 Elise C. Ohki1
Elise C. Ohki1 Kyla R. Rodgers
Kyla R. Rodgers Richard C. Chou
Richard C. Chou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 26 September 2017
Sec. Non-Neuronal Cells
Volume 11 - 2017 | https://doi.org/10.3389/fncel.2017.00301
Amyloid beta (Aβ) is a peptide cleaved from amyloid precursor protein that contributes to the formation of senile plaques in Alzheimer’s disease (AD). The relationship between Aβ and astrocyte proliferation in AD remains controversial. Despite pathological findings of increased astrocytic mitosis in AD brains, in vitro studies show an inhibitory effect of Aβ on astrocyte proliferation. In this study, we determined the effect of an active fragment of Aβ (Aβ25-35) on the cell cycle progression of primary rat astrocytes. We found that Aβ25-35 (0.3–1.0 μg/ml) enhanced astrocyte proliferation in vitro in a time- and concentration-dependent manner. Increased DNA synthesis by Aβ25-35 was observed during the S phase of the astrocyte cell cycle, as indicated by proliferation kinetics and bromodeoxyuridine immunocytochemical staining. Aggregation of Aβ25-35 abolished the upregulatory effect of Aβ on astrocyte proliferation. Further examination indicated that Aβ25-35 affected astrocyte proliferation during early or mid-G1 phase but had no effect on DNA synthesis at the peak of S phase. These results provide insight into the relationship between Aβ25-35 and astrocyte cell cycling in AD.
Alzheimer’s disease (AD), the most common form of neurodegenerative disease (Alzheimer’s Association, 2015), is characterized by two pathological hallmarks: senile plaques composed of amyloid beta (Aβ) and neurofibrillary tangles made up of hyperphosphorylated tau (Scheltens et al., 2016). Deposition of Aβ fibrils is an early finding in AD brains (Roth et al., 1966; Pike et al., 1995a; Rama Rao and Kielian, 2015). Aβ is a peptide that is proteolytically cleaved from transmembrane amyloid precursor protein (APP) by alpha, beta, or gamma secretases and can vary in length (LaFerla et al., 2007; Chow et al., 2010). Mutations in either APP or presenilins, which are the catalytic subunits of the gamma secretase complex, increase the risk for AD, possibly by elevating the production of abnormal APP cleavage products that are more prone to forming pathogenic fibrils (Iwatsubo et al., 1994, 1995; Gu and Guo, 2013). Several different Aβ fragments alter the functions of neurons and glia, including Aβ25-35 (Iversen et al., 1995; Millucci et al., 2010; Abeti et al., 2011; Dal Pra et al., 2011), Aβ1-42 (Iversen et al., 1995; Dal Pra et al., 2011; Blennow et al., 2015), Aβ22-35 (Takadera et al., 1993; Iversen et al., 1995), and Aβ12-28 (Iversen et al., 1995; Giulian et al., 1996). Each of these species has different propensities for forming aggregates and senile plaques (Kummer and Heneka, 2014). Among these Aβ25-35 was also shown to induce the production of Aβ1-42 in cultured human astrocytes (Dal Pra et al., 2011). Although Aβ is believed to be critical for the development of AD, the biology of different peptide species and their aggregates, as well as the mechanisms by which they drive pathogenesis remain largely unknown (De Strooper and Karran, 2016).
Interestingly, amyloid deposits and senile plaques are surrounded by reactive astrocytes (Pike et al., 1995a; Rama Rao and Kielian, 2015), a phenomenon noted by Alzheimer himself in his initial pathological analysis and recently confirmed by others (Alzheimer et al., 1995; Santillo et al., 2011). Although remaining in a state of quiescence under normal circumstances, mammalian astrocytes play critical roles in the biochemical and physiological functions of the central nervous system (Langan, 1993), including shaping synaptic transmission (Murphy-Royal et al., 2015), regulating cerebral blood flow (Zonta et al., 2003), and maintaining proper levels of extracellular ions and neurotransmitters (Simard and Nedergaard, 2004; Olsen et al., 2015). However, in AD and other pathological conditions, astrocytes undergo metabolic and phenotypic transformations, re-entering the cell cycle and actively proliferating, a state known as reactive astrogliosis (Amaducci et al., 1981; Pekny et al., 2016; Rodriguez-Arellano et al., 2016). Reactivation of astrocytes is thought to be a crucial early step in the pathogenesis of AD, and prevention of astrogliosis has been proposed as a potential therapeutic strategy (Pekny et al., 2014, 2016; Scholl et al., 2015; Rodriguez-Arellano et al., 2016). The mechanisms underlying cell-cycle re-entry in astrocytes and the relationship between different Aβ species, aggregation states, and reactive astrogliosis remain a matter of intense debate, and further investigation is necessary before novel therapeutic strategies can be proposed (Jordan-Sciutto et al., 2002; Gartner et al., 2003; Yang et al., 2006).
The goal of the present study was to gain a better understanding of the functional relationship between Aβ and proliferation of astrocytes. We hypothesized that soluble Aβ, but not aggregated (agAβ), promotes astrocyte reactivation regardless of cell-cycle phase. To test this hypothesis, we treated serum-deprived and non-serum-deprived rat astrocytes with Aβ25-35 and measured their rates of proliferation after varying lengths of time in vitro. We further investigated if aggregation of soluble Aβ25-35 alters its effects on astrocytes. Our results provide insight into the functional roles of Aβ25-35, as well as results of its aggregation, in reactive astrocytosis.
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the SUNY at Buffalo, and all experiments were carried out in accordance with the approved procedures. Newborn pups were purchased from Harlan Sprague-Dawley and housed briefly in the Animal Research Center, SUNY at Buffalo. The cerebral cortices of newborn (<48 h) Sprague-Dawley rat pups (Harlan Sprague Dawley, Indianapolis, IN, United States) were harvested and mechanically dissociated. Astrocytes were isolated by filtration through nylon mesh with 20 μm pore size to remove tissue debris (Langan and Slater, 1992; Langan and Chou, 2011). Primary astrocyte cultures were maintained in 10% fetal bovine serum (Hyclone, Logan, UT, United States) and 1% penicillin–streptomycin (v/v; Sigma, St. Louis, MO, United States) in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco/Life Technologies, Inc., Grand Island, NY, United States) at 37°C in a CO2 incubator (5% CO2/95% humidified air) until in vitro studies. A previous study shows that the initial astrocyte cultures generated by this method are of ≥95% purity (Langan and Chou, 2011; Langan et al., 2017).
Primary astrocytes were harvested by trypsinization and passaged into 6-well plates at a concentration of 1 × 104 cells/cm2 in 10% bovine calf serum (BCS; Hyclone)/DMEM (v/v). After an initial 2-h incubation at 37°C in 5% CO2/95% humidified air, the medium was removed, and cell cultures were washed once with phosphate-buffered saline (PBS, pH 7.4) to remove cell debris and non-adherent cells; 3 ml of 10% BCS/DMEM was then added to adherent cells. Astrocytes were allowed to proliferate at 37°C in 5% CO2/95% humidified air for 48 h. At the end of incubation, the supernatant was replaced with 2 ml fresh 10% BCS/DMEM in the presence of varying concentrations of agAβ25-35 (in PBS, pH 7.4), freshly prepared non-agAβ25-35 (in PBS, pH 7.4), or control peptide (CP; Cal-Biochem, La Jolla, CA, United States) (in PBS, pH 7.4), which was the Aβ fragment of interest with the amino acids in reverse order (i.e., Aβ35-25). This step represented the start of the experiment (i.e., T0).
Astrocytes were passaged into 6-well plates at a concentration of 1 × 104 cells/cm2 in 10% BCS/DMEM. Cells were then allowed to grow to 30–50% confluence in 10% BCS/DMEM at 37°C in 5% CO2/95% humidified air. At the end of incubation, the medium was removed, and cells were rinsed with PBS (pH 7.4). Cells were then overlaid with 3 ml 0.1% BCS/DMEM and incubated at 37°C in 5% CO2/95% humidified air for 48 h, such that by the end of the incubation period, 85–90% of cells entered cell-cycle arrest (Langan and Slater, 1991; Chou and Langan, 2003; Langan and Chou, 2011; Langan et al., 2017). At the end of the serum-deprivation process, the medium was replaced with 2 ml 10% BCS/DMEM. This serum up-shift allowed astrocytes to re-enter the cell cycle in a first-order manner and represented the start of the cell-cycle entry experiment (i.e., T0) (Langan and Volpe, 1987; Kniss and Burry, 1988; Chou and Langan, 2003; Langan and Chou, 2011; Langan et al., 2017). Varying concentrations of agAβ25-35, non-aggregated (freshly prepared) Aβ25-35, or CP were added to the cultures concurrently with the medium, allowing astrocytes to re-enter the cell cycle under defined experimental conditions.
The Aβ25-35 peptide is a truncated product of Aβ that was shown to be physiologically active, and was widely published in articles using rat models of AD (Soto et al., 1998; Arif et al., 2009; Fedotova et al., 2016; Nell et al., 2016; Soultanov et al., 2016). Others have shown that the effects of Aβ25-35 on different cellular functions in both astrocytes and neurons are similar to those of Aβ1-42 (Chiarini et al., 2010; Kaminsky et al., 2010); additionally, although its physiologic role remains unclear, histopathological studies have shown that Aβ25-35 is present in senile plaques of Alzheimer’s brains (Kubo et al., 2003). For these reasons, Aβ25-35 is the focus of the present studies using rat astrocytes. Quantitative analysis of Aβ25-35 aggregation by sedimentation assay was performed according to an established procedure (Pike et al., 1994, 1995b). In brief, the lysine residues in the non-agAβ25-35 fragment react with the fluorescent marker fluorescamine (FLCN; Molecular Probes, Inc., Eugene, OR, United States). Aggregation of the Aβ25-35 fragments is measured by a decrease in the number of lysine residues available to bind to FLCN, resulting in a reduction in fluorescence intensity proportional to the extent of aggregation. Different concentrations of Aβ25-35 and CP were solubilized in 1 ml PBS (pH 7.4) at room temperature (RT) for 2 or 48 h. Aliquots of the FLCN solution in acetonitrile were added to each set of samples to achieve a 1:1 mol/ml ratio of peptide to FLCN. Following the addition of FLCN, half of the samples were measured for FLCN fluorescence without centrifugation, and half were centrifuged at 100,000 × g for 1 h, and the supernatant was used to measure FLCN fluorescence (Pike et al., 1995b). Fluorescence was measured at 478 nm by exciting the peptide at 383 nm with a fluorescence (LS-5) spectrophotometer (Perkin-Elmer, Norwalk, CT, United States) (De Bernardo et al., 1974; Alavi et al., 2013).
The incorporation of tritiated [methyl-3H]-thymidine into primary astrocytes was used to quantify cell proliferation according to established procedures (Langan and Volpe, 1987; Langan and Slater, 1992; Chou and Langan, 2003; Langan and Chou, 2011; Langan et al., 2017). Radio-labeled [methyl-3H]-thymidine (25 Ci/mmol; Amersham, Arlington Heights, IL, United States) was added to each well 1 h prior to the termination of the experiment at a final activity of 1.0 μCi/ml (37°C, 5% CO2/95% humidified air). At the end of the incubation, cultures were washed with 2 ml Tris–EDTA buffer (pH 7.4) twice to remove any excess 3H-thymidine. DNA and total cellular protein were extracted using the trichloroacetic acid precipitation method (Langan and Volpe, 1987; Langan and Slater, 1992). Cell proliferation was measured as the incorporation of radioactivity per microgram of protein present in the acid-precipitated portion (cpm/μg protein). Tritium was quantified in the samples with a beta counter (LKB Wallac, Gaithersburg, MD, United States) for 10 min using an Ecoscint-A liquid scintillation cocktail (National Diagnostics, Manville, NJ, United States), and the total cellular protein in the samples was determined by Bradford assay (BioRad, Hercules, CA, United states) using a microplate reader (model 3550-UV; BioRad, Hercules, CA, United states) at a wavelength of 595 nm.
The number of cells undergoing active DNA synthesis was quantified by immunocytochemical staining for bromodeoxyuridine (BrdU) (Sigma) (Yong et al., 1988; Langan and Slater, 1991). Primary astrocyte cultures were passaged onto 4.9 mm2 glass cover slips coated with poly-lysine (Sigma) in 12-well plates, grown to 30–50% confluency, and rendered into cell cycle arrest by serum deprivation as described above. Serum-deprived astrocytes were then allowed to re-enter the cell cycle under experimental conditions. To identify cells undergoing active DNA synthesis, BrdU was added to cell cultures at a final concentration of 15 μM for a period of 2.5 h prior to the termination of experiments. At the end of incubation, the cover slips were removed and rinsed with PBS (pH 7.4) followed by fixation with 4% paraformaldehyde (PFA; Sigma) in PBS (pH 7.4) for 10 min at RT. After fixation, the 4% PFA was aspirated, and cells were washed twice with PBS (pH 7.4). Fixed cells were then treated with 2 M HCl (10 min, RT) to permeabilize the nuclear membrane. The HCl was removed, and cells were again washed twice with PBS (pH 7.4) followed by incubation with 0.1 M NaB4O7 to neutralize the HCl (10 min, RT). After acid neutralization, the NaB4O7 was removed, and cells were washed three times with PBS (pH 7.4). Next, cells were blocked with 5% bovine serum albumin in PBS (20 min, RT). Monoclonal antibody for BrdU (Sigma) was added at a dilution of 1:200 for 1 h at RT followed by the addition of goat anti-rat rhodamine-labeled secondary antibody (Sigma) at a dilution of 1:200 for 30 min. The percent of BrdU+ nuclei was calculated based on the cell count in each of the randomly selected high powered fields using a phase-fluorescent microscope (Langan and Slater, 1991; Li et al., 1996).
In parallel to the cell proliferation assays, an identical set of cells was subcultured and treated as described above for each set of experiments. At the end of each time point, cells were harvested by trypsinization, centrifuged, and resuspended in PBS. A 0.4% trypan blue solution (Sigma) was added to the cell suspension in a 1:1 v/v ratio. A manual cell count was performed using a hemocytometer.
All experiments were repeated at least three times, and all conditions were performed in triplicate. Data were expressed as mean ± standard error of the mean (SEM) and analyzed using Student’s t-tests for unpaired comparisons and one-way ANOVA for multiple group comparisons. Statistical significance was defined as p < 0.05.
We first examined the effect of Aβ25-35 on astrocyte proliferation by measuring DNA synthesis of non-serum-deprived primary astrocytes treated with freshly prepared and graded concentrations of Aβ25-35 (0.01–3.0 μg/ml). Astrocytes proliferated in an exponential-like manner over a 48-h period, with a lag phase in the initial 24 h, a log phase in the next 12 h (24–36 h), and a slowed rate of proliferation in the last 12 h tested (36–48 h). We found that Aβ25-35 increased the rate of DNA synthesis in primary astrocytes in a time- and concentration-dependent manner (Figure 1A). At a concentration of 0.3 μg/ml, Aβ25-35 only caused a significant increase in DNA synthesis after 48 h of incubation compared with control. The greatest effect on astrocyte proliferation was obtained using a concentration of 1.0 μg/ml Aβ25-35, which caused significant increases in DNA synthesis after 24, 36, and 48 h of incubation. A further increase in the concentration of Aβ25-35 to 3.0 μg/ml did not upregulate the rate of astrocyte proliferation at any time point, which therefore followed a classical concentration-effect response (Figure 1A). Based on these findings, we utilized Aβ25-35 at a concentration of 1.0 μg/ml in subsequent experiments.
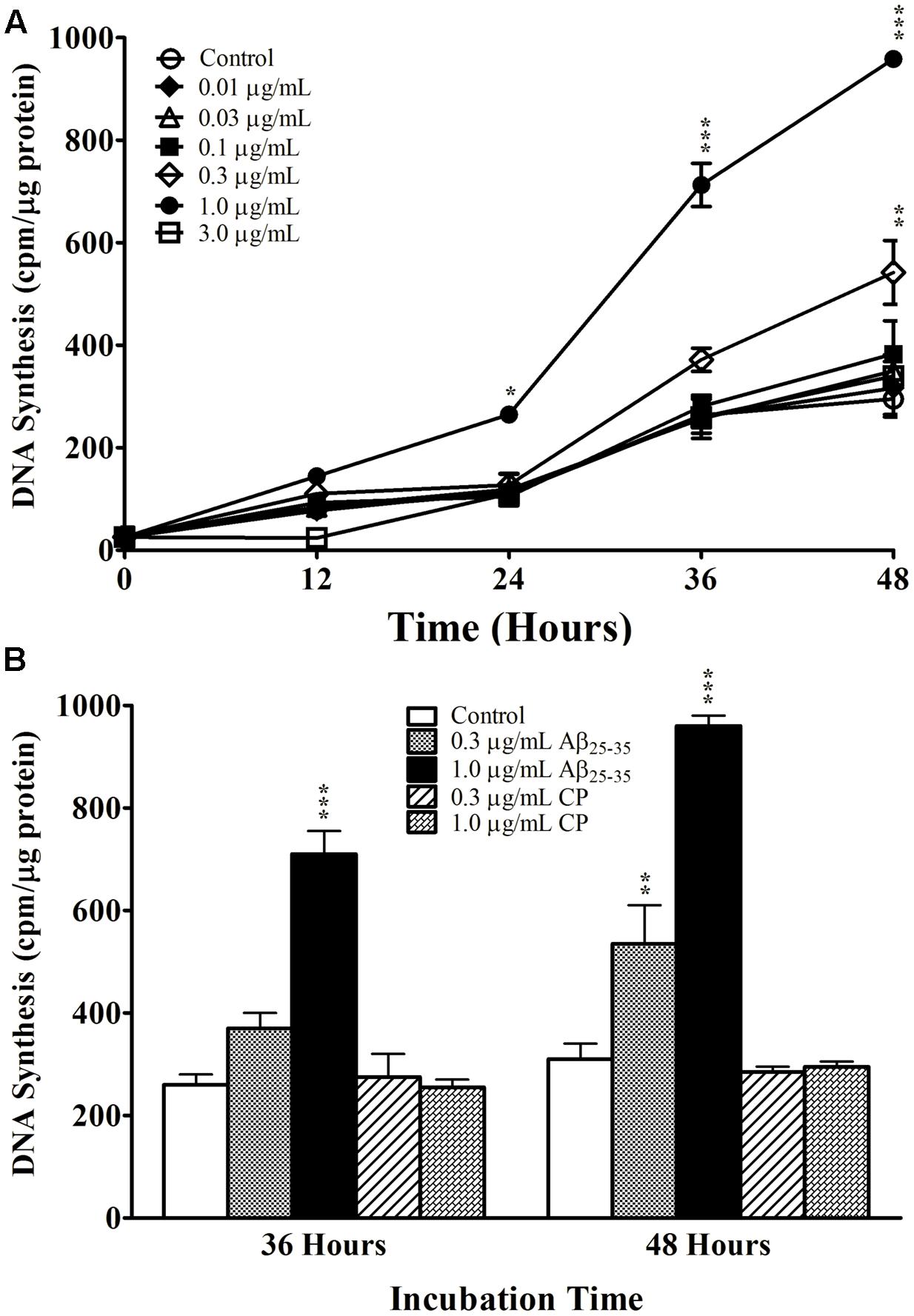
FIGURE 1. The effect and specificity of Aβ25-35 on proliferation of non-serum-deprived primary astrocytes. (A) Subcultured astrocytes were incubated with graded concentrations of freshly prepared and solubilized Aβ25-35 or control. 3H-thymidine was added to cultures 1 h prior to termination. Cultures were terminated every 12 h and processed for 3H-thymidine uptake to measure rate of DNA synthesis. (B) Subcultured astrocytes were incubated with freshly prepared Aβ25-35 or CP at various concentrations for 36 or 48 h. Rate of DNA synthesis was measured using 3H-thymidine incorporation assay. ∗p < 0.05; ∗∗p < 0.02; ∗∗∗p < 0.001 (results were average values of three representative experiments).
We next examined the specificity of the response to Aβ25-35 by treating primary astrocytes with either Aβ25-35 or CP consisting of the reversed amino acid sequence (i.e., Aβ35-25) at varying concentrations for 36 or 48 h. CP did not upregulate DNA synthesis in primary astrocyte cultures compared with control at either time point or either concentration tested (Figure 1B), supporting the notion that the increased proliferation of astrocytes in response to Aβ25-35 depends on its amino acid sequence.
We next addressed whether Aβ25-35 affects cell-cycle kinetics of astrocytes in vitro. Astrocytes were first subcultured in serum-deprived medium that rendered cell-cycle arrest. Astrocytes were then exposed to serum up-shift (Langan and Chou, 2011; Langan et al., 2017), resulting in re-entry into the cell cycle with highly replicable kinetics (Figure 2). Consistent with previous studies (Langan and Slater, 1992; Langan, 1993), following serum up-shift, astrocytes remained in G1 phase for 12 h and then entered the S phase for the next 12 h, as shown by a first-order increase in DNA synthesis (Figure 2). When astrocytes re-entered the cell cycle in the presence of Aβ25-35 (1 μg/ml), the rate of proliferation was significantly upregulated compared with CP-treated astrocytes (Figure 2). Aβ25-35 did not impact the length of G1 or increase the rate of proliferation of astrocytes during this time, suggesting that the effect of Aβ25-35 on astrocytes is restricted to S phase. Additionally, altered proliferation in serum-deprived astrocytes was specifically mediated by Aβ25-35, as CP did not significantly impact DNA synthesis compared with control (Figure 2).
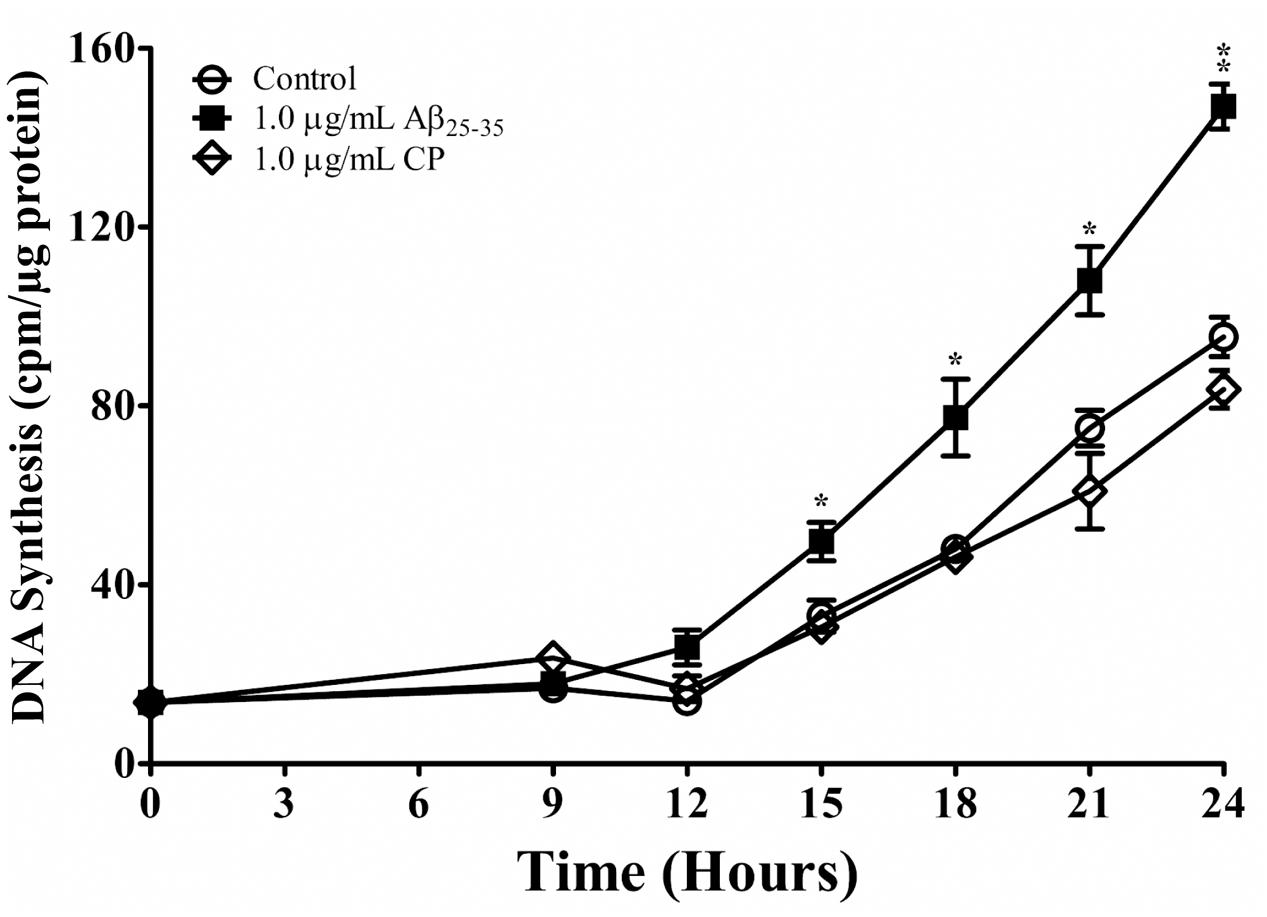
FIGURE 2. The effect of Aβ25-35 on proliferation of serum-deprived primary astrocytes. Newborn astrocytes were rendered into G0 phase by serum deprivation and then allowed to re-enter the cell cycle via serum up-shift in the presence of freshly prepared Aβ25-35 (1.0 μg/ml) or CP (1.0 μg/ml). 3H-thymidine incorporation assay was performed to measure rate of DNA synthesis. ∗p < 0.05; ∗∗p < 0.02; ∗∗∗p < 0.001 (results were average values of three representative experiments).
We confirmed these findings using immunocytochemical staining for BrdU (Figure 3 and Table 1) and quantifying the percent of BrdU+ cells at T0 and T24 in the presence or absence of Aβ25-35 (1 μg/ml). Following 48 h of serum deprivation (i.e., T0), more than 81% of cells were in a state of quiescence (Figure 3 and Table 1). At T24, the peak of S phase, about 65% of the cells were BrdU+, indicating that they had re-entered the cell cycle and undergone active proliferation (Figure 3 and Table 1). In the presence of Aβ25-35, significantly more astrocytes re-entered the cell cycle, as indicated by BrdU+ staining (Figure 3 and Table 1). This increase in the percentage of BrdU+ cells was specific to Aβ25-35 treatment, as there was no significant change in the percentage of BrdU+ cells after 24 h of incubation with CP (Figure 3 and Table 1) compared to control.
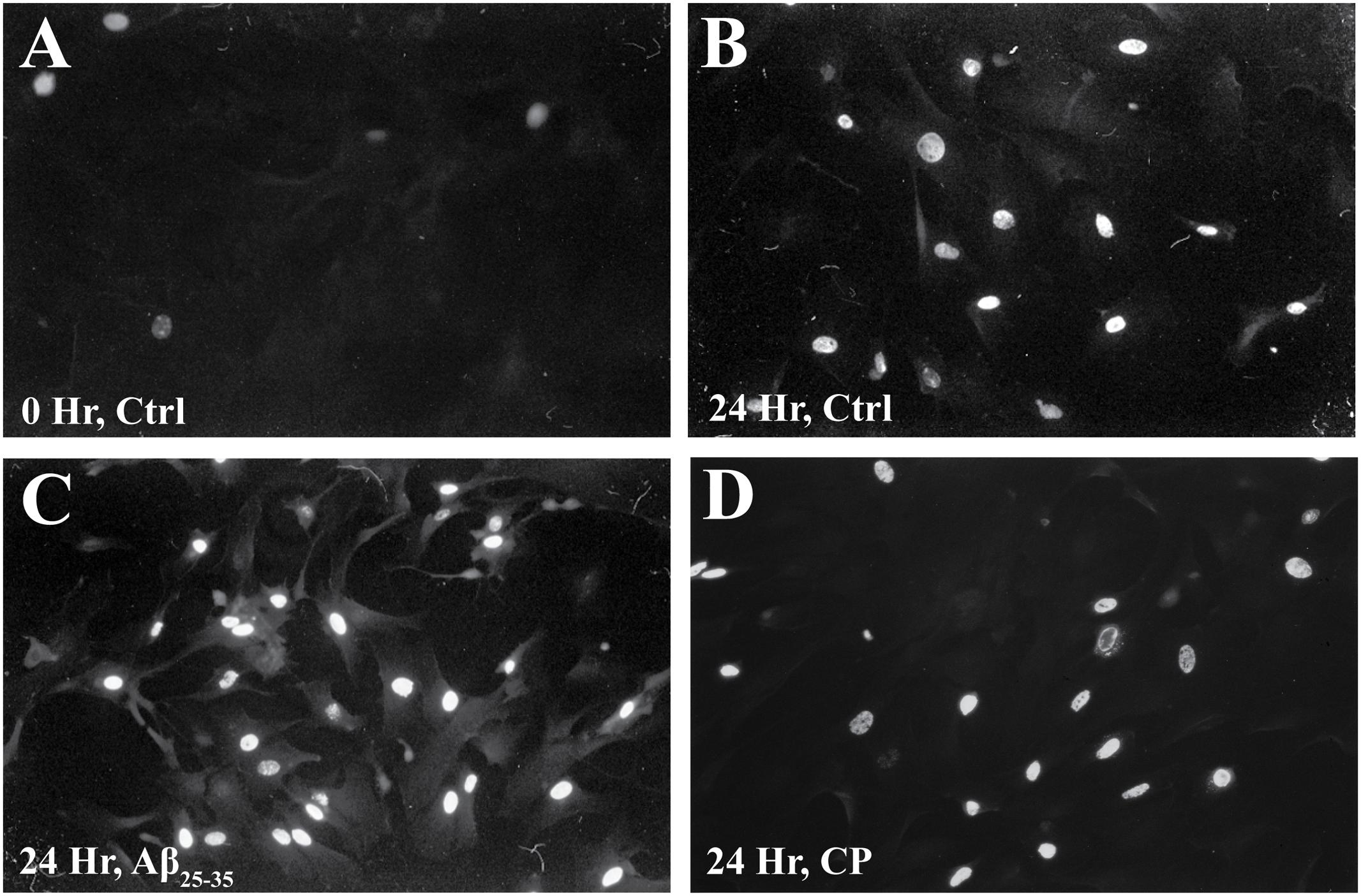
FIGURE 3. The effect of Aβ25-35 on BrdU incorporation in serum-deprived primary astrocytes. Newborn astrocytes were serum deprived for 48 h, resulting in cell cycle arrest, as demonstrated by low BrdU+ cell count (A; 18.38 ± 0.02%, n = 9, total cell count = 102). Following serum deprivation, cells were stimulated to re-enter the cell cycle in the presence of either freshly prepared Aβ25-35 (1.0 μg/ml) or CP (1.0 μg/ml). BrdU was added to the cultures 2.5 h prior to termination. After 24 h of incubation, samples were processed for immunocytochemical staining of BrdU. There was no significant difference in the number of BrdU+ cells between the control (B; 65.42 ± 0.02%, n = 11, total cell count = 104) and CP-treated astrocytes (D; 62.44 ± 0.02%, n = 11, total cell count = 100). Significantly more Aβ25-35-treated astrocytes were BrdU+ (C; 87.35 ± 0.02%; n = 11, total cell count = 102, p < 0.001) compared to controls. Images were taken with fluorescence microscopy.

TABLE 1. Percent of BrdU+ astrocytes in response to various treatments for 24 h.
In order to generate agAβ25-35, we allowed graded concentrations of Aβ25-35 to aggregate for 2 or 48 h; aggregation was confirmed by measuring FLCN incorporation. Aggregation was compared in two different manners: (1) time-dependent effect in the non-centrifugation portion, i.e., 2 vs. 48 h, as represented by the statistical symbol “∗” and (2) concentration-dependent effect in each time point, i.e., suspension vs. supernatant, as represented by the statistical symbol “†”. We observed a significant decrease in fluorescence after 48 h at all concentrations of Aβ25-35 (0.5–10.0 μg/ml) when compared with 2 h incubation (Table 2), both in the non-centrifuged solution and in the supernatant of the centrifuged solution. These data suggest that incubating the peptide at RT for 48 h results in the formation of aggregates, as evidenced by the decreased FLCN absorbance in the supernatants of samples that were solubilized for 48 h as compared to those that were solubilized for 2 h prior to centrifugation. Based on our findings, non-centrifuged solutions were used for downstream experiments in order to ensure that astrocytes were exposed to aggregates of varying sizes. To address the question of whether oligomerization of Aβ25-35 affects astrocyte proliferation, we treated non-serum-deprived primary astrocyte cultures for 12, 24, 36, or 48 h with graded concentrations of agAβ25-35 or non-agAβ25-35. In contrast to our earlier findings using Aβ25-35 (Figure 1A), agAβ25-35 only significantly increased the rate of proliferation of non-serum-deprived astrocytes compared with control after 24 h of stimulation at a concentration of 3.0 μg/ml (Figure 4).
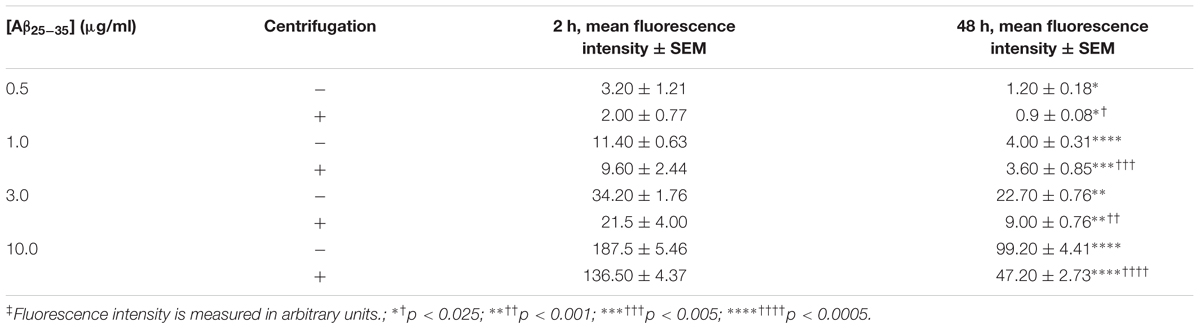
TABLE 2. Effect of time and fraction on Aβ25-35 aggregation in solution‡.
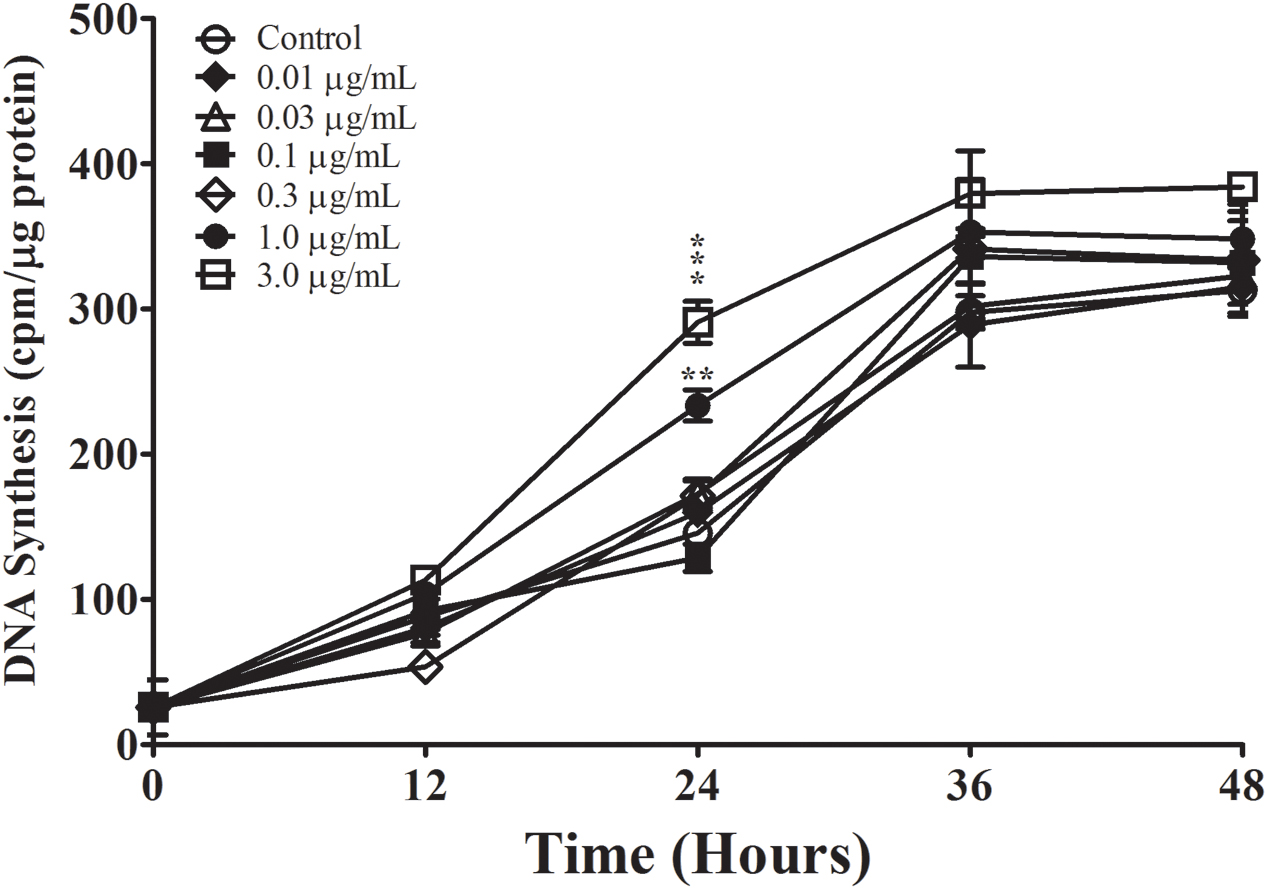
FIGURE 4. The effect of Aβ25-35 aggregation on the proliferation of non-serum-deprived primary astrocytes. Freshly prepared Aβ25-35 was allowed to aggregate in PBS (pH 7.4) for 48 h; aggregation was confirmed by FLCN incorporation assay (Table 2). Non-serum-deprived astrocytes were treated with varying concentrations of agAβ25-35 (0.01–3.0 μg/ml), and 3H-thymidine incorporation assay was performed to measure rate of DNA synthesis. ∗p < 0.05; ∗∗p < 0.02; ∗∗∗p < 0.001 (results were average values of three representative experiments).
Finally, we examined whether the effect of Aβ25-35 on astrocyte proliferation is phase-specific. Aβ25-35 or CP (1.0 μg/ml) was added to serum-deprived astrocyte cultures at the time of serum up-shift or various times after serum up-shift (T6, T12, and T16). All cultures were terminated at the peak of the S phase (T24), and the rates of proliferation were determined using 3H-thymidine incorporation. Consistent with our earlier results, we found that adding Aβ25-35 at T0 significantly increased the rate of DNA synthesis at the peak of S phase (T24) compared with control or CP (p < 0.001; Figure 5A). When Aβ25-35 was added in the middle of G1 phase at T6, we observed a similar increase in the rate of DNA synthesis compared with CP or control (p < 0.02). However, when addition was delayed until the G1/S phase transition point (T12) or until the initiation of S phase, the rates of astrocyte proliferation were not significantly different among the Aβ25-35, CP, and control groups (Figure 5A), suggesting that the effect of Aβ25-35 on astrocyte proliferation is phase-specific, and initiated during the G1 phase and before G1/S phase transition point.
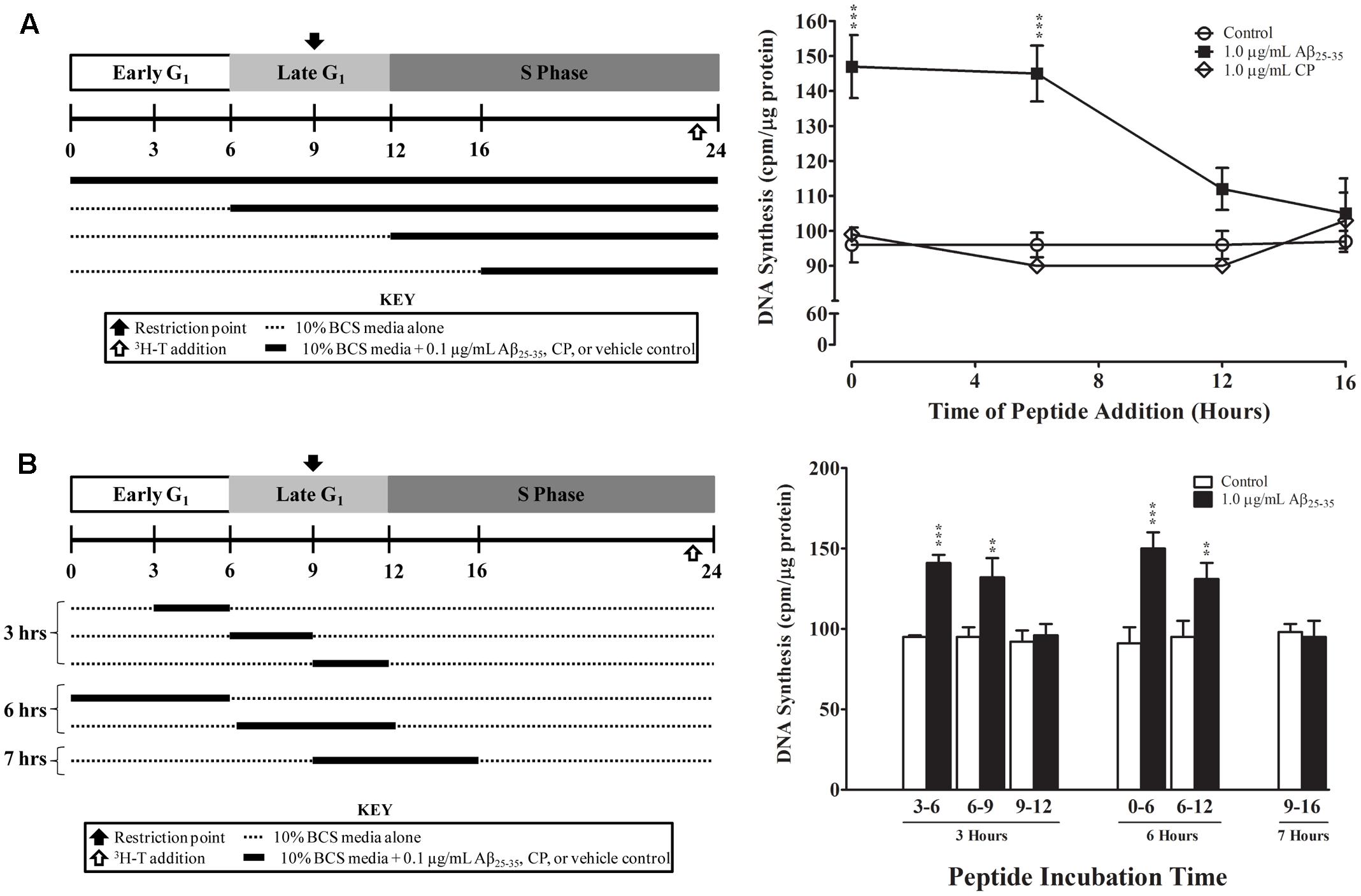
FIGURE 5. Temporal effects of Aβ25-35 on the proliferation of serum-deprived primary astrocytes. Newborn astrocytes were first serum-deprived and then re-entered into the cell cycle by serum up-shift. (A) Left, experimental approach; Right, Aβ25-35 (1.0 μg/ml) or CP (1.0 μg/ml) was added to the cultures at varying times after serum up-shift (0, 6, 12, or 16 h). (B) Left, experimental approach; Right, Aβ25-35 (1.0 μg/ml) was added to the cultures at different time points and then removed via medium changes after 3, 6, or 7 h of incubation. All cultures were terminated at T24, and 3H-thymidine incorporation assay was performed to measure rate of DNA synthesis. ∗p < 0.05; ∗∗p < 0.02; ∗∗∗p < 0.001 (results were average values of three representative experiments).
We further defined the critical period of Aβ25-35 exposure by using a “delayed addition and early removal” paradigm. In these experiments, we added Aβ25-35 to the cultures at different points of the cell cycle and then changed the medium to remove Aβ25-35 after varying exposure durations (3, 6, or 7 h); all cultures were terminated at the peak of the S phase (T24), and the rate of proliferation was determined using 3H-thymidine incorporation. We found that Aβ25-35 significantly increased the rate of astrocyte proliferation as compared to control only if it was added to the culture during early to mid-G1 phase (T0, T3, or T6; Figure 5B). When Aβ25-35 was added to the cultures in late G1 phase (T9), there was no significant difference in the rate of DNA synthesis compared with control (Figure 5B), regardless of whether the exposure duration was 3 (T9–T12) or 7 h (T9–T16). CP did not alter the rate of DNA synthesis during any of these exposure periods (data not shown). These results suggest that Aβ25-35 affects the proliferation of astrocytes during early-to-mid-G1 phase.
A definitive diagnosis of AD is based on histopathological evidence at autopsy or brain biopsy, including the presence of Aβ deposits, neuritic changes with formation of paired helical filaments and hyperphosphorylated tau, and reactive astrogliosis (Serrano-Pozo et al., 2011). Astrocytosis is widespread in the early phases of AD but declines as the disease progresses, indicating that gliosis may be an early pathogenic event (Pike et al., 1995a; Santillo et al., 2011; Rodriguez-Vieitez et al., 2015, 2016; Scholl et al., 2015). In vitro studies consistently demonstrate that Aβ is neurotoxic and consequently induces neuritic changes (Sendrowski et al., 2015; Sadleir et al., 2016). Although pathological studies show that Aβ-containing senile plaques are often surrounded by reactive astrocytes, the relationship between different Aβ species and functional changes in astrocytes is less clear. Here, we hypothesize that Aβ25-35 increases astrocyte proliferation, but that aggregation of Aβ25-35 inhibits this effect.
Interestingly, although aberrant expression of mitotic regulators, such as cyclin c and binding partner Cdk8 have been observed in the astrocytes of AD brains (Ueberham et al., 2003), in vitro studies demonstrate highly variable and contrasting effects of Aβ on astrocytes (Agostinho et al., 2010), including induction of apoptosis (Brera et al., 2000; Hou et al., 2011; Saha and Biswas, 2015) and oxidative stress (Brera et al., 2000), as well as changes in morphology (Salinero et al., 1997) and proliferation (Hernandez-Guillamon et al., 2009). Our results show that Aβ25-35 upregulates proliferation in both serum-deprived and non-serum-deprived astrocytes in time- and concentration-dependent manners as measured by 3H-thymidine incorporation assay and BrdU staining (Figures 1, 2); importantly, Aβ25-35 did not induce cellular death in our experiments, as demonstrated by minimal cell death in trypan blue exclusion assay (<2%). We found that CP (Aβ35-25) did not impact astrocyte proliferation, confirming the bio-specific effect of Aβ25-35 on the rate of DNA synthesis in primary astrocytes depends on its amino acid sequence (Figures 1, 2). Although increasing the concentration of Aβ25-35 upregulated the rate of proliferation over a certain range (i.e., 0.01–1.0 μg/ml), showing a clear concentration-effect of Aβ25-35 on DNA synthesis in astroctyes, further increase of the Aβ25-35 concentration to 3.0 μg/ml failed to enhance DNA synthesis (Figure 1). While our data demonstrate a narrow concentration-effect window, this phenomenon has been reported in other biological systems, including that seen in astrocytes. For example, it was shown that just a 1% change in the concentration of isoflurane had a dramatic effect on astrocyte calcium signaling in ferret visual cortex (Schummers et al., 2008). Similarly, it has been shown that mitochondrial function is extremely sensitive to a narrow range of Ca2+ ion concentrations (Schild et al., 2005), as is cellular survival in response to free zinc ions (Bozym et al., 2010). While the mechanisms underlying the concentration effects of each of these phenomena are currently unknown, one potential explanation of our data is that Aβ25-35 follows a classical concentration-effect response via specific receptors; however, further research is required in order to elucidate the molecular basis of this response (Salinero et al., 1997; Brera et al., 2000; Hou et al., 2011; Saha and Biswas, 2015). Our data showing narrow effective concentration range on astrocyte proliferation do suggest a plausible explanation for astrocyte toxicity in advanced AD. In fact, others have shown that Aβ25-35 is cytotoxic to astrocytes, those studies were carried out using concentrations 1000-fold higher than our current study; therefore, our results do not exclude the findings that high doses of Aβ25-35 can be cytotoxic to astrocytes in vitro.
Our findings also demonstrate that the effect of Aβ25-35 is cell cycle phase-specific. First, Aβ25-35 increases the rate of DNA synthesis in serum-deprived astrocytes during the S phase but not during G1 phase (Figure 2). Second, the timing of exposure to Aβ25-35 was critical. To upregulate astrocyte proliferation, Aβ25-35 needed to be added to astrocytes during early-to-mid-G1 phase. If Aβ25-35 was added any later than 6 h into G1, the rate of proliferation was not impacted (Figure 5). These findings offer novel insights into the effect of Aβ25-35 on astrocyte proliferation, and the mechanisms underlying the phase specificity are currently under investigation in our laboratory.
We showed that aggregation of Aβ25-35 impaired its upregulatory effect on astrocyte proliferation, as agAβ25-35 increased DNA synthesis relative to control only at a concentration of 3.0 μg/ml and only at T24 (i.e., 12 h into the S phase, Figure 4). Senile plaques, a hallmark feature of AD pathology, are made up of fibrillar or β-sheet aggregates of amyloid peptide; amyloid deposition has been shown to occur progressively over the course of the disease and has been proposed as a biomarker for disease staging (Jack et al., 2013). However, our results clearly show that aggregation of amyloid peptide limits its ability to stimulate astrocytes. As astrocyte activation precedes plaque formation in AD patients (Rodriguez-Vieitez et al., 2015), it is conceivable that initial Aβ deposition increases astrocyte proliferation and leads to reactive astrocytosis during the early stages of amyloid deposition, but that as senile plaques by amyloid aggregation are formed, the rate of astrocyte reactivation slows down.
Our study is the first to show evidence of the upregulatory effect of Aβ25-35 on DNA synthesis during the S phase of the primary astrocyte cell cycle. Our results demonstrate that Aβ25-35 affected the cell cycle only when astrocytes were exposed during early-to-mid-G1 phase. Exposing primary astrocytes to Aβ25-35 3 h prior to G1/S intersection (T9) failed to increase DNA synthesis in the S phase, irrespective of exposure duration. Therefore, these results suggest the temporal importance of Aβ exposure preceding the late G1 phase restriction point of the astrocyte cell cycle. The time-based specificity of the effect of Aβ25-35 on the astrocyte cell cycle thus points to the possibility that this peptide participates in the complex array of biochemical and molecular events that confer commitment to cell-cycle progression; further investigation is required to determine the exact mechanisms responsible. Additionally, while the present study focuses on the effect of Aβ25-35 on astrocytes, it would also be worthwhile to examine its impact on the cellular functions and rate of proliferation of other cells important for brain function, such as microglia, which have crucial roles in central nervous system immunity, and endothelial cells, which maintain the blood–brain barrier.
Experiments were conceived and designed by EO, TL, and RC. Experiments were performed by EO and RC. Data were analyzed by EO, KR, and RC. Reagents/materials/tools provided by TL. Manuscript was written and prepared by EO, KR, and RC. All authors reviewed and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abeti, R., Abramov, A. Y., and Duchen, M. R. (2011). Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 134(Pt 6), 1658–1672. doi: 10.1093/brain/awr104
Agostinho, P., Cunha, R. A., and Oliveira, C. (2010). Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 16, 2766–2778. doi: 10.2174/138161210793176572
Alavi, P., Yousefi, R., Amirghofran, S., Karbalaei-Heidari, H. R., and Moosavi-Movahedi, A. A. (2013). Structural analysis and aggregation propensity of reduced and nonreduced glycated insulin adducts. Appl. Biochem. Biotechnol. 170, 623–638. doi: 10.1007/s12010-013-0207-1
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Alzheimer’s Association (2015). 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 11, 332–384.
Amaducci, L., Forno, K. I., and Eng, L. F. (1981). Glial fibrillary acidic protein in cryogenic lesions of the rat brain. Neurosci. Lett. 21, 27–32. doi: 10.1016/0304-3940(81)90052-5
Arif, M., Chikuma, T., Ahmed, M. M., Nakazato, M., Smith, M. A., and Kato, T. (2009). Effects of memantine on soluble Alphabeta(25-35)-induced changes in peptidergic and glial cells in Alzheimer’s disease model rat brain regions. Neuroscience 164, 1199–1209. doi: 10.1016/j.neuroscience.2009.08.063
Blennow, K., Mattsson, N., Scholl, M., Hansson, O., and Zetterberg, H. (2015). Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol. Sci. 36, 297–309. doi: 10.1016/j.tips.2015.03.002
Bozym, R. A., Chimienti, F., Giblin, L. J., Gross, G. W., Korichneva, I., Li, Y., et al. (2010). Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp. Biol. Med. 235, 741–750. doi: 10.1258/ebm.2010.009258
Brera, B., Serrano, A., and de Ceballos, M. L. (2000). beta-amyloid peptides are cytotoxic to astrocytes in culture: a role for oxidative stress. Neurobiol. Dis. 7, 395–405. doi: 10.1006/nbdi.2000.0313
Chiarini, A., Whitfield, J., Bonafini, C., Chakravarthy, B., Armato, U., and Dal Pra, I. (2010). Amyloid-beta(25-35), an amyloid-beta(1-42) surrogate, and proinflammatory cytokines stimulate VEGF-A secretion by cultured, early passage, normoxic adult human cerebral astrocytes. J. Alzheimers Dis. 21, 915–926. doi: 10.3233/JAD-2010-100471
Chou, R. C., and Langan, T. J. (2003). In vitro synchronization of mammalian astrocytic cultures by serum deprivation. Brain Res. Brain Res. Protoc. 11, 162–167. doi: 10.1016/S1385-299X(03)00043-6
Chow, V. W., Mattson, M. P., Wong, P. C., and Gleichmann, M. (2010). An overview of APP processing enzymes and products. Neuromolecular Med. 12, 1–12. doi: 10.1007/s12017-009-8104-z
Dal Pra, I., Whitfileld, J. F., Pacchiana, R., Bonafini, C., Talacchi, A., Chakravarthy, B., et al. (2011). The amyloid-beta(4)(2) proxy, amyloid-beta(25-35), induces normal human cerebral astrocytes to produce amyloid-beta(4)(2). J. Alzheimers Dis. 24, 335–347. doi: 10.3233/JAD-2011-101626
De Bernardo, S., Weigele, M., Toome, V., Manhart, K., Leimgruber, W., Bohlen, P., et al. (1974). Studies on the reaction of fluorescamine with primary amines. Arch. Biochem. Biophys. 163, 390–399. doi: 10.1016/0003-9861(74)90490-1
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Fedotova, J., Soultanov, V., Nikitina, T., Roschin, V., Ordyan, N., and Hritcu, L. (2016). Cognitive-enhancing activities of the polyprenol preparation Ropren(R) in gonadectomized beta-amyloid (25-35) rat model of Alzheimer’s disease. Physiol. Behav. 157, 55–62. doi: 10.1016/j.physbeh.2016.01.035
Gartner, U., Bruckner, M. K., Krug, S., Schmetsdorf, S., Staufenbiel, M., and Arendt, T. (2003). Amyloid deposition in APP23 mice is associated with the expression of cyclins in astrocytes but not in neurons. Acta Neuropathol. 106, 535–544. doi: 10.1007/s00401-003-0760-8
Giulian, D., Haverkamp, L. J., Yu, J. H., Karshin, W., Tom, D., Li, J., et al. (1996). Specific domains of beta-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J. Neurosci. 16, 6021–6037.
Gu, L., and Guo, Z. (2013). Alzheimer’s Abeta42 and Abeta40 peptides form interlaced amyloid fibrils. J. Neurochem. 126, 305–311. doi: 10.1111/jnc.12202
Hernandez-Guillamon, M., Delgado, P., Ortega, L., Pares, M., Rosell, A., Garcia-Bonilla, L., et al. (2009). Neuronal TIMP-1 release accompanies astrocytic MMP-9 secretion and enhances astrocyte proliferation induced by beta-amyloid 25-35 fragment. J. Neurosci. Res. 87, 2115–2125. doi: 10.1002/jnr.22034
Hou, L., Liu, Y., Wang, X., Ma, H., He, J., Zhang, Y., et al. (2011). The effects of amyloid-beta42 oligomer on the proliferation and activation of astrocytes in vitro. In Vitro Cell Dev. Biol. Anim. 47, 573–580. doi: 10.1007/s11626-011-9439-y
Iversen, L. L., Mortishire-Smith, R. J., Pollack, S. J., and Shearman, M. S. (1995). The toxicity in vitro of beta-amyloid protein. Biochem. J. 311(Pt 1), 1–16. doi: 10.1042/bj3110001
Iwatsubo, T., Mann, D. M., Odaka, A., Suzuki, N., and Ihara, Y. (1995). Amyloid beta protein (A beta) deposition: a beta 42(43) precedes A beta 40 in Down syndrome. Ann. Neurol. 37, 294–299. doi: 10.1002/ana.410370305
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., and Ihara, Y. (1994). Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13, 45–53. doi: 10.1016/0896-6273(94)90458-8
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013). Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/S1474-4422(12)70291-0
Jordan-Sciutto, K. L., Malaiyandi, L. M., and Bowser, R. (2002). Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J. Neuropathol. Exp. Neurol. 61, 358–367. doi: 10.1093/jnen/61.4.358
Kaminsky, Y. G., Marlatt, M. W., Smith, M. A., and Kosenko, E. A. (2010). Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Abeta(25-35). Exp. Neurol. 221, 26–37. doi: 10.1016/j.expneurol.2009.09.005
Kniss, D. A., and Burry, R. W. (1988). Serum and fibroblast growth factor stimulate quiescent astrocytes to re-enter the cell cycle. Brain Res. 439, 281–288. doi: 10.1016/0006-8993(88)91485-0
Kubo, T., Kumagae, Y., Miller, C. A., and Kaneko, I. (2003). Beta-amyloid racemized at the Ser26 residue in the brains of patients with Alzheimer disease: implications in the pathogenesis of Alzheimer disease. J. Neuropathol. Exp. Neurol. 62, 248–259. doi: 10.1093/jnen/62.3.248
Kummer, M. P., and Heneka, M. T. (2014). Truncated and modified amyloid-beta species. Alzheimers Res. Ther. 6, 28. doi: 10.1186/alzrt258
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509. doi: 10.1038/nrn2168
Langan, T. J. (1993). Deterministic role of the cell cycle in newborn brain development and in brain injury. Semin. Neurol. 13, 92–99. doi: 10.1055/s-2008-1041112
Langan, T. J., and Chou, R. C. (2011). Synchronization of mammalian cell cultures by serum deprivation. Methods Mol. Biol. 761, 75–83. doi: 10.1007/978-1-61779-182-6_5
Langan, T. J., Rodgers, K. R., and Chou, R. C. (2017). Synchronization of mammalian cell cultures by serum deprivation. Methods Mol. Biol. 1524, 97–105. doi: 10.1007/978-1-4939-6603-5_6
Langan, T. J., and Slater, M. C. (1991). Cell cycling of astrocytes and their precursors in primary cultures: a mevalonate requirement identified in late G1, but before the G1/S transition, involves polypeptides. J. Neurochem. 56, 1058–1068. doi: 10.1111/j.1471-4159.1991.tb02029.x
Langan, T. J., and Slater, M. C. (1992). Astrocytes derived from long-term primary cultures recapitulate features of astrogliosis as they re-enter the cell division cycle. Brain Res. 577, 200–209. doi: 10.1016/0006-8993(92)90275-E
Langan, T. J., and Volpe, J. J. (1987). Cell cycle-specific requirement for mevalonate, but not for cholesterol, for DNA synthesis in glial primary cultures. J. Neurochem. 49, 513–521. doi: 10.1111/j.1471-4159.1987.tb02894.x
Li, V., Kelly, K., Schrot, R., and Langan, T. J. (1996). Cell cycle kinetics and commitment in newborn, adult, and tumoral astrocytes. Brain Res. Dev. Brain Res. 96, 138–147. doi: 10.1016/0165-3806(96)00109-5
Millucci, L., Ghezzi, L., Bernardini, G., and Santucci, A. (2010). Conformations and biological activities of amyloid beta peptide 25-35. Curr. Protein Pept. Sci. 11, 54–67. doi: 10.2174/138920310790274626
Murphy-Royal, C., Dupuis, J. P., Varela, J. A., Panatier, A., Pinson, B., Baufreton, J., et al. (2015). Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 18, 219–226. doi: 10.1038/nn.3901
Nell, H. J., Au, J. L., Giordano, C. R., Terlecky, S. R., Walton, P. A., Whitehead, S. N., et al. (2016). The targeted antioxidant, catalase-SKL, reduces beta-amyloid toxicity in the rat brain. Brain Pathol. 27, 86–94. doi: 10.1111/bpa.12368
Olsen, M. L., Khakh, B. S., Skatchkov, S. N., Zhou, M., Lee, C. J., and Rouach, N. (2015). New insights on astrocyte ion channels: critical for homeostasis and neuron-glia signaling. J. Neurosci. 35, 13827–13835. doi: 10.1523/JNEUROSCI.2603-15.2015
Pekny, M., Pekna, M., Messing, A., Steinhauser, C., Lee, J. M., Parpura, V., et al. (2016). Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345. doi: 10.1007/s00401-015-1513-1
Pekny, M., Wilhelmsson, U., and Pekna, M. (2014). The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 565, 30–38. doi: 10.1016/j.neulet.2013.12.071
Pike, C. J., Cummings, B. J., and Cotman, C. W. (1995a). Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp. Neurol. 132, 172–179.
Pike, C. J., Cummings, B. J., Monzavi, R., and Cotman, C. W. (1994). Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience 63, 517–531. doi: 10.1016/0306-4522(94)90547-9
Pike, C. J., Walencewicz-Wasserman, A. J., Kosmoski, J., Cribbs, D. H., Glabe, C. G., and Cotman, C. W. (1995b). Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J. Neurochem. 64, 253–265.
Rama Rao, K. V., and Kielian, T. (2015). Neuron-astrocyte interactions in neurodegenerative diseases: role of neuroinflammation. Clin. Exp. Neuroimmunol. 6, 245–263. doi: 10.1111/cen3.12237
Rodriguez-Arellano, J. J., Parpura, V., Zorec, R., and Verkhratsky, A. (2016). Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 323, 170–182. doi: 10.1016/j.neuroscience.2015.01.007
Rodriguez-Vieitez, E., Ni, R., Gulyas, B., Toth, M., Haggkvist, J., Halldin, C., et al. (2015). Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur. J. Nucl. Med. Mol. Imaging 42, 1119–1132. doi: 10.1007/s00259-015-3047-0
Rodriguez-Vieitez, E., Saint-Aubert, L., Carter, S. F., Almkvist, O., Farid, K., Scholl, M., et al. (2016). Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 139(Pt 3), 922–936. doi: 10.1093/brain/awv404
Roth, M., Tomlinson, B. E., and Blessed, G. (1966). Correlation between scores for dementia and counts of ‘senile plaques’ in cerebral grey matter of elderly subjects. Nature 209, 109–110. doi: 10.1038/209109a0
Sadleir, K. R., Kandalepas, P. C., Buggia-Prevot, V., Nicholson, D. A., Thinakaran, G., and Vassar, R. (2016). Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 132, 235–256. doi: 10.1007/s00401-016-1558-9
Saha, P., and Biswas, S. C. (2015). Amyloid-beta induced astrocytosis and astrocyte death: implication of FoxO3a-Bim-caspase3 death signaling. Mol. Cell. Neurosci. 68, 203–211. doi: 10.1016/j.mcn.2015.08.002
Salinero, O., Moreno-Flores, M. T., Ceballos, M. L., and Wandosell, F. (1997). beta-Amyloid peptide induced cytoskeletal reorganization in cultured astrocytes. J. Neurosci. Res. 47, 216–223. doi: 10.1002/(SICI)1097-4547(19970115)47:2<216::AID-JNR10>3.0.CO;2-0
Santillo, A. F., Gambini, J. P., Lannfelt, L., Langstrom, B., Ulla-Marja, L., Kilander, L., et al. (2011). In vivo imaging of astrocytosis in Alzheimer’s disease: an 11C-L-deuteriodeprenyl and PIB PET study. Eur. J. Nucl. Med. Mol. Imaging 38, 2202–2208. doi: 10.1007/s00259-011-1895-9
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Schild, L., Plumeyer, F., and Reiser, G. (2005). Ca2+ rise within a narrow window of concentration prevents functional injury of mitochondria exposed to hypoxia/reoxygenation by increasing antioxidative defence. FEBS J. 272, 5844–5852. doi: 10.1111/j.1742-4658.2005.04978.x
Scholl, M., Carter, S. F., Westman, E., Rodriguez-Vieitez, E., Almkvist, O., Thordardottir, S., et al. (2015). Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Sci. Rep. 5:16404. doi: 10.1038/srep16404
Schummers, J., Yu, H., and Sur, M. (2008). Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320, 1638–1643. doi: 10.1126/science.1156120
Sendrowski, K., Sobaniec, W., Stasiak-Barmuta, A., Sobaniec, P., and Popko, J. (2015). Study of the protective effects of nootropic agents against neuronal damage induced by amyloid-beta (fragment 25-35) in cultured hippocampal neurons. Pharmacol. Rep. 67, 326–331. doi: 10.1016/j.pharep.2014.09.013
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Simard, M., and Nedergaard, M. (2004). The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 129, 877–896. doi: 10.1016/j.neuroscience.2004.09.053
Soto, C., Sigurdsson, E. M., Morelli, L., Kumar, R. A., Castano, E. M., and Frangione, B. (1998). Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer’s therapy. Nat. Med. 4, 822–826. doi: 10.1038/nm0798-822
Soultanov, V., Fedotova, J., Nikitina, T., Roschin, V., Ordyan, N., and Hritcu, L. (2016). Antidepressant-like effect of Ropren®in β-amyloid-(25-35) rat model of Alzheimer’s disease with altered levels of androgens. Mol. Neurobiol. 54, 2611–2621. doi: 10.1007/s12035-016-9848-8
Takadera, T., Sakura, N., Mohri, T., and Hashimoto, T. (1993). Toxic effect of a beta-amyloid peptide (beta 22-35) on the hippocampal neuron and its prevention. Neurosci. Lett. 161, 41–44. doi: 10.1016/0304-3940(93)90135-8
Ueberham, U., Hessel, A., and Arendt, T. (2003). Cyclin C expression is involved in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 24, 427–435. doi: 10.1016/S0197-4580(02)00132-X
Yang, Y., Varvel, N. H., Lamb, B. T., and Herrup, K. (2006). Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J. Neurosci. 26, 775–784. doi: 10.1523/JNEUROSCI.3707-05.2006
Yong, V. W., Kim, S. U., and Pleasure, D. E. (1988). Growth factors for fetal and adult human astrocytes in culture. Brain Res. 444, 59–66. doi: 10.1016/0006-8993(88)90913-4
Keywords: astrocyte, amyloid beta, cell cycle, primary cultures, Alzheimer’s disease
Citation: Ohki EC, Langan TJ, Rodgers KR and Chou RC (2017) Non-aggregated Aβ25-35 Upregulates Primary Astrocyte Proliferation In Vitro. Front. Cell. Neurosci. 11:301. doi: 10.3389/fncel.2017.00301
Received: 11 May 2017; Accepted: 11 September 2017;
Published: 26 September 2017.
Edited by:
Juan Andrés Orellana, Pontificia Universidad Católica de Chile, ChileReviewed by:
Markel Olabarria, Columbia University, United StatesCopyright © 2017 Ohki, Langan, Rodgers and Chou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard C. Chou, cmljaGFyZC5jLmNob3VAaGl0Y2hjb2NrLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.