 Rommy von Bernhardi
Rommy von Bernhardi Francisca Cornejo
Francisca Cornejo Guillermo E. Parada1
Guillermo E. Parada1 Jaime Eugenín
Jaime Eugenín
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Cell. Neurosci., 28 October 2015
Sec. Cellular Neuropathology
Volume 9 - 2015 | https://doi.org/10.3389/fncel.2015.00426
This article is part of the Research TopicGlial Cells: Managers of Neuro-immunityView all 18 articles
Aging is the main risk factor for Alzheimer’s disease (AD); being associated with conspicuous changes on microglia activation. Aged microglia exhibit an increased expression of cytokines, exacerbated reactivity to various stimuli, oxidative stress, and reduced phagocytosis of β-amyloid (Aβ). Whereas normal inflammation is protective, it becomes dysregulated in the presence of a persistent stimulus, or in the context of an inflammatory environment, as observed in aging. Thus, neuroinflammation can be a self-perpetuating deleterious response, becoming a source of additional injury to host cells in neurodegenerative diseases. In aged individuals, although transforming growth factor β (TGFβ) is upregulated, its canonical Smad3 signaling is greatly reduced and neuroinflammation persists. This age-related Smad3 impairment reduces protective activation while facilitating cytotoxic activation of microglia through several cellular mechanisms, potentiating microglia-mediated neurodegeneration. Here, we critically discuss the role of TGFβ-Smad signaling on the cytotoxic activation of microglia and its relevance in the pathogenesis of AD. Other protective functions, such as phagocytosis, although observed in aged animals, are not further induced by inflammatory stimuli and TGFβ1. Analysis in silico revealed that increased expression of receptor scavenger receptor (SR)-A, involved in Aβ uptake and cell activation, by microglia exposed to TGFβ, through a Smad3-dependent mechanism could be mediated by transcriptional co-factors Smad2/3 over the MSR1 gene. We discuss that changes of TGFβ-mediated regulation could at least partially mediate age-associated microglia changes, and, together with other changes on inflammatory response, could result in the reduction of protective activation and the potentiation of cytotoxicity of microglia, resulting in the promotion of neurodegenerative diseases.
Homeostasis of the nervous system is maintained by the finely tuned interaction of glial cells and neurons, involving a complex network of signaling pathways. Inflammation, a primarily beneficial process mediated by the activation of glia in response to injury, illness or infection, allows for the elimination of harmful stimuli and the repair of damaged tissue. However, this process can become dysregulated, when the activating stimulus cannot be removed, or in the context of a maintained inflammatory environment, as observed in aging (von Bernhardi et al., 2010). Thus, neuroinflammation can also be a self-perpetuating deleterious response, with persistent activation of glia, sustained release of inflammatory mediators, and increased oxidative and nitrosative stress; becoming a source of additional injury to host cells. Chronic neuroinflammation plays a role in a number of neurodegenerative diseases (Block and Hong, 2005; von Bernhardi et al., 2007; Gao and Hong, 2008), inducing neuronal injury.
Microglia are the brain resident innate immune system (Hemmer et al., 2002; Ransohoff and Perry, 2009; Rivest, 2009). When stimulated, microglia activate and change their functional properties (Liu et al., 2001; von Bernhardi and Eugenin, 2004; Lue et al., 2010). They can sense and respond to a wide range of stimuli, including central nervous system (CNS) trauma, ischemia, infection, toxic, and autoimmune insults (Kreutzberg, 1996; Streit, 2002; Kim and de Vellis, 2005; Schwab and McGeer, 2008; Lue et al., 2010; von Bernhardi et al., 2010). In fact, microglia are activated in virtually all CNS diseases (Kreutzberg, 1996; Hanisch and Kettenmann, 2007; Neumann et al., 2009). They are the main producers of a broad spectrum of inflammatory mediators, such as eicosanoids, cytokines (Nakamura, 2002; Kim and de Vellis, 2005; Tichauer et al., 2007), chemokines, reactive oxygen species (ROS), nitric oxide (NO)·, small metabolite mediators, and proteases (α-antichymotrypsin and α-antitrypsin) (Benveniste et al., 2001; Nakamura, 2002; Streit, 2002; Li et al., 2007; Tichauer et al., 2007; Neumann et al., 2009; Lue et al., 2010).
Other glial cell type, astrocytes, which will not be discussed in this work, share several functions with microglia. They are important for neurotrophic support and metabolism, synaptic regulation and several other functions, in addition to their participation in β-amyloid (Aβ) clearance (Rossner et al., 2005; Murgas et al., 2012). Inflammatory mediators regulate the innate immune defense, induce bystander damage, and modify synaptic function (Aldskogius et al., 1999; Selkoe, 2002; Di Filippo et al., 2008) according to environmental conditions (Li et al., 2007; von Bernhardi, 2007). Depending on the activation context, microglia secrete inflammatory cytokines such as interleukin 1β (IL1β), tumor necrosis factor α (TNFα) and interferon gamma (IFNγ), and reactive species (Kettenmann et al., 2011), as well as regulatory cytokines like interleukin 10 (IL10) and transforming growth factor β (TGFβ1; Nakajima et al., 2007; Sierra et al., 2007; Welser-Alves and Milner, 2013). Inflammatory cytokines trigger the production of several inflammatory factors that could affect neuronal function. In response to IFNγ, for example, glia produce NO· by up-regulation of inducible nitric oxide synthase (iNOS) and release superoxide radicals (O2·−) by a nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase mediated mechanism (Hu et al., 1995; Calabrese et al., 2007). Neuroinflammation affects neuron-glia crosstalk and establishes interactions with oxidizing agents through redox sensors in enzymes, receptors, and transcription factors, all of which can affect neuronal function (Liu et al., 2012), inducting neurodegeneration (Raj et al., 2014). Oxidative stress, in turn, further increases inflammatory cytokines, creating a vicious cycle (Rosales-Corral et al., 2010), with profound impact in cell homeostasis and survival (Satoh and Lipton, 2007).
Astrocyte and microglia activation occur through the phosphorylation of MAPKs and the activation of nuclear factor kappa B (NFκB) pathway, inducing the expression of inflammatory mediators (Van Eldik et al., 2007; Glass et al., 2010; Heneka et al., 2010). MAPKs include extracellular signal-regulated protein kinases (ERKs) and stress activated protein kinases c-Jun NH2-terminal kinase (JNK) and P38. Activated MAPKs exert their actions both in the cytoplasm and translocating into the nucleus, phosphorylating transcription factors. Noteworthy, ERK and P38 appear to be key actors in the production of free radicals (Bhat et al., 1998; Marcus et al., 2003; Qian et al., 2008). The ERK pathway is regulated by pro- and anti-inflammatory cytokines, determining the timing of microglia activation (Saud et al., 2005; Glass et al., 2010). In addition, P38 is involved in the production of NO· by up-regulating iNOS (Saha et al., 2007; Munoz and Ammit, 2010), and enhances the expression of inflammatory cytokines, such as TNFα, through both transcriptional and post-transcriptional mechanisms. P38 can induce transcription of the TNFα gene by increasing activator protein-1 (AP-1) activity (Spriggs et al., 1992) and enhances its production by increasing the stability and translation of TNFα mRNA (Dean et al., 2004).
Activation of microglia shows a broad functional spectrum associated with specific expression patterns of cytokines and their receptors (Town et al., 2005). Depending on the stimuli they receive, they show different activation profiles (Gordon, 2003; Martinez et al., 2008; Mosser and Edwards, 2008), including: (i) classical activation (M1 activation), which under certain conditions will be cytotoxic; (ii) alternative phagocytic/neuroprotective (M2 activation; Gordon, 2003; Martinez et al., 2008); or (iii) regulatory activation (Mosser and Edwards, 2008). Activation of interferon-regulatory factor 5 (IRF5), defines commitment to the M1 macrophage lineage (Satoh et al., 2010), while IRF4 controls M2 polarization (Satoh et al., 2010; Krausgruber et al., 2011). In M2 macrophages, activation of NFκB-p50 appears to be associated with the inhibition of M1-polarizing genes (Porta et al., 2009). M2-type induction, through secretion of IL4, IL10 and TGFβ, promotes humoral immune responses and down-regulates M1 responses, inhibiting many macrophage inflammatory functions (Town et al., 2005). A third group, regulatory macrophages, arises at later stages and have a primary role limiting inflammation (Mosser, 2003; Lucas et al., 2005; Mosser and Edwards, 2008).
The term “inflamm-aging” was coined in reference to the state of mild chronic inflammation (Franceschi et al., 2000) observed in aged individuals, functionally characterized by a reduced capability to deal with stressing stimuli. The age-related immune changes, known as immune-senescence (Larbi et al., 2008), would be also induced by cumulative low-level inflammation, which induces changes in gene expression related to inflammation and immune response (Lee et al., 2000; de Magalhães et al., 2009), increases plasmatic levels of inflammatory cytokines (Singh and Newman, 2011), and activates inflammatory intracellular pathways (Helenius et al., 1996).
In aged animals, protein homeostasis is impaired at multiple levels, including chaperone-mediated protein folding and stability, protein trafficking, protein degradation and autophagy. A major consequence of these impairments is the aggregation of abnormal proteins, which are related to neurodegenerative diseases, such as Parkinson’s disease (PD) and Alzheimer’s disease (AD; Taylor and Dillin, 2011). Aged microglia undergo multiple functional changes (reviewed in von Bernhardi et al., 2015), impacting the neuronal environment and promoting development of cognitive impairments (Figure 1). Among these changes, microglial cell production of ROS and inflammatory cytokines could contribute to the onset of chronic neurodegenerative diseases (von Bernhardi, 2010). Decline of lysosomal and mitochondrial functions results in an exacerbated generation of ROS and inflammatory mediators by microglia. Moreover, aged microglia show a decreased ability to phagocytose Aβ in comparison with young microglia (Floden and Combs, 2011).
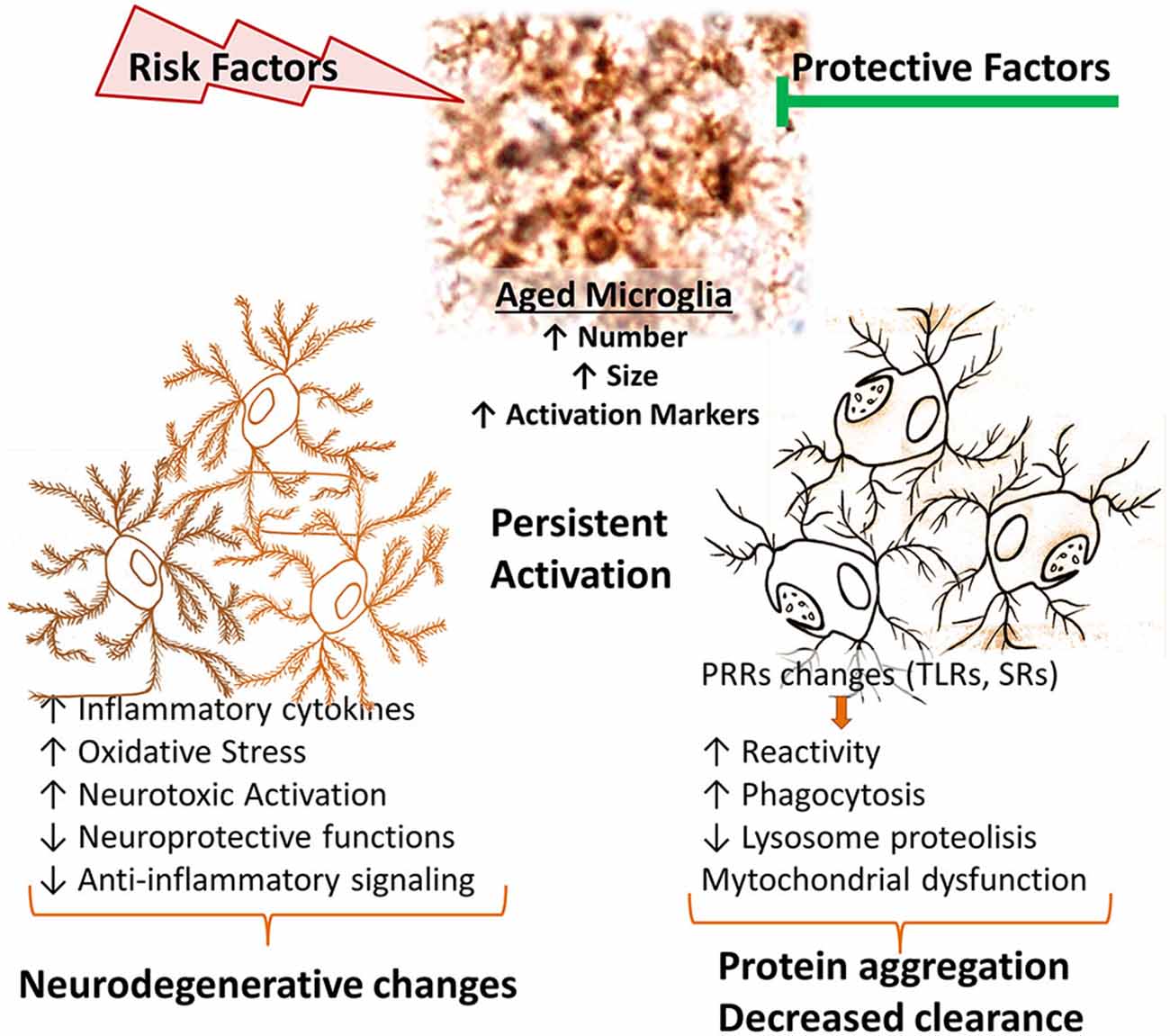
Figure 1. Age-related changes of microglial cell function. In aged brains, there is an increased number, size and activation of microglia. Age-related microglia changes depend both on gained and lost functions. Diverse stimuli or injury processes can further promote an inflammatory environment, promoting cytotoxic microglial cell activation. Aged microglia show increased basal phagocytic activity, although a reduced capacity to induce phagocytosis when stimulated, together with reduced lysosomal activity, resulting in a decreased clearance activity. They also have increased production of inflammatory cytokines and reactive species. Those changes result in a shift of balance towards decreased protective functions and an increased neurotoxicity. PRRs, pattern recognition receptors; SRs, scavenger receptors; TLRs, Toll-like receptors.
In aging, activated microglia remain as the principal source of inflammatory molecules and oxidative products of the CNS (Pawate et al., 2004; Qin et al., 2005; Hayashi et al., 2008). Both basal production of IL-6 and lipopolysaccharide (LPS)-induced secretion of IL-6 and IL1β are higher in aged microglia than in younger cells (Ye and Johnson, 1999; Sierra et al., 2007). In fact, mild stimulatory events or minor injuries, otherwise innocuous, could induce a robust and potentially damaging response. Thus, a stimulus that normally would trigger a protective response, in presence of age-related impairment of regulation could determine a persistent activation, associated, for example, to oxidative stress (von Bernhardi, 2007; Herrup, 2010). Similarly, aged microglia become more inflammatory than their younger counterparts upon systemic inflammatory stimulation, exacerbating damage (Combrinck et al., 2002; Cunningham et al., 2005; Godbout et al., 2005; Sierra et al., 2007). Accordingly, when exposed to endotoxin, microglia derived from adult mice secrete high amounts of ROS, whereas those from young animals mostly produce NO (Tichauer et al., 2014). Those effects depend, at least partly, on the upregulation of Toll-like receptors (TLRs), increased expression of the TLR4 co-receptor CD14 (Letiembre et al., 2007), changes in TLR4 signaling, and changes on the expression profile of scavenger receptors (SRs; Yamamoto et al., 2002; Hickman et al., 2008).
The activation of TLRs, CD14, and SRs by specific ligands is associated with microglial cell activation (Godoy et al., 2012; Murgas et al., 2012; Nakajima et al., 2007), production of inflammatory mediators, and uptake of macromolecules, including Aβ (Alarcón et al., 2005). There is conflicting evidence regarding the effect of age on phagocytosis. In contrast with reports indicating that microglia from aged mice have a decreased ability to phagocytose Aβ compared with young mice (Floden and Combs, 2011), we observed that basal phagocytosis of aged microglia is slightly increased compared with that from young mice, but phagocytosis fails to be induced by TGFβ (Tichauer et al., 2014) or LPS (Cornejo et al., 2014). Class A SR (SR-A) appears to play a key role for Aβ internalization by microglia (Chung et al., 2001) and degradation by cathepsin B (Yang et al., 2011), and for activation of microglia (Cornejo and Von Bernhardi, 2013). SR-A participates in the phagocytosis of Aβ and other anionic molecules, leading to the production of ROS (El Khoury et al., 1996). In AD, microglia expressing SR-A have been observed in close association with senile plaques (Honda et al., 1998; Bornemann et al., 2001). SR-A inhibition appears to increase Aβ burden in the brain of AD patients, potentially promoting neurotoxic effects and disease progression (Frenkel et al., 2013). The expression of these receptors decreases in the brain of aging animal models of AD (Hickman et al., 2008). Age-related changes in the expression of receptors involved in inflammatory activation could account for part of the function impairment of microglia, and provide insight regarding cell phenotypes that could play a role in the pathophysiological changes leading to neurodegenerative diseases. Given the various protective functions served by microglia, rather than seeking the inhibition of microglia, the regulation of those receptors involved in microglia activation could reduce some of the deleterious effects secondary to age-related microglial cell dysfunction.
Neuropathology in AD is characterized by the deposition of Aβ plaques and neurofibrillary tangles, constituted by hyper-phosphorylated tau, in the brain parenchyma (Hardy and Selkoe, 2002), intimately associated with activated microglia and astrocytes (Kim and de Vellis, 2005; Jellinger, 2006; Heneka and O’banion, 2007; von Bernhardi, 2007; von Bernhardi et al., 2010), and loss of synapses and neurons (Uylings and de Brabander, 2002). Worth mentioning, Aloise Alzheimer already stated in the early 1900’s that plaques and tangles probably were markers of an upstream process rather than the disease cause (Davis and Chisholm, 1999).
Microglial cell reactivity to Aβ and phagocytic activity are modulated by astrocytes, attenuating the cytotoxic response of microglia (DeWitt et al., 1998; von Bernhardi and Ramírez, 2001). However, modulation is abolished when microglia exposed to Aβ was previously primed (von Bernhardi and Eugenin, 2004), condition in which microglia show increased cytotoxicity, Aβ precursor protein (APP) synthesis, Aβ aggregation, and impairment of the uptake and degradation of Aβ compared with non-activated microglia (Rogers et al., 2002; von Bernhardi et al., 2007; Ramírez et al., 2008).
P38 appears to be involved in several pathological processes of AD. P38 becomes activated at early stages of the disease (Pei et al., 2001; Sun et al., 2003), being one of the kinases that phosphorylates specific sites in tau (Feijoo et al., 2005; Churcher, 2006). Inhibition of P38 abolishes Aβ-induced neuronal death in vitro (Zhu et al., 2005). P38 and NFκB appear to have a critical role for glial cell activation. Activation of those pathways are involved in Aβ-mediated induction of NO and TNFα production by glia (O’Neill and Kaltschmidt, 1997; Akama et al., 1998; Saha et al., 2007; Munoz and Ammit, 2010), which correlates with Aβ-induced cognitive impairment (Tran et al., 2001; Wang et al., 2005; He et al., 2007; Medeiros et al., 2007). Stimulation by Aβ induces a transient phosphorylation of P38, and a slower activation of NFκB (Flores and von Bernhardi, 2012) depending on the up-regulation of the transcriptional activity of NFκB by P38 (Saha et al., 2007), which contributes to neuroinflammation by activating AP-1 and by stabilizing mRNA and enhancing activity of NFκB. Production of TNFα and NO have different temporal profiles, in agreement with the early induction of cytokines by Aβ that appears to be needed for the subsequent induction of iNOS expression (Akama and Van Eldik, 2000).
The consideration that brain innate immune response can be involved in the genesis of neurodegenerative diseases (Nguyen et al., 2002; Bjorkqvist et al., 2009; von Bernhardi et al., 2010), lead to re-consider the role of Aβ and propose glia as a leading factor in AD pathology (Figure 2; von Bernhardi, 2007). However, for most scientists who adhere to the “amyloid cascade hypothesis”, Aβ is viewed as the cause of AD and neuroinflammation is considered just a consequence of glia activation (Akiyama et al., 2000; Heneka and O’banion, 2007; Hirsch and Hunot, 2009).
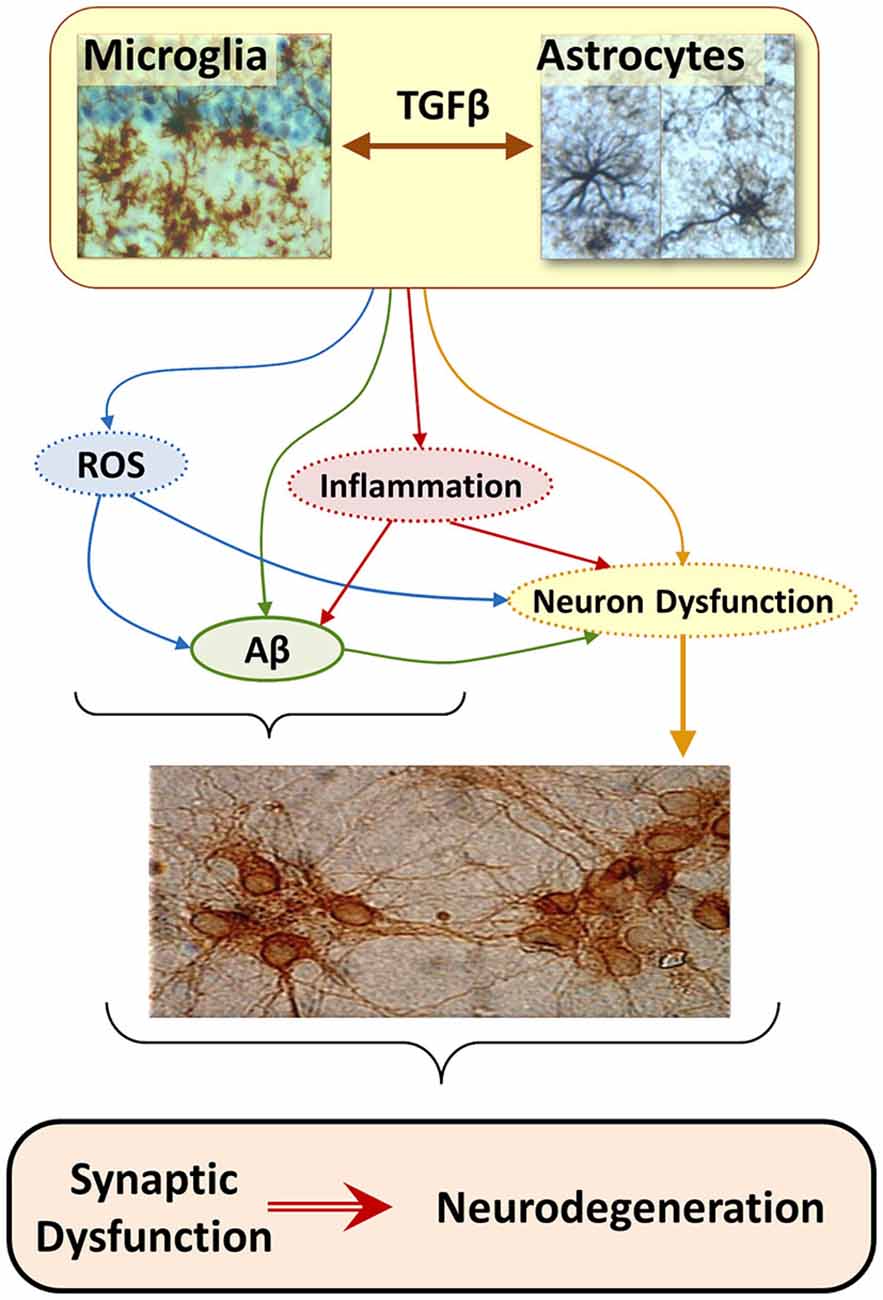
Figure 2. The “Glial Cell Dysregulation Hypothesis” for Alzheimer’s disease (AD). The glial cell dysregulation hypothesis proposes that AD has its cause on changes on the activation and impaired regulation of microglia, which become increasingly cytotoxic decreasing their protective functions. Microglia is under the regulation of astrocytes which, among other factors, secrete TGFβ. Inflammatory activation, secondary to aging and to certain forms of pathological stimuli, can result in glial cell dysregulation. Dysregulated glia, though the abnormal release of cytokines, reactive species, and other mediators, contributes to the increased expression of Aβ precursor protein (APP) and aggregation of Aβ, as well to functional and degenerative changes of neurons, perpetuating abnormal activation of glia, synaptic dysfunction and cell damage.
Astrocytes modulate microglia cytotoxicity and phagocytosis of Aβ (von Bernhardi and Ramírez, 2001). TGFβ1, secreted by astrocytes and neurons among other cells, regulates microglia activation, reducing release of inflammatory cytokines and reactive species (Chen et al., 2002; Mittaud et al., 2002; Herrera-Molina and Von Bernhardi, 2005; Herrera-Molina et al., 2012), protecting neuronal cells in vitro (Hu et al., 1995; Lieb et al., 2003; Herrera-Molina and Von Bernhardi, 2005) and promoting phagocytosis (Wyss-Coray et al., 2001). However, chronically activated microglia show a reduced response to such a modulation (von Bernhardi and Eugenin, 2004), showing instead an increased cytotoxicity and impaired uptake of Aβ (von Bernhardi et al., 2007; Ramírez et al., 2008). Regulation by TGFβ1 depends on a Smad3-mediated mechanism (Flores and von Bernhardi, 2012; Tichauer and von Bernhardi, 2012). Age-related inhibition on the activation of Smad has a profound effect on the regulation of microglia by TGFβ (Tichauer et al., 2014).
In the context of the “glial cell dysregulation hypothesis” neurotoxicity is not viewed as a consequence of hyperactive but rather of “mis-active”, dysfunctional microglia (von Bernhardi, 2007). Solid evidence show that adequately activated microglia are needed as scavenger cells in the CNS, participating for example in Aβ clearance (Paresce et al., 1996; Alarcón et al., 2005). However, lost response to normal regulatory feedback and/or an impaired ability to clear Aβ (Paresce et al., 1997; von Bernhardi, 2007), could lead microglia to develop predominantly cytotoxic features, establishing an inflammatory environment with increased oxidative stress, conditions that are amyloidogenic (Gabuzda et al., 1994; Wang et al., 2004), and promote neuron dysfunction (Figure 2). Thus, microglia, initially protective, would become chronically activated and show an exacerbated reactivity, contributing to brain cytotoxicity and neurodegeneration (Nguyen et al., 2002; Wyss-Coray and Mucke, 2002; Saud et al., 2005).
Regulation of glial cell activation appears to be impaired under sustained inflammatory stimulation (Ramírez et al., 2008), as those observed in the aged brain (Tichauer et al., 2014). Whereas inflammatory activation of glia by Aβ is relatively mild in culture, it is markedly potentiated in primed glia (von Bernhardi et al., 2007). Likewise, attenuation of microglia reactivity by astrocytes is greatly reduced when glia are exposed to inflammatory conditions (von Bernhardi and Eugenin, 2004). The priming of glia, rather than Aβ, could be the main trigger for abnormal glia activation in response to a stimulus that normally would not produce a sustained robust activation, a condition we named “dysregulated glia” (von Bernhardi, 2007). In that sense, in contrast to microglia normally reacting mildly when exposed to Aβ, microglia have an enhanced activation under chronic inflammatory conditions. Enhanced activation in turn could result in an increased cytotoxicity (von Bernhardi et al., 2015).
TGFβ is present in three isoforms, TGFβ1, TGFβ2 and TGFβ3. Astrocytes secrete preferentially TGFβ1. Increased production of TGFβ1 in response to inflammatory conditions is one of the regulatory mechanisms secondary to cell activation (Herrera-Molina and Von Bernhardi, 2005) that limits the temporal and spatial extent of neuroinflammation (Ramírez et al., 2005; Saud et al., 2005), and neurotoxicity (Eyupoglu et al., 2003). The modulation exerted by TGFβ1 is mediated by the activation of Smad3, which is down regulated in AD patients (Colangelo et al., 2002) and aged mice (Tichauer et al., 2014), and the activation of ERK (Saud et al., 2005), which also appears to be neuroprotective under certain conditions (Zhu et al., 2002, 2004). In addition to Smad, dynamic regulation of PI3K and MAPK, which are activated as part of the TGFβ signaling pathway (Figure 3) as well as with other inflammatory cytokines, can be key factors for cell viability and the regulation of inflammation. In the following sections, we will discuss how inhibition of Smad3 pathway and increased levels of TGFβ, as observed in aging, could modify regulatory signals, leading to glia dysregulation. The lack of inhibition of microglia inflammatory activation by TGFβ could result in cytotoxicity and neurodegenerative changes as those observed in AD. Impairment of TGFβ-Smad3 signaling could reduce the capability of microglia to deal with injury, inhibiting beneficial responses while inducing progression towards a more inflammatory state in the aging brain (Franceschi et al., 2000). This neuroinflammatory state could favor the development of age-related neurodegenerative diseases (Larbi et al., 2008), affecting the regulation of several inflammatory signaling pathways as we will discuss on the following sections.
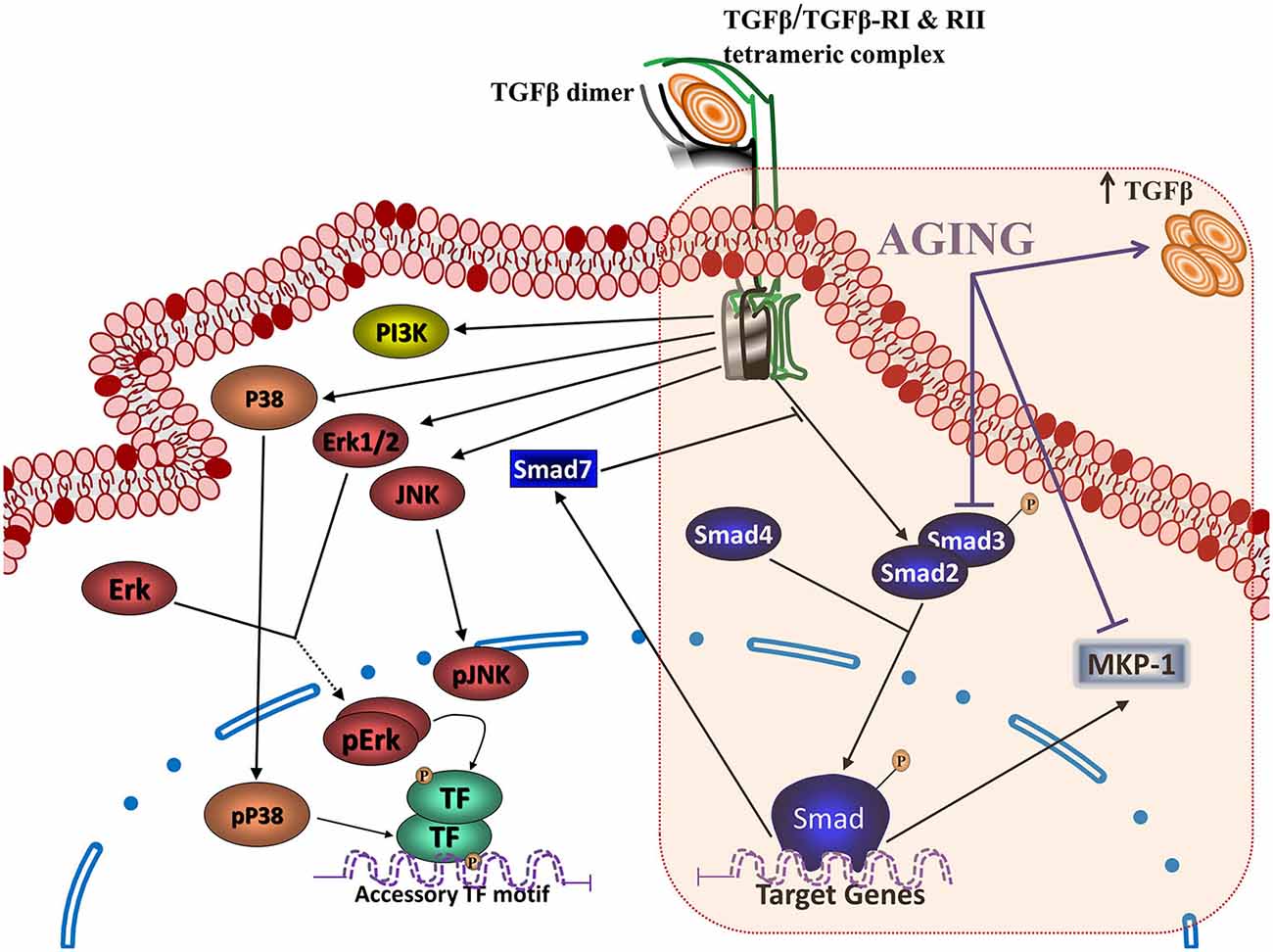
Figure 3. Transforming growth factor β (TGFβ) signaling pathways and aging. Binding of TGFβ to the type II TGFβ receptor dimer (TGFβRII) triggers recruitment of type I receptor dimer (TGFβRI), generating the heterotetrameric TGFβ receptor capable of activating the intracellular signaling pathways for TGFβ action. The activation of this complex activates (a) the canonical TGFβ signaling, with the phosphorylation of Smads 2 and/or 3 dimers, which bind to Smad 4 and translocate into the nucleus to regulate gene transcription, together with the activation of (b) non-canonical TGFβ signaling, which includes activation of MAPKs (ERK, extracellular signal-regulated protein kinase; JNK, c-Jun NH2-terminal kinase; and P38) and PI3K. Aging results in several changes on TGFβ signaling, including an increased production of TGFβ, as well as inhibition of the Smad pathway and activation of the phosphatase MAPK phosphatase (MKP-1).
TGFβ1 is a pleiotropic cytokine and a potent regulator of neuroinflammation and cytotoxicity. Many beneficial effects depend on the regulation of microglial cell activity by TGFβ1 (Hu et al., 1995; Lieb et al., 2003; Herrera-Molina and Von Bernhardi, 2005). In the brain, TGFβ1 is associated with neuroprotection in excitotoxicity, hypoxia, and ischemia, as well as with interfering with cell death cascades induced by compounds such as Aβ (Caraci et al., 2011). TGFβ1 secreted at the injury site promotes microglia recruitment, allowing for an efficient removal of the noxious stimulus. Stimulation of hippocampal cultures with LPS and IFNγ increases the secretion and activation of TGFβ1 (Uribe-San Martén et al., 2009). TGFβ1 secreted by hippocampal neurons and astrocytes in vitro (Ramírez et al., 2005; Tichauer et al., 2007) and microglia (Welser-Alves and Milner, 2013) decreases release of inflammatory mediators, and NO by microglia (Chen et al., 2002; Mittaud et al., 2002; Herrera-Molina and Von Bernhardi, 2005; Saud et al., 2005; Herrera-Molina et al., 2012), and increases viability of neurons (Hu et al., 1995; Lieb et al., 2003; Herrera-Molina and Von Bernhardi, 2005). Inhibition of LPS-induced macrophage and microglia activation by TGFβ1 is regulated in a Smad3-dependent manner (Werner et al., 2000; Le et al., 2004). The same mechanism is also involved in astrocyte-mediated neuroprotection against N-methyl-D-aspartate (NMDA)-induced neuronal injury (Docagne et al., 2002; Katayama et al., 2010) that results from the sustained activation of the ERK pathway, playing also a pivotal role in astrogliosis (Chu et al., 2004).
TGFβ signaling pathway (Figure 3) is activated when TGFβ interacts and induces assembly of an heterotetrameric receptor, containing two serine/threonine kinase receptors, type II and type I (Rahimi and Leof, 2007). In mammals, there are five type II receptors, TβRII, ActR-II, ActR-IIB, BMPR-II, AMHR-II, and seven type I receptors, the activin receptor-like kinases 1-7 (ALK1-7; Rahimi and Leof, 2007). In canonical TGFβ signaling pathway, ligand binding induces type II receptor to phosphorylate and activate type I receptor, which then phosphorylates receptor activated members of the Smad family (R-Smad). TGFβ activates the phosphorylation of Smad2 and Smad3, their assembly with a Smad common-partner, Smad4, and the nuclear translocation of the heterotrimeric complex (Smad2/Smad2/Smad4, Smad3/Smad3/Smad4 or Smad2/Smad3/Smad4). In the nucleus, the complex interacts with AGAC enriched Smad binding elements (SBE) on the DNA, acting as a transcriptional co-activator (Wrighton et al., 2009). They bind to specific sequences where they can activate or inhibit transcription, regulating gene expression of target genes associated with inflammatory activation (Schmierer and Hill, 2007; Heldin and Moustakas, 2012), including that of Smad 7 (Ross and Hill, 2008) that belongs to a third type, the inhibitory Smads (I-Smads: Smad6/7), which are an endogenous inhibitory system.
Smad pathways act as co-factors coupled to master transcriptional factors to direct gene expression in a cell-specific manner (Mullen et al., 2011). The main regulation of TGFβ signaling occurs on Smad. For example, Smad2/3 can be acetylated by transcriptional co-activators such as p300 and CREB-binding protein (CBP) in a TGFβ-dependent way (Simonsson et al., 2006; Tu and Luo, 2007). This post-transduction modification favors binding of the Smad complex to DNA, and the transcription of its target genes (Simonsson et al., 2006). Also, phosphorylation of Smad3 mediated by CDK2 and CDK4 inhibits its transcriptional activity (Liu, 2006; Buxton and Duan, 2008), and Smad3-PIAS (protein inhibitor of activated STAT) interaction suppresses Smad3 activation by TGFβ and favors its SUMOilation (Imoto et al., 2003). These and others post-transduction modifications, such as MAPKs-mediated phosphorylation (see below), can also regulate Smad activity and therefore TGFβ-mediated transcription (Ross and Hill, 2008).
In addition to TGFβ-mediated Smad activation, TGFβ activates a complex Smad-independent (TGFβ-non canonical pathways) signaling pathway (Weiss and Attisano, 2013; Figure 3). Non canonical signaling includes MAPK pathways ERK, P38 and JNK, and PI3K/Akt (Derynck and Zhang, 2003; Weiss and Attisano, 2013), and participate in many biological processes such as cell cycle inhibition, immunosuppression and neuroprotection, among others (Bosco et al., 2013).
TGFβ orchestrates the expression of numerous genes associated with inflammation and the immune response. In the nervous system, the TGFβ pathway is involved in the regulation of genes associated with cell cycle, cell proliferation, preservation of neural progenitor cells, oligodendroglia and neuronal differentiation, neuron survival and function, and the several neurotransmission-related genes (Kandasamy et al., 2014). TGFβ has a role in adult neurogenesis (He et al., 2007), and in the differentiation of adult neural progenitor cells, inducing the expression of several voltage-dependent channel subunits (Kcnd3, Scn1b, Cacng4, and Accn1) and other neuronal proteins, such as Cadps, Snap25, Grik4, Gria3, Syngr3, Gria4, doublecortin (DCX), Nrxn1, Sept8, and Als2cr3, suggesting that TGFβ participates, at least in part, in the induction of a functional neuronal phenotype, (Kraus et al., 2013). In addition, TGFβ participates in cell migration by modulating the expression of cell adhesion proteins such as nCAM, integrin α3, αV and β1 (Siegenthaler and Miller, 2004; Milner, 2009).
TGFβ regulates MHC class II expression in astrocytes, (Johns et al., 1992). It also regulates the expression of some constituents of its own pathway, including receptors type I TGFβ receptor (TGFβRI) and type II TGFβ receptor (TGFβRII), Smad7 and Smad3 (Ma et al., 2007; Qin et al., 2009). Microglia treated with TGFβ show a reduced expression of the immune mediators CCL3, CCL2, IL1a, IL1rl2, CCR5 and CD11c (Abutbul et al., 2012; Butovsky et al., 2014), and upregulation of CX3CR1, CSF3 and TLR3 expression (Chen et al., 2002; Butovsky et al., 2014), as well as SRs, which will be discussed in a later section.
In the brain, TGFβ favors cell survival, modulating the expression of Bad, Blc-2 and Bcl-x1 as a mechanism of neuroprotection against apoptosis (Dhandapani and Brann, 2003). TGFβ also participates in the regulation of temporal transition between early and late phases of neurogenesis and the regulation of the stem cell potency (Dias et al., 2014). Increased levels of TGFβ have been reported in several brain areas, including the hippocampus and hypothalamus, during aging (Bye et al., 2001; Werry et al., 2010). Cells showing increased production of TGFβ apparently do not include neurons, since TGFβ transcripts level are severely reduced in aged neurons (de Sampaio e Spohr et al., 2002).
Aging also affects the circadian variation of TGFβ expression. It has been reported a loss of the diurnal pattern of TGFβ expression, as well as a loss of the day/night expression of activated Smad3 compared with the pattern observed in young animals (Beynon et al., 2009) that have profound functional effects. In addition, the pedunculopontine (PPT) nucleus of aged rats, a structure related to sleep and cognitive functions, shows an over activation of the TGFβ-Smad signaling pathway that appears to be involved in sleep-related memory impairment in aging (George et al., 2006).
There are age-related changes on TGFβ signaling at several levels. Some depend on changes on the level or the pattern of secretion of TGFβ, or in its canonical signaling pathway, Smad (Figure 3). Others depend on changes on the interaction of TGFβ with other inflammatory mediators or their transcription factors, such as IFNγ and NFκB, or on regulatory components, such as MAPK phosphatases (MKP-1). Finally, there are age-related changes on its regulation on cellular processes, as observed with stem cells. The various changes will be discussed on the following sections.
TGFβ1, produced by astrocytes in vitro, decreases microglial NO and ROS production induced by LPS and IFNγ (Herrera-Molina and Von Bernhardi, 2005). Induction of both NO and ROS is prevented by TGFβ1 in neonatal, but not in adult animals. Therefore, response to inflammatory stimulation appears to become more oxidative, and for that reason, potentially more cytotoxic in aged animals (Tichauer et al., 2014). Moreover, modulation by TGFβ1 also is abolished in microglia obtained from animals previously exposed to inflammatory conditions (Tichauer et al., 2014).
TGFβ-Smad pathway is very important on the regulatory and neuroprotective effect of TGFβ1 (Derynck and Zhang, 2003), being involved in the induction of the quiescent phenotype of microglia (Abutbul et al., 2012). Activation of the TGFβ1-Smad3 pathway induces glial cells to produce MKP-1, a phosphatase exerting negative regulation on inflammatory activation that inhibits Aβ-induced MAPK and NFκB signaling (Figure 4), and decreases production of TNFα and NO (Flores and von Bernhardi, 2012). MKP-1 appears to preferentially dephosphorylate P38 and JNK, but it also dephosphorylates ERK in some cell types (Liu et al., 2007; Boutros et al., 2008).
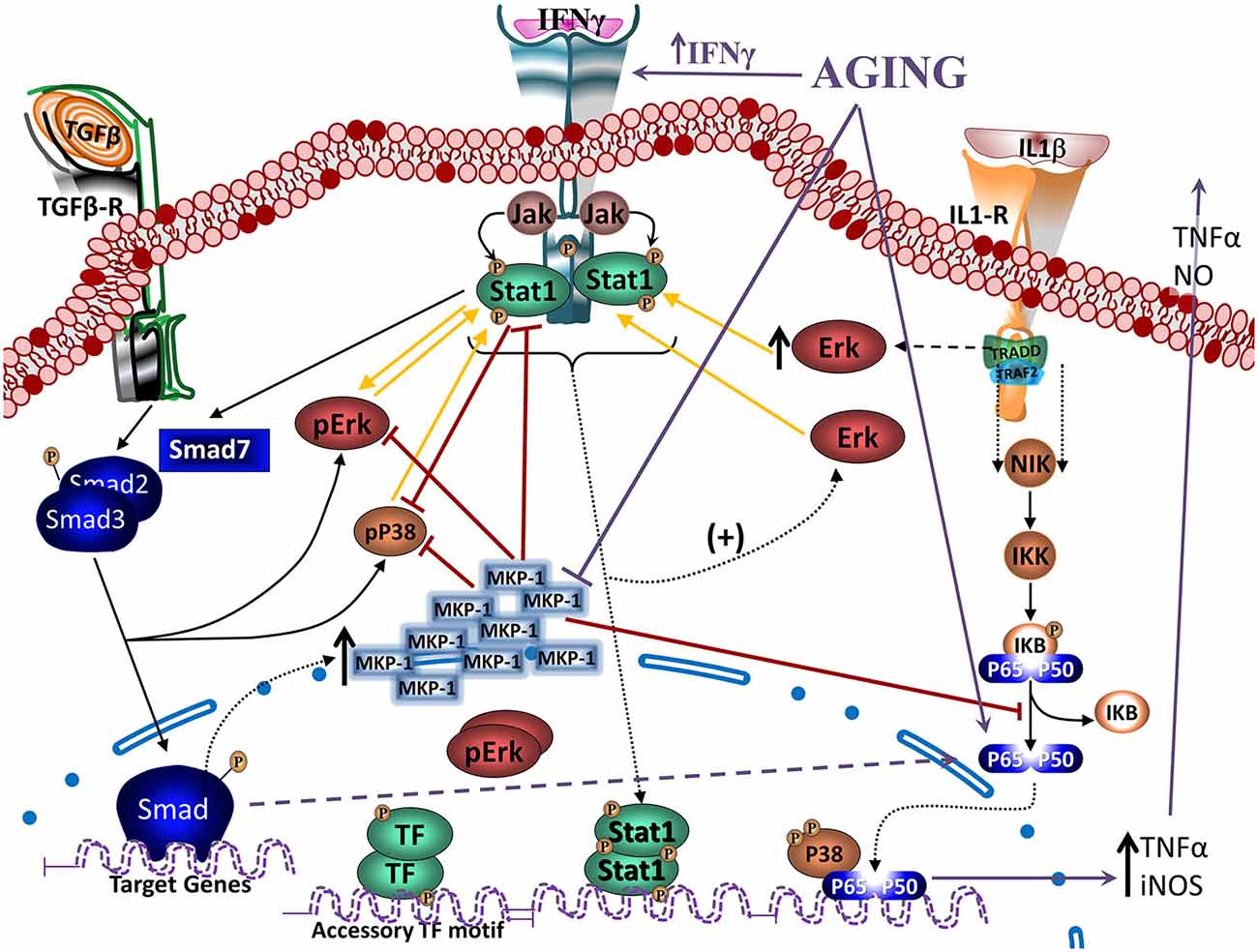
Figure 4. Regulation of Janus activated kinase (JAK)-Stat and nuclear factor kappa B (NFκB) signaling by TGFβ through MKP-1. Modulation of Interferon gamma (IFNγ)- and Interleukin 1β (IL1β)- induced signaling pathways (as models of inflammatory activation) by TGFβ1 reveals, at least partially, the anti-inflammatory effects of TGFβ1. IFNγ activates JAK- signal transducer and activator of transcription-type-1 (STAT1) pathway, increasing pSTAT1tyr, pERKs and to a lesser extent pP38. ERK and P38 MAPKs potentiate STAT1 activation by phosphorylation of a serine residue. IFNγ-induced signaling pathway inhibits Smad signaling by increasing synthesis of its endogenous inhibitor Smad7. TGFβ1 is also able to activate MAPKs. Particularly, TGFβ1 can activate ERK and P38 and also, through the activation of the Smad3 pathway, induces an increase in MKP-1 expression. Particularly in microglia, TGFβ1 inhibits IFNγ-induced STAT1 activation via a MKP-1-mediated inhibition of ERK1/2. Cytokines, such as IL1β induces activation of glial cells and the production of TNFα and nitric oxide (NO) through the activation of P38 and NFκB pathways. TGFβ1, by inducing an increase in MKP-1 levels, inhibits P38 and NFκB pathways, reducing production of TNFα and NO. In aging, there are increased levels of IFNγ, inhibiting activation of the TGFβ1-Smad signaling pathway, as well as reducing induction of MKP-1, which will result in a decreased regulation of MAPKs and NFκB pathways. An alternative pathway observed in aging leads to atypical TGFβ1 signaling that through inducing sequestering of IκB, induces activation of NFκB. Furthermore, age-related inflammation and increased production of reactive oxygen species (ROS) are strong activators of NFκB.
TGFβ1-dependent regulatory mechanisms are impaired in aging. Aged microglia show a basal activated status, which has been linked to neuronal damage, cognitive impairment, and an increased susceptibility to neurodegenerative diseases, such as AD (Block et al., 2007). Age-related alteration of TGFβ pathway includes changes in TGFβ release, Smad3 activation, and on microglial response induced by inflammatory stimuli in the hippocampus of aged mice, as well as abolition of TGFβ-induced phagocytosis (Tichauer and von Bernhardi, 2012) by aged microglia (Tichauer et al., 2014). In addition, Smad2/3 expression pattern is altered, showing increased expression of Smad3 in aging. In contrast, Smad2 (Deltaexon3), a splice form of Smad2 that directly binds to the DNA, is highly expressed prenatally and in early postnatal life, but it diminishes with aging (Ueberham et al., 2009).
Both age and inflammatory status affect the amount and phosphorylation of Smad3 protein in mice hippocampus (Tichauer et al., 2014). Whereas 2-month old mice show a robust increase of Smad3 in the hippocampus after a systemic inflammatory stimulus, 12-month old animals maintain Smad3 at its increased basal level (Figure 3). Similarly, phosphorylation (activation) of Smad is not induced by inflammation in old animals (Tichauer et al., 2014). The activation of the Smad pathway in young animals could depend on the effective elevation of TGFβ1 levels induced by inflammatory stimulation (Wynne et al., 2010). The induction of Smad3 expression could depend on the activation of MAPK1 (Ross et al., 2007). In contrast, in adult mice, increased basal levels of TGFβ1 (Colangelo et al., 2002; Lukiw, 2004) maintains elevated Smad3, becoming unresponsive to new inflammatory stimulation. Increased levels of TGFβ1 with a reduced activation of Smad signaling can result in an unbalance between the various TGFβ1 activated pathways (Schmierer and Hill, 2007). Furthermore, considering that the non-Smad TGFβ1 pathways MAPKs and PI3K, also participate in inflammatory activation signal transduction, and their activation is not abolished in aged mice, inhibition of Smad could abolish the regulatory effect of TGFβ1 on inflammation, facilitating the cytotoxic activation of glia. The partial inhibition of TGFβ1-Smad3 signaling in aging could explain the persistent activation of microglia and mild neuroinflammation, regardless the elevated levels of TGFβ1 in aged mice. The lost ability to modulate microglia activation, together with the increased ROS production by aged animals, could result in a predominantly cytotoxic activation.
The transcription factor NFκB is a robust candidate for showing age-dependent changes due to its role in the regulation of immunity, inflammation, and cell death (Adler et al., 2007). Blockade of NFκB in aged mice has been reported to reverse the gene expression program and cell morphology, “rejuvenating” old mice (Adler et al., 2008). Robust evidence in a variety of cell and animal based experimental systems show that oxidative stress and inflammation are strong inducers of NFκB activation (Muriach et al., 2014). They are frequently associated with aging, and are involved in the pathophysiology of several chronic diseases observed in aged individuals, like diabetes and AD. In fact, Aβ can be a strong inducer of NFκB in neuron cell death via the induction of intracellular ROS (Lee et al., 2005b; Valerio et al., 2006), and through tumor necrosis factor receptor 1 (TNFR1) signaling, which result in neuronal apoptosis (Li et al., 2004; Valerio et al., 2006). Inhibition of these pathways could be beneficial in the treatment of neurodegenerative diseases, including AD (Lee et al., 2005a,b; Munoz et al., 2007; Paris et al., 2007; Wang et al., 2008a). The effect appears to involve attenuation of Aβ-induced activation of ERK1 and P38 MAPKs, which are upstream NFκB signaling pathway (Pannaccione et al., 2005; Valerio et al., 2006).
TGFβ1 can also promote inflammatory activity under certain conditions. Yan et al. showed that TGFβ1 injection to the hypothalamus resulted in inflammatory NFκB signaling. Activation is via the TGFβ-R2 receptors expressed on neurons of the medial basal hypothalamus. It induces formation of RNA stress granules that accelerate the decay of IκBα, resulting in activation of NFκB (Yan et al., 2014). Although the work was oriented to understanding mechanisms of diabetes, similar conditions are also observed in aging.
In addition to this novel mechanism of TGFβ-dependent activation of NFκB, aging also shows reduction of MKP-1 (see subsection “Age-Related Changes on TGFβ and MKP-1”) that impairs inhibitory regulation over NFκB and MPAKs, potentiating cell reactivity, inflammatory activation, and potentially oxidative stress and cytotoxicity. In addition, increased production of ROS and neuroinflammation will result in an independent activation of NFκB.
IFNγ is a potent activator of microglia (Ng et al., 1999; Klegeris et al., 2005). IFNγ increases in the aged brain although its endogenous cell source remains unidentified (Lyons et al., 2011). The main signaling pathways induced by IFNγ are signal transducer and activator of transcription-type-1 (STAT1) and MAPKs (Figure 5; Blanchette et al., 2003; Platanias, 2005; Gough et al., 2008). STAT1 is activated by a Janus activated kinase (JAK)-dependent phosphorylation on tyrosine Y701 (pSTAT1tyr) and then translocates into the nucleus to induce the expression of target genes. Full transcriptional activity requires a second phosphorylation on serine S727 (pSTAT1ser; Wen et al., 1995).
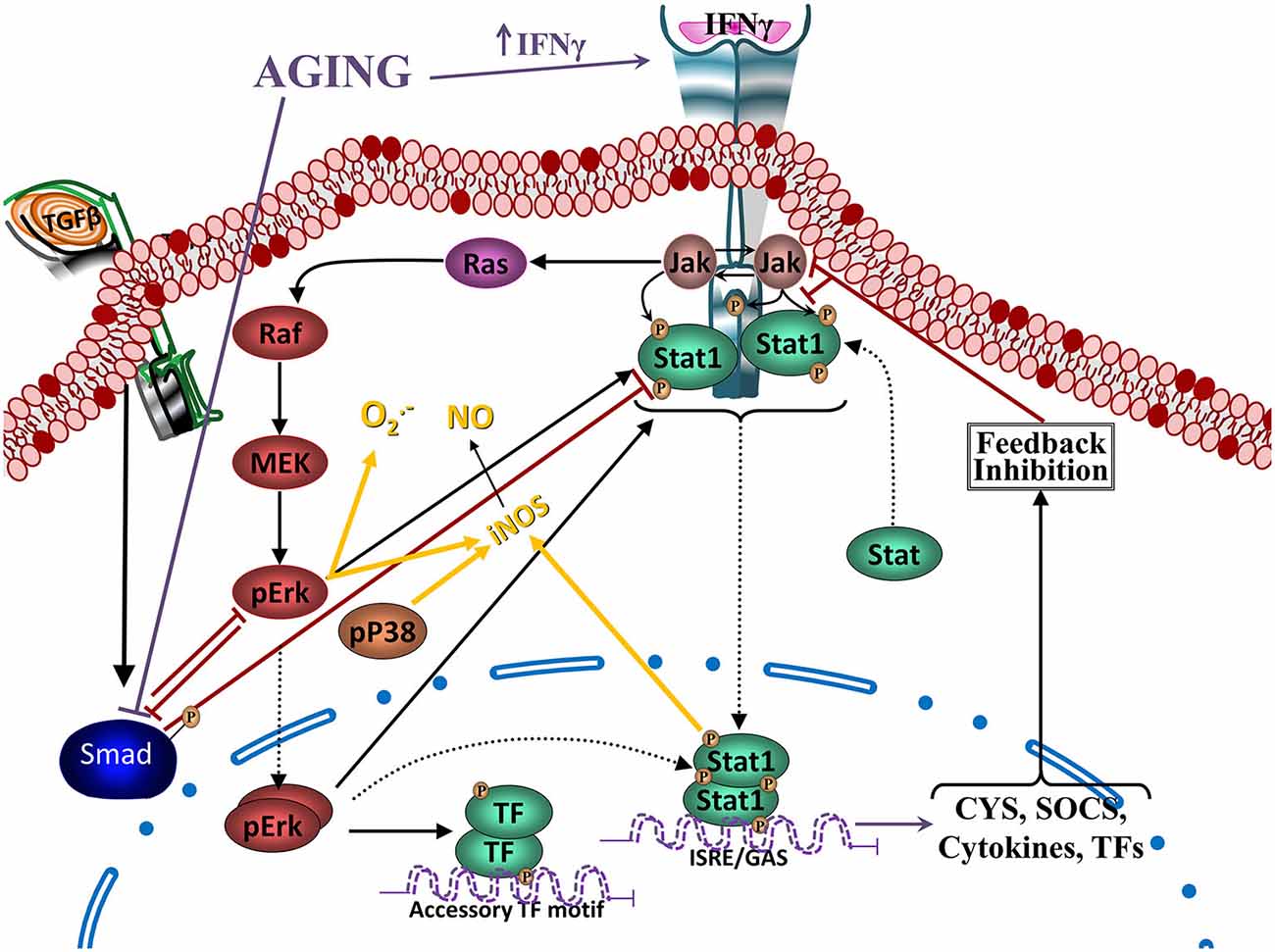
Figure 5. Reciprocal regulation of TGFβ and Jak-Stat signaling. Crosstalk between IFNγ- and TGFβ1- induced signaling pathways. IFNγ activates JAK-STAT1 pathway, increasing pSTAT1tyr, which will translocate to the nucleus and activate transcription of several cytokines and other mediators and receptors involved in inflammatory activation. In addition, it will also activate pERKs and to a lesser extent pP38. ERK and P38 MAPKs potentiate STAT1 activation and induce inducible nitric oxide synthase (iNOS), increasing production of NO, as well as production of . IFNγ-induced signaling pathway inhibits TGFβ1-Smad signaling by inducing its endogenous inhibitor Smad7 (see Figure 4) and through inhibition by ERK. On the other hand, TGFβ1, through the activation of the Smad3 pathway, inhibits IFNγ-induced STAT1 activation via a MKP-1-mediated inhibition of ERK1/2 and iNOS expression, reducing production of NO and . In aging, there are increased levels of IFNγ, inhibiting activation of the TGFβ1-Smad signaling pathway, as well as a reduced activation of TGFβ1-Smad, which will result in a reduced regulation of MAPKs (and NF-κB pathways as shown in the previous figure).
STAT1 is a key signaling pathway involved in the up-regulation of iNOS and NO· production (Dell’Albani et al., 2001; Gough et al., 2008). Inhibition of ERK1/2 and P38 decrease IFNγ-induced pSTAT1ser, which correlates with a reduction in NO· production. Decrease of pSTAT1ser and NO· production is additive when both MAPK are inhibited, indicating that ERK1/2 and P38 are needed for full activation of the STAT1 pathway in glia (Figure 4), and other cell types (Blanchette et al., 2003; Platanias, 2005; Gough et al., 2008). In contrast, O2·− production induced by IFNγ depends on increased levels of pERK1/2, but not pP38 (Bhat et al., 1998; Dang et al., 2003).
IFNγ suppresses TGFβ signaling through up-regulation of the inhibitory Smad7 (Ulloa et al., 1999), and there is a reciprocal regulatory interaction between TGFβ1 and IFNγ activated pathways (Figure 4). TGFβ1 released by hippocampal cells induce a transient increase of pERKs and a persistent increase of pP38, decreasing IFNγ-induced O2·− and NO· production by glia (Herrera-Molina and Von Bernhardi, 2005), decreasing activation of STAT1 and ERK by IFNγ (Figure 5). Also, after persistent stimulation, IFNγ decreases TGFβ1 induced P38 signal transduction (Herrera-Molina et al., 2012). IFNγ-TGFβ1 crosstalk regulates the production of radical species through the modulation of STAT1, ERK1/2 and P38 activation. Co-treatment with TGFβ1 and IFNγ results in decreased IFNγ-induced pERK1/2, pSTAT1ser, pSTAT1tyr, total STAT1 and also reduces induction of P38 activation by TGFβ1. Suppression of pSTAT1ser appears to be mediated by a TGFβ1-induced decrease of pERKs. In contrast, inhibition of pSTAT1tyr depends on the decrease of total STAT1 mediated by TGFβ1 (Figure 5). Increased MKP-1 activity appears to be responsible for the reduction of IFNγ-induced activation of glia induced by TGFβ1 co-treatment. In fact, transfection with MKP-1 siRNA significantly reduces modulation of IFNγ-induced NO· production by TGFβ1. Thus, MKP-1 induction appears to be responsible of the effects on MAPK pathways and link them with those observed on the STAT1 pathway (Figure 4; Flores and von Bernhardi, 2012; Herrera-Molina et al., 2012). The regulatory interaction between TGFβ1 and IFNγ has been well described in tissue repair in vivo. IFNγ null mice show increased amounts of TGFβ1 and activation of TGFβ1 signaling, indicating that IFNγ exerts negative regulation of TGFβ1 activity (Ishida et al., 2004). On the other hand, TGFβ1 null mice has elevated levels of IFNγ and STAT1 activation, and iNOS and NO· production, indicating that absence of TGFβ1 results in the deregulation of IFNγ pathway and its target genes (McCartney-Francis and Wahl, 2002).
Age-related increase of IFNγ and changes on MPK-1 regulation could be key elements for the increased activation of microglia in aged animals. Aged animals show increased levels of IFNγ, directly potentiating inflammatory signaling and further inhibiting the Smad pathway through the induction of Smad7. On the other hand, decreased activation of TGFβ-Smad3 by both age-related changes and increased IFNγ, further reduce MKP-1 induction, suppressing the regulatory effect of TGFβ on inflammatory activation.
Among the molecular mechanism underlying the anti-inflammatory and neuroprotective effects of TGFβ1, negative regulation of MAPK signaling, key inducer of glial cell activation, is exerted by a group of MKP. In the brain, induction of MKP-1 expression in response to anti-inflammatory molecules has been demonstrated for both astrocytes and microglia (Eljaschewitsch et al., 2006; Lee et al., 2008). TGFβ1 increases MKP-1 in glia, and other cells (Jono et al., 2003; Tong and Hamel, 2007), induction that is not affected by the presence of inflammatory conditions. Increased MKP-1 reduces the activation of P38 and NFκB pathways and decreases the NO and TNFα production induced by Aβ (Flores and von Bernhardi, 2012). siRNA transfection targeting MKP-1 attenuates the effects of TGFβ1, causing a significant amelioration of the modulation of Aβ-induced TNFα and NO production by TGFβ1 (Flores and von Bernhardi, 2012). Furthermore, MKP-1 null mice show increased P38 and JNK activity and cytokine and NO production, suggesting that this phosphatase serves as an immune regulator (Liu et al., 2007; Boutros et al., 2008).
Induction of MKP-1 is mediated by the Smad3 pathway (Figure 4). Smad3 inhibition greatly reduces TGFβ1-mediated MKP-1 induction, suggesting that it is a transcriptional target for Smad3, and results in a significant amelioration of the inhibition of TNFα and NO production. MKP-1 stability and enzymatic activity can be regulated through phosphorylation and acetylation, respectively (Liu et al., 2007; Boutros et al., 2008). Although other mechanisms are involved in the regulation of the production of inflammatory mediators by TGFβ1, Smad3-mediated MKP-1 induction is a novel manner of TGFβ1 action on glia that supports its anti-inflammatory role. Increased MKP-1 levels appears also to be the mechanism of action for other anti-inflammatory molecules, such as glucocorticoids (Kassel et al., 2001; Jang et al., 2007; King et al., 2009), and it has been demonstrated that this phosphatase participates in STAT1 dephosphorylation (Venema et al., 1998).
Age-related decrease on MKP-1 secondary to the inhibition of the Smad pathway results in the impairment of the inhibitory regulation on NFκB and MAPK pathways, potentiating inflammatory activation and cytotoxicity.
As the brain ages, TGFβ has important roles both in neuronal survival and in the promotion of stem cell quiescence (Kandasamy et al., 2014). In the hippocampus, TGFβ appears to potentiate the survival and proliferation of intermediate progenitor cells in the dentate gyrus of aged mice, by a Smad3-dependent mechanism (Tapia-González et al., 2013). Regarding regulation of neural stem cells in the aged brain (Dias et al., 2014), TGFβ lengthens G1 phase of the cell cycle in activated stem cells, impairing cell cycle progression of neural progenitors and neurogenesis (Daynac et al., 2014). Because of those effects, blockade of TGFβ signaling could improve neurogenesis in the aged brain (Pineda et al., 2013).
Although TGFβ serves key role in neuronal surveillance and stem cell proliferation, most of the cellular changes induced by aging have been described in glial cells. Expression of TGFβ by oligodendrocytes is reduced in aging, condition that interferes with oligodendrocytes recruitment and reduces remyelination (Hinks and Franklin, 2000). In aged microglia and astrocytes, TGFβ expression shows a regional specificity. TGFβ signaling increases after brain infarct in aged individuals (Doyle et al., 2010). In contrast, microglia and astrocytes located close to the leptomeninges show reduced TGFβ expression with age, reduction that could have a role on the increased permeability of leptomeninges during systemic inflammation (Wu et al., 2008).
There is evidence that impaired TGFβ signaling could be involved in the pathogenesis of AD. AD patients show decreased plasmatic levels of TGFβ1 (Mocali et al., 2004; Juraskova et al., 2010), but increased levels in cerebrospinal fluid (Blobe et al., 2000; Tarkowski et al., 2002), and within Aβ plaques (Burton et al., 2002). Brains of AD patients have reduced levels of TGFβRII (Tesseur et al., 2006). Reduced levels of Smad3 and impairment of Smad3 signaling have been observed in the AD brain, associated with increased Aβ accumulation, Aβ-induced neurodegeneration and neurofibrillary tangle formation (Luterman et al., 2000; Colangelo et al., 2002; Katsel et al., 2005; Tesseur et al., 2006). In addition to decreased expression of Smad3 in hippocampi of AD patients, hippocampal neurons show increased levels of activated Smad2 (Lee et al., 2006), along with alterations in the subcellular localization of phosphorylated Smad2/3 (Colangelo et al., 2002; Lee et al., 2006), which remains in the cytoplasm of neurons, instead of translocating into the nucleus (Lee et al., 2006; Ueberham et al., 2006). The ectopic localization of activated Smads in AD could be attributed to another pathological feature observed in AD, the hyperphosphorylation of tau. Hyperphosphorylated tau is associated with the sequestration of activated Smad2/3, and the disruption of TGFβ signaling (Baig et al., 2009). Both reduced presence of TGFβRII and defects on Smad are associated with inhibition of TGFβ signaling.
The TGFβ pathway exerts direct regulation over some of the pathological features of AD, since it upregulates the expression of the APP in normal human astrocytes by a Smad4-dependent mechanism (Burton et al., 2002). TGFβ also favors stabilization of the APP mRNA by binding it to a RNA-protein complex that reduces the rate of APP mRNA decay (Amara et al., 1999). In addition, TGFβ1 has been implicated on the promotion of amyloid angiopathy in frontal cortex and meninges (Wyss-Coray et al., 1997, 2000; Mazur-Kolecka et al., 2003), and on the increased production of Aβ by astrocytes in APP/TGFβ1 transgenic mice (Lesne et al., 2003). Notoriously, this amyloid angiopathy appears at a younger age when the overexpression of TGFβ1 in astrocytes occurs in the absence of SR-A (TGFβ1/SR-A−/− mice) (Lifshitz et al., 2013), which lead us to further inquire on the expression of that receptor, as discussed on the next two sections. Furthermore, those effects appear to depend on the activation of astrocytes that stimulate the production of APP due to the presence of a TGFβ1 response element in the 5’UTR of APP. Moreover, Tg2576 mice with a dominant negative TGFβ1-receptor II that blocks Smad2/3 signaling show a conspicuous reduction of amyloid deposits in the brain (Town et al., 2008).
In contrast with those potentially deleterious effects of TGFβ1, increased TGFβ1 has been also associated with a lower burden of Aβ in the parenchyma, which correlates with an increased microglia activation. Several reports show that TGFβ1 has anti-amyloidogenic roles, reducing the Aβ burden and inhibiting the formation of neuritic plaques, effects that appear to be mediated by the promotion of microglia-mediated Aβ degradation (Wyss-Coray et al., 2001). The neuroprotective role of TGFβ1 against Aβ toxicity has been studied in vitro and in vivo models of AD (Prehn et al., 1996; Caraci et al., 2008). Furthermore, TGFβ1 Smad3 also inhibits the production of radical species induced by inflammatory stimuli, and induces phagocytosis of Aβ in vitro (Tichauer and von Bernhardi, 2012).
However, induction of phagocytosis is lost as animals age (von Bernhardi et al., 2011). Smad3 pathway is altered in microglia from adult mice, affecting the induction of Aβ phagocytosis and the modulation of radical species production by TGFβ1.
The uncoupling of TGFβ1 signal transduction pathway could result in an altered pattern of microglial activation and reduced clearance of amyloid; effects that in fact are observed in aging and in AD. Impairment of TGFβ signaling can potentiate neuroinflammation, favoring neuronal dysfunction and neurodegenerative changes (Tesseur and Wyss-Coray, 2006). Reduced TGFβ-Smad3 signaling results in age-related neuroinflammation and neurodegeneration and in increased accumulation of Aβ (Tesseur et al., 2006). The accumulation of Aβ could depend on a reduction of its clearance, and be mediated by the reduced expression of SR-A by glia (Tichauer and von Bernhardi, 2012). These changes could facilitate cytotoxic inflammation and neurodegenerative diseases in aging (von Bernhardi et al., 2011). If accumulation of Aβ depends indeed on impaired clearance, it could situate Aβ as the result of disease progression instead of being its primary cause, as we propose in the “microglial cell dysregulation” hypothesis for AD.
The effect of TGFβ is cell and tissue specific. Whereas TGFβ reduces expression of some SRs in circulating macrophages, TGFβ increases expression of SR-A, while decreases expression of SR-BI by microglia (Tichauer and von Bernhardi, 2012). Given the relevance of SRs as well as other pattern recognition receptors (PRRs) on the inflammatory activation and the scavenger function of microglia, changes on the expression of these receptors could have a profound effect on microglial cell activation (von Bernhardi, 2007).
In addition to phagocytosis, SR-A is also involved in the regulation of glia activation (Murgas et al., 2014). Accordingly, the use of SR-A antagonists appears to improve the phenotypic features of AD (Handattu et al., 2009) by reducing microglial activation (Handattu et al., 2013). These results support the idea that SR-A activity could be part of the molecular mechanism involved in glial cell activation. In contrast to most SRs, SR-A expression is not necessarily downregulated, but can be increased by its ligands (Nikolic et al., 2011). In addition, binding of ligands to SRA recruits SRs to the membrane surface by a PI3K-activated mechanism (Cholewa et al., 2010). In macrophage cell lines, like THP-1 and J774A.1 (Bottalico et al., 1991; Nishimura et al., 1998; Draude and Lorenz, 2000; Argmann et al., 2001; Michael et al., 2012) and human monocytes (Draude and Lorenz, 2000), TGFβ1 reduces SR-A expression, through a mechanism that depends on Smad-2 (Michael et al., 2012). In addition, it has been also reported that induction of TGFβ1 by statin treatment, abolish induction of SR-A by inflammatory stimuli, in a mechanism mediated by ERK activation (Baccante et al., 2004).
In addition of increasing their expression of SR-A, TGFβ1 also increases Aβ uptake by microglia, an effect that is prevented by the Smad3 inhibitor SIS3 (Tichauer and von Bernhardi, 2012). Studies by our laboratory have also shown that induction of Aβ phagocytosis by TGFβ1 is decreased in aged mice (Tichauer et al., 2014). Reduction of induction of Aβ phagocytosis, together with decreased expression of SR-A in the brain of aged APP/PS1 mice (Hickman et al., 2008), suggest the existence of an altered regulation of SR-A expression in aging. Given its participation in Aβ uptake, SR-A impairment could be involved in the accumulation of Aβ during aging, with a mechanism associated with TGFβ1-Smad3 signaling (Tichauer and von Bernhardi, 2012), and could be related with AD pathogenesis.
Given our observation of increased protein expression of SR-A in microglia exposed to TGFβ through a Smad3-dependent mechanism (Tichauer et al., 2014), we performed in silico analyses of human genomic data to determine if the effect could be mediated by the transcriptional co-factors Smad2/3 over the MSR1 gene coding SR-A. As shown in Figure 6A, MSR1 gene is located at chromosome 8 and its transcription commonly generates three main splicing variants (Figure 6B). Analysis of ChIP-Seq data generated by Kim et al. revealed a significant peak of binding of Smad2/3 and Smad4 at the intron 7 of the MSR1 gene when human embryonic stem cells (hESC) were treated with activin (Figure 6C), a TGFβ receptor agonist (Kim et al., 2011). In addition, the MSR1 gene is ubiquitously bound by CTCF (Figure 6D), a protein factor having a key role in chromatin topological regulation (Ong and Corces, 2014) and previously described as a robust transcriptional repressor (Holwerda and De Laat, 2013) in various cell lines. The chromatin state segmentation data shows that the CTCF binding site is located in a genomic insulator region that is surrounded by highly repressed chromatin (Figure 6E), and is correlated with the presence of a topological domain predicted from Hi-C experiments (Figure 6F; Dixon et al., 2012). The presence of a CTCF peak in the MSR1 gene and the fact this gene is in a topologic domain suggest that MSR1 is highly repressed in most cell types (Figures 6D–F). This could explain the tissue specificity of this gene, which is almost exclusively present on the monocyte-macrophage lineage (Christie et al., 1996; Godoy et al., 2012), and astrocytes (Alarcón et al., 2005).
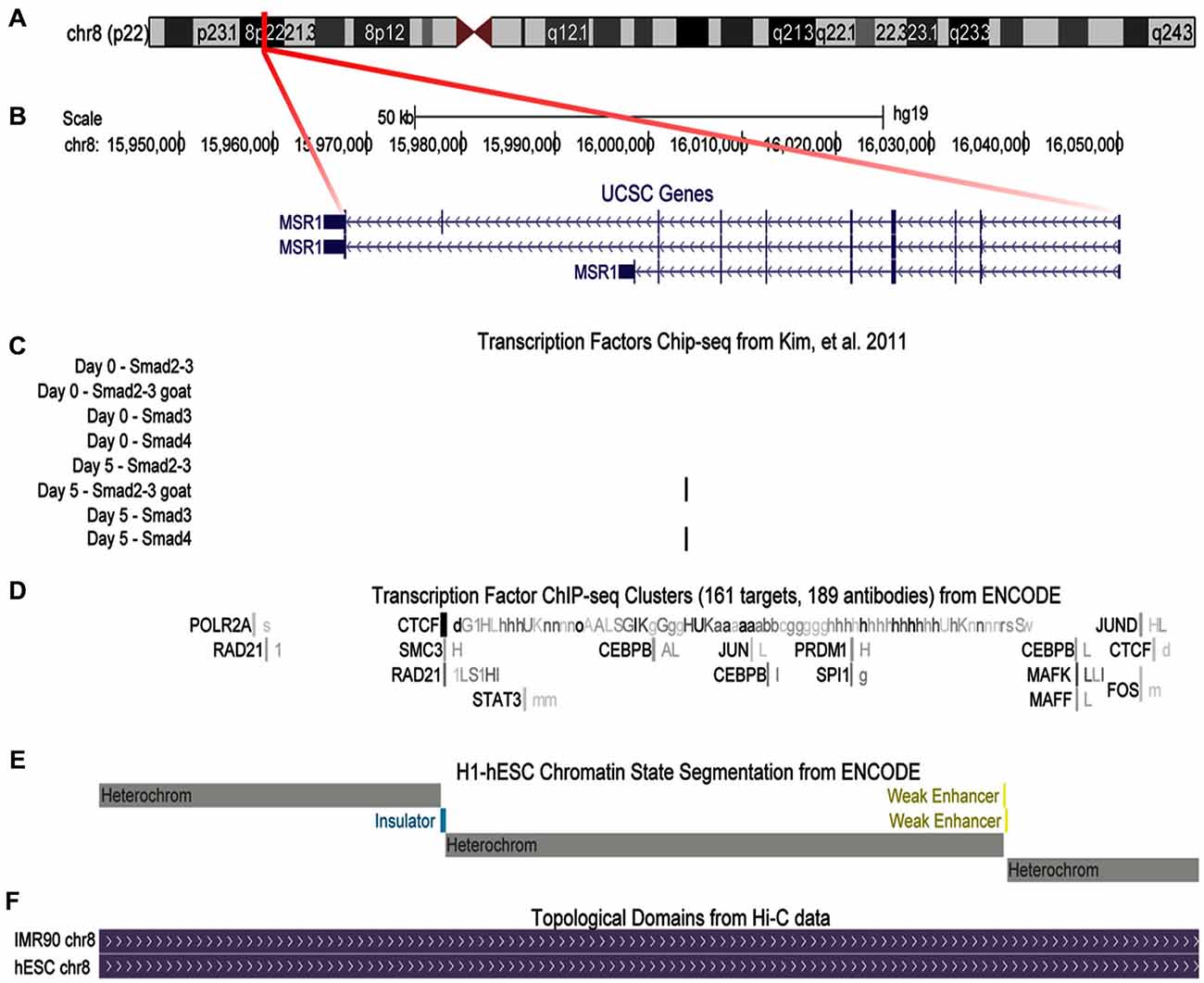
Figure 6. Binding of transcriptional co-factors Smad2/3 and 4 to the MSR1 gene. SR-A gene structure is shown using UCSC gene annotation, ChIP-Seq available data, and chromatin structure associated to this gene. (A) Ideogram of chromosome 8. The region labeled in red shows the MSR1 gene location. (B) MSR1 gene structure, indicating the three most common isoforms. Arrows indicate the direction of transcription and boxes symbolize exons. (C) ChIP-Seq data for Smads using four different antibodies before activin treatment (day 0) and 5 days after treatment of human embryonic stem cells (hESC; Kim et al., 2011); black bars show significant signals of ChIP-Seq indicating Smad2/3 and Smad 4 binding to MSR1 gene after activin treatment (day 5). (D) ChIP-Seq results provided by ENCODE project for 161 transcription factors from various cell lines. Boxes represent ChIP-Seq significant signal for a specific transcription factor. The darkness of the box is proportional to the maximal signal intensity of ChIP-Seq observed in a cell line, shown next to the box, in a lowercase letter, cells where this signal was found significant. (E) Chromatin segmentation state data generated by ENCODE project for H1-hESC (Aad et al., 2011); gray boxes represent heterochromatin sites, yellow is used for enhancer sites, and blue are for insulators. (F) Genomic topological domains detected by using Hi-C data in hESC and IMR90 cell lines (Dixon et al., 2012).
We also analyzed data of DNase sensitivity and ChIP-Seq available at WashU Epigenome Browser (Zhou et al., 2013), finding that H1-hESC cells differentiated into mesenchymatic cells present a hypersensitivity peak to DNase coincidental to the CTCF binding site (Figure 7A). In contrast, after treatment of H1-hESC cells with activin to differentiate them into mesoderm, the DNase hyper-sensitivity peak related to CTCF disappears, but a new DNase hypersensitivity peak is observed, which match with the Smad2/3 and Smad 4 binding site detected by the previous ChIP-Seq data analysis (Figures 7A,B; Kim et al., 2011). This shift between those two DNase hypersensitivity peaks after activin treatment leads us to infer that TGFβ pathway activation interferes with the binding of CTCF and might promote a topological rearrangement that induces MSR1 transcription by a mechanism mediated by Smad2/3/4. This hypothesis is currently been experimentally tested by our group.
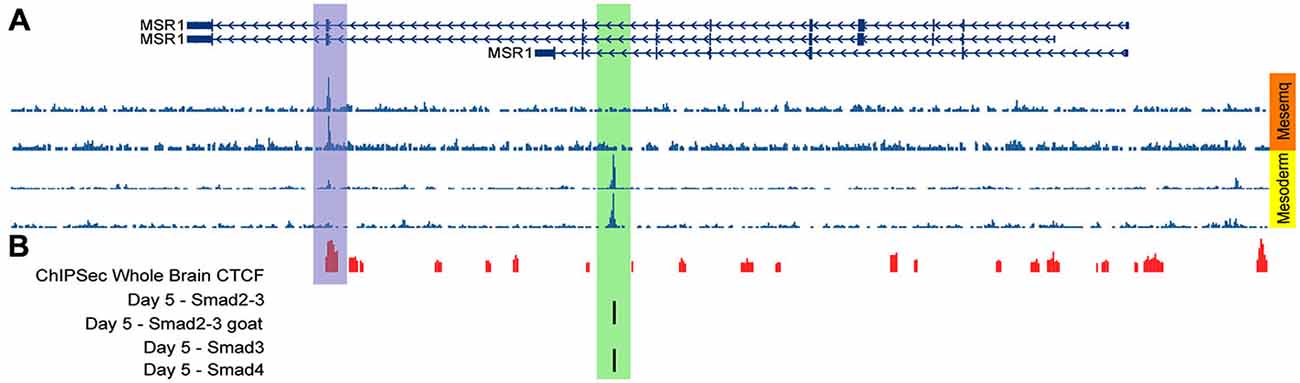
Figure 7. DNase sensitive sites matching with CTCF binding sites after activin treatment. (A) DNase hypersensitivity data in H1-hESC cells differentiated into mesenchymal cells (orange block) and treated with activin to differentiate H1-hESC cells into mesoderm (yellow block); different rows correspond to different experiments. (B) ChIP-Seq data for CTCF in the whole brain (red bars) and for Smad 2/3 and 4 (black bars; Kim et al., 2011).
The brain of AD patients reveal changes on the methylation pattern of some genes described as susceptible to be involved in AD, such as PSEN1 y APOE, and in genes related to homeostasis of gene methylation, like MTHRF and DNMT1 (Wang et al., 2008b). This study also identified age-specific epigenetic changes, suggesting that epigenetics could have a role in the development of AD (Wang et al., 2008b). Given that possibility, a future challenge is to assess the existence of epigenetic changes on the MSR1 gene or on genes related with the TGFβ pathway during aging and in AD. Changes potentially affecting SR-A expression or function as the brain ages would affect inflammatory activation and Aβ uptake by glia.
Age dependent changes such as microglia mis-activation, production of ROS, and decreased proteasome activity could establish the grounds for microglial cell dysfunction, leading to cytotoxicity and accumulation of Aβ or other protein aggregates. The combined effect of various age-associated changes, in addition to the individual endophenotypic condition and diverse environmental stimuli can initiate the vicious circle of cytotoxic activation of microglia.
Innate immune response, with microglia as the pivotal player, is recognized to have a profound immune-modulatory and reparative potential. However, chronic activation and dysregulation of microglia can lead to deleterious effects, inducing malfunction and damage of the CNS. Microglia activation appears to undergo different phases depending on their environmental and functional context. Whereas inhibition of microglia can be beneficial at a certain phase of disease progression it can become detrimental at another. A critical area of research would be to understand their activation process, developing pharmacologic tools directed towards selected properties of microglia. That would be a major improvement respect the present approach of turning off microglial cell activation as a whole, which likely has a major bearing in the limitations of past thinking about immunoinhibitory drugs for neurodegenerative diseases.
Our interest in identifying protective and regulatory pathways, to potentiate them while inhibiting cytotoxic activation of microglia, lead us to study the effect of TGFβ on microglia function in aging and various inflammatory conditions. TGFβ-Smad3 is involved in many protective functions of microglia, and shows major changes with aging. Our working model (Figure 8) shows that upon activation by various stimuli, TGFβ binds to its receptor activating a complex signaling pathways that includes activation of both the canonical Smad pathway as well as the non-canonical MAPKs and PI3K. As result of the activation of those pathways, among many changes, there will be changes on the expression pattern of SRs, the phagocytic activity and the production of inflammatory cytokines and other inflammatory and oxidative stress mediators. In aged microglia, increased amounts of TGFβ will act upon its receptor. However, secondary to age-related changes or chronic inflammation, the activation of Smad3 pathway is inhibited. Inhibition of Smad3 activation in the context of increased TGFβ levels shifts the regulatory signaling towards a dysregulated inflammatory activation, potentially leading to the impairment of protective response, development of an increased cytotoxicity and to neurodegenerative changes. Thus, increased neuroinflammation, decreased Aβ clearance and impaired cell viability could be consequence of the impaired TGFβ signaling.
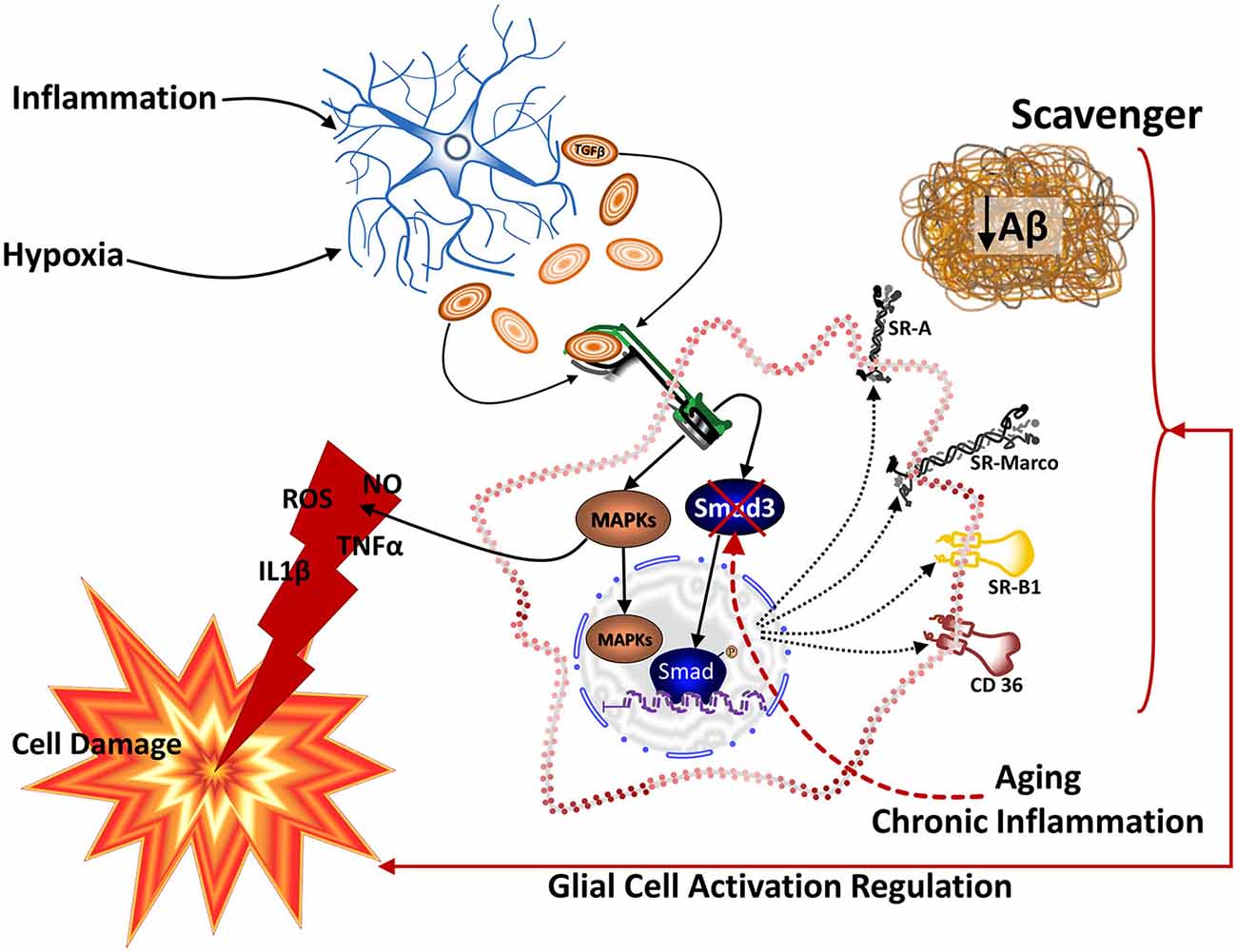
Figure 8. Model for TGFβ1-Smad3 pathway regulation in aged microglia. Diverse stimuli, including inflammatory stimulation and hypoxia, induce astrocytes to secrete TGFβ1. Binding of TGFβ1 to its receptor results in the activation of the Smad3 pathway, as well as MAPKs and PI3K signaling in microglia. Thus, TGFβ1 regulates the inflammatory activation of microglia in addition to modifying the SRs profile expressed by the cell. SRs appear to be involved both in the uptake of Aβ and in the activation of glial cells. Several of the effects of TGFβ1 on cell viability, reduction of inflammatory cytokines and reactive species, and expression of SRs depend on the activation of Smad3. In aging or after exposure to chronic inflammatory conditions, canonical activation of Smad3 is greatly reduced, whereas MAPKs remains activatible. As result of this change on TGFβ1 signaling, microglia show increased cytotoxicity, undergo changes on their expression of SRs and decrease their Aβ clearance. Thus, reduced TGFβ1-Smad3 activity on aged microglia appears to impair the beneficial effect of TGFβ1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Support by grant FONDECYT 1131025 and 1130874, and CONICYT fellowship 21120013 are acknowledged. We acknowledge the excellent technical help of Ms. Laura Eugenín-von Bernhardi with the figures.
Aad, G., Abbott, B., Abdallah, J., Abdelalim, A. A., Abdesselam, A., Abdinov, O., et al. (2011). Measurement of the W+ W- cross section in sqrt(s) = 7 TeV pp collisions with ATLAS. Phys. Rev. Lett. 107:041802. doi: 10.2172/982232
Abutbul, S., Shapiro, J., Szaingurten-Solodkin, I., Levy, N., Carmy, Y., Baron, R., et al. (2012). TGF-beta signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia 60, 1160–1171. doi: 10.1002/glia.22343
Adler, A. S., Kawahara, T. L., Segal, E., and Chang, H. Y. (2008). Reversal of aging by NFkappaB blockade. Cell Cycle 7, 556–559. doi: 10.4161/cc.7.5.5490
Adler, A. S., Sinha, S., Kawahara, T. L., Zhang, J. Y., Segal, E., and Chang, H. Y. (2007). Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 21, 3244–3257. doi: 10.1101/gad.1588507
Akama, K. T., Albanese, C., Pestell, R. G., and Van Eldik, L. J. (1998). Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc. Natl. Acad. Sci. U S A 95, 5795–5800. doi: 10.1073/pnas.95.10.5795
Akama, K. T., and Van Eldik, L. J. (2000). Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J. Biol. Chem. 275, 7918–7924. doi: 10.1074/jbc.275.11.7918
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/S0197-4580(00)00124-X
Alarcón, R., Fuenzalida, C., Santibanez, M., and von Bernhardi, R. (2005). Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J. Biol. Chem. 280, 30406–30415. doi: 10.1074/jbc.m414686200
Aldskogius, H., Liu, L., and Svensson, M. (1999). Glial responses to synaptic damage and plasticity. J. Neurosci. Res. 58, 33–41. doi: 10.1002/(sici)1097-4547(19991001)58:1<33::aid-jnr5>3.0.co;2-m
Amara, F. M., Junaid, A., Clough, R. R., and Liang, B. (1999). TGF-beta(1), regulation of alzheimer amyloid precursor protein mRNA expression in a normal human astrocyte cell line: mRNA stabilization. Brain Res. Mol. Brain Res. 71, 42–49. doi: 10.1016/s0169-328x(99)00158-8
Argmann, C. A., Van Den Diepstraten, C. H., Sawyez, C. G., Edwards, J. Y., Hegele, R. A., Wolfe, B. M., et al. (2001). Transforming growth factor-beta1 inhibits macrophage cholesteryl ester accumulation induced by native and oxidized VLDL remnants. Arterioscler. Thromb. Vasc. Biol. 21, 2011–2018. doi: 10.1161/hq1201.099426
Baccante, G., Mincione, G., Di Marcantonio, M. C., Piccirelli, A., Cuccurullo, F., and Porreca, E. (2004). Pravastatin up-regulates transforming growth factor-beta1 in THP-1 human macrophages: effect on scavenger receptor class A expression. Biochem. Biophys. Res. Commun. 314, 704–710. doi: 10.1016/j.bbrc.2003.12.150
Baig, S., van Helmond, Z., and Love, S. (2009). Tau hyperphosphorylation affects Smad 2/3 translocation. Neuroscience 163, 561–570. doi: 10.1016/j.neuroscience.2009.06.045
Benveniste, E. N., Nguyen, V. T., and O’Keefe, G. M. (2001). Immunological aspects of microglia: relevance to Alzheimer’s disease. Neurochem. Int. 39, 381–391. doi: 10.1016/s0197-0186(01)00045-6
Beynon, A. L., Thome, J., and Coogan, A. N. (2009). Age and time of day influences on the expression of transforming growth factor-beta and phosphorylated SMAD3 in the mouse suprachiasmatic and paraventricular nuclei. Neuroimmunomodulation 16, 392–399. doi: 10.1159/000228914
Bhat, N. R., Zhang, P., Lee, J. C., and Hogan, E. L. (1998). Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J. Neurosci. 18, 1633–1641.
Bjorkqvist, M., Wild, E. J., and Tabrizi, S. J. (2009). Harnessing immune alterations in neurodegenerative diseases. Neuron 64, 21–24. doi: 10.1016/j.neuron.2009.09.034
Blanchette, J., Jaramillo, M., and Olivier, M. (2003). Signalling events involved in interferon-gamma-inducible macrophage nitric oxide generation. Immunology 108, 513–522. doi: 10.1046/j.1365-2567.2003.01620.x
Blobe, G. C., Schiemann, W. P., and Lodish, H. F. (2000). Role of transforming growth factor beta in human disease. N. Engl. J. Med. 342, 1350–1358. doi: 10.1056/NEJM200005043421807
Block, M. L., and Hong, J. S. (2005). Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog. Neurobiol. 76, 77–98. doi: 10.1016/j.pneurobio.2005.06.004
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Bornemann, K. D., Wiederhold, K. H., Pauli, C., Ermini, F., Stalder, M., Schnell, L., et al. (2001). Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am. J. Pathol. 158, 63–73. doi: 10.1016/s0002-9440(10)63945-4
Bosco, P., Ferri, R., Salluzzo, M. G., Castellano, S., Signorelli, M., Nicoletti, F., et al. (2013). Role of the transforming-growth-factor-beta1 gene in late-onset Alzheimer’s disease: implications for the treatment. Curr. Genomics 14, 147–156. doi: 10.2174/1389202911314020007
Bottalico, L. A., Wager, R. E., Agellon, L. B., Assoian, R. K., and Tabas, I. (1991). Transforming growth factor-beta 1 inhibits scavenger receptor activity in THP-1 human macrophages. J. Biol. Chem. 266, 22866–22871.
Boutros, T., Chevet, E., and Metrakos, P. (2008). Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death and cancer. Pharmacol. Rev. 60, 261–310. doi: 10.1124/pr.107.00106
Burton, T., Liang, B., Dibrov, A., and Amara, F. (2002). Transforming growth factor-beta-induced transcription of the Alzheimer beta-amyloid precursor protein gene involves interaction between the CTCF-complex and Smads. Biochem. Biophys. Res. Commun. 295, 713–723. doi: 10.1016/s0006-291x(02)00725-8
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Buxton, I. L., and Duan, D. (2008). Cyclic GMP/protein kinase G phosphorylation of Smad3 blocks transforming growth factor-beta-induced nuclear Smad translocation: a key antifibrogenic mechanism of atrial natriuretic peptide. Circ. Res. 102, 151–153. doi: 10.1161/CIRCRESAHA.107.170217
Bye, N., Zieba, M., Wreford, N. G., and Nichols, N. R. (2001). Resistance of the dentate gyrus to induced apoptosis during ageing is associated with increases in transforming growth factor-beta1 messenger RNA. Neuroscience 105, 853–862. doi: 10.1016/s0306-4522(01)00236-6
Calabrese, V., Guagliano, E., Sapienza, M., Panebianco, M., Calafato, S., Puleo, E., et al. (2007). Redox regulation of cellular stress response in aging and neurodegenerative disorders: role of vitagenes. Neurochem. Res. 32, 757–773. doi: 10.1007/s11064-006-9203-y
Caraci, F., Battaglia, G., Bruno, V., Bosco, P., Carbonaro, V., Giuffrida, M. L., et al. (2011). TGF-beta1 Pathway as a new target for neuroprotection in Alzheimer’s disease. CNS Neurosci. Ther. 17, 237–249. doi: 10.1111/j.1755-5949.2009.00115.x
Caraci, F., Battaglia, G., Busceti, C., Biagioni, F., Mastroiacovo, F., Bosco, P., et al. (2008). TGF-beta 1 protects against Abeta-neurotoxicity via the phosphatidylinositol-3-kinase pathway. Neurobiol. Dis. 30, 234–242. doi: 10.1016/j.nbd.2008.01.007
Chen, S., Luo, D., Streit, W. J., and Harrison, J. K. (2002). TGF-beta1 upregulates CX3CR1 expression and inhibits fractalkine-stimulated signaling in rat microglia. J. Neuroimmunol. 133, 46–55. doi: 10.1016/s0165-5728(02)00354-5
Cholewa, J., Nikolic, D., and Post, S. R. (2010). Regulation of class A scavenger receptor-mediated cell adhesion and surface localization by PI3K: identification of a regulatory cytoplasmic motif. J. Leukoc. Biol. 87, 443–449. doi: 10.1189/jlb.0509318
Christie, R. H., Freeman, M., and Hyman, B. T. (1996). Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer’s disease. Am. J. Pathol. 148, 399–403.
Chu, C. T., Levinthal, D. J., Kulich, S. M., Chalovich, E. M., and DeFranco, D. B. (2004). Oxidative neuronal injury. The dark side of ERK1/2. Eur. J. Biochem. 271, 2060–2066. doi: 10.1111/j.1432-1033.2004.04132.x
Chung, H., Brazil, M. I., Irizarry, M. C., Hyman, B. T., and Maxfield, F. R. (2001). Uptake of fibrillar beta-amyloid by microglia isolated from MSR-A (type I and type II) knockout mice. Neuroreport 12, 1151–1154. doi: 10.1097/00001756-200105080-00020
Churcher, I. (2006). Tau therapeutic strategies for the treatment of Alzheimer’s disease. Curr. Top. Med. Chem. 6, 579–595. doi: 10.2174/156802606776743057
Colangelo, V., Schurr, J., Ball, M. J., Pelaez, R. P., Bazan, N. G., and Lukiw, W. J. (2002). Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 70, 462–473. doi: 10.1002/jnr.10351
Combrinck, M. I., Perry, V. H., and Cunningham, C. (2002). Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 112, 7–11. doi: 10.1016/s0306-4522(02)00030-1
Cornejo, F., and Von Bernhardi, R. (2013). Role of scavenger receptors in glia-mediated neuroinflammatory response associated with Alzheimer’s disease. Mediators Inflamm. 2013:895651. doi: 10.1155/2013/895651
Cornejo, F. A., Vruwink, M., Metz, C., Ramírez, G., and Von Bernhardi, R. (2014). Role of Scavenger Receptor Class A in glial cell activation in Alzheimer’s disease. Social Neuroscience Abstract 44th Annual Meeting, Washington, DC.
Cunningham, C., Wilcockson, D. C., Campion, S., Lunnon, K., and Perry, V. H. (2005). Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 25, 9275–9284. doi: 10.1523/jneurosci.2614-05.2005
Dang, P. M., Morel, F., Gougerot-Pocidalo, M. A., and El Benna, J. (2003). Phosphorylation of the NADPH oxidase component p67(PHOX) by ERK2 and P38MAPK: selectivity of phosphorylated sites and existence of an intramolecular regulatory domain in the tetratricopeptide-rich region. Biochemistry 42, 4520–4526. doi: 10.1021/bi0205754
Davis, J. N., 2nd, and Chisholm, J. C. (1999). Alois alzheimer and the amyloid debate. Nature 400:810. doi: 10.1192/pb.24.12.480-a
Daynac, M., Pineda, J. R., Chicheportiche, A., Gauthier, L. R., Morizur, L., Boussin, F. D., et al. (2014). TGFbeta lengthens the G1 phase of stem cells in aged mouse brain. Stem Cells 32, 3257–3265. doi: 10.1002/stem.1815
de Magalhães, J. P., Curado, J., and Church, G. M. (2009). Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 25, 875–881. doi: 10.1093/bioinformatics/btp073
de Sampaio e Spohr, T. C., Martinez, R., da Silva, E. F., Neto, V. M., and Gomes, F. C. (2002). Neuro-glia interaction effects on GFAP gene: a novel role for transforming growth factor-beta1. Eur. J. Neurosci. 16, 2059–2069. doi: 10.1046/j.1460-9568.2002.02283.x
Dean, J. L., Sully, G., Clark, A. R., and Saklatvala, J. (2004). The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 16, 1113–1121. doi: 10.1016/j.cellsig.2004.04.006
Dell’Albani, P., Santangelo, R., Torrisi, L., Nicoletti, V. G., de Vellis, J., and Giuffrida Stella, A. M. (2001). JAK/STAT signaling pathway mediates cytokine-induced iNOS expression in primary astroglial cell cultures. J. Neurosci. Res. 65, 417–424. doi: 10.1002/jnr.1169
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi: 10.1038/nature02006
DeWitt, D. A., Perry, G., Cohen, M., Doller, C., and Silver, J. (1998). Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp. Neurol. 149, 329–340. doi: 10.1006/exnr.1997.6738
Dhandapani, K. M., and Brann, D. W. (2003). Transforming growth factor-beta: a neuroprotective factor in cerebral ischemia. Cell Biochem. Biophys. 39, 13–22. doi: 10.1385/cbb:39:1:13
Di Filippo, M., Sarchielli, P., Picconi, B., and Calabresi, P. (2008). Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol. Sci. 29, 402–412. doi: 10.1016/j.tips.2008.06.005
Dias, J. M., Alekseenko, Z., Applequist, J. M., and Ericson, J. (2014). Tgfbeta signaling regulates temporal neurogenesis and potency of neural stem cells in the CNS. Neuron 84, 927–939. doi: 10.1016/j.neuron.2014.10.033
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. doi: 10.1038/nature11082
Docagne, F., Nicole, O., Gabriel, C., Fernandez-Monreal, M., Lesne, S., Ali, C., et al. (2002). Smad3-dependent induction of plasminogen activator inhibitor-1 in astrocytes mediates neuroprotective activity of transforming growth factor-beta 1 against NMDA-induced necrosis. Mol. Cell. Neurosci. 21, 634–644. doi: 10.1006/mcne.2002.1206
Doyle, K. P., Cekanaviciute, E., Mamer, L. E., and Buckwalter, M. S. (2010). TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J. Neuroinflammation 7:62. doi: 10.1186/1742-2094-7-62
Draude, G., and Lorenz, R. L. (2000). TGF-beta1 downregulates CD36 and scavenger receptor A but upregulates LOX-1 in human macrophages. Am. J. Physiol. Heart Circ. Physiol. 278, H1042–H1048.
El Khoury, J., Hickman, S. E., Thomas, C. A., Cao, L., Silverstein, S. C., and Loike, J. D. (1996). Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 382, 716–719. doi: 10.1038/382716a0
Eljaschewitsch, E., Witting, A., Mawrin, C., Lee, T., Schmidt, P. M., Wolf, S., et al. (2006). The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 49, 67–79. doi: 10.1016/j.neuron.2005.11.027
Eyupoglu, I. Y., Bechmann, I., and Nitsch, R. (2003). Modification of microglia function protects from lesion-induced neuronal alterations and promotes sprouting in the hippocampus. FASEB J. 17, 1110–1111. doi: 10.1096/fj.02-0825fje
Feijoo, C., Campbell, D. G., Jakes, R., Goedert, M., and Cuenda, A. (2005). Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J. Cell Sci. 118, 397–408. doi: 10.1242/jcs.01655
Floden, A. M., and Combs, C. K. (2011). Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J. Alzheimers. Dis. 25, 279–293. doi: 10.3233/JAD-2011-101014
Flores, B., and von Bernhardi, R. (2012). Transforming growth factor beta1 modulates amyloid beta-induced glial activation through the Smad3-dependent induction of MAPK phosphatase-1. J. Alzheimers Dis. 32, 417–429. doi: 10.3233/JAD-2012-120721
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N Y Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Frenkel, D., Wilkinson, K., Zhao, L., Hickman, S. E., Means, T. K., Puckett, L., et al. (2013). Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 4:2030. doi: 10.1038/ncomms3030
Gabuzda, D., Busciglio, J., Chen, L. B., Matsudaira, P., and Yankner, B. A. (1994). Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 269, 13623–13628.
Gao, H. M., and Hong, J. S. (2008). Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365. doi: 10.1016/j.it.2008.05.002
George, O., Parducz, A., Dupret, D., Kharouby, M., Le Moal, M., Piazza, P. V., et al. (2006). Smad-dependent alterations of PPT cholinergic neurons as a pathophysiological mechanism of age-related sleep-dependent memory impairments. Neurobiol. Aging 27, 1848–1858. doi: 10.1016/j.neurobiolaging.2005.10.014
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Godbout, J. P., Chen, J., Abraham, J., Richwine, A. F., Berg, B. M., Kelley, K. W., et al. (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 19, 1329–1331. doi: 10.1096/fj.05-3776fje
Godoy, B., Murgas, P., Tichauer, J., and Von Bernhardi, R. (2012). Scavenger receptor class A ligands induce secretion of IL1beta and exert a modulatory effect on the inflammatory activation of astrocytes in culture. J. Neuroimmunol. 251, 6–13. doi: 10.1016/j.jneuroim.2012.06.004
Gordon, S. (2003). Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35. doi: 10.1038/nri978
Gough, D. J., Levy, D. E., Johnstone, R. W., and Clarke, C. J. (2008). IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. 19, 383–394. doi: 10.1016/j.cytogfr.2008.08.004
Handattu, S. P., Garber, D. W., Monroe, C. E., Van Groen, T., Kadish, I., Nayyar, G., et al. (2009). Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 34, 525–534. doi: 10.1016/j.nbd.2009.03.007
Handattu, S. P., Monroe, C. E., Nayyar, G., Palgunachari, M. N., Kadish, I., Van Groen, T., et al. (2013). In vivo and in vitro effects of an apolipoprotein e mimetic peptide on amyloid-beta pathology. J. Alzheimers Dis. 36, 335–347. doi: 10.3233/JAD-122377
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hayashi, Y., Yoshida, M., Yamato, M., Ide, T., Wu, Z., Ochi-Shindou, M., et al. (2008). Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J. Neurosci. 28, 8624–8634. doi: 10.1523/jneurosci.1957-08.2008
He, P., Zhong, Z., Lindholm, K., Berning, L., Lee, W., Lemere, C., et al. (2007). Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 178, 829–841. doi: 10.1084/jem2048oia23
Heldin, C. H., and Moustakas, A. (2012). Role of Smads in TGFbeta signaling. Cell Tissue Res. 347, 21–36. doi: 10.1007/s00441-011-1190-x
Helenius, M., Hanninen, M., Lehtinen, S. K., and Salminen, A. (1996). Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-kappa B. Biochem. J. 318, 603–608. doi: 10.1042/bj3180603
Hemmer, B., Archelos, J. J., and Hartung, H. P. (2002). New concepts in the immunopathogenesis of multiple sclerosis. Nat. Rev. Neurosci. 3, 291–301. doi: 10.1038/nrn784
Heneka, M. T., and O’banion, M. K. (2007). Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 184, 69–91. doi: 10.1016/j.jneuroim.2006.11.017
Heneka, M. T., O’banion, M. K., Terwel, D., and Kummer, M. P. (2010). Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 117, 919–947. doi: 10.1007/s00702-010-0438-z
Herrera-Molina, R., and Von Bernhardi, R. (2005). Transforming growth factor-beta 1 produced by hippocampal cells modulates microglial reactivity in culture. Neurobiol. Dis. 19, 229–236. doi: 10.1016/j.nbd.2005.01.003
Herrera-Molina, R., Flores, B., Orellana, J. A., and Von Bernhardi, R. (2012). Modulation of interferon-gamma-induced glial cell activation by transforming growth factor beta1: a role for STAT1 and MAPK pathways. J. Neurochem. 123, 113–123. doi: 10.1111/j.1471-4159.2012.07887.x
Herrup, K. (2010). Reimagining Alzheimer’s disease—an age-based hypothesis. J. Neurosci. 30, 16755–16762. doi: 10.1523/jneurosci.4521-10.2010
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/jneurosci.0616-08.2008
Hinks, G. L., and Franklin, R. J. (2000). Delayed changes in growth factor gene expression during slow remyelination in the CNS of aged rats. Mol. Cell. Neurosci. 16, 542–556. doi: 10.1006/mcne.2000.0897
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. doi: 10.1016/s1474-4422(09)70062-6
Holwerda, S. J., and De Laat, W. (2013). CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20120369. doi: 10.1098/rstb.2012.0369
Honda, M., Akiyama, H., Yamada, Y., Kondo, H., Kawabe, Y., Takeya, M., et al. (1998). Immunohistochemical evidence for a macrophage scavenger receptor in Mato cells and reactive microglia of ischemia and Alzheimer’s disease. Biochem. Biophys. Res. Commun. 245, 734–740. doi: 10.1006/bbrc.1998.8120
Hu, S., Sheng, W. S., Peterson, P. K., and Chao, C. C. (1995). Cytokine modulation of murine microglial cell superoxide production. Glia 13, 45–50. doi: 10.1002/glia.440130106
Imoto, S., Sugiyama, K., Muromoto, R., Sato, N., Yamamoto, T., and Matsuda, T. (2003). Regulation of transforming growth factor-beta signaling by protein inhibitor of activated STAT, PIASy through Smad3. J. Biol. Chem. 278, 34253–34258. doi: 10.1074/jbc.m304961200
Ishida, Y., Kondo, T., Takayasu, T., Iwakura, Y., and Mukaida, N. (2004). The essential involvement of cross-talk between IFN-gamma and TGF-beta in the skin wound-healing process. J. Immunol. 172, 1848–1855. doi: 10.4049/jimmunol.172.3.1848
Jang, B. C., Lim, K. J., Suh, M. H., Park, J. G., and Suh, S. I. (2007). Dexamethasone suppresses interleukin-1beta-induced human beta-defensin 2 mRNA expression: involvement of p38 MAPK, JNK, MKP-1 and NF-kappaB transcriptional factor in A549 cells. FEMS Immunol. Med. Microbiol. 51, 171–184. doi: 10.1111/j.1574-695x.2007.00293.x
Jellinger, K. A. (2006). Alzheimer 100—highlights in the history of Alzheimer research. J. Neural Transm. 113, 1603–1623. doi: 10.1007/s00702-006-0578-3
Johns, L. D., Babcock, G., Green, D., Freedman, M., Sriram, S., and Ransohoff, R. M. (1992). Transforming growth factor-beta 1 differentially regulates proliferation and MHC class-II antigen expression in forebrain and brainstem astrocyte primary cultures. Brain Res. 585, 229–236. doi: 10.1016/0006-8993(92)91211-v
Jono, H., Xu, H., Kai, H., Lim, D. J., Kim, Y. S., Feng, X. H., et al. (2003). Transforming growth factor-beta-Smad signaling pathway negatively regulates nontypeable Haemophilus influenzae-induced MUC5AC mucin transcription via mitogen-activated protein kinase (MAPK) phosphatase-1-dependent inhibition of p38 MAPK. J. Biol. Chem. 278, 27811–27819. doi: 10.1074/jbc.m301773200
Juraskova, B., Andrys, C., Holmerova, I., Solichova, D., Hrnciarikova, D., Vankova, H., et al. (2010). Transforming growth factor beta and soluble endoglin in the healthy senior and in Alzheimer’s disease patients. J. Nutr. Health Aging 14, 758–761. doi: 10.1007/s12603-010-0325-1
Kandasamy, M., Lehner, B., Kraus, S., Sander, P. R., Marschallinger, J., Rivera, F. J., et al. (2014). TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons. J. Cell. Mol. Med. 18, 1444–1459. doi: 10.1111/jcmm.12298
Kassel, O., Sancono, A., Kratzschmar, J., Kreft, B., Stassen, M., and Cato, A. C. (2001). Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 20, 7108–7116. doi: 10.1093/emboj/20.24.7108
Katayama, T., Sakaguchi, E., Komatsu, Y., Oguma, T., Uehara, T., and Minami, M. (2010). Sustained activation of ERK signaling in astrocytes is critical for neuronal injury-induced monocyte chemoattractant protein-1 production in rat corticostriatal slice cultures. Eur. J. Neurosci. 31, 1359–1367. doi: 10.1111/j.1460-9568.2010.07160.x
Katsel, P. L., Davis, K. L., and Haroutunian, V. (2005). Large-scale microarray studies of gene expression in multiple regions of the brain in schizophrenia and Alzheimer’s disease. Int. Rev. Neurobiol. 63, 41–82. doi: 10.1016/s0074-7742(05)63003-6
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kim, S. U., and de Vellis, J. (2005). Microglia in health and disease. J. Neurosci. Res. 81, 302–313. doi: 10.1002/jnr.20562
Kim, S. W., Yoon, S. J., Chuong, E., Oyolu, C., Wills, A. E., Gupta, R., et al. (2011). Chromatin and transcriptional signatures for Nodal signaling during endoderm formation in hESCs. Dev. Biol. 357, 492–504. doi: 10.1016/j.ydbio.2011.06.009
King, E. M., Holden, N. S., Gong, W., Rider, C. F., and Newton, R. (2009). Inhibition of NF-kappaB-dependent transcription by MKP-1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J. Biol. Chem. 284, 26803–26815. doi: 10.1074/jbc.M109.028381
Klegeris, A., Bissonnette, C. J., and McGeer, P. L. (2005). Modulation of human microglia and THP-1 cell toxicity by cytokines endogenous to the nervous system. Neurobiol. Aging 26, 673–682. doi: 10.1016/j.neurobiolaging.2004.06.012
Kraus, S., Lehner, B., Reichhart, N., Couillard-Despres, S., Wagner, K., Bogdahn, U., et al. (2013). Transforming growth factor-beta1 primes proliferating adult neural progenitor cells to electrophysiological functionality. Glia 61, 1767–1783. doi: 10.1002/glia.22551
Krausgruber, T., Blazek, K., Smallie, T., Alzabin, S., Lockstone, H., Sahgal, N., et al. (2011). IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238. doi: 10.1038/ni.1990
Kreutzberg, G. W. (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318. doi: 10.1016/0166-2236(96)10049-7
Larbi, A., Franceschi, C., Mazzatti, D., Solana, R., Wikby, A., and Pawelec, G. (2008). Aging of the immune system as a prognostic factor for human longevity. Physiology (Bethesda) 23, 64–74. doi: 10.1152/physiol.00040.2007
Le, Y., Iribarren, P., Gong, W., Cui, Y., Zhang, X., and Wang, J. M. (2004). TGF-beta1 disrupts endotoxin signaling in microglial cells through Smad3 and MAPK pathways. J. Immunol. 173, 962–968. doi: 10.4049/jimmunol.173.2.962
Lee, C. K., Weindruch, R., and Prolla, T. A. (2000). Gene-expression profile of the ageing brain in mice. Nat. Genet. 25, 294–297. doi: 10.1038/77046
Lee, H. G., Ueda, M., Zhu, X., Perry, G., and Smith, M. A. (2006). Ectopic expression of phospho-Smad2 in Alzheimer’s disease: uncoupling of the transforming growth factor-beta pathway? J. Neurosci. Res. 84, 1856–1861. doi: 10.1002/jnr.21072
Lee, J. H., Woo, J. H., Woo, S. U., Kim, K. S., Park, S. M., Joe, E. H., et al. (2008). The 15-deoxy-delta 12,14-prostaglandin J2 suppresses monocyte chemoattractant protein-1 expression in IFN-gamma-stimulated astrocytes through induction of MAPK phosphatase-1. J. Immunol. 181, 8642–8649. doi: 10.4049/jimmunol.181.12.8642
Lee, S. Y., Ha, T. Y., Son, D. J., Kim, S. R., and Hong, J. T. (2005a). Effect of sesaminol glucosides on beta-amyloid-induced PC12 cell death through antioxidant mechanisms. Neurosci. Res. 52, 330–341. doi: 10.1016/j.neures.2005.04.003
Lee, S. Y., Lee, J. W., Lee, H., Yoo, H. S., Yun, Y. P., Oh, K. W., et al. (2005b). Inhibitory effect of green tea extract on beta-amyloid-induced PC12 cell death by inhibition of the activation of NF-kappaB and ERK/p38 MAP kinase pathway through antioxidant mechanisms. Brain Res. Mol. Brain Res. 140, 45–54. doi: 10.1016/j.molbrainres.2005.07.009
Lesne, S., Docagne, F., Gabriel, C., Liot, G., Lahiri, D. K., Bueé, L., et al. (2003). Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. J. Biol. Chem. 278, 18408–18418. doi: 10.1074/jbc.m300819200
Letiembre, M., Hao, W., Liu, Y., Walter, S., Mihaljevic, I., Rivest, S., et al. (2007). Innate immune receptor expression in normal brain aging. Neuroscience 146, 248–254. doi: 10.1016/j.neuroscience.2007.01.004
Li, L., Lu, J., Tay, S. S., Moochhala, S. M., and He, B. P. (2007). The function of microglia, either neuroprotection or neurotoxicity, is determined by the equilibrium among factors released from activated microglia in vitro. Brain Res. 1159, 8–17. doi: 10.1016/j.brainres.2007.04.066
Li, R., Yang, L., Lindholm, K., Konishi, Y., Yue, X., Hampel, H., et al. (2004). Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J. Neurosci. 24, 1760–1771. doi: 10.1523/jneurosci.4580-03.2004
Lieb, K., Engels, S., and Fiebich, B. L. (2003). Inhibition of LPS-induced iNOS and NO synthesis in primary rat microglial cells. Neurochem. Int. 42, 131–137. doi: 10.1016/s0197-0186(02)00076-1
Lifshitz, V., Weiss, R., Levy, H., and Frenkel, D. (2013). Scavenger receptor A deficiency accelerates cerebrovascular amyloidosis in an animal model. J. Mol. Neurosci. 50, 198–203. doi: 10.1007/s12031-012-9909-z
Liu, B., Wang, K., Gao, H. M., Mandavilli, B., Wang, J. Y., and Hong, J. S. (2001). Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J. Neurochem. 77, 182–189. doi: 10.1046/j.1471-4159.2001.00216.x
Liu, F. (2006). Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 17, 9–17. doi: 10.1016/j.cytogfr.2005.09.010
Liu, X., Wu, Z., Hayashi, Y., and Nakanishi, H. (2012). Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience 216, 133–142. doi: 10.1016/j.neuroscience.2012.04.050
Liu, Y., Shepherd, E. G., and Nelin, L. D. (2007). MAPK phosphatases–regulating the immune response. Nat. Rev. Immunol. 7, 202–212. doi: 10.1038/nri2035
Lucas, M., Zhang, X., Prasanna, V., and Mosser, D. M. (2005). ERK activation following macrophage FcgammaR ligation leads to chromatin modifications at the IL-10 locus. J. Immunol. 175, 469–477. doi: 10.4049/jimmunol.175.1.469
Lue, L. F., Kuo, Y. M., Beach, T., and Walker, D. G. (2010). Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Mol. Neurobiol. 41, 115–128. doi: 10.1007/s12035-010-8106-8
Lukiw, W. J. (2004). Gene expression profiling in fetal, aged and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem. Res. 29, 1287–1297. doi: 10.1023/b:nere.0000023615.89699.63
Luterman, J. D., Haroutunian, V., Yemul, S., Ho, L., Purohit, D., Aisen, P. S., et al. (2000). Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch. Neurol. 57, 1153–1160. doi: 10.1001/archneur.57.8.1153
Lyons, A., Murphy, K. J., Clarke, R., and Lynch, M. A. (2011). Atorvastatin prevents age-related and amyloid-beta-induced microglial activation by blocking interferon-gamma release from natural killer cells in the brain. J. Neuroinflammation 8:27. doi: 10.1186/1742-2094-8-27
Ma, Y. P., Ma, M. M., Ge, S., Guo, R. B., Zhang, H. J., Frey, W. H., II, et al. (2007). Intranasally delivered TGF-beta1 enters brain and regulates gene expressions of its receptors in rats. Brain Res. Bull. 74, 271–277. doi: 10.1016/j.brainresbull.2007.06.021
Marcus, J. S., Karackattu, S. L., Fleegal, M. A., and Sumners, C. (2003). Cytokine-stimulated inducible nitric oxide synthase expression in astroglia: role of Erk mitogen-activated protein kinase and NF-κB. Glia 41, 152–160. doi: 10.1002/glia.10168
Martinez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461. doi: 10.2741/2692
Mazur-Kolecka, B., Frackowiak, J., Le Vine, H., III, Haske, T., Evans, L., Sukontasup, T., et al. (2003). TGFbeta1 enhances formation of cellular Abeta/apoE deposits in vascular myocytes. Neurobiol. Aging 24, 355–364. doi: 10.1016/s0197-4580(02)00095-7
McCartney-Francis, N. L., and Wahl, S. M. (2002). Dysregulation of IFN-gamma signaling pathways in the absence of TGF-beta 1. J. Immunol. 169, 5941–5947. doi: 10.4049/jimmunol.169.10.5941
Medeiros, R., Prediger, R. D., Passos, G. F., Pandolfo, P., Duarte, F. S., Franco, J. L., et al. (2007). Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J. Neurosci. 27, 5394–5404. doi: 10.1523/jneurosci.5047-06.2007
Michael, D. R., Salter, R. C., and Ramji, D. P. (2012). TGF-beta inhibits the uptake of modified low density lipoprotein by human macrophages through a Smad-dependent pathway: a dominant role for Smad-2. Biochim. Biophys. Acta 1822, 1608–1616. doi: 10.1016/j.bbadis.2012.06.002
Milner, R. (2009). Microglial expression of alphavbeta3 and alphavbeta5 integrins is regulated by cytokines and the extracellular matrix: beta5 integrin null microglia show no defects in adhesion or MMP-9 expression on vitronectin. Gila 57, 714–723. doi: 10.1002/glia.20799
Mittaud, P., Labourdette, G., Zingg, H., and Guenot-Di Scala, D. (2002). Neurons modulate oxytocin receptor expression in rat cultured astrocytes: involvement of TGF-beta and membrane components. Glia 37, 169–177. doi: 10.1002/glia.10029
Mocali, A., Cedrola, S., Della Malva, N., Bontempelli, M., Mitidieri, V. A., Bavazzano, A., et al. (2004). Increased plasma levels of soluble CD40, together with the decrease of TGF beta 1, as possible differential markers of Alzheimer disease. Exp. Gerontol. 39, 1555–1561. doi: 10.1016/j.exger.2004.07.007
Mosser, D. M. (2003). The many faces of macrophage activation. J. Leukoc. Biol. 73, 209–212. doi: 10.1189/jlb.0602325
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Mullen, A. C., Orlando, D. A., Newman, J. J., Lovén, J., Kumar, R. M., Bilodeau, S., et al. (2011). Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 147, 565–576. doi: 10.1016/j.cell.2011.08.050
Munoz, L., and Ammit, A. J. (2010). Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 58, 561–568. doi: 10.1016/j.neuropharm.2009.11.010
Munoz, L., Ralay Ranaivo, H., Roy, S. M., Hu, W., Craft, J. M., McNamara, L. K., et al. (2007). A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflammation 4:21. doi: 10.1186/1742-2094-4-21
Murgas, P., Cornejo, F. A., Merino, G., and Von Bernhardi, R. (2014). SR-A regulates the inflammatory activation of astrocytes. Neurotox. Res. 25, 68–80. doi: 10.1007/s12640-013-9432-1
Murgas, P., Godoy, B., and Von Bernhardi, R. (2012). Abeta potentiates inflammatory activation of glial cells induced by scavenger receptor ligands and inflammatory mediators in culture. Neurotox. Res. 22, 69–78. doi: 10.1007/s12640-011-9306-3
Muriach, M., Flores-Bellver, M., Romero, F. J., and Barcia, J. M. (2014). Diabetes and the brain: oxidative stress, inflammation and autophagy. Oxid. Med. Cell. Longev. 2014:102158. doi: 10.1155/2014/102158
Nakajima, K., Tohyama, Y., Maeda, S., Kohsaka, S., and Kurihara, T. (2007). Neuronal regulation by which microglia enhance the production of neurotrophic factors for GABAergic, catecholaminergic and cholinergic neurons. Neurochem. Int. 50, 807–820. doi: 10.1016/j.neuint.2007.02.006
Nakamura, Y. (2002). Regulating factors for microglial activation. Biol. Pharm. Bull. 25, 945–953. doi: 10.1248/bpb.25.945
Neumann, H., Kotter, M. R., and Franklin, R. J. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–295. doi: 10.1093/brain/awn109
Ng, Y. K., Yong, V. W., and Ling, E. A. (1999). Microglial reaction in some CNS nuclei following nerves transection in BALB/c and interferon-gamma gene knockout mice. Neurosci. Lett. 262, 207–210. doi: 10.1016/s0304-3940(99)00076-2
Nguyen, M. D., Julien, J. P., and Rivest, S. (2002). Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 3, 216–227. doi: 10.1038/nrn752
Nikolic, D., Calderon, L., Du, L., and Post, S. R. (2011). SR-A ligand M-CSF dynamically regulate SR-A expression and function in primary macrophages via p38 MAPK activation. BMC Immunol. 12:37. doi: 10.1186/1471-2172-12-37
Nishimura, N., Harada-Shiba, M., Tajima, S., Sugano, R., Yamamura, T., Qiang, Q. Z., et al. (1998). Acquisition of secretion of transforming growth factor-beta 1 leads to autonomous suppression of scavenger receptor activity in a monocyte-macrophage cell line, THP-1. J. Biol. Chem. 273, 1562–1567. doi: 10.1074/jbc.273.3.1562
O’Neill, L. A., and Kaltschmidt, C. (1997). NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 20, 252–258. doi: 10.1016/s0166-2236(96)01035-1
Ong, C. T., and Corces, V. G. (2014). CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 15, 234–246. doi: 10.1038/nrg3663
Pannaccione, A., Secondo, A., Scorziello, A., Cali, G., Taglialatela, M., and Annunziato, L. (2005). Nuclear factor-kappaB activation by reactive oxygen species mediates voltage-gated K+ current enhancement by neurotoxic beta-amyloid peptides in nerve growth factor-differentiated PC-12 cells and hippocampal neurones. J. Neurochem. 94, 572–586. doi: 10.1111/j.1471-4159.2005.03075.x
Paresce, D. M., Chung, H., and Maxfield, F. R. (1997). Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J. Biol. Chem. 272, 29390–29397. doi: 10.1074/jbc.272.46.29390
Paresce, D. M., Ghosh, R. N., and Maxfield, F. R. (1996). Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 17, 553–565. doi: 10.1016/s0896-6273(00)80187-7
Paris, D., Patel, N., Quadros, A., Linan, M., Bakshi, P., Ait-Ghezala, G., et al. (2007). Inhibition of Abeta production by NF-kappaB inhibitors. Neurosci. Lett. 415, 11–16. doi: 10.1016/j.neulet.2006.12.029
Pawate, S., Shen, Q., Fan, F., and Bhat, N. R. (2004). Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J. Neurosci. Res. 77, 540–551. doi: 10.1002/jnr.20180
Pei, J. J., Braak, E., Braak, H., Grundke-Iqbal, I., Iqbal, K., Winblad, B., et al. (2001). Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimers. Dis. 3, 41–48.
Pineda, J. R., Daynac, M., Chicheportiche, A., Cebrian-Silla, A., Sii Felice, K., Garcia-Verdugo, J. M., et al. (2013). Vascular-derived TGF-beta increases in the stem cell niche and perturbs neurogenesis during aging and following irradiation in the adult mouse brain. EMBO Mol. Med. 5, 548–562. doi: 10.1002/emmm.201202197
Platanias, L. C. (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. doi: 10.1038/nri1604
Porta, C., Rimoldi, M., Raes, G., Brys, L., Ghezzi, P., Di Liberto, D., et al. (2009). Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. U S A 106, 14978–14983. doi: 10.1073/pnas.0809784106
Prehn, J. H., Bindokas, V. P., Jordan, J., Galindo, M. F., Ghadge, G. D., Roos, R. P., et al. (1996). Protective effect of transforming growth factor-beta 1 on beta-amyloid neurotoxicity in rat hippocampal neurons. Mol. Pharmacol. 49, 319–328.
Qian, J. J., Cheng, Y. B., Yang, Y. P., Mao, C. J., Qin, Z. H., Li, K., et al. (2008). Differential effects of overexpression of wild-type and mutant human α-synuclein on MPP+-induced neurotoxicity in PC12 cells. Neurosci Lett. 435, 142–146. doi: 10.1016/j.neulet.2008.02.021
Qin, H., Chan, M. W., Liyanarachchi, S., Balch, C., Potter, D., Souriraj, I. J., et al. (2009). An integrative ChIP-chip and gene expression profiling to model SMAD regulatory modules. BMC Syst. Biol. 3:73. doi: 10.1186/1752-0509-3-73
Qin, L., Li, G., Qian, X., Liu, Y., Wu, X., Liu, B., et al. (2005). Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 52, 78–84. doi: 10.1002/glia.20225
Rahimi, R. A., and Leof, E. B. (2007). TGF-beta signaling: a tale of two responses. J. Cell. Biochem. 102, 593–608. doi: 10.1002/jcb.21501
Raj, D. D., Jaarsma, D., Holtman, I. R., Olah, M., Ferreira, F. M., Schaafsma, W., et al. (2014). Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol. Aging 35, 2147–2160. doi: 10.1016/j.neurobiolaging.2014.03.025
Ramírez, G., Rey, S., and Von Bernhardi, R. (2008). Proinflammatory stimuli are needed for induction of microglial cell-mediated AbetaPP_244-C and Abeta-neurotoxicity in hippocampal cultures. J. Alzheimers. Dis. 15, 45–59.
Ramírez, G., Toro, R., Döbeli, H., and Von Bernhardi, R. (2005). Protection of rat primary hippocampal cultures from A beta cytotoxicity by pro-inflammatory molecules is mediated by astrocytes. Neurobiol. Dis. 19, 243–254. doi: 10.1016/j.nbd.2005.01.007
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Rivest, S. (2009). Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439. doi: 10.1038/nri2565
Rogers, J., Strohmeyer, R., Kovelowski, C. J., and Li, R. (2002). Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 40, 260–269. doi: 10.1002/glia.10153
Rosales-Corral, S., Reiter, R. J., Tan, D. X., Ortiz, G. G., and Lopez-Armas, G. (2010). Functional aspects of redox control during neuroinflammation. Antioxid. Redox Signal. 13, 193–247. doi: 10.1089/ars.2009.2629
Ross, K. R., Corey, D. A., Dunn, J. M., and Kelley, T. J. (2007). SMAD3 expression is regulated by mitogen-activated protein kinase kinase-1 in epithelial and smooth muscle cells. Cell. Signal. 19, 923–931. doi: 10.1016/j.cellsig.2006.11.008
Ross, S., and Hill, C. S. (2008). How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 40, 383–408. doi: 10.1016/j.biocel.2007.09.006
Rossner, S., Lange-Dohna, C., Zeitschel, U., and Perez-Polo, J. R. (2005). Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J. Neurochem. 92, 226–234. doi: 10.1111/j.1471-4159.2004.02857.x
Saha, R. N., Jana, M., and Pahan, K. (2007). MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 179, 7101–7109. doi: 10.4049/jimmunol.179.10.7101
Satoh, T., and Lipton, S. A. (2007). Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 30, 37–45. doi: 10.1016/j.tins.2006.11.004
Satoh, T., Takeuchi, O., Vandenbon, A., Yasuda, K., Tanaka, Y., Kumagai, Y., et al. (2010). The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 11, 936–944. doi: 10.1038/ni.1920
Saud, K., Herrera-Molina, R., and Von Bernhardi, R. (2005). Pro- and anti-inflammatory cytokines regulate the ERK pathway: implication of the timing for the activation of microglial cells. Neurotox. Res. 8, 277–287. doi: 10.1007/bf03033981
Schmierer, B., and Hill, C. S. (2007). TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982. doi: 10.1038/nrm2297
Schwab, C., and McGeer, P. L. (2008). Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J. Alzheimers Dis. 13, 359–369.
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Siegenthaler, J. A., and Miller, M. W. (2004). Transforming growth factor beta1 modulates cell migration in rat cortex: effects of ethanol. Cereb. Cortex 14, 791–802. doi: 10.1093/cercor/bhh039
Sierra, A., Gottfried-Blackmore, A. C., Mcewen, B. S., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi: 10.1002/glia.20468
Simonsson, M., Kanduri, M., Gronroos, E., Heldin, C. H., and Ericsson, J. (2006). The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. J. Biol. Chem. 281, 39870–39880. doi: 10.1074/jbc.m607868200
Singh, T., and Newman, A. B. (2011). Inflammatory markers in population studies of aging. Ageing Res. Rev. 10, 319–329. doi: 10.1016/j.arr.2010.11.002
Spriggs, D. R., Deutsch, S., and Kufe, D. W. (1992). Genomic structure, induction and production of TNF-alpha. Immunol. Ser. 56, 3–34.
Streit, W. J. (2002). Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40, 133–139. doi: 10.1002/glia.10154
Sun, A., Liu, M., Nguyen, X. V., and Bing, G. (2003). P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 183, 394–405. doi: 10.1016/s0014-4886(03)00180-8
Tapia-González, S., Muñoz, M. D., Cuartero, M. I., and Sánchez-Capelo, A. (2013). Smad3 is required for the survival of proliferative intermediate progenitor cells in the dentate gyrus of adult mice. Cell Commun. Signal. 11:93. doi: 10.1186/1478-811x-11-93
Tarkowski, E., Issa, R., Sjögren, M., Wallin, A., Blennow, K., Tarkowski, A., et al. (2002). Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 23, 237–243. doi: 10.1016/s0197-4580(01)00285-8
Taylor, R. C., and Dillin, A. (2011). Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3:a004440. doi: 10.1101/cshperspect.a004440
Tesseur, I., and Wyss-Coray, T. (2006). A role for TGF-beta signaling in neurodegeneration: evidence from genetically engineered models. Curr. Alzheimer Res. 3, 505–513. doi: 10.2174/156720506779025297
Tesseur, I., Zou, K., Esposito, L., Bard, F., Berber, E., Can, J. V., et al. (2006). Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Invest. 116, 3060–3069. doi: 10.1172/jci27341
Tichauer, J., Saud, K., and von Bernhardi, R. (2007). Modulation by astrocytes of microglial cell-mediated neuroinflammation: effect on the activation of microglial signaling pathways. Neuroimmunomodulation 14, 168–174. doi: 10.1159/000110642
Tichauer, J. E., and von Bernhardi, R. (2012). Transforming growth factor-beta stimulates beta amyloid uptake by microglia through Smad3-dependent mechanisms. J. Neurosci. Res. 90, 1970–1980. doi: 10.1002/jnr.23082
Tichauer, J. E., Flores, B., Soler, B., Eugenin-von Bernhardi, L., Ramírez, G., and von Bernhardi, R. (2014). Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 37, 187–196. doi: 10.1016/j.bbi.2013.12.018
Tong, X. K., and Hamel, E. (2007). Transforming growth factor-beta 1 impairs endothelin-1-mediated contraction of brain vessels by inducing mitogen-activated protein (MAP) kinase phosphatase-1 and inhibiting p38 MAP kinase. Mol. Pharmacol. 72, 1476–1483. doi: 10.1124/mol.107.039602
Town, T., Laouar, Y., Pittenger, C., Mori, T., Szekely, C. A., Tan, J., et al. (2008). Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 14, 681–687. doi: 10.1038/nm1781
Town, T., Nikolic, V., and Tan, J. (2005). The microglial “activation” continuum: from innate to adaptive responses. J. Neuroinflammation 2:24. doi: 10.1186/1742-2094-2-24
Tran, M. H., Yamada, K., Olariu, A., Mizuno, M., Ren, X. H., and Nabeshima, T. (2001). Amyloid beta-peptide induces nitric oxide production in rat hippocampus: association with cholinergic dysfunction and amelioration by inducible nitric oxide synthase inhibitors. FASEB . 15, 1407–1409. doi: 10.1096/fj.00-0719fje
Tu, A. W., and Luo, K. (2007). Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J. Biol. Chem. 282, 21187–21196. doi: 10.1074/jbc.m700085200
Ueberham, U., Lange, P., Ueberham, E., Brückner, M. K., Hartlage-Rubsamen, M., Pannicke, T., et al. (2009). Smad2 isoforms are differentially expressed during mouse brain development and aging. Int. J. Dev. Neurosci. 27, 501–510. doi: 10.1016/j.ijdevneu.2009.04.001
Ueberham, U., Ueberham, E., Gruschka, H., and Arendt, T. (2006). Altered subcellular location of phosphorylated Smads in Alzheimer’s disease. Eur. J. Neurosci. 24, 2327–2334. doi: 10.1111/j.1460-9568.2006.05109.x
Ulloa, L., Doody, J., and Massagué, J. (1999). Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 397, 710–713. doi: 10.1038/17826
Uribe-San Martén, R., Herrera-Molina, R., Olavarréa, L., Ramérez, G., and von Bernhardi, R. (2009). Reduction of beta-amyloid-induced neurotoxicity on hippocampal cell cultures by moderate acidosis is mediated by transforming growth factor beta. Neuroscience 158, 1338–1347. doi: 10.1016/j.neuroscience.2008.11.002
Uylings, H. B., and de Brabander, J. M. (2002). Neuronal changes in normal human aging and Alzheimer’s disease. Brain Cogn. 49, 268–276. doi: 10.1006/brcg.2001.1500
Valerio, A., Boroni, F., Benarese, M., Sarnico, I., Ghisi, V., Bresciani, L. G., et al. (2006). NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur. J. Neurosci. 23, 1711–1720. doi: 10.1111/j.1460-9568.2006.04722.x
Van Eldik, L. J., Thompson, W. L., Ralay Ranaivo, H., Behanna, H. A., and Martin Watterson, D. (2007). Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int. Rev. Neurobiol. 82, 277–296. doi: 10.1016/s0074-7742(07)82015-0
Venema, R. C., Venema, V. J., Eaton, D. C., and Marrero, M. B. (1998). Angiotensin II-induced tyrosine phosphorylation of signal transducers and activators of transcription 1 is regulated by Janus-activated kinase 2 and Fyn kinases and mitogen-activated protein kinase phosphatase 1. J. Biol. Chem. 273, 30795–30800. doi: 10.1074/jbc.273.46.30795
von Bernhardi, R. (2007). Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotox. Res. 12, 215–232. doi: 10.1007/bf03033906
von Bernhardi, R. (2010). Immunotherapy in Alzheimer’s disease: where do we stand? Where should we go? J. Alzheimers Dis. 19, 405–421. doi: 10.3233/JAD-2010-1248
von Bernhardi, R., and Eugenin, J. (2004). Microglial reactivity to beta-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res. 1025, 186–193. doi: 10.1016/j.brainres.2004.07.084
von Bernhardi, R., and Ramírez, G. (2001). Microglia-astrocyte interaction in Alzheimer’s disease: friends or foes for the nervous system? Biol. Res. 34, 123–128. doi: 10.4067/s0716-97602001000200017
von Bernhardi, R., Eugenin-von Bernhardi, L., and Eugenín, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
von Bernhardi, R., Ramírez, G., Toro, R., and Eugenín, J. (2007). Pro-inflammatory conditions promote neuronal damage mediated by Amyloid Precursor Protein and decrease its phagocytosis and degradation by microglial cells in culture. Neurobiol. Dis. 26, 153–164. doi: 10.1016/j.nbd.2006.12.006
von Bernhardi, R., Tichauer, J., and Eugenín-von Bernhardi, L. (2011). Proliferating culture of aged microglia for the study of neurodegenerative diseases. J. Neurosci. Methods 202, 65–69. doi: 10.1016/j.jneumeth.2011.08.027
von Bernhardi, R., Tichauer, J. E., and Eugenín, J. (2010). Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 112, 1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x
Wang, Q., Wu, J., Rowan, M. J., and Anwyl, R. (2005). Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur. J. Neurosci. 22, 2827–2832. doi: 10.1111/j.1460-9568.2005.04457.x
Wang, Q., Zhang, J. Y., Liu, S. J., and Li, H. L. (2008a). Overactivated mitogen-activated protein kinase by anisomycin induces tau hyperphosphorylation. Sheng Li Xue Bao 60, 485–491.
Wang, S. C., Oelze, B., and Schumacher, A. (2008b). Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One 3:e2698. doi: 10.1371/journal.pone.0002698
Wang, T., Qin, L., Liu, B., Liu, Y., Wilson, B., Eling, T. E., et al. (2004). Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J. Neurochem. 88, 939–947. doi: 10.1046/j.1471-4159.2003.02242.x
Weiss, A., and Attisano, L. (2013). The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2, 47–63. doi: 10.1002/wdev.86
Welser-Alves, J. V., and Milner, R. (2013). Microglia are the major source of TNF-alpha and TGF-beta1 in postnatal glial cultures; regulation by cytokines, lipopolysaccharide and vitronectin. Neurochem. Int. 63, 47–53. doi: 10.1016/j.neuint.2013.04.007
Wen, Z., Zhong, Z., and Darnell, J. E., Jr. (1995). Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250. doi: 10.1016/0092-8674(95)90311-9
Werner, F., Jain, M. K., Feinberg, M. W., Sibinga, N. E., Pellacani, A., Wiesel, P., et al. (2000). Transforming growth factor-beta 1 inhibition of macrophage activation is mediated via Smad3. J. Biol. Chem. 275, 36653–36658. doi: 10.1074/jbc.m004536200
Werry, E. L., Enjeti, S., Halliday, G. M., Sachdev, P. S., and Double, K. L. (2010). Effect of age on proliferation-regulating factors in human adult neurogenic regions. J. Neurochem. 115, 956–964. doi: 10.1111/j.1471-4159.2010.06992.x
Wrighton, K. H., Lin, X., and Feng, X. H. (2009). Phospho-control of TGF-beta superfamily signaling. Cell Res. 19, 8–20. doi: 10.1038/cr.2008.327
Wu, Z., Tokuda, Y., Zhang, X. W., and Nakanishi, H. (2008). Age-dependent responses of glial cells and leptomeninges during systemic inflammation. Neurobiol. Dis. 32, 543–551. doi: 10.1016/j.nbd.2008.09.002
Wynne, A. M., Henry, C. J., Huang, Y., Cleland, A., and Godbout, J. P. (2010). Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 24, 1190–1201. doi: 10.1016/j.bbi.2010.05.011
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35, 419–432. doi: 10.1016/s0896-6273(02)00794-8
Wyss-Coray, T., Lin, C., Sanan, D. A., Mucke, L., and Masliah, E. (2000). Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 156, 139–150. doi: 10.1016/s0002-9440(10)64713-x
Wyss-Coray, T., Lin, C., Yan, F., Yu, G. Q., Rohde, M., McConlogue, L., et al. (2001). TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 7, 612–618. doi: 10.1038/87945
Wyss-Coray, T., Masliah, E., Mallory, M., McConlogue, L., Johnson-Wood, K., Lin, C., et al. (1997). Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 389, 603–606. doi: 10.1038/39321
Yamamoto, K., Shimokawa, T., Yi, H., Isobe, K., Kojima, T., Loskutoff, D. J., et al. (2002). Aging accelerates endotoxin-induced thrombosis : increased responses of plasminogen activator inhibitor-1 and lipopolysaccharide signaling with aging. Am. J. Pathol. 161, 1805–1814. doi: 10.1016/S0002-9440(10)64457-4
Yan, J., Zhang, H., Yin, Y., Li, J., Tang, Y., Purkayastha, S., et al. (2014). Obesity- and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat. Med. 20, 1001–1008. doi: 10.1038/nm.3616
Yang, C. N., Shiao, Y. J., Shie, F. S., Guo, B. S., Chen, P. H., Cho, C. Y., et al. (2011). Mechanism mediating oligomeric Abeta clearance by naive primary microglia. Neurobiol. Dis. 42, 221–230. doi: 10.1016/j.nbd.2011.01.005
Ye, S. M., and Johnson, R. W. (1999). Increased interleukin-6 expression by microglia from brain of aged mice. J. Neuroimmunol. 93, 139–148. doi: 10.1016/s0165-5728(98)00217-3
Zhou, X., Lowdon, R. F., Li, D., Lawson, H. A., Madden, P. A., Costello, J. F., et al. (2013). Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods 10, 375–376. doi: 10.1038/nmeth.2440
Zhu, X., Mei, M., Lee, H. G., Wang, Y., Han, J., Perry, G., et al. (2005). P38 activation mediates amyloid-beta cytotoxicity. Neurochem. Res. 30, 791–796. doi: 10.1007/s11064-005-6872-x
Zhu, Y., Culmsee, C., Klumpp, S., and Krieglstein, J. (2004). Neuroprotection by transforming growth factor-beta1 involves activation of nuclear factor-kappaB through phosphatidylinositol-3-OH kinase/Akt and mitogen-activated protein kinase-extracellular-signal regulated kinase1,2 signaling pathways. Neuroscience 123, 897–906. doi: 10.1016/j.neuroscience.2003.10.037
Keywords: aging, cytokines, Glia, MAPK, NFκB, neurodegenerative diseases, neuroinflammation, transforming growth factor-β
Citation: von Bernhardi R, Cornejo F, Parada GE and Eugenín J (2015) Role of TGFβ signaling in the pathogenesis of Alzheimer’s disease. Front. Cell. Neurosci. 9:426. doi: 10.3389/fncel.2015.00426
Received: 16 August 2015; Accepted: 09 October 2015;
Published: 28 October 2015.
Edited by:
James Francis Curtin, Dublin Institute of Technology, IrelandCopyright © 2015 von Bernhardi, Cornejo, Parada and Eugenín. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rommy von Bernhardi, cnZvbmJAbWVkLnB1Yy5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.