.0493; 14 days, p = 0.0004; 21 days, p = 0.0004; 28 days, p = 0.0004, two-way ANOVA followed by Tukey’s post hoc test. (B) Representative micrograph of longitudinal spinal cord sections stained with H & E in both Apoe KO and WT mice at 7 days post-injury. The boundary of the lesion is outlined with a dotted line. (C) Quantitative analysis of the lesion area that determined from the length from injury site rostral and caudal 0.5 cm and expressed as percentage of lesion area per spinal cord analyzed. Four representative sections per animal (one of every ten serial sections) were stained. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). #p < 0.05, WT-SCI vs. KO-SCI, p = 0.0392, Student’s t-test, Scale Bars = 500 μm. (D) Representative sections showing normal neurons that contained prominent nucleoli, loose chromatin, and Nissl body (arrow). (E) Quantitative analysis of motor neurons in the ventral horn at 7 days post-injury. Four transverse sections were chosen at ±300 μm, ±600 μm from rostral to caudal around the epicenter were obtained from each animal. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). ***p < 0.001, WT-Sham vs. WT-SCI, p < 0.001; &&&p < 0.001, KO-Sham vs. KO-SCI, p < 0.001; ##p < 0.01, WT-SCI vs. KO-SCI, p = 0.0055, one-way ANOVA followed by Tukey’s post hoc test, Scale Bars = 100 μm.
Inflammatory cytokines, including IL-6 and IL-1β, aggravate cell death and inflammation after SCI (Keane et al., 2006; Oyinbo, 2011). We thus explored whether Apoe deficiency altered expression of these cytokines after SCI. As shown in Figure 4A, when compared with sham mice, the expression of lL-6 and IL-1β were increased in both WT and Apoe KO mice at 7 dpi (WT-Sham vs. WT-SCI, IL-6: 0.11 ± 0.05 vs. 0.38 ± 0.07, p < 0.05, IL-1β: 0.26 ± 0.02 vs. 0.93 ± 011, p < 0.05; KO-Sham vs. KO-SCI, IL-1β: 0.60 ± 0.09 vs. 1.77 ± 0.31, p < 0.001; IL-6: 0.13 ± 0.03 vs. 0.69 ± 0.11, p < 0.001, Figures 4A–C). Strikingly, the expression of these pro-inflammatory cytokines was higher in Apoe KO than in WT mice (WT-SCI vs. KO-SCI, IL-6: 0.38 ± 0.07 vs. 0.69 ± 0.11, p < 0.05, IL-1β: 0.93 ± 0.12 vs. 1.78 ± 0.31, p < 0.01, Figures 4A–C). Because activation of NF-κB promotes overproduction of inflammatory cytokines and further causes neuronal death after SCI, we examined the levels of p-NF-κB were higher in spinal cords of Apoe KO than WT mice at 7 dpi (WT-SCI vs. KO-SCI, 0.50 ± 0.08 vs. 0.82 ± 0.06, p < 0.05, Figures 4A,D). The other two representative WBs for IL-6, IL-1β and p-NF-κB are attached on Supplementary Figure S4. Taken together, these results support the hypothesis that Apoe deficiency induces larger production of inflammatory cytokines through activation of NF-κB following SCI.
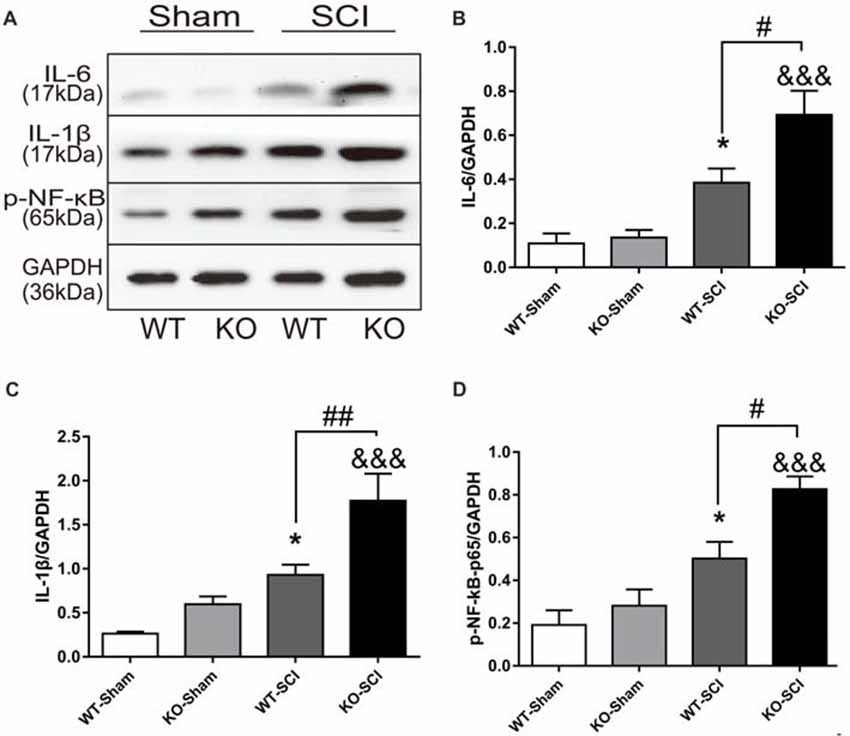
Figure 4. Apoe deficiency aggravates inflammatory response after SCI. (A) Representative WBs of inflammation-related proteins interleukin-6 (IL-6) and interleukin-1β (IL-1β), p-NF-κB and the loading control (GAPDH) in WT and Apoe KO groups at 7 days after SCI. (B–D) Quantitative analysis of IL-6, IL-1β and p-NF-κB expression. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). *p < 0.05, WT-Sham vs. WT-SCI, IL-6, p = 0.0459, IL-1β, p = 0.0465, p-NF-κB, p = 0.0223; &&&p < 0.001, KO-Sham vs. KO-SCI; IL-6, p < 0.001, IL-1β, p = 0.0002, p-NF-κB, p < 0.001; #p < 0.05, ##p < 0.01, WT-SCI vs. KO-SCI, IL-6, p = 0.0215, IL-1β, p = 0.0086, p-NF-κB, p = 0.0162, one-way ANOVA followed by Tukey’s post hoc test.
Oxidative stress plays a crucial role in defining neuronal cell damage after SCI (Visavadiya et al., 2016). Nrf2 is a transcription factor with a strong anti-oxidative activity and anti-inflammation effects that prevents neurons the death (Campolo et al., 2017). We examined the expression of Nrf2 and its downstream signaling protein HO-1 and NQO1 by WB in mice with SCI. As is shown in Figure 5A, there were no between-group differences in either sham group. In contrast, the ablation of Apoe was obviously associated with a decline in Nrf2 and HO-1 expression (WT-SCI vs. KO-SCI, Nrf2: 0.39 ± 0.04 vs. 0.03 ± 0.01, p < 0.001; HO-1: 0.59 ± 0.06 vs. 0.18 ± 0.02, p < 0.001, Figures 5A–C), although the expressed of NQO1 was not significant in Apoe KO and WT mice at 7 dpi. There are other two representative WBs for Nrf2, HO-1 and NQO-1 presented on Supplementary Figure S5. Based on these results, the deficiency of Apoe appears to decrease anti-oxidative response after SCI.
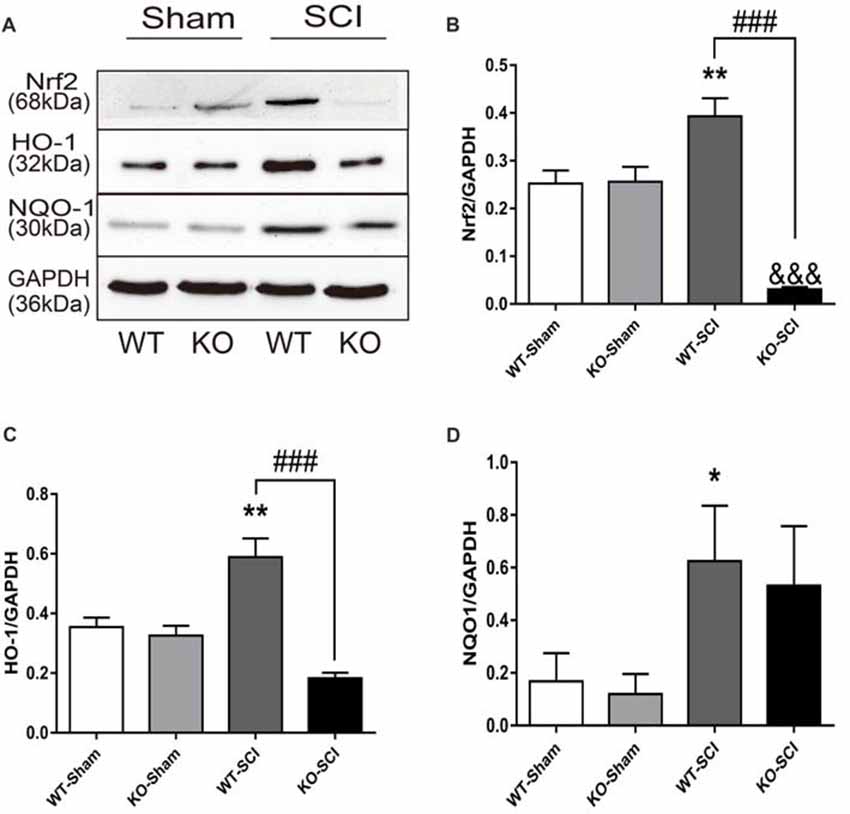
Figure 5. Apoe deficiency increases oxidative stress after SCI. (A) Representative WBs of antioxidative proteins Nrf2, HO-1, NQO1 and the loading control (GAPDH) in WT and Apoe KO groups at 7 days after SCI. (B–D) Quantitative analysis of Nrf2, HO-1 and NQO1 expression. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). *p < 0.05, **p < 0.01, WT-Sham vs. WT-SCI, Nrf2: p = 0.054, HO-1: p = 0.0013, NQO1: p = 0.0486; &&&p < 0.001, KO-Sham vs. KO-SCI, Nrf2: p < 0.0001, HO-1: p = 0.0768, NQO1: p = 0.6424; ###p < 0.001, WT-SCI vs. KO-SCI, Nrf2, p < 0.0001, HO-1, p < 0.0001, NQO1: p > 0.9999; one-way ANOVA followed by Tukey’s post hoc test. NQO1, Kruskal-Wallis test by Dunn’s post hoc test.
The overproduction of inflammatory mediators regulates neuronal survival, cell apoptosis, and locomotor functions after SCI (Yu and Fehlings, 2011; Cheng et al., 2016). Apoptosis occurs following SCI as a result of down-regulation of anti-apoptotic Bcl-2 and up-regulation of pro-apoptotic Bax, as well as activation of caspase-3 (Emery et al., 1998; Sastry and Rao, 2001). We observed that the levels of Bcl-2 did not differ in the spinal cord of two sham groups but was much lower in Apoe KO than in WT mice at 7 dpi (WT-SCI vs. KO-SCI, 0.45 ± 0.09 vs. 0.04 ± 0.02, p < 0.05, Figures 6A,B). Conversely, mice having Apoe deficiency has significantly enhanced Bax expression compared with the WT group at 7 dpi, while expression of Bax was weak in the two sham genotype groups (WT-SCI vs. KO-SCI, 0.94 ± 0.09 vs. 1.87 ± 0.26, p < 0.001, Figures 6A,D). To further assess the effects of APOE on neuronal apoptosis, double immunofluorescent staining was performed for Cleaved-caspase-3 and NeuN (Figure 6C). Compared with the WT group, stronger immunoreactivity of Cleaved-caspase 3-positive neurons was observed in the anterior horn of the injured spinal cord in Apoe KO group 7 days after injury (WT-SCI vs. KO-SCI, 0.11 ± 0.00 vs. 0.13 ± 0.00, p < 0.001, Figure 6E). Additional two representative WBs of Bcl2 and Bax are provided on Supplementary Figure S6. Thus, these results suggest that Apoe deficiency augments SCI-induced neuronal apoptosis.
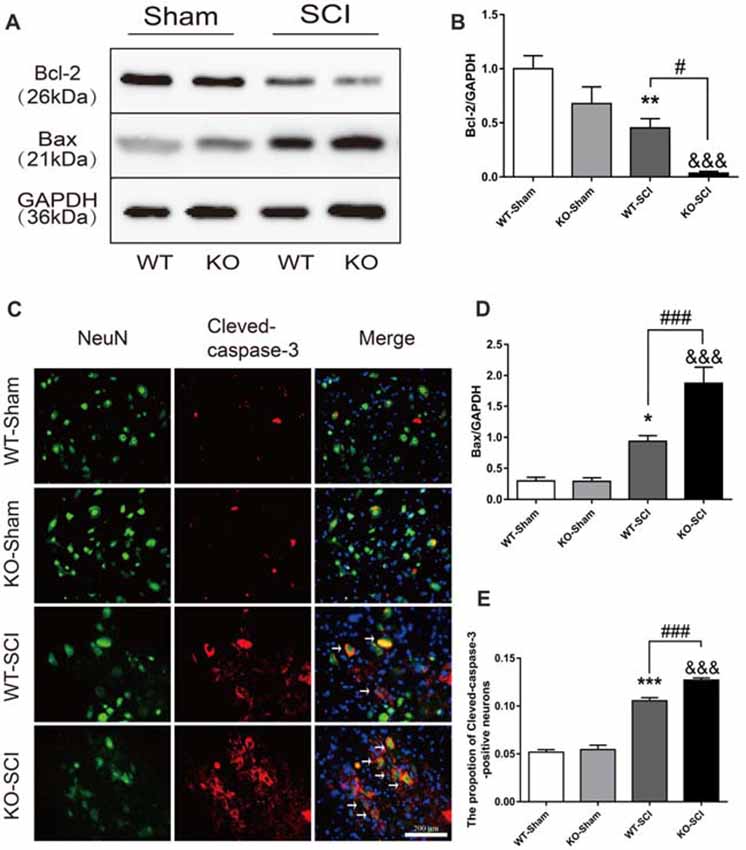
Figure 6. Apoe deficiency exacerbates neuronal apoptosis after SCI. (A) Representative WBs for Bcl-2 and Bax expressions and the loading control (GAPDH) in Apoe KO and WT group at 7 days post-injury. (B,D) Quantification of the expression levels of Bcl-2 and Bax. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). *p < 0.05, **p < 0.01, WT-Sham vs. WT-SCI, Bcl-2: p = 0.005, Bax: p = 0.0204, &&&p < 0.001, KO-Sham vs. KO-SCI, Bcl-2: p = 0.0008, Bax: p < 0.001, #p < 0.05, ###p < 0.001, WT-SCI vs. KO-SCI, Bcl-2: p = 0.0436, Bax: p = 0.0005, one-way ANOVA followed by Tukey’s post hoc test. (C) Representative double immunofluorescent staining for Cleaved-caspase-3-positive cells (red), NeuN (green), and DAPI (blue) in injured spinal cord of Apoe KO and WT mice at 7 days post-injury. (E) Quantification of Cleaved-caspase-3-positive cells co-labeled with NeuN to reveal apoptotic neurons. Four transverse sections were chosen at ±500 μm, ±800 μm rostral to caudal to the epicenter were obtained from each animal. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). ***p < 0.001, WT-Sham vs. WT-SCI, p < 0.0001, &&&p < 0.001, KO-Sham vs. KO-SCI, p < 0.0001, ###p < 0.001, WT-SCI vs. KO-SCI, p = 0.0003, one-way ANOVA followed by Tukey’s post hoc test. Scale bar = 100 μm.
To further investigate whether Apoe deficiency worsen secondary injury after SCI was dependent on the activation of NF-κB. PDTC, an inhibitor of the NF-κB signaling pathway, was used to block activator of NF-κB (Jiménez-Garza et al., 2005; Bell et al., 2014). WB analysis revealed that p-NF-κB protein level in both WT and KO groups with PDTC treatment was lower than in both the WT and KO groups with vehicle treatment (WT-SCI-Veh vs. WT-SCI-PDTC, 0.79 ± 0.08 vs. 0.39 ± 0.11, p < 0.05; KO-SCI-Veh vs. KO-SCI-PDTC, 1.26 ± 0.09 vs. 0.88 ± 0.1, p < 0.05, Figures 7A,B). In addition, the NF-κB protein level in WT-SCI-PDTC group was less than it in KO-SCI-PDTC (WT-SCI-PDTC vs. KO-SCI-PDTC, 0.39 ± 0.11 vs. 0.88 ± 0.11, p < 0.01 Figures 7A,B). Also, the expression of IL-6 was less in WT-SCI-PDTC than KO-SCI-PDTC, although the expression of IL-1β was not significance in both WT-SCI-PDTC and KO-SCI-PDTC (WT-SCI-PDTC vs. KO-SCI-PDTC, IL-6: 0.11 ± 0.01 vs. 0.28 ± 0.05, p < 0.01, IL-1β: 0.32 ± 0.03 vs. 0.60 ± 0.07, p > 0.05, Figures 7A,C,D). Because activation of Nrf2/ARE pathway also down-regulates NF-κB regulating inflammation (Mao et al., 2012; Wang et al., 2012), we analyzed whether Apoe deficiency increased inflammation by directly activation of NF-κB or indirectly via inactivation Nrf2-HO-1 pathway. Administration of PDTC treatment after SCI, the expression of Nrf2 and HO-1 in WT mice more increased than KO mice, but the expression of NQO-1 was no difference (WT-SCI-PDTC vs. KO-SCI-PDTC, Nrf2: 0.52 ± 0.03 vs. 0.18 ± 0.05, p < 0.001, HO-1: 0.89 ± 0.05 vs. 0.53 ± 0.07, p < 0.05; NQO-1: 0.61 ± 0.17 vs. 0.40 ± 0.13, p > 0.05, Figures 7E,H). The other twice representative WBs for related proteins are shown in Supplementary Figure S7. These data suggesting activation of NF-κB was indispensable for Apoe deficiency to enhance the release of pro-inflammation cytokines after SCI.
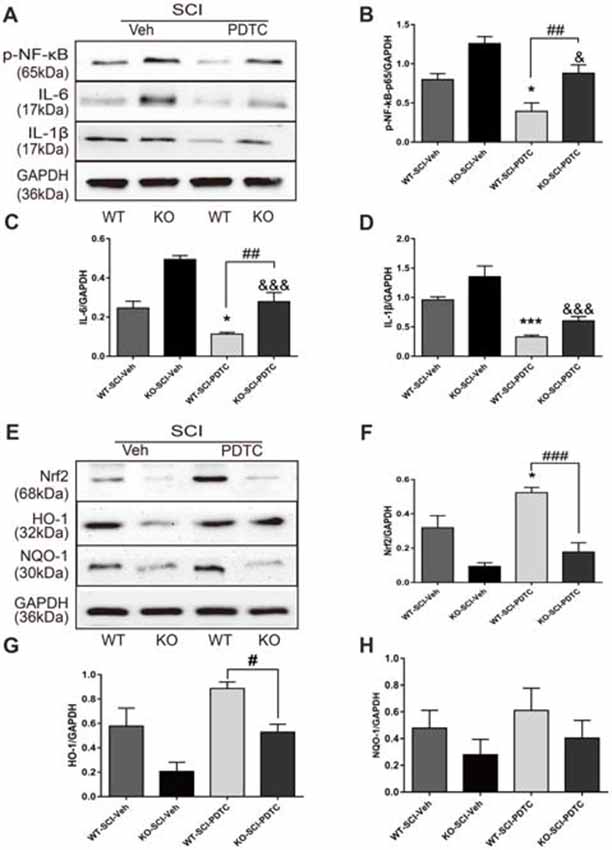
Figure 7. Apoe deficiency exacerbates inflammation by activating of NF-κB and inactivating of Nrf2 signaling pathways following SCI. (A) Representative immunoblots for p-NF–κB, IL-6, IL-1β and the loading control (GAPDH) in WT and Apoe KO groups at 7 days after SCI. (B–D) Quantitative levels of p-NF-κB, IL-6 and IL-1β in lesion area of Apoe KO and WT mice at 7 days post-injury. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). *p < 0.05, ***p < 0.001, WT-SCI-Veh vs. WT-SCI-PDTC, p-NF-κB: p = 0.0273, IL-6: p = 0.0193, IL-1β, p = 0.0005; &p < 0.05, &&&p < 0.001, KO-SCI-Veh vs. KO-SCI-PDTC, p-NF-κB: p = 0.0425, IL-6: p < 0.001, IL-1β: p < 0.0001; ##p < 0.01, ###p < 0.001, WT-SCI-PDTC vs. KO-SCI-PDTC, p-NF-κB: p = 0.0063, IL-6: p = 0.0022, IL-1β: p = 0.2549, one-way ANOVA followed by Tukey’s post hoc test. (E) Representative immunoblots for Nrf2, HO-1, NQO1 and the loading control (GAPDH) in WT and Apoe KO groups at 7 days after SCI. (F–H) Quantitative levels of Nrf2, HO-1, NQO1 in lesion area of Apoe KO and WT mice at 7 days post-injury. Data are expressed as mean ± SEM of three independent experiments (n = 6 per group). *p < 0.05, ***p < 0.001, WT-SCI-Veh vs. WT-SCI-PDTC, Nrf2: p = 0.0238, HO-1: p = 0.0234, NQO1: p > 0.05; &p < 0.05, KO-SCI-Veh vs. KO-SCI-PDTC, Nrf2: p = 0.6152, HO-1: p = 0.0455, NQO1: p > 0.05; #p < 0.05, ###p < 0.001, WT-SCI-PDTC vs. KO-SCI-PDTC, Nrf2: p < 0.001, HO-1: p = 0.0406, NQO1: p > 0.05, one-way ANOVA followed by Tukey’s post hoc test. WT-SCI and vehicle-treated group (WT-SCI-Veh), KO-SCI and vehicle-treated group (KO-SCI-Veh), WT-SCI and PDTC-treated (WT-SCI-PDTC), KO-SCI and PDTC-treated group (KO-SCI-PDTC).
To reveal the potential mechanisms through which APOE regulates SCI-induced inflammation and apoptosis in an extracellular dependent manner, we examined the activation of MAPK pathways (ERK, p-38MAPK, JNK), which are documented to regulate neuronal proliferation, inflammation, and apoptosis (Esposito et al., 2009). Interestingly, compared with sham group, p-EKR, p-p38 and p-JNK level were more elevated in WT mice at 7 days following injury (WT-Sham vs. WT-SCI, p-ERK: 0.59 ± 0.13 vs. 1.42 ± 0.19, p < 0.01; p-p38: 0.40 ± 0.14 vs. 0.77 ± 0.08, p < 0.01; p-JNK: 0.59 ± 0.07 vs. 1.19 ± 0.12, p < 0.05, Supplementary Figure S2). While the levels of p-JNK and p-p38 demonstrated not statistically significant in Apoe KO and WT mice at 7 days, Apoe KO mice displayed a tendency of increased levels compared with WT mice at 7 dpi (Supplementary Figure S8). Overall, these results demonstrate that MAPK pathways appear not to mediate the increased expression of inflammatory cytokines and apoptosis in Apoe KO mice after SCI.
In the present study, based on analyses of Apoe KO and WT mice with contused SCI, we demonstrate that Apoe deficiency exaggerates inflammation by increasing expression of NF-κB and inflammatory cytokines such as IL-6, IL-1β. In addition, Apoe deficiency reduces Nrf2-HO-1 pathway by activation of NF-κB signaling pathways. Furthermore, the depletion of Apoe increases neuronal apoptosis along with worsening functional outcome and increased lesion size. Here, we provide clear evidence that the mechanism of ablation of Apoe activated NF-κB and inactivated Nrf2-HO-1 pathway, which aggravates post-injury inflammation and oxidative stress. Our findings are important for traumatic SCI, in which Apoe deficiency triggers a more severe secondary cascade in the pathological process.
It is well-known that SCI is a destructive condition that irreversibly aggravates the primary injury, subsequently exacerbates the secondary damage, and leads to permanent neurological deficits (Zhao et al., 2017). This pathophysiology of secondary damage includes a series of biochemical events, including inflammation, oxidative stress, apoptosis and astroglial scar formation (Silva et al., 2014; Ahuja et al., 2017). The concept of early secondary injury mechanism is divided into immediate (<2 h), early acute (<48 h) and sub-acute (<2 weeks) stage (Rowland et al., 2008). Although there is no cure for initial traumatic injury directly damaged to spinal cord tissues, effective restraint of secondary injury plays a key role in reducing disability (Sun et al., 2016). Therefore, early therapies directed against multiple secondary damage mechanisms can be more effective to promote functional recovery after SCI. The function of APOE has drawn increasing attention, because its involvement in anti-inflammatory responses, anti-oxidative stress, and anti-apoptotic effects (Laskowitz et al., 1997; Kitagawa et al., 2002; Jiang et al., 2015), we hypothesized that its functions may block or reduce the secondary damage and improve functional recovery by NF-κB activation after SCI.
APOE plays a crucial role in modulating neuronal repair, remodeling, and protection after CNS trauma and disease (Yin et al., 2012; Saul and Wirths, 2017; Teng et al., 2017). During nerve regeneration, APOE is involved in the mobilization and reutilization of myelin-derived cholesterol for axon repair (Mahley, 1988; Li et al., 2010). Strikingly, the APOE is overexpressed in the spinal cord after SCI (Resnick et al., 2004) and serum after chronic constriction injury (Bellei et al., 2017). We found that after acute SCI in mice, APOE levels first decreased in the first 3 days, but then increased to a peak expression at 7 dpi (Figure 1A). It is well-acknowledged that activated macrophages and glia released pro-inflammatory cytokines were elevated between 30 min and 48 h following early acute SCI (Donnelly and Popovich, 2008). Several lines of evidence suggest pro-inflammatory responses decrease APOE synthesis in macrophages and glia (Gafencu et al., 2007; Yang et al., 2016). We found the APOE production gradually increased and reached the highest levels at 7 dpi. One possible function of the increased APOE production could probably suppress the inflammation in the pathological process of acute sub-stages of SCI. Endogenous APOE and exogenous APOE mimetic peptides reduced glial activation and inflammatory cytokines release after CNS disease. Interestingly, blocking inflammatory signaling effectively increases APOE expression in neuroinflammatory processes (Pocivavsek and Rebeck, 2009; Mailleux et al., 2017). These evidences suggest that APOE production and inflammation are in a negative feedback loop, with APOE suppressing inflammation and inflammation suppressing APOE production. Therefore, APOE affects changes in the inflammatory response in the pathogenesis of SCI.
Similar to findings that Apoe KO limits functional recovery and enlarges tissue damage in various types of nerve trauma (Herz et al., 2014; Pang et al., 2016), we and Cheng et al. (2018) have demonstrated that Apoe deficiency leads to more worsening anatomical and functional deficits in a mouse model of SCI. Moreover, accumulating studies have demonstrated that APOE mimic peptide could reduce apoptosis and neuroinflammation in many neurological diseases (Laskowitz et al., 2006; Wei et al., 2013; Wu et al., 2016). In addition, APOE4, is well known as the strongest genetic risk factor of Alzheimer’s disease (Shi et al., 2017; Underwood, 2017). Also, APOE4 is linked with a worse neurological outcome and longer rehabilitation in patients with SCI (Jha et al., 2008). We had found APOE4 mice have poorer locomotion performance than WT mice after SCI (Data were unshown). Notably, we found changes in the profile of endogenous APOE protein are closely related to the impaired neurological function following SCI. The behavioral function of Apoe KO mice was significantly poorer than that of WT mice when the APOE reached its high levels at 7 dpi (Figure 3A). Consistent with the findings that functional recovery after SCI correlates with the lesion area in the injury epicenter (Basso et al., 1996), we also observed that Apoe KO mice harbored larger lesion area and lower number of motor neurons than WT mice after SCI (Figures 3B–E). These histological outcomes were consistent with the functional deterioration, which indicates that APOE is important in neurologic recovery after SCI.
Although complex mechanisms mediate the severity of secondary injury after SCI, several studies have shown that overproduction of inflammatory cytokines, including lL-6 and IL-1β secreted by activated microglia and macrophages is harmful to the injured spinal cord, because they induce neuronal cell death and hinder recovery of motor function (Anwar et al., 2016; Rust and Kaiser, 2017). It is well-known that inhibiting NF-κB activation could regulate locomotive recovery through reduced lesion volume and alteration of the inflammatory environment (La Rosa et al., 2004; Brambilla et al., 2005; Bracchi-Ricard et al., 2013). We show that the absence of Apoe significantly increased the release of inflammatory cytokines (IL-6 and IL-1β) and activator of NF-κB after SCI. Previous studies have also shown the Apoe-deficiency and APOE4 up-regulated greater proinflammatory cytokines after stimulation with LPS (Lynch et al., 2001, 2003; Ali et al., 2005). Furthermore, several evidences reported that APOE4 affected NF-κB-mediated gene transcription that triggered a significant elevation of inflammation cytokines (Ophir et al., 2005; Shih et al., 2015; Theendakara et al., 2016). To analyze the role of the NF-κB signaling pathway in Apoe deficiency-induced inflammation of injured spinal cord, PDTC, an inhibitor of NF-κB nuclear translocation, was used (Bell et al., 2014). PDTC treatment resulted in a decrease of activation of NF-κB and release of inflammatory mediators in both WT-SCI-PDTC and KO-SCI-PDTC groups, indicating PDTC succeeded in suppressing activation of NF-κB. In addition, Apoe deficiency more increase the expression of p-NF-κB than WT mice with PDTC treatment after SCI (Figure 7). These findings support previous studies that APOE may reduce inflammation at the transcriptional level in NFκB-dependent manner. Thus, our results find that the ablation of Apoe increases inflammatory response by NF-κB pathway after SCI.
Also, a neuroprotective role of Nrf2, a redox-sensitive transcription factor, decrease inflammation, oxidative stress and apoptosis after SCI (Pomeshchik et al., 2014). However, studies of associations between APOE and Nrf2 in CNS have not generated clear conclusions. Pomeshchik et al. (2014) has shown that Nrf2 expression reached a peak in WT mice after SCI 7 days. And this study found APOE was significantly up-regulated and reached the highest level at the same time with Nrf2 expression. One possible reason is the increased APOE with the anti-oxidant properties may enhance the Nrf2 production. Apoe KO mice with diabetic gastroparesis demonstrated that decreased expression of Nrf2 elevated oxidative stress (Ravella et al., 2012). In addition, Graeser et al. (2011) have shown that Nrf2 has different regulatory effects on Apoe genotypes, such as APOE4 reduces the protective role of Nrf2 in cardiovascular disease. Strikingly, as our results show in Figure 5, deficiency of Apoe decreased the Nrf2 signaling pathway after SCI. Furthermore, accumulating evidences shown that activation of NF-κB could directly repress Nrf2 signaling at the transcriptional level (Li et al., 2008; Yu et al., 2011). Previous researchers pointed that PDTC is a potent inducer of Nrf2 and its anti-oxidative targets in astrocytes but not neurons (Liddell et al., 2016). But the inactivation of NF-κB regulating Nrf2 pathway is elusive and rarely addressed in SCI. In the current study, PDTC-mediated NF-κB inhibition elevated Nrf2 and HO-1 level in WT mice after SCI. Of note, Apoe−/− mice treated with PDTC administration expressed Nrf2-HO-1 still lower than WT mice after SCI. On the other hand, PDTC had no apparent effect on Nrf2 and HO-1 in Apoe−/− mice after SCI (Figure 7). Nrf2 signaling pathways may not be directly associated with NF-κB activating inflammation induced by Apoe deficiency in SCI, but various signaling pathways regulate the Apoe is associated with Nrf2 which needs to be further investigated.
An increase in the levels of detrimental inflammatory mediators contributes to neuronal apoptosis and neurologic insult after SCI (Brambilla et al., 2005). Recent publications demonstrate that reducing neuronal apoptosis after SCI may be a favorable therapeutic strategy (Coll-Miró et al., 2016; Pei et al., 2017). It has been reported Bcl-2 protein possesses anti-apoptosis, and Bax is involved in inducing cell apoptosis (Wei et al., 2001; Bai et al., 2017). Also, Caspase-3, apoptosis-related protein markers, is activated by apoptotic stimuli and essential for apoptotic chromatin condensation and DNA (Cheng et al., 2001; Jiang et al., 2016; Xu X. et al., 2017). It is well-known that APOE was linked to neuronal apoptosis (Mahley et al., 2006; Krasemann et al., 2017). For example, APOE-containing lipoproteins binding to the low-density lipoprotein receptor-related protein prevents neuronal apoptosis (Hayashi et al., 2007). Our results show that mice lacking Apoe had increased apoptosis-related proteins Bax and Cleaved-caspase-3 and decreased anti-apoptotic protein Bcl-2 after SCI (Figure 6). Therefore, our conclusion is consistent with previous observations that Apoe deficiency up-regulates neuronal apoptosis after SCI. Yet, the exact mechanism by which Apoe is associated with SCI-induced neuronal apoptosis through increased inflammation needs further evaluation.
In addition, the linkage between MAPK signaling pathway and neuroprotective effects of APOE in SCI remains uncertain. A recent study shown that SCI induces activated phosphorylation of MAPK (Riego et al., 2018). Inhibition of JNK, ERK and P38 have shown that inflammation is alleviated in different animal models of pain (Ji et al., 2010). Of note, the ERK1/2 plays an important role in the activation of ApoE receptors and purified APOE3 and a tandem APOE peptide -mediated neurite outgrowth (Qiu et al., 2004). APOE receptor interaction promotes neural stem cell survival by ERK signaling, and APOE is required for oligodendrogenesis (Gan et al., 2011). However, our results of p-ERK1/2, p-p38 and p-JNK has no apparent difference in Apoe KO and WT mice after (Supplementary Figure S2), which seem not to support previous findings that APOE isoform-dependent inflammatory responses include NF-κB and MAPK pathways (Dose et al., 2016). A possible functional discrepancy between Apoe and its isoforms may explain these results. We will therefore explore the effects of Apoe isoforms on inflammation post-SCI in our future studies. Besides, several pieces of evidence demonstrate that APOE plays a key role in suppressing inflammation and promoting neural cell proliferation and survival by binding to its ligand (Safina et al., 2016). Because secondary damage alleviation by APOE binding to its receptor was not investigated in this study, further studies should be conducted to address this issue using appropriate techniques.
We also noted that inhibition of NF-κB leads to the histological improvement and function recovery after SCI (Church et al., 2017; He et al., 2017; Machova Urdzikova et al., 2017; Xu J. et al., 2017). For instance, selective inhibition of NF-κB signaling in astrocytes contributes to protective effects after SCI (Brambilla et al., 2005; Bracchi-Ricard et al., 2013). La Rosa et al. (2004) suggested that treatment with PDTC (30 mg/kg) significantly reduced the motor disturbance at SCI 7 days. Therefore, one limitation of the present study is that function recovery by which PDTC treatment for inhibiting the NF-κB regulated inflammation in Apoe−/− mice after SCI, was not elaborated. Further study is crucial to elucidate this issue.
In conclusion, these findings demonstrate that: (i) Apoe deficiency could aggravate locomotor loss and tissue damage following SCI in mice; (ii) Apoe deficiency could up-regulate inflammatory cascades by NF-κB activation, and inhibiting Nrf2-HO-1 anti-oxidative pathway; and (iii) Apoe deficiency could exacerbate neuronal apoptosis following SCI. Although the complex mechanism by which Apoe knockout worsen secondary injury is not clear in this study, and at least in part through inflammatory regulation of NF-κB signaling. Apoe-mediated NF-κB signaling may offer a new approach to various pharmacological targets for SCI.
XY and SC wrote the main manuscript. ZS and XL performed research. SC, XL and HW collected samples. ZS and YL analyzed data. YL and CL discussed results and prepared figures. XY, CL and XM designed experiment. LM, ZZ and LB revised manuscript.
This work was supported by the National Natural Science Foundation of China (NSFC; Grant Nos. 31471138, 81671907, 81701898).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We sincerely thank all the researchers for their valuable technical assistance and the writing of the article. We also thank Department of Neurobiology and Key Laboratory of Neurodegenerative Diseases of Liaoning Province, Jinzhou Medical University, for experimental equipment.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2018.00142/full#supplementary-material
Ahuja, C. S., Nori, S., Tetreault, L., Wilson, J., Kwon, B., Harrop, J., et al. (2017). Traumatic spinal cord injury—repair and regeneration. Neurosurgery 80, S9–S22. doi: 10.1093/neuros/nyw080
Ali, K., Middleton, M., Puré, E., and Rader, D. J. (2005). Apolipoprotein E suppresses the type I inflammatory response in vivo. Circ. Res. 97, 922–927. doi: 10.1161/01.res.0000187467.67684.43
Anwar, M. A., Al Shehabi, T. S., and Eid, A. H. (2016). Inflammogenesis of secondary spinal cord injury. Front. Cell. Neurosci. 10:98. doi: 10.3389/fncel.2016.00098
Bai, L., Mei, X., Wang, Y., Yuan, Y., Bi, Y., Li, G., et al. (2017). The role of netrin-1 in improving functional recovery through autophagy stimulation following spinal cord injury in rats. Front. Cell. Neurosci. 11:350. doi: 10.3389/fncel.2017.00350
Bartus, K., Galino, J., James, N. D., Hernandez-Miranda, L. R., Dawes, J. M., Fricker, F. R., et al. (2016). Neuregulin-1 controls an endogenous repair mechanism after spinal cord injury. Brain 139, 1394–1416. doi: 10.1093/brain/aww039
Basso, T., Beattie, M. S., and Bresnahan, J. C. (1996). Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp. Neurol. 256, 244–256. doi: 10.1006/exnr.1996.0098
Basso, D. M., Fisher, L. C., Anderson, A. J., Jakeman, L. Y. N. B., Tigue, D. M. M. C., and Popovich, P. G. (2006). Basso mouse scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma 23, 635–659. doi: 10.1089/neu.2006.23.635
Bell, R. D., Winkler, E. A., Singh, I., Sagare, A. P., Deane, R., Wu, Z., et al. (2014). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 21, 59–69. doi: 10.1038/nature11087
Bellei, E., Vilella, A., Monari, E., Bergamini, S., Tomasi, A., Cuoghi, A., et al. (2017). Serum protein changes in a rat model of chronic pain show a correlation between animal and humans. Sci. Rep. 7:41723. doi: 10.1038/srep41723
Bethea, J. R., Castro, M., Keane, R. W., Lee, T. T., Dietrich, W. D., and Yezierski, R. P. (1998). Traumatic spinal cord injury induces nuclear factor-κB activation. J. Neurosci. 18, 3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998
Bracchi-Ricard, V., Lambertsen, K. L., Ricard, J., Nathanson, L., Karmally, S., Johnstone, J., et al. (2013). Inhibition of astroglial NF-κB enhances oligodendrogenesis following spinal cord injury. J. Neuroinflammation 10:92. doi: 10.1186/1742-2094-10-92
Brambilla, R., Bracchi-Ricard, V., Hu, W.-H., Frydel, B., Bramwell, A., Karmally, S., et al. (2005). Inhibition of astroglial nuclear factor δB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 202, 145–156. doi: 10.1084/jem.20041918
Bu, G. (2010). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Campolo, M., Casili, G., Biundo, F., Crupi, R., Cordaro, M., Cuzzocrea, S., et al. (2017). The neuroprotective effect of dimethyl fumarate in an MPTP-mouse model of Parkinson’s disease: involvement of reactive oxygen species/nuclear factor-κB/nuclear transcription factor related to NF-E2. Antioxid. Redox Signal. 27, 453–471. doi: 10.1089/ars.2016.6800
Cheng, P., Kuang, F., and Ju, G. (2016). Aescin reduces oxidative stress and provides neuroprotection in experimental traumatic spinal cord injury. Free Radic. Biol. Med. 99, 405–417. doi: 10.1016/j.freeradbiomed.2016.09.002
Cheng, E. H. A.-Y. A., Wei, M. C., Weiler, S., Flavell, R. A., Mak, T. W., Lindsten, T., et al. (2001). BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8, 705–711. doi: 10.1016/s1097-2765(01)00320-3
Cheng, X., Zheng, Y., Bu, P., Qi, X., Fan, C., Li, F., et al. (2018). Apolipoprotein E as a novel therapeutic neuroprotection target after traumatic spinal cord injury. Exp. Neurol. 299, 97–108. doi: 10.1016/j.expneurol.2017.10.014
Church, J. S., Milich, L. M., Lerch, J. K., Popovich, P. G., and McTigue, D. M. (2017). E6020, a synthetic TLR4 agonist, accelerates myelin debris clearance, Schwann cell infiltration, and remyelination in the rat spinal cord. Glia 65, 883–899. doi: 10.1002/glia.23132
Coll-Miró, M., Francos-Quijorna, I., Santos-Nogueira, E., Torres-Espin, A., Bufler, P., Dinarello, C. A., et al. (2016). Beneficial effects of IL-37 after spinal cord injury in mice. Proc. Natl. Acad. Sci. U S A 113, 1411–1416. doi: 10.1073/pnas.1523212113
Donnelly, D. J., and Popovich, P. G. (2008). Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp. Neurol. 209, 378–388. doi: 10.1016/j.expneurol.2007.06.009
Dose, J., Huebbe, P., Nebel, A., and Rimbach, G. (2016). APOE genotype and stress response—a mini review. Lipids Health Dis. 15:121. doi: 10.1186/s12944-016-0288-2
Elliott, D. A., Kim, W. S., Jans, D. A., and Garner, B. (2007). Apoptosis induces neuronal apolipoprotein-E synthesis and localization in apoptotic bodies. Neurosci. Lett. 416, 206–210. doi: 10.1016/j.neulet.2007.02.014
Emery, E., Aldana, P., Bunge, M. B., Puckett, W., Srinivasan, A., Keane, R. W., et al. (1998). Apoptosis after traumatic human spinal cord injury. J. Neurosurg. 89, 911–920. doi: 10.3171/jns.1998.89.6.0911
Esposito, E., Genovese, T., Caminiti, R., Bramanti, P., Meli, R., and Cuzzocrea, S. (2009). Melatonin reduces stress-activated/mitogen-activated protein kinases in spinal cord injury. J. Pineal Res. 46, 79–86. doi: 10.1111/j.1600-079X.2008.00633.x
Gafencu, A. V., Robciuc, M. R., Fuior, E., Zannis, V. I., Kardassis, D., and Simionescu, M. (2007). Inflammatory signaling pathways regulating ApoE gene expression in macrophages. J. Biol. Chem. 282, 21776–21785. doi: 10.1074/jbc.M611422200
Gallardo, G., Schlüter, O. M., and Südhof, T. C. (2008). A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat. Neurosci. 11, 301–308. doi: 10.1038/nn2058
Gan, H. T., Tham, M., Hariharan, S., Ramasamy, S., Yu, Y. H., and Ahmed, S. (2011). Identification of ApoE as an autocrine/paracrine factor that stimulates neural stem cell survival via MAPK/ERK signaling pathway. J. Neurochem. 117, 565–578. doi: 10.1111/j.1471-4159.2011.07227.x
Graeser, A. C., Boesch-Saadatmandi, C., Lippmann, J., Wagner, A. E., Huebbe, P., Storm, N., et al. (2011). Nrf2-dependent gene expression is affected by the proatherogenic apoE4 genotype-studies in targeted gene replacement mice. J. Mol. Med. 89, 1027–1035. doi: 10.1007/s00109-011-0771-1
Hayashi, H., Campenot, R. B., Vance, D. E., and Vance, J. E. (2007). Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J. Neurosci. 27, 1933–1941. doi: 10.1523/JNEUROSCI.5471-06.2007
Hayashi, H., Campenot, R. B., Vance, D. E., and Vance, J. E. (2009). Protection of neurons from apoptosis by Apolipoprotein E-containing lipoproteins does not require lipoprotein uptake and involves activation of phospholipase Cγ1 and inhibition of calcineurin. J. Biol. Chem. 284, 29605–29613. doi: 10.1074/jbc.M109.039560
He, Z., Zhou, Y., Lin, L., Wang, Q., Khor, S., Mao, Y., et al. (2017). Dl-3-n-butylphthalide attenuates acute inflammatory activation in rats with spinal cord injury by inhibiting microglial TLR4/NF-κB signalling. J. Cell. Mol. Med. 21, 3010–3022. doi: 10.1111/jcmm.13212
Herz, J., Hagen, S. I., Bergmüller, E., Sabellek, P., Göthert, J. R., Buer, J., et al. (2014). Exacerbation of ischemic brain injury in hypercholesterolemic mice is associated with pronounced changes in peripheral and cerebral immune responses. Neurobiol. Dis. 62, 456–468. doi: 10.1016/j.nbd.2013.10.022
Hong, L. T. A., Kim, Y. M., Park, H. H., Hwang, D. H., Cui, Y., Lee, E. M., et al. (2017). An injectable hydrogel enhances tissue repair after spinal cord injury by promoting extracellular matrix remodeling. Nat. Commun. 8:533. doi: 10.1038/s41467-017-00583-8
Iwata, A., Browne, K. D., Chen, X.-H., Yuguchi, T., and Smith, D. H. (2005). Traumatic brain injury induces biphasic upregulation of ApoE and ApoJ protein in rats. J. Neurosci. Res. 82, 103–114. doi: 10.1002/jnr.20607
Jha, A., Lammertse, D. P., Coll, J. R., Charlifue, S., Coughlin, C. T., Whiteneck, G. G., et al. (2008). Apolipoprotein E epsilon4 allele and outcomes of traumatic spinal cord injury. J. Spinal Cord Med. 31, 171–176. doi: 10.1080/10790268.2008.11760708
Jiang, X., Li, L., Ying, Z., Pan, C., Huang, S., Li, L., et al. (2016). A small molecule that protects the integrity of the electron transfer chain blocks the mitochondrial apoptotic pathway. Mol. Cell 63, 229–239. doi: 10.1016/j.molcel.2016.06.016
Jiang, L., Zhong, J., Dou, X., Cheng, C., Huang, Z., and Sun, X. (2015). Effects of ApoE on intracellular calcium levels and apoptosis of neurons after mechanical injury. Neuroscience 301, 375–383. doi: 10.1016/j.neuroscience.2015.06.005
Ji, R. R., Gereau, R. W., IV., Malcangio, M., and Strichartz, G. R. (2010). MAP kinase and pain. Brain Res. Rev. 60, 135–148. doi: 10.1016/j.brainresrev.2008.12.011
Jiménez-Garza, O., Camacho, J., Ibarra, A., Martínez, A., and Guízar-Sahagún, G. (2005). Early effects of modulating nuclear factor-δB activation on traumatic spinal cord injury in rats. Ann. N Y Acad. Sci. 1053, 148–150. doi: 10.1196/annals.1344.012
Kanninen, K. M., Pomeshchik, Y., Leinonen, H., Malm, T., Koistinaho, J., and Levonen, A. L. (2015). Applications of the Keap1-Nrf2 system for gene and cell therapy. Free Radic. Biol. Med. 88, 350–361. doi: 10.1016/j.freeradbiomed.2015.06.037
Keane, R. W., Davis, A. R., and Dietrich, W. D. (2006). Inflammatory and apoptotic signaling after spinal cord injury. J. Neurotrauma 23, 335–344. doi: 10.1089/neu.2006.23.335
Kim, E., Woo, M.-S., Qin, L., Ma, T., Beltran, C. D., Bao, Y., et al. (2015). Daidzein augments cholesterol homeostasis via ApoE to promote functional recovery in chronic stroke. J. Neurosci. 35, 15113–15126. doi: 10.1523/JNEUROSCI.2890-15.2015
Kitagawa, K., Matsumoto, M., Kuwabara, K., Takasawa, K., Tanaka, S., Sasaki, T., et al. (2002). Protective effect of apolipoprotein E against ischemic neuronal injury is mediated through antioxidant action. J. Neurosci. Res. 68, 226–232. doi: 10.1002/jnr.10209
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566.e9–581.e9. doi: 10.1016/j.immuni.2017.08.008
Lang, B. T., Cregg, J. M., DePaul, M. A., Tran, A. P., Xu, K., Dyck, S. M., et al. (2015). Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature 518, 404–408. doi: 10.1038/nature13974
La Rosa, G., Cardali, S., Genovese, T., Conti, A., Di Paola, R., La Torre, D., et al. (2004). Inhibition of the nuclear factor-κB activation with pyrrolidine dithiocarbamate attenuating inflammation and oxidative stress after experimental spinal cord trauma in rats. J. Neurosurg. 3, 311–321. doi: 10.3171/spi.2004.1.3.0311
Laskowitz, D. T., Goel, S., Bennett, E. R., and Matthew, W. D. (1997). Apolipoprotein E suppresses glial cell secretion of TNFα. J. Neuroimmunol. 76, 70–74. doi: 10.1016/s0165-5728(97)00021-0
Laskowitz, D. T., Horsburgh, K., and Roses, A. D. (1998). Apolipoprotein E and the CNS response to injury. J. Cereb. Blood Flow Metab. 18, 465–471. doi: 10.1097/00004647-199805000-00001
Laskowitz, D. T., Fillit, H., Yeung, N., Toku, K., and Vitek, M. P. (2006). Apolipoprotein E-derived peptides reduce CNS inflammation: implications for therapy of neurological disease. Acta Neurol. Scand. 114, 15–20. doi: 10.1111/j.1600-0404.2006.00680.x
Li, J., Chen, S., Zhao, Z., Luo, Y., Hou, Y., and Li, H. (2017). Effect of VEGF on inflammatory regulation, neural survival, and functional improvement in rats following a complete spinal cord transection. Front. Cell. Neurosci. 11:381. doi: 10.3389/fncel.2017.00381
Li, X.-Z., Dinga, Y.-Z., Wu, H.-F., Bian, Z. P., Xu, J. D., Gu, C. R., et al. (2017). Astragaloside IV prevents cardiac remodeling in the Apolipoprotein E-deficient mice by regulating cardiac homeostasis and oxidative stress. Cell. Physiol. Biochem. 44, 2422–2438. doi: 10.1159/000486166
Li, F.-Q., Fowler, K. A., Neil, J. E., Colton, C. A., and Vitek, M. P. (2010). An apolipoprotein E-mimetic stimulates axonal regeneration and remyelination after peripheral nerve injury. J. Pharmacol. Exp. Ther. 334, 106–115. doi: 10.1124/jpet.110.167882
Li, W., Khor, T. O., Xu, C., Shen, G., Jeong, W., Yu, S., et al. (2008). Activation of Nrf2-antioxidant signaling attenuates NFδB-inflammatory response and elicits apoptosis §. Biochem. Pharmacol. 76, 1485–1489. doi: 10.1016/j.bcp.2008.07.017
Li, L., Oppenheim, R. W., Lei, M., and Houenou, L. J. (1994). Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J. Neurobiol. 25, 759–766. doi: 10.1002/neu.480250702
Liu, C.-C., Zhao, N., Fu, Y., Wang, N., Linares, C., Tsai, C.-W., et al. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron 96, 1024.e3–1032.e3. doi: 10.1016/j.neuron.2017.11.013
Liddell, J. R., Lehtonen, S., Duncan, C., Keksa-Goldsteine, V., Levonen, A.-L., Goldsteins, G., et al. (2016). Pyrrolidine dithiocarbamate activates the Nrf2 pathway in astrocytes. J. Neuroinflammation 13:49. doi: 10.1186/s12974-016-0515-9
Lu, M., Wang, S., Han, X., and Lv, D. (2013). Butein inhibits NF-κB activation and reduces infiltration of inflammatory cells and apoptosis after spinal cord injury in rats. Neurosci. Lett. 542, 87–91. doi: 10.1016/j.neulet.2013.03.004
Lynch, J. R., Morgan, D., Mance, J., Matthew, W. D., and Laskowitz, D. T. (2001). Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J. Neuroimmunol. 114, 107–113. doi: 10.1016/s0165-5728(00)00459-8
Lynch, J. R., Tang, W., Wang, H., Vitek, M. P., Bennett, E. R., Sullivan, P. M., et al. (2003). APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 278, 48529–48533. doi: 10.1074/jbc.m306923200
Ma, M., Basso, D. M., Walters, P., Stokes, B. T., and Jakeman, L. B. (2001). Behavioral and histological outcomes following graded spinal cord contusion injury in the C57Bl/6 mouse. Exp. Neurol. 169, 239–254. doi: 10.1006/exnr.2001.7679
Machova Urdzikova, L., Ruzicka, J., Karova, K., Kloudova, A., Svobodova, B., Amin, A., et al. (2017). A green tea polyphenol epigallocatechin-3-gallate enhances neuroregeneration after spinal cord injury by altering levels of inflammatory cytokines. Neuropharmacology 126, 213–223. doi: 10.1016/j.neuropharm.2017.09.006
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630. doi: 10.1126/science.3283935
Mahley, R. W., and Huang, Y. (2012). Apolipoprotein E sets the stage: response to injury triggers neuropathology. Neuron 76, 871–885. doi: 10.1016/j.neuron.2012.11.020
Mahley, R. W., Weisgraber, K. H., and Huang, Y. (2006). Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 103, 5644–5651. doi: 10.1073/pnas.0600549103
Mailleux, J., Timmermans, S., Nelissen, K., Vanmol, J., Vanmierlo, T., van Horssen, J., et al. (2017). Low-density lipoprotein receptor deficiency attenuates neuroinflammation through the induction of Apolipoprotein E. Front. Immunol. 8:1701. doi: 10.3389/fimmu.2017.01701
Mao, L., Wang, H., Qiao, L., and Wang, X. (2010). Disruption of Nrf2 enhances the upregulation of nuclear factor-κB activity, tumor necrosis factor- α, and matrix metalloproteinase-9 after spinal cord injury in mice. Mediators Inflamm. 2010:238321. doi: 10.1155/2010/238321
Mao, L., Wang, H., Wang, X., Tian, L., and Xu, J. Y. (2012). Disruption of Nrf2 exacerbated the damage after spinal cord injury in mice. J. Trauma Acute Care Surg. 72, 189–198. doi: 10.1097/TA.0b013e31821bf541
Narang, A., Qiao, F., Atkinson, C., Zhu, H., Yang, X., Kulik, L., et al. (2017). Natural IgM antibodies that bind neoepitopes exposed as a result of spinal cord injury, drive secondary injury by activating complement. J. Neuroinflammation 14:120. doi: 10.1186/s12974-017-0894-6
Nuriel, T., Peng, K. Y., Ashok, A., Dillman, A. A., Mathews, H. Y., Apuzzo, J., et al. (2017). The endosomal—lysosomal pathway is dysregulated by APOE4 expression in vivo. Front. Neurosci. 11:702. doi: 10.3389/fnins.2017.00702
Ophir, G., Amariglio, N., Jacob-Hirsch, J., Elkon, R., Rechavi, G., and Michaelson, D. M. (2005). Apolipoprotein E4 enhances brain inflammation by modulation of the NF-κB signaling cascade. Neurobiol. Dis. 20, 709–718. doi: 10.1016/j.nbd.2005.05.002
Oyinbo, C. A. (2011). Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol. Exp. 71, 281–299.
Pang, J., Wu, Y., Peng, J., Yang, P., Kuai, L., Qin, X., et al. (2016). Potential implications of Apolipoprotein E in early brain injury after experimental subarachnoid hemorrhage: involvement in the modulation of blood-brain barrier integrity. Oncotarget 7, 56030–56044. doi: 10.18632/oncotarget.10821
Pei, J., Fan, L., Nan, K., Li, J., Dang, X., and Wang, K. (2017). HSYA alleviates secondary neuronal death through attenuating oxidative stress, inflammatory response, and neural apoptosis in SD rat spinal cord compression injury. J. Neuroinflammation 14:97. doi: 10.1186/s12974-017-0870-1
Pocivavsek, A., and Rebeck, G. W. (2009). Inhibition of c-Jun N-terminal kinase increases apoE expression in vitro and in vivo. Biochem. Biophys. Res. Commun. 387, 516–520. doi: 10.1016/j.bbrc.2009.07.048
Pomeshchik, Y., Kidin, I., Savchenko, E., Rolova, T., Yamamoto, M., Levonen, A.-L., et al. (2014). Does Nrf2 gene transfer facilitate recovery after contusion spinal cord injury? Antioxid. Redox Signal. 20, 1313–1323. doi: 10.1089/ars.2013.5453
Qiu, Z., Hyman, B. T., and Rebeck, G. W. (2004). Apolipoprotein E receptors mediate neurite outgrowth through activation of p44/42 mitogen-activated protein kinase in primary neurons. J. Biol. Chem. 279, 34948–34956. doi: 10.1074/jbc.M401055200
Rafati, D. S., Geissler, K., Johnson, K., Unabia, G., Hulsebosch, C., Nesic-Taylor, O., et al. (2008). Nuclear factor-κB decoy amelioration of spinal cord injury-induced inflammation and behavior outcomes. J. Neurosci. Res. 86, 566–580. doi: 10.1002/jnr.21508
Ramer, L. M., Ramer, M. S., and Bradbury, E. J. (2014). Restoring function after spinal cord injury: towards clinical translation of experimental strategies. Lancet Neurol. 13, 1241–1256. doi: 10.1016/s1474-4422(14)70144-9
Ravella, K., Yang, H., and Gangula, P. R. R. (2012). Impairment of gastric nitrergic and NRF2 system in Apolipoprotein E knockout mice. Dig. Dis. Sci. 57, 1504–1509. doi: 10.1007/s10620-012-2070-2
Resnick, D. K., Schmitt, C., Miranpuri, G. S., Dhodda, V. K., Isaacson, J., and Vemuganti, R. (2004). Molecular evidence of repair and plasticity following spinal cord injury. Neuroreport 15, 837–839. doi: 10.1097/00001756-200404090-00020
Riego, G., Redondo, A., Leánez, S., and Pol, O. (2018). Mechanism implicated in the anti-allodynic and anti-hyperalgesic effects induced by the activation of heme oxygenase 1/carbon monoxide signaling pathway in the central nervous system of mice with neuropathic pain. Biochem. Pharmacol. 148, 52–63. doi: 10.1016/j.bcp.2017.12.007
Rowland, J. W., Hawryluk, G. W., Kwon, B., and Fehlings, M. G. (2008). Current status of acute spinal cord injury pathophysiology and emerging therapies: promise on the horizon. Neurosurg. Focus 25:E2. doi: 10.3171/FOC.2008.25.11.E2
Rust, X. R., and Kaiser, J. (2017). Insights into the dual role of inflammation after spinal cord injury. J. Neurosci. 37, 4658–4660. doi: 10.1523/JNEUROSCI.0498-17.2017
Safina, D., Schlitt, F., Romeo, R., Pflanzner, T., Pietrzik, C. U., Narayanaswami, V., et al. (2016). Low-density lipoprotein receptor-related protein 1 is a novel modulator of radial glia stem cell proliferation, survival, and differentiation. Glia 64, 1363–1380. doi: 10.1002/glia.23009
Sastry, P. S., and Rao, K. S. (2001). Apoptosis and the nervous system. J. Neurochem. 74, 1–20. doi: 10.1046/j.1471-4159.2000.0740001.x
Saul, A., and Wirths, O. (2017). Endogenous Apolipoprotein E (ApoE) fragmentation is linked to amyloid pathology in transgenic mouse models of Alzheimer’s disease. Mol. Neurobiol. 54, 319–327. doi: 10.1007/s12035-015-9674-4
Seitz, A., Kragol, M., Aglow, E., Showe, L., and Heber-Katz, E. (2003). Apolipoprotein E expression after spinal cord injury in the mouse. J. Neurosci. Res. 71, 417–426. doi: 10.1002/jnr.10482
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Shih, R.-H., Wang, C.-Y., and Yang, C.-M. (2015). NF-κB signaling pathways in neurological inflammation: a mini review. Front. Mol. Neurosci. 8:77. doi: 10.3389/fnmol.2015.00077
Silva, N. A., Sousa, N., Reis, R. L., and Salgado, A. J. (2014). From basics to clinical: a comprehensive review on spinal cord injury. Prog. Neurobiol. 114, 25–57. doi: 10.1016/j.pneurobio.2013.11.002
Sun, X., Jones, Z. B., Chen, X., Zhou, L., So, K.-F., and Ren, Y. (2016). Multiple organ dysfunction and systemic inflammation after spinal cord injury: a complex relationship. J. Neuroinflammation 13:260. doi: 10.1186/s12974-016-0736-y
Teng, Z., Guo, Z., Zhong, J., Cheng, C., Huang, Z., Wu, Y., et al. (2017). ApoE influences the blood-brain barrier through the NF-κB/MMP-9 pathway after traumatic brain injury. Sci. Rep. 7:6649. doi: 10.1038/s41598-017-06932-3
Theendakara, V., Peters-libeu, C. A., Spilman, X. P., Poksay, K. S., Bredesen, D. E., and Rao, R. V. (2016). Direct transcriptional effects of Apolipoprotein E. J. Neurosci. 36, 685–700. doi: 10.1523/JNEUROSCI.3562-15.2016
Underwood, E. (2017). How ApoE4 endangers brains. Science 357:1224. doi: 10.1126/science.357.6357.1224
Visavadiya, N. P., Patel, S. P., VanRooyen, J. L., Sullivan, P. G., and Rabchevsky, A. G. (2016). Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol. 8, 59–67. doi: 10.1016/j.redox.2015.12.011
Wang, X., de Rivero Vaccari, J. P., Wang, H., Diaz, P., German, R., Marcillo, A. E., et al. (2012). Activation of the nuclear factor E2-related factor 2/antioxidant response element pathway is neuroprotective after spinal cord injury. J. Neurotrauma 29, 936–945. doi: 10.1089/neu.2011.1922
Wang, R., Hong, J., Lu, M., Neil, J. E., Vitek, M. P., Liu, X., et al. (2014). ApoE mimetic ameliorates motor deficit and tissue damage in rat spinal cord injury. J. Neurosci. Res. 92, 884–892. doi: 10.1002/jnr.23371
Wang, L., Yao, Y., He, R., Meng, Y., Li, N., Zhang, D., et al. (2017). Methane ameliorates spinal cord ischemia-reperfusion injury in rats: antioxidant, anti-inflammatory and anti-apoptotic activity mediated by Nrf2 activation. Free Radic. Biol. Med. 103, 69–86. doi: 10.1016/j.freeradbiomed.2016.12.014
Wei, J., Zheng, M., Liang, P., Wei, Y., Yin, X., Tang, Y., et al. (2013). Apolipoprotein E and its mimetic peptide suppress Th1 and Th17 responses in experimental autoimmune encephalomyelitis. Neurobiol. Dis. 56, 59–65. doi: 10.1016/j.nbd.2013.04.009
Wei, M. C., Zong, W., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., et al. (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. doi: 10.1126/science.1059108
Wu, Y., Pang, J., Peng, J., Cao, F., Vitek, M. P., Li, F., et al. (2016). An ApoE-derived mimic peptide, COG1410, alleviates early brain injury via reducing apoptosis and neuroinflammation in a mouse model of subarachnoid hemorrhage. Neurosci. Lett. 627, 92–99. doi: 10.1016/j.neulet.2016.05.058
Xu, J., He, J., He, H., Peng, R., and Xi, J. (2017). TWEAK-Fn14 influences neurogenesis status via modulating NF-κB in mice with spinal cord injury. Mol. Neurobiol. 54, 7497–7506. doi: 10.1007/s12035-016-0248-x
Xu, X., Huang, E., Tai, Y., Zhao, X., Chen, X., Chen, C., et al. (2017). Nupr1 modulates dopaminergic neuronal apoptosis and autophagy through CHOP-Trib3-mediated endoplasmic reticulum stress signaling pathway. Front. Mol. Neurosci. 10:203. doi: 10.3389/fnmol.2017.00203
Yang, L., Liu, C.-C., Zheng, H., Kanekiyo, T., Atagi, Y., Jia, L., et al. (2016). LRP1 modulates the microglial immune response via regulation of JNK and NF-κB signaling pathways. J. Neuroinflammation 13:304. doi: 10.1186/s12974-016-0772-7
Yin, C., Zhou, S., Jiang, L., and Sun, X. (2012). Mechanical injured neurons stimulate astrocytes to express apolipoprotein E through ERK pathway. Neurosci. Lett. 515, 77–81. doi: 10.1016/j.neulet.2012.03.023
Yu, W. R., and Fehlings, M. G. (2011). Fas/FasL-mediated apoptosis and inflammation are key features of acute human spinal cord injury: implications for translational, clinical application. Acta Neuropathol. 122, 747–761. doi: 10.1007/s00401-011-0882-3
Yu, M., Li, H., Liu, Q., Liu, F., Tang, L., Li, C., et al. (2011). Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 23, 883–892. doi: 10.1016/j.cellsig.2011.01.014
Zhao, H., Chen, S., Gao, K., Zhou, Z., Wang, C., Shen, Z., et al. (2017). Resveratrol protects against spinal cord injury by activating autophagy and inhibiting apoptosis mediated by the SIRT1/AMPK signaling pathway. Neuroscience 348, 241–251. doi: 10.1016/j.neuroscience.2017.02.027
Keywords: contusive spinal cord injury, Apolipoprotein E, neuroinflammation, oxidative stress, apoptosis
Citation: Yang X, Chen S, Shao Z, Li Y, Wu H, Li X, Mao L, Zhou Z, Bai L, Mei X and Liu C (2018) Apolipoprotein E Deficiency Exacerbates Spinal Cord Injury in Mice: Inflammatory Response and Oxidative Stress Mediated by NF-κB Signaling Pathway. Front. Cell. Neurosci. 12:142. doi: 10.3389/fncel.2018.00142
Received: 08 December 2017; Accepted: 09 May 2018;
Published: 23 May 2018.
Edited by:
Alexandre Henriques, Neuro-Sys, FranceReviewed by:
Gourav Roy Choudhury, Texas Biomedical Research Institute, United StatesCopyright © 2018 Yang, Chen, Shao, Li, Wu, Li, Mao, Zhou, Bai, Mei and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xifan Mei, bWVpeGlmYW4xOTcxQDE2My5jb20=
Chang Liu, bGl1Y2hhbmcxOTcxbWVpQDE2My5jb20=
† Co-first authors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.