 Nataša Kopitar-Jerala
Nataša Kopitar-Jerala
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 08 December 2015
Sec. Non-Neuronal Cells
Volume 9 - 2015 | https://doi.org/10.3389/fncel.2015.00458
This article is part of the Research Topic Glial Cells: Managers of Neuro-immunity View all 18 articles
Stefin B (cystatin B) is an endogenous cysteine cathepsin inhibitor localized in the cytosol, mitochondria and nucleus. Its expression is upregulated upon macrophage activation and cellular stress. Mutations in the gene of stefin B are associated with the neurodegenerative disease known as Unverricht-Lundborg disease (EPM1). It was reported that early microglial activation precedes neuronal loss in the brain of the stefin B-deficient mice, implying a role of the inhibitor at the cross-talk between microglia and cerebellar cells. Detailed analysis of microglial activation in stefin B-deficient microglia showed a significantly higher proportion of both pro-inflammatory M1 and anti-inflammatory M2 microglia in stefin B-deficient mouse brain compared with control mice. In our recent work, we demonstrated that stefin B-deficient mice were significantly more sensitive to the lethal lipopolysaccharide (LPS)-induced sepsis, due to increased caspase-11 expression and secreted higher amounts of pro-inflammatory cytokines IL-1β and IL-18. Upon LPS stimulation, stefin B was targeted into the mitochondria, and the lack of stefin B resulted in the increased destabilization of the mitochondrial membrane potential and mitochondrial superoxide generation. The increased caspase-11 gene expression and better pro- inflammatory caspase-1 and -11 activation determined in stefin B deficient bone marrow-derived macrophages resulted in enhanced non-canonical inflammasome activation. Since signaling pathways in macrophages could be compared to the ones in microglia we propose that inflammasome activation could play an important role in the pathogenesis of EPM1.
Inflammation is a protective and tightly regulated immune response to tissue damage or pathogen invasion (Chovatiya and Medzhitov, 2014). In the central nervous system (CNS), this process is known as neuroinflammation and is characterized by the activation of the microglia and astrocytes population (Aguzzi et al., 2013). The innate immune response is triggered upon the recognition of pathogen-associated molecular patterns (PAMPs), derived from invading pathogens, and danger-associated molecular patterns (DAMPs), induced as a result of endogenous stress, by pattern-recognition receptors (PRRs; Akira et al., 2006). Activation of PRRs by PAMPs or DAMPs triggers signaling cascades that promote gene transcription by nuclear factor-κB (NF-κB), activator protein 1 (AP1), and interferon regulatory factors (IRFs) and results in the production of pro-inflammatory cytokines, interferons, and other pro-inflammatory proteins (Akira et al., 2006; Kawai and Akira, 2009). DAMPs correspond to endogenous ligands that are released by dying or damaged cells after cellular stress and can be recognized by PRRs such as membrane-bound toll-like receptors (TLRs) or cytosolic nucleotide-binding domain and leucine-rich repeat-containing (NLR), the RIG-I-like receptor (RLR), the AIM2-like receptor (ALR; Medzhitov, 2007; Moresco et al., 2011; Franchi et al., 2012).
In the CNS, PRRs are primarily expressed by microglia, macrophages and astrocytes. These receptors are either membrane-bound and sense extracellular or endosomally located signals (TLRs) or are located within the cytoplasm and sense intracellular signals (NLRs). Recently, it was proposed that TLRs have an important role in the crosstalk between neurons and glial cells in the CNS. TLR signaling was linked to neurogenesis, it was also found to be involved in the pathogenesis of neurodegenerative diseases (Heneka et al., 2014). Only cytosolic receptors are involved in the formation of inflammasomes. The inflammasome is an intracellular multimolecular complex for the activation of inflammatory caspases-1 and -11 which leads into the cleavage and secretion of IL-1β and IL-18 and cell death called – pyroptosis (Martinon and Tschopp, 2007; Lamkanfi and Dixit, 2014). Caspases-1 and -11 both induce pyroptosis, but only caspase-1 processes IL-1β and IL-18 (Kayagaki et al., 2011). The nucleotide binding and oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, which is composed of NLRP3, the adaptor molecule apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and the cysteine protease caspase-1, is one of the most studied inflammasomes with responses to various endogenous and exogenous danger signals (Latz et al., 2013). The priming step, that up-regulates NLPR3 pro-IL-1β gene expression, provides TLR signaling (Bauernfeind, 2009). Once primed, NLRP3 can respond to its stimuli and assemble the NLRP3 inflammasome. Stimuli that induce NLRP3 inflammasome assembly include ATP, pore-forming toxins, crystalline substances, nucleic acids, hyaluronan, and fungal, bacterial, or viral pathogens (Latz et al., 2013; Lamkanfi and Dixit, 2014). It has been proposed that phagocytosis of crystalline or particulate structures triggers lysosomal destabilization and subsequent release of the lysosomal cathepsins into the cytosol, and subsequently activates NLRP3 inflammasome (Halle et al., 2008). However, it is not known yet if the cathepsins interact directly with the inflammasome or the process involves molecules activated by the cathepsins. Recent studies have revealed a role for reactive oxygen species (ROS) of mitochondrial origin in the promotion of NLRP3 inflammasome activation (Nakahira et al., 2011; Zhou et al., 2011). Several reports showed that caspase-8 localizes and binds to ASC specks, indicating that caspase-8 is an important component of the inflammasome complex (Man, 2013; Sagulenko, 2013). In addition to the canonical [lipopolysaccharide (LPS) and ATP] NLRP3 inflammasome activation, a non-canonical inflammasome activation was described (Kayagaki et al., 2011; Rathinam et al., 2012; Broz and Monack, 2013). Canonical inflammasomes convert procaspase-1 into the catalytically active enzyme, whereas an undefined non-canonical inflammasome promotes activation of procaspase-11 (Lamkanfi and Dixit, 2014). The mouse caspase-11 (gene name Casp4) has 46% similarities to caspase-1 and is orthologous to human caspases-4 and -5 (Wang et al., 1996; Kajiwara, 2014). Non canonical inflammasomes could be activated by Gram-negative, but not by Gram-positive, bacteria, indicating that a specific factor from Gram-negative bacteria – LPS is required (Broz et al., 2012; Rathinam et al., 2012). In addition, caspase-11 detected intracellular LPS and some intracellular bacteria, directly mediate cell death and IL-1α secretion by a TLR4-independent mechanism (Hagar et al., 2013; Lamkanfi and Dixit, 2014). The non-canonical inflammasome pathway caspase-11 can interact with caspase-1 and forms a heterodimeric complex. It could induce a lytic cell death similarly to caspase-1; however, it cannot by itself trigger IL-1β/-18 processing (Wang et al., 1998; Kayagaki et al., 2011). Only caspase-11-deficient mice, but not caspase-1-deficient mice were protected from endotoxic shock (Wang et al., 1998; Kayagaki et al., 2011). The CNS is particularly sensitive to IL-1β and IL-18 signaling because multiple neural cell types in the CNS express receptors for these cytokines (Allan et al., 2005; Alboni et al., 2010).
The goal of the present review is to describe recent advances in neuroinflammation and the role of stefin B in the process.
Microglia are CNS resident myeloid cells of embryonic hematopoietic origin and comprise approximately 12% of cells in the brain (Aguzzi et al., 2013). Other CNS resident cells descend from neuroepithelial stem cells and are categorized as neurons and macroglia, with glia further subdivided into astrocytes and oligodendrocytes.
Astrocytes maintain CNS homeostasis and provide neuronal support in healthy conditions; moreover, astrocytes can undergo diverse phenotypic changes that may be protective or causative with regard to pathology (Sofroniew and Vinters, 2010). Astrocytes can produce numerous inflammatory molecules like cytokines, chemokines, growth factors, and nitric oxide (NO). Analysis of astrocyte transcriptome profiles indicates that astrocyte exposure either in vivo or in vitro to PAMPs such as LPS turns astrocyte transcriptome changes toward pro-inflammatory and potentially cytotoxic profiles (Hamby, 2012; Zamanian, 2012). Although astrocytes may undergo diverse phenotypic changes and secrete pro-inflammatory molecules, a recent study reported that NLRP3 inflammasome was expressed and functional only in mouse brain microglia, but not in astrocytes (Gustin et al., 2015). However, microglial–astrocyte interactions are important in the CNS innate immunity.
Microglia is a unique myeloid cell population, derived from primitive myeloid progenitors that arise before embryonic day 8, before vascularization or definitive hematopoiesis in the embryo (Ginhoux et al., 2010). Its density varies by brain region, they are localized mostly in the grey matter, with the highest concentrations being found in the hippocampus, olfactory telencephalon, basal ganglia, and substantia nigra (Lawson et al., 1990). Upon localization, microglia acquires a compact or ramified phenotype (Lawson et al., 1990; Block et al., 2007). In their resting state microglia have ramified morphology, and monitor the brain environment. In response to immunological stimuli or brain injury the cells are activated (Saijo and Glass, 2011). Activated microglia acquire a compact phenotype and up-regulate several surface molecules like major histocompatibility complex (MHC) molecules, chemokine receptors and several other markers (Rock, 2004). Under other circumstances, however, microglia become over-activated and can induce significant and highly detrimental neurotoxic effects by the excess production of a large array of cytotoxic factors such as superoxide (Colton and Gilbert, 1987), NO (Moss and Bates, 2001), and tumor necrosis factor-α (TNFα; Lee et al., 1993). In some cases, microglial responses could also be protective to the CNS (Lalancette-Hebert et al., 2007; Vinet et al., 2012). Gene expression and morphological changes associated with microglial activation have been extensively studied (Prinz and Priller, 2014). Several TLRs are expressed on the microglial membrane and signaling induced by TLR activation results in production of neurotoxicity and could contribute to the microglial response to neuronal damage. Activation of TLR2, TLR4, and TLR9 induces microglial production of NO through multiple ligands (Ebert, 2005). TLR9 recognizes single-stranded unmethylated CpG-DNA (bacterial DNA), which stimulates an increase in the production of microglial NO and TNFα (Olson and Miller, 2004; Ebert, 2005). TLR4 together with CD14 is implicated in brain inflammation and microglial activation in response to endotoxemia (Chakravarty and Herkenham, 2005). Monocyte-derived macrophages are classified as M1, M2a, M2b, and M2c subsets (Gordon and Taylor, 2005; Geissmann et al., 2010). It is possible that microglia also transcribe activation-dependent genes, like macrophages. Both microglia and macrophages share several similarities, they are both myeloid-derived cells; however, there are also some differences between the two cell types. Some common markers used for microglial identification such as CD11b, CD11c, and CX3CR1, could be found in microglial cells as well as in monocytes, macrophages, and dendritic cells. The difference in the expression level of cell membrane tyrosine phosphatase CD45 may be used to discriminate CD45low microglia from CD45high blood-derived cells by flow cytometry (Sedgwick et al., 1991; Ford et al., 1995). However, the signaling pathways in NLRP3 inflammasome activation are comparable between macrophages and microglia (Halle et al., 2008). Microglia clear apoptotic cells and are involved in both elimination and maintenance of synapses, they use their fine processes to monitor for dysfunctional synapses, which they are able to eliminate by phagocytosis (Wake et al., 2009). They also promote synaptic activity by secretion of brain-derived neurotrophic factor (BDNF), a molecule that is essential for learning-dependent synapse formation (Parkhurst, 2013). Moreover, microglia could modulate adult neurogenesis in the brain (Vukovic et al., 2012). Some studies suggested that microglial cells not only have a scavenger role during development but can also promote the death of some neuronal populations (Marin-Teva, 2004). Several studies have reported NLRP3 activation in microglia or CNS macrophages, although NLRP3 has also been proposed to function in neurons (Compan, 2012; Ramos, 2012). The activation mechanisms reported for NLRP3 activation in macrophages, such as ROS production, K+ efflux and endosomal rupture, also apply to NLRP3 activation within microglia (Halle et al., 2008; Hoegen, 2011; Heneka, 2013). Not only caspase-1, but also caspase-11 is expressed in microglial cells and could contribute to inflammasome activation (Lee et al., 2001; Kim et al., 2003).
Cystatins were initially characterized as inhibitors of lysosomal cysteine cathepsins, however, in recent years some alternative functions for cystatins have been proposed. Cystatins possessing inhibitory function are members of three families, family I (stefins), family II (cystatins), and family III (kininogens; Kopitar-Jerala, 2006; Turk et al., 2008).
The cystatins (cysteine proteinase inhibitor) are reversible and tight-binding inhibitors of the papain (C1) and legumain (C13) families of cysteine proteases and have significant similarities in amino acid sequence (Barrett, 1981; Barrett et al., 1986). The inhibitory profile of a particular cystatin is rather specific, despite significant sequence homologies (Turk et al., 2008). Type 1 cystatins – stefins are mostly intracellular cystatins present in the cytosol and the nuclei (Abrahamson et al., 1986). They are single-chain polypeptides ∼100 amino acid residues long, are synthesized without signal peptides and do not possess any disulfide bonds or carbohydrate side-chains. Recently, we reported the mitochondrial localization of stefin B (Maher et al., 2014a). Type 2 cystatins are mainly extracellular, secreted proteins. They are synthesized with 20–26 residue long signal peptides and most of them are found in physiologically relevant concentrations in body fluids (Abrahamson et al., 1986; Kopitar-Jerala, 2006; Turk et al., 2008). They contain disulphide bridges and may be phosphorylated (Laber et al., 1989). Type II cystatins also possess a second reactive site for inhibition of the C13 family of cysteine proteases (legumain; Alvarez-Fernandez et al., 1999). Cystatin C was found upregulated in the serum of patients with autoimmune diseases like systemic lupus erythematosus (Lertnawapan et al., 2012). Moreover, cystatin F was found abundant in the cells of the immune system: macrophages and dendritic cells and the cells involved in target cell killing (NK cells and cytotoxic T cells (CTLs; Halfon, 1998; Ni et al., 1998; Obata-Onai et al., 2002). It was also found in the microglial cells and monocyte/macrophages in the CNS. Cystatin F is expressed as a di-sulfide-linked dimer (Cappello et al., 2004) and translocated to endolysosomes where it regulates cathepsin activity. Cystatin F transport to endolysosomes depends on its N-linked glycosylation and it was reported that the secreted dimeric cystatin F could be internalized and activated by the mannose-6-phosphate receptor system (Colbert et al., 2009). After proteolytic removal of its N-terminal part, cystatin F becomes a potent inhibitor of cathepsin C with the potential to regulate pro-granzyme processing and cell cytotoxicity (Hamilton et al., 2008). Recently, we demonstrated that cathepsin V in IL-2 stimulated NK cells could process cystatin F (Maher et al., 2014b). In cytotoxic cells, cystatin F, therefore, appears as a key regulator of granzyme processing and consequently cell cytotoxicity.
Type 3 cystatins are high molecular weight (60–120 kDa) multidomain proteins and have three tandemly repeated type 2-like cystatin domains (Salvesen et al., 1986). The mammalian cystatins belonging to this type are the kininogens (Ohkubo et al., 1984). Cystatins in immune cells have been reported to participate in the release of nitric oxide, phagocytosis, and expression of cytokines (Kopitar-Jerala, 2006; Magister and Kos, 2013; Maher et al., 2014a).
Stefin B belongs to the type one cystatins and is located in the cytosol, mitochondria, and nucleus where it protects cells from the detrimental release of the lysosomal cysteine cathepsins. In the nucleus, stefin B interacts with nucleosomes, specifically with histones H2A.Z, H2B, and H3 and cathepsin L (Ceru et al., 2010). Goulet et al. (2004) has shown that only shorter procathepsin L isoforms translocate to the nucleus and stimulate processing of the CUX1 transcription factor at the G1/S transition of the cell cycle. Stefin B-deficient mouse embryonic fibroblasts entered S phase earlier than wild type mouse embryonic fibroblasts. In contrast, increased expression of stefin B in the nucleus delayed cell cycle progression in T98G cells. The delay in cell cycle progression was associated with the inhibition of cathepsin L in the nucleus, as judged from the decreased cleavage of the CUX1 transcription factor (Ceru et al., 2010). Moreover, we have shown that stefin B overexpression in the nucleus delayed not only cell cycle progression, but also caspase activation (Sun et al., 2012). Mutations in the gene encoding stefin B (either through a multiplied repeat unit in the promoter or through point mutations in the structural gene) are present in both alleles of the gene in patients with a form of progressive myoclonus epilepsy of Unverricht-Lundborg type (EPM1; Pennacchio et al., 1996; Lalioti et al., 1997; Pennacchio et al., 1998). EPM1 is an autosomal recessively inherited neurodegenerative disease, characterized by the cerebellar granule neurons apoptosis, progressive ataxia and myoclonic epilepsy (Joensuu et al., 2008). In lymphoid cells of EPM1 patients, increased cathepsin activity, due to reduced expression of stefin B was reported (Rinne et al., 2002), we determined increased overall cathepsin activity in untreated, as well as in classically activated stefin B-deficient bone marrow-derived macrophages (BMDMs) compared to WT cells (Maher et al., 2014c).
Stefin B-deficient mice develop myoclonic seizures by one month of age and progressive ataxia by six months of age (Pennacchio et al., 1998). Houseweart et al. (2003) reported that the removal of cathepsin B from stefin B-deficient mice greatly reduced the neuronal apoptosis, but did not rescue the ataxia and seizure phenotype. Moreover, stefin B deficiency was implicated in the impaired redox homeostasis, resulting in a pronounced oxidative stress-induced cell death and neurodegeneration (Lehtinen et al., 2009). Thymocytes from stefin B deficient mice were significantly more sensitive to apoptosis induced with the inhibitor of protein kinase C, staurosporin (Kopitar-Jerala et al., 2005). Manninen et al. examined in detail the spatiotemporal dynamics of the brain atrophy in stefin B-deficient mice (Manninen et al., 2014). They showed progressive but non-uniform volume loss of the stefin B-deficient mouse brains, indicating that different neuronal populations possess distinct sensitivity to the damage, as a consequence of stefin B deficiency. The authors suggested that the white matter damage in the brain of stefin B-deficient mice was secondary to glial activation and neurodegeneration (Manninen et al., 2014). Another report showed that the early microglial activation precedes neuronal loss in the brain of the stefin B deficient mice, implying a role of the inhibitor at the cross-talk between microglia and cerebellar cells (Tegelberg et al., 2012).
Joensuu et al. (2014) analyzed the gene expression changes in the cerebellum of pre-symptomatic and symptomatic stefin B -deficient mice and in cultured stefin B-deficient cerebellar granule cells. Already in the cerebellum of pre-symptomatic stefin B-deficient mice (7 days after the birth), multiple changes in gene expression related to synapse maturation, development, and function during postnatal maturation were observed. More prominent changes were reported in the GABAergic signaling pathway (Joensuu et al., 2014). GABA plays a central role in controlling neuronal development and connectivity and defective GABAergic signaling in the cerebellum of stefin B deficient mice underlines a mechanism for ataxia in these mice (Grusser-Cornehls and Baurle, 2001). At a later stage (30 days after the birth), in symptomatic stefin B-deficient mice, the authors reported the upregulation of immune response genes, in line with the results showing early glial activation that preceded neuronal degeneration (Tegelberg et al., 2012). Moreover, Joensuu et al. (2014), reported the upregulation of the genes involved in cell cycle progression, in stefin B-deficient granule neurons. We have shown that the interactions of stefin B with cathepsin L in the nucleus influence cell cycle progression into the S phase (Ceru et al., 2010). We cannot exclude the possibility that the impaired cathepsin regulation in the synapses could lead to morphological and functional changes observed in stefin B-deficient mice (Joensuu et al., 2014). For example, cathepsin B-like immunoreactivity was observed at synaptic sites and myristoylated-alanine-rich C-kinase substrate (MARCKS), a known substrate of cathepsin B, was specifically degraded in response to intense NMDA receptor stimulation (Graber et al., 2004). Previously, we reported the increased cleavage of MARCKS in the brains and macrophages of stefin B-deficient mice, when compared to cells and tissue from control wild-type animals (Kopitar-Jerala and Turk, 2007).
The increased expression of inflammatory genes indicates that neuro-inflammation, together with neuronal dysfunction, plays a crucial role in pathology of EPM1. Pro-inflammatory chemokines and cytokines, highly expressed in symptomatic stefin B-deficient mice were reported to lower the seizure threshold and may thus contribute to recurrent excitation in epilepsy (Devinsky et al., 2013). Okuneva et al. (2015) reported significantly higher stefin B mRNA expression in microglia than in neurons or astrocytes, which is in line with our observation that stefin B is highly upregulated in activated macrophages (Maher et al., 2014c). In pre-symptomatic stefin B-deficient mice compared to control animals the ratio of M1/M2 microglia is skewed towards M2 type, but towards M1 type in symptomatic mice. In addition, a heightened expression of both pro-inflammatory inducible nitric oxide synthase (iNOS), anti-inflammatory arginase 1 (ARG1) and chemokine release was detected (Okuneva et al., 2015). Interestingly, MHCII surface expression was suppressed. We have reported that IFN-γ and LPS-activated stefin B-deficient BMDMs produced higher amounts of NO, and expressed more iNOS than WT BMDMs. IL-10 is a potent anti-inflammatory cytokine that is crucial for dampening the inflammatory response after pathogen invasion and acts to protect the host from excessive inflammation (Fiorentino et al., 1991). We showed decreased expression of IL-10 in BMDMs of stefin B deficient mice, due to impaired STAT3 signaling (Maher et al., 2014c). IL-10 plays an essential role in mediating inflammatory processes not only in the cells of immune system, but also in the brain (Zocchia et al., 1997). It has been demonstrated that it increases the survival of cerebellar granule cells by blocking caspase-3-like activity (Bachis et al., 2001). It is tempting to speculate that the decreased IL-10 expression in stefin B-deficient mice could contribute to the increased apoptosis in the cerebellum in EPM1.
In the developing mouse cerebellum, Purkinje cells die and a majority of these neurons are engulfed by microglial cells. Interestingly, apoptosis of Purkinje cells in the cerebellum was strongly reduced by selective elimination of microglia and superoxide produced by microglia cells (Marin-Teva, 2004). In our recent work we showed that stefin B-deficient mice were significantly more sensitive to the lethal LPS-induced endotoxemia due to increased caspase-11 expression. The increased caspase-11 gene expression and better caspase-1 and -11 processing determined in stefin B-deficient BMDMs resulted in enhanced IL-1β and IL-18 processing and secretion (Figure 1). The increased cathepsin activity determined in stefin B deficient BMDMs was not essential for inflammasome activation, since treatment of BMDMs with the cathepsin inhibitor E-64d did not influence caspase-1 activation and IL-1β secretion. Upon LPS stimulation, stefin B was targeted to mitochondria, and the lack of stefin B resulted in the increased destabilization of mitochondrial membrane potential and mitochondrial ROS generation (Maher et al., 2014a). The induction of ROS in microglia may therefore play an important role in non-canonical inflammasome activation and cell death in the cerebellum in disease.
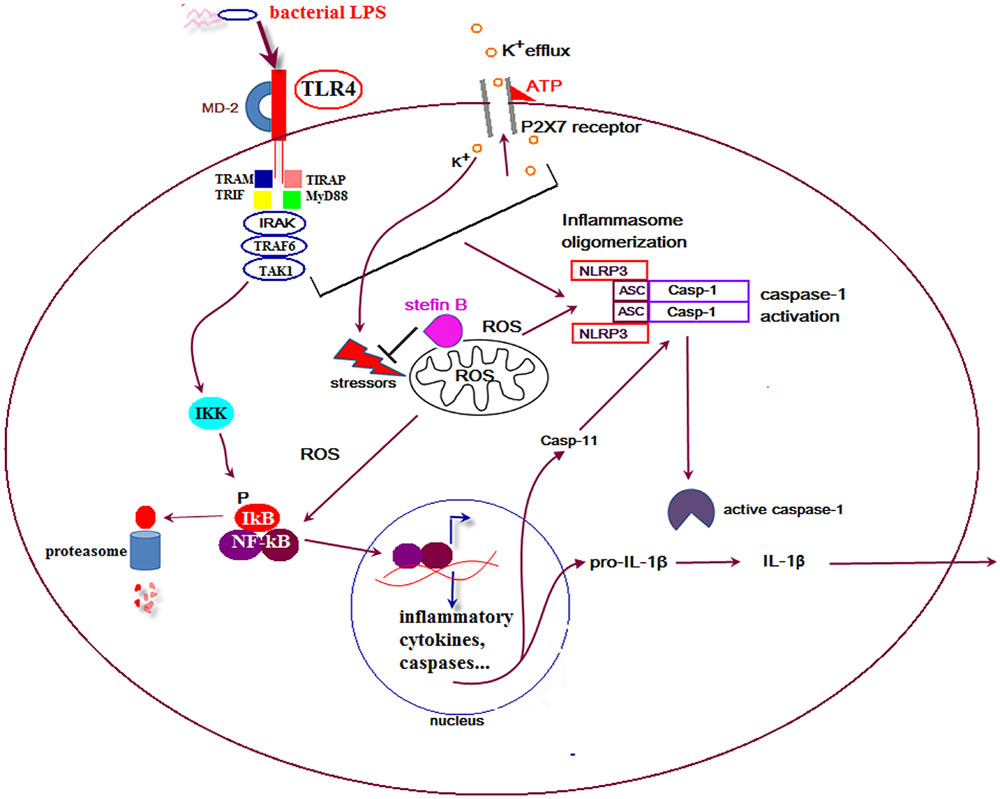
FIGURE 1. Proposed model for the role of stefin B in non canonical inflammasome activation. Upon lipopolysaccharide (LPS) stimulation stefin B is translocated from cytosol into mitochondria and protects mitochondrial membrane integrity. Stefin B deficiency resulted in the breakdown of mitochondria membrane potential and increased mtROS generation. The consequence of the increased mtROS detected in stefin B-deficient bone marrow-derived macrophages (BMDMs) upon LPS stimulation, was the increased nuclear factor-κB (NF-κB) activation and caspase-11 expression. Increased caspase-11 expression resulted in increased inflammasome activation and pro-inflammatory IL-1β and IL-18 secretion.
This review summarizes recent discoveries that may contribute to the understanding of the role of stefin B in neuro-inflammation. Several studies, each from a different angle, have contributed a piece of the puzzle, a process we are trying to understand. Stefin B-deficient mice have proven to be a valuable tool to explore the function of the protein in the pathology of disease. During the past couple of years, several new data from microarray experiments, histology, as well as magnetic resonance imaging have revealed that neuro-inflammation is an essential process in the pathology of EPM1. Biochemical experiments in macrophages have contributed some hints regarding the signaling pathways in inflammasome activation. Signaling pathways in macrophages were compared to the ones in microglia and the expression of caspase-11 was strongly induced by activation of rat glial cells, as well as in astrocytes, with interferon-γ and LPS (Hur et al., 2001). The expression of caspase-11 in microglia may play an important role in non-canonical inflammasome activation and cell death in the cerebellum in disease. However, some questions still remain and some more pieces need to be added to complete the whole picture. Additional experiments will reveal if the inflammasome activation and caspase-11 expression are part of the pathology of EPM1.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Slovenian Research Agency Grants: P-0140.
Abrahamson, M., Barrett, A. J., Salvesen, G., and Grubb, A. (1986). Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J. Biol. Chem. 261, 11282–11289.
Aguzzi, A., Barres, B. A., and Bennett, M. L. (2013). Microglia: scapegoat, saboteur, or something else? Science 339, 156–161. doi: 10.1126/science.1227901
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Alboni, S., Cervia, D., Sugama, S., and Conti, B. (2010). Interleukin 18 in the CNS. J. Neuroinflammation 7:9. doi: 10.1186/1742-2094-7-9
Allan, S. M., Tyrrell, P. J., and Rothwell, N. J. (2005). Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 5, 629–640. doi: 10.1038/nri1664
Alvarez-Fernandez, M., Barrett, A. J., Gerhartz, B., Dando, P. M., Ni, J., and Abrahamson, M. (1999). Inhibition of mammalian legumain by some cystatins is due to a novel second reactive site. J. Biol. Chem. 274, 19195–19203. doi: 10.1074/jbc.274.27.19195
Bachis, A., Colangelo, A. M., Vicini, S., Doe, P. P., De Bernardi, M. A., Brooker, G., et al. (2001). Interleukin-10 prevents glutamate-mediated cerebellar granule cell death by blocking caspase-3-like activity. J. Neurosci. 21, 3104–3112.
Barrett, A. J. (1981). Alpha 2-macroglobulin. Methods Enzymol. 80, 737–754. doi: 10.1016/S0076-6879(81)80056-0
Barrett, A. J., Fritz, H., Grubb, A., Isemura, S., Jarvinen, M., Katunuma, N., et al. (1986). Nomenclature and classification of the proteins homologous with the cysteine-proteinase inhibitor chicken cystatin. Biochem. J. 236:312. doi: 10.1042/bj2360312
Bauernfeind, F. G. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. doi: 10.4049/jimmunol.0901363
Block, M. L., Zecca, L., and Hong, J.-S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Broz, P., and Monack, D. M. (2013). Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog. 9:e1003144. doi: 10.1371/journal.ppat.1003144
Broz, P., Ruby, T., Belhocine, K., Bouley, D. M., Kayagaki, N., Dixit, V. M., et al. (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291. doi: 10.1038/nature11419
Cappello, F., Gatti, E., Camossetto, V., David, A., Lelouard, H., and Pierre, P. (2004). Cystatin F is secreted, but artificial modification of its C-terminus can induce its endocytic targeting. Exp. Cell Res. 297, 607–618. doi: 10.1016/j.yexcr.2004.03.048
Ceru, S., Konjar, S., Maher, K., Repnik, U., Krizaj, I., Bencina, M., et al. (2010). Stefin B interacts with histones and cathepsin L in the nucleus. J. Biol. Chem. 285, 10078–10086. doi: 10.1074/jbc.M109.034793
Chakravarty, S., and Herkenham, M. (2005). Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J. Neurosci. 25, 1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005
Chovatiya, R., and Medzhitov, R. (2014). Stress, inflammation, and defense of homeostasis. Mol. Cell. 54, 281–288. doi: 10.1016/j.molcel.2014.03.030
Colbert, J. D., Plechanovová, A., and Watts, C. (2009). Glycosylation directs targeting and activation of cystatin f from intracellular and extracellular sources. Traffic 10, 425–437. doi: 10.1111/j.1600-0854.2009.00881.x
Colton, C. A., and Gilbert, D. L. (1987). Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 223, 284–288. doi: 10.1016/0014-5793(87)80305-8
Compan, V. (2012). Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37, 487–500. doi: 10.1016/j.immuni.2012.06.013
Devinsky, O., Vezzani, A., Najjar, S., De Lanerolle, N. C., and Rogawski, M. A. (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36, 174–184. doi: 10.1016/j.tins.2012.11.008
Ebert, S. (2005). Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J. Neuroimmunol. 159, 87–96. doi: 10.1016/j.jneuroim.2004.10.005
Fiorentino, D. F., Zlotnik, A., Mosmann, T. R., Howard, M., and O’garra, A. (1991). IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147, 3815–3822.
Ford, A. L., Goodsall, A. L., Hickey, W. F., and Sedgwick, J. D. (1995). Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 154, 4309–4321.
Franchi, L., Munoz-Planillo, R., and Nunez, G. (2012). Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332. doi: 10.1038/ni.2231
Geissmann, F., Gordon, S., Hume, D. A., Mowat, A. M., and Randolph, G. J. (2010). Unravelling mononuclear phagocyte heterogeneity. Nat. Rev. Immunol. 10, 453–460. doi: 10.1038/nri2784
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gordon, S., and Taylor, P. R. (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964. doi: 10.1038/nri1733
Goulet, B., Baruch, A., Moon, N. S., Poirier, M., Sansregret, L. L., Erickson, A., et al. (2004). A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol. Cell. 14, 207–219. doi: 10.1016/S1097-2765(04)00209-6
Graber, S., Maiti, S., and Halpain, S. (2004). Cathepsin B-like proteolysis and MARCKS degradation in sub-lethal NMDA-induced collapse of dendritic spines. Neuropharmacology 47, 706–713. doi: 10.1016/j.neuropharm.2004.08.004
Grusser-Cornehls, U., and Baurle, J. (2001). Mutant mice as a model for cerebellar ataxia. Prog. Neurobiol. 63, 489–540. doi: 10.1016/S0301-0082(00)00024-1
Gustin, A., Kirchmeyer, M., Koncina, E., Felten, P., Losciuto, S., Heurtaux, T., et al. (2015). NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS ONE 10:e0130624. doi: 10.1371/journal.pone.0130624
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K., and Miao, E. A. (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. doi: 10.1126/science.1240988
Halfon, S. (1998). Leukocystatin, a new class II cystatin expressed selectively by hematopoietic cells. J. Biol. Chem. 273, 16400–16408. doi: 10.1074/jbc.273.26.16400
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hamby, M. E. (2012). Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J. Neurosci. 32, 14489–14510. doi: 10.1523/JNEUROSCI.1256-12.2012
Hamilton, G., Colbert, J.D., Schuettelkopf, A.W., and Watts, C. (2008). Cystatin F is a cathepsin C-directed protease inhibitor regulated by proteolysis. EMBO J. 27, 499–508. doi: 10.1038/sj.emboj.7601979
Heneka, M. T. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heneka, M. T., Kummer, M. P., and Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. doi: 10.1038/nri3705
Hoegen, T. (2011). The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J. Immunol. 187, 5440–5451. doi: 10.4049/jimmunol.1100790
Houseweart, M. K., Pennacchio, L. A., Vilaythong, A., Peters, C., Noebels, J. L., and Myers, R. M. (2003). Cathepsin B but not cathepsins L or S contributes to the pathogenesis of Unverricht-Lundborg progressive myoclonus epilepsy (EPM1). J. Neurobiol. 56, 315–327. doi: 10.1002/neu.10253
Hur, J., Kim, S. Y., Kim, H., Cha, S.-H., Lee, M.-S., and Suk, K. (2001). Induction of caspase-11 by inflammatory stimuli in rat astrocytes: lipopolysaccharide induction through p38 mitogen-activated protein kinase pathway1. FEBS Lett. 507, 157–162. doi: 10.1016/S0014-5793(01)02975-1
Joensuu, T., Lehesjoki, A. E., and Kopra, O. (2008). Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia 49, 557–563. doi: 10.1111/j.1528-1167.2007.01422.x
Joensuu, T., Tegelberg, S., Reinmaa, E., Segerstråle, M., Hakala, P., Pehkonen, H., et al. (2014). Gene expression alterations in the cerebellum and granule neurons of Cstb(-/-) mouse are associated with early synaptic changes and inflammation. PLoS ONE 9:e89321. doi: 10.1371/journal.pone.0089321
Kajiwara, Y. (2014). A critical role for human caspase-4 in endotoxin sensitivity. J. Immunol. 193, 335–343. doi: 10.4049/jimmunol.1303424
Kawai, T., and Akira, S. (2009). The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21, 317–337. doi: 10.1093/intimm/dxp017
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Kim, N.-G., Lee, H., Son, E., Kwon, O.-Y., Park, J.-Y., Park, J.-H., et al. (2003). Hypoxic induction of caspase-11/caspase-1/interleukin-1β in brain microglia. Mol. Brain Res. 114, 107–114. doi: 10.1016/S0169-328X(03)00135-9
Kopitar-Jerala, N. (2006). The role of cystatins in cells of the immune system. FEBS Lett. 580, 6295–6301. doi: 10.1016/j.febslet.2006.10.055
Kopitar-Jerala, N., Schweiger, A., Myers, R. M., Turk, V., and Turk, B. (2005). Sensitization of stefin B-deficient thymocytes towards staurosporin-induced apoptosis is independent of cysteine cathepsins. FEBS Lett. 579, 2149–2155. doi: 10.1016/j.febslet.2005.03.002
Kopitar-Jerala, N., and Turk, B. (2007). Cleavage of the myristoylated alanine-rich C kinase substrate (MARCKS) by cysteine cathepsins in cells and tissues of stefin B-deficient mice. Biol. Chem. 388, 847–852. doi: 10.1515/BC.2007.092
Laber, B., Krieglstein, K., Henschen, A., Kos, J., Turk, V., Huber, R., et al. (1989). The cysteine proteinase inhibitor chicken cystatin is a phosphoprotein. FEBS Lett. 248, 162–168. doi: 10.1016/0014-5793(89)80453-3
Lalancette-Hebert, M., Gowing, G., Simard, A., Weng, Y. C., and Kriz, J. (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596 –2605. doi: 10.1523/JNEUROSCI.5360-06.2007
Lalioti, M. D., Scott, H. S., Buresi, C., Rossier, C., Bottani, A., Morris, M. A., et al. (1997). Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 386, 847–851. doi: 10.1038/386847a0
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. doi: 10.1016/j.cell.2014.04.007
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-W
Lee, J., Hur, J., Lee, P., Kim, J. Y., Cho, N., Kim, S. Y., et al. (2001). Dual role of inflammatory stimuli in activation-induced cell death of mouse microglial cells: initiation of two separate apoptotic pathways via induction of interferon regulatory factor-1 and caspase-11. J. Biol. Chem. 276, 32956–32965. doi: 10.1074/jbc.M104700200
Lee, S. C., Liu, W., Dickson, D. W., Brosnan, C. F., and Berman, J. W. (1993). Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 [beta]. J. Immunol. 150, 2659–2667.
Lehtinen, M. K., Tegelberg, S., Schipper, H., Su, H., Zukor, H., Manninen, O., et al. (2009). Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 29, 5910–5915. doi: 10.1523/JNEUROSCI.0682-09.2009
Lertnawapan, R., Bian, A., Rho, Y. H., Raggi, P., Oeser, A., Solus, J. F., et al. (2012). Cystatin C is associated with inflammation but not atherosclerosis in systemic lupus erythematosus. Lupus 21, 279–287. doi: 10.1177/0961203311425527
Magister, S., and Kos, J. (2013). Cystatins in immune system. J. Cancer 4, 45–56. doi: 10.7150/jca.5044
Maher, K., Jeric Kokelj, B., Butinar, M., Mikhaylov, G., Mancek-Keber, M., Stoka, V., et al. (2014a). A role for stefin B (cystatin B) in inflammation and endotoxemia. J. Biol. Chem. 289, 31736–31750. doi: 10.1074/jbc.M114.609396
Maher, K., Konjar, S., Watts, C., Turk, B., and Kopitar-Jerala, N. (2014b). Cystatin F regulates proteinase activity in IL-2-activated natural killer cells. Protein Pept. Lett. 21, 957–965. doi: 10.2174/0929866521666140403124146
Maher, K., Završnik, J., Jeriè-Kokelj, B., Vasiljeva, O., Turk, B., and Kopitar-Jerala, N. (2014c). Decreased IL-10 expression in stefin B-deficient macrophages is regulated by the MAP kinase and STAT-3 signaling pathways. FEBS Lett. 588, 720–726. doi: 10.1016/j.febslet.2014.01.015
Man, S. M. (2013). Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1[beta] production. J. Immunol. 191, 5239–5246. doi: 10.4049/jimmunol.1301581
Manninen, O., Laitinen, T., Lehtimaki, K. K., Tegelberg, S., Lehesjoki, A. E., Grohn, O., et al. (2014). Progressive volume loss and white matter degeneration in cstb-deficient mice: a diffusion tensor and longitudinal volumetry MRI study. PLoS ONE 9:e90709 doi: 10.1371/journal.pone.0090709
Marin-Teva, J. L. (2004). Microglia promote the death of developing purkinje cells. Neuron 41, 535–547. doi: 10.1016/S0896-6273(04)00069-8
Martinon, F., and Tschopp, J. (2007). Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 14, 10–22. doi: 10.1038/sj.cdd.4402038
Medzhitov, R. (2007). Recognition of microorganisms and activation of the immune response. Nature 449, 819–826. doi: 10.1038/nature06246
Moresco, E. M., Lavine, D., and Beutler, B. (2011). Toll-like receptors. Curr. Biol. 21:39. doi: 10.1016/j.cub.2011.05.039
Moss, D. W., and Bates, T. E. (2001). Activation of murine microglial cell lines by lipopolysaccharide and interferon-[gamma] causes NO-mediated decreases in mitochondrial and cellular function. Eur. J. Neurosci. 13, 529–538. doi: 10.1046/j.1460-9568.2001.01418.x
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Ni, J., Fernandez, A., Danielsson, L., Chillakuru, R. A., Zhang, J., Grub, A., et al. (1998). Cystatin F is a glycosylated human low molecular weight cystein proteinase inhibitor. J. Biol. Chem. 273, 24797–24804. doi: 10.1074/jbc.273.38.24797
Obata-Onai, A., Hashimoto, S., Onai, N., Kurachi, M., Nagai, S., Shizuno, K., et al. (2002). Comprehensive gene expression analysis of human NK cells and CD8(+) T lymphocytes. Int. Immunol. 14, 1085–1098. doi: 10.1093/intimm/dxf086
Ohkubo, I., Kurachi, K., Takasawa, T., Shiokawa, H., and Sasaki, M. (1984). Isolation of a human cDNA for alpha 2-thiol proteinase inhibitor and its identity with low molecular weight kininogen. Biochemistry 23, 5691–5697. doi: 10.1021/bi00319a005
Okuneva, O., Körber, I., Li, Z., Tian, L., Joensuu, T., Kopra, O., et al. (2015). Abnormal microglial activation in the Cstb-/- mouse, a model for progressive myoclonus epilepsy, EPM1. Glia 63, 400–411. doi: 10.1002/glia.22760
Olson, J. K., and Miller, S. D. (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 173, 3916–3924. doi: 10.4049/jimmunol.173.6.3916
Parkhurst, C. N. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Pennacchio, L. A., Bouley, D. M., Higgins, K. M., Scott, M. P., Noebels, J. L., and Myers, R. M. (1998). Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice. Nat. Genet. 20, 251–258. doi: 10.1038/3059
Pennacchio, L. A., Lehesjoki, A. E., Stone, N. E., Willour, V. L., Virtaneva, K., Miao, J., et al. (1996). Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) [see comments]. Science 271, 1731–1734. doi: 10.1126/science.271.5256.1731
Prinz, M., and Priller, J. (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312. doi: 10.1038/nrn3722
Ramos, H. J. (2012). IL-1[beta] signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 8:e1003039. doi: 10.1371/journal.ppat.1003039
Rathinam, V. A., Vanaja, S. K., Waggoner, L., Sokolovska, A., Becker, C., Stuart, L. M., et al. (2012). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619. doi: 10.1016/j.cell.2012.07.007
Rinne, R., Saukko, P., Jarvinen, M., and Lehesjoki, A. E. (2002). Reduced cystatin B activity correlates with enhanced cathepsin activity in progressive myoclonus epilepsy. Ann. Med. 34, 380–385. doi: 10.1080/078538902320772124
Rock, R. B. (2004). Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 17, 942–964. doi: 10.1128/CMR.17.4.942-964.2004
Sagulenko, V. (2013). AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160. doi: 10.1038/cdd.2013.37
Saijo, K., and Glass, C. K. (2011). Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 11, 775–787. doi: 10.1038/nri3086
Salvesen, G., Parkes, C., Abrahamson, M., Grubb, A., and Barrett, A. J. (1986). Human low-Mr kininogen contains three copies of a cystatin sequence that are divergent in structure and in inhibitory activity for cysteine proteinases. Biochem. J. 234, 429–434. doi: 10.1042/bj2340429
Sedgwick, J. D., Schwender, S., Imrich, H., Dorries, R., Butcher, G. W., and Ter Meulen, V. (1991). Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc. Natl. Acad. Sci. U.S.A. 88, 7438–7442. doi: 10.1073/pnas.88.16.7438
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Sun, T., Turk, V., Turk, B., and Kopitar-Jerala, N. (2012). Increased expression of stefin B in the nucleus of T98G astrocytoma cells delays caspase activation. Front. Mol. Neurosci. 5:93. doi: 10.3389/fnmol.2012.00093
Tegelberg, S., Kopra, O., Joensuu, T., Cooper, J. D., and Lehesjoki, A.-E. (2012). Early microglial activation precedes neuronal loss in the brain of the Cstb-/- mouse model of progressive myoclonus epilepsy, EPM1. J. Neuropathol. Exp. Neurol. 71, 40–53. doi: 10.1097/NEN.0b013e31823e68e1
Turk, V., Stoka, V., and Turk, D. (2008). Cystatins: biochemical and structural properties, and medical relevance. Front. Biosci. 13:5406–5420. doi: 10.2741/3089
Vinet, J., Weering, H. R., Heinrich, A., Kalin, R. E., Wegner, A., Brouwer, N., et al. (2012). Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J. Neuroinflamm. 9:27. doi: 10.1186/1742-2094-9-27
Vukovic, J., Colditz, M. J., Blackmore, D. G., Ruitenberg, M. J., and Bartlett, P. F. (2012). Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J. Neurosci. 32, 6435–6443. doi: 10.1523/JNEUROSCI.5925-11.2012
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009
Wang, S., Miura, M., Jung, Y., Zhu, H., Gagliardini, V., Shi, L., et al. (1996). Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J. Biol. Chem. 271, 20580–20587. doi: 10.1074/jbc.271.34.20580
Wang, S., Miura, M., Jung, Y. K., Zhu, H., Li, E., and Yuan, J. (1998). Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92, 501–509. doi: 10.1016/S0092-8674(00)80943-5
Zamanian, J. L. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi: 10.1038/nature09663
Keywords: cystatins, EPM1, inflammation, microglia, TLR, NLRP3 inflammasome, ROS
Citation: Kopitar-Jerala N (2015) The Role of Stefin B in Neuro-inflammation. Front. Cell. Neurosci. 9:458. doi: 10.3389/fncel.2015.00458
Received: 15 July 2015; Accepted: 11 November 2015;
Published: 08 December 2015.
Edited by:
Jeffrey Michael Zirger, Centers for Disease Control and Prevention, USAReviewed by:
Jiawei Zhou, Chinese Academy of Sciences, ChinaCopyright © 2015 Kopitar-Jerala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nataša Kopitar-Jerala, bmF0YXNhLmtvcGl0YXJAaWpzLnNp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.