 Rowan A. Radford1
Rowan A. Radford1 Marco Morsch1
Marco Morsch1 Stephanie L. Rayner1
Stephanie L. Rayner1 Nicholas J. Cole1
Nicholas J. Cole1 Dean L. Pountney2
Dean L. Pountney2 Roger S. Chung1*
Roger S. Chung1*- 1Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, NSW, Australia
- 2Menzies Health Institute Queensland, Griffith University, Gold Coast, QLD, Australia
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are two progressive, fatal neurodegenerative syndromes with considerable clinical, genetic and pathological overlap. Clinical symptoms of FTD can be seen in ALS patients and vice versa. Recent genetic discoveries conclusively link the two diseases, and several common molecular players have been identified (TDP-43, FUS, C9ORF72). The definitive etiologies of ALS and FTD are currently unknown and both disorders lack a cure. Glia, specifically astrocytes and microglia are heavily implicated in the onset and progression of neurodegeneration witnessed in ALS and FTD. In this review, we summarize the current understanding of the role of microglia and astrocytes involved in ALS and FTD, highlighting their recent implications in neuroinflammation, alterations in waste clearance involving phagocytosis and the newly described glymphatic system, and vascular abnormalities. Elucidating the precise mechanisms of how astrocytes and microglia are involved in ALS and FTD will be crucial in characterizing these two disorders and may represent more effective interventions for disease progression and treatment options in the future.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal and rapidly progressing multisystem neurodegenerative syndrome, characterized by the degeneration of the motor neurons (MNs) in the motor cortex, brainstem and spinal cord (Hardiman et al., 2011). Symptoms present first as focal upper and/or lower MN dysfunction of a skeletal muscle group which progressively deteriorates, ultimately spreading to other muscle groups (Ravits, 2014). Disease progression is rapid, with 50% of patients dying due to respiratory complications within 3 years of symptom onset (Kiernan et al., 2011). Over the last 25 years, it has become increasingly apparent that ALS shares significant overlap with another progressive and fatal neurodegenerative syndrome Frontotemporal dementia (FTD). Up to 50% of patients with ALS develop FTD symptoms and approximately 15% of FTD patients display MN dysfunction typical of ALS (Ng et al., 2015). Besides this clinical connection, ALS and FTD also share significant genetic and pathological overlap (Bennion Callister and Pickering-Brown, 2014), represented in Figure 1. However, the causal mechanism/s of both syndromes are currently unknown and treatment is largely symptomatic (Hardiman et al., 2011; Piguet et al., 2011).
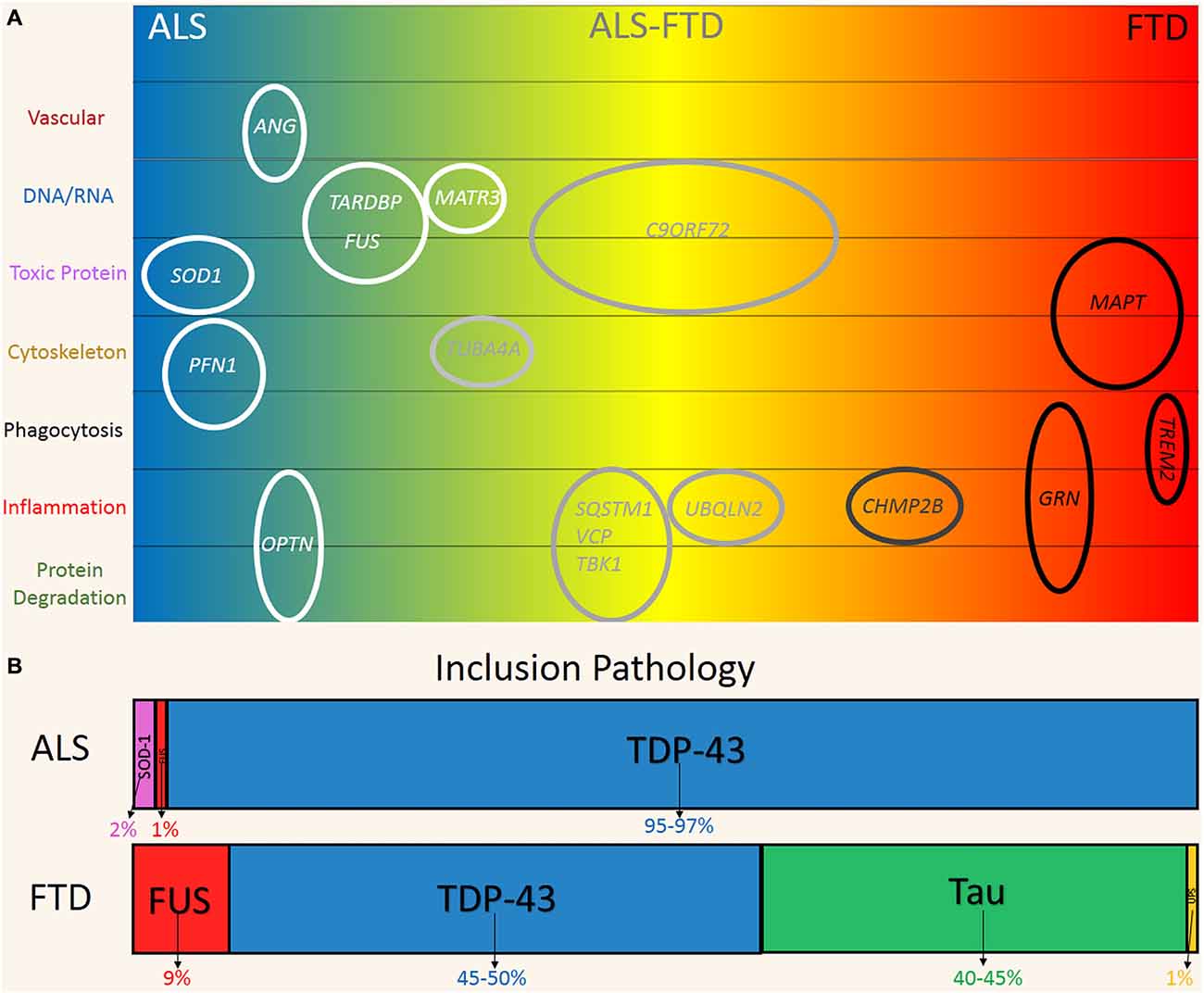
Figure 1. Genetic and pathological overlap between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). (A) Familial and sporadic genetic mutations were linked to the clinical phenotypes on the ALS (blue) and FTD (red) spectrum. Genes are plotted according to their hypothesized mechanism in relation to disease (top to bottom). (B) Pathological protein inclusions are a hallmark of ALS and FTD, reflecting the significant overlap on the disease spectrum. FUS (Red) and TDP-43 (Blue) inclusions are found in both ALS and FTD. Predominate SOD1 (Pink) and Tau (Green) is more indicative of ALS and FTD respectively. FTD-UPS (Yellow) is found in ~1% of cases and represent cases of familial CHMP2B mutations.
Five years ago, large hexanucleotide repeat expansions (~100–1600 G4C2 repeats) of intronic regions of the C9ORF72 gene were discovered in sporadic and familial forms of ALS and FTD (Renton et al., 2014). These studies provided seminal evidence for a direct molecular link between these two conditions. The repeat expansions are now recognized as the most common known mutation in both familial and sporadic ALS and FTD. Expansions have been identified in up to 40% and 25% of familial cases and ~6% and 7% of sporadic or seemingly non-inherited forms of ALS and FTD respectively (Robberecht and Philips, 2013; Renton et al., 2014). Multiple other genes have also been linked to ALS and FTD. Mutations in the genes TARDBP and fused in sarcoma (FUS), which respectively encode for the proteins TDP-43 and FUS, are associated with ~9% of familial, 2% of sporadic cases of ALS and rarely in FTD (Renton et al., 2014). The MAPT gene encodes microtubule associated protein tau (tau) and mutations have been identified in ~2–11% of familial FTD cases (Sieben et al., 2012). SOD1 is another gene that is strongly associated with ALS with mutations found in 12–20% of familial and 1–2% of sporadic cases (Al-Chalabi et al., 2012). With the exception of MAPT these genes are not segregated to neurons and are expressed by glia and various other cell types, which suggests a multicellular pathogenesis.
The presence of ubiquitinated, cytoplasmic inclusions in neurons and some glia is a pathological hallmark shared by the two disorders (Ng et al., 2015). Figure 1B shows the distribution of inclusion pathology seen in both ALS and FTD. In ~95% of ALS and 50% of FTD cases, these inclusions are predominately comprised of TDP-43. FUS protein inclusions are found in ~1% and 10% of ALS and FTD cases respectively (Mackenzie et al., 2010). Tau inclusion pathology is more characteristic of FTD (~40% of cases) and is only found rarely in cases of ALS (Dickson et al., 2011; Ng et al., 2015). At the other end of the spectrum, SOD-1 inclusion pathology is seen in ~2% of ALS cases and is incredibly rare in FTD with only one case reported (Bennion Callister and Pickering-Brown, 2014). Taken together, the pathogenic and genetic features represent a clear commonality between ALS and FTD, which are now believed to exist on a phenotypic continuum (Ling et al., 2013).
In addition to protein inclusions, another feature of ALS and FTD neuropathology is reactive gliosis, which is characterized by astrocytic hypertrophy and microglial proliferation (Al-Chalabi et al., 2012; Ng et al., 2015). Reactive gliosis is an indicator of neuroinflammation (Streit et al., 2004) and occurs in areas of neuronal loss and inclusion pathology in ALS and FTD (Brettschneider et al., 2012). Studies over the past 15 years have strongly indicated that ALS and FTD propagate via multiple cell types, with reactive gliosis being heavily implicated (Ilieva et al., 2009). Astrocytes and microglia in particular have been shown to be associated with disease progression and spreading (Philips and Robberecht, 2011).
Various pathways have been implicated to contribute to ALS and FTD neurodegeneration, including inflammation, RNA toxicity and altered splicing/expression (DNA/RNA homeostasis), and cytoskeletal, vascular and protein dysfunction (Lagier-Tourenne and Cleveland, 2009; Garbuzova-Davis et al., 2012; Ravits et al., 2013). Both microglia and astrocytes can be compromised through a variety of these signaling pathways that result in deregulated glia–motor neuron communication. However, the precise contribution of glial cells and their exact involvement in ALS and FTD pathology is currently under intense investigation. Here we aim to summarize the established and novel implications of astrocyte and microglia in ALS and FTD, identifying key aspects of the neuroinflammatory involvement, microglia phagocytosis, defective waste clearance and circulatory dysfunction.
The Pathogenic Role of Neuroinflammatory Glia in ALS
Inflammatory glia have been repeatedly reported in animal models of FTD (see Roberson, 2012). Yet, current models of tau pathology describe clinically a very heterogeneous group, including FTD and Parkinsonism, and our focus in this review will therefore be on the neuropathological characterization of inflammatory glia in ALS. Significant insight into the pathogenic role of glia in disease progression has been revealed through allografted chimeric and conditional knock out studies using ALS mutant SOD-1 rodent models (see Robberecht and Philips, 2013). These studies have convincingly shown that both astrocytes and microglia/myeloid progenitors significantly influence the progression of neurodegeneration in these models. More recent studies have added weight to the existing evidence that astrocytes contribute to ALS progression by utilizing mice xenografted with human glial progenitors generated from induced pluripotent stem cells (iPSCs). Grafted glial progenitors from patient iPSCs with familial SOD1 mutations differentiated into astrocytes and induced MN degeneration and motor deficits in WT mice. Progenitors from healthy individuals without ALS linked mutations did not contribute to an ALS phenotype (Chen et al., 2015). In another study, iPSC derived glial progenitors from healthy individuals (lacking ALS mutations) formed astrocytes that increased the survival of MNs when transplanted at disease onset in a mutant SOD-1 mouse model (Kondo et al., 2014). Such in vivo studies provide important insight into the pathogenic role of ALS patient glia and demonstrate a potential mechanism of how glia can influence the progression of neurodegeneration (i.e., modifying the molecular phenotype and function).
Notably, astrocytes retrieved from post-mortem central nervous system (CNS) of familial (SOD1 and unidentified) and sporadic cases were also neurotoxic to co-cultured MNs (Haidet-Phillips et al., 2011; Re et al., 2014). While the necrotic environment of post-mortem tissue has to be considered, another study reported that astrocytes generated from sporadic and familial (C9ORF72 and SOD1) ALS iPSCs were also toxic to co-cultured MNs (Meyer et al., 2014). These findings correlate with studies that report the neurotoxicity of glia derived from SOD-1 transgenic mice (Di Giorgio et al., 2007; Nagai et al., 2007). Collectively, this glia-induced neurotoxicity suggests a common mechanism in both sporadic and familial ALS.
Different experimental approaches further suggest that astrocytes are neurotoxic to MNs in the context of ALS. A recently reported ubiquitous RNAi knockdown of TDP-43 in mice led to severe neurodegeneration and an ALS phenotype. The study revealed a greater knockdown of TDP-43 in astrocytes compared to MNs, significant astrogliosis and marked upregulation of lipocalin-2 expression in reactive astrocytes (Yang et al., 2014). Lipocalin-2 is a feature of inflammatory astrocytes (Zamanian et al., 2012) and can enhance reactive astrogliosis via autocrine signaling (Lee et al., 2009). Specific knockout of TDP-43 in cortical and MNs in mice produced a less severe phenotype, which further highlights the contribution of multiple cell types in ALS and FTD (Wu et al., 2012b; Iguchi et al., 2013).
Interestingly, overexpression of ALS associated mutant TDP-43 driven by an astrocytic promoter was sufficient to cause MN degeneration in rats and was also associated with marked up-regulation of lipocalin-2 in reactive astrocytes (Tong et al., 2013). Overexpression of mutant TDP-43 in rat neurons also induced gliosis and lipocalin-2 upregulation in surrounding reactive astrocytes. The analysis of post-mortem frontal cortex of FTD patients likewise revealed an increase in lipocalin-2, and that recombinant lipocalin-2 was exclusively toxic to cultured neurons (Bi et al., 2013). Huang et al. (2014) reported that inducible overexpression of mutant TDP-43 can increase lipocalin-2 expression in cultured astrocytes and while pathological mutations can lead to alterations in RNA homeostasis similar to those seen in knockdown studies. Further studies are clearly needed to understand the mechanism of pathological TDP-43 and the ambiguous role of lipocalin-2 in ALS and FTD pathogenesis and its potential as a therapeutic target or biomarker for assessing neuroinflammation.
Recent advances in patient neuroimaging have allowed direct visualization of neuroinflammation such as gliosis. Positron Emission Tomography (PET) and Single Positron Emission Computed Tomography (SPECT) or Magnetic Resonance Imaging (MRI) in patients targeting activated microglial receptors or astrocytic metabolites have shown gliosis throughout various symptomatic stages of ALS and FTD while absent in non-disease controls (Cagnin et al., 2004; Chiò et al., 2014). This, along with data from animal models, strongly indicates that gliosis is unlikely to represent a specific event only seen in post-mortem tissue at the end stage of ALS and FTD. More recently, modalities that specifically evaluate astrocyte metabolism (e.g., radiopharmaceutical acetate derivatives; Marik et al., 2009; Ouyang et al., 2014) could be used to monitor astrogliosis in patients more accurately. New imaging ligands such as the modern translocator protein ligands allow for tracking microglia activation with higher specificity and reduced radiation dosage (Corcia et al., 2012). Collectively, these rapidly improving technologies are revealing important information regarding the involvement of astrocytes and microglia in various stages of degeneration in ALS and FTD patients.
The Role of Glial Phagocytosis in ALS and FTD
Astrocytes have been found to highly express an array of phagocytic receptors and actively contribute to this process by phagocytosing synapses and axonal mitochondria in the developing and adult CNS (Chung et al., 2013; Davis et al., 2014). Nonetheless, microglial cells have been shown to be the main culprit for phagocytosis and synaptic pruning that is crucial to CNS function by removing potentially toxic debris and the reorganization of the CNS connectome (Neumann et al., 2009; Xavier et al., 2014). The altered phagocytic activity of microglia has been implicated in multiple neurodegenerative disorders. This link has been highlighted through three discoveries of genetic mutations in phagocytosis-related genes in ALS and FTD patients (Figure 2A). TREM2 is exclusively expressed by microglia in the CNS (Colonna, 2003; Thrash et al., 2009) and missense variants have been recognized as a risk factor for ALS, FTD, Alzheimer’s and Parkinson’s disease (Rayaprolu et al., 2013; Cady et al., 2014; Harms et al., 2014). Furthermore, recessive mutations in TREM2 are also associated to an orphan neurodegenerative condition known as Nasu-Hakola Syndrome with patients presenting with lytic bone cysts, atypical FTD and psychiatric dysfunction (Kaneko et al., 2010). Interestingly homozygous and compound heterozygous mutations are linked to a familial FTD-like disorder without bone involvement (Kaneko et al., 2010; Guerreiro et al., 2013; Borroni et al., 2014). These mutations in TREM2 are proposed to confer loss of TREM2 protein function, which causes decreased microglial phagocytosis and altered inflammatory responses (Kleinberger et al., 2014; Wang et al., 2015). Additionally, dysfunctional microglial phagocytosis is directly linked to FTD via mutations in GRN (progranulin) and may confer elevated risk of developing Alzheimer’s disease and ALS (Petkau and Leavitt, 2014). Progranulin is expressed by neurons and microglia and following secretion can act as a neuroinflammatory modulator (Petkau et al., 2010), and facilitate microglial recognition of apoptotic cells and potentially toxic elements such as amyloid beta (Aβ; Pickford et al., 2011; Minami et al., 2014). Also, loss of function mutations in PFN1 (profilin 1) have been identified in familial ALS (Wu et al., 2012a) and profilin has been shown to be essential in regulating actin dynamics necessary for phagocytosis, phagosome formation and is upregulated in microglia following insult (Pearson et al., 2003; Dong et al., 2004; Kim et al., 2012). Research into how these PFN1 mutations influence microglia function will be crucial to understanding the pathogenicity of those mutations. While progranulin and profilin 1 mutations are likely to impact multiple cell types (especially neurons), cell specific transcriptome analysis of the mouse cortex indicates that all three genes are highly transcribed in microglia (Zhang et al., 2014). Taken together, these studies highlight a potential link between reduced microglial phagocytic capacity and the development of neurodegeneration, ALS and particularly FTD and is represented in Figure 2A.
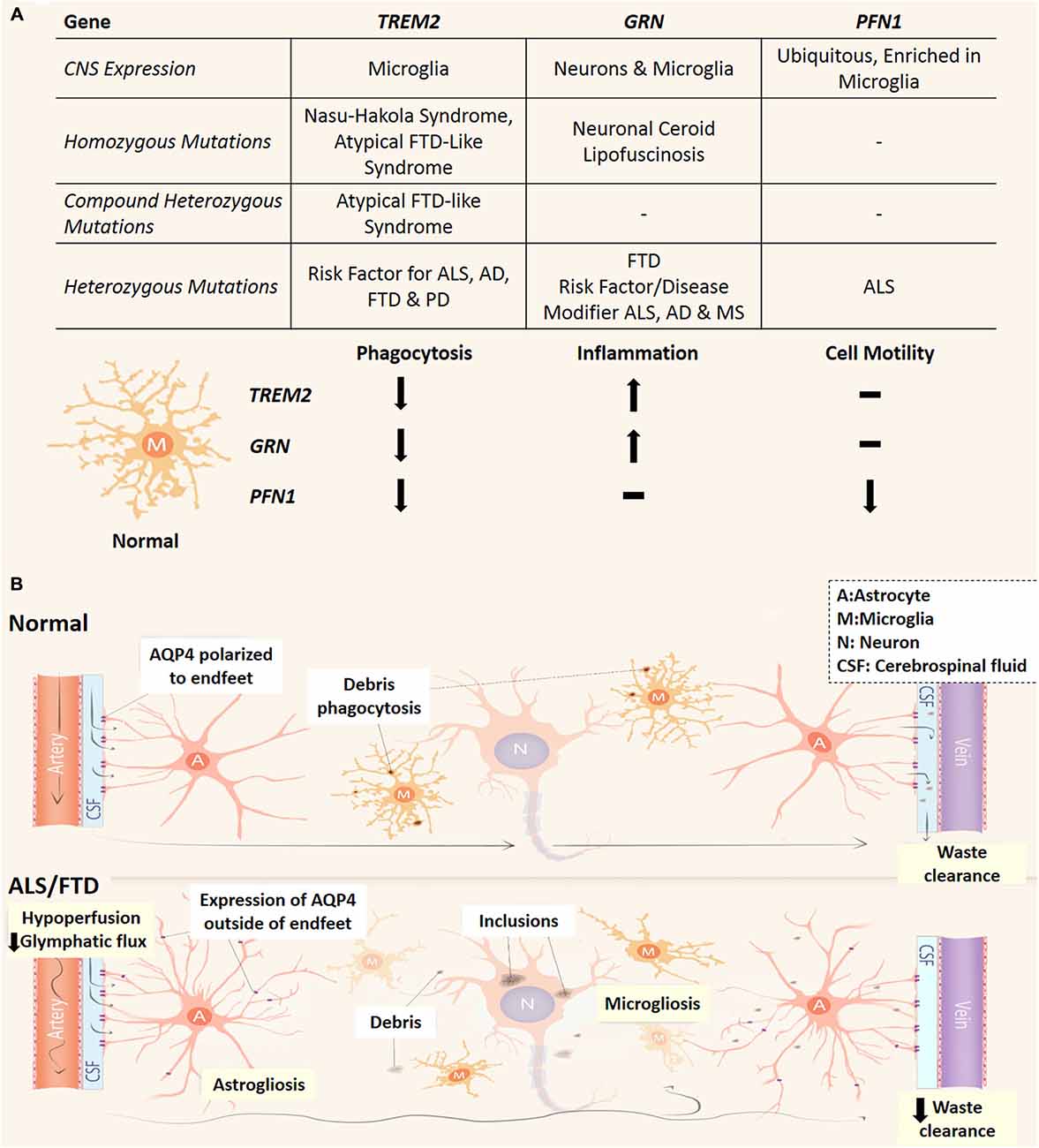
Figure 2. Phagocytic dysfunction and the glymphatic pathway and its (potential) involvement in ALS and FTD. (A) Three genes (TREM2, GRN and PFN1) which link microglial phagocytic dysfunction to ALS and FTD and their effect on microglial phenotype. All three are predicted loss of function mutations which decrease the phagocytic capacity of microglia. Depending on the type of mutation/s to these genes different neurodegenerative conditions can arise, while variants cause increase risk of developing neurodegenerative conditions or worsen prognosis. Neuronal Ceroid Lipofuscinosis is a type of neurodegenerative lysosomal disorder which has been reported in patients PRGN null patients (Petkau and Leavitt, 2014). AD, Alzheimer’s disease; PD, Parkinson’s disease; MS, multiple sclerosis. (B) In the normal CNS, CSF circulates in a perivascular compartment driven by arterial pulse pressure. Astrocytic endfeet cover the perivascular space and facilitate movement of CSF into the parenchyma largely via AQP4. This fluid flow through the interstitial space allows the removal of debris from the extracellular space before draining into venous perivascular compartments. Microglia also remove potentially toxic waste via phagocytosis and dysfunctional microglial phagocytosis is linked to ALS and FTD pathogenesis (see A). In the CNS of ALS and FTD patients, glymphatic function and microglial phagocytosis may be compromised and contribute to neurodegeneration. Reactive astrocytes conceivably lose AQP4 polarization and express it elsewhere. This may lead to turbulent flow through the interstitium. Cerebral vascular function is reduced in patients which could potentially lead to decreased pressure for glymphatic function. A, astrocytes; M, microglia; N, neuron; CSF, cerebrospinal fluid.
Phagocytosis is incomplete without the intracellular breakdown of engulfed material. Autophagy is an essential component of this internal degradation inside phagocytes as autophagosome-lysosome fusion is crucial to break down this debris. Alterations to this pathway are directly implicated in ALS, ALS-FTD, FTD and/or Multisystem Proteinopathy pathogenesis through mutations to OPTN, SQST1, VCP and the recently discovered TBK1 (Renton et al., 2014; Freischmidt et al., 2015). These genes are vital to autophagosome formation, maturation and therefore crucial in LC3-assisted phagocytosis and intracellular waste clearance (Tresse et al., 2010; Seto et al., 2013). Interestingly these genes are also involved in inflammation as they can regulate Nuclear Factor-kappaB (NF-κB) signaling (Pomerantz and Baltimore, 1999; Asai et al., 2002; Zhu et al., 2007; Duran et al., 2008; Tresse et al., 2010; Seto et al., 2013). NF-κB is one of the major regulators of neuroinflammatory activation of glia (Zamanian et al., 2012) and its induction is seen in post-mortem ALS tissue, mutant SOD-1 and TDP-43 models (Migheli et al., 1997; Swarup et al., 2011a,b; Frakes et al., 2014). Any defects to these genes would potentially impact upon the function of innate immune cells (Deretic et al., 2013), particularly those segregated in the CNS like microglia and astrocytes. Determining how these genes are expressed, regulated and function in astrocytes and microglia will provide important insights into the neurodegenerative mechanisms underlying ALS and FTD.
The Emerging Role of the Glymphatic System and Vascular Function in ALS and FTD
While phagocytosis of apoptotic neurons and cellular debris is a major pathway for removal of toxic substances within the CNS, the glymphatic system has recently emerged as a different clearance pathway with important immune functions (Iliff et al., 2012). The glymphatic system mediates circulation of cerebrospinal fluid (CSF) and exchange of interstitial fluid to remove extracellular waste (such as Aβ and tau proteins) and distribute compounds such as glucose, lipids, and neuromodulators to the CNS (Thrane et al., 2013; Xie et al., 2013; Iliff et al., 2014). The glymphatics run parallel to the CNS vasculature in a paravascular space enclosed by astrocytic endfeet (Figure 2B). Accordingly, the glymphatic system (reviewed in Jessen et al., 2015) relies heavily upon the vasculature in order to function as pressure differentials between arteries and veins propel the CSF through the CNS parenchyma (Iliff et al., 2013). The bulk of glymphatic flow through the CNS is facilitated by aquaporin-4 (AQP4), a water transporter specific to astrocytes in the CNS and polarized to their endfeet (Iliff et al., 2012; Papadopoulos and Verkman, 2013). Interestingly, elevated AQP4 with loss of astrocytic endfeet depolarization has been reported in transgenic mutant SOD-1 rat models and reactive astrocytes have been shown to up-regulate AQP4 elsewhere in the astrocytic arbor apart from the endfeet (Bataveljić et al., 2012; Papadopoulos and Verkman, 2013). Glymphatic flow significantly increased during non-rapid eye movement sleep and was largely controlled by norepinephrine (which modulates arousal) acting upon astrocytic α-adrenoceptors (Xie et al., 2013; Paukert et al., 2014). Increased levels of norepinephrine have been observed in the CSF, plasma and spinal cord tissue of ALS patients (Brooks et al., 1980; Bertel et al., 1991) and norepinephrine CSF levels were positively correlated with the severity of dementia in FTD (Engelborghs et al., 2008). It has been hypothesized that increased CSF levels of norepinephrine could lead to decreased glymphatic function, while any aberrant expression of AQP4 could potentially create turbulent convective flux through the CNS interstitium, ultimately leading to decreased removal of neurotoxic metabolites (Kress et al., 2014; Jessen et al., 2015). MRI imaging techniques allow live-imaging of the glymphatic system (Iliff et al., 2013; Yang et al., 2013) and are a novel approach to detect flow abnormalities in the glymphatic system in ALS and FTD patients. Recent studies have identified lymphatic vessels present in the dura mater, which drains CNS interstitial fluid via the glymphatic system and CSF from the subarachnoid space (Aspelund et al., 2015; Louveau et al., 2015). As T cells are implicated in the progression of ALS patients and animal models (Philips and Robberecht, 2011) this would reflect a novel way for lymphocytes to monitor and interact with CNS tissue via the glymphatic system and potentially influence neuroinflammatory events in ALS and FTD.
Glymphatic function is intimately linked to vascular flow via the parallel anatomy and requirement of pressure differentials created by blood flow. During development and in the mature CNS, astrocytes and microglia are crucial to complex signaling cascades and angiogenesis necessary for cerebrovascular function (see Abbott et al., 2006). Interestingly, two genes involved in vascular function have been linked to ALS. Mutations in ANG (angiogenin) have been found to segregate with both familial and sporadic forms of ALS and Parkinson’s disease (Greenway et al., 2006; van Es et al., 2011). Angiogenin was enriched and secreted by MNs with paracrine effects exclusively on astrocytes in vitro (Skorupa et al., 2012, 2013). VEGFa promoter haplotypes causing decreased expression also infer a greater risk of ALS. VEGFa is predominately expressed by astrocytes in the CNS (Zhang et al., 2014) and decreased VEGFa levels significantly reduce survival in mutant SOD-1 mice (Lambrechts et al., 2003). While decreased expression is a greater risk for ALS and can cause MN degeneration due to reduced ischemic tolerance (Oosthuyse et al., 2001), increased VEGFa expression by reactive astrocytes due to NF-κB-dependent pathways leads to greater infiltration of peripheral immune cells and blood brain barrier (BBB) breakdown in multiple sclerosis (MS) mouse models (Argaw et al., 2012; Chapouly et al., 2015). This highlights a potential dual role for VEGFa in neurodegeneration and how inappropriate control of VEGFa expression is associated with various forms of neurodegeneration. Further implicating the involvement of vascular defects, compromised blood brain and spinal cord barriers have been observed in post-mortem ALS and FTD tissue (De Reuck et al., 2012; Garbuzova-Davis et al., 2012). Early dysfunction of the blood spinal cord barrier has been shown to contribute to early MN damage in transgenic ALS-mutant SOD-1 mice (Zhong et al., 2008; Winkler et al., 2014). Further, patient cerebral perfusion neuroimaging studies have noticed hypoperfusion abnormalities in areas that correlate with neurodegeneration in ALS and FTD (Martin et al., 2001; Du et al., 2006; Zhong et al., 2008; Chiò et al., 2014; Winkler et al., 2014). Altogether these studies implicate vascular dysfunction in the pathogenesis of ALS and FTD.
Conclusion
Research over the last decades has established that astrocytes and microglia play crucial roles in the development and/or progression of ALS and FTD through their complex interactions. Recent advances in iPSC technology have highlighted that glia secrete toxic factors that can trigger neurodegeneration. New gene discoveries have implicated that defects in glial phagocytic and neuroinflammatory activity are associated with neurodegeneration. There is now emerging evidence suggesting that non-inflammatory glial properties associated with vascular fluid flow and waste clearance have important roles in disease pathogenesis. Collectively, an emerging body of recent literature highlights the critical role of microglia and astrocytes in the etiology of ALS and FTD.
Funding
ARC Discovery Grants (DP120100180, DP140103233), NHMRC Project Grant (APP1034816), The Snow Foundation and BitFury.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jennifer Mann for proofreading. Funding: ARC Discovery Grants (DP120100180, DP140103233).
Abbreviations
ALS, Amyotrophic lateral sclerosis; Aβ, Amyloid beta; AQP4, Aquaporin 4; CNS, Central Nervous System; CSF, Cerebrospinal Fluid; FTD, Frontotemporal dementia; FUS, Fused in Sarcoma; GRN, Progranulin; iPSC, Inducible Pluripotent Stem Cell; MRI, Magnetic Resonance Imaging; MN, Motor Neuron; NF, κB, Nuclear Factor-kappa B; OPTN, Optineurin; PET, Positron Emission Tomography; PFN1, Profilin 1; RNAi, RNA interference; SOD1, Superoxide Dismutase 1; SPECT, Single Positron Emission Computed Tomography; SQTSM1, Sequestosome 1; Tau, Microtubule Associated Protein Tau; TBK1, Tank Binding Kinase 1; TREM2, Triggering Receptor Expressed on Myeloid cells 2; TDP, 43-TAR DNA-binding 43; VCP, Valosin-Containing Protein.
References
Abbott, N. J., Ronnback, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi: 10.1007/978-1-61779-452-0_34
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and Van Den Berg, L. (2012). The genetics and neuropathology of amyotrophic lateral sclerosis. Acta. Neuropathol. 124, 339–352. doi: 10.1007/s00401-012-1022-4
Argaw, A. T., Asp, L., Zhang, J., Navrazhina, K., Pham, T., Mariani, J. N., et al. (2012). Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Invest. 122, 2454–2468. doi: 10.1172/JCI60842
Asai, T., Tomita, Y., Nakatsuka, S. I., Hoshida, Y., Myoui, A., Yoshikawa, H., et al. (2002). VCP (p97) Regulates NFKB Signaling Pathway, Which Is Important for Metastasis of Osteosarcoma Cell Line. Jpn. J. Cancer Res. 93, 296–304. doi: 10.1111/j.1349-7006.2002.tb02172.x
Aspelund, A., Antila, S., Proulx, S. T., Karlsen, T. V., Karaman, S., Detmar, M., et al. (2015). A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 212, 991–999. doi: 10.1084/jem.20142290
Bataveljić, D., Nikolić, L., Milosević, M., Todorović, N., and Andjus, P. R. (2012). Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1G93A rat model. Glia 60, 1991–2003. doi: 10.1002/glia.22414
Bennion Callister, J., and Pickering-Brown, S. M. (2014). Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp. Neurol. 262, 84–90. doi: 10.1016/j.expneurol.2014.06.001
Bertel, O., Malessa, S., Sluga, E., and Hornykiewicz, O. (1991). Amyotrophic lateral sclerosis: changes of noradrenergic and serotonergic transmitter systems in the spinal cord. Brain Res. 566, 54–60. doi: 10.1016/0006-8993(91)91680-y
Bi, F., Huang, C., Tong, J., Qiu, G., Huang, B., Wu, Q., et al. (2013). Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. U S A 110, 4069–4074. doi: 10.1073/pnas.1218497110
Borroni, B., Ferrari, F., Galimberti, D., Nacmias, B., Barone, C., Bagnoli, S., et al. (2014). Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging. 35, 934.e7–934.e10. doi: 10.1016/j.neurobiolaging.2013.09.017
Brettschneider, J., Libon, D., Toledo, J., Xie, S., Mccluskey, L., Elman, L., et al. (2012). Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta. Neuropathol. 123, 395–407. doi: 10.1007/s00401-011-0932-x
Brooks, B. R., Zielger, M. G., Lake, C. R., Wood, J. H., Enna, S. J., and Engel, W. K. (1980). Cerebrospinal fluid norepinephrine and free γ-aminobutyric acid in amyotrophic lateral sclerosis. Brain Res. Bull. 5, 765–768. doi: 10.1016/0361-9230(80)90126-4
Cady, J., Koval, E. D., Benitez, B. A., Zaidman, C., Jockel-Balsarotti, J., Allred, P., et al. (2014). The TREM2 variant p.R47H is a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 71, 449–453. doi: 10.1001/jamaneurol.2013.6237
Cagnin, A., Rossor, M., Sampson, E. L., Mackinnon, T., and Banati, R. B. (2004). In vivo detection of microglial activation in frontotemporal dementia. Ann. Neurol. 56, 894–897. doi: 10.1002/ana.20332
Chapouly, C., Tadesse Argaw, A., Horng, S., Castro, K., Zhang, J., Asp, L., et al. (2015). Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain 138, 1548–1567. doi: 10.1093/brain/awv077
Chen, H., Qian, K., Chen, W., Hu, B., Blackbourn, L. W., Du, Z., et al. (2015). Human-derived neural progenitors functionally replace astrocytes in adult mice. J. Clin. Invest. 125, 1033–1042. doi: 10.1172/jci69097
Chiò, A., Pagani, M., Agosta, F., Calvo, A., Cistaro, A., and Filippi, M. (2014). Neuroimaging in amyotrophic lateral sclerosis: insights into structural and functional changes. Lancet. Neurol. 13, 1228–1240. doi: 10.1016/S1474-4422(14)70167-X
Chung, W. S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi: 10.1038/nature12776
Colonna, M. (2003). TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453. doi: 10.1038/nri1106
Corcia, P., Tauber, C., Vercoullie, J., Arlicot, N., Prunier, C., Praline, J., et al. (2012). Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 7:e52941. doi: 10.1371/journal.pone.0052941
Davis, C. H. O., Kim, K. Y., Bushong, E. A., Mills, E. A., Boassa, D., Shih, T., et al. (2014). Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. U S A 111, 9633–9638. doi: 10.1073/pnas.1404651111
De Reuck, J., Deramecourt, V., Cordonnier, C., Auger, F., Durieux, N., Bordet, R., et al. (2012). Detection of microbleeds in post-mortem brains of patients with frontotemporal lobar degeneration: a 7.0-Tesla magnetic resonance imaging study with neuropathological correlates. Eur. J. Neurol. 19, 1355–1360. doi: 10.1111/j.1468-1331.2012.03776.x
Deretic, V., Saitoh, T., and Akira, S. (2013). Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737. doi: 10.1016/b978-0-12-801043-3.00031-5
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10, 608–614. doi: 10.3410/f.1087760.540674
Dickson, D. W., Kouri, N., Murray, M. E., and Josephs, K. A. (2011). Neuropathology of frontotemporal lobar degeneration-tau (FTLD-Tau). J. Mol. Neurosci. 45, 384–389.
Dong, J. H., Ying, G. X., and Zhou, C. F. (2004). Entorhinal deafferentation induces the expression of profilin mRNA in the reactive microglial cells in the hippocampus. Glia 47, 102–108. doi: 10.1002/glia.10355
Du, A. T., Jahng, G. H., Hayasaka, S., Kramer, J. H., Rosen, H. J., Gorno-Tempini, M. L., et al. (2006). Hypoperfusion in frontotemporal dementia and Alzheimer disease by arterial spin labeling MRI. Neurology 67, 1215–1220. doi: 10.1212/01.wnl.0000238163.71349.78
Duran, A., Linares, J. F., Galvez, A. S., Wikenheiser, K., Flores, J. M., Diaz-Meco, M. T., et al. (2008). The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell 13, 343–354. doi: 10.1016/j.ccr.2008.02.001
Engelborghs, S., Vloeberghs, E., Le Bastard, N., Van Buggenhout, M., Mariën, P., Somers, N., et al. (2008). The dopaminergic neurotransmitter system is associated with aggression and agitation in frontotemporal dementia. Neurochem. Int. 52, 1052–1060. doi: 10.1016/j.jalz.2008.05.1962
Frakes, A. E., Ferraiuolo, L., Haidet-Phillips, A. M., Schmelzer, L., Braun, L., Miranda, C. J., et al. (2014). Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81, 1009–1023. doi: 10.1016/j.neuron.2014.01.013
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Muller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Garbuzova-Davis, S., Hernandez-Ontiveros, D. G., Rodrigues, M. C. O., Haller, E., Frisina-Deyo, A., Mirtyl, S., et al. (2012). Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 1469, 114–128. doi: 10.1016/j.brainres.2012.05.056
Greenway, M. J., Andersen, P. M., Russ, C., Ennis, S., Cashman, S., Donaghy, C., et al. (2006). ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 38, 411–413. doi: 10.3410/f.1031679.373763
Guerreiro, R., Lohmann, E. and Brás, J. (2013). Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 70, 78–84. doi: 10.1001/jamaneurol.2013.579
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from Familial and Sporadic ALS Patients are Toxic to Motor Neurons. Nat. Biotechnol. 29, 824–828. doi: 10.1038/nbt.1957
Hardiman, O., Van Den Berg, L. H., and Kiernan, M. C. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639–649. doi: 10.1038/nrneurol.2011.153
Harms, M., Cady, J., Koval, E., Benitez, B., Zaidman, C., Jockel-Balsarotti, J., et al. (2014). The trem2 variant p.R47h is a risk factor for sporadic amyotrophic lateral sclerosis (I5-2.002). Neurol. 82, I5-2.002.
Huang, C., Huang, B., Bi, F., Yan, L. H., Tong, J., Huang, J., et al. (2014). Profiling the genes affected by pathogenic TDP-43 in astrocytes. J. Neurochem. 129, 932–939. doi: 10.1111/jnc.12660
Iguchi, Y., Katsuno, M., Niwa, J. I., Takagi, S., Ishigaki, S., Ikenaka, K., et al. (2013). Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 136, 1371–1382. doi: 10.1093/brain/awt029
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772. doi: 10.1083/jcb.200908164
Iliff, J. J., Chen, M. J., Plog, B. A., Zeppenfeld, D. M., Soltero, M., Yang, L., et al. (2014). Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 34, 16180–16193. doi: 10.1523/jneurosci.3020-14.2014
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4, 147ra111. doi: 10.1126/scitranslmed.3003748
Iliff, J. J., Wang, M., Zeppenfeld, D. M., Venkataraman, A., Plog, B. A., Liao, Y., et al. (2013). Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 33, 18190–18199. doi: 10.1523/jneurosci.1592-13.2013
Jessen, N., Munk, A., Lundgaard, I., and Nedergaard, M. (2015). The glymphatic system: a beginner’s guide. Neurochem. Res. doi: 10.1007/s11064-015-1581-6
CrossRef Full Text | Google Scholar [Epub ahead of print].
Kaneko, M., Sano, K., Nakayama, J., and Amano, N. (2010). Nasu-Hakola disease: The first case reported by Nasu and review: The 50th anniversary of japanese society of neuropathology. Neuropathol. 30, 463–470. doi: 10.1111/j.1440-1789.2010.01127.x
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, J. G., Moon, M. Y., Kim, H. J., Li, Y., Song, D. K., Kim, J. S., et al. (2012). Ras-related GTPases Rap1 and RhoA Collectively Induce the Phagocytosis of Serum-opsonized Zymosan Particles in Macrophages. J. Biol. Chem. 287, 5145–5155. doi: 10.1074/jbc.m111.257634
Kleinberger, G., Yamanishi, Y., Suárez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6, 243ra286. doi: 10.1126/scitranslmed.3009093
Kondo, T., Funayama, M., Tsukita, K., Hotta, A., Yasuda, A., Nori, S., et al. (2014). Focal Transplantation of Human iPSC-Derived Glial-Rich Neural Progenitors Improves Lifespan of ALS Mice. Stem Cell Reports 3, 242–249. doi: 10.1016/j.stemcr.2014.05.017
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, H. S., Zeppenfeld, D., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
Lagier-Tourenne, C., and Cleveland, D. W. (2009). Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004. doi: 10.1016/j.cell.2009.03.006
Lambrechts, D., Storkebaum, E., Morimoto, M., Del-Favero, J., Desmet, F., Marklund, S. L., et al. (2003). VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat. Genet. 34, 383–394. doi: 10.3410/f.1014424.193513
Lee, S., Park, J. Y., Lee, W. H., Kim, H., Park, H. C., Mori, K., et al. (2009). Lipocalin-2 Is an Autocrine Mediator of Reactive Astrocytosis. J. Neurosci. 29, 234–249. doi: 10.1523/jneurosci.5273-08.2009
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., et al. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. doi: 10.3410/f.725528187.793507666
Mackenzie, I. R. A., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/s1474-4422(10)70195-2
Marik, J., Ogasawara, A., Martin-Mcnulty, B., Ross, J., Flores, J. E., Gill, H. S., et al. (2009). PET of glial metabolism using 2-18F-fluoroacetate. J. Nucl. Med. 50, 982–990. doi: 10.2967/jnumed.108.057356
Martin, J. A., Craft, D. K., Su, J. H., Kim, R. C., and Cotman, C. W. (2001). Astrocytes degenerate in frontotemporal dementia: possible relation to hypoperfusion. Neurobiol. Aging. 22, 195–207. doi: 10.1016/s0197-4580(00)00231-1
Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., Mcelroy, S., Renusch, S., et al. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. U S A 111, 829–832. doi: 10.1073/pnas.1314085111
Migheli, A., Piva, R., Atzori, C., Troost, D., and Schiffer, D. (1997). c-Jun, JNK/SAPK kinase and transcription factor NF-κB are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 56, 1314–1322. doi: 10.1097/00005072-199712000-00006
Minami, S. S., Min, S. W., Krabbe, G., Wang, C., Zhou, Y., Asgarov, R., et al. (2014). Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 20, 1157–1164. doi: 10.1038/nm.3672
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Neumann, H., Kotter, M. R., and Franklin, R. J. M. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–295. doi: 10.1093/brain/awn109
Ng, A. S. L., Rademakers, R., and Miller, B. L. (2015). Frontotemporal dementia: a bridge between dementia and neuromuscular disease. Ann. N. Y. Acad. Sci. 1338, 71–93. doi: 10.1111/nyas.12638
Oosthuyse, B., Moons, L., Storkebaum, E., Beck, H., Nuyens, D., Brusselmans, K., et al. (2001). Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 28, 131–138. doi: 10.1038/88842
Ouyang, Y., Tinianow, J. N., Cherry, S. R., and Marik, J. (2014). Evaluation of 2-[lsqb]18F[rsqb]fluoroacetate kinetics in rodent models of cerebral hypoxia-ischemia. J. Cereb. Blood Flow. Metab. 34, 836–844. doi: 10.1038/jcbfm.2014.22
Papadopoulos, M. C., and Verkman, A. S. (2013). Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 14, 265–277. doi: 10.1038/nrn3468
Paukert, M., Agarwal, A., Cha, J., Doze, V. A., Kang, J. U., and Bergles, D. E. (2014). Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 82, 1263–1270. doi: 10.1016/j.neuron.2014.04.038
Pearson, A. M., Baksa, K., Rämet, M., Protas, M., Mckee, M., Brown, D., et al. (2003). Identification of cytoskeletal regulatory proteins required for efficient phagocytosis in Drosophila. Microbes Infect. 5, 815–824. doi: 10.1016/s1286-4579(03)00157-6
Petkau, T. L., and Leavitt, B. R. (2014). Progranulin in neurodegenerative disease. Trends Neurosci. 37, 388–398. doi: 10.1016/j.tins.2014.04.003
Petkau, T. L., Neal, S. J., Orban, P. C., Macdonald, J. L., Hill, A. M., Lu, G., et al. (2010). Progranulin expression in the developing and adult murine brain. J. Comp. Neurol. 518, 3931–3947. doi: 10.1002/cne.22430
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet. Neurol. 10, 253–263. doi: 10.1016/s1474-4422(11)70015-1
Pickford, F., Marcus, J., Camargo, L. M., Xiao, Q., Graham, D., Mo, J. R., et al. (2011). Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am. J. Pathol. 178, 284–295. doi: 10.1016/j.ajpath.2010.11.002
Piguet, O., Hornberger, M., Mioshi, E., and Hodges, J. R. (2011). Behavioural-variant frontotemporal dementia: diagnosis, clinical staging and management. Lancet. Neurol. 10, 162–172. doi: 10.1016/S1474-4422(10)70299-4
Pomerantz, J. L., and Baltimore, D. (1999). NF-kappaB; activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO. J. 18, 6694–6704. doi: 10.1093/emboj/18.23.6694
Ravits, J. (2014). Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exp. Neurol. 262, 121–126. doi: 10.1016/j.expneurol.2014.07.021
Ravits, J., Appel, S., Baloh, R. H., Barohn, R., Brooks, B. R., Elman, L., et al. (2013). Deciphering amyotrophic lateral sclerosis: What phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 5–18. doi: 10.3109/21678421.2013.778548
Rayaprolu, S., Mullen, B., Baker, M., Lynch, T., Finger, E., Seeley, W., et al. (2013). TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 8, 19. doi: 10.1186/1750-1326-8-19
Re, D. B., Le verche, V., Yu, C., Amoroso, M. W., Politi, K. A., Phani, S., et al. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 81, 1001–1008. doi: 10.1016/j.neuron.2014.01.011
Renton, A. E., Chio, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Roberson, E. D. (2012). Mouse models of frontotemporal dementia. Ann. Neurol. 72, 837–849. doi: 10.1002/ana.23722
Seto, S., Tsujimura, K., Horii, T., and Koide, Y. (2013). Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome formation in response to mycobacterium tuberculosis infection in dendritic cells. PLoS ONE 8:e86017. doi: 10.1371/journal.pone.0086017
Sieben, A., Van Langenhove, T., Engelborghs, S., Martin, J. J., Boon, P., Cras, P., et al. (2012). The genetics and neuropathology of frontotemporal lobar degeneration. Acta. Neuropathol. 124, 353–372. doi: 10.1007/s00401-012-1029-x
Skorupa, A., King, M. A., Aparicio, I. M., Dussmann, H., Coughlan, K., Breen, B., et al. (2012). Motoneurons secrete angiogenin to induce RNA cleavage in astroglia. J. Neurosci. 32, 5024–5038. doi: 10.1523/jneurosci.6366-11.2012
Skorupa, A., Urbach, S., Vigy, O., King, M. A., Chaumont-Dubel, S., Prehn, J. H. M., et al. (2013). Angiogenin induces modifications in the astrocyte secretome: Relevance to amyotrophic lateral sclerosis. J. Proteomics 91, 274–285. doi: 10.1016/j.jprot.2013.07.028
Streit, W., Mrak, R., and Griffin, W. S. (2004). Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 1, 14. doi: 10.1385/1-59259-297-x:003
Swarup, V., Phaneuf, D., Bareil, C., Robertson, J., Rouleau, G. A., Kriz, J., et al. (2011a). Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 134, 2610–2626. doi: 10.1093/brain/awr159
Swarup, V., Phaneuf, D., Dupré, N., Petri, S., Strong, M., Kriz, J., et al. (2011b). Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J. Exp. Med. 208, 2429–2447. doi: 10.1084/jem.20111313
Thrane, V. R., Thrane, A. S., Plog, B. A., Thiyagarajan, M., Iliff, J. J., Deane, R., et al. (2013). Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci. Rep. 3, 2582. doi: 10.1038/srep02582
Thrash, J. C., Torbett, B. E., and Carson, M. J. (2009). Developmental Regulation of TREM2 and DAP12 Expression in the Murine CNS: Implications for Nasu-Hakola Disease. Neurochem. Res. 34, 38–45. doi: 10.1007/s11064-008-9657-1
Tong, J., Huang, C., Bi, F., Wu, Q., Huang, B., Liu, X., et al. (2013). Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. Embo. J. 32, 1917–1926. doi: 10.1038/emboj.2013.122
Tresse, E., Salomons, F. A., Vesa, J., Bott, L. C., Kimonis, V., Yao, T. P., et al. (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. doi: 10.4161/auto.6.2.11014
van Es, M. A., Schelhaas, H. J., Van Vught, P. W. J., Ticozzi, N., Andersen, P. M., Groen, E. J. N., et al. (2011). Angiogenin variants in Parkinson disease and amyotrophic lateral Sclerosis-like. Ann. Neurol. 70, 964–973. doi: 10.1002/ana.22611
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease modelling. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Winkler, E. A., Sengillo, J. D., Sagare, A. P., Zhao, Z., Ma, Q., Zuniga, E., et al. (2014). Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model Mice. Proc. Natl. Acad. Sci. USA 111, E1035–E1042. doi: 10.1073/pnas.1401595111
Wu, C. H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012a). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Wu, L. S., Cheng, W. C., and Shen, C. K. J. (2012b). Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J. Biol. Chem. 287, 27335–27344. doi: 10.1074/jbc.m112.359000
Xavier, A. L., Menezes, J. R. L., Goldman, S. A., and Nedergaard, M. (2014). Fine-tuning the central nervous system: microglial modelling of cells and synapses. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369, 20130593. doi: 10.1098/rstb.2013.0593
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep Drives Metabolite Clearance from the Adult Brain. Science 342, 373–377. doi: 10.1126/science.1241224
Yang, C., Wang, H., Qiao, T., Yang, B., Aliaga, L., Qiu, L., et al. (2014). Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A 111, E1121–E1129. doi: 10.1073/pnas.1322641111
Yang, L., Kress, B. T., Weber, H. J., Thiyagarajan, M., Wang, B., Deane, R., et al. (2013). Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J. Transl. Med. 11, 107–107. doi: 10.1186/1479-5876-11-107
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’keeffe, S., et al. (2014). An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons and Vascular Cells of the Cerebral Cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhong, Z., Deane, R., Ali, Z., Parisi, M., Shapovalov, Y., O’banion, K., et al. (2008). ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 11, 420–422. doi: 10.1038/nn2073
Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, astrocyte, microglia, neuroinflammation, phagocytosis, glymphatic, vasculature
Citation: Radford RA, Morsch M, Rayner SL, Cole NJ, Pountney DL and Chung RS (2015) The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell. Neurosci. 9:414. doi: 10.3389/fncel.2015.00414
Received: 15 July 2015; Accepted: 01 October 2015;
Published: 27 October 2015.
Edited by:
James Francis Curtin, Dublin Institute of Technology, IrelandReviewed by:
Björn Spittau, Albert-Ludwigs-University Freiburg, GermanyDennis Qing Wang, The Third Affiliated Hospital of Sun Yat-Sen University, China
Copyright © 2015 Radford, Morsch, Rayner, Cole, Pountney and Chung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger S. Chung, cm9nZXIuY2h1bmdAbXEuZWR1LmF1