 Alessandro Guidotti
Alessandro Guidotti Dennis R. Grayson
Dennis R. Grayson Hector J. Caruncho2
Hector J. Caruncho2- 1Department of Psychiatry, The Psychiatric Institute, College of Medicine, University of Illinois at Chicago, Chicago, IL, USA
- 2College of Pharmacy and Nutrition, University of Saskatchewan, Saskatoon, SK, Canada
REELIN (RELN) is a large (420 kDa) glycoprotein that in adulthood is mostly synthesized in GABAergic neurons of corticolimbic structures. Upon secretion in the extracellular matrix (ECM), RELN binds to VLDL, APOE2, and α3β2 Integrin receptors located on dendritic shafts and spines of postsynaptic pyramidal neurons. Reduced levels of RELN expression in the adult brain induce cognitive impairment and dendritic spine density deficits. RELN supplementation recovers these deficits suggesting a trophic action for RELN in synaptic plasticity. We and others have shown that altered RELN expression in schizophrenia (SZ) and bipolar (BP) disorder patients is difficult to reconcile with classical Mendelian genetic disorders and it is instead plausible to associate these disorders with altered epigenetic homeostasis. Support for the contribution of altered epigenetic mechanisms in the down-regulation of RELN expression in corticolimbic structures of psychotic patients includes the concomitant increase of DNA-methyltransferases and the increased levels of the methyl donor S-adenosylmethionine (SAM). It is hypothesized that these conditions lead to RELN promoter hypermethylation and a reduction in RELN protein amounts in psychotic patients. The decreased synthesis and release of RELN from GABAergic corticolimbic neurons could serve as a model to elucidate the epigenetic pathophysiological mechanisms acting at pyramidal neuron dendrites that regulate synaptic plasticity and cognition in psychotic and non-psychotic subjects.
Introduction
REELIN (RELN) is an extracellular matrix (ECM) glycoprotein that controls neuronal cell migration and the lamination of the corticolimbic structures during embryonic development (D’Arcangelo et al., 1995). RELN also plays a role in controlling dendritic spines, and synapse structure and function in adulthood (Costa et al., 2001). Research in the last 20 years, has suggested that abnormal brain RELN expression is a feature that associates with major neuropsychiatric disorders including schizophrenia (SZ), bipolar (BP) disorder (Impagnatiello et al., 1998; Fatemi et al., 2000; Guidotti et al., 2000), autism (Fatemi, 2002), depression (Lussier et al., 2009, 2011, 2013), and Alzheimer’s disease (Herz and Chen, 2006). While the role of RELN in dendritic spine structure, synapse plasticity, and cognitive function in adulthood has been extensively studied, considerably less research has focused on the mechanisms whereby RELN expression is altered in neuropsychiatric conditions. Here, we review evidence for a role of the epigenetic control of the expression of RELN in the regulation of neuronal plasticity and behavior in SZ and BP disorder patients compared with controls devoid of major psychiatric disorders.
RELN in the Adult Mammalian Brain
Neuronal Location
In the cortex and hippocampus of adult rodents and primates, RELN is predominantly synthesized and secreted by GABAergic interneurons (Alcántara et al., 1998; Impagnatiello et al., 1998; Pesold et al., 1998, 1999; Guidotti et al., 2000; Rodriguez et al., 2000; Kadriu et al., 2012). Immunohistochemistry coupled to in situ hybridization studies distinguishes at least two sets of GABAergic interneurons based on their ability to synthesize and secrete RELN. The first synthesizes and secretes RELN onto apical and basal dendrites of pyramidal neurons and includes GABAergic horizontal, double bouquet, multipolar and Martinotti neurons in layers 1 and 2 of the mammalian cortices (Figure 1). The second set of GABAergic neurons, which do not usually express RELN, include chandelier and basket interneurons that innervate the axon initial segment or somata of pyramidal neurons, respectively (Pesold et al., 1998, 1999). In contrast, in cerebellum, RELN is predominately synthesized by glutamatergic granule neurons, and is secreted by their parallel fiber axon terminals into the ECM surrounding the dendrites of GABAergic Purkinje cells (Pesold et al., 1998). Studies in primary cultures of rat cerebellar granule cells suggest that RELN is secreted in the extracellular medium in a manner that is blocked by the constitutive secretory pathway inhibitor brefeldin. Moreover, secretion of RELN is independent of neuronal activity (Lacor et al., 2000). These findings suggest the possibility that secretory pathway activators might be useful in facilitating RELN secretion when RELN expression is compromised.
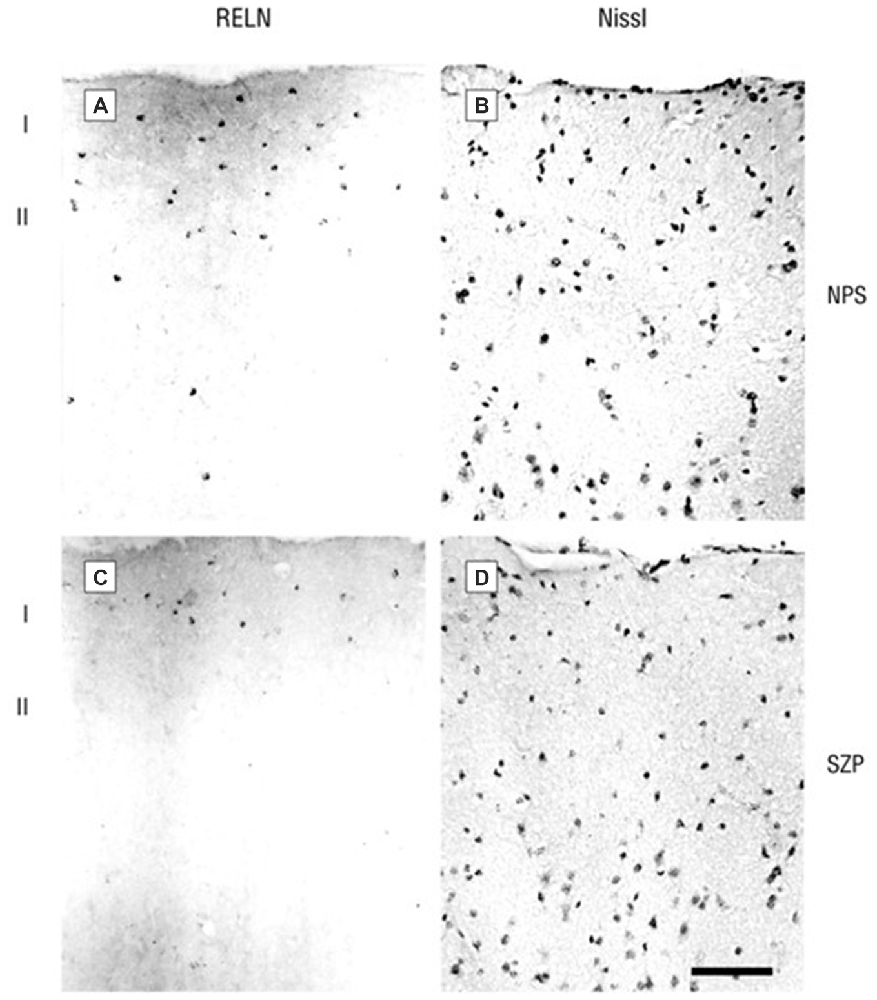
Figure 1. Photomicrographs of 20 μm sections of prefrontal cortex (PFC) of a non-psychiatric subject (NPS) and of a schizophrenia patient (SZP) immunolabeled for RELN (A,C, left side) or Nissl-stained (B,D, right side). RELN positive neurons are mostly localized in layer 1. Note that the NPS has a higher density of RELN-positive cells and also a stronger extracellular diffuse RELN immunostaining halo. Reprinted with permission from Guidotti et al. (2000).
Extracellular Location
Once released in the extracellular space, RELN binds to VLDL, APOE and α3β1 integrin receptors activating the signal transduction system in the effector cells including apical and basilar dendrites of pyramidal neurons in the neocortex or Purkinje cells in the cerebellum (D’Arcangelo et al., 1999; Hiesberger et al., 1999; Dong et al., 2003; Strasser et al., 2004).
Using electron microscopic techniques, Costa et al. (2001) demonstrated the presence of RELN-like immunoreactivity decorating the dendritic shafts and spines of distal apical dendrites of pyramidal neurons in the frontal cortex. This area, as well as the hippocampal fissure, is characterized by strong diffuse RELN–immunoreactivity (Pesold et al., 1998). The colocalization of RELN with the α3 subunit of the integrin receptor at post-synaptic densities of adult rat and primate brains suggests that a RELN signaling mechanism involving integrin and VLDL and APOE2 receptors may be operative in modulating the strength of synaptic function (Rodriguez et al., 2000; Dong et al., 2003; Niu et al., 2004). It has been shown that RELN interacting with VLDL, APOE2, or integrin receptors results in activation of the Src-tyrosine kinase family Fyn-kinase, leading to tyrosine phosphorylation and recruitment of the cytoplasmic adaptor protein DAB1 (Figure 2; Jossin et al., 2003; Bock et al., 2004; Kuo et al., 2005). Studies suggest that DAB1 phosphorylation is a crucial step in the activation of RELN signal transduction pathways (Rice et al., 1998; Trommsdorff et al., 1999; Niu et al., 2004; Howell and Pillai, 2015). DAB1 is frequently expressed in proximity of synapses located on dendritic spines or shafts of cortical pyramidal neurons (Rodriguez et al., 2000). Hence, phosphorylated DAB1 may regulate cytoskeletal protein synthesis at dendrites by activating the translation of dendritic resident mRNAs (see Figure 2, from Costa et al., 2001).
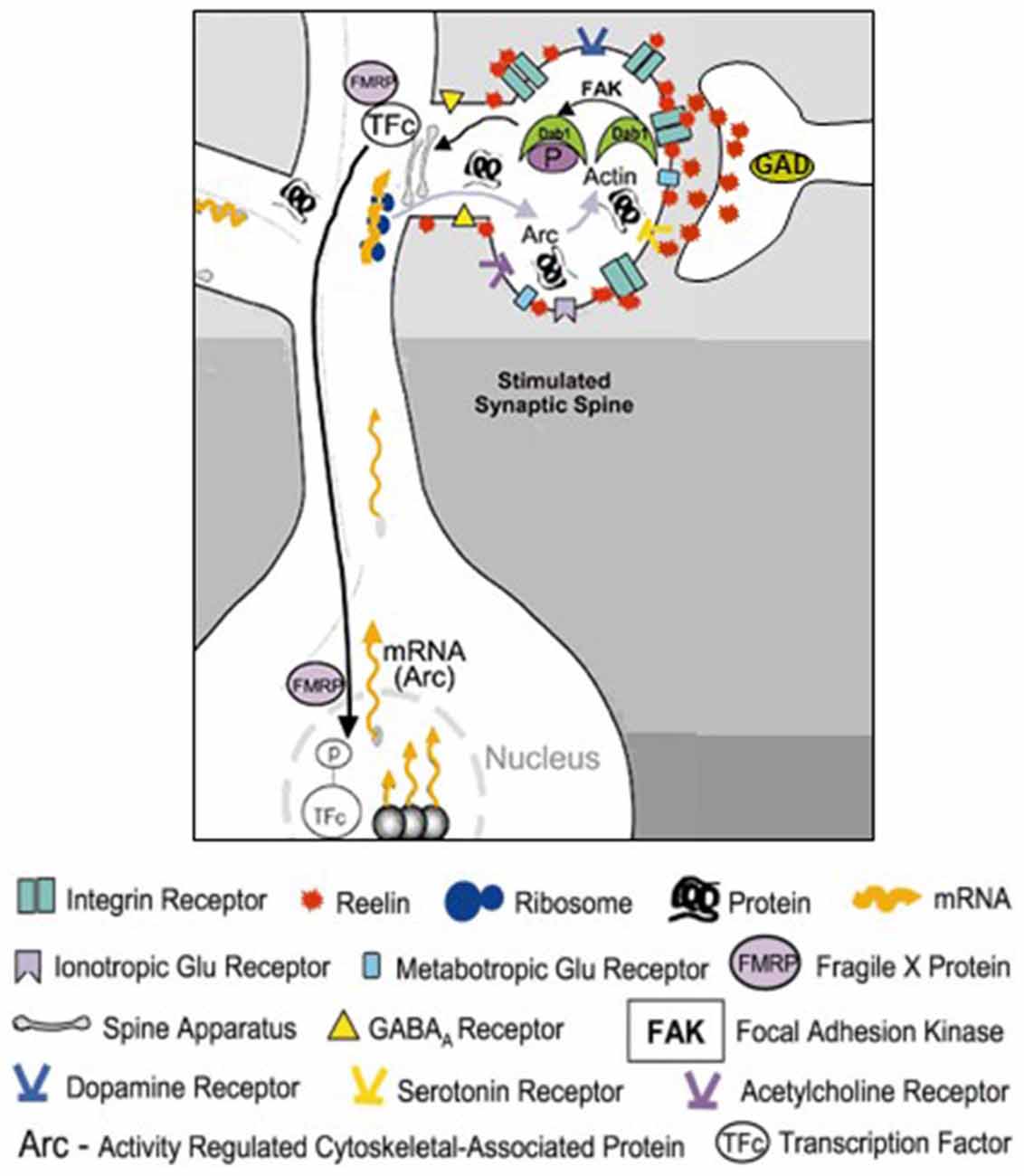
Figure 2. Schematic representation of the RELN signaling pathways in dendritic spines. RELN released in the extracellular matrix (ECM) from the terminal of cortical GABAergic neuron binds to dendritic shafts and spines and modulates the transcriptional function of activated dendritic synapses. Modified from Costa et al. (2001). RELN binding to Integrin receptors located on dendritic spines mediates activation of focal adhesion kinase (FAK) that directly or indirectly, via activation of Fyn kinase, phosphorylates DAB1 adaptor protein resident in dendrites. Phosphorylated dendritic DAB1 may recruit or activate ribosomal structures and induce the synthesis of ARC (Activity regulated cytoskeletal-associated protein) or cytoskeletal proteins like β-actin.
RELN Regulates Spine Density and Excitatory Synaptic Function
Animal models in which RELN expression is genetically reduced provide important information on the impact of RELN on synaptic plasticity and cognition. Reeler mice display altered LTP and deficits in active avoidance tasks (Goldowitz and Koch, 1986; Marrone et al., 2006). Young adult heterozygous reeler mice (HRM) which exhibit a 50% reduction in RELN content have significantly reduced dendritic spine densities and also show a deficit in LTP (Tueting et al., 1999, 2006; Liu et al., 2001; Levenson et al., 2008; Niu et al., 2008; Iafrati et al., 2014). Adult HRM also have a defective molecular composition of the synaptic structure (Ventruti et al., 2011), as well as deficits in excitatory postsynaptic responses to glutamate receptor agonists and reduced LTP (Levenson et al., 2006). Addition of recombinant RELN to hippocampal slices or direct injection of RELN into the cerebral ventricles enhances hippocampal LTP (Beffert et al., 2006; Pujadas et al., 2010; Rogers et al., 2011).
HRM also display deficits in cognitive function (Krueger et al., 2006; Stranahan et al., 2011), executive function (Brigman et al., 2006), fear conditioning learning (Ammassari-Teule et al., 2009), anxiety and motor impulsivity (Ognibene et al., 2007). Importantly, RELN supplementation recovers sensory motor gating, synaptic plasticity, and associative learning deficits in HRM (Rogers et al., 2013). In addition to the HRM data described above, Lussier et al. (2013) reported that hippocampal RELN deficiency elicited by chronic stress (repeated corticosterone treatment) can impair adult hippocampal neurogenesis and lead to the development of a depression-like phenotype. Co-treatment with antidepressant drugs prevents both RELN deficit and the development of the depression-like phenotype (Fenton et al., 2015).
Addition of recombinant RELN to cortical synaptosomes in vitro induces the expression of activity-regulated cytoskeleton-associated protein (Arc; Dong et al., 2003), and augments the density and clustering of proteins in postsynaptic membranes (i.e., neurotransmitter receptors), which provides further evidence of a functional role for RELN in regulating the synaptic strength of glutamatergic inputs onto dendritic spines (Caruncho et al., 2004).
RELN in the Brains of Schizophrenia (SZ) and Bipolar (BP) Disorder Patients
A number of molecular, anatomical (dendritic spine density), behavioral, and cognitive deficits associated with reduced RELN expression (mRNA and protein) are observed in subjects with SZ and BP disorder. In different post-mortem brain cohorts, we and others have demonstrated that RELN expression is reduced by approximately 50% in the prefrontal cortex (PFC), temporal cortex, hippocampus, and caudate nucleus of patients with SZ and BP disorder (Impagnatiello et al., 1998; Fatemi et al., 2000; Guidotti et al., 2000). In successive studies we found that the expression of RELN was paralleled by decreases in the levels of GAD67 but not DAB1 or GAD65. Slices from the same samples immunostained for RELN and counterstained for Nissl or NeuN to recognize neurons showed that RELN-positive neurons were significantly decreased by 30–50% in patients with SZ or BP disorder with psychosis but not in those with unipolar depression when compared to non-psychiatric subject (NPSs; Figure 1). Differences were absent for GAD65, and NeuN expression implying that RELN and GAD67 down-regulation is unrelated to neuronal damage (Guidotti et al., 2000). The RELN and GAD67 downregulation is also unrelated to postmortem interval, dose, duration, or presence of antipsychotic medication. Similar to HRM, RELN deficiency in the neocortex of SZ and BP disorder patients is associated with a decrease in GAD67, reduced prepulse inhibition to startle, and loss of dendritic spines, all features associated with SZ pathology (Tueting et al., 1999; Glantz and Lewis, 2001; Liu et al., 2001; Grayson and Guidotti, 2013).
Like the RELN deficiency in the cerebellar cortex of HRM, the RELN deficiency in cerebellar cortex of SZ and BP disorder patients is associated with a 20% decrease of GABAergic Purkinje neurons (Hadj-Sahraoui et al., 1996; Maloku et al., 2010). Collectively these data suggest that RELN plays a central role in inducing and maintaining the structure and function of GABAergic and glutamatergic neurons and their reciprocal interactions (Grayson and Guidotti, 2013).
Since SZ and BP disorder have a neurodevelopmental origin (Folsom and Fatemi, 2013) and RELN is a major player in brain development and maturation (D’Arcangelo et al., 1995), an important question raised by these studies is whether the altered epigenetic (promoter hypermethylation) regulation of RELN in brains of SZ and BP patients is initiated early in embryonic or perinatal life or develops later in life as the consequence of the GABAergic neuropathology related to the development of SZ morbidity. To address this question, the extent of methylation of the RELN promoter was measured in offspring born from mice stressed during pregnancy. These offspring, at adulthood, display SZ-like behavioral endophenotypes (increased locomotor activity, PPI, social recognition deficits), and a decrease of RELN, GAD67, and BDNF expression associated with an increase in methylation at their respective promoters. We also found that the amount of Methyl CpG Binding Protein 2 (MECP2) binding to the RELN promoter at birth was higher than that observed in the adult (Matrisciano et al., 2013). These data suggest that RELN promoter hypermethylation is likely initiated early in life, including during embryonic life, and is then maintained throughout adulthood.
Is an Altered Epigenetic Regulation of Gene Expression the Molecular Mechanism Mediating RELN Expression Down-Regulation in SZ and BP Disorder?
Mutations in the RELN gene are associated with a form of autosomal recessive lissencephaly with abnormal axonal connectivity, and cerebellar hypoplasia (Hong et al., 2000). Human subjects with RELN gene mutations exhibit marked ventricular dilation, mental retardation, and epilepsy and a marked decrease in muscle tone that appears of neurogenic origin (Hourihane et al., 1993). Heterozygous RELN mutations have been shown to cause autosomal-dominant lateral temporal epilepsy (Dazzo et al., 2015).
A highly conserved single nucleotide polymorphism (SNP) has been identified in the vicinity of the regulatory region of the RELN gene (Shifman et al., 2008; Wedenoja et al., 2010). This polymorphism is associated with an increased risk of psychotic symptoms. Although these studies highlight the importance of RELN gene variants as risk factors in the etiopathogenesis of psychiatric disorders, it is important to note that variants in the RELN gene are rare and cannot explain the high frequency of RELN expression downregulation observed in the general population of SZ, BP disorder and autism spectrum disorder patients (Zhang et al., 2002; Lintas and Persico, 2010; Grayson and Guidotti, 2013; Wang et al., 2014; Zhubi et al., 2014).
The epidemiological and clinical evidence that SZ and BP disorders do not follow the rules expected for a Mendelian-genetic disorder led to the proposal that environmental insults may influence RELN gene expression by altering epigenetic regulatory mechanisms and led to the hypothesis that epigenetic factors are operative in mediating changes in the expression of RELN and other SZ candidate genes in psychotic patients (Costa et al., 2003). To better understand the rules governing the epigenetic regulation of RELN, we cloned the human gene and experimentally examined its regulation in both neuroprogenitor NT2 cells (Chen et al., 2002; Mitchell et al., 2005) and mouse cortical neurons in vitro (Dong et al., 2003; Noh et al., 2005). Data from these studies support the concept that the RELN promoter is regulated epigenetically through changes in DNA methylation. Furthermore, we have reported that the down-regulation of RELN expression in GABAergic neurons of SZ and BP patients is associated with an overexpression of DNA methyltransferase 1 (DNMT1) and DNA methyltransferase 3a (DNMT3a) in neocortical and striatal GABAergic neurons (Veldic et al., 2004, 2007; Ruzicka et al., 2007). DNMTs are a family of enzymes that catalyze the transfer of a methyl group from the methyl donor S-adenosylmethionine (SAM) to the 5′ carbon of cytosine of many gene promoters (Grayson and Guidotti, 2013). Increased promoter methylation generally leads to decreased gene expression. Interestingly, the inhibitory action of DNMTs on RELN expression also likely occur through the formation of chromatin repressor complexes which include, in addition of DNMTs, also the methyl CpG binding domain proteins, SIN3A, and histone deacetylases (see Grayson and Guidotti, 2013 for review).
The hypothesis that an epigenetic pathology of the Reln promoter is operative in the transcriptional down-regulation of the corresponding gene in SZ or BP disorder patients is supported by the evidence that there is an increased level of SAM in the PFC of these patients (Guidotti et al., 2007), and that hypermethylation of the RELN promoter (Abdolmaleky et al., 2005; Grayson et al., 2005, 2006; Lintas and Persico, 2010) is associated with the down-regulation of the corresponding protein in the PFC of psychotic patients (Guidotti et al., 2000), although negative findings for RELN promoter hypermethylation have also been reported (Mill et al., 2008).
In other studies a decreased histone methylation at GABAergic gene promoters (Huang et al., 2007), and an increased histone deacetylase -1 expression and down regulation of GABAergic gene expression in PFC and hippocampus of SZ patients have been reported (Benes et al., 2007; Sharma et al., 2008). A summary of many of the methylation studies of RELN in neuropsychiatric patients, SZ-like epigenetic mouse models, and neuronal culture systems are summarized in Table 1. These data are consistent with the epigenetic GABAergic theory of major psychosis (Costa et al., 2003; Grayson and Guidotti, 2013) and suggest that RELN promoter methylation should be further studied to establish its temporal and casual association with the etiopathogenesis of SZ and BP disorder.
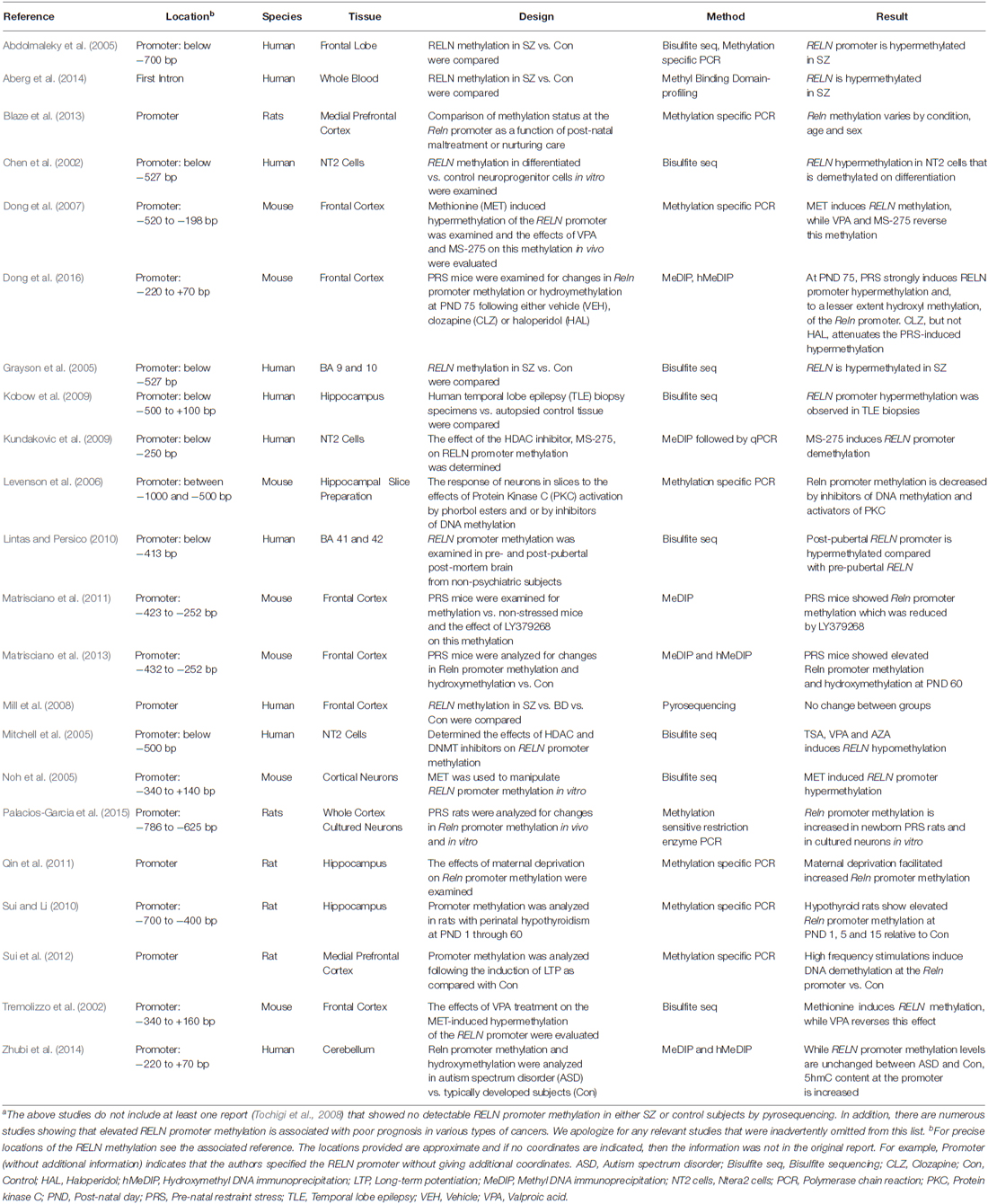
Table 1. Summary showing studies of RELN methylation relevant to neurobiologya.
RELN, Spine Density Down Regulation and Cognitive Performance Deficits Induced by L-Methionine Treatment
Support for the hypothesis that an increase of DNA methylation contributes to the down-regulation of RELN and other GABAergic or glutamatergic genes in psychotic patients is sustained by clinical studies conducted in the early 1970s (for review see Wyatt et al., 1971; Cohen et al., 1974; Grayson et al., 2009). In these studies L-methionine (MET, the precursor of SAM), administered in large doses (10–20 g/day) for 3–4 weeks to SZ patients was reported to exacerbate psychotic symptomatology (Cohen et al., 1974; Grayson et al., 2009). Patients were administered large doses of L-methionine either with or without a monoamine oxidase inhibitor in an attempt to reduce the levels of putative bioactive psychedelic compounds. Interestingly, many of the treated patients responded with a worsening of their symptoms (Cohen et al., 1974).
In both mouse FC and neuronal cultures, the administration of large doses of L-methionine increases SAM levels and facilitates the hypermethylation of GABAergic gene promoters, including Reln, and GAD67 and the reduced expression of these genes (Tremolizzo et al., 2002, 2005; Mitchell et al., 2005; Noh et al., 2005; Chen et al., 2007). Similar to the HRM, spine density is also decreased in the dendrites of mice treated with L-methionine (Figure 3, Tueting et al., 2010). Furthermore, L-methionine treated mice display SZ-like behavioral abnormalities (Tremolizzo et al., 2005). Collectively, these data suggest that the reduction of dendritic spines observed in brain of L-methionine-treated mice are likely due to MET-induced altered epigenetic mechanisms that lead to decreased expression of RELN (Tremolizzo et al., 2005; Tueting et al., 2010).
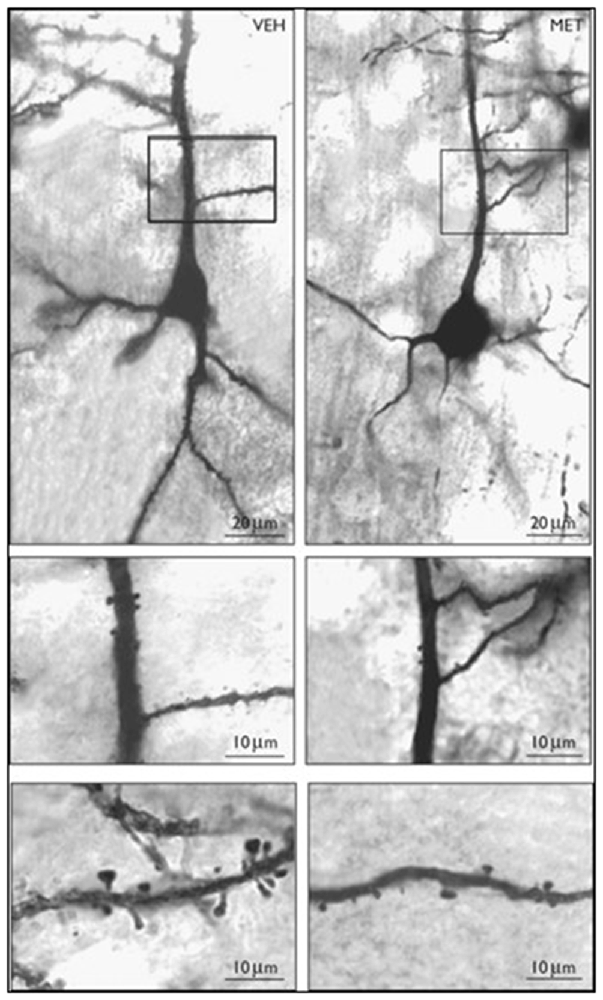
Figure 3. Photomicrographs of Golgi stained layer III pyramidal neurons in vehicle (VEH) and methionine (MET) treated mouse frontal cortex. Top panel: 10× objective; Middle panel: 20×; Bottom panel: 100× Vehicle or L-methionine (5.2 mmol/kg/twice a day) was administered for 14 days. Reprinted with permission from Tueting et al. (2010).
RELN Promoter Methylation in Neurons is a Dynamic Process that can be Targeted by Environmental Factors and Drugs
The dogma that in post-mitotic neurons DNA methylation patterns are established during development and remain stable thereafter (Razin and Shemer, 1995) has been challenged by convincing evidence that in post-mitotic neurons, methylation patterns of specific cytosine/guanine (CpG) dinucleotide-rich promoters, change rapidly. Thus, in neurons, promoter methylation provides a series of targets on which the environment, drugs, and/or toxins can modify transcription and affect neuronal phenotype profiles without altering the genotype (Szyf, 2009). To verify this hypothesis, we treated (Tremolizzo et al., 2002, 2005; Tueting et al., 2010) mice protractedly with L-methionine (as described above) and measured the ratio of 5 methyl cytosine (5mC) to unmethylated cytosine (C) of the murine RELN CpG-enriched promoter region from −340 to +160 bp (Tremolizzo et al., 2005) or the murine GAD67 CpG-enriched promoter region from −760 to −311 bp (Satta et al., 2008) by measuring the fraction of promoters immunoprecipitated by specific anti-5mC or anti-MeCP2 antibodies with competitive RT-PCR and internal standards (Dong et al., 2005). We found that (Dong et al., 2005; Tremolizzo et al., 2005) methionine induces an increase of brain RELN and GAD67 promoter methylation (Dong et al., 2005), and downregulation of RELN and GAD67 mRNA and cognate protein expression associated with decreased spine density (Figure 3), and SZ-like behavioral modifications (Tremolizzo et al., 2002, 2005; Tueting et al., 2010). The effects of methionine on the RELN promoter, RELN protein level, dendritic spine density, and SZ-like behavioral modifications are reversed by the administration of Valproic acid (VPA) and other HDAC inhibitors (Dong et al., 2005; Tremolizzo et al., 2005). These findings, together with data obtained in the HRM, suggest the working hypothesis that the down-regulation of spine density and SZ-like behavioral modifications in L-methionine treated mice may be, in part, due to decreased expression of RELN.
Concluding Remarks
SZ and BP are neurodevelopmental disorders with genetic risk load and behavioral and neurochemical SZ-like phenotypes triggered by exposure to prenatal or perinatal environmental insults: stress, toxins, infection, trauma. In mice exposed prenatally to restraint stress, we found increased DNMT levels that are associated with RELN promoter hypermethylation, RELN expression downregulation, SZ-like epigenetic behavioral modifications, and decreased dendritic spine density in adultood (Tremolizzo et al., 2005; Tueting et al., 2010; Dong et al., 2016). L-Methionine supplementation in rats induces epigenetic variations including RELN promoter hypermethylation in offspring (Weaver et al., 2005). Further, there is an epigenomic reprogramming of RELN and glucocorticoid receptors in hippocampal pyramidal neurons after methionine administration (Weaver et al., 2006). Our studies in cultured mouse cortical neurons (Noh et al., 2005) and human neuronal progenitors (Kundakovic et al., 2007, 2009) not only show that the hypermethylation of promoters induced by L-methionine is blocked by siRNA-mediated DNMT-KO or by reduction of DNMT activity with small molecule antagonists but also that this blockade induces the overexpression of RELN, GAD67, or BDNF proteins (Kundakovic et al., 2007, 2009).
Collectively, these data challenge the classic concept that 5-methylcytosine patterns in DNA remain stable in post-mitotic neurons and strongly suggest that by increasing brain SAM content, L-methionine facilitates the promoter methylation mediated by DNMT1 or DNMT3a in the central nervous system (Grayson and Guidotti, 2013). Unlike the DNA sequence of a cell, which is stable and strongly conserved, epigenetic processes that impact DNA methylation and chromatin architecture are highly dynamic. That is, they can be tissue-specific, developmentally-regulated, and modified by a wide range of drugs and other environmental factors (Szyf, 2009; Ptak and Petronis, 2010; Grayson and Guidotti, 2013; Dong et al., 2016).
Studies using the L-methionine mouse model or offspring of restraint stressed mothers may be aimed at determining whether antipsychotics capable of reducing RELN promoter methylation (e.g., clozapine), enhance spine density, and relieve SZ-like epigenetic behaviors (Tremolizzo et al., 2005; Dong et al., 2016). These models should provide useful preclinical tools for screening small molecules for their capacity to reverse SZ candidate gene promoter methylation and the associated neuronal and behavioral deficits.
Author Contributions
All authors contributed equally to the ideas and editing of the manuscript.
Funding
AG is supported by the following NIH grants AA022538, R01 MH093348, R01 MH101043, DRG is supported by AA022538. HJC is supported by a SHRF Establishment Grant, and a NSERC Discovery grant.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdolmaleky, H. M., Cheng, K. H., Russo, A., Smith, C. L., Faraone, S. V., Wilcox, M., et al. (2005). Hypermethylation of the reelin (Reelin) promoter in the brain of schizophrenic patients: a preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 134B, 60–66. doi: 10.1002/ajmg.b.30140
Aberg, K. A., McClay, J. L., Nerella, S., Clark, S., Kumar, G., Chen, W., et al. (2014). Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 71, 255–264. doi: 10.1001/jamapsychiatry.2013.3730
Alcántara, S., Ruiz, M., D’Arcangelo, G., Ezan, F., de Lecea, L., Curran, T., et al. (1998). Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 18, 7779–7799.
Ammassari-Teule, M., Sgobio, C., Biamonte, F., Marrone, C., Mercuri, N. B., and Keller, F. (2009). Reelin haploinsufficiency reduces the density of PV+ neurons in circumscribed regions of the striatum and selectively alters striatal-based behaviors. Psychopharmacology (Berl) 204, 511–521. doi: 10.1007/s00213-009-1483-x
Beffert, U., Durudas, A., Weeber, E. J., Stolt, P. C., Giehl, K. M., Sweatt, J. D., et al. (2006). Functional dissection of Reelin signaling by site-directed disruption of Disabled-1 adaptor binding to apolipoprotein E receptor 2: distinct roles in development and synaptic plasticity. J. Neurosci. 26, 2041–2052. doi: 10.1523/jneurosci.4566-05.2006
Benes, F. M., Lim, B., Matzilevich, D., Walsh, J. P., Subburaju, S., and Minns, M. (2007). Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. U S A 104, 10164–10169. doi: 10.1073/pnas.0703806104
Blaze, J., Scheuing, L., and Roth, T. L. (2013). Differential methylation of genes in the medial prefrontal cortex of developing and adult rats following exposure to maltreatment or nurturing care during infancy. Dev. Neurosci. 35, 306–316. doi: 10.1159/000350716
Bock, H. H., Jossin, Y., May, P., Bergner, O., and Herz, J. (2004). Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. J. Biol. Chem. 279, 33471–33479. doi: 10.1074/jbc.m401770200
Brigman, J. L., Padukiewicz, K. E., Sutherland, M. L., and Rothblat, L. A. (2006). Executive functions in the heterozygous reeler mouse model of schizophrenia. Behav. Neurosci. 120, 984–988. doi: 10.1037/0735-7044.120.4.984
Caruncho, H. J., Dopeso-Reyes, I. G., Loza, M. I., and Rodríguez, M. A. (2004). A GABA, reelin and the neurodevelopmental hypothesis of schizophrenia. Crit. Rev. Neurobiol. 16, 25–32. doi: 10.1615/CritRevNeurobiol.v16.i12.20
Chen, Y., Kundakovic, M., Agis-Balboa, R. C., Pinna, G., and Grayson, D. R. (2007). Induction of the reelin promoter by retinoic acid is mediated by Sp1. J. Neurochem. 103, 650–665. doi: 10.1111/j.1471-4159.2007.04797.x
Chen, Y., Sharma, R. P., Costa, R. H., Costa, E., and Grayson, D. R. (2002). On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 30, 2930–2939. doi: 10.1093/nar/gkf401
Cohen, S. M., Nichols, A., Wyatt, R., and Pollin, W. (1974). The administration of methionine to chronic schizophrenic patients: a review of ten studies. Biol. Psychiatry 8, 209–225.
Costa, E., Chen, Y., Davis, J., Dong, E., Noh, J. S., Tremolizzo, L., et al. (2003). REELIN and schizophrenia: a disease at the interface of the genome and the epigenome. Mol. Interv. 2, 47–57. doi: 10.1124/mi.2.1.47
Costa, E., Davis, J., Grayson, D. R., Guidotti, A., Pappas, G. D., and Pesold, C. (2001). Dendritic spine hypoplasticity and downregulation of reelin and GABAergic tone in schizophrenia vulnerability. Neurobiol. Dis. 8, 723–742. doi: 10.1006/nbdi.2001.0436
D’Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D. S., Sheldon, M., and Curran, T. (1999). Reelin is a ligand for lipoprotein receptors. Neuron 24, 471–479. doi: 10.1016/S0896-6273(00)80860-0
D’Arcangelo, G., Miao, G. G., Chen, S. C., Soares, H. D., Morgan, J. I., and Curran, T. (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723. doi: 10.1038/374719a0
Dazzo, E., Fanciulli, M., Serioli, E., Minervini, G., Pulitano, P., and Binelli, S. (2015). Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am. J. Hum. Genet. 96, 992–1000. doi: 10.1016/j.ajhg.2015.04.020
Dong, E., Agis-Balboa, R. C., Simonini, M. V., Grayson, D. R., Costa, E., and Guidotti, A. (2005). Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine- induced mouse model of schizophrenia. Proc. Natl. Acad. Sci. U S A 102, 12578–12583. doi: 10.1073/pnas.0505394102
Dong, E., Caruncho, H., Liu, W. S., Smalheiser, N. R., Grayson, D. R., Costa, E., et al. (2003). A reelin-integrin receptor interaction regulates Arc mRNA translation in synaptoneurosomes. Proc. Natl. Acad. Sci. U S A 100, 5479–5484. doi: 10.1073/pnas.1031602100
Dong, E., Guidotti, A., Grayson, D. R., and Costa, E. (2007). Histone hyperacetylation induces demethylation of reelin and 67-kDa glutamic acid decarboxylase promoters. Proc. Natl. Acad. Sci. U S A 104, 4676–4681. doi: 10.1073/pnas.0700529104
Dong, E., Tueting, P., Matrisciano, F., Grayson, D. R., and Guidotti, A. (2016). Behavioral and molecular neuroepigenetic alterations in prenatally stressed mice: relevance for the study of chromatin remodeling properties of antipsychotic drugs. Transl. Psychiatry 6:e711. doi: 10.1038/tp.2015.191
Fatemi, S. H. (2002). The role of Reelin in pathology of autism. Mol. Psychiatry 7, 919–920. doi: 10.1038/sj.mp.4001248
Fatemi, S. H., Earle, J. A., and McMenomy, T. (2000). Hippocampal CA4 Reelin-positive neurons. Mol. Psychiatry 5:571. doi: 10.1038/sj.mp.4000794
Fenton, E. Y., Fournier, N. M., Lussier, A. L., Romay-Tallon, R., Caruncho, H. J., and Kalynchuk, L. E. (2015). Imipramine protects against the deleterious effects of chronic corticosterone on depression-like behavior, hippocampal reelin expression and neuronal maturation. Prog. Neuropsychopharmacol. Biol. Psychiatry 60, 52–59. doi: 10.1016/j.pnpbp.2015.02.001
Folsom, T. D., and Fatemi, S. H. (2013). The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 68, 122–135. doi: 10.1016/j.neuropharm.2012.08.015
Glantz, L. A., and Lewis, D. A. (2001). Dendritic spine density in schizophrenia and depression. Arch. Gen. Psychiatry 58:203. doi: 10.1001/archpsyc.58.2.203
Goldowitz, D., and Koch, J. (1986). Performance of normal and neurological mutant mice on radial arm maze and active avoidance tasks. Behav. Neural Biol. 46, 216–226. doi: 10.1016/s0163-1047(86)90696-5
Grayson, D. R., Chen, Y., Costa, E., Dong, E., Guidotti, A., Kundakovic, M., et al. (2006). The human reelin gene: transcription factors (+), repressors (−) and the methylation switch (+/−) in schizophrenia. Pharmacol. Ther. 111, 272–286. doi: 10.1016/j.pharmthera.2005.01.007
Grayson, D. R., Chen, Y., Dong, E., Kundakovic, M., and Guidotti, A. (2009). From trans-methylation to cytosine methylation: evolution of the methylation hypothesis of schizophrenia. Epigenetics 4, 144–149. doi: 10.4161/epi.4.3.8534
Grayson, D. R., and Guidotti, A. (2013). The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology 38, 138–166. doi: 10.1038/npp.2012.125
Grayson, D. R., Jia, X., Chen, Y., Sharma, R. P., Mitchell, C. P., Guidotti, A., et al. (2005). Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. U S A 102, 9341–9346. doi: 10.1073/pnas.0503736102
Guidotti, A., Auta, J., Davis, J. M., Di-Giorgi-Gerevni, V., Dwivedi, Y., Grayson, D. R., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 57, 1061–1069. doi: 10.1001/archpsyc.57.11.1061
Guidotti, A., Ruzicka, W., Grayson, D. R., Veldic, M., Pinna, G., Davis, J. M., et al. (2007). S-adenosyl methionine and DNA methyltransferase-1 mRNA overexpression in psychosis. Neuroreport 18, 57–60. doi: 10.1097/wnr.0b013e32800fefd7
Hadj-Sahraoui, N., Frédéric, F., Delhaye-Bouchaud, N., and Mariani, J. (1996). Gender effect on Purkinje cell loss in the cerebellum of the heterozygous reeler mouse. J. Neurogenet. 11, 45–58. doi: 10.3109/01677069609107062
Herz, J., and Chen, Y. (2006). Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 7, 850–859. doi: 10.1038/nrn2009
Hiesberger, T., Trommsdorff, M., Howell, B. W., Goffinet, A., Mumby, M. C., Cooper, J. A., et al. (1999). Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–489. doi: 10.1016/s0896-6273(00)80861-2
Hong, S. E., Shugart, Y. Y., Huang, D. T., Shahwan, S. A., Grant, P. E., Hourihane, J. O., et al. (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human Reelin mutations. Nat. Genet. 26, 93–96. doi: 10.1038/79246
Hourihane, J. O., Bennett, C. P., Chaudhuri, R., Robb, S. A., and Martin, N. D. (1993). A sibship with a neuronal migration defect, cerebellar hypoplasia and congenital lymphedema. Neuropediatrics 24, 43–46. doi: 10.1055/s-2008-1071511
Howell, K. R., and Pillai, A. (2015). Long-term effects of prenatal hypoxia on schizophrenia-like phenotype in heterozygous reeler mice. Mol. Neurobiol. doi: 10.1007/s12035-015-9265-4 [Epub ahead of print].
Huang, H. S., Matevossian, A., Whittle, C., Kim, S. Y., Schumacher, A., Baker, S. P., et al. (2007). Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J. Neurosci. 27, 11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007
Iafrati, J., Orejarena, M. J., Lassalle, O., Bouamrane, L., Gonzalez-Campo, C., and Chavis, P. (2014). Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 19, 417–426. doi: 10.1038/mp.2013.66
Impagnatiello, F., Guidotti, A. R., Pesold, C., Dwivedi, Y., Caruncho, H., Pisu, M. G., et al. (1998). A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U S A 95, 15718–15723. doi: 10.1073/pnas.95.26.15718
Jossin, Y., Bar, I., Ignatova, N., Tissir, F., De Rouvroit, C. L., and Goffinet, A. M. (2003). The reelin signaling pathway: some recent developments. Cereb. Cortex 13, 627–633. doi: 10.1093/cercor/13.6.627
Kadriu, B., Guidotti, A., Chen, Y., and Grayson, D. R. (2012). DNA methyltransferases1 (DNMT1) and 3a (DNMT3a) colocalize with GAD67-positive neurons in the GAD67-GFP mouse brain. J. Comp. Neurol. 520, 1951–1964. doi: 10.1002/cne.23020
Kobow, K., Jeske, I., Hildebrandt, M., Hauke, J., Hahnen, E., Buslei, R., et al. (2009). Increased Reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 68, 356–364. doi: 10.1097/NEN.0b013e31819ba737
Krueger, D. D., Howell, J. L., Hebert, B. F., Olausson, P., Taylor, J. R., and Nairn, A. C. (2006). Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology (Berl) 189, 95–104. doi: 10.1007/s00213-006-0530-0
Kundakovic, M., Chen, Y., Costa, E., and Grayson, D. R. (2007). DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Mol. Pharmacol. 71, 644–653. doi: 10.1124/mol.106.030635
Kundakovic, M., Chen, Y., Guidotti, A., and Grayson, D. R. (2009). The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol. Pharmacol. 75, 342–354. doi: 10.1124/mol.108.051763
Kuo, G., Arnaud, L., Kronstad-O’Brien, P., and Cooper, J. A. (2005). Absence of Fyn and Src causes a reeler-like phenotype. J. Neurosci. 25, 8578–8586. doi: 10.1523/JNEUROSCI.1656-05.2005
Lacor, P. N., Grayson, D. R., Auta, J., Sugaya, I., Costa, E., and Guidotti, A. (2000). Reelin secretion from glutamatergic neurons in culture is independent from neurotransmitter regulation. Proc. Natl. Acad. Sci. U S A 97, 3556–3561. doi: 10.1073/pnas.97.7.3556
Levenson, J. M., Qiu, S., and Weeber, E. J. (2008). The role of reelin in adult synaptic function and the genetic and epigenetic regulation of the reelin gene. Biochim. Biophys. Acta 1779, 422–431. doi: 10.1016/j.bbagrm.2008.01.001
Levenson, J. M., Roth, T. L., Lubin, F. D., Miller, C. A., Huang, I. C., Desai, P., et al. (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 281, 15763–15773. doi: 10.1074/jbc.m511767200
Lintas, C., and Persico, A. M. (2010). Neocortical Reelin promoter methylation increases significantly after puberty. Neuroreport 21, 114–118. doi: 10.1097/WNR.0b013e328334b343
Liu, W. S., Pesold, C., Rodriguez, M. A., Carboni, G., Auta, J., Lacor, P., et al. (2001). Down-regulation of dendritic spine and glutamic acid decarboxylase 67 expressions in the reelin haploinsufficient heterozygous reeler mouse. Proc. Natl. Acad. Sci. U S A 98, 3477–3482. doi: 10.1073/pnas.051614698
Lussier, A. L., Caruncho, H. J., and Kalynchuk, L. E. (2009). Repeated exposure to corticosterone, but not restraint, decreases the number of reelin-positive cells in the adult rat hippocampus. Neurosci. Lett. 460, 170–174. doi: 10.1016/j.neulet.2009.05.050
Lussier, A. L., Lebedeva, K., Fenton, E. Y., Guskjolen, A., Caruncho, H. J., and Kalynchuk, L. L. E. (2013). The progressive development of depression-like behavior in corticosterone-treated rats is paralleled by slowed granule cell maturation and decreased reelin expression in the adult dentate gyrus. Neuropharmacology 71, 174–183. doi: 10.1016/j.neuropharm.2013.04.012
Lussier, A. L., Romay-Tallón, R., Kalynchuk, L. E., and Caruncho, H. J. (2011). Reelin as a putative vulnerability factor for depression: examining the depressogenic effects of repeated corticosterone in heterozygous reeler mice. Neuropharmacology 60, 1064–1074. doi: 10.1016/j.neuropharm.2010.09.007
Maloku, E., Covelo, I. R., Hanbauer, I., Guidotti, A., Kadriu, B., Hu, Q., et al. (2010). Lower number of cerebellar Purkinje neurons in psychosis is associated with reduced reelin expression. Proc. Natl. Acad. Sci. U S A 107, 4407–4411. doi: 10.1073/pnas.0914483107
Marrone, M. C., Marinelli, S., Biamonte, F., Keller, F., Sgobio, C. A., Ammassari-Teule, M., et al. (2006). Altered cortico-striatal synaptic plasticity and related behavioural impairments in reeler mice. Eur. J. Neurosci. 24, 2061–2070. doi: 10.1111/j.1460-9568.2006.05083.x
Matrisciano, F., Dong, E., Gavin, D. P., Nicoletti, F., and Guidotti, A. (2011). Activation of group II metabotropic glutamate receptors promotes DNA demethylation in the mouse brain. Mol. Pharmacol. 80, 174–182. doi: 10.1124/mol.110.070896
Matrisciano, F., Tueting, P., Dalal, I., Kadriu, B., Grayson, D. R., Davis, J. M., et al. (2013). Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 68, 184–194. doi: 10.1016/j.neuropharm.2012.04.013
Mill, J., Tang, T., Kaminsky, Z., Khare, T., Yazdanpanah, S., and Bouchard, L. (2008). Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 82, 696–711. doi: 10.1016/j.ajhg.2008.01.008
Mitchell, C. P., Chen, Y., Kundakovic, M., Costa, E., and Grayson, D. R. (2005). Histone deacetylase inhibitors decrease reelin promoter methylation in vitro. J. Neurochem. 93, 483–492. doi: 10.1111/j.1471-4159.2005.03040.x
Niu, S., Renfro, A., Quattrocchi, C. C., Sheldon, M., and D’Arcangelo, G. (2004). Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 41, 71–84. doi: 10.1016/s0896-6273(03)00819-5
Niu, S., Yabut, O., and D’Arcangelo, G. (2008). The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 28, 10339–10348. doi: 10.1523/JNEUROSCI.1917-08.2008
Noh, J. S., Sharma, R. P., Veldic, M., Salvacion, A. A., Jia, X., Chen, Y., et al. (2005). DNA methyltransferase 1 regulates reelin mRNA expression in mouse primary cortical cultures. Proc. Natl. Acad. Sci. U S A 102, 1749–1754. doi: 10.1073/pnas.0409648102
Ognibene, E., Adriani, W., Macrì, S., and Laviola, G. (2007). Neurobehavioural disorders in the infant reeler mouse model: interaction of genetic vulnerability and consequences of maternal separation. Behav. Brain Res. 177, 142–149. doi: 10.1016/j.bbr.2006.10.027
Palacios-Garcia, I., Lara-Vásquez, A., Montiel, J. F., Díaz-Véliz, G. F., Sepúlveda, H., Utreras, E., et al. (2015). Prenatal stress down-regulates reelin expression by methylation of its promoter and induces adult behavioral impairments in rats. PLoS One 10:e0117680. doi: 10.1371/journal.pone.0117680
Pesold, C., Impagnatiello, F., Pisu, M. G., Uzunov, D. P., Costa, E., Guidotti, A., et al. (1998). Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. U S A 95, 3221–3226. doi: 10.1073/pnas.95.6.3221
Pesold, C., Liu, W. S., Guidotti, A., Costa, E., and Caruncho, H. J. (1999). Cortical bitufted, horizontal and Martinotti cells preferentially express and secrete reelin into perineuronal nets, nonsynaptically modulating gene expression. Proc. Natl. Acad. Sci. U S A 96, 3217–3222. doi: 10.1073/pnas.96.6.3217
Ptak, C., and Petronis, A. (2010). Epigenetic approaches to psychiatric disorders. Dialogues Clin. Neurosci. 12, 25–35.
Pujadas, L., Gruart, A., Bosch, C., Delgado, L., Teixeira, C. M., Rossi, D., et al. (2010). Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 30, 4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010
Qin, L., Tu, W., Sun, W., Zhang, J., Chen, Y., and Zhao, H. (2011). Retardation of neurobehavioral development and reelin down-regulation regulated by further DNA methylation in the hippocampus of the rat pups are associated with maternal deprivation. Behav. Brain Res. 217, 142–147. doi: 10.1016/j.bbr.2010.10.018
Razin, A., and Shemer, R. (1995). DNA methylation in early development. Hum. Mol. Genet. 4, 1751–1755.
Rice, D. S., Sheldon, M., D’Arcangelo, G., Nakajima, K., Goldowitz, D., and Curran, T. (1998). Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 125, 3719–3729.
Rodriguez, M. A., Pesold, C., Liu, W. S., Kriho, V., Guidotti, A., Pappas, G. D., et al. (2000). Colocalization of integrin receptors and reelin in dendritic spine postsynaptic densities of adult nonhuman primate cortex. Proc. Natl. Acad. Sci. U S A 97, 3550–3555. doi: 10.1073/pnas.97.7.3550
Rogers, J. T., Rusiana, I., Trotter, J., Zhao, L., Donaldson, E., Pak, D. T., et al. (2011). Reelin supplementation enhances cognitive ability, synaptic plasticity and dendritic spine density. Learn. Mem. 18, 558–564. doi: 10.1101/lm.2153511
Rogers, J. T., Zhao, L., Trotter, J. H., Rusiana, I., Peters, M. M., Li, Q., et al. (2013). Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 27, 386–395. doi: 10.1177/0269881112463468
Ruzicka, W. B., Zhubi, A., Veldic, M., Grayson, D. R., Costa, E., and Guidotti, A. (2007). Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 12, 385–397. doi: 10.1038/sj.mp.4001954
Satta, R., Maloku, E., Zhubi, A., Pibiri, F., Hajos, M., Costa, E., et al. (2008). Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc. Natl. Acad. Sci. U S A 105, 16356–16361. doi: 10.1073/pnas.0808699105
Sharma, R. P., Grayson, D. R., and Gavin, D. P. (2008). Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr. Res. 98, 111–117. doi: 10.1016/j.schres.2007.09.020
Shifman, S., Johannesson, M., Bronstein, M., Chen, S. X., Collier, D. A., Craddock, N. J., et al. (2008). Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 4:e28. doi: 10.1371/journal.pgen.0040028
Stranahan, A. M., Salas-Vega, S., Jiam, N. T., and Gallagher, M. (2011). Interference with reelin signaling in the lateral entorhinal cortex impairs spatial memory. Neurobiol. Learn. Mem. 96, 150–155. doi: 10.1016/j.nlm.2011.03.009
Strasser, V., Fasching, D., Hauser, C., Mayer, H., Bock, H. H., Hiesberger, T., et al. (2004). Receptor clustering is involved in Reelin signaling. Mol. Cell Biol. 24, 1378–1386. doi: 10.1128/mcb.24.3.1378-1386.2004
Sui, L., and Li, B.-M. (2010). Effects of perinatal hypothyroidism on regulation of reelin and brain-derived neurotrophic factor gene expression in rt hippocampus: role of DNA methylation and histone acetylation. Steroids 75, 988–997. doi: 10.1016/j.steroids.2010.06.005
Sui, L., Wang, Y., Ju, L.-H., and Chen, M. (2012). Epigenetic regulation of reelin and brain-derived neurotrophic factor genes in long term potentiation in rat medial prefrontal cortex. Neurobiol. Learn. Mem. 97, 425–440. doi: 10.1016/j.nlm.2012.03.007
Szyf, M. (2009). Epigenetics, DNA methylation and chromatin modifying drugs. Annu. Rev. Pharmacol. Toxicol. 49, 243–263. doi: 10.1146/annurev-pharmtox-061008-103102
Tochigi, M., Iwamoto, K., Bundo, M., Komori, A., Sasaki, T., Kato, N., et al. (2008). Methylation status of the reelin promoter region in the brain of schizophrenia subjects. Biol. Psychiatry 63, 530–533. doi: 10.1016/j.biopsych.2007.07.003
Tremolizzo, L., Carboni, G., Ruzicka, W. B., Mitchell, C. P., Sugaya, I., Tueting, P., et al. (2002). An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc. Natl. Acad. Sci. U S A 99, 17095–17100. doi: 10.1073/pnas.262658999
Tremolizzo, L., Doueiri, M. S., Dong, E., Grayson, D. R., Davis, J., Pinna, G., et al. (2005). Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol. Psychiatry 57, 500–509. doi: 10.1016/j.biopsych.2004.11.046
Trommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger, W., Nimpf, J., et al. (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701. doi: 10.1016/s0092-8674(00)80782-5
Tueting, P., Costa, E., Dwivedi, Y., Guidotti, A., Impagnatiello, F., Manev, R., et al. (1999). The phenotypic characteristics of heterozygous reeler mouse. Neuroreport 10, 1329–1334. doi: 10.1097/00001756-199904260-00032
Tueting, P., Davis, J. M., Veldic, M., Pibiri, F., Kadriu, B., Guidotti, A., et al. (2010). L-methionine decreases dendritic spine density in mouse frontal cortex. Neuroreport 21, 543–548. doi: 10.1097/WNR.0b013e3283373126
Tueting, P., Doueiri, M. S., Guidotti, A., Davis, J. M., and Costa, E. (2006). Reelin down-regulation in mice and psychosis endophenotypes. Neurosci. Biobehav. Rev. 30, 1065–1077. doi: 10.1016/j.neubiorev.2006.04.001
Veldic, M., Caruncho, H. J., Liu, W. S., Davis, J., Satta, R., Grayson, D. R., et al. (2004). DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc. Natl. Acad. Sci. U S A 101, 348–353. doi: 10.1073/pnas.2637013100
Veldic, M., Kadriu, B., Maloku, E., Agis-Balboa, R. C., Guidotti, A., Davis, J. M., et al. (2007). Costa E. Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr. Res. 91, 51–61. doi: 10.1016/j.schres.2006.11.029
Ventruti, A., Kazdoba, T. M., Niu, S., and D’Arcangelo, G. (2011). Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 189, 32–42. doi: 10.1016/j.neuroscience.2011.05.050
Wang, Z., Hong, Y., Zou, L., Zhong, R., Zhu, B., Shen, N., et al. (2014). Reelin gene variants and risk of autism spectrum disorders: an integrated meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 165B, 192–200. doi: 10.1002/ajmg.b.32222
Weaver, I. C., Champagne, F. A., Brown, S. E., Dymov, S., Sharma, S., Meaney, M. J., et al. (2005). Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J. Neurosci. 25, 11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005
Weaver, I. C., Meaney, M. J., and Szyf, M. (2006). Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc. Natl. Acad. Sci. U S A 103, 3480–3485. doi: 10.1073/pnas.0507526103
Wedenoja, J., Tuulio-Henriksson, A., Suvisaari, J., Loukola, A., Paunio, T., Partonen, T., et al. (2010). Replication of association between working memory and Reelin, a potential modifier gene in schizophrenia. Biol. Psychiatry 67, 983–991. doi: 10.1016/j.biopsych.2009.09.026
Wyatt, R. J., Benedict, A., and Davis, J. (1971). Biochemical and sleep studies of schizophrenia: a review of the literature-1960–1970. Schiz. Bull. 4, 10–44. doi: 10.1093/schbul/1.4.10
Zhang, H., Liu, X., Zhang, C., Mundo, E., Macciardi, F., Grayson, D. R., et al. (2002). Reelin gene alleles and susceptibility to autism spectrum disorders. Mol. Psychiatry 7, 1012–1017. doi: 10.1038/sj.mp.4001124
Keywords: RELN, synaptic plasticity, promoter methylation, schizophrenia, bipolar disorder, Dab1
Citation: Guidotti A, Grayson DR and Caruncho HJ (2016) Epigenetic RELN Dysfunction in Schizophrenia and Related Neuropsychiatric Disorders Front. Cell. Neurosci. 10:89. doi: 10.3389/fncel.2016.00089
Received: 22 January 2016; Accepted: 21 March 2016;
Published: 05 April 2016.
Edited by:
Gabriella D’Arcangelo, Rutgers, The State Univesity of New Jersey, USACopyright © 2016 Guidotti, Grayson and Caruncho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Guidotti, YWd1aWRvdHRpQHBzeWNoLnVpYy5lZHU=