 Yutaka Koyama
Yutaka Koyama- Laboratory of Pharmacology, Faculty of Pharmacy, Osaka Ohtani University, Tondabayashi, Osaka, Japan
Astrocytes play an essential role in supporting brain functions in physiological and pathological states. Modulation of their pathophysiological responses have beneficial actions on nerve tissue injured by brain insults and neurodegenerative diseases, therefore astrocytes are recognized as promising targets for neuroprotective drugs. Recent investigations have identified several astrocytic mechanisms for modulating synaptic transmission and neural plasticity. These include altered expression of transporters for neurotransmitters, release of gliotransmitters and neurotrophic factors, and intercellular communication through gap junctions. Investigation of patients with mental disorders shows morphological and functional alterations in astrocytes. According to these observations, manipulation of astrocytic function by gene mutation and pharmacological tools reproduce mental disorder-like behavior in experimental animals. Some drugs clinically used for mental disorders affect astrocyte function. As experimental evidence shows their role in the pathogenesis of mental disorders, astrocytes have gained much attention as drug targets for mental disorders. In this paper, I review functional alterations of astrocytes in several mental disorders including schizophrenia, mood disorder, drug dependence, and neurodevelopmental disorders. The pharmacological significance of astrocytes in mental disorders is also discussed.
Introduction
Astrocytes are the most numerous glial cell in the brain and play an essential role in maintaining efficient neurotransmission through the supply of energy metabolites, turnover of neurotransmitters, and establishment of the blood–brain barrier. In earlier studies, astrocytes were not thought to be actively involved in synaptic transmission, but this perception was revised when astrocytes were shown to express receptors for most neurotransmitters, and by which, astrocytic actions are regulated in response to receptor activation. Recent studies have confirmed that astrocytes are more actively involved in synaptic transmission than previously predicted (Perea et al., 2009). Astrocytic mechanisms that regulate synaptic transmission include release of astrocyte-derived neuroactive substances and dynamic regulation of neurotransmitter turnover in response to nerve excitation. Moreover, accumulating evidence has revealed specialized actions of astrocytes in the injured brain. One well-studied feature of astrocytes in neuropathological conditions (including acute brain insults and neurodegenerative diseases) is their phenotypic conversion to reactive astrocytes. Following phenotypic conversion, astrocytes function is altered to affect viability and repair of damaged nerve tissue (Sofroniew, 2009; Buffo et al., 2010; Koyama, 2014). Supported by these findings, modulation of astrocytic pathophysiological function was predicted to have beneficial actions on protection and repair of injured nerve tissue. Subsequent experiments demonstrated the effectiveness of this strategy using neuroprotective drugs (Acarin et al., 2001; Cifra et al., 2011; Tsuda et al., 2011; Carbone et al., 2012), and the pharmacological significance of astrocytes as a drug target for acute brain insults and neurodegenerative diseases is now accepted.
Dysfunction of monoamine- or L-glutamate (L-Glu)-mediated synaptic transmission in particular brain regions is known to be a primary pathogenic cause of many mental disorders (Herberg and Rose, 1990; Lee et al., 2007; Laruelle, 2014; Perez and Lodge, 2014). Prompted by the concept that astrocytes are more actively involved in synaptic transmission, many studies have been carried out to relate astrocyte dysfunction with mental disorders. Nervous tissue dysplasia during embryonic and postnatal brain development has also been suggested to induce mental disorders in adults. Although neuronal degeneration is not a common pathological feature in mental disorder patients, morphological and functional observations reveal alterations in astrocyte density and gene expression in several disorders (Cotter et al., 2002; Stockmeier et al., 2004; Choudary et al., 2005; Madeira et al., 2008; Habl et al., 2009; Beardsley and Hauser, 2014). Moreover, many observations have shown that modulation of astrocyte function using gene manipulation and pharmacological tools affects mental disorder-like behavior in experimental animals (Ballas et al., 2009; Basu et al., 2009; Labrie et al., 2010; Sun et al., 2012; Yang et al., 2012; Kong et al., 2014). Additionally, studies on the therapeutic mechanisms of drugs currently used to treat mental disorders found that their beneficial effects are mediated via astrocytic mechanisms (Table 1). From these findings, astrocytes were suggested to play an important role in the etiology of mental disorders. In the present clinical field, several types of effective drugs are used for care of patients with mental disorders. However, further development of psychiatric drugs will be required. Thus, the use of astrocytic cell pathways was proposed as a novel strategy in mental disorder etiology, as well as in the mechanisms of neuroprotective drugs.
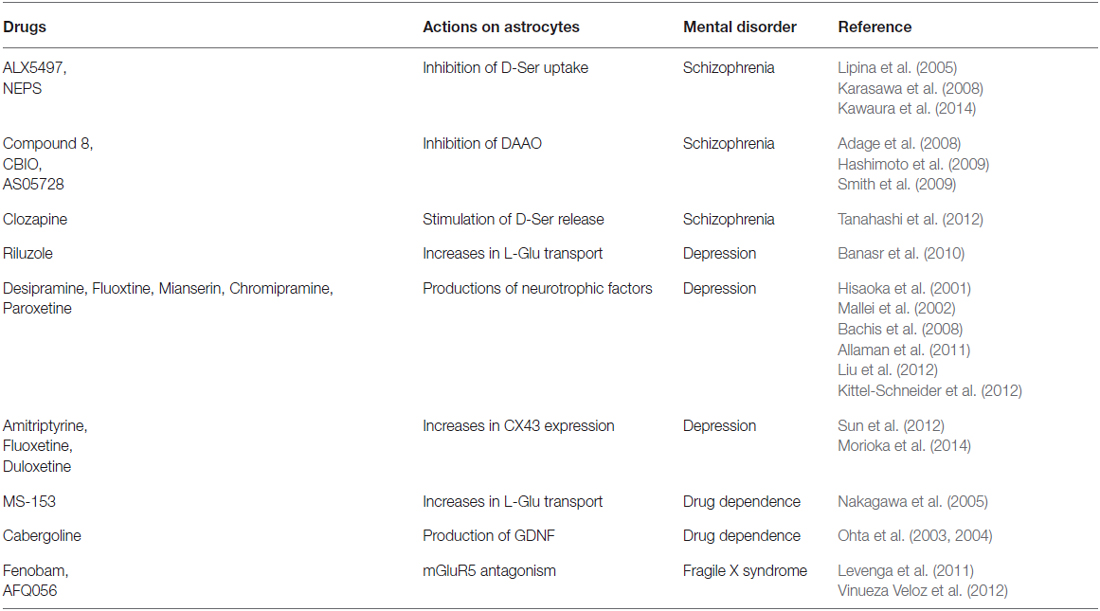
Table 1. Drugs showing therapeutic effects on mental disorders by modulation of astrocytic functions.
This paper reviews recent studies on the possible roles of astrocytes in the pathogenesis of mental disorders i.e., schizophrenia, mood disorders, drug dependence, and mental retardation (Rett syndrome and fragile X syndrome, FXS). The pharmacological significance of astrocytes as drug targets for mental disorders is also discussed.
Novel Concepts of Astrocyte Function
Astrocytes are known to play a supporting role in synaptic transmission including maintenance of the ionic balance in extracellular fluid, supply of energy metabolites to neurons, and reducing transmitters released into the synaptic cleft (Parpura et al., 2012). To undertake these supporting roles, astrocytes have many specific transporters and neurotransmitter metabolizing enzymes. During regulation of synaptic transmission by L-Glu, astrocytes take up synaptic L-Glu through highly expressed excitatory amino acid transporters (EAAT-1 and EAAT-2). Subsequently, L-Glu is metabolized to L-glutamine by an astrocyte-specific enzyme, glutamine synthetase (GS). Release of astrocytic L-glutamine is used as a neuronal L-Glu precursor, and this interplay between neurons and astrocytes is known as the glutamine cycle. Specific transporters and metabolizing enzymes for other neurotransmitters are also expressed in astrocytes. Expression levels of astrocytic transporters and metabolizing enzymes are not static, but are dynamically regulated in response to synaptic activity. This enables astrocytes to effectively support synaptic transmission. Aside from their supporting role, the concept that astrocytes are more actively involved in synaptic transmission is being recognized. This concept involves the “tripartite synapse”, with astrocytes surrounding the synaptic cleft as an essential component of the synapse, as well as pre- and post-synaptic neurons, and with part of the pre-synaptic signal circumvented via astrocytes to modulate the direct signal to the post-synaptic neuron (Perea et al., 2009). Evidence to support this includes the discovery of “gliotransmitters”. The term “gliotransmitter” is used to describe neuroactive substances released from astrocytes in response to a pre-synaptic signal. Astrocytes excited by L-Glu and adenosine triphosphate (ATP), release L-Glu, ATP, adenosine, D-serine (D-Ser), and eicosanoids in a Ca2+ dependent mechanism. Because of this excitation-induced release and modulatory action on synaptic transmission, these substances are thought to be putative gliotransmitters (Araque et al., 2014). However, regulation of synaptic transmission by gliotransmitters is still controversial in physiological states. While release of gliotransmitters is stimulated in a Ca2+-dependent manner, experimental manipulation of increased astrocytic Ca2+ failed to affect excitatory synaptic activity in the hippocampus (Fiacco et al., 2007; Petravicz et al., 2008). Moreover, increased astrocytic Ca2+ levels in response to pre-synaptic activations were obtained after excitation of post-synaptic neurons (Agulhon et al., 2012). From these findings, Agulhon et al. (2012) proposed that the gliotransmitter role is less significant in physiological states.
In pathological states, astrocyte function is remarkably altered. Specifically, astrocytes are converted to a reactive phenotype in response to brain injury, which is characterized by cell body hypertrophy and increased expression of glial fibrillary acidic protein (GFAP), an astrocyte-specific intermediate filament protein (Sofroniew, 2009; Koyama, 2014). Phenotypic conversion to reactive astrocytes is accompanied by altered expression of various functional molecules, such as transporters and neurotransmitter metabolizing enzymes (Buffo et al., 2010). Altered activities of these astrocytic molecules may result in disturbed synaptic transmission and aggravate excitoxicity-induced nerve injury. Several types of soluble factors (e.g., cytokines, chemokines, and neurotrophic factors) that regulate pathophysiological responses in nerve tissue are produced by reactive astrocytes (Hamby and Sofroniew, 2010; Colangelo et al., 2014). Excess production of cytokines and chemokines causes microglial activation, infiltration of blood cells, neural apoptosis, and breakdown of the blood–brain barrier, which exacerbates neuroinflammation in the injured brain. However, reactive astrocytes also produce neurotrophic factors, including brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF; Koyama et al., 2003). These astrocyte-derived neurotrophic factors prevent neuronal damage and stimulate neurogenesis, both of which improve dysfunction of the injured brain. By releasing these soluble factors, reactive astrocytes play prominent roles in regulating pathophysiological responses in injured nerve tissue, and suggest that modulation of astrocyte function may be a promising target for neuroprotective drugs, which can treat acute brain insults and neurodegenerative diseases. The neuroprotective action of some drugs in modulating astrocyte function have been observed in animal models of brain ischemia, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS; Acarin et al., 2001; Cifra et al., 2011; Tsuda et al., 2011; Carbone et al., 2012). There have been some excellent review papers on the pharmacological significance of astrocytes as a target for neuroprotective drugs (Darlington, 2005; Hamby and Sofroniew, 2010; Colangelo et al., 2014).
In addition to the release of gliotransmitters and neurotrophic factors, studies have shown novel roles for astrocytic connexin-43 (CX43) and aquaporin-4 (AQP4) in regulating nerve function in both the pathological and physiological state. CX43 is a main component of the astrocytic gap junction (Koulakoff et al., 2008). Intracellular communication through gap junctions enables sharing of cytosolic messengers and excitability between adjacent cells (Scemes and Spray, 2012). In astrocytes, CX43 expression is altered by brain injury (Rouach et al., 2002), which affects the neuroprotective actions and proliferation of astrocytes (Tabernero et al., 2006; Gangoso et al., 2012; Theodoric et al., 2012). Therefore, modulation of CX43-mediated gap junction communication may be a target for neuroprotective drugs. Besides these pathophysiological roles, gap junction activity stimulates the release of various gliotransmitters. Stehberg et al. (2012) found that administration of CX43 inhibitors to the rat basolateral amygdala prevents fear memory consolidation, suggesting CX43 involvement in physiological nerve function. Reduced CX43 expression was observed in patients with major depressive disorder (MDD) and alcohol dependence (Bernard et al., 2011; Miguel-Hidalgo et al., 2014). From these findings, CX43 was proposed to be a target of drugs for mental disorders (Sun et al., 2012; Morioka et al., 2014). AQP4 is a water channel highly expressed in astrocytes. With regards the functional role of astrocytic AQP4, its relationship to brain edema etiology has been investigated (Manley et al., 2000). AQP4 is thought to be involved in glial scar formation at injured nerve tissue and in brain edema, because AQP4 inhibitors stimulate migration of reactive astrocytes (Saadoun et al., 2005; Verkman et al., 2006). Besides these pathological roles, recent studies have suggested novel roles for astrocytic AQP4 in synaptic plasticity and mental disorder pathogenesis. Skucas et al. (2011) found that induction of long term-potentiation was attenuated in the hippocampus of AQP4 null mice. In addition, deletion of astrocytic AQP4 decreased morphine dependence and the anti-depressant actions of fluoxetine (Kong et al., 2009; Yan et al., 2013).
Because of the identification of these astrocytic functions, the relationship between astrocytes and higher brain functions, including regulation of emotion and mentality, has gained greater attention. Many studies have since been performed to determine the involvement of astrocyte dysfunction in mental disorders (Figure 1), and have shown that astrocytes contribute to the pathogenesis of some disorders.
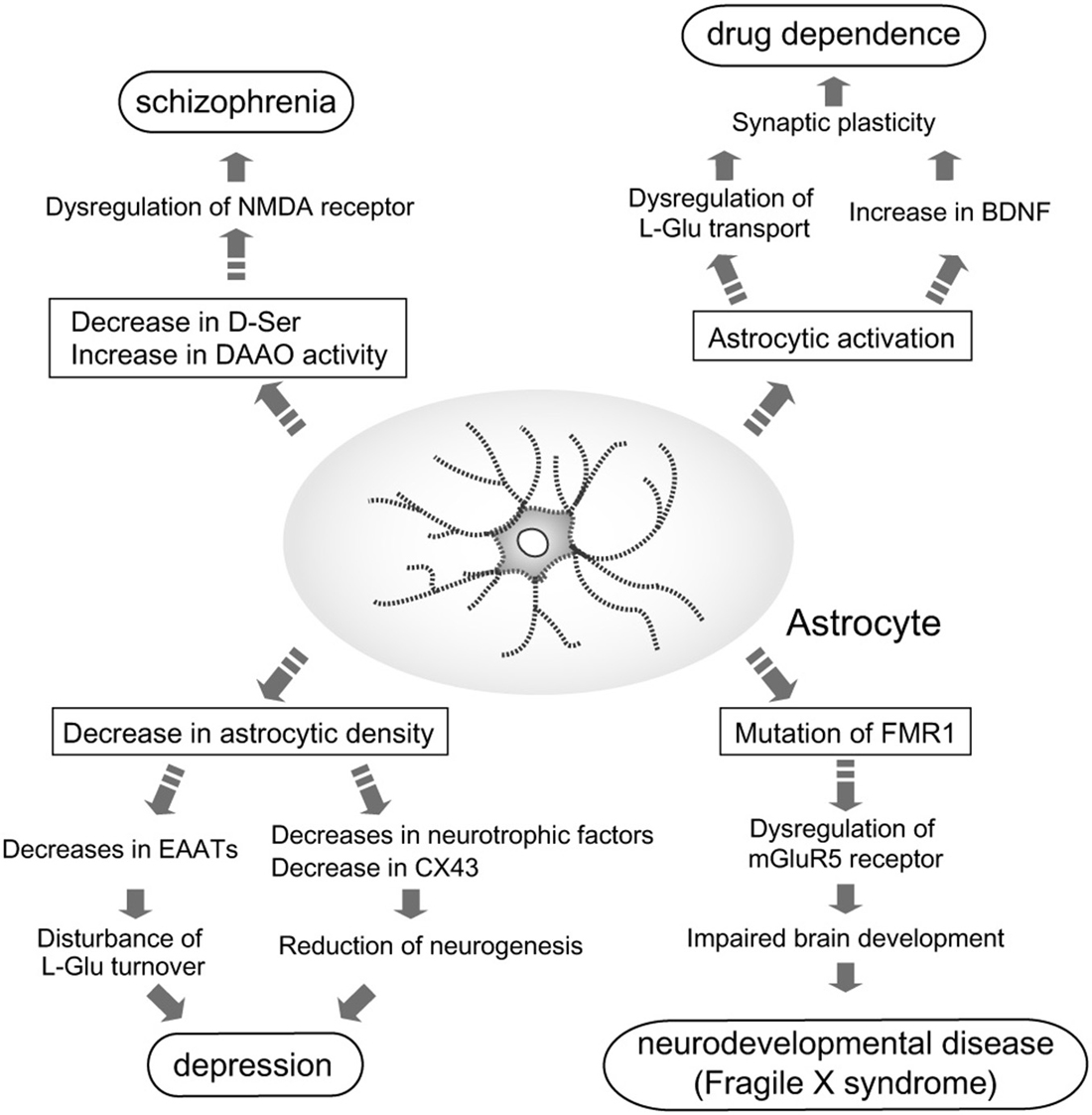
Figure 1. Astrocyte roles in the pathogenesis of mental disorders. Pathological roles for astrocytes in induction and/or aggravation of schizophrenia, depression, drug dependence, and fragile X syndrome (FXS) are proposed. In patients with schizophrenia, D-Ser content in the brain is decreased, while D-amino acid oxidase (DAAO) expression is increased. These alterations in D-Ser may cause schizophrenia via dysfunction of N-methyl-D-aspartate (NMDA) receptor-mediated signaling. Decreased astrocyte cell number is found in patients with depression. Reduction of astrocytes leads to leads to decreases in neurotrophic factors, CX43 and excitatory amino acid transporters (EAATs). Decreases in these astrocytic molecules cause disturbance of L-Glu turnover and neurogenesis, which may aggravate depression. Many dependence-producing drugs activate astrocytes. Production of brain-derived neurotrophic factor (BDNF) by reactive astrocytes enhances the rewarding effects of psychostimulants. fragile X mental retardation 1 (FMR1) is the gene responsible for FXS. Mutations in FMR1 cause dysfunction of mGluR5 signaling in neurons and astrocytes, which impairs normal brain development.
Astrocytes in Mental Disorders
Schizophrenia
Schizophrenia is a mental disease that affects approximately 1% of the population. Its symptoms are hallucination, delusions, thought disorder, flat affect, social withdrawal, and cognitive disorder. Genetic and environmental factors are involved in schizophrenia, although its detailed mechanisms are not fully understood. Drugs with antagonistic potency against dopamine D2 receptors are widely used for treating schizophrenia. These antagonists effectively manage the abnormal behavior, and thus dysfunction of midbrain dopamine transmission is generally accepted to underlie the symptoms of schizophrenia. Further studies have shown involvement of L-Glu-mediated excitatory transmission in schizophrenia pathogenesis (Coyle, 2006; Laruelle, 2014). In experimental animals, N-methyl-D-aspartate (NMDA) receptor antagonists cause schizophrenia-like behavioral abnormalities, accompanied by dopamine system hyperactivation (Lipina et al., 2005; Karasawa et al., 2008; Bado et al., 2011; Kawaura et al., 2014). Moreover, administration of NMDA antagonists to schizophrenic patients aggravates their symptoms (Javitt and Zukin, 1991; Krystal et al., 1994), suggesting that inhibition of NMDA receptor-mediated transmission facilitates induction of schizophrenia. NMDA receptors have an allosteric site that regulates L-Glu-mediated receptor activation. D-serine is a necessary co-factor for NMDA receptor/channel gating, and enhances the excitatory signal (Balu et al., 2012; Van Horn et al., 2013). The D-Ser biosynthetic enzyme, serine racemase (SR), and D-Ser degradation enzyme, D-amino acid oxidase (DAAO), are both present in brain regions with high NMDA receptor expression (Van Horn et al., 2013). Immunohistochemical observations show that SR locates to astrocytes (Wolosker et al., 1999; Panatier et al., 2006), while D-Ser release from astrocytes is stimulated by excitatory amino acids (Martineau et al., 2014), indicating that D-Ser serves as a gliotransmitter. In schizophrenia patients, D-Ser levels are decreased in cerebrospinal fluid (Hashimoto et al., 2003; Bendikov et al., 2007), whereas DAAO protein and its activity are increased in the hippocampus and cerebrum (Madeira et al., 2008; Habl et al., 2009). Human genetic analysis shows that several polymorphic variants of SR and DAAO are related to increased risk of schizophrenia (Labrie et al., 2009; Caldinelli et al., 2013). Concurrent with these observations, manipulation of brain D-Ser levels induces schizophrenia-like behavior in experimental animals. Basu et al. (2009) reported that genetic deletion of SR causes hyperactivity and impaired memory in mice, accompanied by altered NMDA responses. Further observations of the SR null mouse found morphological and neurochemical abnormalities in the brain, similar to those in schizophrenia (Puhl et al., 2014). In contrast, DAAO deletion reverses schizophrenia-like abnormal behavior in mice with impaired NMDA receptor function (Labrie et al., 2010). The effect of D-Ser and related drugs has been examined in animal models of schizophrenia. Administration of D-Ser, D-Ser reuptake inhibitors (ALX5407 and (R)-(N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl] sarcosine (NFPS)) and DAAO inhibitors ([4H-thieno [3, 2-b]pyrrole-5-carboxylic acid] (compound 8), 5-chloro-benzo[d]isoxazol-3-ol (CBIO) and AS057278) improve impaired pre-pulse inhibition and cognitive defects induced by NMDA antagonists (Lipina et al., 2005; Adage et al., 2008; Karasawa et al., 2008; Hashimoto et al., 2009; Smith et al., 2009; Bado et al., 2011; Kawaura et al., 2014). Therapeutic effects of D-Ser and glycine on negative symptoms of schizophrenia patients have been reported, and more effective drugs for enhancing NMDA receptor signaling should be explored (Tuominen et al., 2005; Tsai and Lin, 2010). Currently, atypical antipsychotics, which improve both positive and negative symptoms, are used for the treatment of schizophrenia. Some atypical antipsychotics (clozapine, olanzapine, and risperidone), but not haloperidol, enhance L-Glu transmission in the prefrontal cortex via NMDA receptors (Ninan et al., 2003; Kargieman et al., 2007). Recently, Tanahashi et al. (2012) showed that clozapine, but not haloperidol, stimulates D-Ser release from astrocytes, suggesting a novel mechanism of atypical antipsychotics in schizophrenia treatment.
Mood Disorders (Major Depressive Disorder)
Among the mood disorders, morphological and functional alterations of astrocytes are apparent in patients with MDD (Sanacora and Banasr, 2013). Postmortem brain examination of MDD patients shows decreased astrocyte cell number and GFAP protein in the hippocampus (Stockmeier et al., 2004), frontal cortex (Ongür et al., 1998; Cotter et al., 2002), and amygdala (Hamidi et al., 2004). Decreased astrocyte cell number and GFAP protein are reproduced in animal models subjected to chronic unpredictable stress (Heine et al., 2004; Czéh et al., 2006). Administration of L-α-aminoadipate (an astrogliotoxin used as a tool to induce specific astrocytic degeneration) into the rat prefrontal cortex causes depressive-like behavior (Banasr and Duman, 2008). Based on these findings, possible involvement of impaired astrocyte function in the pathogenesis of depression has been investigated. While therapeutic mechanisms of clinically used antidepressants can be explained by the “monoamine hypothesis”, L-Glu transmission has also been considered as a therapeutic target for depression. In rat social interaction and sucrose intake tests, administration of L-Glu transport inhibitors leads to depressive-like behavior (Lee et al., 2007; John et al., 2012), suggesting that impaired L-Glu turnover between astrocytes and neurons causes depression. As well as GFAP, expressions of astrocyte-specific molecules (e.g., EAAT-1, EAAT-2, and GS) are decreased in MDD, along with the reduction in astrocyte cell number (Choudary et al., 2005). As EAAT-1 and EAAT-2 are the main uptake pathways for extracellular L-Glu into astrocytes, decreased EAAT-1 and EAAT-2 expression may cause impaired L-Glu turnover and result in depression. Involvement of impaired L-Glu turnover in depression pathogenesis is supported by the beneficial effect of riluzole in animal models of depression. Riluzole, which is clinically used for ALS treatment, activates L-Glu transporters (Fumagalli et al., 2008). Furthermore, Banasr et al. (2010) found that riluzole reverses decreased GFAP expression in the rat prefrontal cortex and improves depressive-like behavior after chronic unpredicted stress. Although the mechanisms underlying morphological and functional alterations of astrocytes remain to be clarified, the beneficial action of riluzole suggests that modulating L-Glu turnover in astrocytes is a novel strategy for treatment of depression.
Neuronal and glial cell genesis is not limited to the developing brain and can occur in restricted areas of the adult brain, mainly the hippocampus and sub-ventricular zone (SVZ). Many studies have attempted to show correlation between the pathology of neurological disorders and deregulation of cellular genesis in the adult brain. Reduced hippocampal neurogenesis is implicated in the pathogenesis of depression, and as a possible target of antidepressants (Santarelli et al., 2003; Banasr and Duman, 2007). Moreover, recent animal model studies implicate astrogliogenesis in depression pathogenesis. Olfactory bulb dissection can induce depressive-like behavioral changes in rats. Keilhoff et al. (2006) showed that olfactory bulb dissection decreases neural precursor proliferation in the hippocampus and SVZ, which can be rescued by the antidepressant, imipramine. Similarly, chronic social stress decreases astrocyte number and cell volume in the rat hippocampus, which can be reversed by fluoxetine (Czéh et al., 2006). In contrast, electroconvulsive seizures, an effective treatment for severe depression, stimulates astrocyte proliferation in the rat hippocampus and prefrontal cortex (Ongür et al., 2007; Jansson et al., 2009). These findings support the involvement of astrogliogenesis in the pathogenesis of depression. Recently, Kong et al. (2014) found that deletion of AQP4, a water channel protein expressed in astrocytes, aggravates depressive-like behavior and is accompanied by a further reduction in astrocyte cell number and hippocampal neurogenesis. This suggests that astrocytic AQP4 may be a novel target for antidepressants.
Increased production of neurotrophic factors is predicted to be an effective treatment strategy for mood disorders, because they promote neurogenesis, gliogenesis, and synaptic structure remodeling. Levels of BDNF (Dwivedi et al., 2003), GDNF (Otsuki et al., 2008; Zhang et al., 2008), and basic fibroblast growth factor (bFGF; Evans et al., 2004) are decreased in patients with depression, and relates to the reduced hippocampal neurogenesis. Astrocytes are a main source of these neurotrophic factors in pathological brain conditions (Koyama et al., 2003). Administration of antidepressants (e.g., desipramine, fluoxetine, mianserin, clomipramine, and paroxetine) increases production of BDNF, GDNF, and bFGF in the rat hippocampus (Mallei et al., 2002; Martínez-Turrillas et al., 2005; Bachis et al., 2008; Liu et al., 2012), while in vitro studies using cultured astrocytes treated with antidepressants shows production of these neurotrophic factors (Hisaoka et al., 2001; Allaman et al., 2011; Kittel-Schneider et al., 2012). Thus, up-regulation of astrocytic trophic factor production may partially underlie the therapeutic actions of presently used antidepressants.
A relationship between CX43, a main component of astrocytic gap junctions, and MDD has been suggested. Reduced brain CX43 expression is observed in MDD patients (Bernard et al., 2011; Miguel-Hidalgo et al., 2014). Inhibition of CX43-mediated gap junction communication causes depressive-like behavior in rodents (Sun et al., 2012). Besides neurotrophic factor production, increased CX43 expression is proposed as a novel mechanism for clinically used antidepressants. Sun et al. (2012) found that fluoxetine and duloxetine increase CX43 expression in rat brain. Moreover, amitriptyline increases CX43 expression by a monoamine-independent mechanism in cultured astrocytes (Morioka et al., 2014).
Drug Dependence
Repeated abuse of opiates, hypnotics, and psychostimulants leads to drug dependence. It is known that drug-induced alterations in synaptic strength in the mesocorticolimbic dopamine system and modulatory glutamatergic neuronal circuits, both part of the brain reward system, underlie drug dependence (van Huijstee and Mansvelder, 2015). Dependence-producing drugs commonly activate the main pathway of the brain reward system, with dopamine released from neurons in the ventral tegmental area (VTA) to the nucleus accumbens (NAcc) and prefrontal cortex. Studies on the mechanisms underlying drug dependence show a possible role for astrocytes in modulating neurotransmission in the brain reward system (Beardsley and Hauser, 2014). Administration of amphetamine, methamphetamine, cocaine, and morphine induces astrocyte activation and increases GFAP expression in rodent brain (Hebert and O’Callaghan, 2000; Fattore et al., 2002; Pubill et al., 2003; Alonso et al., 2007). Although these astrocytic alterations are not necessarily a common pathological feature shared by other drugs, these observations facilitate examination of the mechanisms underlying drug dependence in the context of astrocyte function.
The L-Glu-mediated neural circuit from the prefrontal cortex to NAcc plays an important regulatory role in the brain reward system (van Huijstee and Mansvelder, 2015). Nakagawa et al. (2005) examined the role of astrocytic L-Glu transporters in mice by co-administrating MS-153, a glutamate transport activator, with morphine, cocaine, or methamphetamine. They found that activation of L-Glu transport attenuates conditioned place preference (CPP) to these drugs. Administration of an adenoviral vector carrying the glutamate transporter 1 (GLT1; EAAT-2) gene into the NAcc also attenuated CPP induction by morphine and methamphetamine (Fujio et al., 2005). Together, these findings suggest there is inhibitory regulation from astrocytic L-Glu transporters on the rewarding effect of dependence-producing drugs.
Astrocyte-derived soluble factors have important roles in regulating synaptic strength and plasticity. The effect of astrocyte-derived factors on susceptibility to drug dependence was examined using conditioned medium from cultured astrocytes. Administration of astrocytic conditioned medium into mouse NAcc caused sensitization of rewarding behavior elicited by methamphetamine and morphine (Narita et al., 2005, 2006), suggesting that astrocytes produce soluble factors that enhance drug dependence. As astrocyte-derived factors affect susceptibility of drug-dependence, the modulatory roles of BDNF and GDNF on rewarding effects of psychostimulants were examined (Ghitza et al., 2010). Enhancement of a rewarding effect by BDNF was first shown by Horger et al. (1999), with chronic BDNF administration into rat NAcc increasing CPP to cocaine. Overexpression of exogenous BDNF and its receptor (TrkB) in rat NAcc also increased CPP to cocaine (Bahi et al., 2008), while mouse BDNF null mutants show reduced CPP (Hall et al., 2003). Positive regulatory roles of BDNF were also suggested from the rewarding effects of morphine and amphetamine (Shen et al., 2006; Vargas-Perez et al., 2009). As had been predicted from animal experiments (Pu et al., 2006; Hatami et al., 2007), a recent study showed that serum BDNF levels in heroin-dependent patients are still higher than those of control groups, even after drug withdrawal (Zhang et al., 2014). The results from viral vector-mediated gene transfer experiments in rodents (Vargas-Perez et al., 2014) propose that enhancement of the BDNF signal in the VTA is related to drug withdrawal aversion.
GDNF was originally discovered as a survival and developmental factor for mesencephalon dopaminergic neurons, and modulates nerve excitation in many brain regions, including the VTA and NAcc (Carnicella and Ron, 2009). In contrast to BDNF, GDNF serves as a negative reinforcement modulator of the rewarding effects of psychostimulants. Administration of GDNF into the rat VTA reduced CPP enhancement to cocaine, while an anti-GDNF neutralizing antibody increased it (Messer et al., 2000). Heterozygous GDNF deletion in mice caused higher sensitivity in CPP and seeking behaviors to methamphetamine than those of wild-type mice (Niwa et al., 2007; Yan et al., 2007). Taking these observations into consideration, a therapeutic effect for drugs enhancing GDNF production in patients with psychostimulant dependence can be expected. Cabergoline, a dopamine D2 agonist used for the treatment of hyperprolactinemia and parkinsonism, increases GDNF production in cultured astrocytes (Ohta et al., 2003, 2004) and rat VTA (Carnicella et al., 2009). Cabergoline-induced GDNF production in rat VTA reduced reinforcement of seeking and drinking behavior for alcohol (Carnicella et al., 2009).
Neurodevelopmental Diseases
Dysplasia of nerve tissue during embryonic and postnatal development underlies some neurological diseases with mental retardation and cognitive defects. During development of the embryonic brain, astrocytes support proliferation and migration of neural precursors, neuronal differentiation, and synaptic formation, although neurogenesis generally precedes maturation of astrocytes from glial precursors (Freeman and Rowitch, 2013). Because of the important role of astrocytes in the developing brain, investigations to explain the etiology of neurodevelopmental diseases by astrocyte dysfunction have been performed (Molofsky et al., 2012; Parpura et al., 2012). A number of studies on two inherited developmental diseases with mental retardation, Rett syndrome and FXS, show that mutations in single genes are responsible for astrocyte dysfunction and impaired brain development.
Rett syndrome, an X-linked neurological disease characterized clinically by distinctive hand movements, seizures, delayed brain and head growth, autism, and mental retardation (Weng et al., 2011), is caused by mutations in a transcription factor, methyl-CpG-binding protein 2 (MeCP2; Samaco and Neul, 2011). In studies using MeCP2 null mutant mice as a model of Rett syndrome (Chen et al., 2001), conditional MeCP2 expression in postnatal neurons partly reversed behavioral abnormalities (Giacometti et al., 2007; Guy et al., 2007), indicating involvement of reduced neural MeCP2 in pathogenesis of the model. In addition, reduced function of astrocytic MeCP2 is also related to Rett syndrome pathogenesis. In vitro experiments by Ballas et al. (2009) found that hippocampal neurons cultured with MeCP2 deleted astrocytes or their conditioned medium, failed to show normal dendritic development. Impaired dendrite formation by astrocytic MeCP2 occurs independent of the presence of neural MeCP2, suggesting that dysregulation of astrocytic soluble factors induced by MeCP2 deletion may relate to induction of Rett syndrome-like phenotypes. Maezawa et al. (2009) reported impairments in BDNF, interleukin-1β, and interleukin-6 production in astrocytes from MeCP2 deleted mutant mice.
FXS is a neurodevelopmental disease characterized by mental retardation, autism, attention deficit, social anxiety, and specific physical features. One of the genes responsible for FXS, fragile X mental retardation 1 (FMR1), is on an X-linked chromosome. Mutations in FMR1, with GCC expansions repeats in the promoter region, decrease production of fragile X mental retardation 1 protein (FMRP), which serves as a regulator of local protein translation. Reduced FMRP activity in neurons leads to dysregulation of synaptic protein expression and affects dendrite formation (Bassell and Warren, 2008). FMR1 gene deletion induces abnormal dendrite elongation and increases spine density in the developing cerebral cortex (Comery et al., 1997; Nimchinsky et al., 2001). Besides neuronal reduction, reduced FMRP activity in astrocytes affects their function during brain development (Jacobs and Doering, 2010; Jacobs et al., 2010). The mechanisms by which reduced FMRP in astrocytes induces abnormal dendrite development were investigated. Yang et al. (2012) found that FMRP deletion increases neurotrophin-3 production in astrocytes, which suggests that excess neurotrophic actions underlie abnormalities in dendrite development. The metabotropic glutamate receptor 5 (mGluR5) is predicted to be a therapeutic drug target for FXS (Levenga et al., 2011; Vinueza Veloz et al., 2012; Pop et al., 2014; Scharf et al., 2015). Higashimori et al. (2013) proposed that down-regulation of astrocytic mGluR5 and GLT-1 (EAAT-2) by FMRP deletion may cause enhanced neuronal excitation and lead to abnormal dendritic development in FXS mouse models.
A Perspective of Astrocytes as a Drug Target for Mental Disorders
Supported by considerable experimental evidence, the importance of astrocytic functions during acute brain insults and neurodegenerative diseases is established. Because modulation of astrocytic function has several beneficial actions, astrocytes are a promising target of neuroprotective drugs (Darlington, 2005; Hamby and Sofroniew, 2010; Colangelo et al., 2014). Although neuronal degeneration is generally not observed, disturbance of neurotransmission, abnormal brain development, and remodeling of synaptic structure are found in the brains of patients with mental disorders. Furthermore, morphological and functional alterations of astrocytes are observed in patients with certain mental disorders (Cotter et al., 2002; Stockmeier et al., 2004; Choudary et al., 2005; Madeira et al., 2008; Habl et al., 2009; Beardsley and Hauser, 2014). Besides their role in neurogenesis and synaptic formation during brain development, accumulating evidence shows that astrocytes are an essential component of synaptic transmission (Parpura et al., 2012; Araque et al., 2014). In addition, involvement of astrocyte-specific molecules such as CX43 and AQP4 in higher brain functions is reported (Sun et al., 2012; Xiao and Hu, 2014). Prompted by these findings, many studies have attempted to clarify the role of astrocytes in mental disorders. As described in this review, involvement of astrocytic dysfunction in the pathogenesis of mental disorders is becoming increasingly studied (Figure 1). Thus, the pharmacological significance of astrocytes as a novel drug target for schizophrenia, mood disorders, drug dependence, and neurodevelopmental disorders has been proposed (Table 1). However, despite the accumulating evidence, compared with neurons, there are still many astrocyte-related issues that need to be clarified. These include classification of astrocyte sub-types, differences in properties among brain regions, astrogliogenesis in the developing and adult brain, and the associated regulatory factors. Further investigation of these issues may lead to novel drugs for the treatment for mental disorders.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (15K07981).
Abbreviations
L-Glu, L-glutamate; D-Ser, D-serine; GS, glutamine synthetase; GFAP, glial fibrillary acidic protein; BDNF, brain-derived neurotrophic factor; GDNF, glial cell line-derived neurotrophic factor; SR, serine racemase; DAAO, D-amino acid oxidase; SVZ, sub-ventricular zone; bFGF, basic fibroblast growth factor; CX43, connexin-43; AQP4, aquaporin-4; MDD, major depressive disorder; NAcc, nucleus accumbens; VTA, ventral tegmental area; CPP, conditioned place preference; FXS, fragile X syndrome; MeCP2, methyl-CpG-binding protein 2; FMRP, fragile X mental retardation 1 protein.
References
Acarin, L., González, B., and Castellano, B. (2001). Triflusal posttreatment inhibits glial nuclear factor-kappaB, downregulates the glial response and is neuroprotective in an excitotoxic injury model in postnatal brain. Stroke 32, 2394–2402. doi: 10.1161/hs1001.097243
Adage, T., Trillat, A. C., Quattropani, A., Perrin, D., Cavarec, L., Shaw, J., et al. (2008). In vitro and in vivo pharmacological profile of AS057278, a selective d-amino acid oxidase inhibitor with potential anti-psychotic properties. Eur. Neuropsychopharmacol. 18, 200–214. doi: 10.1016/j.euroneuro.2007.06.006
Agulhon, C., Sun, M. Y., Murphy, T., Myers, T., Lauderdale, K., and Fiacco, T. A. (2012). Calcium signaling and gliotransmission in normal vs. reactive astrocytes. Front. Pharmacol. 3:139. doi: 10.3389/fphar.2012.00139
Allaman, I., Fiumelli, H., Magistretti, P. J., and Martin, J. L. (2011). Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology (Berl) 216, 75–84. doi: 10.1007/s00213-011-2190-y
Alonso, E., Garrido, E., Díez-Fernández, C., Pérez-García, C., Herradón, G., Ezquerra, L., et al. (2007). Yohimbine prevents morphine-induced changes of glial fibrillary acidic protein in brainstem and alpha2-adrenoceptor gene expression in hippocampus. Neurosci. Lett. 412, 163–167. doi: 10.1016/j.neulet.2006.11.002
Araque, A., Carmignoto, G., Haydon, P. G., Oliet, S. H., Robitaille, R., and Volterra, A. (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. doi: 10.1016/j.neuron.2014.02.007
Bachis, A., Mallei, A., Cruz, M. I., Wellstein, A., and Mocchetti, I. (2008). Chronic antidepressant treatments increase basic fibroblast growth factor and fibroblast growth factor-binding protein in neurons. Neuropharmacology 55, 1114–1120. doi: 10.1016/j.neuropharm.2008.07.014
Bado, P., Madeira, C., Vargas-Lopes, C., Moulin, T. C., Wasilewska-Sampaio, A. P., Maretti, L., et al. (2011). Effects of low-dose D-serine on recognition and working memory in mice. Psychopharmacology (Berl) 218, 461–470. doi: 10.1007/s00213-011-2330-4
Bahi, A., Boyer, F., Chandrasekar, V., and Dreyer, J. L. (2008). Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference and reinstatement in rats. Psychopharmacology (Berl) 199, 169–182. doi: 10.1007/s00213-008-1164-1
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Balu, D. T., Basu, A. C., Corradi, J. P., Cacace, A. M., and Coyle, J. T. (2012). The NMDA receptor co-agonists, D-serine and glycine, regulate neuronal dendritic architecture in the somatosensory cortex. Neurobiol. Dis. 45, 671–682. doi: 10.1016/j.nbd.2011.10.006
Banasr, M., Chowdhury, G. M., Terwilliger, R., Newton, S. S., Duman, R. S., Behar, K. L., et al. (2010). Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol. Psychiatry 15, 501–511. doi: 10.1038/mp.2008.106
Banasr, M., and Duman, R. S. (2007). Regulation of neurogenesis and gliogenesis by stress and antidepressant treatment. CNS Neurol. Disord. Drug. Targets 6, 311–320. doi: 10.2174/187152707783220929
Banasr, M., and Duman, R. S. (2008). Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 64, 863–870. doi: 10.1016/j.biopsych.2008.06.008
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Basu, A. C., Tsai, G. E., Ma, C. L., Ehmsen, J. T., Mustafa, A. K., Han, L., et al. (2009). Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol. Psychiatry 14, 719–727. doi: 10.1038/mp.2008.130
Beardsley, P. M., and Hauser, K. F. (2014). Glial modulators as potential treatments of psychostimulant abuse. Adv. Pharmacol. 69, 1–69. doi: 10.1016/b978-0-12-420118-7.00001-9
Bendikov, I., Nadri, C., Amar, S., Panizzutti, R., De Miranda, J., Wolosker, H., et al. (2007). A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr. Res. 90, 41–51. doi: 10.1016/j.schres.2006.10.010
Bernard, R., Kerman, I. A., Thompson, R. C., Jones, E. G., Bunney, W. E., Barchas, J. D., et al. (2011). Altered expression of glutamate signaling, growth factor and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 16, 634–646. doi: 10.1038/mp.2010.44
Buffo, A., Rolando, C., and Ceruti, S. (2010). Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 79, 77–89. doi: 10.1016/j.bcp.2009.09.014
Caldinelli, L., Sacchi, S., Molla, G., Nardini, M., and Pollegioni, L. (2013). Characterization of human DAAO variants potentially related to an increased risk of schizophrenia. Biochim. Biophys. Acta 1832, 400–410. doi: 10.1016/j.bbadis.2012.11.019
Carbone, M., Duty, S., and Rattray, M. (2012). Riluzole neuroprotection in a Parkinson’s disease model involves suppression of reactive astrocytosis but not GLT-1 regulation. BMC Neurosci. 13:38. doi: 10.1186/1471-2202-13-38
Carnicella, S., Ahmadiantehrani, S., He, D. Y., Nielsen, C. K., Bartlett, S. E., Janak, P. H., et al. (2009). Cabergoline decreases alcohol drinking and seeking behaviors via glial cell line-derived neurotrophic factor. Biol. Psychiatry 66, 146–153. doi: 10.1016/j.biopsych.2008.12.022
Carnicella, S., and Ron, D. (2009). GDNF-a potential target to treat addiction. Pharmacol. Ther. 122, 9–18. doi: 10.1016/j.pharmthera.2008.12.001
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG-binding protein 2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Choudary, P. V., Molnar, M., Evans, S. J., Tomita, H., Li, J. Z., Vawter, M. P., et al. (2005). Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. U S A 102, 15653–15658. doi: 10.1073/pnas.0507901102
Cifra, A., Nani, F., and Nistri, A. (2011). Riluzole is a potent drug to protect neonatal rat hypoglossal motoneurons in vitro from excitotoxicity due to glutamate uptake block. Eur. J. Neurosci. 33, 899–913. doi: 10.1111/j.1460-9568.2010.07579.x
Colangelo, A. M., Alberghina, L., and Papa, M. (2014). Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci. Lett. 565, 59–64. doi: 10.1016/j.neulet.2014.01.014
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., et al. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U S A 94, 5401–5404. doi: 10.1073/pnas.94.10.5401
Cotter, D., Mackay, D., Chana, G., Beasley, C., Landau, S., and Everall, I. P. (2002). Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 12, 386–394. doi: 10.1093/cercor/12.4.386
Coyle, J. T. (2006). Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 26, 365–384. doi: 10.1007/s10571-006-9062-8
Czéh, B., Simon, M., Schmelting, B., Hiemke, C., and Fuchs, E. (2006). Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 31, 1616–1626. doi: 10.1038/sj.npp.1300982
Darlington, C. L. (2005). Astrocytes as targets for neuroprotective drugs. Curr. Opin. Investig. Drugs 6, 700–703.
Dwivedi, Y., Rizavi, H. S., Conley, R. R., Roberts, R. C., Tamminga, C. A., and Pandey, G. N. (2003). Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 60, 804–815. doi: 10.1001/archpsyc.60.8.804
Evans, S. J., Choudary, P. V., Neal, C. R., Li, J. Z., Vawter, M. P., Tomita, H., et al. (2004). Dysregulation of the fibroblast growth factor system in major depression. Proc. Natl. Acad. Sci. U S A 101, 15506–15511. doi: 10.1073/pnas.0406788101
Fattore, L., Puddu, M. C., Picciau, S., Cappai, A., Fratta, W., Serra, G. P., et al. (2002). Astroglial in vivo response to cocaine in mouse dentate gyrus: a quantitative and qualitative analysis by confocal microscopy. Neuroscience 110, 1–6. doi: 10.1016/s0306-4522(01)00598-x
Fiacco, T. A., Agulhon, C., Taves, S. R., Petravicz, J., Casper, K. B., Dong, X., et al. (2007). Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626. doi: 10.1016/j.neuron.2007.04.032
Freeman, M. R., and Rowitch, D. H. (2013). Evolving concepts of gliogenesis: a look way back and ahead to the next 25 years. Neuron 80, 613–623. doi: 10.1016/j.neuron.2013.10.034
Fujio, M., Nakagawa, T., Sekiya, Y., Ozawa, T., Suzuki, Y., Minami, M., et al. (2005). Gene transfer of GLT-1, a glutamate transporter, into the nucleus accumbens shell attenuates methamphetamine- and morphine-induced conditioned place preference in rats. Eur. J. Neurosci. 22, 2744–2754. doi: 10.1111/j.1460-9568.2005.04467.x
Fumagalli, E., Funicello, M., Rauen, T., Gobbi, M., and Mennini, T. (2008). Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur. J. Pharmacol. 578, 171–176. doi: 10.1016/j.ejphar.2007.10.023
Gangoso, E., Ezan, P., Valle-Casuso, J. C., Herrero-González, S., Koulakoff, A., Medina, J. M., et al. (2012). Reduced connexin43 expression correlates with c-Src activation, proliferation and glucose uptake in reactive astrocytes after an excitotoxic insult. Glia 60, 2040–2049. doi: 10.1002/glia.22418
Ghitza, U. E., Zhai, H., Wu, P., Airavaara, M., Shaham, Y., and Lu, L. (2010). Role of BDNF and GDNF in drug reward and relapse: a review. Neurosci. Biobehav. Rev. 35, 157–171. doi: 10.1016/j.neubiorev.2009.11.009
Giacometti, E., Luikenhuis, S., Beard, C., and Jaenisch, R. (2007). Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc. Natl. Acad. Sci. U S A 104, 1931–1936. doi: 10.1073/pnas.0610593104
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Habl, G., Zink, M., Petroianu, G., Bauer, M., Schneider-Axmann, T., von Wilmsdorff, M., et al. (2009). Increased D-amino acid oxidase expression in the bilateral hippocampal CA4 of schizophrenic patients: a post-mortem study. J. Neural Transm. 116, 1657–1665. doi: 10.1007/s00702-009-0312-z
Hall, F. S., Drgonova, J., Goeb, M., and Uhl, G. R. (2003). Reduced behavioral effects of cocaine in heterozygous brain-derived neurotrophic factor (BDNF) knockout mice. Neuropsychopharmacology 28, 1485–1490. doi: 10.1038/sj.npp.1300192
Hamby, M. E., and Sofroniew, M. V. (2010). Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7, 494–506. doi: 10.1016/j.nurt.2010.07.003
Hamidi, M., Drevets, W. C., and Price, J. L. (2004). Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol. Psychiatry 55, 563–569. doi: 10.1016/j.biopsych.2003.11.006
Hashimoto, K., Fujita, Y., Horio, M., Kunitachi, S., Iyo, M., Ferraris, D., et al. (2009). Co-administration of a D-amino acid oxidase inhibitor potentiates the efficacy of D-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol. Psychiatry 65, 1103–1106. doi: 10.1016/j.biopsych.2009.01.002
Hashimoto, K., Fukushima, T., Shimizu, E., Komatsu, N., Watanabe, H., Shinoda, N., et al. (2003). Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch. Gen. Psychiatry 60, 572–576. doi: 10.1001/archpsyc.60.6.572
Hatami, H., Oryan, S., Semnanian, S., Kazemi, B., Bandepour, M., and Ahmadiani, A. (2007). Alterations of BDNF and NT-3 genes expression in the nucleus paragigantocellularis during morphine dependency and withdrawal. Neuropeptides 41, 321–328. doi: 10.1016/j.npep.2007.04.007
Hebert, M. A., and O’Callaghan, J. P. (2000). Protein phosphorylation cascades associated with methamphetamine-induced glial activation. Ann. N. Y. Acad. Sci. 914, 238–262. doi: 10.1111/j.1749-6632.2000.tb05200.x
Heine, V. M., Maslam, S., Zareno, J., Joëls, M., and Lucassen, P. J. (2004). Suppressed proliferation and apoptotic changes in the rat dentate gyrus after acute and chronic stress are reversible. Eur. J. Neurosci. 19, 131–144. doi: 10.1046/j.1460-9568.2003.03100.x
Herberg, L. J., and Rose, I. C. (1990). Excitatory amino acid pathways in brain-stimulation reward. Behav. Brain Res. 39, 230–239. doi: 10.1016/0166-4328(90)90029-e
Higashimori, H., Morel, L., Huth, J., Lindemann, L., Dulla, C., Taylor, A., et al. (2013). Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum. Mol. Genet. 22, 2041–2054. doi: 10.1093/hmg/ddt055
Hisaoka, K., Nishida, A., Koda, T., Miyata, M., Zensho, H., Morinobu, S., et al. (2001). Antidepressant drug treatments induce glial cell line-derived neurotrophic factor (GDNF) synthesis and release in rat C6 glioblastoma cells. J. Neurochem. 79, 25–34. doi: 10.1046/j.1471-4159.2001.00531.x
Horger, B. A., Iyasere, C. A., Berhow, M. T., Messer, C. J., Nestler, E. J., and Taylor, J. R. (1999). Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J. Neurosci. 19, 4110–4122.
Jacobs, S., and Doering, L. C. (2010). Astrocytes prevent abnormal neuronal development in the fragile x mouse. J. Neurosci. 30, 4508–4514. doi: 10.1523/JNEUROSCI.5027-09.2010
Jacobs, S., Nathwani, M., and Doering, L. C. (2010). Fragile X astrocytes induce developmental delays in dendrite maturation and synaptic protein expression. BMC Neurosci. 11:132. doi: 10.1186/1471-2202-11-132
Jansson, L., Wennström, M., Johanson, A., and Tingström, A. (2009). Glial cell activation in response to electroconvulsive seizures. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 1119–1128. doi: 10.1016/j.pnpbp.2009.06.007
Javitt, D. C., and Zukin, S. R. (1991). Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308. doi: 10.1176/ajp.148.10.1301
John, C. S., Smith, K. L., Van’t Veer, A., Gompf, H. S., Carlezon, W. A. Jr., Cohen, B. M., et al. (2012). Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology 37, 2467–2475. doi: 10.1038/npp.2012.105
Karasawa, J., Hashimoto, K., and Chaki, S. (2008). D-Serine and a glycine transporter inhibitor improve MK-801-induced cognitive deficits in a novel object recognition test in rats. Behav. Brain Res. 186, 78–83. doi: 10.1016/j.bbr.2007.07.033
Kargieman, L., Santana, N., Mengod, G., Celada, P., and Artigas, F. (2007). Antipsychotic drugs reverse the disruption in prefrontal cortex function produced by NMDA receptor blockade with phencyclidine. Proc. Natl. Acad. Sci. U S A 104, 14843–14848. doi: 10.1073/pnas.0704848104
Kawaura, K., Koike, H., Kinoshita, K., Kambe, D., Kaku, A., Karasawa, J. I., et al. (2014). Effects of a glycine transporter-1 inhibitor and D-serine on MK-801-induced immobility in the forced swimming test in rats. Behav. Brain Res. 278, 186–192. doi: 10.1016/j.bbr.2014.09.046
Keilhoff, G., Becker, A., Grecksch, G., Bernstein, H. G., and Wolf, G. (2006). Cell proliferation is influenced by bulbectomy and normalized by imipramine treatment in a region-specific manner. Neuropsychopharmacology 31, 1165–1176. doi: 10.1038/sj.npp.1300924
Kittel-Schneider, S., Kenis, G., Schek, J., van den Hove, D., Prickaerts, J., Lesch, K. P., et al. (2012). Expression of monoamine transporters, nitric oxide synthase 3 and neurotrophin genes in antidepressant-stimulated astrocytes. Front. Psychiatry 3:33. doi: 10.3389/fpsyt.2012.00033
Kong, H., Sha, L. L., Fan, Y., Xiao, M., Ding, J. H., Wu, J., et al. (2009). Requirement of AQP4 for antidepressive efficiency of fluoxetine: implication in adult hippocampal neurogenesis. Neuropsychopharmacology 34, 1263–1276. doi: 10.1038/npp.2008.185
Kong, H., Zeng, X. N., Fan, Y., Yuan, S. T., Ge, S., Xie, W. P., et al. (2014). Aquaporin-4 knockout exacerbates corticosterone-induced depression by inhibiting astrocyte function and hippocampal neurogenesis. CNS Neurosci. Ther. 20, 391–402. doi: 10.1111/cns.12222
Koulakoff, A., Ezan, P., and Giaume, C. (2008). Neurons control the expression of connexin 30 and connexin 43 in mouse cortical astrocytes. Glia 56, 1299–1311. doi: 10.1002/glia.20698
Koyama, Y. (2014). Signaling molecules regulating phenotypic conversions of astrocytes and glial scar formation in damaged nerve tissues. Neurochem. Int. 78, 35–42. doi: 10.1016/j.neuint.2014.08.005
Koyama, Y., Tsujikawa, K., Matsuda, T., and Baba, A. (2003). Intracerebroventricular administration of an endothelin ETB receptor agonist increases expressions of GDNF and BDNF in rat brain. Eur. J. Neurosci. 18, 887–894. doi: 10.1046/j.1460-9568.2003.02797.x
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., et al. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive and neuroendocrine responses. Arch. Gen. Psychiatry 51, 199–214. doi: 10.1001/archpsyc.1994.03950030035004
Labrie, V., Fukumura, R., Rastogi, A., Fick, L., Wang, W., Boutros, P. C., et al. (2009). Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum. Mol. Genet. 18, 3227–3243. doi: 10.1093/hmg/ddp261
Labrie, V., Wang, W., Barger, S. W., Baker, G. B., and Roder, J. C. (2010). Genetic loss of D-amino acid oxidase activity reverses schizophrenia-like phenotypes in mice. Genes Brain Behav. 9, 11–25. doi: 10.1111/j.1601-183x.2009.00529.x
Laruelle, M. (2014). Schizophrenia: from dopaminergic to glutamatergic interventions. Curr. Opin. Pharmacol. 14, 97–102. doi: 10.1016/j.coph.2014.01.001
Lee, Y., Gaskins, D., Anand, A., and Shekhar, A. (2007). Glia mechanisms in mood regulation: a novel model of mood disorders. Psychopharmacology (Berl) 191, 55–65. doi: 10.1007/s00213-006-0652-4
Levenga, J., Hayashi, S., de Vrij, F. M., Koekkoek, S. K., van der Linde, H. C., Nieuwenhuizen, I., et al. (2011). AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol. Dis. 42, 311–317. doi: 10.1016/j.nbd.2011.01.022
Lipina, T., Labrie, V., Weiner, I., and Roder, J. (2005). Modulators of the glycine site on NMDA receptors, D-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology (Berl) 179, 54–67. doi: 10.1007/s00213-005-2210-x
Liu, Q., Zhu, H. Y., Li, B., Wang, Y. Q., Yu, J., and Wu, G. C. (2012). Chronic clomipramine treatment restores hippocampal expression of glial cell line-derived neurotrophic factor in a rat model of depression. J. Affect. Disord. 141, 367–372. doi: 10.1016/j.jad.2012.03.018
Madeira, C., Freitas, M. E., Vargas-Lopes, C., Wolosker, H., and Panizzutti, R. (2008). Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr. Res. 101, 76–83. doi: 10.1016/j.schres.2008.02.002
Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., and Jin, L. W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009
Mallei, A., Shi, B., and Mocchetti, I. (2002). Antidepressant treatments induce the expression of basic fibroblast growth factor in cortical and hippocampal neurons. Mol. Pharmacol. 61, 1017–1024. doi: 10.1124/mol.61.5.1017
Manley, G. T., Fujimur, M., Ma, T., Noshita, N., Filiz, F., Bollen, A. W., et al. (2000). Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 6, 159–163. doi: 10.1038/72256
Martineau, M., Parpura, V., and Mothet, J. P. (2014). Cell-type specific mechanisms of D-serine uptake and release in the brain. Front. Synaptic Neurosci. 6:12. doi: 10.3389/fnsyn.2014.00012
Martínez-Turrillas, R., Del Río, J., and Frechilla, D. (2005). Sequential changes in BDNF mRNA expression and synaptic levels of AMPA receptor subunits in rat hippocampus after chronic antidepressant treatment. Neuropharmacology 49, 1178–1188. doi: 10.1016/j.neuropharm.2005.07.006
Messer, C. J., Eisch, A. J., Carlezon, W. A. Jr., Whisler, K., Shen, L., Wolf, D. H., et al. (2000). Role for GDNF in biochemical and behavioral adaptations to drugs of abuse. Neuron 26, 247–257. doi: 10.1016/s0896-6273(00)81154-x
Miguel-Hidalgo, J. J., Wilson, B. A., Hussain, S., Meshram, A., Rajkowska, G., and Stockmeier, C. A. (2014). Reduced connexin 43 immunolabeling in the orbitofrontal cortex in alcohol dependence and depression. J. Psychiatr. Res. 55, 101–109. doi: 10.1016/j.jpsychires.2014.04.007
Molofsky, A. V., Krencik, R., Ullian, E. M., Tsai, H. H., Deneen, B., Richardson, W. D., et al. (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 26, 891–907. doi: 10.1101/gad.188326.112
Morioka, N., Suekama, K., Zhang, F. F., Kajitani, N., Hisaoka-Nakashima, K., Takebayashi, M., et al. (2014). Amitriptyline up-regulates connexin43-gap junction in rat cultured cortical astrocytes via activation of the p38 and c-Fos/AP-1 signalling pathway. Br. J. Pharmacol. 171, 2854–2867. doi: 10.1111/bph.12614
Nakagawa, T., Fujio, M., Ozawa, T., Minami, M., and Satoh, M. (2005). Effect of MS-153, a glutamate transporter activator, on the conditioned rewarding effects of morphine, methamphetamine and cocaine in mice. Behav. Brain Res. 156, 233–239. doi: 10.1016/j.bbr.2004.05.029
Narita, M., Miyatake, M., Narita, M., Shibasaki, M., Shindo, K., Nakamura, A., et al. (2006). Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology 31, 2476–2488. doi: 10.1038/sj.npp.1301007
Narita, M., Miyatake, M., Shibasaki, M., Tsuda, M., Koizumi, S., Narita, M., et al. (2005). Long-lasting change in brain dynamics induced by methamphetamine: enhancement of protein kinase C-dependent astrocytic response and behavioral sensitization. J. Neurochem. 93, 1383–1392. doi: 10.1111/j.1471-4159.2005.03097.x
Nimchinsky, E. A., Oberlander, A. M., and Svoboda, K. (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21, 5139–5146.
Ninan, I., Jardemark, K. E., and Wang, R. Y. (2003). Differential effects of atypical and typical antipsychotic drugs on N-methyl-D-aspartate- and electrically evoked responses in the pyramidal cells of the rat medial prefrontal cortex. Synapse 48, 66–79. doi: 10.1002/syn.10189
Niwa, M., Nitta, A., Yamada, Y., Nakajima, A., Saito, K., Seishima, M., et al. (2007). An inducer for glial cell line-derived neurotrophic factor and tumor necrosis factor-alpha protects against methamphetamine-induced rewarding effects and sensitization. Biol. Psychiatry 61, 890–901. doi: 10.1016/j.biopsych.2006.06.016
Ohta, K., Fujinami, A., Kuno, S., Sakakimoto, A., Matsui, H., Kawahara, Y., et al. (2004). Cabergoline stimulates synthesis and secretion of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor by mouse astrocytes in primary culture. Pharmacology 71, 162–168. doi: 10.1159/000077451
Ohta, K., Kuno, S., Mizuta, I., Fujinami, A., Matsui, H., and Ohta, M. (2003). Effects of dopamine agonists bromocriptine, pergolide, cabergoline and SKF-38393 on GDNF, NGF and BDNF synthesis in cultured mouse astrocytes. Life Sci. 73, 617–626. doi: 10.1016/s0024-3205(03)00321-7
Ongür, D., Drevets, W. C., and Price, J. L. (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U S A 95, 13290–13295. doi: 10.1073/pnas.95.22.13290
Ongür, D., Pohlman, J., Dow, A. L., Eisch, A. J., Edwin, F., Heckers, S., et al. (2007). Electroconvulsive seizures stimulate glial proliferation and reduce expression of Sprouty2 within the prefrontal cortex of rats. Biol. Psychiatry 62, 505–512. doi: 10.1016/j.biopsych.2006.11.014
Otsuki, K., Uchida, S., Watanuki, T., Wakabayashi, Y., Fujimoto, M., Matsubara, T., et al. (2008). Altered expression of neurotrophic factors in patients with major depression. J. Psychiatr. Res. 42, 1145–1153. doi: 10.1016/j.jpsychires.2008.01.010
Panatier, A., Theodosis, D. T., Mothet, J. P., Touquet, B., Pollegioni, L., Poulain, D. A., et al. (2006). Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784. doi: 10.1016/j.cell.2006.02.051
Parpura, V., Heneka, M. T., Montana, V., Oliet, S. H., Schousboe, A., Haydon, P. G., et al. (2012). Glial cells in (patho)physiology. J. Neurochem. 121, 4–27. doi: 10.1111/j.1471-4159.2012.07664.x
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431. doi: 10.1016/j.tins.2009.05.001
Perez, S. M., and Lodge, D. J. (2014). New approaches to the management of schizophrenia: focus on aberrant hippocampal drive of dopamine pathways. Drug Des. Devel. Ther. 8, 887–896. doi: 10.2147/DDDT.s42708
Petravicz, J., Fiacco, T. A., and McCarthy, K. D. (2008). Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 28, 4967–4973. doi: 10.1523/JNEUROSCI.5572-07.2008
Pop, A. S., Gomez-Mancilla, B., Neri, G., Willemsen, R., and Gasparini, F. (2014). Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology (Berl) 231, 1217–1226. doi: 10.1007/s00213-013-3330-3
Pu, L., Liu, Q. S., and Poo, M. M. (2006). BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat. Neurosci. 9, 605–607. doi: 10.1038/nn1687
Pubill, D., Canudas, A. M., Pallàs, M., Camins, A., Camarasa, J., and Escubedo, E. (2003). Different glial response to methamphetamine- and methylenedioxymethamphetamine-induced neurotoxicity. Naunyn Schmiedebergs Arch. Pharmacol. 367, 490–499. doi: 10.1007/s00210-003-0747-y
Puhl, M. D., Mintzopoulos, D., Jensen, J. E., Gillis, T. E., Konopaske, G. T., Kaufman, M. J., et al. (2014). In vivo magnetic resonance studies reveal neuroanatomical and neurochemical abnormalities in the serine racemase knockout mouse model of schizophrenia. Neurobiol. Dis. 73C, 269–274. doi: 10.1016/j.nbd.2014.10.009
Rouach, N., Avignone, E., Même, W., Koulakoff, A., Venance, L., Blomstrand, F., et al. (2002). Gap junctions and connexin expression in the normal and pathological central nervous system. Biol. Cell. 94, 457–475. doi: 10.1016/s0248-4900(02)00016-3
Saadoun, S., Papadopoulos, M. C., Watanabe, H., Yan, D., Manley, G. T., and Verkman, A. S. (2005). Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J. Cell Sci. 118(Pt. 24), 5691–5698. doi: 10.1242/jcs.02680
Samaco, R. C., and Neul, J. L. (2011). Complexities of Rett syndrome and MeCP2. J. Neurosci. 31, 7951–7959. doi: 10.1523/JNEUROSCI.0169-11.2011
Sanacora, G., and Banasr, M. (2013). From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol. Psychiatry 73, 1172–1179. doi: 10.1016/j.biopsych.2013.03.032
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., et al. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809. doi: 10.1126/science.1083328
Scemes, E., and Spray, D. C. (2012). Extracellular K+; and astrocyte signaling via connexin and pannexin channels. Neurochem. Res. 37, 2310–2316. doi: 10.1007/s11064-012-0759-4
Scharf, S. H., Jaeschke, G., Wettstein, J. G., and Lindemann, L. (2015). Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharmacol. 20C, 124–134. doi: 10.1016/j.coph.2014.11.004
Shen, F., Meredith, G. E., and Napier, T. C. (2006). Amphetamine-induced place preference and conditioned motor sensitization requires activation of tyrosine kinase receptors in the hippocampus. J. Neurosci. 26, 11041–11051. doi: 10.1523/jneurosci.2898-06.2006
Skucas, V. A., Mathews, I. B., Yang, J., Cheng, Q., Treister, A., Duffy, A. M., et al. (2011). Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J. Neurosci. 31, 6392–6397. doi: 10.1523/JNEUROSCI.6249-10.2011
Smith, S. M., Uslaner, J. M., Yao, L., Mullins, C. M., Surles, N. O., Huszar, S. L., et al. (2009). The behavioral and neurochemical effects of a novel D-amino acid oxidase inhibitor compound 8 [4H-thieno [3,2-b]pyrrole-5-carboxylic acid] and D-serine. J. Pharmacol. Exp. Ther. 328, 921–930. doi: 10.1124/jpet.108.147884
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
Stehberg, J., Moraga-Amaro, R., Salazar, C., Becerra, A., Echeverría, C., Orellana, J. A., et al. (2012). Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 26, 3649–3657. doi: 10.1096/fj.11-198416
Stockmeier, C. A., Mahajan, G. J., Konick, L. C., Overholser, J. C., Jurjus, G. J., Meltzer, H. Y., et al. (2004). Cellular changes in the postmortem hippocampus in major depression. Biol. Psychiatry 56, 640–650. doi: 10.1016/j.biopsych.2004.08.022
Sun, J. D., Liu, Y., Yuan, Y. H., Li, J., and Chen, N. H. (2012). Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology 37, 1305–1320. doi: 10.1038/npp.2011.319
Tabernero, A., Medina, J. M., and Giaume, C. (2006). Glucose metabolism and proliferation in glia: role of astrocytic gap junctions. J. Neurochem. 99, 1049–1061. doi: 10.1111/j.1471-4159.2006.04088.x
Tanahashi, S., Yamamura, S., Nakagawa, M., Motomura, E., and Okada, M. (2012). Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 165, 1543–1555. doi: 10.1111/j.1476-5381.2011.01638.x
Theodoric, N., Bechberger, J. F., Naus, C. C., and Sin, W. C. (2012). Role of gap junction protein connexin43 in astrogliosis induced by brain injury. PLoS One 7:e47311. doi: 10.1371/journal.pone.0047311
Tsai, G. E., and Lin, P. Y. (2010). Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr. Pharm. Des. 16, 522–537. doi: 10.2174/138161210790361452
Tsuda, M., Kohro, Y., Yano, T., Tsujikawa, T., Kitano, J., Tozaki-Saitoh, H., et al. (2011). , JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain. 134(Pt. 4), 1127–1139. doi: 10.1093/brain/awr025
Tuominen, H. J., Tiihonen, J., and Wahlbeck, K. (2005). Glutamatergic drugs for schizophrenia: a systematic review and meta-analysis. Schizophr. Res. 72, 225–234. doi: 10.1016/j.schres.2004.05.005
Van Horn, M. R., Sild, M., and Ruthazer, E. S. (2013). D-serine as a gliotransmitter and its roles in brain development and disease. Front. Cell. Neurosci. 7:39. doi: 10.3389/fncel.2013.00039
van Huijstee, A. N., and Mansvelder, H. D. (2015). Glutamatergic synaptic plasticity in the mesocorticolimbic system in addiction. Front. Cell. Neurosci. 8:466. doi: 10.3389/fncel.2014.00466
Vargas-Perez, H., Bahi, A., Bufalino, M. R., Ting-A-Kee, R., Maal-Bared, G., Lam, J., et al. (2014). BDNF signaling in the VTA links the drug-dependent state to drug withdrawal aversions. J. Neurosci. 34, 7899–7909. doi: 10.1523/JNEUROSCI.3776-13.2014
Vargas-Perez, H., Ting-A Kee, R., Walton, C. H., Hansen, D. M., Razavi, R., Clarke, L., et al. (2009). Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science 324, 1732–1734. doi: 10.1126/science.1168501
Verkman, A. S., Binder, D. K., Bloch, O., Auguste, K., and Papadopoulos, M. C. (2006). Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim. Biophys. Acta 1758, 1085–1093. doi: 10.1016/j.bbamem.2006.02.018
Vinueza Veloz, M. F., Buijsen, R. A., Willemsen, R., Cupido, A., Bosman, L. W., Koekkoek, S. K., et al. (2012). The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 11, 325–331. doi: 10.1111/j.1601-183x.2011.00763.x
Weng, S. M., Bailey, M. E., and Cobb, S. R. (2011). Rett syndrome: from bed to bench. Pediatr. Neonatol. 52, 309–316. doi: 10.1016/j.pedneo.2011.08.002
Wolosker, H., Blackshaw, S., and Snyder, S. H. (1999). Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. U S A 96, 13409–13414. doi: 10.1073/pnas.96.23.13409
Xiao, M., and Hu, G. (2014). Involvement of aquaporin 4 in astrocyte function and neuropsychiatric disorders. CNS Neurosci. Ther. 20, 385–390. doi: 10.1111/cns.12267
Yan, H. T., Wu, N., Lu, X. Q., Su, R. B., Zheng, J. Q., and Li, J. (2013). Aquaporin-4 deficiency attenuates opioid dependence through suppressing glutamate transporter-1 down-regulation and maintaining glutamate homeostasis. CNS Neurosci. Ther. 19, 12–19. doi: 10.1111/cns.12012
Yan, Y., Yamada, K., Niwa, M., Nagai, T., Nitta, A., and Nabeshima, T. (2007). Enduring vulnerability to reinstatement of methamphetamine-seeking behavior in glial-cell-line-derived neurotrophic factor mutant mice. FASEB J. 21, 1994–2004. doi: 10.1096/fj.06-7772com
Yang, Q., Feng, B., Zhang, K., Guo, Y. Y., Liu, S. B., Wu, Y. M., et al. (2012). Excessive astrocyte-derived neurotrophin-3 contributes to the abnormal neuronal dendritic development in a mouse model of fragile X syndrome. PLoS Genet. 8:e1003172. doi: 10.1371/journal.pgen.1003172
Zhang, J., Zhang, X., Su, H., Tao, J., Xie, Y., Han, B., et al. (2014). Increased serum brain-derived neurotrophic factor levels during opiate withdrawal. Neurosci. Lett. 571, 61–65. doi: 10.1016/j.neulet.2014.04.048
Keywords: astrocyte, schizophrenia, mood disease, drug dependence, neurodevelopmental disorder
Citation: Koyama Y (2015) Functional alterations of astrocytes in mental disorders: pharmacological significance as a drug target. Front. Cell. Neurosci. 9:261. doi: 10.3389/fncel.2015.00261
Received: 23 March 2015; Accepted: 23 June 2015;
Published: 06 July 2015.
Edited by:
Takahiro A. Kato, Kyushu University, JapanReviewed by:
Amit Agarwal, Johns Hopkins University, USAGrant Robert Gordon, University of Calgary, Canada
Copyright © 2015 Koyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yutaka Koyama, Laboratory of Pharmacology, Faculty of Pharmacy, Osaka Ohtani University, 3-11-1 Nishikiori-Kita, Tondabayashi, Osaka 584-8540, Japan,a295YW1heUBvc2FrYS1vaHRhbmkuYWMuanA=