 Marcos J. Guerrero-Muñoz
Marcos J. Guerrero-Muñoz Julia Gerson
Julia Gerson Diana L. Castillo-Carranza
Diana L. Castillo-Carranza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 02 December 2015
Sec. Cellular Neuropathology
Volume 9 - 2015 | https://doi.org/10.3389/fncel.2015.00464
This article is part of the Research TopicRecent advances in synapse biology and its implication in neurodevelopmental and neurodegenerative diseasesView all 13 articles
Alzheimer’s disease (AD) is a progressive disorder in which the most noticeable symptoms are cognitive impairment and memory loss. However, the precise mechanism by which those symptoms develop remains unknown. Of note, neuronal loss occurs at sites where synaptic dysfunction is observed earlier, suggesting that altered synaptic connections precede neuronal loss. The abnormal accumulation of amyloid-β (Aβ) and tau protein is the main histopathological feature of the disease. Several lines of evidence suggest that the small oligomeric forms of Aβ and tau may act synergistically to promote synaptic dysfunction in AD. Remarkably, tau pathology correlates better with the progression of the disease than Aβ. Recently, a growing number of studies have begun to suggest that missorting of tau protein from the axon to the dendrites is required to mediate the detrimental effects of Aβ. In this review we discuss the novel findings regarding the potential mechanisms by which tau oligomers contribute to synaptic dysfunction in AD.
Alzheimer’s disease (AD) is a devastating progressive neurodegenerative condition and the most common cause of dementia among the elderly. The disease is characterized by memory loss and cognitive impairment, and eventually the inability to perform daily life activities. Currently, available treatments for AD only provide relief of symptoms with no effect on the course of the disease. As the longevity of the worldwide population increases, the amount of people susceptible to AD will continue to rise (Reitz and Mayeux, 2014). After decades of research, the precise underlying cause or causes of sporadic AD remain unknown. Therefore, there is an urgent need to understand the pathological mechanisms involved in AD to develop effective treatments.
The profound neuropathological changes to synaptic communication seem to be responsible for cognitive decline and memory dysfunction, the most striking symptoms of AD. However, a great deal of research is needed to come to a complete understanding of the mechanism by which these symptoms develop. Analysis of AD brain cases have revealed synaptic degeneration, neuronal loss and accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs) composed mainly of fibrillar amyloid β peptide (Aβ) and tau protein, respectively (Serrano-Pozo et al., 2011a). For over two decades amyloid plaques were considered to be the primary cause of AD (Hardy and Allsop, 1991). However, amyloid plaque deposition does not correlate with cognitive impairment observed in AD patients.
While amyloid pathology lies upstream of tau pathology (Oddo et al., 2003; Small and Duff, 2008), growing evidence indicates that tau pathology drives cognitive decline in AD (Murray et al., 2015), providing an explanation for the lack of connection between the staging of amyloid plaques and disease symptoms. Tau pathology has thus been considered a secondary amyloidosis in the progression of AD, but the relationship between Aβ and tau is still contentious. The main function of tau is to stabilize microtubules within the neurons. In AD, tau undergoes post-translational modifications that affect the affinity of tau to the microtubule, leading to tau self-association and the eventual formation of NFTs. It has been established that soluble forms of Aβ, but not fibrils (Kayed et al., 2003; Baglioni et al., 2006; Haass and Selkoe, 2007; Shankar et al., 2007; Walsh and Selkoe, 2007), correlate with the onset of the disease only in the presence of tau, suggesting that the latter mediates Aβ toxicity (Lue et al., 1999; McLean et al., 1999; Wang et al., 1999). However, though NFTs correlate better with cognitive decline and neuronal loss (Braak and Braak, 1991b; Arriagada et al., 1992; Gomez-Isla et al., 1997; Giannakopoulos et al., 2003) than amyloid deposits, they do not seem to be the most toxic tau aggregates in disease, with many studies supporting the hypothesis that soluble oligomers drive tau toxicity (Maeda et al., 2006, 2007; Patterson et al., 2011; Lasagna-Reeves et al., 2012b). The deleterious effects of tau pathology may be partly due to a gain of toxic function. The discovery that mutations in the gene encoding tau (MAPT) lead to neurodegeneration strongly supports this hypothesis. Although no mutations in the MAPT gene have been found in AD, they are associated with familial frontotemporal dementia (FTDP-17) reviewed by Goedert et al. (1999), resulting in tau’s inability to bind microtubules and subsequent aggregation into oligomers and NFT’s. Since microtubules are important components of axonal processes, the loss of tau function affects neuronal stability and impairs axonal transport. However ablation of tau in mice does not induce neurological deficits or cell death but instead makes the tau knockout more resistant to seizures (Roberson et al., 2007), suggesting that the pathogenesis of tau is not due solely to a loss of function.
Whether or not the loss of tau function leads to neuronal dysfunction is still in debate. However, there is a large body of evidence demonstrating that aggregated tau acquires a toxic function in which tau oligomers are clearly implicated as driving the mechanism.
In this review, we summarize novel findings regarding the role of tau oligomers at the synapse and their interaction with other amyloid proteins in mediating cognitive decline in AD.
Synaptic plasticity is thought to be the route by which learning and the acquisition of new memories occurs. In AD, marked synapse loss underlies cognitive deficits that appear to depend upon neurodegenerative processes induced by Aβ and tau. Postmortem human brain samples have been found to show gliosis and oxidative stress in the vicinity of amyloid plaques and NFT’s that may contribute to synaptic changes (McLellan et al., 2003; Ingelsson et al., 2004; Serrano-Pozo et al., 2011b). The overexpression of Aβ in mice revealed neurite degeneration after plaque formation (Meyer-Luehmann et al., 2008). However, therapeutic approaches in AD mouse models suggested that plaques are inert and an increase in this metastable aggregate is not associated with neurological deficits (Cheng et al., 2007), but rather is beneficial since cognitive function was improved in mice (Jankowsky et al., 2003, 2005; Lesne et al., 2008). Using the Tg2576 mouse model, we found that removal of tau oligomers by immunotherapy shifted the Aβ aggregation pathway to amyloid plaques, while improving cognition in mice (Castillo-Carranza et al., 2015). These findings could explain the presence of amyloid plaques in individuals without clinical symptoms of AD, thus termed high pathology controls or non-demented with AD neuropathology (NDAN) subjects (Bjorklund et al., 2012) and unsuccessful clinical trials even after removing amyloid plaques (Cappai and Barnham, 2008; Hardy, 2009).
Neurites surrounding plaques often contain phosphorylated tau aggregates (Woodhouse et al., 2005; Serrano-Pozo et al., 2011a). During the course of AD, tau is hyperphosphorylated and accumulates into fibrillar aggregates in the somatodendritic compartment (Spillantini and Goedert, 2013). NFTs have historically been considered the main hallmark in tauopathies, including AD (Braak and Braak, 1991a,b, 1996). However, NFT-containing neurons have been shown to be functionally intact in vivo (Kuchibhotla et al., 2014). A comparative analysis of AD cases versus high-pathology control or NDAN subjects revealed no significant differences in levels of NFTs, rather showing that increased levels of phosphorylated tau in the synaptic compartment were associated with dementia (Perez-Nievas et al., 2013). While signaling cascades involved in long-term potentiation and memory are not affected by NFTs (Kuchibhotla et al., 2014) postmortem analysis of brains from people with mild cognitive impairment showed that cognitive symptoms correlate with pre-fibrillar tau rather than NFT’s (Vana et al., 2011; Mufson et al., 2014). Further supporting ex vivo evidence for the importance of a tau aggregation intermediate in neurodegeneration, tau transgenic animal models acquire behavioral deficits, synaptic dysfunction, and cell death in the absence of NFT formation (Wittmann et al., 2001; Andorfer et al., 2003; SantaCruz et al., 2005; Spires et al., 2006; Berger et al., 2007; Yoshiyama et al., 2007; Cowan et al., 2010). Furthermore, upon suppression of tau in tauopathy transgenic models, mice show cognitive improvement in spite of continued presence of NFTs (SantaCruz et al., 2005; Sydow et al., 2011). Moreover, electrophysiological impairment and structural degeneration to neurons do not depend on the presence of NFTs (Rocher et al., 2010; Crimins et al., 2012). The observation that cell death occurs in disease prior to the formation of NFTs, suggests that pre-filamentous forms of tau confer toxicity before fibrillization (Gomez-Isla et al., 1997; Terry, 2000; van de Nes et al., 2008).
Protein misfolding is the initial step in the aggregation pathway of both Aβ and tau. Post-translational modifications and the formation of disulfide bridges increase the ability of both proteins to self-aggregate into oligomers (Chirita et al., 2005; Sahara et al., 2007). Evidence suggests that tau monomer is first converted to an oligomeric state prior to the formation of fibrils (Ruschak and Miranker, 2009; Lasagna-Reeves et al., 2010; Lee et al., 2011). In vitro, tau aggregation does not occur spontaneously but the addition of polyanionic compounds and free fatty acids induce fibril formation (King et al., 2000; Barghorn and Mandelkow, 2002; Chirita et al., 2003; von Bergen et al., 2005). These various structures differ not only in aggregation state, but also in their toxic effects.
Growing data suggest that prefilamentous forms of tau, specifically oligomers, are neurotoxic (Patterson et al., 2011; Lasagna-Reeves et al., 2012a). Tau oligomers have been isolated at very early stages of the disease, prior to the onset of the clinical symptoms (Maeda et al., 2006; Lasagna-Reeves et al., 2012b). By atomic force microscopy (AFM), tau oligomers display a spherical morphology that corresponds with two or more molecules of tau, ranging between 6 and 20 nm (Sahara et al., 2008). These are dynamic structures that become β-sheet rich (Lasagna-Reeves et al., 2010). In brain samples from AD cases, tau oligomers were found at a fourfold higher concentration compared to healthy control samples (Himmelstein et al., 2012). In AD, tau is abnormally phosphorylated at multiple positions. However this may not be a requirement for tau to be able to form oligomers and become toxic.
In addition to AD, tau oligomers were identified in progressive supranuclear palsy (PSP), dementia with Lewy bodies (DLB) as well as Huntington’s diseases cases (Gerson et al., 2014; Sengupta et al., 2015; Vuono et al., 2015). Thus, the presence of tau oligomers in several tauopathies prompted the hypothesis that tau oligomers follow a common mechanism of toxicity between diseases. However, little is known about the properties of tau oligomers and the mechanism by which they lead to cell loss.
Many studies have demonstrated the toxicity of tau oligomers when applied extracellularly to cultured neuronal cells, leading to tau uptake into the cell and increased intracellular calcium levels. In mice, the injection of tau oligomers induces mitochondrial abnormalities and synaptic dysfunction (Berger et al., 2007; Lasagna-Reeves et al., 2011).
Growing evidence suggests that the accumulation of tau oligomers at the synapse may be critical for neurodegeneration. We have shown that recombinant tau oligomers display amnesic effects and synaptic dysfunction when administered intracranially to wild-type mice (Lasagna-Reeves et al., 2011, 2012b). It appears as though a redistribution of pathological tau from the axon to the cell body and dendrites is responsible for spine loss observed in disease (Zempel et al., 2010). In AD patients, Aβ binds preferentially to neuronal dendrites promoting tau missorting (Zempel and Mandelkow, 2012). It is well-established that aggregated Aβ contributes to tau phosphorylation and mislocalization (Gotz et al., 2001; Ferrari et al., 2003; De Felice et al., 2008; Ittner et al., 2010; Chabrier et al., 2012). However, a growing body of evidence suggests that Aβ induces tau pathology by direct interaction in a prion-like manner. In the prion field, the acquisition of β-sheet conformation by a prion protein allows it to seed the misfolding and aggregation of other prion molecules, reviewed by Jucker and Walker (2011). The pathologic similarities between prion disease and AD suggest that it might also be inducible in a prion-like manner. In vitro assays have shown that Aβ oligomers can seed tau oligomerization, providing evidence that this phenomenon may occur in vivo (Lasagna-Reeves et al., 2010). The induction of tau misfolding in transgenic mice overexpressing amyloid precursor protein (APP) (Castillo-Carranza et al., 2015) and mice infused with brain extract from aged APP23 transgenic mice (Bolmont et al., 2007), suggest that Aβ can seed tau oligomerization in vivo as well (Figure 1). Importantly, it is possible that a direct interaction between the two proteins may be involved in the induction of synaptic dysfunction as tau and Aβ coexist within synaptic compartments in AD brain (Hoover et al., 2010; Ittner et al., 2010; Zempel et al., 2010; Tai et al., 2012; Miller et al., 2014). However, the complexity and diversity of amyloid aggregates has made the elucidation of the interaction between the two proteins difficult. In humans, a 56-kDa Aβ aggregate, called Aβ∗56 seems to have a pathogenic role specifically during the asymptomatic phase of AD (Handoko et al., 2013). Notably, Aβ∗56 correlates with the pathological form of tau (Lesne, 2013) whereas Aβ dimers isolated from AD samples trigger endogenous tau hyperphosphorylation followed by neuritic degeneration of cells in culture (Jin et al., 2011). Different Aβ assemblies including Aβ∗56 are prominent in the synaptic terminals of AD patients (Sokolow et al., 2012). Recently, we provided evidence for the interaction of tau oligomers and Aβ peptide in the Tg2576 mouse. The reduction of tau oligomers by immunotherapy ameliorated memory deficits, implying a role for tau in mediating cognitive decline in aged mice overexpressing APP. Moreover, a marked reduction of Aβ∗56 and increase of trimers suggest that the removal of tau oligomers modulates Aβ levels (Castillo-Carranza et al., 2015). It seems likely that the increase in trimers in treated mice may be a consequence of Aβ∗56 disassembly which has been suggested to be comprised of four Aβ trimers (Lesne et al., 2006). However, reduction of Aβ alone by immunotherapeutic approaches was not sufficient to improve cognition in mice displaying tau pathology, whereas reduction of both pathologies did confer benefits (Oddo et al., 2006) providing support for a synergistic relationship between Aβ and tau in which tau induces toxicity downstream of Aβ.
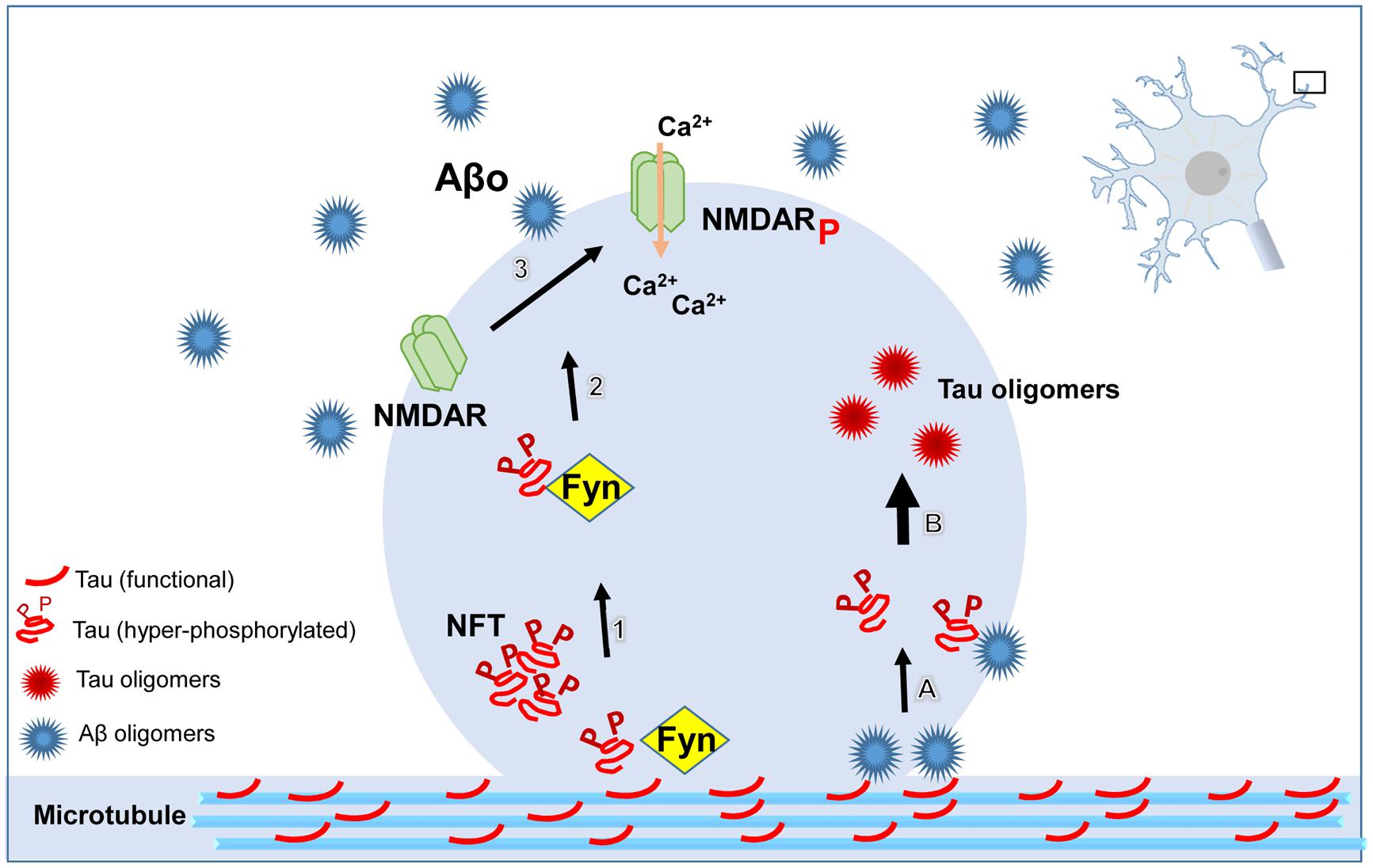
FIGURE 1. Schematic illustrating the pathological role of tau at dendritic spines. ß-amyloid (Aβ) oligomers directly or indirectly lead to the dystrophic changes in neurites mediated by tau. Hyperphosphorylated tau targets the kinase, Fyn, to the postsynaptic compartment. Fyn phosphorylates NR2B, a subunit of the N-methyl-D-aspartate receptor (NMDAR), resulting in the over-activation of NMDAR, followed by increased concentration of Ca2+ in the cytoplasmic compartment. Aβ oligomers (Aβo) seed tau misfolding and aggregation by direct interaction resulting in tau oligomer formation at dendritic spines. All of these pathways converge in the aggregation of tau protein, spine loss, and consequently, cognitive impairment.
While Aβ and tau aggregates are the two main pathological hallmarks of AD, Lewy bodies comprised of α-synuclein protein are found in more than half of sporadic AD cases studied (Hamilton, 2000). Importantly, a recent study showed that toxic, non-fibrillar α-synuclein is significantly elevated in AD cases absent Lewy body pathology (Larson et al., 2012). In its native state, α-synuclein is found at the synapse where it promotes neurotransmitter release (Burré et al., 2010), highlighting its potential importance in synaptotoxicity in AD. Moreover, elevated soluble α-synuclein was associated with a decrease in presynaptic vesicle proteins in AD brains (Larson et al., 2012). These results combined with evidence that oligomeric tau and α-synuclein interact and co-aggregate in disease (Sengupta et al., 2015) suggests that the two proteins may act in a toxic synergistic mechanism at the synapse in AD.
Tau protein promotes neurite outgrowth and is differentially expressed and phosphorylated in the developing brain. During periods of neurite growth, high levels of tau phosphorylated at Ser202 and Thr205 (recognized by AT8 antibody) are seen similarly to during Alzheimer’s conditions, while levels are dramatically reduced when neurites are stabilized and synaptogenesis occurs, corresponding to tau levels and phosphorylation state in healthy adult brain (Brion et al., 1994; Rösner et al., 1995; Riederer, 2001). Collectively, these results suggest that study into the normal function of tau protein may be critical to understanding the synaptic dysfunction due to tau abnormalities in AD. The decrease in synaptic function may be due partially to an overall decrease in synaptogenesis in AD. Cell adhesion molecules such as Nectin-3 are important for both synaptic plasticity and synaptogenesis. Expression of human tau protein as well as tau injections in mice were shown to be associated with a decrease in levels of Nectin-3 in brain regions of importance to memory and cognition (Maurin et al., 2013). Moreover, neurons expressing neuronal nitric oxide synthase that acts as a messenger for synaptogenesis are particularly prone to neurodegeneration in AD brains and the protein was found to colocalize with NFTs (Thorns et al., 1998). A recent study showed that levels of neurogenesis are significantly lowered in the Htau mouse model overexpressing human wild-type tau in a mouse tau knockout background, providing evidence that tau aggregation alone may decrease neurogenesis and synaptogenesis (Komuro et al., 2015).
The characterization of tau aggregates and potential routes of tau spreading has led to important results indicating that tau oligomers can be found in a large percentage of pre-synaptic and post-synaptic compartments in AD, suggesting a toxic role for tau oligomers in synaptic transmission (Tai et al., 2014). Synaptic communication occurs at dendritic spines. Thereby, reductions in spine number or morphological changes would be expected to contribute to synaptic dysfunction and cognitive deficits. Studies have shown that Aβ oligomers interact with tau, inducing its translocation to synaptic spines (Frandemiche et al., 2014). Dendritic spines present with various morphologies can drastically affect their functionality. Spines found to have particularly large post-synaptic densities are believed to provide for stable synaptic connections for memory formation. We have found that a reduction of tau oligomers in the Tg2576 AD mouse model is associated with a significant increase in mushroom-shaped spines with large postsynaptic densities (Castillo-Carranza et al., 2015). Moreover, in Htau mice the levels of tau correlate with cognitive deficits, decrease in long term potentiation, lowering of synaptic proteins, and a reduction in the level of mushroom-shaped spines as well as an increase in amount of thin spines (Polydoro et al., 2009; Dickstein et al., 2010; Alldred et al., 2012). A mouse model overexpressing both Aβ and wildtype tau exhibited a synergistic toxic effect to dendritic spines that was greater than effects of the expression of either protein alone (Chabrier et al., 2014). While these results highlight the toxic effect of tau pathology at the synapse, some studies have shown that Aβ can induce synaptic dysfunction, spine loss, and changes to spine morphology independently of tau (Shahani et al., 2006; Tackenberg and Brandt, 2009; Tackenberg et al., 2013). On the other hand, tau aggregation alone has been shown to be sufficient to cause synaptic detriment. The injection of human tau blocked synaptic transmission of squid axons (Moreno et al., 2011). A comparative analysis of synapses showed that only AD brains but not controls, contained tau phosphorylated at serines 396/404 in a greater number of postsynaptic than presynaptic sites (Tai et al., 2012). In contrast, this type of tau is accumulated in the presynapse of the entorhinal cortex from aged P301L mice, a transgenic overexpressing human mutant tau (Harris et al., 2012). The accumulation of phosphorylated P301L mutated tau is accompanied by disruption of synaptic transmission and impaired glutamate receptor subunit GluA1, GluA2/3, and NR1 trafficking to the postsynaptic density (Hoover et al., 2010). Overexpression of P301L tau in rTg4510 mice causes synaptic dysfunction and loss of synapses (Rocher et al., 2010; Crimins et al., 2011, 2012, 2013; Kopeikina et al., 2011, 2013). A recent study by Decker et al., showed that pre-NFTs—likely comprised largely of tau oligomers—cause pre- and postsynaptic morphological changes (a gain of toxic function) at the mossy fibers located in the CA3 brain region in transgenic mice expressing the aggregation prone ΔK280 (TauRDΔ) mutant human Tau (Decker et al., 2015). Interestingly, examination of tau knockout mice revealed normal synaptic plasticity, but weak synaptic transmission comparable to mice TauRDΔ. Previous studies have shown that missorting of tau to the somatodendritic compartment leads to retraction of mossy fibers from CA3 in hibernating ground squirrels, in a process which seems to be reversible suggesting a physiological role of Tau in mossy fiber plasticity (Arendt et al., 2003).
Taken together, these studies suggest an important role for tau within both the pre- and postsynapse, suggesting that when tau misfolds and aggregates into oligomers in disease it may cause synaptic dysfunction.
Recently, the function of tau has been expanded to include a role in synaptic plasticity. Studies showed that knocking out tau in vivo eliminates long term depression (LTD) in the CA1 of the hippocampus. LTD describes an activity-dependent reduction or weakening of synaptic contacts. Tau phosphorylation induced by Aβ-mediated NMDA receptor activation is associated with an increase in LTD (Mondragón-Rodríguez et al., 2012; Kimura et al., 2014), likely dependent upon AMPA receptor internalization mediated by tau (Regan et al., 2015). These results suggest a physiologically important role for tau in synaptic transmission, as well as highlight a potential route of toxicity if the misprocessing of tau leads to signaling cascades inducing increased LTD in the hippocampus in AD. Furthermore, mutated tau was found to be associated with misprocessing of glutamate signaling and excitotoxicity in vivo, further supporting a role for tau in regulation of synaptic transmission (Hunsberger et al., 2015). Tau transgenic mice show alterations in spine and post-synaptic density volume, as well as basal plasticity changes measured by electrophysiology. The detection of defective myelination in axons from tauopathy mice suggests that signaling detriments may also be partially dependent on myelination errors (Maurin et al., 2014).
It is postulated that tau mediates the NMDA receptor through the tyrosine kinase, Fyn, a member of the Src family (Larson et al., 2012). The function of Fyn is to phosphorylate NR2B, a subunit of the NMDA receptor. Tau seems to have a crucial function at dendrites by targeting the kinase Fyn to postsynaptic compartments, resulting in the over-activation of NMDA receptors (Lee et al., 1998; Reynolds et al., 2008; Ittner et al., 2010). Tau or Aβ stabilization of NR2B with PSD95 enhances glutamatergic toxicity. This interaction seems to be mediated by phosphorylation of tau (Bhaskar et al., 2005; Reynolds et al., 2008; Usardi et al., 2011). However, dephosphorylated tau is able to cause cell death by activating muscarinic acetylcholine receptors with a higher affinity than acetylcholine (Gomez-Ramos et al., 2008, 2009; Diaz-Hernandez et al., 2010). In APP mice, reduction of Fyn prevented Aβ-mediated toxicity (Chin et al., 2004, 2005). The absence of tau or tau lacking the microtubule binding domain abolished Fyn targeting to dendritic spines, thus preventing memory deficits associated with Aβ. Moreover, inhibiting Fyn in a mouse model of AD led to a decrease in tau phosphorylation and reversal of memory deficits associated with a restoration of synapse density (Kaufman et al., 2015). Further, a double transgenic mouse generated by crossing mice overexpressing human APP (hAPP) and Htau mice exhibit accelerated cognitive impairment, enhanced aggregation of soluble and insoluble tau, as well as reduction of dendritic spines. Interestingly Fyn is upregulated only in the double transgenic but not in single transgenic Htau or hAPP, suggesting that the presence of Aβ and tau is required in order for Fyn to promote dendritic abnormalities(Chabrier et al., 2014). We have recently shown that reduction of tau oligomers by immunotherapy does not affect Fyn protein levels after treatment, but it does have effects on spine plasticity, suggesting that tau oligomers mediate a different pathway in dendritic spines (Castillo-Carranza et al., 2015). This may be true for tau oligomers specifically since it was previously reported that tau binding to Fyn is increased upon phosphorylation of tau (Mondragón-Rodríguez et al., 2012) particularly at AT8 or PHF1 sites, markers of NFTs, that seem to enhance Fyn SH3 binding to the proline-rich domain of tau (Bhaskar et al., 2005).
Functional tau has been detected in small concentrations at dendrites (Ittner et al., 2010). Under physiological conditions a small proportion of full-length tau is secreted to the extracellular space (Yamada et al., 2011) raising the question of whether extracellular tau has a physiological function. Tau has been found in cerebrospinal fluid (CSF) from healthy people (Ittner et al., 2010). In AD the levels of hyperphosphorylated tau in CSF correlated well with the progression of the disease. Moreover, tau was found in subcellular compartments responsible for protein trafficking and secretion such as autophagic vacuoles, endoplasmic reticulum, and Golgi apparatus (Tang et al., 2015). Microvesicle shedding and exosome release are some of the possible mechanisms that have been proposed to involve tau secretion from neurons. Exosomes refer to vesicles formed through budding of the endosomal membrane into larger vesicles termed multivesicular bodies (MVBs). Thus it is possible that once released from neurons, tau can eventually misfold, acquire a toxic function and become a potential source of seeds that can propagate throughout the brain. Extracellular tau released from ghost tangles or damaged neurons may become toxic to neighboring cells. However, recently Pooler et al. (2013) showed that propagation of tau pathology is an active process associated with synapses rather than release due to cell death. In vivo, microdialysis of mouse brains suggests that increasing neuronal and synaptic activity correlates with higher levels of extracellular tau (Yamada et al., 2011). Therefore, trans-synaptic communication is a possible avenue by which misfolded tau oligomers spread and compromise functional tau.
One of the most critical mechanisms under investigation in the field of neurodegeneration today is how tau pathology spreads from affected to unaffected brain regions. Many studies have suggested that oligomeric tau may be capable of propagating in the brain, inducing the misfolding of functional tau, reviewed by Gerson and Kayed (2013). While the mechanism is currently unknown and many different hypotheses exist, there has been some reported evidence for the spread of tau oligomers through the synapse. In combination with stereotypic staging of NFT pathology in AD that follows a transsynaptic pattern of spreading of pathology (Braak and Braak, 1991b), studies in animal models have provided direct evidence for this pathway.
Using a transgenic mouse that conditionally expresses tau in the area of the brain where Alzheimer’s disease pathology first arises—the entorhinal cortex—researchers have shown that tau appears to spread between synaptically connected brain regions, suggesting a potential role of the synapse in pathological tau transport (de Calignon et al., 2012; Liu et al., 2012). After thorough analysis of mice injected with tau aggregates in the hippocampus, it was found that tau spreading occurred in none of 20 neighboring brain regions analyzed and was only found in synaptically connected areas (Ahmed et al., 2014). In order to determine whether tau oligomeric aggregates are capable of transporting between the axonal and somatodendritic compartments of the neuron to allow for synaptic transport of tau taken up in the cell in disease, neurons were cultured in microfluidic chambers and treated with tau oligomers. Researchers found that oligomeric tau was able to spread both anterogradely and retrogradely between cellular compartments, though tau monomers and fibrils were not able to enter the cell when administered in the media (Wu et al., 2013). Further evidence of the trans-synaptic mechanism for the spread of tau aggregates is the presence of phosphorylated tau at the synapse in AD brain that correlates with dysfunction of the ubiquitin proteasome system. This accumulation of tau unable to be degraded may account for the spread of tau through the synapse (Tai et al., 2012). Moreover, stimulation of neuronal firing through the activation of AMPA receptors led to an increase in extracellular tau in cultured cells, while levels were decreased when pharmacological agents reducing synaptic vesicle release and neuronal activity were applied (Pooler et al., 2013). In order to confirm the importance of synaptic contacts in the transfer of tau aggregates between cells, neurons expressing synaptogenic adhesion proteins were cultured to induce the formation of synapses. When compared to control cells, neurons with heightened synaptic formation also increased the uptake and spreading of tau aggregates after treatment. Moreover, inhibiting the formation of synapses, as well as neuronal activity led to a decrease in the ability of tau aggregates administered to cells to transport between neurons (Calafate et al., 2015).
In spite of all the evidence suggesting a toxic role of tau, therapeutic interventions have been focused on targeting Aβ in preclinical and clinical studies. Although preclinical studies showed very exciting results, clinical trials did not prevent the progression of cognitive decline. It seems likely that Aβ initiates a cascade of events that at a certain stage becomes irreversible, thereby making Aβ removal insufficient to avert cognitive decline. A secondary pathological event that causes dementia and has the potential to become independent of Aβ pathology is the formation of toxic tau oligomers. These structures are able to self-propagate, spread through synapses and induce synaptic dysfunction. Thus, understanding the mechanisms by which tau oligomers spread throughout the synapse may be critical for the design of novel therapeutic strategies to treat AD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by The Mitchell Center for Neurodegenerative Diseases.
Ahmed, Z., Cooper, J., Murray, T., Garn, K., McNaughton, E., Clarke, H., et al. (2014). A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127, 667–683. doi: 10.1007/s00401-014-1254-6
Alldred, M. J., Duff, K. E., and Ginsberg, S. D. (2012). Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol. Dis. 45, 751–762. doi: 10.1016/j.nbd.2011.10.022
Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K. L., Barde, Y. A., et al. (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590. doi: 10.1046/j.1471-4159.2003.01879.x
Arendt, T., Stieler, J., Strijkstra, A. M., Hut, R. A., Rudiger, J., Van der Zee, E. A., et al. (2003). Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 23, 6972–6981.
Arriagada, P. V., Marzloff, K., and Hyman, B. T. (1992). Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42, 1681–1688. doi: 10.1212/WNL.42.9.1681
Baglioni, S., Casamenti, F., Bucciantini, M., Luheshi, L. M., Taddei, N., Chiti, F., et al. (2006). Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J. Neurosci. 26, 8160–8167. doi: 10.1523/JNEUROSCI.4809-05.2006
Barghorn, S., and Mandelkow, E. (2002). Toward a Unified Scheme for the Aggregation of Tau into Alzheimer Paired Helical Filaments†. Biochemistry 41, 14885–14896. doi: 10.1021/bi026469j
Berger, Z., Roder, H., Hanna, A., Carlson, A., Rangachari, V., Yue, M., et al. (2007). Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 27, 3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007
Bhaskar, K., Yen, S. H., and Lee, G. (2005). Disease-related modifications in tau affect the interaction between Fyn and Tau. J. Biol. Chem. 280, 35119–35125. doi: 10.1074/jbc.M505895200
Bjorklund, N. L., Sadagoparamanujam, R. L., Ghirardi, V.-M., Woltjer, V., and Taglialatela, G. (2012). Absence of amyloid ββ oligomers at the postsynapse and regulated synaptic Zn2++ in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol. Neurodegener. 7:23. doi: 10.1186/1750-1326-7-23
Bolmont, T., Clavaguera, F., Meyer-Luehmann, M., Herzig, M. C., Radde, R., Staufenbiel, M., et al. (2007). Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am. J. Pathol. 171, 2012–2020. doi: 10.2353/ajpath.2007.070403
Braak, H., and Braak, E. (1991a). Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1, 213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x
Braak, H., and Braak, E. (1991b). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Braak, H., and Braak, E. (1996). Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol. Scand. Suppl. 165, 3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x
Brion, J. P., Octave, J. N., and Couck, A. M. (1994). Distribution of the phosphorylated microtubule-associated protein tau in developing cortical neurons. Neuroscience 63, 895–909. doi: 10.1016/0306-4522(94)90533-9
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M., and Südhof, T. C. (2010). αα-Synuclein Promotes SNARE-Complex Assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Calafate, S., Buist, A., Miskiewicz, K., Vijayan, V., Daneels, G., de Strooper, B., et al. (2015). Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 11, 1176–1183. doi: 10.1016/j.celrep.2015.04.043
Cappai, R., and Barnham, K. J. (2008). Delineating the mechanism of Alzheimer’s disease A beta peptide neurotoxicity. Neurochem. Res. 33, 526–532. doi: 10.1007/s11064-007-9469-8
Castillo-Carranza, D. L., Guerrero-Munoz, M. J., Sengupta, U., Hernandez, C., Barret, A., Dineley, K. T., et al. (2015). Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an AD mouse model. J. Neurosci. 35, 4857–4868. doi: 10.1523/JNEUROSCI.4989-14.2015
Chabrier, M. A., Blurton-Jones, M., Agazaryan, A. A., Nerhus, J. L., Martinez-Coria, H., and LaFerla, F. M. (2012). Soluble abeta promotes wild-type tau pathology in vivo. J. Neurosci. 32, 17345–17350. doi: 10.1523/JNEUROSCI.0172-12.2012
Chabrier, M. A., Cheng, D., Castello, N. A., Green, K. N., and LaFerla, F. M. (2014). Synergistic effects of amyloid-beta and wild-type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer’s disease. Neurobiol. Dis. 64, 107–117. doi: 10.1016/j.nbd.2014.01.007
Cheng, I. H., Scearce-Levie, K., Legleiter, J., Palop, J. J., Gerstein, H., Bien-Ly, N., et al. (2007). Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 282, 23818–23828. doi: 10.1074/jbc.M701078200
Chin, J., Palop, J. J., Puolivali, J., Massaro, C., Bien-Ly, N., Gerstein, H., et al. (2005). Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 25, 9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005
Chin, J., Palop, J. J., Yu, G. Q., Kojima, N., Masliah, E., and Mucke, L. (2004). Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 24, 4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004
Chirita, C. N., Congdon, E. E., Yin, H., and Kuret, J. (2005). Triggers of full-length tau aggregation:? a role for partially folded intermediates†. Biochemistry 44, 5862–5872. doi: 10.1021/bi0500123
Chirita, C. N., Necula, M., and Kuret, J. (2003). Anionic micelles and vesicles induce tau fibrillization in vitro. J. Biol. Chem. 278, 25644–25650. doi: 10.1074/jbc.M301663200
Cowan, C. M., Bossing, T., Page, A., Shepherd, D., and Mudher, A. (2010). Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 120, 593–604. doi: 10.1007/s00401-010-0716-8
Crimins, J. L., Pooler, A., Polydoro, M., Luebke, J. I., and Spires-Jones, T. L. (2013). The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer’s disease. Ageing Res. Rev. 12, 757–763. doi: 10.1016/j.arr.2013.03.002
Crimins, J. L., Rocher, A. B., and Luebke, J. I. (2012). Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 124, 777–795. doi: 10.1007/s00401-012-1038-9
Crimins, J. L., Rocher, A. B., Peters, A., Shultz, P., Lewis, J., and Luebke, J. I. (2011). Homeostatic responses by surviving cortical pyramidal cells in neurodegenerative tauopathy. Acta Neuropathol. 122, 551–564. doi: 10.1007/s00401-011-0877-0
de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
Decker, J. M., Kruger, L., Sydow, A., Zhao, S., Frotscher, M., Mandelkow, E., et al. (2015). Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca(++++) dysregulation. Acta Neuropathol. Commun. 3:23. doi: 10.1186/s40478-015-0193-3
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 29, 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
Diaz-Hernandez, M., Gomez-Ramos, A., Rubio, A., Gomez-Villafuertes, R., Naranjo, J. R., Miras-Portugal, M. T., et al. (2010). Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 285, 32539–32548. doi: 10.1074/jbc.M110.145003
Dickstein, D., Brautigam, H., Stockton, S. Jr., Schmeidler, J., and Hof, P. (2010). Changes in dendritic complexity and spine morphology in transgenic mice expressing human wild-type tau. Brain Struct. Funct. 214, 161–179. doi: 10.1007/s00429-010-0245-1
Ferrari, A., Hoerndli, F., Baechi, T., Nitsch, R. M., and Gotz, J. (2003). beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J. Biol. Chem. 278, 40162–40168. doi: 10.1074/jbc.M308243200
Frandemiche, M. L., De Seranno, S., Rush, T., Borel, E., Elie, A., Arnal, I., et al. (2014). Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 34, 6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014
Gerson, J., Sengupta, U., Lasagna-Reeves, C., Guerrero-Munoz, M., Troncoso, J., and Kayed R.. (2014). Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol. Commun. 2:73 doi: 10.1186/2051-5960-2-73
Gerson, J. E., and Kayed, R. (2013). Formation and propagation of tau oligomeric seeds. Front. Neurol. 4:93. doi: 10.3389/fneur.2013.00093
Giannakopoulos, P., Herrmann, F. R., Bussiere, T., Bouras, C., Kovari, E., Perl, D. P., et al. (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500. doi: 10.1212/01.WNL.0000063311.58879.01
Goedert, M., Jakes, R., and Crowther, R. A. (1999). Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 450, 306–311. doi: 10.1016/S0014-5793(99)00508-6
Gomez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Diaz-Hernandez, J. I., Miras-Portugal, M. T., and Avila, J. (2009). Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 19, 708–717. doi: 10.1016/j.euroneuro.2009.04.006
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681. doi: 10.1016/j.mcn.2007.12.010
Gotz, J., Chen, F., van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. doi: 10.1126/science.1062097
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hamilton, R. L. (2000). Lewy bodies in alzheimer’s disease: a neuropathological review of 145 cases using αα-synuclein immunohistochemistry. Brain Pathol. 10, 378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x
Handoko, M., Grant, M., Kuskowski, M., Zahs, K. R., Wallin, A., Blennow, K., et al. (2013). Correlation of specific amyloid-beta oligomers with tau in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol. 70, 594–599. doi: 10.1001/jamaneurol.2013.48
Hardy, J. (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 110, 1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388. doi: 10.1016/0165-6147(91)90609-V
Harris, J. A., Koyama, A., Maeda, S., Ho, K., Devidze, N., Dubal, D. B., et al. (2012). Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS ONE 7:e45881. doi: 10.1371/journal.pone.0045881
Himmelstein, D. S., Ward, S. M., Lancia, J. K., Patterson, K. R., and Binder, L. I. (2012). Tau as a therapeutic target in neurodegenerative disease. Pharmacol. Ther. 136, 8–22. doi: 10.1016/j.pharmthera.2012.07.001
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. doi: 10.1016/j.neuron.2010.11.030
Hunsberger, H. C., Rudy, C. C., Batten, S. R., Gerhardt, G. A., and Reed, M. N. (2015). P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J. Neurochem. 132, 169–182. doi: 10.1111/jnc.12967
Ingelsson, M., Fukumoto, H., Newell, K. L., Growdon, J. H., Hedley-Whyte, E. T., Frosch, M. P., et al. (2004). Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62, 925–931. doi: 10.1212/01.WNL.0000115115.98960.37
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jankowsky, J. L., Melnikova, T., Fadale, D. J., Xu, G. M., Slunt, H. H., Gonzales, V., et al. (2005). Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neurosci. 25, 5217–5224. doi: 10.1523/JNEUROSCI.5080-04.2005
Jankowsky, J. L., Xu, G., Fromholt, D., Gonzales, V., and Borchelt, D. R. (2003). Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 62, 1220–1227.
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824. doi: 10.1073/pnas.1017033108
Jucker, M., and Walker, L. C. (2011). Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 70, 532–540. doi: 10.1002/ana.22615
Kaufman, A. C., Salazar, S. V., Haas, L. T., Yang, J., Kostylev, M. A., Jeng, A. T., et al. (2015). Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 77, 953–971. doi: 10.1002/ana.24394
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kimura, T., Whitcomb, D. J., Jo, J., Regan, P., Piers, T., Heo, S., et al. (2014). Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2:369.
King, M. E., Gamblin, T. C., Kuret, J., and Binder, L. I. (2000). Differential assembly of human tau isoforms in the presence of arachidonic acid. J. Neurochem. 74, 1749–1757. doi: 10.1046/j.1471-4159.2000.0741749.x
Komuro, Y., Xu, G., Bhaskar, K., and Lamb, B. T. (2015). Human tau expression reduces adult neurogenesis in a mouse model of tauopathy. Neurobiol. Aging 36, 2034–2042. doi: 10.1016/j.neurobiolaging.2015.03.002
Kopeikina, K. J., Carlson, G. A., Pitstick, R., Ludvigson, A. E., Peters, A., Luebke, J. I., et al. (2011). Tau Accumulation Causes Mitochondrial Distribution Deficits in Neurons in a Mouse Model of Tauopathy and in Human Alzheimer’s Disease Brain. Am. J. Pathol. 179, 2071–2082. doi: 10.1016/j.ajpath.2011.07.004
Kopeikina, K. J., Wegmann, S., Pitstick, R., Carlson, G. A., Bacskai, B. J., Betensky, R. A., et al. (2013). Tau causes synapse loss without disrupting calcium homeostasis in the rTg4510 model of tauopathy. PLoS ONE 8:e80834. doi: 10.1371/journal.pone.0080834
Kuchibhotla, K. V., Wegmann, S., Kopeikina, K. J., Hawkes, J., Rudinskiy, N., Andermann, M. L., et al. (2014). Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, 510–514. doi: 10.1073/pnas.1318807111
Larson, M. E., Sherman, M. A., Greimel, S., Kuskowski, M., Schneider, J. A., Bennett, D. A., et al. (2012). Soluble αα-synuclein is a novel modulator of alzheimer’s disease pathophysiology. J. Neurosci. 32, 10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Guerrero-Muoz, M. J., Jackson, G. R., and Kayed, R. (2010). Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041. doi: 10.1021/bi1016233
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Clos, A. L., Jackson, G. R., and Kayed, R. (2011). Tau Oligomers Impair Memory and Induce Synaptic and Mitochondrial Dysfunction in Wild-type Mice. Mol. Neurodegener. 6:39. doi: 10.1186/1750-1326-6-39
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Guerrero-Munoz, M. J., Kiritoshi, T., Neugebauer, V., et al. (2012a). Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2:700. doi: 10.1038/srep00700
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Sarmiento, J., Troncoso, J., Jackson, G. R., et al. (2012b). Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 26, 1946–1959. doi: 10.1096/fj.11-199851
Lee, G., Newman, S. T., Gard, D. L., Band, H., and Panchamoorthy, G. (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 111(Pt 21), 3167–3177.
Lee, J., Culyba, E. K., Powers, E. T., and Kelly, J. W. (2011). Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 7, 602–609. doi: 10.1038/nchembio.624
Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Lesne, S., Kotilinek, L., and Ashe, K. H. (2008). Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience 151, 745–749. doi: 10.1016/j.neuroscience.2007.10.054
Lesne, S. E. (2013). Breaking the code of amyloid- oligomers. Int. J. Cell Biol. 2013:950783. doi: 10.1155/2013/950783
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7:e31302. doi: 10.1371/journal.pone.0031302
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., et al. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862. doi: 10.1016/S0002-9440(10)65184-X
Maeda, S., Sahara, N., Saito, Y., Murayama, S., Ikai, A., and Takashima, A. (2006). Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 54, 197–201. doi: 10.1016/j.neures.2005.11.009
Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., et al. (2007). Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. doi: 10.1021/bi061359o
Maurin, H., Chong, S.-A., Kraev, I., Davies, H., Kremer, A., Seymour, C. M., et al. (2014). Early structural and functional defects in synapses and myelinated axons in stratum lacunosum moleculare in two preclinical models for tauopathy. PLoS ONE 9:e87605. doi: 10.1371/journal.pone.0087605
Maurin, H., Seymour, C. M., Lechat, B., Borghgraef, P., Devijver, H., Jaworski, T., et al. (2013). Tauopathy differentially affects cell adhesion molecules in mouse brain: early down-regulation of nectin-3 in stratum lacunosum moleculare. PLoS ONE 8:e63589. doi: 10.1371/journal.pone.0063589
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Beyreuther, K., et al. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M
McLellan, M. E., Kajdasz, S. T., Hyman, B. T., and Bacskai, B. J. (2003). In vivo imaging of reactive oxygen species specifically associated with thioflavine S-positive amyloid plaques by multiphoton microscopy. J. Neurosci. 23, 2212–2217.
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, Rozkalne, A., Koenigsknecht-Talboo, J., et al. (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724. doi: 10.1038/nature06616
Miller, E. C., Teravskis, P. J., Dummer, B. W., Zhao, X., Huganir, R. L., and Liao, D. (2014). Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 39, 1214–1224. doi: 10.1111/ejn.12507
Mondragón-Rodríguez, S., Trillaud-Doppia, E., Dudilot, A., Bourgeois, C., Lauzon, M., Leclerc, N., et al. (2012). Interaction of endogenous tau protein with synaptic proteins is regulated by n-methyl-d-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 287, 32040–32053. doi: 10.1074/jbc.M112.401240
Moreno, H., Choi, S., Yu, E., Brusco, J., Avila, J., Moreira, J. E., et al. (2011). Blocking effects of human tau on squid giant synapse transmission and its prevention by T-817 MA. Front Synaptic Neurosci 3:3. doi: 10.3389/fnsyn.2011.00003
Mufson, E. J., Ward, S., and Binder, L. (2014). Prefibrillar tau oligomers in mild cognitive impairment and Alzheimer’s disease. Neurodegener. Dis. 13, 151–153. doi: 10.1159/000353687
Murray, M. E., Lowe, V. J., Graff-Radford, N. R., Liesinger, A. M., Cannon, A., Przybelski, S. A., et al. (2015). Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain. 138, 1370–1381. doi: 10.1093/brain/awv050
Oddo, S., Caccamo, A., Kitazawa, M., Tseng, B. P., and LaFerla, F. M. (2003). Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 24, 1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012
Oddo, S., Vasilevko, V., Caccamo, A., Kitazawa, M., Cribbs, D. H., and LaFerla, F. M. (2006). Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J. Biol. Chem. 281, 39413–39423. doi: 10.1074/jbc.M608485200
Patterson, K. R., Remmers, C., Fu, Y., Brooker, S., Kanaan, N. M., Vana, L., et al. (2011). Characterization of prefibrillar tau oligomers in vitro and in Alzheimers disease. J. Biol. Chem. 286, 23063–23076. doi: 10.1074/jbc.M111.237974
Perez-Nievas, B. G., Stein, T. D., Tai, H. C., Dols-Icardo, O., Scotton, T. C., Barroeta-Espar, I., et al. (2013). Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136(Pt 8), 2510–2526. doi: 10.1093/brain/awt171
Polydoro, M., Acker, C. M., Duff, K., Castillo, P. E., and Davies, P. (2009). Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 29, 10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009
Pooler, A. M., Phillips, E. C., Lau, D. H. W., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Regan, P., Piers, T., Yi, J.-H., Kim, D.-H., Huh, S., Park, S. J., et al. (2015). Tau phosphorylation at serine 396 residue is required for hippocampal ltd. J. Neurosci. 35, 4804–4812. doi: 10.1523/JNEUROSCI.2842-14.2015
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
Reynolds, C. H., Garwood, C. J., Wray, S., Price, C., Kellie, S., Perera, T., et al. (2008). Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 283, 18177–18186. doi: 10.1074/jbc.M709715200
Riederer, B. (2001). Differential phosphorylation of tau proteins during kitten brain development and Alzheimer’s disease. J. Neurocytol. 30, 145–158. doi: 10.1023/A:1011991207942
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rocher, A. B., Crimins, J. L., Amatrudo, J. M., Kinson, M. S., Todd-Brown, M. A., Lewis, J., et al. (2010). Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp. Neurol. 223, 385–393. doi: 10.1016/j.expneurol.2009.07.029
Rösner, H., Rebhan, M., Vacun, G., and Vanmechelen, E. (1995). Developmental expression of tau proteins in the chicken and rat brain: rapid down-regulation of a paired helical filament epitope in the rat cerebral cortex coincides with the transition from immature to adult tau isoforms. Int. J. Dev. Neurosci. 13, 607–617. doi: 10.1016/0736-5748(95)00042-F
Ruschak, A. M., and Miranker, A. D. (2009). The role of prefibrillar structures in the assembly of a peptide amyloid. J. Mol. Biol. 393, 214–226. doi: 10.1016/j.jmb.2009.06.026
Sahara, N., Maeda, S., Murayama, M., Suzuki, T., Dohmae, N., Yen, S.-H., et al. (2007). Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 25, 3020–3029. doi: 10.1111/j.1460-9568.2007.05555.x
Sahara, N., Maeda, S., and Takashima, A. (2008). Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration. Curr. Alzheimer Res. 5, 591–598. doi: 10.2174/156720508786898442
SantaCruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. doi: 10.1126/science.1113694
Sengupta, U., Guerrero-Muñoz, M. J., Castillo-Carranza, D. L., Lasagna-Reeves, C. A., Gerson, J. E., Paulucci-Holthauzen, A. A., et al. (2015). Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol. Psychiatry 78, 672–683. doi: 10.1016/j.biopsych.2014.12.019
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011a). Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Serrano-Pozo, A., Mielke, M. L., Gomez-Isla, T., Betensky, R. A., Growdon, J. H., Frosch, M. P., et al. (2011b). Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 179, 1373–1384. doi: 10.1016/j.ajpath.2011.05.047
Shahani, N., Subramaniam, S., Wolf, T., Tackenberg, C., and Brandt, R. (2006). Tau aggregation and progressive neuronal degeneration in the absence of changes in spine density and morphology after targeted expression of alzheimer’s disease-relevant tau constructs in organotypic hippocampal slices. J. Neurosci. 26, 6103–6114. doi: 10.1523/JNEUROSCI.4245-05.2006
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Small, S. A., and Duff, K. (2008). Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron 60, 534–542. doi: 10.1016/j.neuron.2008.11.007
Sokolow, S., Henkins, K. M., Bilousova, T., Miller, C. A., Vinters, H. V., Poon, W., et al. (2012). AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol. Aging 33, 1545–1555. doi: 10.1016/j.neurobiolaging.2011.05.011
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi: 10.1016/S1474-4422(13)70090-5
Spires, T. L., Orne, J. D., SantaCruz, K., Pitstick, R., Carlson, G. A., Ashe, K. H., et al. (2006). Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607. doi: 10.2353/ajpath.2006.050840
Sydow, A., Van der Jeugd, A., Zheng, F., Ahmed, T., Balschun, D., Petrova, O., et al. (2011). Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 31, 2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011
Tackenberg, C., and Brandt, R. (2009). Divergent pathways mediate spine alterations and cell death induced by amyloid-ββ, wild-type tau, and R406W Tau. J. Neurosci. 29, 14439–14450. doi: 10.1523/JNEUROSCI.3590-09.2009
Tackenberg, C., Grinschgl, S., Trutzel, A., Santuccione, A. C., Frey, M. C., Konietzko, U., et al. (2013). NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis. 4:e608. doi: 10.1038/cddis.2013.129
Tai, H. C., Serrano-Pozo, A., Hashimoto, T., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435. doi: 10.1016/j.ajpath.2012.06.033
Tai, H.-C., Wang, B. Y., Serrano-Pozo, A., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2014). Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol. Commun. 2:146. doi: 10.1186/s40478-014-0146-2
Tang, Z., Ioja, E., Bereczki, E., Hultenby, K., Li, C., Guan, Z., et al. (2015). mTor mediates tau localization and secretion: Implication for Alzheimer’s disease. Biochim. Biophys. Acta 1853, 1646–1657. doi: 10.1016/j.bbamcr.2015.03.003
Terry, R. D. (2000). Do neuronal inclusions kill the cell? J. Neural Transm. Suppl. 59, 91–93. doi: 10.1007/978-3-7091-6781-6_12
Thorns, V., Hansen, L., and Masliah, E. (1998). nNOS expressing neurons in the entorhinal cortex and hippocampus are affected in patients with Alzheimer’s Disease. Exp. Neurol. 150, 14–20. doi: 10.1006/exnr.1997.6751
Usardi, A., Pooler, A. M., Seereeram, A., Reynolds, C. H., Derkinderen, P., Anderton, B., et al. (2011). Tyrosine phosphorylation of tau regulates its interactions with Fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau. FEBS J. 278, 2927–2937. doi: 10.1111/j.1742-4658.2011.08218.x
Vana, L., Kanaan, N. M., Ugwu, I. C., Wuu, J., Mufson, E. J., and Binder, L. I. (2011). Progression of tau pathology in cholinergic Basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. Am. J. Pathol. 179, 2533–2550. doi: 10.1016/j.ajpath.2011.07.044
van de Nes, J. A., Nafe, R., and Schlote, W. (2008). Non-tau based neuronal degeneration in Alzheimer’s disease – an immunocytochemical and quantitative study in the supragranular layers of the middle temporal neocortex. Brain Res. 1213, 152–165. doi: 10.1016/j.brainres.2008.03.043
von Bergen, M., Barghorn, S., Biernat, J., Mandelkow, E.-M., and Mandelkow, E. (2005). Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim. Biophys. Acta 1739, 158–166. doi: 10.1016/j.bbadis.2004.09.010
Vuono, R., Winder-Rhodes, S., de Silva, R., Cisbani, G., Drouin-Ouellet, J., REGISTRY Investigators of the European Huntington’s Disease Network, et al. (2015). The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain 138(Pt 7), 1907–1918. doi: 10.1093/brain/awv107
Walsh, D. M., and Selkoe, D. J. (2007). A beta oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x
Wang, J., Dickson, D. W., Trojanowski, J. Q., and Lee, V. M. (1999). The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp. Neurol. 158, 328–337. doi: 10.1006/exnr.1999.7085
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., et al. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. doi: 10.1126/science.1062382
Woodhouse, A., West, A. K., Chuckowree, J. A., Vickers, J. C., and Dickson, T. C. (2005). Does beta-amyloid plaque formation cause structural injury to neuronal processes? Neurotox Res. 7, 5–15. doi: 10.1007/BF03033772
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Yamada, K., Cirrito, J. R., Stewart, F. R., Jiang, H., Finn, M. B., Holmes, B. B., et al. (2011). In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci. 31, 13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Zempel, H., and Mandelkow, E. M. (2012). Linking amyloid-beta and tau: amyloid-beta induced synaptic dysfunction via local wreckage of the neuronal cytoskeleton. Neurodegener. Dis. 10, 64–72. doi: 10.1159/000332816
Keywords: Alzheimer’s disease, tau oligomers, Aβ oligomers, synapsis, dendrites
Citation: Guerrero-Muñoz MJ, Gerson J and Castillo-Carranza DL (2015) Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front. Cell. Neurosci. 9:464. doi: 10.3389/fncel.2015.00464
Received: 17 May 2015; Accepted: 16 November 2015;
Published: 02 December 2015.
Edited by:
Pier Giorgio Mastroberardino, Erasmus University Medical Center Rotterdam, NetherlandsReviewed by:
Irving E. Vega, Michigan State University, USACopyright © 2015 Guerrero-Muñoz, Gerson and Castillo-Carranza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana L. Castillo-Carranza, ZGxjYXN0aWxAdXRtYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.