 Joseph V. Raimondo
Joseph V. Raimondo Richard J. Burman
Richard J. Burman Arieh A. Katz
Arieh A. Katz Colin J. Akerman
Colin J. Akerman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 21 October 2015
Sec. Cellular Neuropathology
Volume 9 - 2015 | https://doi.org/10.3389/fncel.2015.00419
This article is part of the Research Topic Ion homeostasis in neurological disorders: novel insights on mechanisms of operation, physiological role and interplay between ion transporters, exchangers and channels. View all 10 articles
Changes in membrane voltage brought about by ion fluxes through voltage and transmitter-gated channels represent the basis of neural activity. As such, electrochemical gradients across the membrane determine the direction and driving force for the flow of ions and are therefore crucial in setting the properties of synaptic transmission and signal propagation. Ion concentration gradients are established by a variety of mechanisms, including specialized transporter proteins. However, transmembrane gradients can be affected by ionic fluxes through channels during periods of elevated neural activity, which in turn are predicted to influence the properties of on-going synaptic transmission. Such activity-induced changes to ion concentration gradients are a feature of both physiological and pathological neural processes. An epileptic seizure is an example of severely perturbed neural activity, which is accompanied by pronounced changes in intracellular and extracellular ion concentrations. Appreciating the factors that contribute to these ion dynamics is critical if we are to understand how a seizure event evolves and is sustained and terminated by neural tissue. Indeed, this issue is of significant clinical importance as status epilepticus—a type of seizure that does not stop of its own accord—is a life-threatening medical emergency. In this review we explore how the transmembrane concentration gradient of the six major ions (K+, Na+, Cl−, Ca2+, H+and ) is altered during an epileptic seizure. We will first examine each ion individually, before describing how multiple interacting mechanisms between ions might contribute to concentration changes and whether these act to prolong or terminate epileptic activity. In doing so, we will consider how the availability of experimental techniques has both advanced and restricted our ability to study these phenomena.
Epilepsy is a common, debilitating neurological condition which is characterized by recurrent seizures. These striking events often involve severe disruption of brain function due to increased excitation and synchronization in underlying brain networks. In this review we focus on how seizure-associated changes in transmembrane ion concentration gradients might affect on-going pathological activity within brain networks. We explore how the concentration gradients of six major ions (Na+, K+, Cl−, Ca2+, H+ and ) are regulated under baseline conditions, and how these homeostatic mechanisms may be temporarily overwhelmed in the face of intense ionic fluxes during epileptic seizures.
The term “epileptic seizure” refers to a diverse array of epileptic phenomena from short focal events to generalized tonic-clonic seizures (McCormick and Contreras, 2001). As a result, different seizure activity patterns will be associated with significant variations in the extent and dynamics of the underlying ionic shifts. Brain area and cell type differences contribute to further heterogeneity in ionic responses. In this review we aim to provide a “refresher” on key concepts in the field by integrating findings across different seizure models, neuronal cell types and brain areas to make general points about ion fluxes associated with seizure activity. Unfortunately the format and scope of this review preclude a complete analysis of all the pertinent details associated with this large topic. As we explore ion dynamics during seizure activity, we identify how experimental technologies have either enabled or limited the study of seizure-associated ion dynamics. In our review, we treat each ion in isolation before examining how multiple interacting mechanisms between ions might contribute to concentration changes and whether these act to sustain or terminate epileptic activity. Finally, we discuss how different cell types and subcellular compartments might experience varying seizure-induced changes in ion concentration gradients.
Electrochemical signaling in the brain is mediated by ions moving into or out of cells according to their transmembrane electrochemical gradient. The ionic current generated by opening a membrane conductance for a particular ion is proportional to the difference between the membrane potential at that point in time and the reversal potential for the ion in question (this is termed the ionic driving force). The ionic reversal potential in turn is a function of the transmembrane concentration gradient. Ionic concentration gradients are established by both active and passive mechanisms. Impermeable ion species, particularly impermeable intracellular anions such as proteins, result in an unequal distribution of permeable anions and cations across the cell membrane even in the absence of active ion regulation (the Gibbs-Donnan effect). Further to this, the bulk of cerebral metabolism and energy utilization are spent on active mechanisms, which establish the final transmembrane electrochemical ion gradients that sub serve synaptic transmission and action potential generation. The central player in this process is the Na+ and K+ ATPase active transporter (Na+/K+ ATPase), which utilizes ATP to establish opposite transmembrane gradients for Na+ and K+ (Morth et al., 2007). A multitude of channels then utilize these gradients to modulate the electrical potential across the neuronal membrane (see Table 1). For example, K+ leak channels contribute to the resting membrane potential while voltage-gated Na+ and K+ channels underpin the propagating waves of depolarization and repolarization that constitute action potentials (Goldstein et al., 2001; Wood, 2001; Yellen, 2002).
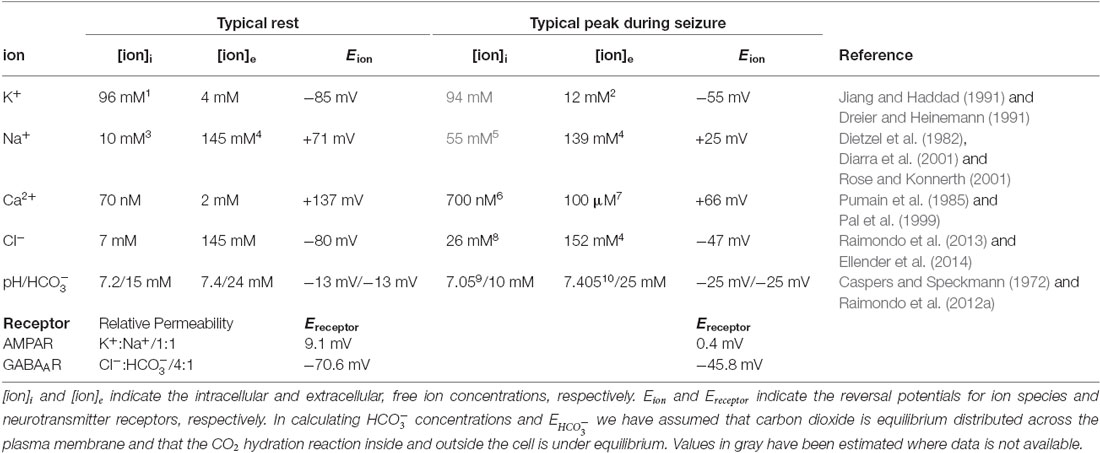
Table 1. Typical ion concentrations at rest and during an epileptic seizure.
The Na+ and K+ gradients generated by the Na+/K+ ATPase also provide the predominant energy substrate for secondary transport of the other ions addressed in this review. For example, in the mature nervous system the intracellular Cl− concentration is kept at lower levels than would be predicted by passive distribution at a given membrane potential due to the action of the cation-chloride cotransporter KCC2. This transporter uses the K+ gradient as the energy source for Cl− extrusion (Kaila et al., 2014a). Interestingly, the transmembrane gradient of Cl− is modulated as a function of development. NKCC1, an alternative cation-chloride cotransporter, which harnesses the Na+ gradient to move Cl− intracellularly, plays a more prominent role in setting the intracellular Cl− concentration gradient in young as opposed to mature tissue (Ben-Ari, 2002; Farrant and Kaila, 2007; Kaila et al., 2014a). Sodium-calcium exchangers (NCXs) employ the Na+ gradient to extrude Ca2+ (Quednau et al., 2004). However, although these low-affinity high-capacity transporters play an important role in Ca2+ extrusion, to achieve the sub 100 nM cytoplasmic Ca2+ concentrations typical of neurons, additional high affinity, ATP driven mechanisms, (namely plasma-membrane and sarcoplasmic reticulum associated Ca2+-ATPase pumps: PMCA, SERCA) are employed (Berridge et al., 2003). Similarly, in the majority of neurons H+ export or import relies on the Na+ gradient to extrude H+ (Na+–H+ exchangers, NHEs) or import (Na+– cotransporters, NBCEs or NDCBEs; Chesler, 2003). The final concentration of is then intimately linked to that of H+ by the reversible reaction of H2O and CO2 to and H+, which is catalyzed by the ubiquitous presence of intracellular and extracellular carbonic anhydrases (Chesler, 2003). Hence, alkaline intracellular environments result in relatively high intracellular concentrations and it is interesting that the expression and activity of different carbonic anydrases can also be developmentally regulated (Ruusuvuori et al., 2004).
The transport mechanisms described above are arguably the most critical in determining transmembrane ion concentrations at rest. In Table 1 we provide a summary of typical resting intracellular and extracellular, free, neuronal ionic concentrations as well as computed reversal potentials for the major ion species addressed in this review (K+, Na+, Cl−, Ca2+, H+ and ). At this stage it may be relevant to point out that in determining the functional effect of ion concentration gradients, it is the free concentration, as opposed to the absolute concentration of the ion, which is important. And the free concentration of an ion can be affected differently between the intracellular and extracellular compartments, as a result of differences in dissociation constants caused by the presence of buffers and organic ions. In addition to resting free ion concentrations, Table 1 also provides values for ion concentrations that could occur during an epileptic seizure. These values are averages that have been derived from different cell-types, brain areas and seizure models. As a result, they are intended to provide only an indication of potential ion concentration dynamics and do not capture the full range of values that are likely to be present in the brain.
In this section we will examine how the transmembrane concentration gradients for each of the six major ion species can change during an epileptic seizure (K+, Na+, Cl−, Ca2+, H+ and ), and how this relates to ongoing seizure dynamics.
Changes in extracellular K+ concentration represent one of the earliest and most thoroughly described examples of activity-induced ion dynamics (Fertziger and Ranck, 1970; Fröhlich et al., 2008). The development of K+ selective electrodes in the early 1970s (Walker, 1971; Singer and Lux, 1975) enabled measurements of extracellular concentrations of K+ ([K+]e) during both physiological and pathological activity. Although early K+ selective electrodes based on liquid ion membrane exchange were also sensitive to various neurotransmitters (Kuramoto and Haber, 1981), the development of liquid membrane electrodes using valinomycin are extremely specific for K+. In addition, these electrodes can be made with tip sizes below 2 μm, which prevents the formation of large dead spaces within the tissue and allows for accurate sensing of K+ in the interstitial space (Ransom et al., 1987; Kaila et al., 1997). Using K+ sensitive elctrodes, Singer and Lux (1975) demonstrated increases in [K+]e that accompanied physiological stimuli in visual cortex in vivo (Singer and Lux, 1975), whilst fluctuations in [K+]e have been shown to be coupled to 1 Hz slow oscillations in ketamine-xylazine anaesthetized cats (Amzica and Steriade, 2000). With regard to pathological activity, in vivo seizures and in vitro seizure-like activity both result in dynamic increases in [K+]e, reaching peak levels of between 9 and 12 mM (Pumain et al., 1985; Yaari et al., 1986; Avoli et al., 1987; Dreier and Heinemann, 1991; Gnatkovsky et al., 2008). It is worth noting that as the extracellular space accounts for only a fifth of total brain volume with the rest being intracellular (Syková and Nicholson, 2008), it is possible that estimates of changes to extracellular ionic concentration might be underestimated in experiments performed in vitro. This is because extracellular ionic changes occurring in vitro can be ameliorated by the large volumes of slice perfusate.
Experiments using K+ selective electrodes have demonstrated that baseline [K+]e is approximately 25-fold lower than [K+]i (typically 4 mM vs. 96 mM, Table 1), which means that even small fluxes of K+ into the extracellular space results in relatively large changes to the transmembrane K+ gradient and consequently the reversal potential for K+. Due to the fact that constitutively activated K+ conductances (Lesage, 2003) are the major determinant in setting the resting membrane potential, extracellular K+ accumulation and accompanying changes to EK+ result in significant membrane depolarization. Network activity and epileptic seizures result in K+ efflux predominantly via intense and prolonged activation of synaptic conductances, particularly by opening glutamate receptors. In addition, membrane depolarization increases K+ flux via both voltage-gated and constitutively active K+ channels. The K+, Cl− cotransporter KCC2, is also responsible for K+ extrusion during intense network activity as intracellular Cl− accumulation provides the thermodynamic gradient for outward K+ transport (Viitanen et al., 2010). Extracellular K+ accumulation is mitigated by a variety of mechanisms including K+ transporters on both neurons and astrocytes (Na+/K+ ATPase, KCC2, NKCC1) as well as passive uptake mechanisms (inward-rectifying K+ channels, Kir) on astrocytes (Kofuji and Newman, 2004; Butt and Kalsi, 2006). What is currently still under considerable debate is whether K+ is shunted between sites of high to low [K+]e through the glial syncytium, a concept termed spatial buffering (Kofuji and Newman, 2004). It is clear however, that under conditions of enhanced activity such as those associated with a seizure, these mechanisms can be overwhelmed and K+ is able to accumulate in the extracellular space. For this reason the “potassium accumulation hypothesis” has long been an attractive, albeit contentious, mechanistic explanation for various aspects of seizure activity (Fröhlich et al., 2008). In this scheme, epileptogenic changes result in enhanced extracellular K+ accumulation during physiological activity, which triggers the onset of a seizure and a further increase in the [K+]e. This results in neurons becoming more depolarized, firing more action potentials and releasing further K+ into the extracellular space. This runaway, positive-feedback cycle is then thought to be arrested when depolarization is so severe that voltage-gated Na+ channels become inactivated and neurons are unable to fire action potentials, a state referred to as depolarization block. Therefore, in this arrangement, extracellular K+ accumulation contributes to the initiation, maintenance and termination of a seizure event.
Although the [K+]e has been observed to increase during seizures, considerable uncertainty still exists as to whether this is a cause, rather than simply an effect of epileptiform activity. Indeed, many aspects of the potassium accumulation hypothesis have remained contentious. Firstly, there has been little experimental verification for a [K+]e threshold for seizure initiation. Secondly, if the potassium accumulation hypothesis were correct, one would expect that K+ would increase during seizure activity and reach a peak as the seizure terminates. In contrast, K+ sensitive microelectrode recordings in vivo and in vitro appear to show that during seizure activity, [K+]e peaks mid seizure, that is, at the end of the tonic phase, before decreasing during the clonic phase (Sypert and Ward, 1974; Dreier and Heinemann, 1991; see Figure 1). Similarly, depolarization block of neurons and a cessation of action potential firing does not appear to occur in tandem with seizure termination (Sypert and Ward, 1974). Although, in at least some cases, seizure cessation has been observed to be associated with the onset of spreading depression, depolarization block and a dramatic increase in [K+]e (Bragin et al., 1997). For these reasons, the potassium accumulation hypothesis has perhaps been viewed as an oversimplification in terms of accounting for seizure termination. However, some of the general conceptual principles involved in the hypothesis are valid. For example, it is likely that any process that results in increasing levels of depolarization may ultimately become inhibitory by causing depolarization block. Furthermore, considerable experimental and computational data has demonstrated the importance of K+ dynamics in the evolution of epileptic seizures. For example, it is well known that raising the extracellular K+ concentration of in vitro preparations, particularly to the 7.5–8.5 mM range, results in epileptiform activity which can closely resemble seizures with distinct tonic and clonic stages (Traynelis and Dingledine, 1988; Jensen et al., 2014). Importantly, recent computational models incorporating K+ ion dynamics have demonstrated that changes to [K+]e can explain the transition between tonic and clonic phases of seizures. Indeed, as is observed experimentally, increases in [K+]e beyond a certain level can result in the network shifting from a tonic to burst firing mode (Fröhlich et al., 2006, 2010). During the tonic firing phase, extracellular K+ once again decreases. In this scheme, extracellular K+ still plays an important role in controling the length of epileptiform activity, albeit in a more subtle fashion.
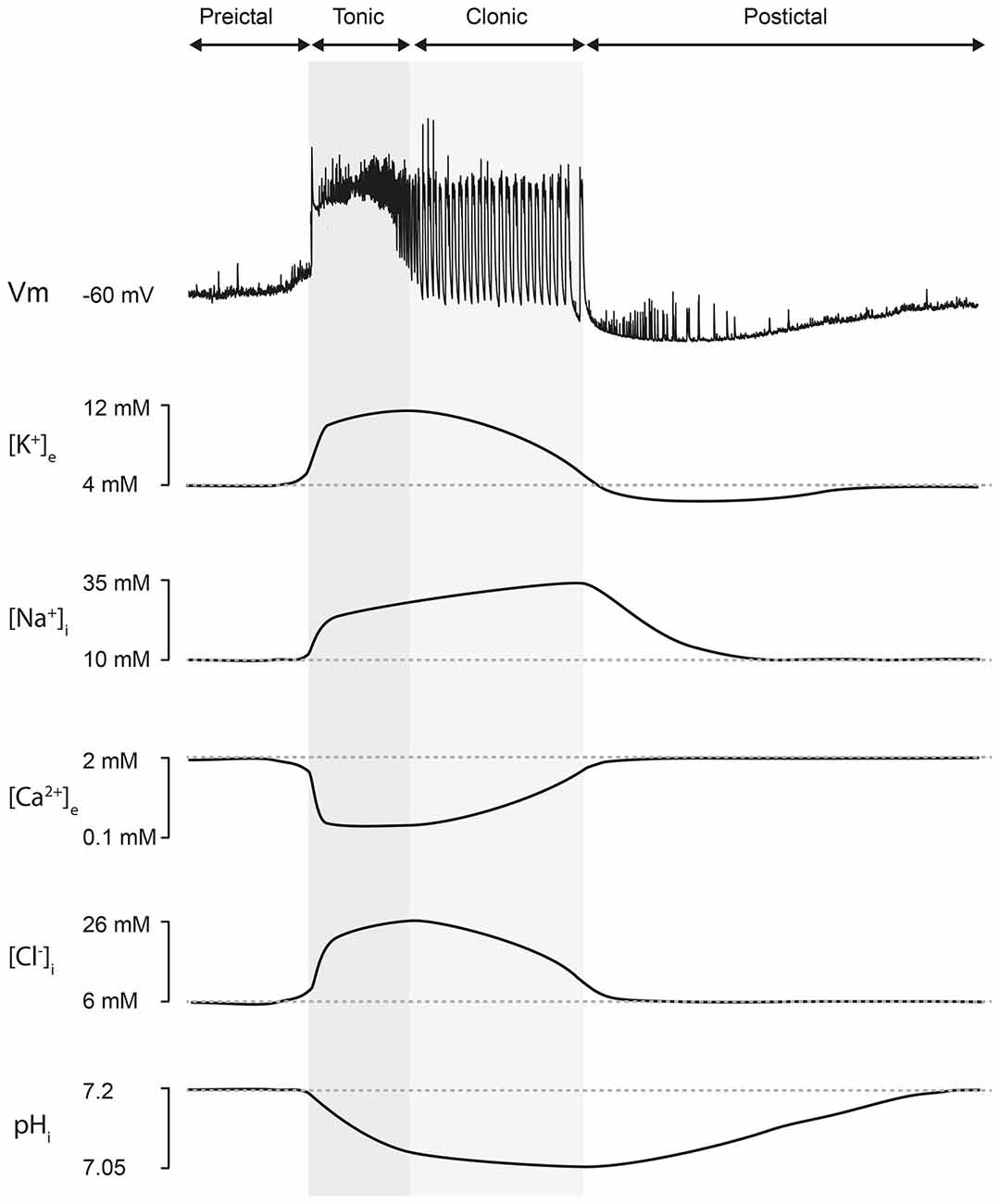
Figure 1. Ion concentration changes during seizures. Top, an intracellular recording from a CA3 hippocampal neuron during a seizure-like event in vitro demonstrates typical neuronal membrane potential changes which accompany seizure episodes. Seizures consist of a “preictal” phase, a “tonic” phase (characterized by membrane potential depolarization upon which occur high-frequency, low-amplitude discharges, dark gray), a “clonic” or “after-discharge phase” (composed of rhythmic bursts of activity that emerge from a relatively hyperpolarized membrane potential background, light gray) and lastly a “postictal” phase (characterized by a hyperpolarized membrane potential lasting for several minutes before slowly recovering to preseizure values). Bottom, representative changes in ion concentration associated with seizures. The accompanying traces should be viewed as “best approximations” based on the available literature. [K+]e is thought to peak at the end of the tonic phase with an undershoot accompanying the postictal period (Dreier and Heinemann, 1991; Fröhlich et al., 2008). The [Na+]i trace is inferred from experimental measurements and supported by computational modeling work, which suggests that [Na+]i is likely to peak at the end of the seizure (Krishnan and Bazhenov, 2011). [Ca2+]e has been shown to drop rapidly to approximately 100 μM during an ictal event (Pumain et al., 1985). [Cl−]i increases during seizures and is highest during the clonic phase (Raimondo et al., 2013; Ellender et al., 2014). Neuronal pHi decreases during the seizure, reaching its minimum around the time of seizure offset, and then slowly recovering during the postictal phase (Xiong et al., 2000; Raimondo et al., 2012a).
In summary, the relationship between K+ dynamics and neural activity is clearly a complex one. Understanding the precise role of K+ in seizure maintenance and termination will no doubt require combining such theoretical work with more sophisticated means of experimentally measuring changes in K+ concentration. This might include the further development of K+ sensitive dyes (Rimmele and Chatton, 2014) and genetically-encoded K+ sensors, akin to those available for Ca2+, Cl− and pH.
The potential for, and relevance of, changes to the transmembrane concentration gradient of Na+ during epileptic seizures has not been extensively investigated. This is largely due to the technical difficulties associated with recording the concentration of this ion within nervous tissue. Although Na+ sensitive electrodes have existed for several decades, their size has largely limited their use to extracellular measurements. In addition, the high baseline extracellular concentration of Na+ ([Na+]e) reduces the signal to noise of recorded changes in extracellular Na+ concentration. That said, the data available indicates that epileptiform activity is associated with a peak reduction in [Na+]e of approximately 4–7 mM (a typical resting [Na+]e is 145 mM, see Table 1; Dietzel et al., 1982). This measurement was made without volume correction, which is important to note as the volume of the extracellular space has been reported to contract by approximately 30% during seizures (Dietzel and Heinemann, 1986). Without this volume contraction and its “concentrating effect”, the reduction in [Na+]e would be expected to be greater.
Decreases in extracellular Na+ occur in tandem with an increase in the concentration of intracellular Na+. The development of Na+ sensitive dyes, the most popular being the ratiometric Na+ indicator SBFI (sodium binding benzofuran isophthalate) allows the quantitative measurement of [Na+]i utilising epifluorescence or more effectively, 2-photon microscopy (Minta and Tsien, 1989; Rose, 2003; Meier et al., 2006). A number of groups have used SBFI to quantify intracellular Na+ accumulation during synaptically-evoked and epileptiform activity (Rose and Konnerth, 2001; Langer and Rose, 2009; Fleidervish et al., 2010; Karus et al., 2015). This influx of Na+ appears to be predominantly mediated by dendritic NMDA receptors (NMDARs), although Na+ fluxes via other Na+ conductances, particularly voltage-gated Na+ channels, are also expected to contribute (Rose and Konnerth, 2001; Langer and Rose, 2009). It is therefore likely that significant increases in [Na+]i follow the intense neural activity and activation of NMDARs that accompanies epileptic seizures. This [Na+]i increase is likely to be most pronounced within dendritic spines as glutamatergic synapses on the spine head are the site of predominant influx, and the small, restricted volume of the spine will result in enhanced concentration increases for a given flux of Na+ (Rose and Konnerth, 2001). However, the exact temporal and spatial quantification of these seizure-associated Na+ fluxes remain to be shown.
It is expected that a progressive breakdown in the transmembrane Na+ gradient should serve to reduce neural excitability and network activity. Inhibitory effects following seizure-associated changes in the Na+ gradient are likely to occur via a number of possible mechanisms. One such mechanism is through a reduction in the driving force for voltage gated Na+ channels. This would alter action potential kinetics and could reduce the ability of neighboring voltage-gated Na+ channels to recruit each other during the course of action potential generation. Based on a peak intracellular concentration of 55 mM and an extracellular concentration of 139 mM, it is estimated that the Na+ reversal would shift from +71 mV at rest to a minimum of +25 mV during a seizure (Dietzel et al., 1982; Rose and Konnerth, 2001). Similarly, intracellular accumulations of Na+ would reduce the Na+ driving force at glutamatergic receptors, reducing the size of excitatory postsynaptic currents. However, as glutamate receptors are equally permable to K+, this effect is somewhat counterbalanced by simultaneous reductions in the driving for K+ (see EAMPA in Table 1). Computational modeling, which accounts for the dynamics of multiple ions in the context of epileptiform activity, has shown that the progressive intracellular increase of Na+ is likely to be an important factor in triggering the termination of seizure activity. Compatible with changes in Na+ altering excitability, in these simulations intracellular Na+ peaks immediately prior to seizure offset (Krishnan and Bazhenov, 2011; see Figure 1).
Another mechanism by which increases in [Na+]i might have an inhibitory effect is via activation of the electrogenic Na+/K+ ATPase. Repetitive action-potential generation and increases in Na+ are well known to stimulate the activity of this transporter (Ritchie and Straub, 1957; McDougal and Osborn, 1976; Thompson and Prince, 1986). Due to the fact that the transporter pumps two K+ ions into the cell for every three Na+ ions extruded, enhanced activity will have a hyperpolarizing effect on the neuronal membrane potential (Krishnan and Bazhenov, 2011; Krishnan et al., 2015). Lastly, the relatively recent discovery of the Na+ -activated K+ family of channels, “Slick” and “Slack” offer a further important mechanism by which increases in [Na+]i could terminate epileptic seizures (Igelstrom, 2013). These channels, which are activated by intracellular Na+ accumulation, have been shown to result in powerful and sustained membrane hyperpolarization following periods of spiking activity (Bhattacharjee et al., 2003; Yuan et al., 2003). As such, Slick and Slack are well placed to play a role in the spontaneous termination of epileptic seizures (Igelstrom, 2013).
Finally, indirect evidence supporting the role of Na+ dynamics in seizure cessation comes from data on the effects of common antiepileptic drugs that target Na+ channels. For example, the anticonvulsant effect of phenytoin is linked to its inhibitory action on voltage-gated Na+ channels. Whilst this anticonvulsant agent effectively raises the threshold for seizure generation, it has also been shown to extend the duration of seizures (Ebert et al., 1997). It is possible that this effect is due to slower seizure-associated intracellular Na+ accumulation on account of reduced voltage-gated Na+ currents.
Under typical baseline conditions the free Ca2+ concentration within neurons is below 100 nM. As a result, activity-dependent Ca2+ influx causes large changes in free [Ca2+]i, constituting a powerful and reliable intracellular signal (Collingridge and Bliss, 1987; Collins et al., 1991; Franklin and Johnson, 1992; Ghosh and Greenberg, 1995). Due to the involvement of Ca2+ in so many fundamental cellular processes, and the large relative changes in its concentration, considerable attention has been devoted to developing tools for monitoring changes in [Ca2+]i. Indeed, observing changes in [Ca2+]i is commonly and reliably used as a proxy for neural activity. Although Ca2+ sensitive electrodes do exist, the small molecule Ca2+ dyes (indicators like Oregon Green BAPTA-1, fura-2 and fluo-4) have until recently been the workhorses of intracellular Ca2+ measurement. These indicators are now gradually being superseded by genetically-encoded Ca2+ reporters, of which the GCaMP family of proteins is the most popular. Targeted mutagenesis has resulted in iterative functional improvements in these reporters, which now exhibit kinetic properties approaching those of the Ca2+ dyes. The major advantage of these sensors is that they can be genetically targeted to specific cell-populations or subcellular locations (Looger and Griesbeck, 2012; Chen et al., 2013). An important caveat however, is that the majority of the Ca2+ reporters described above are non-ratiometric and, where ratiometric reporters have been used in epilepsy studies (e.g., Fura-2 and Twitch), these have rarely been used in their calibrated form. This means that recorded Ca2+ signals are qualitative and provide a read out of neuronal activity, rather than quantitative measurements of Ca2+ concentration. In addition, many of the reporters are ultimately limited in their ability to resolve subcellular Ca2+ signaling processes, which can occur in microdomains close to sites of Ca2+ influx. Therefore studies to date have tended to focus on the more widespread increases in intracellular Ca2+ that are observed during seizure-activity.
During the course of an epileptic seizure, Ca2+ influx occurs predominantly via voltage-gated Ca2+ channels and NMDARs. Membrane depolarization simultaneously activates the voltage-gated channels and releases the voltage-dependent Mg2+ block of the NMDAR. This combines with high levels of glutamate released during the seizure to generate a pronounced Ca2+ influx and rapid accumulation of intracellular Ca2+. As is the case for Na+, seizure-associated increases in [Ca2+]i are likely to be very large within dendritic spines due to their small volume and the strong activation of NMDARs at spine-associated glutamatergic synapses. Calcium imaging during the course of seizure activity demonstrates that Ca2+ influx occurs in almost all neurons and astrocytes during an ictal event (Trevelyan et al., 2007). As described above, the vast majority of studies have used relative, as opposed to absolute reporters of Ca2+. This means that surprisingly little quantitative data exists describing Ca2+ concentration changes during seizures. However, the data that does exist suggests that the extracellular concentration of Ca2+ drops significantly in tandem with the rapid and widespread flow of Ca2+ into cells during an ictal event (Benninger et al., 1980; Somjen, 1980; Pumain et al., 1985; Pal et al., 1999; also see Figure 1). Indeed, in vivo measurements of extracellular Ca2+ concentration in primates during seizures has shown that Ca2+ drops to within the 100 μM range (Pumain et al., 1985; Figure 1). At this concentration, synaptic transmission is significantly compromized (Feng and Durand, 2003), as there is too little extracellular Ca2+ available to sustain the Ca2+ influxes necessary for vesicle fusion and synaptic release of neurotransmitter. In addition, the dissipation of the transmembrane Ca2+ gradient would cause a negative shift in the reversal potential for Ca2+ (see Table 1). Furthermore, Ca2+ entry also results in the opening of Ca2+-activated K+ conductances (Vergara, 1998). One would predict all of these processes to play a negative feedback or inhibitory role, and thereby serve to terminate a seizure.
Despite this prediction, it is also known that the experimental removal of Ca2+ from the extracellular space reliably evokes seizure-like activity both in vitro and in vivo (Haas and Jefferys, 1984; Feng and Durand, 2003). Indeed, reduced extracellular Ca2+ is an established model of non-synaptic epileptiform activity. The reduction in extracellular Ca2+ is believed to reduce surface charge screening, which shifts the conductance-voltage relationship of voltage-gated channels. Voltage-gated channels are therefore more likely to be activated and thus cause enhanced excitability (McLaughlin et al., 1971). Furthermore, with the desynchronising effect of synaptic noise removed, local field effects are able to result in ephaptic coupling and synchronization of neurons (Taylor and Dudek, 1982). These phenomena are particularly prevalent in the rodent hippocampus where the somata of pyramidal cells are tightly packed, although this aspect of hippocampal anatomy is much less pronounced in the human (West and Gundersen, 1990). Rodent studies have shown that experimental manipulation of extracellular osmolality, which in turn effects cell swelling, the size of the extracellular space and hence ephaptic coupling, directly controls the duration of zero Ca2+ induced epileptiform bursts (Dudek et al., 1990). It has been suggested that zero Ca2+ induced epileptiform activity may represent a useful analog of the ictal phase of epileptic seizures. The reduced extracellular Ca2+ concentration and cell swelling associated with seizures (Olsson et al., 2006) mean that the non-synaptic mechanisms contributing to zero Ca2+ epileptiform activity, could play an important role in extending the ictal phase of epileptic seizures.
The transmembrane Cl− gradient has a critical influence on network activity due to the fact that the channels responsible for fast synaptic inhibition, GABAA receptors (GABAARs), are primarily permeable to Cl−. Open GABAARs are approximately 4 times more permeable to Cl− than to (Kaila and Voipio, 1987). Therefore at rest, EGABAA (typically around −70 mV) is much closer to the very negative Cl− reversal (ECl−; typically −85 mV) than the considerably more positive reversal (E; typically −13 mV; Bormann et al., 1987; Kaila et al., 1993). EGABAA′s proximity to the neuronal resting membrane potential means that relatively small changes in [Cl−]i, and hence EGABAA can greatly influence the functional effect of GABAAR activation and consequently neuronal output.
Inhibitory synapses are particularly prone to short-term activity-dependent changes in [Cl−]i. Indeed, Cl− accumulation is often used as the definitive example of what is referred to as short-term ionic plasticity (Rivera et al., 2004; Raimondo et al., 2012b; Kaila et al., 2014b). This is any process that results in short-lasting changes to the ionic driving force for post-synaptic receptors. The study of Cl− dynamics in the context of network activity has been aided by experimental tools which are either able to determine [Cl−]i indirectly, via the measurement of EGABAA using the gramicidin perforated patch clamp technique (Kyrozis and Reichling, 1995) or single channel recordings in cell attached mode (Tyzio et al., 2008), or directly by using fluorescent Cl− reporters (Kuner and Augustine, 2000; Markova et al., 2008; Arosio et al., 2010; Raimondo et al., 2013). It should be noted that genetically encoded Cl− indicators, whilst enabling subcellular and cell-type specific targeting, suffer from intrinsic pH sensitivity. This is especially problematic in the context of measuring Cl− fluxes during seizures, which are also expected to generate significant pH shifts (Raimondo et al., 2012a). However, the latest Cl− reporters based on a novel fusion protein (Arosio et al., 2010; Raimondo et al., 2013) afford simultaneous measurements of pH, which allows the Cl− signal to be corrected for concurrent shifts in pH. Although this is a relatively complex correction procedure with the potential for measurement artifacts to occur if performed incorrectly. Experiments using these techniques have definitively demonstrated that intense activation of GABAARs, particularly in tandem with concurrent membrane depolarization, can overwhelm endogenous Cl− extrusion systems such as the cation-chloride cotransporter KCC2 and result in rapid intracellular Cl− accumulation (Kaila and Voipio, 1987; Kaila et al., 1989; Thompson and Gähwiler, 1989; Staley et al., 1995; Staley and Proctor, 1999; Ellender et al., 2014). Intracellular Cl− accumulation during intense activation of GABAARs can occur even in the absence of concurrent depolarization mediated by other conductances. This is because GABAARs are also permeable to . A corresponding collapse of the gradient is prevented by the activity of intra- and extracellular carbonic anhydrases, which use CO2 as a substrate to rapidly regenerate intracellular (Kaila et al., 1990; Rivera et al., 2005). As a result, the relatively stable and more positive E maintains the driving force for continued intracellular Cl− accumulation during repeated activation of GABAARs. Interestingly this process appears to be developmentally controled, at least in rats, where the appropriate isoforms of carbonic anhydrase are up regulated at postnatal day 12 (Ruusuvuori et al., 2004). Intracellular Cl− accumulation not only reduces the size of subsequent GABAAR mediated hyperpolarizing potentials, but can ultimately render GABAergic transmission depolarising or even excitatory. Epileptic seizures are characterized by widespread membrane depolarization and powerful activation of neurotransmitter receptors including GABAARs. It is therefore unsurprising that significant Cl− accumulation has been shown to accompany epileptiform activity in a wide array of in vitro seizure models (Lamsa and Kaila, 1997; Isomura et al., 2003; Fujiwara-Tsukamoto et al., 2010; Ilie et al., 2012; Raimondo et al., 2013; Ellender et al., 2014). Gramicidin perforated patch recordings indicate seizure-associated depolarising shifts in EGABAA in the region of 30 mV (Ellender et al., 2014). Similarly, dynamic readout of [Cl−]i using genetically encoded Cl− sensors have observed seizure induced Cl− accumulation between 10 and 20 mM (Raimondo et al., 2013). Ion sensitive microelectrode recordings of [Cl−]e have demonstrated that although Cl− moves into the intracellular space, [Cl−]e actually increases by about 7 mM during seizures. This is because the extracellular space contracts by about 30% “concentrating” the remaining extracellular Cl−. Nonetheless, these changes to the electrochemical gradient for Cl− result in GABAergic transmission being rendered excitatory and hence serves to exacerbate rather than abort epileptiform activity (see Table 1 and Figure 1). Indeed, computational models have suggested that the extent of intracellular Cl− accumulation is a pivotal determinant of seizure duration (Krishnan and Bazhenov, 2011). Experimental evidence has confirmed that excitatory GABAergic signaling maintains the clonic phase of seizure-like events in vitro (Fujiwara-Tsukamoto et al., 2010; Ellender et al., 2014). Although Cl− accumulation and seizure-induced excitatory shifts in GABAergic signaling remain to be demonstrated in vivo, it is likely that Cl− dynamics play a role in determining when seizures terminate.
Due to the effect of pH on protein folding and enzymatic activity, it is a critical parameter that is tightly regulated for optimum cell function. pH is fundamental to a variety of cellular processes including cell division, metabolism, apoptosis and migration (Opie, 1965; Denker and Barber, 2002; Putney and Barber, 2003; Abad et al., 2004). Within the nervous system, the control of pH has particular relevance for synaptic transmission and the modulation of network excitability (Drapeau and Nachshen, 1988; Tabb et al., 1992; Dulla et al., 2005). In general, alkalosis increases neuronal excitability whilst acidosis reduces neural excitability (Balestrino and Somjen, 1988; Sparing et al., 2007; Tolner et al., 2011). Multiple endogenous buffer and transport systems exist to control both intracellular and extracellular pH. One of the most important is the buffer system, which utilizes intra- and extracellular carbonic anhydrases to catalyze the reversible reaction of H2O and CO2 to and H+. As a result, pH and concentration are closely linked (see Figure 2).
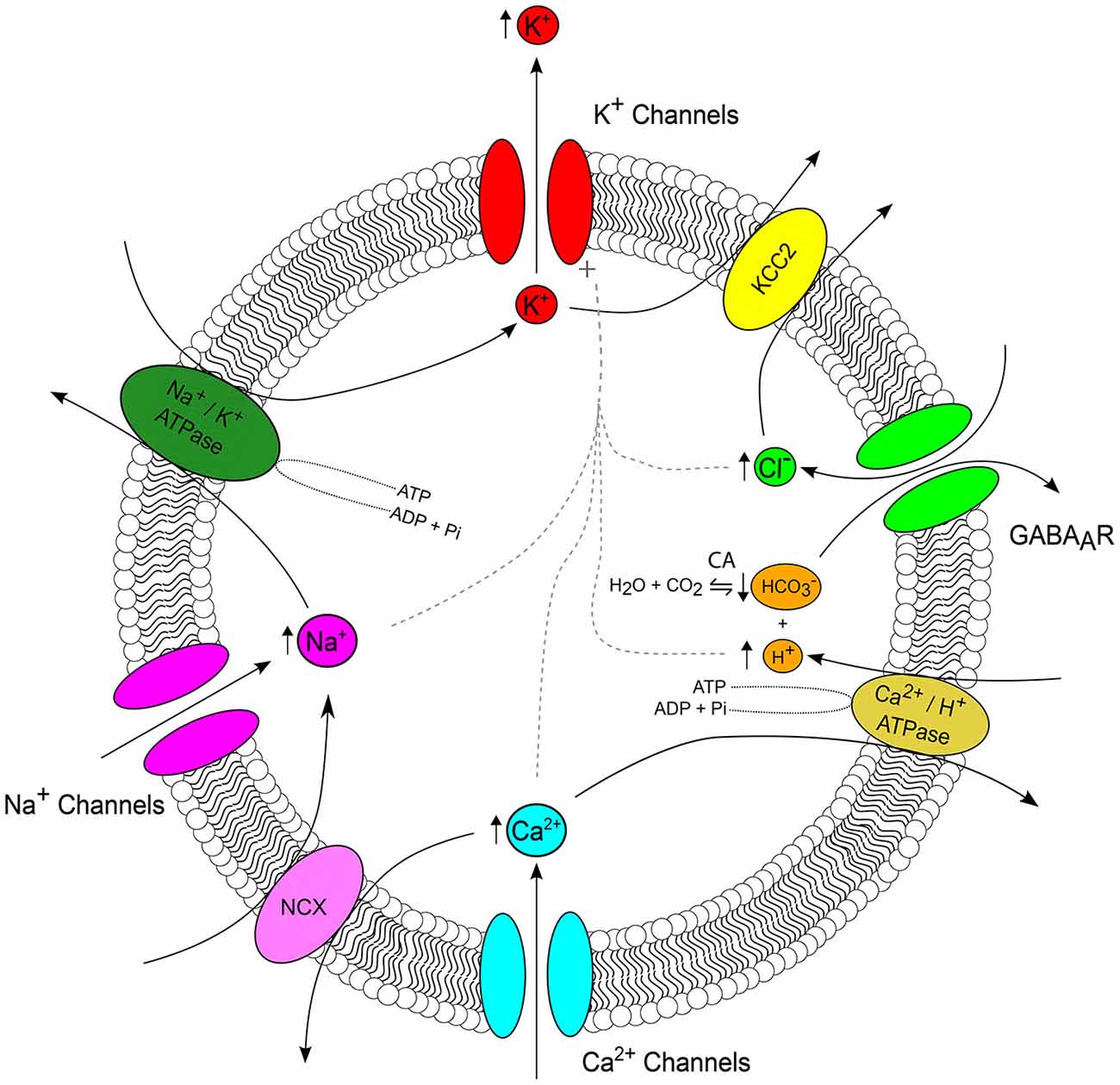
Figure 2. Ion interactions during seizures. The major ion channels and transporters, which serve as important nodes of interaction between ions are depicted, with arrows highlighting the typical direction of ionic flux. Arrows adjacent to an ion reflect the direction of seizure induced concentration change (see Figure 1). During a seizure, K+ efflux via a host of K+ channels (red) results in accumulation of extracellular K+. The cation-chloride transporter KCC2 (yellow) serves as an important link between the seizure-associated reduction in the transmembrane K+ gradient and intracellular Cl− accumulation via GABAA receptors (GABAARs) (green). GABAARs, which are permeable to both Cl− and connect the regulation of Cl−, and pH via carbonic anhydrases which catalyze the reversible reaction of H2O and CO2 to and H+ (orange). Seizures are associated with intracellular acidification which is due, in part, to the activity of Ca2+/H+ ATPase as it imports H+ and extrudes Ca2+ in attempt to restore baseline Ca2+ concentrations following activity-induced Ca2+ influx (cyan). Na+/Ca2+ exchangers (NCX, pink) connect Ca2+ and Na+ concentration. Seizure-associated Na+ influx via voltage and ligand gated Na+ channels (magenta) up regulates the activity of Na+/K+ ATPases (forest green). Finally, increased intra-neuronal concentrations of Cl−, H+, Ca2+ and Na+ all activate K+ channels (dashed gray line). The number and complexity of possible ionic interactions highlights the importance of computational models for determining the relevance of these continuously evolving variables, which are often difficult to study experimentally.
Multiple experimental tools exist for the measurement of pH including pH-sensitive micro-electrodes, pH-sensitive fluorescent dyes and genetically encoded pH sensors (Rose and Deitmer, 1995; Boyarsky et al., 1988; Buckler and Vaughan-Jones, 1990; Kneen et al., 1998). Despite the presence of intrinsic buffering systems, the use of these tools has demonstrated a plethora of activity-dependent pH changes in multiple systems (Chesler, 2003). Measurements of intraneuronal pH in the context of in vitro seizure models has revealed intracellular acidification which increases as a function of seizure duration (Xiong et al., 2000; Raimondo et al., 2012a). For seizure-like events longer that 30 s, peak shifts often exceeded 0.2 pH units and are closely correlated with the termination of epileptiform activity (Xiong et al., 2000; Raimondo et al., 2012a; Figure 1). At least three major processes are likely to be involved in seizure-induced acidification. Firstly, a fall in pH is linked to the activity-induced entry of Ca2+ which then requires extrusion via the function of Ca2+/H+ ATPases located in the plasma membrane and endoplasmic reticulum (Schwiening et al., 1993; Makani and Chesler, 2010). Secondly, prolonged neural activity will increase the production of metabolic acids such as CO2 and lactate (Wang et al., 1994). And thirdly, the intense GABAAR activation that accompanies seizure activity has been shown to result in considerable efflux and a resulting intracellular acidification (Pasternack et al., 1993; Trapp et al., 1996). With regard to the extracellular space, seizures or electrically stimulated activity are associated with more complex pH transients, which may be biphasic (an initial extracellular alkaline shift followed by a prolonged acidosis; Caspers and Speckmann, 1972; Urbanics et al., 1978; Xiong and Stringer, 2000). It is likely that this complexity is due to the fact that pH transients observed in the extracellular space must somehow reflect the combined consequence of seizure-associated intracellular pH transients in both neurons and other cell types, such as astrocytes (Chesler, 2003).
Progressive intraneuronal acidification during a seizure has been suggested to be an important factor influencing seizure termination. Although the precise mechanisms remain unknown, it is likely that acidification could alter excitability by modulating the conductance of ligand and voltage-gated channels. For example, acidification has been shown to reduce the conductance of NMDARs (Tang et al., 1990; Traynelis and Cull-Candy, 1990; Vyklický et al., 1990), enhance the conductance of GABAARs (Takeuchi and Takeuchi, 1967; Smart and Constanti, 1982; Pasternack et al., 1996) and reduce voltage gated Ca2+ currents (Iijima et al., 1986; Barnes and Bui, 1991; Tombaugh and Somjen, 1997; Tombaugh, 1998). Furthermore, intracellular acidification is known to reduce gap junction coupling (Spray et al., 1981), which would serve to reduce the neuronal synchronization that accompanies seizures (de Curtis et al., 1998). Indeed, metabolic acidosis, which would lower intracellular pH, could be relevant for the mechanism of action of some antiepileptic treatments including the ketogenic diet (Neal et al., 2008) and carbonic anhydrase inhibitors (Velisek and Veliskova, 1993).
The above discussion has highlighted the fact that epileptic seizures are associated with marked changes to the transmembrane concentration gradient for the six major ions (K+, Na+, Cl−, Ca2+, H+ and ). What adds a significant layer of complexity to this picture, is the fact that these ionic fluxes interact in multiple ways during the evolution of a seizure. In this section we briefly highlight several important nodes of interaction between these ion species (see Figure 2).
Increased K+ conductance and K+ efflux is a common consequence of the seizure-associated accumulation of Na+, Cl−, Ca2+ and H+. Increases in [Na+]i and [Ca2+]i trigger the activation of specialized families of K+ channels, which are gated by either Na+ (Igelstrom, 2013) or Ca2+ (Vergara, 1998). Similarly, raised intracellular Cl− co-operatively activates both Slick and Slack (Kaczmarek, 2013), and acidic intracellular pH enhances the conductance of inwardly rectifying K+ channels (Yang and Jiang, 1999). Enhanced K+ efflux, whilst having a hyperpolarising effect on the membrane potential, also results in a dissipation of the transmembrane K+ gradient and an accumulation of extracellular K+ with complex effects on network excitability as described in the potassium section above.
The cation-chloride transporter KCC2 serves as an important link between dynamic changes in K+ and Cl− (Figure 2). As a cotransporter, KCC2 relies on the thermodynamic driving force for K+ and Cl− to transport both ions in the same direction across the membrane. Therefore, increases in extracellular K+ during a seizure are predicted to reduce the ability of KCC2 to utilize the K+ gradient to extrude Cl−. Viewed from an alternative perspective, the accumulation in intracellular Cl− that is mediated by intense activation of the GABAAR during a seizure would be expected to enhance K+ extrusion via KCC2 (Viitanen et al., 2010), albeit until extracellular K+ accumulation reaches a level that renders outward transport of both ions energetically unfavorable. This example demonstrates how dynamic changes in the concentration of the principle ions can exert complex forms of cross talk.
ions provide a pivotal node of interaction between neuronal regulation of Cl− and H+. This is due to the fact that GABAARs are permeable to both Cl− and , and because intracellular and extracellular carbonic anhydrases link pH to []i via the reversible reaction of H2O and CO2 to and H+ (see Figure 2). Although carbonic anhydrases are largely able to regenerate intracellular in the face of the efflux associated with intense activation of GABAARs, the generation of intracellular H+ that accompanies epileptic activity will shift the equilibrium set point of the catalyzed reaction of H2O and CO2, to and H+. Therefore, activity induced acidification is predicted to cause a reduction in intracellular and hence a hyperpolarization of E (Table 1). As EGABAA is a product of both E and ECl−, this progressive acidification should reduce the depolarising shift in EGABAA, which occurs following seizure induced Cl− accumulation (Doyon et al., 2011).
Finally, the transmembrane transport of neurotransmitters such as glutamate (Excitatory Amino Acid Transporter—EAAT1–5) and GABA (GABA Transporter—GAT1–3) are intimately linked to the electrochemical gradients of several ion species. Glutamate transport in both neurons and astrocytes is driven by the electrochemical gradients for glutamate, Na+, H+ and K+ (Danbolt, 2001). Interestingly, several of the glutamate transporters (EAAT1,3,4) also act as glutamate sensitive Cl− channels (Fairman et al., 1995). Similarly, GABA transport depends on the electrochemical gradients of GABA, Na+ and Cl−. Whilst the activation of ionotropic receptors for GABA and glutamate, as discussed extensively above, results in alterations to the transmembrane gradient for multiple ion species. These changes to the transmembrane ion gradients in turn affect the direction and rate of neurotransmitter transport across cell membranes. For example, the depolarization and increased intracellular Na+ concentrations that occur during seizures are predicted to result in a reversal in the direction of GABA transport by GABA transporters (Wu et al., 2007), switching from reuptake to extrusion of GABA into the extracellular space (During et al., 1995).
These multifaceted interactions between the different ion concentration dynamics, electrochemical gradients and synaptic transmission remind us of the difficulties involved in investigating these phenomena. Indeed, this is perhaps the greatest challenge for the field, and underscores the potential importance of computational models for determining the relevance and impact of continuously interacting and evolving variables, which can be difficult to tease apart experimentally.
So far we have focussed on ion concentration gradients across the neuronal membrane. It is important to note that astrocytes are interwoven into the fabric of neuronal networks where, like neurons, they express a host of proteins that establish particular ion concentration gradients across their membranes. By affecting ion concentrations within the extracellular space, astrocytes also modulate the ion concentration gradients that are critical for synaptic transmission and network excitability. One of the best described roles for astrocytes entails the control of extracellular K+ as described in the potassium section above. Astrocytes act as a K+ “sink” that prevents excessive extracellular build-up of K+ during neuronal activity. Indeed, in the context of seizure activity, there is accumulating evidence from animal models and tissue excised from patients with temporal lobe epilepsy, that astrocytic dysfunction plays a significant role in epileptogenesis, particularly with regard to extracellular K+ regulation (Hinterkeuser et al., 2000; Steinhäuser and Seifert, 2002; Wallraff et al., 2006; Bedner et al., 2015).
Compared to neuronal studies, measurements of intra-astrocytic ion concentration dynamics during epileptiform activity have received much less attention. Nonetheless, elevated network activity is thought to result in increases in intra-astrocytic Ca2+, Na+ and K+, as well as an intra-astrocytic alkalinization (Chesler and Kraig, 1989; Walz, 2000; Volterra et al., 2014; Karus et al., 2015). The direction of activity-induced astrocytic Cl− flux is still quite unclear, although GABAAR activation has been suggested to result in Cl− efflux from astrocytes (Egawa et al., 2013). If these changes are also evident during a seizure, astrocytic movement of K+ and H+ would be expected to ameliorate the seizure-induced changes in the concentration of these ions in neurons. Meanwhile, the astrocytic movement of Ca2+ and Na+ would be expected to have the opposite effect, and exacerbate the changes in neurons. Determining the relevance of ion dynamics in different cell types for seizure propagation and termination represents an interesting area for future research.
In addition to the cell-type specific control of ion concentration gradients, different subcellular compartments within an individual neuron or astrocyte can also show differences in ion concentration. Such subcellular differences may result from the expression pattern of ion transporters, or the particular influxes of ion species into different regions of a cell. Local concentration changes resulting from ionic fluxes across the membrane are a function of the diffusive and ion transport properties of the compartment concerned, as well as the volume. For example, a given flux of ions will cause a greater concentration change in a small intracellular compartment, compared to a large volume compartment. Indeed, experimental and computational studies have shown that a flux of Cl− will cause a greater change in [Cl−]i and hence EGABAA when Cl− loading occurs within dendrites, than in the soma (Staley and Proctor, 1999; Raimondo et al., 2012b). As discussed in preceding sections, this is also relevant for Na+ and Ca2+ influxes associated with activity-induced glutamatergic conductances on dendritic spines. The ionic changes that occur during a seizure are therefore a function of the site of flux and the properties of the compartment concerned. Recent evidence has emerged that, at least within in vitro models of seizures, strong recruitment of soma-targeting parvalbumin expressing interneurons means that the site of maximal Cl− accumulation occurs within somatic as opposed to dendritic regions of hippocampal pyramidal neurons (Ellender et al., 2014). However, it is fair to say that subcellular differences in seizure-evoked ionic changes and their functional relevance for on-going pathological activity are poorly described. This is therefore an area of research that should benefit greatly from advances in genetically-encoded ion sensors and related optogenetic techniques.
Increased knowledge of seizure-associated ion dynamics will no doubt have important implications for anticonvulsant therapies, particularly in the context of treatments for status epilepticus. In the case of status epilepticus, endogenous inhibitory processes are unable to terminate seizure activity and it seems highly likely that perturbed ion concentration gradients play an important role in this pathological state. It is therefore important to avoid the assumption that agents that have a particular effect on synaptic signaling under resting conditions, will have the same effect during status epilepticus. As a potential example, agents from the benzodiazepine class of drugs, which are positive modulators of GABAAR signaling, are commonly used as the first line therapy in the treatment of status epilepticus. If seizure mediated intracellular Cl− accumulation, as described above, has rendered fast GABAergic transmission excitatory within major brain areas, it is possible that these agents may not be ideally suited for aborting pathological hyperexcitability (Deeb et al., 2013). Although this remains to be demonstrated experimentally, seizure-associated Cl− accumulation may be part of the explanation as to why benzodiazepines often fail to halt status epilepticus (Yasiry and Shorvon, 2014). In this light, novel therapies that take into account the perturbed ion concentration gradients of a seizure may be more appropriate. For instance, the development of strategies to enhance ion transport and the restoration of ion concentration gradients may constitute a valuable strategy for aborting or preventing extended seizure states (Gagnon et al., 2013).
Seizure termination is difficult to study experimentally as the manipulation of potential terminating mechanisms is likely to involve variables that also influence seizure initiation and propagation. Furthermore, during the peak of the ictal state multiple processes occur simultaneously and influence one another in complex ways. Nonetheless, it is clear that seizure-associated changes in transmembrane ion concentration gradients play an important role in the evolution and continuation of pathological network activity. Future research will no doubt take advantage of novel genetically-targeted ion sensors and powerful computing technologies in order to determine how multiple ion species interact in the context of epileptic seizures. It is our hope that a better appreciation of the potential influence of ion dynamics in seizures will stimulate novel avenues of epilepsy research and ultimately result in improved intervention strategies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
JVR was supported by a South African National Research Foundation Innovation Postdoctoral Fellowship, a UCT URC Postdoctoral Fellowship and the Blue Brain Project. The research leading to these results has received funding from the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007–2013), ERC grant agreement number 617670. We thank the reviewers for their constructive comments and would particularly like to acknowledge Juha Voipio’s suggestions on how to improve the Review.
Abad, M. F. C., Di Benedetto, G., Magalhães, P. J., Filippin, L., and Pozzan, T. (2004). Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J. Biol. Chem 279, 11521–11529. doi: 10.1074/jbc.m306766200
Amzica, F., and Steriade, M. (2000). Neuronal and glial membrane potentials during sleep and paroxysmal oscillations in the neocortex. J. Neurosci. 20, 6648–6665.
Arosio, D., Ricci, F., Marchetti, L., Gualdani, R., Albertazzi, L., and Beltram, F. (2010). Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat. Methods 7, 516–518. doi: 10.1038/nmeth.1471
Avoli, M., Louvel, J., Pumain, R., and Olivier, A. (1987). Seizure-like discharges induced by lowering [Mg2+]o in the human epileptogenic neocortex maintained in vitro. Brain Res. 417, 199–203.
Balestrino, M., and Somjen, G. G. (1988). Concentration of carbon dioxide, interstitial pH and synaptic transmission in hippocampal formation of the rat. J. Physiol. 396, 247–266. doi: 10.1113/jphysiol.1988.sp016961
Barnes, S., and Bui, Q. (1991). Modulation of calcium-activated chloride current via pH-induced changes of calcium channel properties in cone photoreceptors. J. Neurosci. 11, 4015–4023.
Bedner, P., Dupper, A., Huttmann, K., Muller, J., Herde, M. K., Dublin, P., et al. (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222. doi: 10.1093/brain/awv067
Ben-Ari, Y. (2002). Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Benninger, C., Kadis, J., and Prince, D. A. (1980). Extracellular calcium and potassium changes in hippocampal slices. Brain Res. 187, 165–182. doi: 10.1016/0006-8993(80)90502-8
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Bhattacharjee, A., Joiner, W. J., Wu, M., Yang, Y., Sigworth, F. J., and Kaczmarek, L. K. (2003). Slick (slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691.
Bormann, J., Hamill, O. P., and Sakmann, B. (1987). Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J. Physiol. 385, 243–286. doi: 10.1113/jphysiol.1987.sp016493
Boyarsky, G., Ganz, M. B., Sterzel, R. B., and Boron, W. F. (1988). pH regulation in single glomerular mesangial cells. I. Acid extrusion in absence and presence of . Am. J. Physiol. 255, C844–C856.
Bragin,A., Penttonen, M., and Buzsáki, G. (1997). Termination of epileptic afterdischarge in the hippocampus. J. Neurosci. 17, 2567–2579.
Buckler, K. J., and Vaughan-Jones, R. D. (1990). Application of a new pH-sensitive fluoroprobe (carboxy-SNARF-1) for intracellular pH measurement in small, isolated cells. Pflugers Arch. 417, 234–239. doi: 10.1007/bf00370705
Butt, A. M., and Kalsi, A. (2006). Inwardly rectifying potassium channels (Kir) in central nervous system glia: A special role for Kir4.1 in glial functions. J. Cell. Mol. Med. 10, 33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x
Caspers, H., and Speckmann, E. (1972). Cerebral pO2, pCO2 and pH: changes during convulsive activity and their significance for spontaneous arrest of seizures. Epilepsia 13, 699–725. doi: 10.1111/j.1528-1157.1972.tb04403.x
Chen, T. W., Wardill, T. J., Sun, Y., Pulver, S. R., Renninger, S. L., Baohan, A., et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. doi: 10.1038/nature12354
Chesler, M. (2003). Regulation and modulation of pH in the brain. Physiol. Rev. 83, 1183–1221. doi: 10.1152/physrev.00010.2003
Chesler, M., and Kraig, R. (1989). Intracellular pH transients of mammalian astrocytes. J. Neurosci. 9, 2011–2019.
Collingridge, G. L., and Bliss, T. V. P. (1987). NMDA receptors - their role in long-term potentiation. Trends Neurosci. 10, 288–293. doi: 10.1016/0166-2236(87)90175-5
Collins, F., Schmidt, M., Guthrie, P., and Kater, S. (1991). Sustained increase in intracellular calcium promotes neuronal survival. J. Neurosci. 11, 2582–2587.
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
de Curtis, M., and Manfridi, A. and Biella, G. (1998). Activity-dependent pH shifts and periodic recurrence of spontaneous interictal spikes in a model of focal epileptogenesis. J. Neurosci. 18, 7543–7551.
Deeb, T. Z., Nakamura, Y., Frost, G. D., and Davies, P. A. and Moss, S. J. (2013). Disrupted Cl(-) homeostasis contributes to reductions in the inhibitory efficacy of diazepam during hyperexcited states. Eur. J. Neurosci. 38, 2453–2467. doi: 10.1111/ejn.12241
Denker, S. P., and Barber, D. L. (2002). Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J. Cell Biol. 159, 1087–1096. doi: 10.1083/jcb.200208050
Diarra, A., Sheldon, C., and Church, J. (2001). In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFI. Am. J. Physiol. Cell Physiol. 280, C1623–C1633. doi: 10.1016/S0165-5876(00)00363-3
Dietzel, I., and Heinemann, U. (1986). Dynamic variations of the brain cell microenvironment in relation to neuronal hyperactivity. Ann. N Y Acad. Sci. 481, 72–86. doi: 10.1111/j.1749-6632.1986.tb27140.x
Dietzel, I., Heinemann, U., Hofmeier, G., and Lux, H. D. (1982). Stimulus-induced Changes in Extracellular Na+ and Cl- Conc entration in Relation to Changes in the Size of the Extracellular Space. Exp. Brain Res. 46, 73–84. doi: 10.1007/bf00238100
Doyon, N., Prescott, S. A., Castonguay, A., Godin, A. G., Kröger, H., and De Koninck, Y. (2011). Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis. PLoS Comput. Biol. 7:e1002149. doi: 10.1371/journal.pcbi.1002149
Drapeau, P., and Nachshen, D. A. (1988). Effects of lowering extracellular and cytosolic pH on calcium fluxes, cytosolic calcium levels and transmitter release in presynaptic nerve terminals isolated from rat. J. Gen. Physiol. 91, 305–315. doi: 10.1085/jgp.91.2.305
Dreier, J., and Heinemann, U. (1991). Regional and time dependent variations of low Mg2+ induced epileptiform activity in rat temporal cortex slices. Exp. Brain Res. 87, 581–596. doi: 10.1007/bf00227083
Dudek, F. E., Obenaus, A., and Tasker, J. G. (1990). Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci. Lett. 120, 267–270. doi: 10.1016/0304-3940(90)90056-f
Dulla, C. G., Dobelis, P., Pearson, T., Frenguelli, B. G., Staley, K. J., and Masino, S. A. (2005). Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron 48, 1011–1023. doi: 10.1016/j.neuron.2005.11.009
During, M. J., Ryder, K. M., and Spencer, D. D. (1995). Hippocampal GABA transporter function in temporal-lobe epilepsy. Nature 376, 174–177. doi: 10.1038/376174a0
Ebert, U., Cramer, S., and Löscher, W. (1997). Phenytoin’s effect on the spread of seizure activity in the amygdala kindling model. Naunyn. Schmiedebergs. Arch. Pharmacol. 356, 341–347. doi: 10.1007/pl00005060
Egawa, K., Yamada, J., Furukawa, T., Yanagawa, Y., and Fukuda, A. (2013). Cl− homeodynamics in gap-junction-coupled astrocytic networks on activation of GABAergic synapses. J. Physiol. 591, 3901–3917. doi: 10.1113/jphysiol.2013.257162
Ellender, T. J., Raimondo, J. V., Irkle, A., Lamsa, K. P., and Akerman, C. J. (2014). Excitatory effects of parvalbumin-expressing interneurons maintain hippocampal epileptiform activity via synchronous afterdischarges. J. Neurosci. 34, 15208–15222. doi: 10.1523/jneurosci.1747-14.2014
Fairman, W. A., Vandenberg, R. J., Arriza, J. L., Kavanaugh, M. P., and Amara, S. G. (1995). An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375, 599–603. doi: 10.1038/375599a0
Farrant, M., and Kaila, K. (2007). The cellular, molecular and ionic basis of GABAA receptor signalling. Prog. Brain Res. 160, 59–87. doi: 10.1016/s0079-6123(06)60005-8
Feng, Z., and Durand, D. M. (2003). Low-calcium epileptiform activity in the hippocampus in vivo. J. Neurophysiol. 90, 2253–2260. doi: 10.1152/jn.00241.2003
Fertziger, A. P., and Ranck, J. B. (1970). Potassium accumulation in interstitial space during epileptiform seizures. Exp. Neurol. 26, 571–585. doi: 10.1016/0014-4886(70)90150-0
Fleidervish, I. A., Lasser-Ross, N., Gutnick, M. J., and Ross, W. N. (2010). Na+ imaging reveals little difference in action potential-evoked Na+ influx between axon and soma. Nat. Neurosci. 13, 852–860. doi: 10.1038/nn.2574
Franklin, J. L., and Johnson, E. M. (1992). Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 15, 501–508. doi: 10.1016/0166-2236(92)90103-f
Fröhlich, F., Bazhenov, M., Iragui-Madoz, V., and Sejnowski, T. J. (2008). Potassium dynamics in the epileptic cortex: new insights on an old topic. Neuroscientist 14, 422–433. doi: 10.1177/1073858408317955
Fröhlich, F., Bazhenov, M., Timofeev, I., Steriade, M., and Sejnowski, T. J. (2006). Slow state transitions of sustained neural oscillations by activity-dependent modulation of intrinsic excitability. J. Neurosci. 26, 6153–6162. doi: 10.1523/jneurosci.5509-05.2006
Fröhlich, F., Sejnowski, T. J., and Bazhenov, M. (2010). Network bistability mediates spontaneous transitions between normal and pathological brain states. J. Neurosci. 30, 10734–10743. doi: 10.1523/jneurosci.1239-10.2010
Fujiwara-Tsukamoto, Y., Isomura, Y., Imanishi, M., Ninomiya, T., Tsukada, M., Yanagawa, Y., et al. (2010). Prototypic seizure activity driven by mature hippocampal fast-spiking interneurons. J. Neurosci. 30, 13679–13689. doi: 10.1523/jneurosci.1523-10.2010
Gagnon, M., Bergeron, M. J., Lavertu, G., Castonguay, A., Tripathy, S., Bonin, R. P., et al. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19, 1524–1528. doi: 10.1038/nm.3356
Ghosh, A., and Greenberg, M. E. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268, 239–247. doi: 10.1126/science.7716515
Gnatkovsky, V., Librizzi, L., Trombin, F., and De Curtis, M. (2008). Fast activity at seizure onset is mediated by inhibitory circuits in the entorhinal cortex in vitro. Ann. Neurol. 64, 674–686. doi: 10.1002/ana.21519
Goldstein, S. A., Bockenhauer, D., O’Kelly, I., and Zilberberg, N. (2001). Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2, 175–184. doi: 10.1038/35058574
Haas, H., and Jefferys, J. (1984). Low-calcium field burst discharges of CA1 pyramidal neurones in rat hippocampal slices. J. Physiol. 354, 185–201. doi: 10.1113/jphysiol.1984.sp015371
Hinterkeuser, S., Schröder, W., Hager, G., Seifert, G., Blümcke, I., Elger, C. E., et al. (2000). Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur. J. Neurosci. 12, 2087–2096. doi: 10.1046/j.1460-9568.2000.00104.x
Igelstrom, K. M. (2013). Is slack an intrinsic seizure terminator? Neuroscientist 19, 248–254. doi: 10.1177/1073858412446311
Iijima, T., Ciani, S., and Hagiwara, S. (1986). Effects of the external pH on Ca channels: experimental studies and theoretical considerations using a two-site, two-ion model. Proc. Natl. Acad. Sci. U S A 83, 654–658. doi: 10.1073/pnas.83.3.654
Ilie, A., Raimondo, J. V., and Akerman, C. J. (2012). Adenosine release during seizures attenuates GABAA receptor-mediated depolarization. J. Neurosci. 32, 5321–5332. doi: 10.1523/jneurosci.5412-11.2012
Isomura, Y., Sugimoto, M., Fujiwara-Tsukamoto, Y., Yamamoto-Muraki, S., Yamada, J., and Fukuda, A. (2003). Synaptically activated Cl-accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. J. Neurophysiol. 90, 2752–2756. doi: 10.1152/jn.00142.2003
Jensen, M. S., Yaari, Y., Jiruska, P., Curtis, M. D., Jefferys, J. G. R., Schevon, C. A., et al. (2014). Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J. Neurophysiol. 77, 1224–1233.
Jiang, C., and Haddad, G. G. (1991). Effect of anoxia on intracellular and extracellular potassium activity in hypoglossal neurons in vitro. J. Neurophysiol. 66, 103–111.
Kaczmarek, L. K. (2013). Slack, slick and sodium-activated potassium channels. ISRN Neurosci. 2013, 1–14. doi: 10.1155/2013/354262
Kaila, K., and Voipio, J. (1987). Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature 330, 163–165. doi: 10.1038/330163a0
Kaila, K., Lamsa, K., Smirnov, S., Taira, T., and Voipio, J. (1997). Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J. Neurosci. 17, 7662–7672.
Kaila, K., Pasternack, M., Saarikoski, J., Voipio, J., and Britain, G. (1989). Influence of GABA-gated bicarbonate conductance on potential, current and intracellular chloride in crayfish muscle fibres. J. Physiol. 416, 161–181. doi: 10.1113/jphysiol.1989.sp017755
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M., and Voipio, J. (2014a). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. doi: 10.1038/nrn3819
Kaila, K., Ruusuvuori, E., Seja, P., Voipio, J., and Puskarjov, M. (2014b). GABA actions and ionic plasticity in epilepsy. Curr. Opin. Neurobiol. 26, 34–41. doi: 10.1016/j.conb.2013.11.004
Kaila, K., Saarikoski, J., and Voipio, J. (1990). Mechanism of action of GABA on intracellular pH and on surface pH in crayfish muscle fibres. J. Physiol. 427, 241–260. doi: 10.1113/jphysiol.1990.sp018170
Kaila, K., Voipio, J., Paalasmaa, P., Pasternack, M., and Deisz, R. (1993). The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J. Physiol. 464, 273–289. doi: 10.1113/jphysiol.1993.sp019634
Karus, C., Mondragão, M. A., Ziemens, D., and Rose, C. R. (2015). Astrocytes restrict discharge duration and neuronal sodium loads during recurrent network activity. Glia 63, 936–957. doi: 10.1002/glia.22793
Kneen, M., Farinas, J., Li, Y., and Verkman, A. S. (1998). Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 74, 1591–1599. doi: 10.1016/s0006-3495(98)77870-1
Kofuji, P., and Newman, E. A. (2004). Potassium buffering in the central nervous system. Neuroscience 129, 1045–1056. doi: 10.1016/j.neuroscience.2004.06.008
Krishnan, G. P., and Bazhenov, M. (2011). Ionic dynamics mediate spontaneous termination of seizures and postictal depression state. J. Neurosci. 31, 8870–8882. doi: 10.1523/jneurosci.6200-10.2011
Krishnan, G. P., Filatov, G., Shilnikov, A., and Bazhenov, M. (2015). Electrogenic properties of the Na+/K+ ATPase control transitions between normal and pathological brain states. J. Neurophysiol. 113, 3356–3374. doi: 10.1152/jn.00460.2014
Kuner, T., and Augustine, G. J. (2000). A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27, 447–459. doi: 10.1016/S0896-6273(00)00056-8
Kuramoto, T., and Haber, B. (1981). The K+ liquid ion exchange electrode system: responses to drugs and neurotransmitters. J. Neurosci. Res. 6, 37–48. doi: 10.1002/jnr.490060105
Kyrozis, A., and Reichling, D. B. (1995). Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J. Neurosci. Methods. 57, 27–35. doi: 10.1016/0165-0270(94)00116-x
Lamsa, K., and Kaila, K. (1997). Ionic mechanisms of spontaneous GABAergic events in rat hippocampal slices exposed to 4-aminopyridine. J. Neurophysiol. 78, 2582–2591.
Langer, J., and Rose, C. R. (2009). Synaptically induced sodium signals in hippocampal astrocytes in situ. J. Physiol. 587, 5859–5877. doi: 10.1113/jphysiol.2009.182279
Lesage, F. (2003). Pharmacology of neuronal background potassium channels. Neuropharmacology 44, 1–7. doi: 10.1016/s0028-3908(02)00339-8
Looger, L. L., and Griesbeck, O. (2012). Genetically encoded neural activity indicators. Curr. Opin. Neurobiol. 22, 18–23. doi: 10.1016/j.conb.2011.10.024
Makani, S., and Chesler, M. (2010). Rapid rise of extracellular pH evoked by neural activity is generated by the plasma membrane calcium ATPase. J. Neurophysiol. 103, 667–676. doi: 10.1152/jn.00948.2009
Markova, O., Mukhtarov, M., Real, E., Jacob, Y., and Bregestovski, P. (2008). Genetically encoded chloride indicator with improved sensitivity. J. Neurosci. Methods. 170, 67–76. doi: 10.1016/j.jneumeth.2007.12.016
McCormick, D. A., and Contreras, D. (2001). On the cellular and network bases of epileptic seizures. Annu. Rev. Physiol. 63, 815–846. doi: 10.1146/annurev.physiol.63.1.815
McDougal, D. B., and Osborn, L. A. (1976). Post-tetanic hyperpolarization, sodium-potassium-activated adenosine triphosphatase and high energy phosphate levels in garfish olfactory nerve. J. Physiol. 256, 41–60. doi: 10.1113/jphysiol.1976.sp011310
McLaughlin, S. G., Szabo, G., and Eisenman, G. (1971). Divalent ions and the surface potential of charged phospholipid membranes. J. Gen. Physiol. 58, 667–687. doi: 10.1085/jgp.58.6.667
Meier, S. D., Kovalchuk, Y., and Rose, C. R. (2006). Properties of the new fluorescent Na+ indicator CoroNa Green: Comparison with SBFI and confocal Na+ imaging. J. Neurosci. Methods 155, 251–259. doi: 10.1016/j.jneumeth.2006.01.009
Minta, A., and Tsien, R. Y. (1989). Fluorescent indicators for cytosolic sodium. J. Biol. Chem. 264, 19449–19457.
Morth, J. P., Pedersen, B. P., Toustrup-Jensen, M. S., Sørensen, T. L. M., Petersen, J., Andersen, J. P., et al. (2007). Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049. doi: 10.1038/nature06419
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2008). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506. doi: 10.1016/S1474-4422(08)70092-9(08)70092-9
Olsson, T., Broberg, M., Pope, K. J., Wallace, A., Mackenzie, L., Blomstrand, F., et al. (2006). Cell swelling, seizures and spreading depression: an impedance study. Neuroscience 140, 505–515. doi: 10.1016/j.neuroscience.2006.02.034
Opie, L. (1965). Effect of extracellular pH on function and metabolism of isolated perfused rat heart. Am. J. Physiol. 209, 1075–1080.
Pal, S., Sombati, S., Limbrick, D. D., and DeLorenzo, R. J. (1999). In vitro status epilepticus causes sustained elevation of intracellular calcium levels in hippocampal neurons. Brain Res. 851, 20–31. doi: 10.1016/s0006-8993(99)02035-1
Pasternack, M., Smirnov, S., and Kaila, K. (1996). Proton modulation of functionally distinct gabaa receptors in acutely isolated pyramidal neurons of rat hippocampus. Neuropharmacology 35, 1279–1288. doi: 10.1016/s0028-3908(96)00075-5
Pasternack, M., Voipio, J., and Kaila, K. (1993). Intracellular carbonic anhydrase activity and its role in GABA-induced acidosis in isolated rat hippocampal pyramidal neurones. Acta. Physiol. Scand. 148, 229–231. doi: 10.1111/j.1748-1716.1993.tb09553.x
Pumain, R., Ménini, C., and Heinemann, U. (1985). Chemical synaptic transmission is not necessary for epileptic seizures to persist in the baboon Papio papio. Exp. Neurol. 89, 250–258. doi: 10.1016/0014-4886(85)90280-8
Putney, L. K., and Barber, D. L. (2003). Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J. Biol. Chem. 278, 44645–44649. doi: 10.1074/jbc.m308099200
Quednau, B. D., Nicoll, D. A., and Philipson, K. D. (2004). The sodium/calcium exchanger family-SLC8. Pflugers. Arch. 447, 543–548. doi: 10.1007/s00424-003-1065-4
Raimondo, J. V., Joyce, B., Kay, L., Schlagheck, T., Newey, S. E., Srinivas, S., et al. (2013). A genetically-encoded chloride and pH sensor for dissociating ion dynamics in the nervous system. Front. Cell. Neurosci. 7:202. doi: 10.3389/fncel.2013.00202
Raimondo, J. V., Irkle, A., Wefelmeyer, W., Newey, S. E., and Akerman, C. J. (2012a). Genetically encoded proton sensors reveal activity-dependent pH changes in neurons. Front. Mol. Neurosci. 5:68. doi: 10.3389/fnmol.2012.00068
Raimondo, J. V., Markram, H., and Akerman, C. J. (2012b). Short-term ionic plasticity at GABAergic synapses. Front. Synaptic Neurosci. 4:5. doi: 10.3389/fnsyn.2012.00005
Ransom, B. R., Carlini, W. G., and Yamate, C. L. (1987). Tip size of ion-exchanger based K+ -selective microelectrodes. II. Effects on measurement of evoked [K+] 0 transients. Can. J. Physiol. Pharmacol. 65, 894–897. doi: 10.1139/y87-144
Rimmele, T. S., and Chatton, J.-Y. (2014). A novel optical intracellular imaging approach for potassium dynamics in astrocytes. PLoS One 9:e109243. doi: 10.1371/journal.pone.0109243
Ritchie, J. M., and Straub, R. W. (1957). The hyperpolarization which follows activity in mammalian non-medullated fibres. J. Physiol. 136, 80–97. doi: 10.1113/jphysiol.1957.sp005744
Rivera, C., Voipio, J., and Kaila, K. (2005). Two developmental switches in GABAergic signalling: the K+-Cl− cotransporter KCC2 and carbonic anhydrase CAVII. J. Physiol. 562, 27–36. doi: 10.1113/jphysiol.2004.077495
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipilä, S., et al. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 24, 4683–4691. doi: 10.1523/jneurosci.5265-03.2004
Rose, C. R. (2003). High-resolution Na+ imaging in dendrites and spines. Pflugers Arch. 446, 317–321. doi: 10.1007/s00424-003-1036-9
Rose, C. R., and Deitmer, J. W. (1995). Stimulus-evoked changes of extra- and intracellular pH in the leech central nervous system. II. Mechanisms and maintenance of pH homeostasis. J. Neurophysiol. 73, 132–140.
Rose, C. R., and Konnerth, A. (2001). NMDA receptor-mediated Na+ signals in spines and dendrites. J. Neurosci. 21, 4207–4214.
Ruusuvuori, E., Li, H., Huttu, K., Palva, J. M., Smirnov, S., Rivera, C., et al. (2004). Carbonic anhydrase isoform VII acts as a molecular switch in the development of synchronous gamma-frequency firing of hippocampal CA1 pyramidal cells. J. Neurosci. 24, 2699–2707. doi: 10.1523/jneurosci.5176-03.2004
Schwiening, C. J., Kennedy, H. J., and Thomas, R. C. (1993). Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proc. R. Soc. Lond. B Biol. Sci. 253, 285–289. doi: 10.1098/rspb.1993.0115
Singer, W., and Lux, H. D. (1975). Extracellular potassium gradients and visual receptive fields in the cat striate cortex. Brain Res. 96, 378–383. doi: 10.1016/0006-8993(75)90751-9
Smart, T. G., and Constanti, A. (1982). A novel effect of zinc on the lobster muscle GABA receptor. Proc. R. Soc. Lond. B Biol. Sci. 215, 327–341. doi: 10.1098/rspb.1982.0045
Somjen, G. G. (1980). Stimulus-evoked and seizure-related responses of extracellular calcium activity in spinal cord compared to those in cerebral cortex. J. Neurophysiol. 44, 617–632.
Sparing, R., Dafotakis, M., Buelte, D., Meister, I. G., and Noth, J. (2007). Excitability of human motor and visual cortex before, during and after hyperventilation. J. Appl. Physiol. 102, 406–411. doi: 10.1152/japplphysiol.00770.2006
Spray, D., Harris, A., and Bennett, M. (1981). Gap junctional conductance is a simple and sensitive function of intracellular pH. Science 211, 712–715. doi: 10.1126/science.6779379
Staley, K. J., and Proctor, W. R. (1999). Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3− transport. J. Physiol. 519, 693–712. doi: 10.1111/j.1469-7793.1999.0693n.x
Staley, K. J., Soldo, B. L., and Proctor, W. R. (1995). Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269, 977–981. doi: 10.1126/science.7638623
Steinhäuser, C., and Seifert, G. (2002). Glial membrane channels and receptors in epilepsy: impact for generation and spread of seizure activity. Eur. J. Pharmacol. 447, 227–237. doi: 10.1016/s0014-2999(02)01846-0
Syková, E., and Nicholson, C. (2008). Diffusion in brain extracellular space. Physiol. Rev. 88, 1277–1340. doi: 10.1152/physrev.00027.2007
Sypert, G. W., and Ward, A. A. (1974). Changes in extracellular potassium activity during neocortical propagated seizures. Exp. Neurol. 45, 19–41. doi: 10.1016/0014-4886(74)90097-1
Tabb, J. S., Kish, P. E., Van Dyke, R., and Ueda, T. (1992). Glutamate transport into synaptic vesicles. Roles of membrane potential, pH gradient and intravesicular pH. J. Biol. Chem. 267, 15412–15418.
Takeuchi, A., and Takeuchi, N. (1967). Anion permeability of the inhibitory post-synaptic membrane of the crayfish neuromuscular junction. J. Physiol. 191, 575–590. doi: 10.1113/jphysiol.1967.sp008269
Tang, C. M., Dichter, M., and Morad, M. (1990). Modulation of the N-Methyl-D-Aspartate Channel by Extracellular H+. Proc. Natl. Acad. Sci. U S A 87, 6445–6449. doi: 10.1073/pnas.87.16.6445
Taylor, C. P., and Dudek, F. E. (1982). Synchronous neural afterdischarges in rat hippocampal slices without active chemical synapses. Science 218, 810–812. doi: 10.1126/science.7134978
Thompson, S. M., and Gähwiler, B. H. (1989). Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J. Neurophysiol. 61, 501–511.
Thompson, S. M., and Prince, D. A. (1986). Activation of electrogenic sodium pump in hippocampal CA1 neurons following glutamate-induced depolarization. J. Neurophysiol. 56, 507–522.
Tolner, E. A., Hochman, D. W., Hassinen, P., Otáhal, J., Gaily, E., Haglund, M. M., et al. (2011). Five percent CO2 is a potent, fast-acting inhalation anticonvulsant. Epilepsia 52, 104–114. doi: 10.1111/j.1528-1167.2010.02731.x
Tombaugh, G. C. (1998). Intracellular pH Buffering Shapes Activity-Dependent Ca2+ Dynamics in Dendrites of CA1 Interneurons. J. Neurophysiol. 80, 1702–1712.
Tombaugh, G. C., and Somjen, G. G. (1997). Differential Sensitivity to Intracellular pH Among High- and Low-Threshold Ca2+ Currents in Isolated Rat CA1 Neurons. J. Neurophysiol. 77, 639–653.
Trapp, S., Lückermann, M., Brooks, P. A., and Ballanyi, K. (1996). Acidosis of rat dorsal vagal neurons in situ during spontaneous and evoked activity. J. Physiol. 496, 695–710. doi: 10.1113/jphysiol.1996.sp021720
Traynelis, S. F., and Cull-Candy, S. G. (1990). Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 345, 347–350. doi: 10.1038/345347a0
Traynelis, S. F., and Dingledine, R. (1988). Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J. Neurophysiol. 59, 259–276.
Trevelyan, A. J., Sussillo, D., and Yuste, R. (2007). Feedforward inhibition contributes to the control of epileptiform propagation speed. J. Neurosci. 27, 3383–3387. doi: 10.1523/jneurosci.0145-07.2007
Tyzio, R., Minlebaev, M., Rheims, S., Ivanov, A., Jorquera, I., Holmes, G. L., et al. (2008). Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus. Eur. J. Neurosci. 27, 2515–2528. doi: 10.1111/j.1460-9568.2008.06234.x
Urbanics, R., Leniger-Follert, E., and Lübbers, D. W. (1978). Time course of changes of extracellular H+ and K+ activities during and after direct electrical stimulation of the brain cortex. Pflugers Arch. 378, 47–53.
Velisek, L., and Veliskova, J. (1993). Anticonvulsant action of carbonic anhydrase inhibition. Sb. Lek. 95, 161–171.
Viitanen, T., Ruusuvuori, E., Kaila, K., and Voipio, J. (2010). The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J. Physiol. 588, 1527–1540. doi: 10.1113/jphysiol.2009.181826
Volterra, A., Liaudet, N., and Savtchouk, I. (2014). Astrocyte Ca(2+) signalling: an unexpected complexity. Nat. Rev. Neurosci. 15, 327–335. doi: 10.1038/nrn3725
Vyklický, L., Vlachová, V., and Krusek, J. (1990). The effect of external pH changes on responses to excitatory amino acids in mouse hippocampal neurones. J. Physiol. 430, 497–517. doi: 10.1113/jphysiol.1990.sp018304
Walker, J. L. (1971). Ion Specific Liquid Ion Exchanger Microelectrodes. Anal. Chem. 43, 89A–93A. doi: 10.1021/ac60298a039
Wallraff, A., Köhling, R., Heinemann, U., Theis, M., Willecke, K., and Steinhäuser, C. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26, 5438–5447. doi: 10.1523/jneurosci.0037-06.2006
Walz, W. (2000). Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 36, 291–300. doi: 10.1016/s0197-0186(99)00137-0
Wang, G. J., Randall, R. D., and Thayer, S. A. (1994). Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J. Neurophysiol. 72, 2563–2569.
West, M. J., and Gundersen, H. J. (1990). Unbiased stereological estimation of the number of neurons in the human hippocampus. J. Comp. Neurol. 296, 1–22. doi: 10.1002/cne.902960102
Wood, J. N. (2001). Voltage-gated sodium channels. Curr. Opin. Pharmacol. 1, 17–21. doi: 10.1016/S1471-4892(01)00007-8
Wu, Y., Wang, W., Díckez-Sampedro, A., and Richerson, G. B. (2007). Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 56, 851–865. doi: 10.1016/j.neuron.2007.10.021
Xiong, Z. Q., and Stringer, J. L. (2000). Regulation of extracellular pH in the developing hippocampus. Brain Res. Dev. Brain Res. 122, 113–117. doi: 10.1016/s0165-3806(00)00057-2
Xiong, Z. Q., Saggau, P., and Stringer, J. L. (2000). Activity-dependent intracellular acidification correlates with the duration of seizure activity. J. Neurosci. 20, 1290–1296.
Yaari, Y., Konnerth, A., and Heinemann, U. (1986). Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. II. Role of extracellular potassium. J. Neurophysiol. 56, 424–438.
Yang, Z., and Jiang, C. (1999). Opposite effects of pH on open-state probability and single channel conductance of kir4.1 channels. J. Physiol. 520, 921–927. doi: 10.1111/j.1469-7793.1999.00921.x
Yasiry, Z., and Shorvon, S. D. (2014). The relative effectiveness of five antiepileptic drugs in treatment of benzodiazepine-resistant convulsive status epilepticus: a meta-analysis of published studies. Seizure 23, 167–174. doi: 10.1016/j.seizure.2013.12.007
Yellen, G. (2002). The voltage-gated potassium channels and their relatives. Nature 419, 35–42. doi: 10.1038/nature00978
Keywords: epilepsy, seizures, ion dynamics, potassium, sodium, calcium, chloride, pH
Citation: Raimondo JV, Burman RJ, Katz AA and Akerman CJ (2015) Ion dynamics during seizures. Front. Cell. Neurosci. 9:419. doi: 10.3389/fncel.2015.00419
Received: 14 July 2015; Accepted: 04 October 2015;
Published: 21 October 2015.
Edited by:
Igor Medina, INMED/INSERM U-901, FranceReviewed by:
Rustem Khazipov, Institut National de la Santé et de la Recherche Médicale, FranceCopyright © 2015 Raimondo, Burman, Katz and Akerman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph V. Raimondo, am9zZXBoLnJhaW1vbmRvQHVjdC5hYy56YQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.