 Yuhui Hu1,2†
Yuhui Hu1,2† Liang Huang
Liang Huang Huanhuan Li
Huanhuan Li Qionglan Yuan
Qionglan Yuan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 19 April 2017
Sec. Cellular Neuropathology
Volume 11 - 2017 | https://doi.org/10.3389/fncel.2017.00110
Cell adhesion molecules (CAMs) are involved in synaptic plasticity and neuronal survival in the adult brain. Neuroplastin 65 (Np65), one member of the immunoglobulin superfamily of CAMs, is brain-specific and highly expressed in rodent forebrain. The roles of Np65 in synaptic plasticity have been confirmed, however, whether Np65 affects neuronal survival remains unknown. To address this gap, we generated, to our knowledge, the first Np65 knockout (KO) mice. By occluding middle cerebral artery to perform ischemic stroke model, we showed that Np65 KO mice exhibited more severe neurological deficits and larger infarction volume measured by TTC staining and more apoptotic cells confirmed by TUNEL staining compared to wild type (WT) mice. Besides, western blot analysis showed that the vesicular glutamate transporter-1(VGluT1), and N-Methyl D-Aspartate receptors, including NR1, NR2A, and NR2B were significantly increased in Np65 KO mice compared with WT mice. In contrast, vesicular gamma amino butyric acid transporter (VGAT) levels were unchanged in two genotypes after stroke. Additionally, phosphorylated-extracellular signal-regulated kinase 1/2 levels were significantly increased in Np65 KO mice compared with WT mice after stroke. Together, these results suggest that Np65 KO mice may be more susceptible to ischemic events in the brain.
Cell adhesion molecules (CAMs) are crucially involved in neuronal differentiation during development and support the maturation and maintenance of synapses in the adult brain. Neuroplastin (Np) is a glycoprotein belonging to the immunoglobulin (Ig) superfamily (IgSF) of CAMs (Langnaese et al., 1997). For alternative splicing of the transcript from a single gene, Np exists in two isoforms, Np55 and Np65 according to molecular weights. The isoforms can be distinguished solely by the presence of the Ig1 module specific to Np65 and absent in Np55 (Langnaese et al., 1997; Smalla et al., 2000). Of the two isoforms, Np65 is brain-specific, whereas Np55 is widely expressed in various tissues (Hill et al., 1988; Langnaese et al., 1997, 1998; Smalla et al., 2000). In the rodent brain, Np65 is predominantly located in forebrain such as the cortex, striatum and hippocampus (Hill et al., 1988; Langnaese et al., 1997, 1998; Smalla et al., 2000).
Accumulating evidence shows that Np65 mediates activity–dependent synaptic plasticity and neuritogenesis and neuronal plasticity (Smalla et al., 2000; Empson et al., 2006; Owczarek et al., 2010). For example, the recombinant ectodomain of Np65 and anti-Np antibodies inhibit long-term potentiation (LTP) in the CA1 region of the hippocampus (Smalla et al., 2000; Empson et al., 2006), probably via p38 MAPK-dependent destabilization and internalization of GluA1-containing glutamate receptors from the synaptic membrane (Empson et al., 2006). In addition, our previous studies have found that Np65 may be involved in modulation of neuritogenesis in vitro (Owczarek et al., 2011). More recently, we found that Np65 knock-out mice exhibit enhanced hippocampus-dependent learning and memory and anxiety-like behaviors (Amuti et al., 2016).
CAMs also play an important role in neuronal survival (Hulley et al., 1998; Ditlevsen et al., 2003, 2007). However, very limited information is available regarding roles of Np65 in neuronal survival, with exception to our observations that the peptides derived from NP Ig1 or Ig2 increase the survival of cerebellar granule neurons induced to undergo apoptosis (Owczarek et al., 2011), suggesting that Np65 interaction protects neurons against apoptosis in vitro.
Here, we performed ischemic stroke model by using Np65 KO mice to investigate the roles of Np65 in neuronal survival in vivo. We identified for the first time Np65 KO mice suffered more severe ischemic injury after stroke and indicate protective effects of Np65 in the brain.
Animal care and experimental protocols complied with National Institutes of Health Guide for the Care and Use of Laboratory Animals, were also approved by the Animal Study Committee at Tongji University School of Medicine. Generation and characterization of Np65 knock-out mice (KO) and their WT littermates were described in our lab (Amuti et al., 2016). In brief, C57BL/6J Np65 heterozygotes (Np65+/−) mice were backcrossed to C57BL/6J mice (Slac Laboratory Animal Center, Shanghai) for at least five generations and resulting offspring were subsequently bred to generate homozygous offspring. Genotyping of the offspring was performed by PCR of tail DNA extracts and by Western blot of Np65 (Amuti et al., 2016).
Focal cerebral ischemia model was performed by transiently occluding middle cerebral artery (MCA) as described (Yuan et al., 2007). In brief, Np65 KO mice and WT mice (2–4 months, 25–30 g) were anesthetized with 1% pentobarbital sodium in 0.9% NaCl. The right common carotid artery, external carotid artery, and internal carotid artery were exposed and a 10-0 monofilament nylon (10 mm long, Beijing Sunbio Biotech Co. Ltd.) was advanced from the common carotid artery into the lumen of the internal carotid artery until it blocked the origin of the MCA. Two hours after ischemia, the MCA ligature was removed and the animals were killed at 1 week after reperfusion. After surgery, mice exhibiting neurological deficits characterized by failure to extend the left forepaw were considered as successful models. The mice were allowed to survive for 1 week. All animals were maintained on a 12 h light-dark cycle in a temperature-controlled room with ad libitum access to water and food. All efforts were made to minimize pain and distress and the methods were carried out in accordance with the relevant guidelines.
Behaviors were tested at 7 days after stroke. Modified Neurological Severity Scale (mNSS) scores were used to evaluate the neurological function according to the previous methods (Yuan et al., 2007). The mNSS was a composite of motor, sensory, reflex, and balance tests. Neurological function was graded on a scale of 0–18 (normal, 0; maximal deficit score, 18). The higher the score, the more severe the injury was (seen in Table 1).
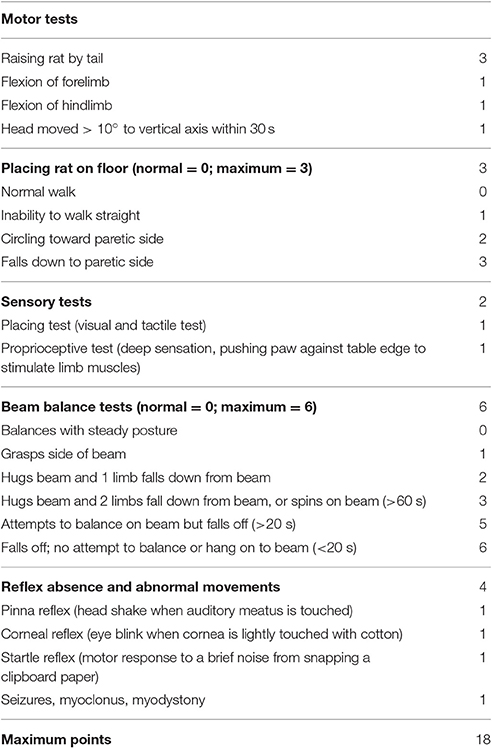
Table 1. Modified Neurological Severity Score Points.
Infarct volume was measured by 2, 3, 5-triphenyltetrazolium chloride (TTC, Sigma) staining as previously described (Horita et al., 2006). Np65 and WT mice (n = 4 per group) were killed by overdose of 1% pentobarbital sodium 7 days after reperfusion and brains were removed carefully and dissected into 6 coronal blocks (1.5 mm thick each). The fresh blocks were immersed in a 2% solution of TTC in saline at 37°C for 30 min and then were imaged by a digital camera (Canon, EOS 40D). The infarct volume was calculated indirectly by subtracting intact area of the ipsilateral hemisphere from the area of the contralateral hemisphere (Swanson et al., 1990; Yang et al., 2011). The results were expressed as percentage of lesion volume compared with contralateral hemisphere area, considering that as 100%.
All mice (n = 4) were killed by overdose of 1% pentobarbital sodium at 7 days after stroke and transcardially perfused with PBS followed by 4% PFA. In addition, untreated WT and Np65 KO mice (n = 4) were considered as control mice. Brains were removed and post-fixed in 4% PFA for 10–16 h, and cryoprotected in 20% sucrose before sectioning with a cryostat (CM 1950, Leica, Heidelberger, Germany). Coronal sections (25 μM thick) were cut and processed for following staining.
Nissl staining was performed to evaluate neurons as described previously (Zhang et al., 2012; Chen et al., 2013). After washing with PBS, the sections were incubated with Nissl Staining Solution (Beyotime Institute of Biotechnology, Nanjing, China) for 20 min at room temperature. Sections were then dehydrated with ethanol and covered with Permount.
Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) staining was used to identify apoptotic cells under light microscope and was performed as described (Matsushita et al., 2000). In situ Cell Death Detection kit (Roche Molecular Biochemicals) was used. Briefly, after sections were treated with 3% H2O2 at room temperature for 15 min and then digested with proteinase K (20 μg/ml) at 37°C for 15 min, each section was incubated with 50 ml of TUNEL reaction mixture (5 ml of enzyme solution and 45 ml of label solution) at 37°C for 60 min, then followed with peroxidase at 37°C for 30 min with diaminobenzidineor for 3–5 min. Negative controls were performed with label solution instead of TUNEL reaction mixture. Sections were counterstained with hematoxylin, cleared and covered with Permount.
For semiquantitative measurements of Nissl and TUNEL staining, four animals per group were analyzed. Three slides with five intervals from sequential hippocampus and striatum sections were chosen in each mouse. Sections were imaged with a light microscopy (Nikon, ECLIPSE, E600, Tokyo, Japan) with three non-overlapping fields under a 200 × magnification. The number of neurons in the hippocampus and striatum (for Nissl staining) or the number of TUNEL-positive cells in the hippocampus and striatum was counted with Image Tool Software (Pro Plus v 6.0). All analyses were performed blinded to the genotype of the animals.
At 7 days after MCAO, Np65 KO, and WT mice (n = 3) were sacrificed under deep anesthesia with pentobarbital sodium. In addition, untreated WT and Np65 KO mice (n = 3) were considered as control mice. Whole brain was immediately removed and right (ipsilateral) forebrain was collected and rapidly frozen in liquid nitrogen and then stored at −80°C until homogenization.
Frozen tissue was weighed, thawed, and homogenized. The samples were centrifuged at 14,000 rpm at 4°C for 20 min and the supernatant was collected. Protein concentrations were measured using a BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Nanjing, China). Proteins were separated by SDS-PAGE and transferred to the nitrocellulose membranes. After blocking with 10% skim milk, the blots were incubated overnight at 4°C with primary antibodies: rabbit anti-Vesicular GABA transporter (VGAT, 1:500), rabbit anti-vesicular glutamate transporter-1 (VGLuT1), phospho-ERK1/2 (Thr 202/Tyr 204, p-ERK1/2) (1:000, Cell Signaling Technology), ERK1/2 (1:1,000, Cell Signaling Technology), mouse anti-GAPDH (1:1,000, Santa Cruz Biotechnology). Subsequently, the membrane was incubated with secondary antibodies against the primary antibody for 1 h at room temperature, and the labeled proteins were detected by using the Bio-Rad system and quantified through Quantity One analysis. The final results were expressed as a ratio of the expression of the protein of interest to that of GAPDH.
Statistical analysis was performed with SPSS13.0 for Windows. All data were expressed as mean ± SEM. Multiple comparisons were performed by using one-way ANOVA followed by Tukey's post hoc test for multiple pair-wise examinations in vivo experiments. The difference was considered significant if p < 0.05.
Np65 gene deletion did not affect locomotor activity of mice (Amuti et al., 2016). Before stroke, both type mice exhibited normal motor movement, sensory, reflex, and balance, the scores were 0. At 7 days after stroke, neurological performance was evaluated by modified neurological severity score (Yang et al., 2011). The higher scores indicate the more severe injury (Yang et al., 2011). As shown in Figure 1, higher scores in Np65 KO mice were observed compared to WT mice after stroke, suggesting that Np65 deficiency exacerbates neurological deficits in mice after stroke.
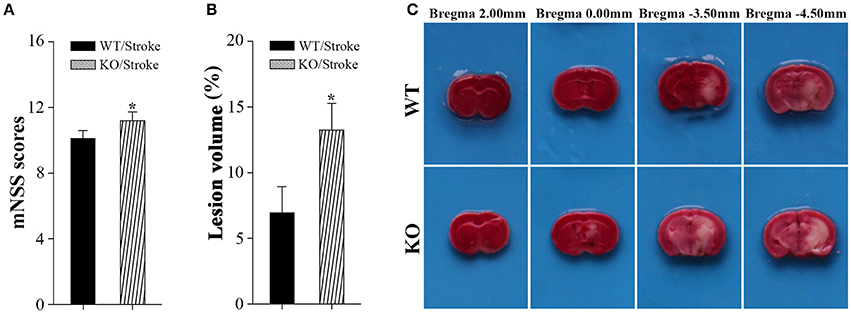
Figure 1. Np65 deficiency exacerbates neurological deficits and increases infarct volume in mice after stroke. (A) Neurological function was determined by modified neurological severity scores (mNSS), summarizing the results of motor, sensory, reflex, and balance tests. Np65 KO mice exhibited higher scores at 7 days after stroke. N = 8. *p < 0.05; (B) Quantitative image analysis of infarct volume. Np65 deficiency increases infarct volume in mice at 7 days after stroke. (C) Representative brain slices were stained by TTC. Normal brain tissue was red and infarct tissue was white; N = 4. *p < 0.05.
In addition, infarct volume, another index suggesting the extent of injury, was calculated by TTC staining at 7 days after stroke. The ischemic volume was indirectly calculated by subtracting intact area of the ipsilateral hemisphere from the area of the contralateral hemisphere (Swanson et al., 1990). The remaining normal brain tissue was typically stained with TTC and appeared red color, and infarct tissue showed no or reduced staining and appeared white color (Bederson et al., 1986). As shown in Figure 1C, the contralateral hemispheres and remaining normal tissues in the ipsilateral hemisphere appeared red color. Meanwhile, ischemic regions in ipsilateral hemisphere appeared white color and were primarily located in hippocampus and striatum. Infarct volume was significantly increased in Np65 KO mice compared to WT mice (Figure 1C). These results indicate that Np65 deficiency increases infarct volume in mice after stroke.
Nissl staining was used to observe the histologic features in mice after stroke. In untreated normal WT and Np65 KO mice, no abnormal neurons were observed and the amounts of neurons in cortex and hippocampus did not differ (Figure 2). In contrast, at 7 days after stroke, the majority of neurons in hippocampus and striatum disappeared and some neurons shrunk with pyknotic nuclei (Figure 2). The numbers of remaining neurons in ischemic hippocampus and striatum were measured and we found that the numbers of remaining neurons were significantly decreased in Np65 KO mice compared to WT mice after stroke for 7 days. These results suggest that Np65 deficiency leads to a more severe injury in mice after stroke.
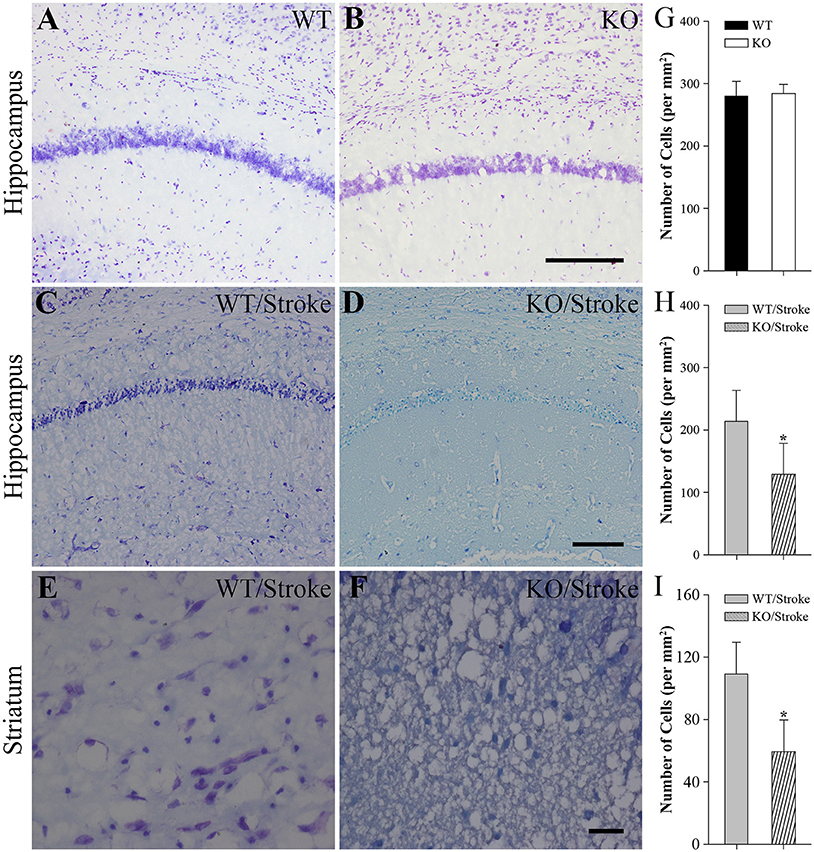
Figure 2. Nissl staining showed fewer neurons of hippocampus and striatum in Np65 KO mice after stroke. (A–F) Representative Nissl staining showed neurons of hippocampus (A–D) and striatum (E,F) in WT and Np65 KO mice in normal (A,B) and after stroke (C–F). Scale bar = 200 μm (A–D) or 50 μm (E,F); (G–I) Histogram showed that the number of neurons of hippocampus was no different between WT and Np65 KO mice (G), but a significant decrease in the ischemic hippocampus (H) and striatum (I) was observed in Np65 KO mice after stroke. N = 4. *p < 0.05.
In addition, the apoptotic cells were measured by using TUNEL staining. In normal Np65 KO and WT mice, very fewer of TUNEL-positive cells were observed and the number of TUNEL-positive cells did not differ (Figure 3). In contrast, many TUNEL-positive cells were present in the hippocampus and striatum at 7 days after stroke. The numbers of apoptotic cells in hippocampus and striatum were significantly increased in Np65 KO mice compared to WT mice (Figure 3).
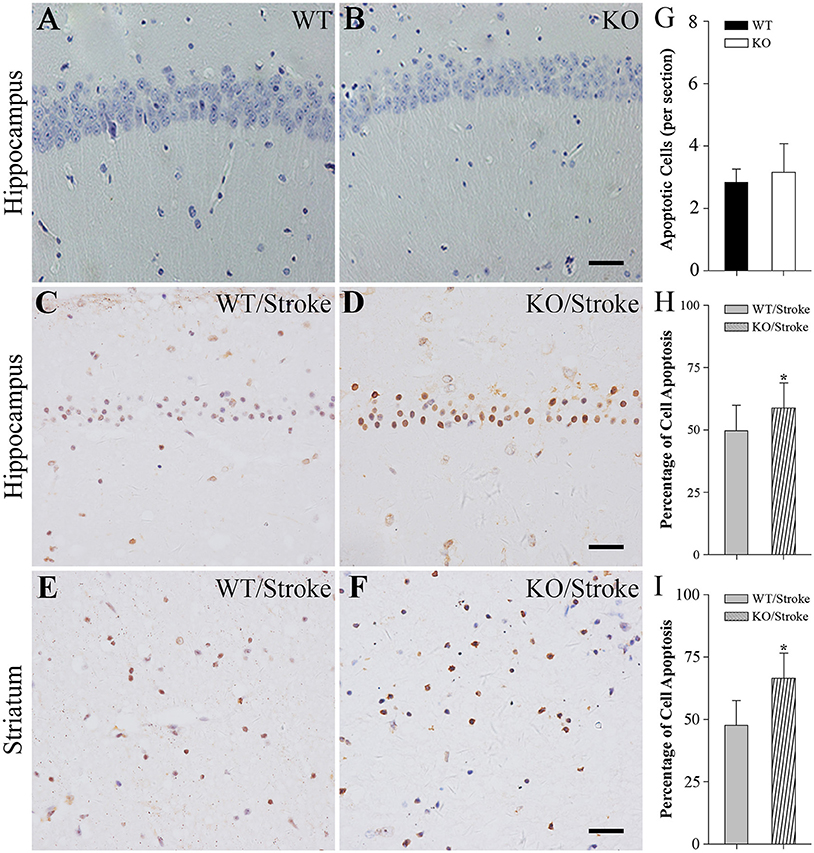
Figure 3. TUNEL staining showed more apoptotic cells in Np65 KO mice after stroke. (A–F) Representative TUNEL staining showed that apoptotic cells (↑) in hippocampus (A–D) and striatum (E,F) in Np65 KO and WT mice in normal (A,B) and after stroke (C–F). Scale bar = 25 μm; (G–I) Histogram showed that the apoptotic cells of hippocampus was no different between WT and Np65 KO mice (G), but a significant increase in the ischemic hippocampus (H) and striatum (I) was observed in Np65 KO mice after stroke. N = 4. *p < 0.05.
Mitogen-activated protein kinases (MAPK) are involved in many physiological and pathological processes. The MAPK family includes extracellular signal-regulated kinase 1/2(ERK1/2), c-Jun N-terminal kinase (JNK), and p38 MAPK (Roux and Blenis, 2004). ERK1/2 signaling has been implicated in synaptic plasticity and memory formation as well as survival (Sweatt, 2004). To explore the underlying mechanisms that Np65 KO mice are susceptibility to ischemia, the ERK1/2 signaling was studied. As shown in Figure 4, p-ERK1/2 levels were significantly increased in Np65 KO mice compared to WT mice although the total ERK1/2 levels were unchanged. At 7 days after stroke, p-ERK1/2 levels were significantly increased in two genotypes. Moreover, far higher p-ERK levels were observed in Np65 KO mice compared to WT mice after stroke. These results suggest that p-ERK1/2 levels are significantly increased in Np65 KO mice after stroke.
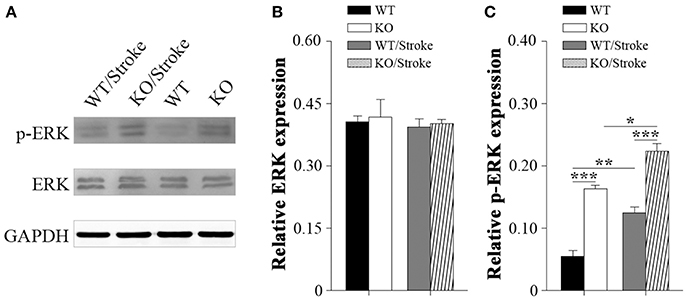
Figure 4. Increased p-ERK1/2 signaling in Np65 KO mice after stroke. (A) Representative immunoblot bands of ERK1/2, phosphorylated-ERK1/2 (p-ERK1/2) from ipsilateral forebrain in Np65 KO and WT mice in normal and after stroke; (B–C) Quantification of immunoreactivity bands normalized against GAPDH showed that the p-ERK1/2 levels were significantly increased in Np65 KO mice compared to WT mice but the total ERK1/2 levels were unchanged. After stroke, p-ERK1/2 levels were significantly increased in two genotypes. Moreover, far higher p-ERK levels were observed in Np65 KO mice compared to WT mice after stroke. N = 3. *p < 0.05; **p < 0.01, ***p < 0.001.
Excitotoxicity has been well accepted to be one of the principal pathogenesis of ischemic stroke. Therefore, inhibitory/excitatory neurotransmitters were explored in mice after stroke. Vesicular gamma amino butyric acid transporter (VGAT), an indirect marker for inhibitory GABA levels, and vesicular glutamate transporter-1 (VGluT1), an indirect marker for glutamate levels, were measured in forebrain by western blot analysis. The results revealed that VGlut1 levels in Np65 KO mice were significantly increased compared to WT mice. In contrast, VGAT protein levels were unchanged in two genotypes. At 7 days after Stroke, VGlut1 expression levels were significantly increased, and much higher levels of VGlut1 in Np65 KO mice were observed in comparison with WT mice (Figures 5A–C). However, VGAT protein levels were unchanged in mice after stroke. In addition, N-Methyl D-Aspartate receptor (NMDAR), a high-affinity of glutamate, was evaluated. The NMDAR is a heterotetrameric complex composed of two obligatory NR1 subunits and two NR2 subunits. The results showed that the expressions of NR2A were apparently higher in Np65 KO mice compared to WT mice. At 7 days after stroke, the NR2A and NR2B expressions were significantly elevated in Np65 KO mice not in WT mice. Notably, NR1, NR2A, and NR2B expressions were significantly elevated in Np65 KO mice compared to WT mice after stroke (Figures 5D–E). These results suggest that elevated glutamate and enhanced NMDAR levels in Np65 KO mice may contribute, at least partially, to more severe damage after stroke.
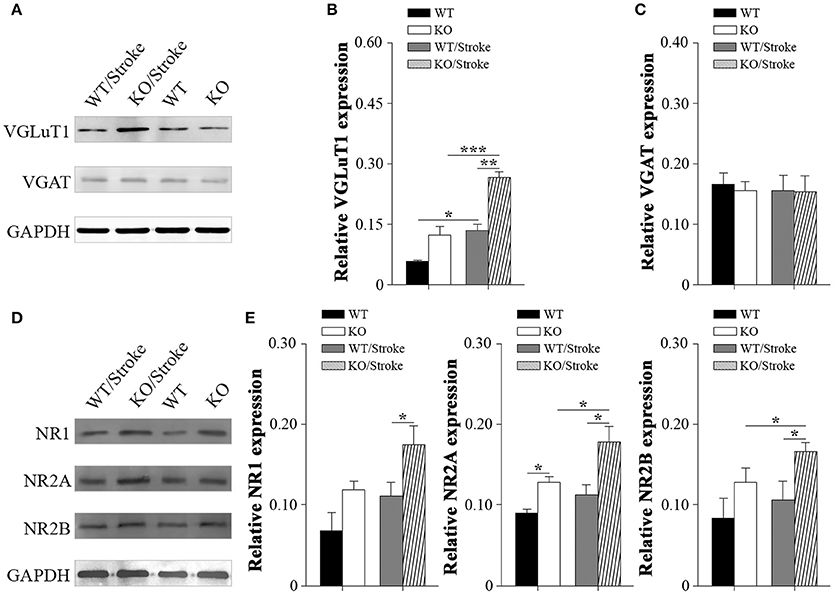
Figure 5. Altered neurotransmitters and NMDA receptors in Np65 KO mice after stroke. (A) Representative immunoblot bands of VGLuT1 and VGAT from ipsilateral forebrain of mice in normal and after stroke; (B–C) Quantification of immunoreactivity bands normalized against GAPDH showed the increased VGlut1 levels and unchanged VGAT levels in Np65 KO mice compared to WT mice. At 7 days after stroke, VGLuT1 expression levels were significantly increased in two genotypes, and much higher levels of VGlut1 appeared in Np65 KO mice. However, VGAT protein levels were unchanged in mice after stroke. N = 3, *p < 0.05, **p < 0.01, ***p < 0.001; (D) Representative immunoblot bands of NMDAR subunits from ipsilateral forebrain of Np65 KO and WT mice in normal and after stroke; (E) Quantification of immunoreactivity bands normalized against GAPDH showed the expressions of NR2A were apparently higher in Np65 KO mice compared to WT mice. At 7 days after stroke, the NR2A and NR2B expressions were significantly elevated in Np65 KO mice but not in WT mice. Notably, NR1, NR2A, and NR2B expressions were significantly elevated in Np65 KO mice compared to WT mice after stroke. N = 3, *p < 0.05.
In the present study, we present evidence that Np65, similar to other CAMs, is also involved in neuronal survival in vivo. We evaluated the role of Np65 in neuronal survival by using Np65 KO mice, and found that Np65 KO mice exposed to ischemic stroke exhibit more severe neurological deficits and larger infarct volume and more apoptotic cells after stroke. Besides, the VGluT1 and NMDA receptors, including NR1 and NR2A, NR2B, p-ERK1/2 levels are significantly increased in Np65 KO mice compared with WT mice after stroke. Together, these results suggest that Np65 KO mice may be susceptible to ischemic events in the brain.
Although previous findings document that CAMs play crucial roles in neural development and are also critically involved in neurite outgrowth and synaptic plasticity in the adult nervous system (Hulley et al., 1998; Kiryushko et al., 2006), the roles of Ig SF CAMs in survival of neurons have only been recognized in different pathological settings (Hulley et al., 1998; Gary and Mattson, 2001; Ditlevsen et al., 2003, 2007). For example, the neural cell adhesion molecule (NCAM) activation, in addition to leading to neurite extension, also leads to an increased survival of primary dopaminergic neurons induced by 6-OHDA to undergo apoptosis (Ditlevsen et al., 2007). L1 neural cell adhesion molecule, another one of IgSF CAMs, promotes survival of fetal dopaminergic neurons by using L1 antibody in vitro (Hulley et al., 1998). Integrin exhibits increased resistance to glutamate-induced apoptosis (Gary and Mattson, 2001). Our previous studies have shown that Np65 gene deletion does not affect locomotor activity and brain framework of organization in adult mice (Amuti et al., 2016). In addition, we showed that peptides derived from NP Ig1 or Ig2 significantly increased the survival of primary cerebellar granule neurons induced to undergo apoptosis by low KCl in the media (Owczarek et al., 2011), suggesting that Np65 protects neurons against apoptosis in vitro. To further explore the possible roles of Np65 in neuronal survival in vivo, we made a stroke model using Np65 KO mice to observe the ischemic damage and neurological behaviors. We found that Np65 KO mice showed more severe neurological deficits, larger infarct and more apoptotic cells compared to WT mice after stroke. To our knowledge, it is the first time to demonstrate that Np 65 KO mice show susceptibility to cerebral ischemia.
Glutamate excitotoxicity is believed to be one of the early events in propagating neurological damage in stroke (Choi, 1992; Iadecola et al., 2001). MK801, a NMDA receptor blocker, has been shown to alleviate ischemic damage in rodent after stroke (Buchan et al., 1992; Gerriets et al., 2003). In our present study, Np65 KO mice were more susceptible to ischemia induced by MCAO than WT mice. To explore the underlying mechanisms, glutamate and NMDA receptors were investigated by western blot analysis. VGluT1, an indirect marker for glutamate levels, and NMDA receptors, including NR1 and NR2A and NR2B, were observed. We found that the expressions of NR2A were significantly increased in Np 65KO mice compared to WT mice. Whether these increases affect brain function needs to be investigated. At 1 week after stroke, VGluT1 and NMDA receptors including NR1 and NR2A and NR2B, were significantly increased in Np65 KO mice compared with WT mice. Contrary to these results, the VGAT, an indirect marker for inhibitory neurotransmitter GABA, was unchanged. Previous studies have confirmed that NP65 trans-homophilic binding activates p38MAPK, which in turn down-regulates surface GluR1 glutamate receptors by internalization in cultured hippocampal neurons (Empson et al., 2006). Moreover, it has shown that Np65 co-localizes with a1 and a2, but not a3 subunits at GABAergic synapses and a5 subunits at extra synaptic sites in cultures (Sarto-Jackson et al., 2012). These results have shown that Np65 affects composition of PSDs, especially transmitter receptors and may involve excitatory and inhibitory synaptic transmission. More recently, Carrott et al. reported that Np65-decicient mice show abnormal synaptogenesis in inner hair cells and causes profound hearing loss (Carrott et al., 2016). Combined our results with these previous studies, it is speculated that the highly increased glutamate and NMDA receptors might render excitotoxicity in Np65 KO mice after stroke and lead to excitotoxic neuronal death. How Np65 deficiency affects the glutamate and its receptors is hitherto unknown in mice after stroke and needs to be further investigated in our next step.
Extracellular signal–regulated kinase 1/2 (ERK1/2), one major member of mitogen-activated protein kinases (MAPK) family, has been implicated in crucial roles in synaptic plasticity and hippocampus-dependent memory (Sweatt, 2004) as well as in cerebral ischemia (Sawe et al., 2008). Our previous study has confirmed that Np65-induced neurite outgrowth involves the activation of ERK1/2 (Owczarek et al., 2011; Beesley et al., 2014). In present study, the ERK1/2 signaling was observed in the ischemic forebrain homogenate at 7 days after stroke. There were no significant differences in total ERK levels between these groups. p-ERK1/2 levels were increased in Np65 KO mice compared to WT mice, which might be related to improved learning and memory in Np65 KO mice (Amuti et al., 2016). Importantly, at 7 days after stroke, a higher increase in p-ERK1/2 levels was observed in Np65 KO mice compared to WT mice. Whether an increase in p-ERK1/2 is protective or detrimental is highly debatable (Sawe et al., 2008). For instance, ERK activity may exaggerate inflammation by up-regulating interleukin 1β (IL-1β), thus leading to necrosis (Wang et al., 2004); it may also block apoptosis by enhancing the level of the antiapoptotic protein Bcl-2 or blocking the proapoptotic protein Bad (Purcell et al., 2003; Roux and Blenis, 2004). In our ischemic settings, Np65 KO mice showed more severe neuronal loss in striatum and hippocampus at 7 days after stroke. Therefore, an increase in p-ERK1/2 might be partially associated with exacerbated damage in Np65 KO mice after stroke. Because p-ERK is differentially expressed in discrete regions of ischemic brains (Hu et al., 2000; Ferrer and Planas, 2003; Zablocka et al., 2003), the limitation of this study is a lack of p-ERK1/2 immunohistological staining to show distribution of p-ERK1/2 positive cells, which helps to understand effects of p-ERK1/2 in this paradigm.
In conclusion, we demonstrated for the first time that Np65 KO mice exhibit more severe neurological deficits and larger infarct volume and more apoptotic cells after ischemic stroke than WT mice. The increased VGluT1 and NMDA receptors and p-ERK1/ 2 may contribute to the increased susceptibility to ischemia in Np65KO mice.
QY designed the work and wrote the manuscript; YH, LH, HZ, and QZ performed the experiments; HL analyzed the data; XL revised the draft. All authors approved the submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by National Natural Science Foundation of China (Grant numbers: 81371213, 81070987, and 30971531) and by grants from the Ministry of Science and Technology of China (2010CB945600 and 2010CB945601).
Amuti, S., Tang, Y., Wu, S., Liu, L., Huang, L., Zhang, H., et al. (2016). Neuroplastin 65 mediates cognitive functions via excitatory/inhibitory synapse imbalance and ERK signal pathway. Neurobiol. Learn. Mem. 127, 72–83. doi: 10.1016/j.nlm.2015.11.020
Bederson, J. B., Pitts, L. H., Germano, S. M., Nishimura, M. C., Davis, R. L., and Bartkowski, H. M. (1986). Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 17, 1304–1308. doi: 10.1161/01.STR.17.6.1304
Beesley, P. W., Herrera-Molina, R., Smalla, K. H., and Seidenbecher, C. (2014). The Neuroplastin adhesion molecules: key regulators of neuronal plasticity and synaptic function. J. Neurochem. 131, 268–283. doi: 10.1111/jnc.12816
Buchan, A. M., Slivka, A., and Xue, D. (1992). The effect of the NMDA receptor antagonist MK-801 on cerebral blood flow and infarct volume in experimental focal stroke. Brain Res. 574, 171–177. doi: 10.1016/0006-8993(92)90814-P
Carrott, L., Bowl, M. R., Aguilar, C., Johnson, S. L., Chessum, L., West, M., et al. (2016). Absence of neuroplastin-65 affects synaptogenesis in mouse inner hair cells and causes profound hearing loss. J. Neurosci. 36, 222–234. doi: 10.1523/JNEUROSCI.1808-15.2016
Chen, N., Liu, C., Yan, N., Hu, W., Zhang, J. G., Ge, Y., et al. (2013). A macaque model of mesial temporal lobe epilepsy induced by unilateral intrahippocampal injection of kainic Acid. PLoS ONE 8:e72336. doi: 10.1371/journal.pone.0072336
Ditlevsen, D. K., Berezin, V., and Bock, E. (2007). Signalling pathways underlying neural cell adhesion molecule-mediated survival of dopaminergic neurons. Eur. J. Neurosci. 25, 1678–1684. doi: 10.1111/j.1460-9568.2007.05436.x
Ditlevsen, D. K., Kohler, L. B., Pedersen, M. V., Risell, M., Kolkova, K., Meyer, M., et al. (2003). The role of phosphatidylinositol 3-kinase in neural cell adhesion molecule-mediated neuronal differentiation and survival. J. Neurochem. 84, 546–556. doi: 10.1046/j.1471-4159.2003.01538.x
Empson, R. M., Buckby, L. E., Kraus, M., Bates, K. J., Crompton, M. R., Gundelfinger, E. D., et al. (2006). The cell adhesion molecule neuroplastin-65 inhibits hippocampal long-term potentiation via a mitogen-activated protein kinase p38- dependent reduction in surface expression of GluR1-containing glutamate receptors. J. Neurochem. 99, 850–860. doi: 10.1111/j.1471-4159.2006.04123.x
Ferrer, I., and Planas, A. M. (2003). Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J. Neuropathol. Exp. Neurol. 62, 329–339. doi: 10.1093/jnen/62.4.329
Gary, D. S., and Mattson, M. P. (2001). Integrin signaling via the PI3-kinase-Akt pathway increases neuronal resistance to glutamate-induced apoptosis. J. Neurochem. 76, 1485–1496. doi: 10.1046/j.1471-4159.2001.00173.x
Gerriets, T., Stolz, E., Walberer, M., Kaps, M., Bachmann, G., and Fisher, M. (2003). Neuroprotective effects of MK-801 in different rat stroke models for permanent middle cerebral artery occlusion: adverse effects of hypothalamic damage and strategies for its avoidance. Stroke 34, 2234–2239. doi: 10.1161/01.STR.0000087171.34637.A9
Hill, I. E., Selkirk, C. P., Hawkes, R. B., and Beesley, P. W. (1988). Characterization of novel glycoprotein components of synaptic membranes and postsynaptic densities, gp65 and gp55, with a monoclonal antibody. Brain Res. 461, 27–43. doi: 10.1016/0006-8993(88)90722-6
Horita, Y., Honmou, O., Harada, K., Houkin, K., Hamada, H., and Kocsis, J. D. (2006). Intravenous administration of glial cell line-derived neurotrophic factor gene-modified human mesenchymal stem cells protects against injury in a cerebral ischemia model in the adult rat. J. Neurosci. Res. 84, 1495–1504. doi: 10.1002/jnr.21056
Hu, B. R., Liu, C. L., and Park, D. J. (2000). Alteration of MAP kinase pathways after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 20, 1089–1095. doi: 10.1097/00004647-200007000-00008
Hulley, P., Schachner, M., and Lubbert, H. (1998). L1 neural cell adhesion molecule is a survival factor for fetal dopaminergic neurons. J. Neurosci. Res. 53, 129–134. doi: 10.1002/(SICI)1097-4547(19980715)53:2<129::AID-JNR1>3.0.CO;2-9
Iadecola, C., Sugimoto, K., Niwa, K., Kazama, K., and Ross, M. E. (2001). Increased susceptibility to ischemic brain injury in cyclooxygenase-1-deficient mice. J. Cereb. Blood Flow Metab. 21, 1436–1441. doi: 10.1097/00004647-200112000-00008
Kiryushko, D., Korshunova, I., Berezin, V., and Bock, E. (2006). Neural cell adhesion molecule induces intracellular signaling via multiple mechanisms of Ca2+ homeostasis. Mol. Biol. Cell 17, 2278–2286. doi: 10.1091/mbc.E05-10-0987
Langnaese, K., Beesley, P. W., and Gundelfinger, E. D. (1997). Synaptic membrane glycoproteins gp65 and gp55 are new members of the immunoglobulin superfamily. J. Biol. Chem. 272, 821–827. doi: 10.1074/jbc.272.2.821
Langnaese, K., Mummery, R., Gundelfinger, E. D., and Beesley, P. W. (1998). Immunoglobulin superfamily members gp65 and gp55: tissue distribution of glycoforms. Febs. Lett. 429, 284–288. doi: 10.1016/S0014-5793(98)00616-4
Matsushita, K., Wu, Y., Qiu, J., Lang-Lazdunski, L., Hirt, L., Waeber, C., et al. (2000). Fas receptor and neuronal cell death after spinal cord ischemia. J. Neurosci. 20, 6879–6887.
Owczarek, S., Kiryushko, D., Larsen, M. H., Kastrup, J. S., Gajhede, M., Sandi, C., et al. (2010). Neuroplastin-55 binds to and signals through the fibroblast growth factor receptor. FASEB J. 24, 1139–1150. doi: 10.1096/fj.09-140509
Owczarek, S., Soroka, V., Kiryushko, D., Larsen, M. H., Yuan, Q., Sandi, C., et al. (2011). Neuroplastin-65 and a mimetic peptide derived from its homophilic binding site modulate neuritogenesis and neuronal plasticity. J. Neurochem. 117, 984–994. doi: 10.1111/j.1471-4159.2011.07269.x
Purcell, J. E., Lenhard, S. C., White, R. F., Schaeffer, T., Barone, F. C., and Chandra, S. (2003). Strain-dependent response to cerebral ischemic preconditioning: differences between spontaneously hypertensive and stroke prone spontaneously hypertensive rats. Neurosci. Lett. 339, 151–155. doi: 10.1016/S0304-3940(02)01476-3
Roux, P. P., and Blenis, J. (2004). ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68, 320–344. doi: 10.1128/MMBR.68.2.320-344.2004
Sarto-Jackson, I., Milenkovic, I., Smalla, K.-H., Gundelfinger, E. D., Kaehne, T., Herrera-Molina, R., et al. (2012). The cell adhesion molecule neuroplastin-65 is a novel interaction partner of γ-aminobutyric acid type A receptors. J. Biol. Chem. 287, 14201–14214. doi: 10.1074/jbc.M111.293175
Sawe, N., Steinberg, G., and Zhao, H. (2008). Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 86, 1659–1669. doi: 10.1002/jnr.21604
Smalla, K. H., Matthies, H., Langnase, K., Shabir, S., Bockers, T. M., Wyneken, U., et al. (2000). The synaptic glycoprotein neuroplastin is involved in long-term potentiation at hippocampal CA1 synapses. Proc. Natl. Acad. Sci. U.S.A. 97, 4327–4332. doi: 10.1073/pnas.080389297
Swanson, R. A., Morton, M. T., Tsao-Wu, G., Savalos, R. A., Davidson, C., and Sharp, F. R. (1990). A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 10, 290–293. doi: 10.1038/jcbfm.1990.47
Sweatt, J. D. (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 14, 311–317. doi: 10.1016/j.conb.2004.04.001
Wang, Z. Q., Wu, D. C., Huang, F. P., and Yang, G. Y. (2004). Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 996, 55–66. doi: 10.1016/j.brainres.2003.09.074
Yang, C., Zhou, L., Gao, X., Chen, B., Tu, J., Sun, H., et al. (2011). Neuroprotective effects of bone marrow stem cells overexpressing glial cell line-derived neurotrophic factor on rats with intracerebral hemorrhage and neurons exposed to hypoxia/reoxygenation. Neurosurgery 68, 691–704. doi: 10.1227/NEU.0b013e3182098a8a
Yuan, Q. L., Yang, C. X., Xu, P., Gao, X. Q., Deng, L., Chen, P., et al. (2007). Neuroprotective effects of ginsenoside Rb1 on transient cerebral ischemia in rats. Brain Res. 1167, 1–12. doi: 10.1016/j.brainres.2007.06.024
Zablocka, B., Dluzniewska, J., Zajac, H., and Domanska-Janik, K. (2003). Opposite reaction of ERK and JNK in ischemia vulnerable and resistant regions of hippocampus: involvement of mitochondria. Brain Res. Mol. Brain Res. 110, 245–252. doi: 10.1016/S0169-328X(02)00653-8
Keywords: neuroplastin 65, stroke, ERK1/2 signaling, NMDA receptor, glutamate
Citation: Hu Y, Zhan Q, Zhang H, Liu X, Huang L, Li H and Yuan Q (2017) Increased Susceptibility to Ischemic Brain Injury in Neuroplastin 65-Deficient Mice Likely via Glutamate Excitotoxicity. Front. Cell. Neurosci. 11:110. doi: 10.3389/fncel.2017.00110
Received: 29 December 2016; Accepted: 31 March 2017;
Published: 19 April 2017.
Edited by:
Laura Cancedda, Fondazione Istituto Italiano di Technologia, ItalyReviewed by:
Alejandro Lopez Tobon, Istituto Europeo di Oncologia, ItalyCopyright © 2017 Hu, Zhan, Zhang, Liu, Huang, Li and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qionglan Yuan, eXFpb25nbEB0b25namkuZWR1LmNu
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.