 Francesco Petrelli
Francesco Petrelli Luca Pucci
Luca Pucci Paola Bezzi
Paola Bezzi- Department of Fundamental Neurosciences, University of Lausanne, Lausanne, Switzerland
The cellular mechanism(s) underlying autism spectrum disorders (ASDs) are not fully understood although it has been shown that various genetic and environmental factors contribute to their etiology. As increasing evidence indicates that astrocytes and microglial cells play a major role in synapse maturation and function, and there is evidence of deficits in glial cell functions in ASDs, one current hypothesis is that glial dysfunctions directly contribute to their pathophysiology. The aim of this review is to summarize microglia and astrocyte functions in synapse development and their contributions to ASDs.
Introduction
Autism spectrum disorders (ASDs) have a worldwide prevalence of about 12-15% and are characterized by significant social, intellectual, behavioral impairment, and sometimes cognitive deficits (Abrahams and Geschwind, 2008; Geschwind, 2009; Quaak et al., 2013). They typically manifest early in development and are associated distinct neurodevelopmental syndromes.
The conceptualisation of autism and related disorders has undergone considerable changes over the last ten years, which are reflected in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V, www.dsm5.org). The proposed revisions of the preceding edition (DSM IV-TR) include combining specific DSM-IV-TR diagnoses into a single broad ASD, and identifying two domains of impairment (social communication and interactions, and restricted repetitive behavior) rather than three (social interaction, communication, and restricted repetitive and stereotyped patterns of behavior, interests, and activities). There are also considerable differences in clinical presentation and disease progression as ASD patient’s present variously severe core symptoms and variable co-morbid conditions such as epilepsy, gastrointestinal problems, intellectual disability, anxiety, and depression (Kim and Lord, 2012). Early estimates that the heritability of ASDs is approximately 90% (Steffenburg et al., 1989; Bailey et al., 1995), and even recently revised estimates that it is about 45%, strongly suggest that genetic mutations are a major cause (Hallmayer et al., 2011; Sandin et al., 2014). However, ASDs are genetically very heterogeneous (State and Levitt, 2011) and seem to be associated with a large number of genetic mutations, including possibly 100s of rare causal variants and common variants with small effects (Murdoch and State, 2013). The genetic heterogeneity of ASD has made it challenging to identify specific genes associated with the disorder, which has thus hindered efforts to dissect disease mechanisms. Recent insights into the genetic pathways that are altered in ASDs have come from studies of syndromic disorders with a high incidence of ASDs caused by mutations of a single gene, including fragile X syndrome (fragile X mental retardation 1 protein, FMR1), Rett syndrome (methyl-CpG-binding protein 2 protein, MECP2), tuberous sclerosis (tuberous sclerosis 1 protein, TSC1), neurofibromatosis type 1 (neurofibromin 1 protein, NF1), and PTEN (phosphatase and tensin homologue) macrocephaly. Recent studies also implicate neurodevelopmental genes in ASDs through the identification of recurrent de novo loss of function mutations in affected individuals (Parikshak et al., 2013; Willsey et al., 2013). Neurodevelopmental genes are indeed an important factor to take into consideration since functional and anatomical insults associated to defects in these genes during brain development can trigger the appearance of ASDs in the childhood. Genome-wide association studies have identified susceptibility genes for ASDs such as forkhead box p2 (FOXP2) (Toma et al., 2013) or the MAM domain containing glycosylphosphatidylinositol anchor (MDGA) genes (Bucan et al., 2009; Pettem et al., 2013; Perez-Garcia and O’Leary, 2016) and have provided evidence supporting the idea that the large numbers of variants associated with ASDs converge toward a core set of dysregulated biological processes (Murdoch and State, 2013). The genes that have been linked to ASDs can be grouped into three broad categories: those involved in synapse structure and activity (Etherton et al., 2011a,b; Peca and Feng, 2012), those involved in protein synthesis (Kelleher and Bear, 2008), and those involved in regulating gene expression (van Bokhoven, 2011). Many of them encode for proteins that have a clear synaptic function, thus making the pathological features of ASDs mainly “neurocentric”. However, as the usefulness of parsing neuronal mechanisms in order to investigate the etiology of ASDs has proved to be limited, alternative biological analyses may help to reveal previously unknown cellular and molecular mechanisms.
Extending the theory of purely genetic causes, it seems likely that genetics alone may not account for all cases of autism. In addition to a certain combination of autism-related genes, exposure to a number of non-heritable environmental factors may significantly affect susceptibility to, and the variable expression of autism and autism-related traits (Pessah and Lein, 2008; Rosenberg et al., 2009; Hallmayer et al., 2011; Estes and McAllister, 2015). In addition to exposure to chemicals or toxins, these include factors such as parental age at the time of conception, and maternal nutrition and infections (including autoimmune diseases) during pregnancy and prematurity (Grabrucker, 2012). One important area of research concerns the way in which environmental influences interact with genetic susceptibility: for example, recent studies have shown that glial cells in the brains of autistic subjects were constantly activated and their inflammation response genes were turned on (Voineagu et al., 2011; Edmonson et al., 2014; Gupta et al., 2014). It is still not clear what role inflammation plays in autism or whether it is beneficial or not, or what causes the activation of glial cells, but these findings and the recent discovery that glial cells may interact with synaptic activity by influencing synapse formation and maturation (Clarke and Barres, 2013; Sahlender et al., 2014; Petrelli and Bezzi, 2016), strongly suggest that glial cells may be involved in the pathogenesis of ASDs. Although some excellent reviews have recently discussed the involvement of glial cells in neuropsychiatric disorders (McGann et al., 2012; Molofsky et al., 2012; Chung et al., 2015), we will give our perspective on the role of microglia and astrocytes in the late step of synapse formation and maturation and in the pathophysiology of ASDs.
Astrocytes and Microglia Influence Synapse Formation and Function
Over the last ten years, it has gradually emerged that glial cells (in particular microglia and astrocytes) influence synapse formation and function (Araque et al., 2014; Chung et al., 2015; Rossi, 2015). Neurons and glial cells are closely associated with each other from an early stage of development, and recent discoveries suggest that the appropriate assembly of neural circuits requires extensive neuron-glia signaling (Ullian et al., 2001; Christopherson et al., 2005; Eroglu, 2009; Allen et al., 2012). In addition to carrying out various homeostatic functions within the central nervous system (CNS), they can also engage in bi-directional communication with neurons by releasing neuroactive substances (Petrelli and Bezzi, 2016). In particular, astrocytes can make contact with multiple neurons and up to 100,000 synapses (Bushong et al., 2002; Halassa et al., 2007), and possess many receptors (mainly G-protein coupled receptors) and ion channels present in neurons, thus enabling them to sense and respond to an array of neuronal signals (Fiacco and McCarthy, 2006). The generation and expansion of astrocytes is largely completed before birth, but the elaboration and maturation of their fine peri-synaptic processes persists during the active period of synaptogenesis (Ullian et al., 2001) in the post-natal period. This suggests that they are in a crucial position to communicate actively with neurons during synaptogenesis and, thus, coordinate the development of neural circuits. The formation of synapses (Clarke and Barres, 2013) and the modulation of synaptic activity and plasticity by astrocytes (Araque et al., 2014) mainly take place as a result of the secretion of neuroactive substances now known as synaptogenic factors. Pfrieger and Barres (1997) were the first to demonstrate the key role of astrocyte-derived soluble molecules in synapse formation, and many subsequent studies by the same group and others have identified the nature of these synaptogenic factors, with the thrombospondins 1- 5, hevin, and glypicans seeming to be crucial substances for the formation of excitatory synapses (Ullian et al., 2001; Christopherson et al., 2005; Kucukdereli et al., 2011; Allen et al., 2012). Overall, these findings are seminally important as they have changed our “neurocentric” view of the astrocytic-dependent processes involved in the formation and maturation of functional synapses. However, we are still only beginning to understand the role of astrocyte-derived synaptogenic molecules in terms of cell biology, and our knowledge of the molecular and cell mechanisms regulating the active release of synaptogenic factors is very limited (Petrelli and Bezzi, 2016). Many of them have only been studied in cultured cells, and the cellular and molecular pathways governing their secretion are still unknown, including whether they are calcium-dependent or not. Interestingly, astrocytes also release many other factors, such as substances regulating metabolism, the energy supply (including cerebral blood flow) and inflammation, and substances regulating synaptic transmission, including neurotransmitters and neuromodulators (i.e., gliotransmitters) (Bezzi and Volterra, 2001; Petrelli and Bezzi, 2016). The cell mechanisms governing the release of gliotransmitters and their effects on synaptic activities have been extensively studied, and there is a large amount of experimental evidence indicating that gliotransmitters play an active role in synaptic physiology (Araque et al., 2014). For example, the tumor necrosis factor alpha (TNFα) released by astrocytes (and possibly also by microglial cells) is required for synaptic scaling (Stellwagen and Malenka, 2006), and can control glutamatergic gliotransmission under both physiological (Santello et al., 2011) and pathological conditions (Bezzi et al., 2001; Habbas et al., 2015). A recent study has shown that pathological concentrations of pro-inflammatory TNFα signal through astrocytes to alter synaptic transmission and impair cognition in a mouse model of multiple sclerosis (Habbas et al., 2015). As inflammation of the CNS is a common feature of nearly all neurological disorders and insults (including ASDs), and cognitive impairments are also common to many neuroinflammatory neurological conditions (including ASDs), it is likely that the activation of glial cells and pathological concentrations of cytokines (notably TNFα) can contribute to the cognitive and behavioral impairments characterizing ASDs.
Microglial cells are resident macrophages of the brain that form the innate defensive system (Hanisch and Kettenmann, 2007; Kettenmann, 2011) and, by acting as sentinels, can detect the first signs of pathogenic invasion or tissue damage. After their conversion from a resting (or surveillance) cell type to an activated form, they remove damaged synapses and their protective or detrimental functions have been extensively studied in various brain pathologies (Kettenmann et al., 2013). During the resting/surveillance phase, microglial processes constantly extend and retract to check the local environment (Nimmerjahn et al., 2005), which includes peri-synaptic astrocytes, pre-synaptic boutons, and post-synaptic spines (Tremblay et al., 2010). Many of their functions are still unclear and there has been much debate about whether they are beneficial or detrimental. Like astrocytes, microglial cells have heterogeneous functions within the brain (Carson et al., 2007; Bailey-Bucktrout et al., 2008; Bulloch et al., 2008; Gowing et al., 2008), and release a wide variety of molecules (e.g., brain-derived neurotrophic factor, BDNF) that can interact with synapses and play important physiological roles in learning and memory (Parkhurst et al., 2013).
In addition to affecting synaptic activity, astrocytes and microglial cells seem to play an important role in removing (‘pruning’) synaptic connections (Stanfield et al., 1982; Nakamura and O’Leary, 1989), a process of structural formation and elimination that is vital for controlling and refining the connectivity of mature neuronal circuits (Steven et al., 2007; Barres, 2008). For example, in the developing brain, astrocytes can physically eliminate synapses through the multiple EGF-like domains 10 (MEGF10) and c-mer proto-oncogene tyrosine kinase (MERTK) phagocytic pathways (Chung et al., 2013) and, in line with this, mice deficient in MEGF10 and MERTK receptors fail to refine their retino-geniculate connections and retain excess functional synapses. Interestingly, the rate of synaptic pruning decreases with age and is regulated by synaptic activity. Like astrocytes, microglia cells are also important in the structural elimination of synapses in developing brain. Microglia processes are highly motile and are well positioned to interact with boutons and spines. However, synaptic pruning particularly involves phagocytic microglial processes that, during the first weeks after birth, can engulf pre- and post-synaptic elements (Paolicelli et al., 2011; Schafer et al., 2012). This early post-natal process depends of different molecules such as CX3C chemokine receptor 1 (CX3CR1), also known as the fractalkine receptor or G-protein coupled receptor 13 (GPR13), and complement receptor 3 (CX3C) (Harrison et al., 1998; Stevens et al., 2007; Zhan et al., 2014; Wu et al., 2015). The importance of the CX3CR1-mediated mechanisms by means of which microglial cells remove synapses has been confirmed by studies of CX3CR1 knockout animals, which have aberrant long-range functional connectivity (Schafer et al., 2012) as well as a deficit in hippocampal LTP and aberrant social behavior (Zhan et al., 2014). Overall, these studies show that physiological interactions of astrocytes and microglia with synapses are crucial for synapse formation and network functioning, and point out that their loss, deviation or functional perturbation might contribute to autism pathogenesis and progression (Figure 1).
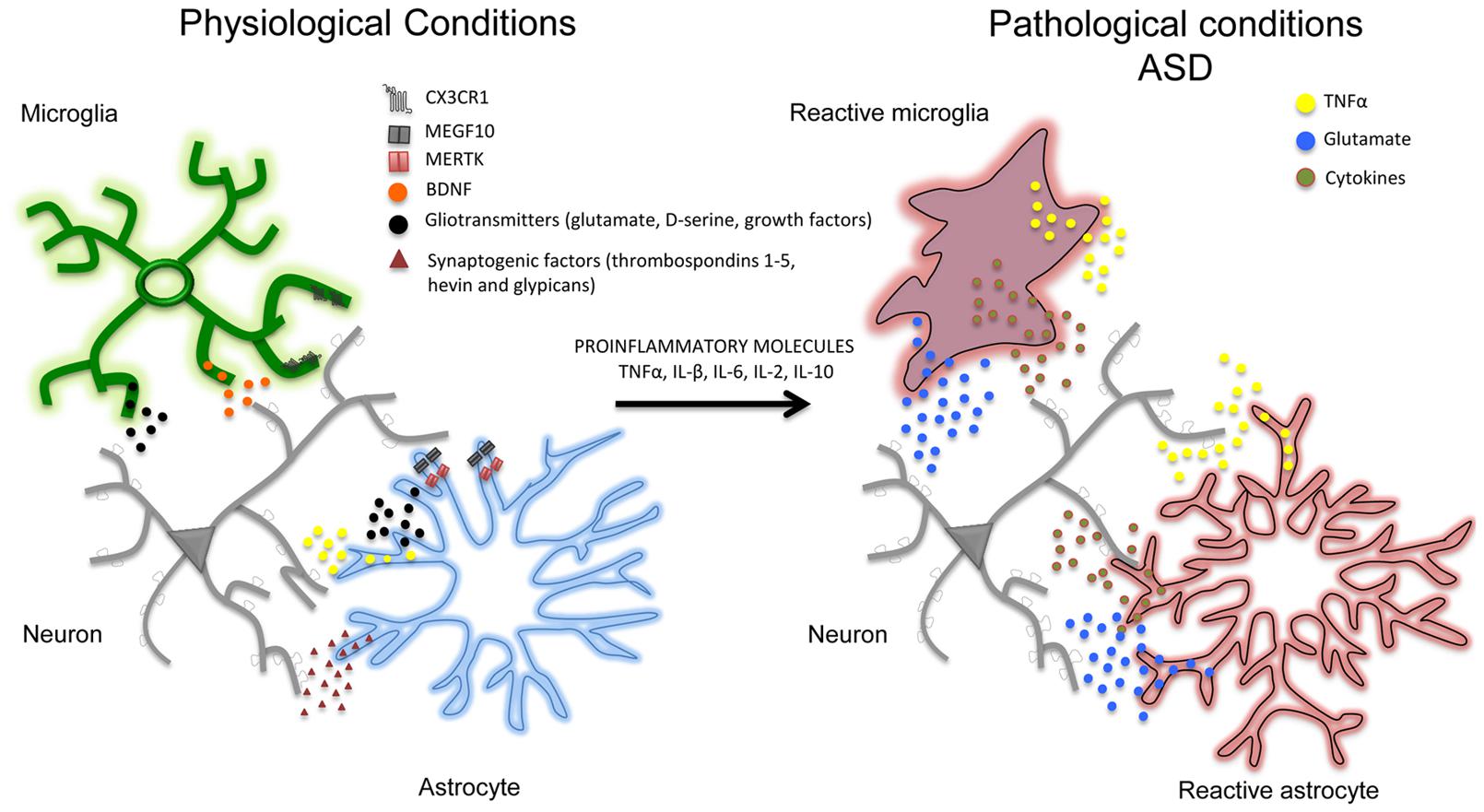
FIGURE 1. In physiological conditions and during postnatal development, neurons (gray) and glial cells (notably astrocytes –blue- and microglia –green-) are closely associated with each other. Astrocytes and microglia release a number of neuroactive substances such as gliotransmitters together with growth factors (such as TNFα, BDNF, glutamate, D-serine; black dots) and synaptogenic factors (such as thrombospondins 1-5, hevin, and glypicans; red dots) that can promote the formation and maturation of synapses. Glial cells can also contribute to synapse remodeling and pruning through CX3CR1, multiple EGF-like domains 10 (MEGF10), c-mer proto-oncogene tyrosine kinase (MERTK) phagocytic pathways. In pathological conditions (i.e., in autistic brains) pro-inflammatory factors (including pro-inflammatory cytokines TNFα, IL-β, IL-6, IL-2, IL-10) may lead to a chronic neuroinflammation in which astrocytes and microglia become reactive (red), release pro-inflammatory mediators (such as TNFα, glutamate and pro-inflammatory cytokines) and may exacerbate the initial inflammatory condition. Although the specific contribution of aberrant glial cells (i.e., reactive gliosis) to the pathophysiology of autism has still to be determined, studies on animal models support a direct association between neuroinflammation and autism, suggesting that reactive gliosis may have a crucial role in many autism phenotypes including the structural and functional alterations of brain connectivity.
The Role of Glial Cells in Autism Spectrum Disorders
The importance of glial cells in the pathophysiology of ASDs is primarily supported by a recent transcriptomic analysis of autistic brains. RNA sequencing revealed a close association between ASDs and the genes related to glial cell activation and genes belonging to immune and inflammatory categories (Voineagu et al., 2011). These transcriptional results are supported by immunohistochemistry data obtained from human post mortem brain samples showing increased reactive gliosis and glial cell proliferation (Purcell et al., 2001; Vargas et al., 2005; Fatemi et al., 2008; Morgan et al., 2012; Tetreault et al., 2012; Edmonson et al., 2014), and by a recent positron emission tomography (PET) functional imaging study showing microglial activation in multiple brain regions of young adults with ASDs (such in the cerebellum, midbrain, pons, fusiform gyri, and the anterior cingulate and orbitofrontal cortices; Suzuki et al., 2013). In line with this, high levels of various pro-inflammatory cytokines, such as interleukin (IL)-6, TNFα, and IL-1β have been reported in the post-mortem brain tissues, (Watkins et al., 2001; Vargas et al., 2005; Li et al., 2009; Wei et al., 2011) and blood of autistic subjects (Gupta et al., 1998; Jyonouchi et al., 2001).
It is now recognized that pro-inflammatory factors play an important role in the etiology of various neurological and neuropsychiatric disorders, including those such as ASDs whose pathogenetic onset occurs during early brain development (Meyer et al., 2011; Voineagu et al., 2011; Gupta et al., 2014; Estes and McAllister, 2015). The developing brain is highly vulnerable to environmental insults such as those associated with strong inflammatory reactions (Dammann and Leviton, 2004) and specific brain lesions (Hagberg et al., 2015). Under normal conditions, inflammatory processes in the developing brain are controlled by a number of homeostatic mechanisms that limit the inflammation induced by an environmental stimulus such as infection (Serhan and Savill, 2005). These surveillance mechanisms are mainly controlled by microglia and astrocytes, and are crucial in ensuring that inflammatory processes efficiently remove invading pathogens and contribute to tissue repair.
It is therefore likely that, under inflammatory conditions, any dysfunction in these mechanisms may lead to deleterious chronic inflammation, and this (together with reports of increased levels of pro-inflammatory cytokines in the post-mortem brains of subjects with ASDs) has led to the hypothesis that chronic neuroinflammation plays a role in the pathogenesis of ASDs, which is supported by the fact that chronic systemic inflammatory conditions such as those associated with autoimmune disorders or infections (together with acute immune activation) are often observed in the mothers of children with ASDs (Atladottir et al., 2009; Keil et al., 2010; Estes and McAllister, 2015). Although the specific contribution of maternal immune dysregulation to the onset of ASDs has yet to be determined in humans, animal models have shown a causal link between the activation of the maternal immune system and altered neurodevelopment. Studies of various animal models of maternal infection during pregnancy support the association between systemic inflammatory processes and ASDs phenotypes (Malkova et al., 2012; Knuesel et al., 2014; Missault et al., 2014), all of which seem to indicate that the ASD-like behavior in the offspring may be caused by altered levels of maternal cytokines, including TNFα, IL1β, IL-2, IL-6, and IL-10 (Ponzio et al., 2007; Smith et al., 2007). It is important to note that there is a specific temporal window in which cytokines confer a risk of developing ASD (i.e., the perinatal period), which suggests that the permeability of the developing blood brain barrier is crucial for the onset of ASD (Meyer et al., 2011). Pro-inflammatory cytokines in the developing brain classically lead to neuroinflammation, a condition in which microglia and astrocytes become reactive (gliosis), proliferate and, depending on the entity of the gliosis, recruit peripheral leukocytes and thus amplify the initial tissue damage (Sofroniew, 2015).
The idea that reactive gliosis may exacerbate the inflammatory conditions caused by immune activation and contribute to the pathogenesis of ASDs is intriguing, but it is still not clear what the changes in glial cell activation tell us about the molecular and cellular mechanisms underlying the disorders. One possibility is that reactive gliosis may perturb the ability of microglia and astrocytes to modulate the maturation, elimination (phagocytosis), or functioning of developing synapses because developing neural networks are highly vulnerable to insults affecting the glial pathways governing the pruning of synapses (Chung et al., 2015). For example, CX3CR1 knockout mice show impaired synaptic pruning, social behavior and functional connectivity (Zhan et al., 2014), all of which are features of ASDs. It can therefore be expected that the inflammatory processes targeting the developing brain have a long-lasting impact on brain and behavioral functions throughout life.
The use of animal models of ASDs has led to substantial advances in our understanding of the role of glial cells, which have been found to be abnormal in mouse models of Rett syndrome (RTT; Maezawa et al., 2009; Yasui et al., 2013), fragile X syndrome (Yuskaitis et al., 2010), and tuberous sclerosis (Uhlmann et al., 2002). In particular, a study of a mouse model of RTT, a devastating neurodevelopmental disorder caused by loss-of-function mutations in the X-linked MECP2 encoding methyl-CpG-binding protein 2 (MeCP2) (Chahrour and Zoghbi, 2007), has shown that MeCP2-deficient microglia cells release an abnormally high level of glutamate, causing excitotoxicity that may contribute to dendritic and synaptic abnormalities (Maezawa and Jin, 2010). MECP2 is a well-known transcription factor that is important in controlling gene expression by interpreting and regulating epigenetic markers (Chao and Zoghbi, 2012). MECP2 is expressed in many tissues but, although the disease is generally attributed to a primary neuronal dysfunction, glial MECP2 seems to play a pathophysiological role as it has been recently shown that MECP2-null astrocytes are unable to support the normal dendritic ramification of wild-type neurons growing in culture (Ballas et al., 2009), and two remarkable studies have found that the expression of wild-type MECP2 protein in the astrocytes or microglia of MECP2-null hosts dramatically improves the pathology (Lioy et al., 2011; Derecki et al., 2012).
Finally, it is interesting to note that a recent RNA-Seq transcriptome and splicing database of glia and neurons (Cahoy et al., 2008) indicates that many of the ASD candidate genes are enriched in glial cells. For example, some genes, such as Homer1 (HOMER1) (Ronesi et al., 2012), 4-aminobutyrate aminotransferase (ABAT; Barnby et al., 2005), fatty acid binding protein 7 (FABP7; Maekawa et al., 2010), and glutathione S-transferase 1 (GSTM1; Ming et al., 2010), are not specific for neurons but, instead, are highly enriched in astrocytes; other genes are equally present in astrocytes and neurons (such as MeCP2), and some are highly enriched in both astrocytes and microglial cells (such as dual-specificity tyrosine-(Y)-phosphorylation- regulated kinase 1A or DYRK1A). Mutations in the genes encoding a number of members of the IL-1 cytokine receptor family are also associated with ASDs, and glial cells are the major contributors to the response of IL-1 signaling pathways to neuroinflammation (Moynagh et al., 1993; Zhang et al., 1996; Molina-Holgado et al., 2000). For example, recent exome-sequencing studies of ASD patients have found a single-nucleotide polymorphism in the gene encoding IL-1 receptor type 2 (IL-1R2) (O’Roak et al., 2011; Sanders et al., 2012) that is highly enriched in microglial cells (Cahoy et al., 2008), and a rare ASD-associated mutation has been identified in the gene encoding IL-1 receptor accessory protein –like 1 (IL-1RAPL1; Bhat et al., 2008), which is highly enriched in astrocytes (Cahoy et al., 2008).
Future Perspectives
Until very recently, the role of glial cells in the onset of ASDs was almost completely overlooked, and so neuropharmacological strategies for treating the symptoms were almost exclusively aimed at neuronal activity and synaptic transmission. However, as accumulating evidence supports the view that astrocytes and microglia are significantly involved in the regulation of synapse formation, function, plasticity, and elimination, the role of astrocyte-derived factors in regulating synapse formation and the role of microglia in synaptic pruning during postnatal development (a period that coincides with the onset of many ASDs) are particularly relevant. These findings may change our “neurocentric” view of the mechanisms involved in the onset and progression of ASDs because that it is unlikely that research solely concentrated on neurons will fully reveal their underlying pathophysiological mechanisms. Recent data suggest that many ASDs are at least partially due to disorders affecting glial cells or neuron-glial interactions, and future pharmacological research should consider the possibility of improving glial cell functions.
Author Contributions
PB wrote the review together with FP and LP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This review was supported by grants from the Swiss National Foundation NCCR “Synapsy” and “Transcure” to PB.
References
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Allen, N. J., Bennett, M. L., Foo, L. C., Wang, G. X., Chakraborty, C., Smith, S. J., et al. (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414. doi: 10.1038/nature11059
Araque, A., Carmignoto, G., Haydon, P. G., Oliet, S. H. R., Robitaille, R., and Volterra, A. (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. doi: 10.1016/j.neuron.2014.02.007
Atladottir, H. O., Pedersen, M. G., Thorsen, P., Mortensen, P. B., Deleuran, B., Eaton, W. W., et al. (2009). Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics 124, 687–694. doi: 10.1542/peds.2008-2445
Bailey, A., Lecouteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., et al. (1995). Autism as a strongly genetic disorder - evidence from a british twin study. Psychol. Med. 25, 63–77. doi: 10.1017/S0033291700028099
Bailey-Bucktrout, S. L., Caulkins, S. C., Goings, G., Fischer, J. A. A., Dzionek, A., and Miller, S. D. (2008). Central nervous system plasmacytoid dendritic cells regulate the severity of relapsing experimental autoimmune encephalomyelitis. J. Immunol. 180, 6457–6461. doi: 10.4049/jimmunol.180.10.6457
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Barnby, G., Abbott, A., Sykes, N., Morris, A., Weeks, D. E., Mott, R., et al. (2005). Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 76, 950–966. doi: 10.1086/430454
Barres, B. A. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. doi: 10.1016/j.neuron.2008.10.013
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq, E., et al. (2001). CXCR4-activated astrocyte glutamate release via TNFa: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710. doi: 10.1038/89490
Bezzi, P., and Volterra, A. (2001). A neuron-glia signalling network in the active brain. Curr. Opin. Neurobiol. 11, 387–394. doi: 10.1016/S0959-4388(00)00223-3
Bhat, S. S., Ladd, S., Grass, F., Spence, J. E., Brasington, C. K., Simensen, R. J., et al. (2008). Disruption of the IL1RAPL1 gene associated with a pericentromeric inversion of the X chromosome in a patient with mental retardation and autism. Clin. Genet. 73, 94–96. doi: 10.1111/j.1399-0004.2007.00920.x
Bucan, M., Abrahams, B. S., Wang, K., Glessner, J. T., Herman, E. I., Sonnenblick, L. I., et al. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5:e1000536. doi: 10.1371/journal.pgen.1000536
Bulloch, K., Miller, M. M., Gal-Toth, J., Milner, T. A., Gottfried-Blackmore, A., Waters, E. M., et al. (2008). CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J. Compar. Neurol. 508, 687–710. doi: 10.1002/cne.21668
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192.
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. doi: 10.1523/JNEUROSCI.4178-07.2008
Carson, M. J., Bilousova, T. V., Puntambekar, S. S., Melchior, B., Doose, J. M., and Ethell, I. M. (2007). A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics 4, 571–579. doi: 10.1016/j.nurt.2007.07.002
Chahrour, M., and Zoghbi, H. Y. (2007). The story of rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chao, H. T., and Zoghbi, H. Y. (2012). MeCP2: only 100% will do. Nat. Neurosci. 15, 176–177. doi: 10.1038/nn.3027
Christopherson, K. S., Ullian, E. M., Stokes, C. C. A., Mullowney, C. E., Hell, J. W., Agah, A., et al. (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. doi: 10.1016/j.cell.2004.12.020
Chung, W., Choi, S. Y., Lee, E., Park, H., Kang, J., Choi, Y., et al. (2015). Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat. Neurosci. 18, 435–443. doi: 10.1038/nn.3927
Chung, W. S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi: 10.1038/nature12776
Clarke, L. E., and Barres, B. A. (2013). Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 14, 311–321. doi: 10.1038/nrn3484
Dammann, O., and Leviton, A. (2004). Inflammatory brain damage in preterm newborns - dry numbers, wet lab, and causal inferences. Early Hum. Dev. 79, 1–15. doi: 10.1016/j.earlhumdev.2004.04.009
Derecki, N. C., Cronk, J. C., Lu, Z. J., Xu, E., Abbott, S. B. G., Guyenet, P. G., et al. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109. doi: 10.1038/nature10907
Edmonson, C., Ziats, M. N., and Rennert, O. M. (2014). Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism 5:9. doi: 10.1186/2040-2392-5-3
Eroglu, C. (2009). The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J. Cell Commun. Signal. 3, 167–176. doi: 10.1007/s12079-009-0078-y
Estes, M. L., and McAllister, A. K. (2015). Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 16, 469–486. doi: 10.1038/nrn3978
Etherton, M., Foldy, C., Sharma, M., Tabuchi, K., Liu, X. R., Shamloo, M., et al. (2011a). Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. U.S.A. 108, 13764–13769. doi: 10.1073/pnas.1111093108
Etherton, M. R., Tabuchi, K., Sharma, M., Ko, J., and Sudhof, T. C. (2011b). An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. Embo J. 30, 2908–2919. doi: 10.1038/emboj.2011.182
Fatemi, S. H., Folsom, T. D., Reutiman, T. J., and Lee, S. (2008). Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse 62, 501–507. doi: 10.1002/syn.20519
Fiacco, T. A., and McCarthy, K. D. (2006). Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia 54, 676–690. doi: 10.1002/glia.20396
Geschwind, D. H. (2009). Advances in Autism. In: Annual Review of Medicine. Palo Alto, CA: Annual Reviews, 367–380.
Gowing, G., Philips, T., Van Wijmeersch, B., Audet, J. N., Dewil, M., Van Den Bosch, L., et al. (2008). Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J. Neurosci. 28, 10234–10244. doi: 10.1523/JNEUROSCI.3494-08.2008
Gupta, S., Aggarwal, S., Rashanravan, B., and Lee, T. (1998). Th1- and Th2-like cytokines in CD4(+) and CD8(+) T cells in autism. J. Neuroimmunol. 85, 106–109. doi: 10.1016/S0165-5728(98)00021-6
Gupta, S., Ellis, S. E., Ashar, F. N., Moes, A., Bader, J. S., Zhan, J. N., et al. (2014). Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 5:5748. doi: 10.1038/ncomms6748
Habbas, S., Santello, M., Becker, D., Stubbe, H., Zappia, G., Liaudet, N., et al. (2015). Neuroinflammatory TNF alpha impairs memory via astrocyte signaling. Cell 163, 1730–1741. doi: 10.1016/j.cell.2015.11.023
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Halassa, M. M., Fellin, T., Takano, H., Dong, J. H., and Haydon, P. G. (2007). Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 27, 6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007
Hallmayer, J., Cleveland, S., Torres, A., Phillips, J., Cohen, B., Torigoe, T., et al. (2011). Genetic heritability and shared environmental factors among twin Pairs with autism. Arch. Genet. Psychiatry 68, 1095–1102. doi: 10.1001/archgenpsychiatry.2011.76
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Harrison, J. K., Jiang, Y., Chen, S. Z., Xia, Y. Y., Maciejewski, D., McNamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U.S.A. 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Jyonouchi, H., Sun, S. N., and Le, H. (2001). Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J. Neuroimmunol. 120, 170–179. doi: 10.1016/S0165-5728(01)00421-0
Keil, A., Daniels, J. L., Forssen, U., Hultman, C., Cnattingius, S., Soderberg, K. C., et al. (2010). Parental autoimmune diseases associated with autism spectrum disorders in offspring. Epidemiology 21, 805–808. doi: 10.1097/EDE.0b013e3181f26e3f
Kelleher, R. J., and Bear, M. F. (2008). The autistic neuron: troubled translation? Cell 135, 401–406. doi: 10.1016/j.cell.2008.10.017
Kettenmann, H., Kirchhoff, F., and Verkhratsky, A. (2013). Microglia: new roles for the synaptic stripper. Neuron 77, 10–18. doi: 10.1016/j.neuron.2012.12.023
Kim, S. H., and Lord, C. (2012). New autism diagnostic interview-revised algorithms for toddlers and young preschoolers from 12 to 47 months of age. J. Autism Dev. Disord. 42, 82–93. doi: 10.1007/s10803-011-1213-1
Knuesel, I., Chicha, L., Britschgi, M., Schobel, S. A., Bodmer, M., Hellings, J. A., et al. (2014). Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 10, 643–660. doi: 10.1038/nrneurol.2014.187
Kucukdereli, H., Allen, N. J., Lee, A. T., Feng, A., Ozlu, M. I., Conatser, L. M., et al. (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proc. Natl. Acad. Sci. U.S.A. 108, E440–E449. doi: 10.1073/pnas.1104977108
Li, X. H., Chauhan, A., Sheikh, A. M., Patil, S., Chauhan, V., Li, X. M., et al. (2009). Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 207, 111–116. doi: 10.1016/j.jneuroim.2008.12.002
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
Maekawa, M., Iwayama, Y., Arai, R., Nakamura, K., Ohnishi, T., Toyota, T., et al. (2010). Polymorphism screening of brain-expressed FABP7, 5 and 3 genes and association studies in autism and schizophrenia in Japanese subjects. J. Hum. Genet. 55, 127–130. doi: 10.1038/jhg.2009.133
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010
Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., and Jin, L. W. (2009). Rett Syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009
Malkova, N. V., Yu, C. Z., Hsiao, E. Y., Moore, M. J., and Patterson, P. H. (2012). Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 26, 607–616. doi: 10.1016/j.bbi.2012.01.011
McGann, J. C., Lioy, D. T., and Mandel, G. (2012). Astrocytes conspire with neurons during progression of neurological disease. Curr. Opin. Neurobiol. 22, 850–858. doi: 10.1016/j.conb.2012.03.009
Meyer, K. A., Ingersoll, B., and Hambrick, D. Z. (2011). Factors influencing adjustment in siblings of children with autism spectrum disorders. Res. Autism Spect. Disord. 5, 1413–1420. doi: 10.1016/j.rasd.2011.01.027
Ming, X., Johnson, W. G., Stenroos, E. S., Mars, A., Lambert, G. H., and Buyske, S. (2010). Genetic variant of glutathione peroxidase 1 in autism. Brain Dev. 32, 105–109. doi: 10.1016/j.braindev.2008.12.017
Missault, S., Van den Eynde, K., Berghe, W. V., Fransen, E., Weeren, A., Timmermans, J. P., et al. (2014). The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav. Immun. 42, 138–146. doi: 10.1016/j.bbi.2014.06.013
Molina-Holgado, F., Toulmond, S., and Rothwell, N. J. (2000). Involvement of interleukin-1 in glial responses to lipopolysaccharide: endogenous versus exogenous interleukin-1 actions. J. Neuroimmunol. 111, 1–9. doi: 10.1016/S0165-5728(00)00344-1
Molofsky, A. V., Krenick, R., Ullian, E., Tsai, H. H., Deneen, B., Richardson, W. D., et al. (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 26, 891–907. doi: 10.1101/gad.188326.112
Morgan, J. T., Chana, G., Abramson, I., Semendeferi, K., Courchesne, E., and Everall, I. P. (2012). Abnormal microglial-neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res. 1456, 72–81. doi: 10.1016/j.brainres.2012.03.036
Moynagh, P. N., Williams, D. C., and Oneill, L. A. J. (1993). Interleukin-1 activates transcription factor nf-kappa-b in glial-cells. Biochem. J. 294, 343–347. doi: 10.1042/bj2940343
Murdoch, J. D., and State, M. W. (2013). Recent developments in the genetics of autism spectrum disorders. Curr. Opin. Genet. Dev. 23, 310–315. doi: 10.1016/j.gde.2013.02.003
Nakamura, H., and O’Leary, D. D. M. (1989). Inaccuracies in initial growth and arborization of chick retinotectal axons followed by course corrections and axon remodeling to develop topographic order. J. Neurosci. 9, 3776–3795.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/ng.835
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Parikshak, N. N., Luo, R., Zhang, A., Won, H., Lowe, J. K., Chandran, V., et al. (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. doi: 10.1016/j.cell.2013.10.031
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R., Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Peca, J., and Feng, G. P. (2012). Cellular and synaptic network defects in autism. Curr. Opin. Neurobiol. 22, 866–872. doi: 10.1016/j.conb.2012.02.015
Perez-Garcia, C. G., and O’Leary, D. D. M. (2016). Formation of the cortical subventricular zone requires MDGA1-mediated aggregation of basal progenitors. Cell Rep. doi: 10.1016/j.celrep.2015.12.066 [Epub ahead of print].
Pessah, I. N., and Lein, P. J. (2008). “Evidence for environmental susceptibility in autism what we need to know about gene x environment interactions,” in Autism: Current Theories and Evidence, ed. A. W. Zimmerman (Totowa, NJ: Humana Press Inc.), 409–428.
Petrelli, F., and Bezzi, P. (2016). Novel insights into gliotransmitters. Curr. Opin. Pharmacol. 26, 138–145. doi: 10.1016/j.coph.2015.11.010
Pettem, K. L., Yokomaku, D., Takahashi, H., Ge, Y., and Craig, A. M. (2013). Interaction between autism-linked MDGAs and neuroligins suppresses inhibitory synapse development. J. Cell Biol. 200, 321–336. doi: 10.1083/jcb.201206028
Pfrieger, F. W., and Barres, B. A. (1997). Synaptic efficacy enhanced by glia cells in vitro. Science 277, 1684–1687. doi: 10.1126/science.277.5332.1684
Ponzio, N. M., Servatius, R., Beck, K., Marzouk, A., and Kreider, T. (2007). Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann. N. Y. Acad. Sci. 1107, 118–128.
Purcell, A. E., Jeon, O. H., Zimmerman, A. W., and Pevsner, J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628. doi: 10.1212/WNL.57.9.1618
Quaak, I., Brouns, M. R., and Van de Bor, M. (2013). The dynamics of autism spectrum disorders: how neurotoxic compounds and neurotransmitters interact. Int. J. Environ. Res. Public Health 10, 3384–3408. doi: 10.3390/ijerph10083384
Ronesi, J. A., Collins, K. A., Hays, S. A., Tsai, N. P., Guo, W. R., Birnbaum, S. G., et al. (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci. 15, 431–440. doi: 10.1038/nn.3033
Rosenberg, R. E., Daniels, A. M., Law, J. K., Law, P. A., and Kaufmann, W. E. (2009). Trends in autism spectrum disorder diagnoses: 1994-2007. J. Autism Dev. Disord. 39, 1099–1111. doi: 10.1007/s10803-009-0723-6
Rossi, D. (2015). Astrocyte physiopathology: at the crossroads of intercellular networking, inflammation and cell death. Prog. Neurobiol. 130, 86–120. doi: 10.1016/j.pneurobio.2015.04.003
Sahlender, D. A., Savtchouk, I., and Volterra, A. (2014). What do we know about gliotransmitter release from astrocytes? Philos. Trans. R. Soc. B Biol. Sci. 369:20130592. doi: 10.1098/rstb.2013.0592
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Sandin, S., Lichtenstein, P., Kuja-Halkola, R., Larsson, H., Hultman, C. M., and Reichenberg, A. (2014). The familial risk of autism. JAMA 311, 1770–1777. doi: 10.1001/jama.2014.4144
Santello, M., Bezzi, P., and Volterra, A. (2011). TNF alpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69, 988–1001. doi: 10.1016/j.neuron.2011.02.003
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Serhan, C. N., and Savill, J. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197. doi: 10.1038/ni1276
Smith, S. E. P., Li, J., Garbett, K., Mirnics, K., and Patterson, P. H. (2007). Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 27, 10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007
Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263. doi: 10.1038/nrn3898
Stanfield, B. B., Oleary, D. D. M., and Fricks, C. (1982). Selective collateral elimination in early postnatal-development restricts cortical distribution of rat pyramidal tract neurons. Nature 298, 371–373. doi: 10.1038/298371a0
State, M. W., and Levitt, P. (2011). The conundrums of understanding genetic risks for autism spectrum disorders. Nat. Neurosci. 14, 1499–1506. doi: 10.1038/nn.2924
Steffenburg, S., Gillberg, C., Hellgren, L., Andersson, L., Gillberg, I. C., Jakobsson, G., et al. (1989). A twin study of autism in denmark, finland, iceland, norway and sweden. J. Child Psychol. Psychiatry. 30, 405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059. doi: 10.1038/nature04671
Steven, M. C., Kiehl, K. A., Pearlson, G. D., and Calhoun, V. D. (2007). Functional neural networks underlying response inhibition in adolescents and adults. Behav. Brain Res. 181, 12–22. doi: 10.1016/j.bbr.2007.03.023
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Suzuki, K., Sugihara, G., Ouchi, Y., Nakamura, K., Futatsubashi, M., Takebayashi, K., et al. (2013). Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 70, 49–58. doi: 10.1001/jamapsychiatry.2013.272
Tetreault, N. A., Hakeem, A. Y., Jiang, S., Williams, B. A., Allman, E., Wold, B. J., et al. (2012). Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 42, 2569–2584. doi: 10.1007/s10803-012-1513-0
Toma, C., Hervas, A., Torrico, B., Balmana, N., Salgado, M., Maristany, M., et al. (2013). Analysis of two language-related genes in autism: a case-control association study of FOXP2 and CNTNAP2. Psychiatr. Genet. 23, 82–85. doi: 10.1097/YPG.0b013e32835d6fc6
Tremblay, M. E., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Uhlmann, E. J., Wong, M., Baldwin, R. L., Bajenaru, M. L., Onda, H., Kwiatkowski, D. J., et al. (2002). Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann. Neurol. 52, 285–296. doi: 10.1002/ana.10283
Ullian, E. M., Sapperstein, S. K., Christopherson, K. S., and Barres, B. A. (2001). Control of synapse number by glia. Science 291, 657–661. doi: 10.1126/science.291.5504.657
van Bokhoven, H. (2011). Genetic and Epigenetic Networks in Intellectual Disabilities. In: Annual Review Genetics, Vol. 45. Palo Alto, CA: Annual Reviews, 81–104.
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W., and Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81. doi: 10.1002/ana.20315
Voineagu, I., Wang, X. C., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi: 10.1038/nature10110
Watkins, L. R., Milligan, E. D., and Maier, S. F. (2001). Glial activation: a driving force for pathological pain. Trends Neurosci. 24, 450–455. doi: 10.1016/S0166-2236(00)01854-3
Wei, H. G., Zou, H., Sheikh, A. M., Malik, M., Dobkin, C., Brown, W. T., et al. (2011). IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflamm. 8:52. doi: 10.1186/1742-2094-8-52
Willsey, A. J., Sanders, S. J., Li, M. F., Dong, S., Tebbenkamp, A. T., Muhle, R. A., et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. doi: 10.1016/j.cell.2013.10.020
Wu, Y. W., Dissing-Olesen, L., MacVicar, B. A., and Stevens, B. (2015). Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 36, 605–613. doi: 10.1016/j.it.2015.08.008
Yasui, D. H., Xu, H. C., Dunaway, K. W., LaSalle, J. M., Jin, L. W., and Maezawa, I. (2013). MeCP2 modulates gene expression pathways in astrocytes. Mol. Autism 4:3. doi: 10.1186/2040-2392-4-3
Yuskaitis, C. J., Beurel, E., and Jope, R. S. (2010). Evidence of reactive astrocytes but not peripheral immune system activation in a mouse model of Fragile X syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 1802, 1006–1012. doi: 10.1016/j.bbadis.2010.06.015
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., et al. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406. doi: 10.1038/nn.3641
Keywords: astrocytes, microglia, neurosciences, autism spectrum disorders (ASD)
Citation: Petrelli F, Pucci L and Bezzi P (2016) Astrocytes and Microglia and Their Potential Link with Autism Spectrum Disorders. Front. Cell. Neurosci. 10:21. doi: 10.3389/fncel.2016.00021
Received: 12 November 2015; Accepted: 19 January 2016;
Published: 12 February 2016.
Edited by:
Johann Steiner, University of Magdeburg, GermanyReviewed by:
Rheinallt Parri, Aston University, UKCarlos Gustavo Perez-Garcia, The Salk Institute, USA
Copyright © 2016 Petrelli, Pucci and Bezzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Bezzi, cGFvbGEuYmV6emlAdW5pbC5jaA==