 Sergio T. Ferreira
Sergio T. Ferreira Mychael V. Lourenco
Mychael V. Lourenco Mauricio M. Oliveira
Mauricio M. Oliveira Fernanda G. De Felice2
Fernanda G. De Felice2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 26 May 2015
Sec. Cellular Neuropathology
Volume 9 - 2015 | https://doi.org/10.3389/fncel.2015.00191
This article is part of the Research Topic Recent advances in synapse biology and its implication in neurodevelopmental and neurodegenerative diseases View all 13 articles
Alzheimer’s disease (AD) is the most common form of dementia in the elderly, and affects millions of people worldwide. As the number of AD cases continues to increase in both developed and developing countries, finding therapies that effectively halt or reverse disease progression constitutes a major research and public health challenge. Since the identification of the amyloid-β peptide (Aβ) as the major component of the amyloid plaques that are characteristically found in AD brains, a major effort has aimed to determine whether and how Aβ leads to memory loss and cognitive impairment. A large body of evidence accumulated in the past 15 years supports a pivotal role of soluble Aβ oligomers (AβOs) in synapse failure and neuronal dysfunction in AD. Nonetheless, a number of basic questions, including the exact molecular composition of the synaptotoxic oligomers, the identity of the receptor(s) to which they bind, and the signaling pathways that ultimately lead to synapse failure, remain to be definitively answered. Here, we discuss recent advances that have illuminated our understanding of the chemical nature of the toxic species and the deleterious impact they have on synapses, and have culminated in the proposal of an Aβ oligomer hypothesis for Alzheimer’s pathogenesis. We also highlight outstanding questions and challenges in AD research that should be addressed to allow translation of research findings into effective AD therapies.
Alzheimer’s disease (AD) is a severe neurodegenerative disorder characterized by memory loss and progressive cognitive disability. It is the most prevalent form of dementia in the elderly, affecting more than 40 million people worldwide (Alzheimer’s Association, 2015). The number of AD cases is reportedly increasing and is expected to almost double by 2030 (Brodaty et al., 2011).
The current scenario is bleak: disease-modifying therapies are still not available, and existing strategies do not target potential causes of AD but rather downstream events in the disease. AD is a disease of complex etiology and, despite intense research efforts in the past few decades, no effective therapeutic approach has translated into clinical application. Meanwhile, costs associated with patient care and symptomatic therapeutics have escalated, making AD a major public health challenge in both developed and developing countries.
Understanding in molecular detail how AD develops represents a critical step towards determining which molecules and pathways should be targeted to efficiently halt or reverse neurological outcomes. AD is primarily a sporadic disorder: age is the main risk factor (LaFerla and Oddo, 2005) and fewer than 5% of the cases appear associated to familial inheritance (less than 1% linked to APP mutations; Tanzi and Bertram, 2001; Herrup, 2010). Thus, identification of toxins that accumulate with aging in the AD brain and the aberrant signaling pathways that lead to synapse dysfunction holds significant potential to pave the way for successful new therapeutic approaches.
Here, we focus on the discovery of soluble Aβ oligomers (AβOs) as toxins that accumulate in the AD brain and target synapses, and how these findings have led to significant advances in our understanding of mechanisms of memory impairment. We address the still unresolved issue of what is/are the receptor(s) to which AβOs bind at synapses, and further discuss recent evidence indicating that different oligomeric assemblies of Aβ may act together to orchestrate the complex pathogenesis of AD. Finally, we offer perspectives on how accumulated evidence from basic research might translate into clinical applications in AD.
Following the initial description of the presence of amyloid plaques in the brain of a severely demented patient (Alzheimer, 1907; English-translated version in Alzheimer et al., 1995), eight decades passed before initial clues on their composition emerged. In 1984, Glenner and Wong isolated a novel peptide derived from AD-linked cerebrovascular pathology (Glenner and Wong, 1984). One year later, Masters and co-workers found that the same peptide, termed amyloid-β peptide (Aβ), is the principal component of amyloid plaque cores isolated from AD brains (Masters et al., 1985).
Aβ is a 4 kDa peptide (with 40- and 42-amino acid residue peptides as the predominant species) derived from proteolytic cleavage of a precursor protein termed amyloid precursor protein (APP; Kang et al., 1987; De Felice et al., 2004; Gralle and Ferreira, 2007). Aβ monomers readily aggregate in aqueous medium, giving rise to various types of assemblies including oligomers, protofibrils and amyloid fibrils. While AβOs are soluble and may spread throughout the brain, amyloid fibrils are larger and insoluble, and assemble into amyloid plaques, forming histological lesions that are characteristic of AD.
The idea that amyloid plaques might account for neurodegeneration (specifically, nerve cell death) and cognitive decline in AD became established in the so-called “amyloid cascade hypothesis”, supported by robust in vitro evidence pointing to a neurotoxic role of Aβ aggregates (e.g., Saitoh et al., 1989; Kowall et al., 1991; Pike et al., 1993; Lorenzo and Yankner, 1994), and put forward in an influential review by Hardy and Higgins (Hardy and Higgins, 1992). The central feature of that proposal was that deposition of aggregated Aβ into plaques would lead to neuronal death, ultimately causing dementia.
Nonetheless, as early as 1991, an intriguing report by Terry and colleagues (Terry et al., 1991) had demonstrated that synapse loss, rather than amyloid plaque load, provided the best pathological correlate of memory decline in AD. That notion was further supported by other studies that did not find direct correlation between plaque deposition and synapse/neuritic loss in postmortem analysis of AD brains (e.g., Masliah et al., 1990, 1993; Dickson et al., 1995). Further indicating a disconnect between plaque pathology and memory impairment, several groups reported that cognitive deficits in transgenic mouse models of AD appeared before plaque deposition or detection of insoluble amyloid aggregates in their brains (e.g., Hsia et al., 1999; Mucke et al., 2000; Westerman et al., 2002). Those observations puzzled researchers for years, and those transgenic animals were not regarded as bona-fide AD models because they exhibited memory impairment in the absence of plaque deposition. Nevertheless, we now understand that the build-up of soluble Aβ species, unknown and undetected at that time, likely underlies memory impairment in such transgenic mice (see below).
The apparent controversy on how amyloid-β toxicity correlated with cognitive decline and memory impairment in AD began to be resolved in 1998. In a seminal study, William L. Klein and colleagues demonstrated that, in addition to the well-known amyloid fibrils involved in plaque formation, Aβ spontaneously forms small, soluble oligomeric assemblies they termed Aβ-derived diffusible ligands (ADDLs). Although oligomeric Aβ had already been described at that time (Podlisny et al., 1995; Kuo et al., 1996; Roher et al., 1996), Lambert et al. (1998) first established that ADDLs could act as neurotoxins in the absence of amyloid fibrils or larger aggregates. ADDLs were shown to inhibit LTP and to promote cell death in a Fyn-dependent mechanism (Lambert et al., 1998). AβOs (more generally referred to as AβOs) are increased in AD brain extracts (Walsh et al., 2002; Gong et al., 2003; Kayed et al., 2003; Lacor et al., 2004; Townsend et al., 2006; Klyubin et al., 2008; Shankar et al., 2009; Xia et al., 2009) and can be detected using oligomer-sensitive antibodies (Lambert et al., 2001, 2007; Kayed et al., 2003; Rasool et al., 2013), but are not detected by conventional histopathological techniques, such as staining with Thioflavin S or Congo Red.
Soon after their identification, oligomers were incorporated into a revised version of the amyloid cascade hypothesis (Klein et al., 2001; Hardy and Selkoe, 2002; Selkoe, 2002a), which posited that soluble AβOs, rather than insoluble fibrils or plaques, would trigger synapse failure and memory impairment. As a corollary, AD would be a disorder of synapses (Selkoe, 2002b), and neuronal loss would only account for impaired brain function at final stages of the disease.
The emergence of AβOs as neurotoxins has considerably modified our understanding of disease mechanisms, not only in the specific case of Alzheimer’s but also of other neurodegenerative disorders in which oligomers formed by other proteins play causal roles (reviewed in Ferreira et al., 2007). Recently, this notion received support from promising results of a Phase 1 clinical trial of the high affinity antibody BIIB037 (Aducanumab), which binds both plaques and soluble oligomers (Clinical Trial ID # NCT01677572). BIIB037 has now been pushed into a Phase 3 trial and results are highly anticipated.
In the following sections, we review multiple synaptotoxic/neurotoxic effects of AβOs, which led to the proposal of an “Aβ oligomer hypothesis” of AD (Ferreira and Klein, 2011) in an attempt to better explain how oligomer accumulation in the brain leads to memory decline and cognitive dysfunction in AD.
AβOs appear to interact with a number of postsynaptic proteins assembled into a putative neuronal receptor complex that may include ionotropic and metabotropic glutamate receptors, the cellular prion protein (PrPC), neuroligin, the Wnt receptor Frizzled and insulin receptors, among other constituents (reviewed in Krafft and Klein, 2010; Ferreira and Klein, 2011; Viola and Klein, 2015). Many neurotoxic effects have been described as resulting from the interaction of AβOs with several receptors or co-receptors. We here provide a brief description of the main components of a putative receptor complex assembled by AβOs.
Glutamate receptors have been closely implicated in the neurotoxicity of AβOs (Ferreira and Klein, 2011). Indeed, AβOs have been reported to interact with AMPA-type and NMDA-type ionotropic receptors (De Felice et al., 2007; Decker et al., 2010a; Zhao et al., 2010), as well as with type 5 metabotropic glutamate receptor (mGluR5; Renner et al., 2010; Um et al., 2013). AβOs appear to target synaptic sites in close proximity to AMPA receptors (AMPARs), and downregulation of AMPAR subunits prevents AβO binding to neurons (Zhao et al., 2010).
We have found that knockdown of NMDA receptors by a viral vector-mediated antisense approach (Decker et al., 2010a) or treatment of neurons with an antibody against the extracellular domain of the constitutive NR1 subunit of NMDA receptors (De Felice et al., 2007) block binding of AβOs to neuronal surface membranes. At the same time, however, presence of NMDA receptors on the neuronal surface is not sufficient for binding of AβOs to hippocampal neurons (Decker et al., 2010a). These results suggest that NMDA receptors are an integral component, likely an organizer of a receptor complex that binds oligomers at the neuronal surface.
Another protein that has received considerable recent attention as a putative oligomer receptor is mGluR5 (Renner et al., 2010; Hamilton et al., 2014; Hu et al., 2014). AβOs are capable of inducing mGluR5 clustering at the membrane, a mechanism responsible for increased calcium influx and neurotoxicity (Renner et al., 2010). Deletion of mGluR5 rescues memory deficits in APP/PS1 mice (Hamilton et al., 2014). Notably, both NMDARs and mGluR5 interact closely with PrPC at the neuronal plasma membrane (Khosravani et al., 2008; Beraldo et al., 2011; Um et al., 2013), further suggesting that they may be part of a PrPC-containing oligomer receptor complex (see below).
Much attention has also been given to the role of PrPC in oligomer binding. Since the original report that PrPC binds AβOs and transduces neurotoxic signals (Laurén et al., 2009), there has been some controversy as to whether PrPC is indeed necessary for oligomer binding and/or oligomer-induced brain dysfunction (Balducci et al., 2010; Kessels et al., 2010; Forloni and Balducci, 2011; Fluharty et al., 2013). Nonetheless, the possibility that PrPC is a key component of an oligomer receptor complex remains attractive. Interestingly, exposure to AβOs recruits PrPC molecules to the neuronal surface (Caetano et al., 2011), consistent with functional interaction between oligomers and PrPC triggering signaling pathways to cause memory impairment (Gimbel et al., 2010; Um et al., 2012, 2013; Peters et al., 2015). Notably, AβO-induced Fyn activation, as previously demonstrated by Lambert et al. (1998), requires a receptor complex containing PrPC and mGluR5 to cause neurotoxicity and memory impairment (Um et al., 2012, 2013). Furthermore, it was recently demonstrated that pharmacological inhibition of Fyn rescued cognitive performance in an AD mouse model (Kaufman et al., 2015).
Wnt signaling has been shown to participate in many aspects of neuronal survival and function (Varela-Nallar and Inestrosa, 2013; Ríos et al., 2014). In the canonical pathway, Wnt binds to Frizzled receptors to stimulate β-catenin nuclear translocation and GSK-3β inhibition. We have reported that AβOs bind to a cysteine-rich domain of Frizzled that is directly involved in Wnt binding, thus impairing neuronal Wnt signaling (Magdesian et al., 2008) and resulting in altered expression of Wnt target genes, possibly including some related to synaptic plasticity mechanisms. Interestingly, Wnt has been found to be protective against the neurotoxicity of AβOs, reducing caspase-3 activation upon Frizzled-1 overexpression. Consistently, deletion of Frizzled-1 potentiated AβO effects (Chacón et al., 2008). Furthermore, activation of Wnt signaling counteracts behavioral impairments in AD mice (De Ferrari et al., 2003; Toledo and Inestrosa, 2010; Silva-Alvarez et al., 2013; Vargas et al., 2014). Thus, it is likely that recruitment of Frizzled to AβO receptor complexes precludes the activation of Wnt signaling and facilitates neuronal damage.
AβOs were reported to interact with the N-terminal tyrosine-rich domain of neuroligin-1. Moreover, in vitro purified N-terminal fragment of neuroligin stabilizes Aβ aggregates (Dinamarca et al., 2011). Intriguingly, neuroligin-1 can be cleaved under conditions of neuronal hyperexcitability, known to develop in AD (Sanchez et al., 2012; Vossel et al., 2013), in a process mediated by NMDARs (Suzuki et al., 2012). This could result in increased free extracellular N-terminal neuroligin fragments, thus facilitating AβO nucleation. Binding of AβOs to neuroligin could directly impact synapses, given that neuroligin associates with several partner proteins to maintain synapse structure and function. On the other hand, it was recently shown that amyloid fibrils suppress neuroligin expression to promote neurodegeneration (Bie et al., 2014). A combination of such events is likely to accelerate synapse damage and bran dysfunction in AD.
Sigma-2/PGRMC1 is the most recently described putative AβO receptor, and appears essential for oligomer neurotoxicity (Izzo et al., 2014a,b). AβOs increase total levels of this receptor in cultured hippocampal neurons (Izzo et al., 2014a), and both inhibition and knockdown of Sigma-2/PGRMC1 abolished AβO binding to synapses. Notably, a small molecule antagonist of Sigma-2/PGRMC1 receptors reversed AD memory impairment in the hAPPSwe/Ldn mouse model of AD (Izzo et al., 2014b). Future studies are warranted to understand how this protein might participate in the oligomer receptor complex, and which downstream signaling mechanisms promote neurotoxicity.
In addition to the ones listed above, several other proteins have been reported to interact with AβOs. These include EphB2, p75 neurotrophin receptor (p75NTR), α7-nicotinic receptor, adrenergic receptors, receptor for advanced glycation end-products (RAGE), calcium channels, and less known components, such as LilRb2, formyl-peptideR2 and FcγRIIb (see Patel and Jhamandas, 2012; Viola and Klein, 2015, for reviews). Sortilin, in association with p75NTR, has also emerged as a candidate AβO binding protein (Takamura et al., 2012). Recently, AβOs have been reported to target ephrin A4 receptors to mediate synapse and cognitive dysfunction in AD mouse models (Fu et al., 2014; Overk and Masliah, 2014; Vargas et al., 2014).
Initial evidence provided by Lambert et al. (1998) indicated that AβO binding is trypsin-sensitive, suggesting that protein receptors are required for AβO interactions. Nevertheless, other types of molecules have been proposed to act as oligomer ligands. The ganglioside GM1 is a non-protein AβO ligand that has attracted considerable interest in recent years. AβOs target GM1-containing regions and lipid rafts (Hong et al., 2014), and bind directly to GM1 to promote neuronal damage (Williams et al., 2011; Calamai and Pavone, 2013). Furthermore, GM1-rich membrane domains are preferential sites for Aβ aggregation (Ogawa et al., 2011; Yamamoto et al., 2012, 2014), suggesting that GM1 could play an important role in the formation of seeds for Aβ oligomerization.
Some evidence also suggests that AβOs can bind directly to lipid membranes (Canale et al., 2013; Yates et al., 2013; Lee et al., 2014a), occasionally forming toxic pores (Arispe et al., 1993, 1996; Tofoleanu and Buchete, 2012; Zhang et al., 2012; Di Scala et al., 2014; Lee et al., 2014a), or serve as ligands to non-protein molecules within the plasma membrane, raising the possibility that direct neurotoxicity could be initiated by membrane interactions of sticky AβOs. A problem with this hypothesis, however, is that it fails to explain how intricate and specific cell-signaling effects could result from non-specific interactions of AβOs with the lipid bilayer at the plasma membrane.
Studies published during the past few years have resulted in a list of more than 15 proteins, some of which are reviewed above, that could act as oligomer receptors at the neuronal plasma membrane (Viola and Klein, 2015). Nonetheless, the nature of such interactions remains elusive in most cases. Few molecules, such as PrPC, neuroligin and Frizzled, have been shown to directly interact with AβOs, but the identity of the putative X receptor that mediates initial binding of oligomers is still unknown. Nevertheless, knockdown or depletion of several candidate receptors significantly reduce AβO binding to neurons or toxicity. This suggests that a multi-protein receptor complex is assembled by oligomers and mediates toxic signal transduction.
It is of interest that PrPC, known to physically bind AβOs, appears to function as a signaling platform at the neuronal plasma membrane, interacting with numerous partners (Linden et al., 2008). Thus, PrPC may be well suited to act as an organizer of a multi-protein receptor complex responsible for the binding and transduction of the multiple neurotoxic effects of AβOs.
Initial interaction between AβOs and their immediate partners could be followed by recruitment of additional components to stabilize AβO binding. Thus, any single member of such a receptor complex would be required but not sufficient to maintain bound AβOs, as is the case for NMDAR (Decker et al., 2010a). Still, given the heterogeneity of oligomers populations, it is possible that oligomers of different sizes bind to different components within the receptor complex, increasing the complexity of signaling mechanisms.
Soluble Aβ (McLean et al., 1999) and AβOs accumulate in the brains and CSF of AD patients (Gong et al., 2003; Kayed et al., 2003; Lacor et al., 2004; Georganopoulou et al., 2005; Haes et al., 2005; Anker et al., 2009; Herskovits et al., 2013; Viola et al., 2015). Interestingly, oligomers are found at elevated levels and in association with synapses in the brains of demented AD individuals (Koffie et al., 2009; Bjorklund et al., 2012; Perez-Nievas et al., 2013), but they have not been detected at synapses in the brains of individuals who lacked pre-mortem cognitive impairment but whose brains presented typical AD neuropathology (Bjorklund et al., 2012). This suggests that the synaptic attack by AβOs could constitute the event leading to memory/cognitive dysfunction in AD. Immunofluorescence co-localization studies have revealed that AβOs target excitatory synapses (Lacor et al., 2004; Takahashi et al., 2004; Kokubo et al., 2005) and promote modifications in synapse structure and composition that disrupt synaptic function (e.g., Lacor et al., 2007; Shankar et al., 2007; Figueiredo et al., 2013). Although most current knowledge on the synaptic impact of AβOs comes from studies in cell culture or in rodent brains, these findings were recently corroborated in a novel non-human primate model of AD (Forny-Germano et al., 2014).
Although some evidence indicates that, at very low (picomolar) concentrations, AβOs may be important for proper synapse function (Puzzo et al., 2008, 2011; Garcia-Osta and Alberini, 2009; Lee et al., 2014b; Ricciarelli et al., 2014), increased levels of AβOs impair synaptic plasticity by inhibiting LTP (e.g., Lambert et al., 1998; Walsh et al., 2002; Klyubin et al., 2005, 2012; Gong et al., 2006; Hu et al., 2008; Jürgensen et al., 2011; An et al., 2013) and facilitating LTD (Wang et al., 2002; Li et al., 2009; Ma et al., 2013; Hu et al., 2014). Importantly, oligomers instigate memory impairment in rodents (Cleary et al., 2005; Lesné et al., 2006; Poling et al., 2008; Shankar et al., 2008; Figueiredo et al., 2013; Ledo et al., 2013; Lourenco et al., 2013), and blocking de novo AβO production acutely reverses synapse loss and memory impairment in APP transgenic mice in the absence of plaque load changes (Fowler et al., 2014).
Many studies have provided evidence (largely from immunofluorescence co-localization and/or co-immunoprecipitation experiments) supporting the notion that AβOs bind to excitatory synapses. However, recent studies have provided evidence that oligomers also bind to axons, albeit less robustly than to dendrites (Baleriola et al., 2014; Gan and Silverman, 2015), and that axonal binding may be, at least in part, responsible for the inhibition of fast axonal transport induced by AβOs (Decker et al., 2010b; Bomfim et al., 2012; Ramser et al., 2013). The specificity of synaptic targeting by AβOs thus deserves further examination.
Once bound to neurons, oligomers trigger multiple aberrant signaling pathways, including abnormal calcium signaling, oxidative stress, tau hyperphosphorylation, removal of plasticity-related receptors (e.g., AMPA and NMDA receptors) from synapses, impaired axonal transport, increased glutamate release from pre-synaptic terminals and elevated D-serine levels, endoplasmic reticulum (ER) stress and inflammation (Roselli et al., 2005; Rowan et al., 2005; Snyder et al., 2005; Hsieh et al., 2006; De Felice et al., 2007, 2008; Shankar et al., 2008; Decker et al., 2010a,b; Kurup et al., 2010; Saraiva et al., 2010; Wu et al., 2010; Brito-Moreira et al., 2011; Jürgensen et al., 2011; Miñano-Molina et al., 2011; Paula-Lima et al., 2011; Zhang et al., 2011; Bomfim et al., 2012; Yoon et al., 2012; Lourenco et al., 2013; Ma et al., 2013; Ramser et al., 2013; Madeira et al., 2015).
Tau hyperphosphorylation and development of neurofibrillary tangles (NFTs) are hallmarks of human AD pathology, and several studies have tried to unveil a potential relationship between amyloid and tau pathologies. Following the initial demonstration that AβOs induce tau hyperphosphorylation in cultured neurons (De Felice et al., 2008), other studies have extended those findings (Ma et al., 2009; Jin et al., 2011), and showed that tau is hyperphosphorylated in mice with increased AβO levels (Chabrier et al., 2012). The specificity of these events is supported by the fact that anti-Aβ antibodies block aberrant tau phosphorylation in vitro (De Felice et al., 2008; Jin et al., 2011) and in vivo (Serrano-Pozo et al., 2010). Moreover, tau hyperphosphorylation and NFT formation were recently demonstrated in a novel monkey model of AD generated by intracereboventricular (i.c.v.) injection of AβOs in cynomolgus monkeys (Forny-Germano et al., 2014). Recent evidence further suggests that amyloid pathology accelerates tau hyperphosphorylation and propagation in vivo (Pooler et al., 2015).
Tau can be phosphorylated by several kinases that are overactive in AD brains (e.g., GSK-3β, Cdk5, Cdc2, PKR, JNK and other MAPKs; reviewed by Mandelkow and Mandelkow, 2012). Significant evidence suggests that AβO-mediated tau phosphorylation leads to cytoskeleton disruption and synaptic loss (Zempel et al., 2010, 2013; Zempel and Mandelkow, 2012). Zempel and collaborators have shown that AβOs induce tau mis-sorting, a phenomenon in which axonal tau moves to dendrites in hyperphosphorylated form, disrupting cytoskeletal organization and axonal transport (Zempel et al., 2010), ultimately instigating neuronal collapse. In accord with this notion, AβOs were shown to impair activity-dependent tau re-localization in neurons (Frandemiche et al., 2014).
Tau pathology has further been shown to cause brain oxidative and ER stress in animal models of AD (Dias-Santagata et al., 2007; Abisambra et al., 2013; Frost et al., 2014) and, of clinical relevance, Aβ-linked cortical volume loss only develops in the presence of p-tau, further connecting amyloid to tau pathology (Desikan et al., 2011). Deletion of tau was further shown to protect hippocampal cultures from the toxicity of fibrillar Aβ and APP23 mice from developing cognitive impairment mediated by Fyn (Ittner et al., 2010).
Successive failures of anti-amyloid therapeutics in promoting cognitive benefits in AD patients has raised the possibility that tau might be a better target for therapeutic development, given its involvement in disease mechanisms. Nonetheless, despite the toxic effects aberrant tau may cause, considerable evidence now indicates that AβOs act upstream of tau to promote neuronal dysfunction. In fact, some aspects of toxicity, such as defective BDNF axonal transport, are not necessarily dependent on tau (Ramser et al., 2013; Takach et al., 2015). Thus, specific targeting of AβOs could be the key for developing effective approaches to prevent both tau-dependent and tau-independent defects in AD.
Of considerable interest are findings that AβOs impair neuronal insulin signaling (Townsend et al., 2007; Zhao et al., 2008; De Felice et al., 2009; Bomfim et al., 2012), providing an explanation at the molecular/cellular level for the connection between AD and diabetes revealed by clinical/epidemiological studies (reviewed in De Felice, 2013). Insulin receptors are widely distributed in the brain (Zhao and Alkon, 2001; Zhao et al., 2004). In addition to its regulatory actions in energy metabolism, insulin has been found to play important roles in synaptic plasticity (Chiu et al., 2008), learning and memory (reviewed in Fernandez and Torres-Alemán, 2012). Following the discovery that AβOs trigger removal of insulin receptors from neuronal plasma membranes (Zhao et al., 2008; De Felice et al., 2009), we have shown that oligomers instigate activation of neuronal stress kinases, including c-Jun N-terminal kinase (JNK), IκB kinase (IKK), and double-stranded RNA-dependent kinase (PKR), resulting in phosphorylation of the insulin receptor substrate 1 (IRS-1) at inhibitory serine residues, and blocking downstream signaling in the insulin pathway (Bomfim et al., 2012). This mechanism is analogous to what takes place in peripheral tissue, leading to insulin resistance in type II diabetes (Hotamisligil et al., 1996; Gregor and Hotamisligil, 2011). Combined removal of insulin receptors from the plasma membrane and serine phosphorylation of IRS-1 effectively blocks neuronal insulin signaling (as verified in AD brain tissue; Talbot et al., 2012), thus impeding plasticity-related actions of insulin at synapses.
Additional AβO-triggered mechanisms of synaptic dysfunction have been elucidated in the past few years. Of particular interest, elevated eIF2α phosphorylation (eIF2α-P) was recently correlated to synapse loss induced by AβOs (Lourenco et al., 2013), consistent with the finding that eIF2α kinases are overactive in brain areas responsible for memory processes in AD (Chang et al., 2002; Hoozemans et al., 2005; Yoon et al., 2012; Ma et al., 2013) and with the known role of eIF2α-P in attenuating global protein translation (Buffington et al., 2014). Importantly, suppression or inhibition of eIF2α kinases improves learning and memory in a physiological context (Costa-Mattioli et al., 2005, 2007; Zhu et al., 2011; Di Prisco et al., 2014) and alleviates memory deficits in AD models (Lourenco et al., 2013; Ma et al., 2013). Synapse damage and loss in AD might further be mediated by a mechanism involving ER stress and activation of the unfolded protein response (UPR), which comprises eIF2α-P as a key player. In fact, elevated levels of UPR markers are found in AD models (Lourenco et al., 2013; Ma et al., 2013), as well as in postmortem AD brains (Chang et al., 2002; Hoozemans et al., 2005, 2009; Chafekar et al., 2007; Yoon et al., 2012). Aberrant activation of the UPR and, in particular, eIF2α-P appears dependent on elevated TNF-α levels and TNFR1 activity, pointing to a central role of neuroinflammation in synapse loss and memory impairment in AD models (Lourenco et al., 2013). Blocking TNFR1 signaling or alleviating brain ER stress through the use of chemical chaperones abrogate AβO-induced cognitive impairment (Lourenco et al., 2013), in accordance with findings in AD transgenic models (He et al., 2007; McAlpine et al., 2009; Ricobaraza et al., 2009, 2012).
As noted above, AβOs trigger multiple neurotoxic/synaptotoxic mechanisms in vitro and in vivo, resulting in impaired cognition. Most of the current knowledge on the neuronal impact of AβOs comes from studies using cell-derived or synthetic oligomer preparations comprising a mixture of species ranging from low-n oligomers (dimers, trimers, tetramers) to higher-order assemblies typically ranging in MW from 50 to 150 kDa.
The complete molecular pathway that leads to Aβ aggregation in its multiple oligomeric forms is still unclear. Although still controversial, significant evidence points to the roles of seed species, including Aβ dimers, trimers and Aβ*56, in the build up of amyloid in the brain (Bitan et al., 2003; Hellstrand et al., 2010; O’Nuallain et al., 2010; Garai and Frieden, 2013; Tsigelny et al., 2014). Aβ monomers rapidly assemble into oligomers that might or not consecutively turn into higher-order aggregates (Rangachari et al., 2007; Velasco et al., 2012), and a stepwise aggregation process has been proposed to generate AβOs in the AD brain (Velasco et al., 2012). Whatever the precise pathway of aggregation of Aβ, high molecular weight oligomers are increased in the CSF of AD patients (Fukumoto et al., 2010) and in AD brain (Gong et al., 2003). In APP/PS1 mice, both low and high molecular weight oligomers are increased in the CSF (Takeda et al., 2013).
Given their molecular heterogeneity, it is possible that different oligomeric species trigger diverse effects in the brain by recruiting distinct signaling mechanisms. This is in line with the complex spectrum of molecular/behavioral outcomes seen in a multifaceted disorder such as AD. Indeed, specific Aβ assemblies (e.g., dimers, trimers and Aβ*56) have been reported to mediate distinct pathological effects in the diseased brain (e.g., Lesné et al., 2006, 2013; Reed et al., 2011).
Recently, we reported that low molecular weight (LMW; dimers-tetramers) and high molecular weight (HMW; ranging from ~50 to ~150 kDa) AβOs have different impacts on synapses, despite the fact that both LMW and HMW oligomers cause memory deficits in mice (Figueiredo et al., 2013). While LMW oligomers alter synaptic composition (revealed by decreased synaptophysin immunoreactivity), HMW oligomers induced neuronal oxidative stress via activation of NMDA receptors. Notably, mice i.c.v. injected with HMW oligomers had recovered from memory impairment 14 days post-injection, while memory damage persisted in mice injected with LMW oligomers (Figueiredo et al., 2013). It is, thus, likely that persistent memory impairment triggered by LMW oligomers results from synapse loss, while the reversible memory loss induced by HMW oligomers may result from aberrant activation of NMDARs, which could be blocked by memantine (Figueiredo et al., 2013). The differential impact on synapses is further supported by a recent report showing that low-n and high-n oligomers have differential binding affinities for neurons, with a population that ranges from decamers to hexadecamers exhibiting preferential synapse targeting (Velasco et al., 2012). HMW species have been recently found to significantly interact with PrPC in AD brains (Dohler et al., 2014), suggesting that PrPC might be important for transducing signals of defined AβO assemblies.
Low-molecular weight oligomers (SDS-stable dimers, trimers and tetramers) were shown to cause synapse loss (Shankar et al., 2007) and acutely impair synaptic plasticity (Chen and Glabe, 2006), with a particularly toxic role for trimers (Townsend et al., 2006). LMW oligomers were also reported to cause tau phosphorylation, cytoskeleton destabilization and cell loss in vivo (Davis et al., 2011; Brouillette et al., 2012).
Elevated Aβ dimers are detected in an age-dependent manner in AD brain (Lesné et al., 2013) and in the brains of J20 APP transgenic mice (Shankar et al., 2009). Dimers isolated from AD brain or CSF impair synapse homeostasis in vivo (Klyubin et al., 2008; Shankar et al., 2008) and their neutralization by monoclonal antibodies corrects synaptic plasticity defects (Klyubin et al., 2008; O’Nuallain et al., 2011). Interestingly, levels of Aβ dimers are increased in AD patients independently of disease progression, suggesting that once a certain level of dimers is attained in the brain, further accumulation is not necessary for disease progression (McDonald et al., 2012). Trimers, on the other hand, correlate with increased in vitro neurotoxicity (Hung et al., 2008) and are thought to act as seeds for generation of high-molecular weight oligomers (Matsumura et al., 2011).
Nonetheless, the actions of dimers/trimers on synapses and cognition remain somewhat controversial as recent reports found that preparations of Aβ dimers did not impact LTP in hippocampal slices (O’Malley et al., 2014) and transgenic brain-extracted trimers did not impact memory even when injected at high concentrations in rats (Reed et al., 2011), findings that are in harmony with in vivo evidence from Lesné et al. (2006). Still, the possibility remains that Aβ dimers/trimers are converted into more toxic species in the presence of additional factors and biophysical conditions that are specific to aged human brains.
Additional low-n species, such as tetramers, have also been proposed to play a distinct role in AD etiology (Bernstein et al., 2009). In a clinical study, Aβ pentamers and decamers were found to correlate with alterations in the cholinergic system, a prominent feature of AD (Bao et al., 2012). Notably, a very recent study has indicated that HMW oligomers, but not LMW oligomers, accumulate in forebrain cholinergic regions in AD, consistent with the deleterious role of AβOs on cholinergic neurotransmission (Baker-Nigh et al., 2015).
Despite our current knowledge of a myriad of detrimental actions of AβOs in neurons, most cellular/synaptic effects of AβOs are still orphan in terms of the identity of the specific oligomeric assemblies involved. In part, and as recently pointed out (Benilova et al., 2012), this is due to the heterogeneity of oligomer preparations used in different studies, which depends on factors including source of the peptide (i.e., synthetic vs. “naturally secreted” by cell lines transfected with human APP, or brain-extracted oligomers), presence of chemical/post-translational modifications in the peptide (e.g., pyroglutamylation, methione oxidation, tyrosine cross-linking), the specific protocol used to prepare oligomers, conditions of storage and aging of oligomer preparations, etc. Intriguingly, for example, recombinant Aβ42 was reported to aggregate faster and to be more neurotoxic than its synthetic version (Finder et al., 2010), and different assembles from synthetic or cell-derived oligomers were found to vary in terms of cognitive outcomes in rats (Reed et al., 2011). This exemplifies some of the complexities involved in comparing results from different groups and using different experimental approaches, and has led to considerable controversy and conflict in the literature. Progress in this area would greatly benefit from identification of the specific assemblies (minimally, in terms of low vs. high molecular weight oligomers) involved in specific effects of AβOs at synapses.
Despite being widely considered neurotoxins that attack synapses with high affinity, evidence has now accumulated that AβOs also elicit responses from glial cells, namely astrocytes and microglia. Indeed, it is plausible that glial responses to AβOs actively participate in neurotoxicity and brain dysfunction. Still, whether the same receptor composition as in neurons is required for AβO binding to glial cells remain unknown.
Astrocytes are known to internalize AβOs (Nielsen et al., 2010), and a recent report suggested that even low concentrations of AβOs induce calcium influx and generation of reactive oxygen species (ROS) in astrocytes, in a mechanism that could even be triggered by a single oligomer particle (Narayan et al., 2014). Through ERK signaling, AβOs were also shown to impair astrocytic metabolism (Tarczyluk et al., 2015) and insulin signaling (Zhang et al., 2015), likely contributing to the observed glial reactivity upon exposure to AβOs (Ledo et al., 2013). Impaired insulin signaling and increased astrogliosis may further facilitate the production of pro-inflammatory cytokines in the brain, as suggested by several reports (Carrero et al., 2012; Ledo et al., 2013; Lourenco et al., 2013). Additional studies are required to further elucidate how AβOs interact with astrocytes, likely disturbing their physiology and propagating neurotoxic signals in the brain.
AβOs also target microglia to promote TNF-α and IL-1β release (Ledo et al., 2013). TNF-α, in turn, stimulates stress kinase activity in neurons, resulting in impaired insulin signaling and cognition in mice (Bomfim et al., 2012; Lourenco et al., 2013). Cytokine profiling in AD brains suggests that microglial cells exhibit a phenotype often referred to as M1-like, characterized by augmented expression of pro-inflammatory cytokines, including IL-1β and TNF-α (Saijo and Glass, 2011).
Still, it is not clear whether pro-inflammatory responses are a result of direct or indirect effects of oligomers on microglia. A number of reports indicate that microglial receptors such as CD36, RAGE, scavenger receptor B-1 (SCARB-1), scavenger receptor A1 (SCARA-1) and TLRs 2 and 4, could directly mediate AβO actions (reviewed by Saijo and Glass, 2011). On the other hand, AβO neurotoxicity and activation of aberrant signaling pathways in neurons may result in secretion of alarmin-like molecules to induce microglial activation. Indeed, a mechanism of this type has been described involving CX3CR1, a chemokine receptor essential for neuron-microglia communication (Fuhrmann et al., 2010).
A complete picture of the contributions of each group of cytokines and chemokines to AβO toxicity is still not available. For example, knock out of interleukin-10 (IL-10), an anti-inflammatory cytokine, ameliorated cognitive impairment in APP/PS1 mice, consistent with enhanced IL-10 signaling in AD brains (Guillot-Sestier et al., 2015). Administration of IL-10 in APP/PS1 mice reduced Aβ phagocytosis, contrary to observations in IL-10-depleted animals (Guillot-Sestier et al., 2015). Suppression of interleukin receptor-associated kinase 4 (IRAK4), a downstream effector of TLR activity, resulted in improved clearance and reduced Aβ levels in APP/PS1 mice (Cameron et al., 2012). Similarly, overexpression of interleukin-1β, a pro-inflammatory cytokine, reduced amyloid plaque burden in 3 × Tg mice, although tau pathology was worsened (Ghosh et al., 2013).
Upon activation, microglia act as potent phagocytic cells, and this may be important as a defense mechanism to prevent early Aβ deposition. However, significant accumulation of AβOs appears to impair phagocytic activity of microglia. Stimulation of the PPARy/RXRa heterodimer by selective agonists intensifies Aβ phagocytosis by microglial cells and improves memory performance in APP/PS1 mice (Yamanaka et al., 2012). This appears to be mediated by CD36, a molecule that has reduced expression in APP/PS1 mice, in a mechanism that could be mediated by TNF-α (Hickman et al., 2008).
In conclusion, AβOs impact astrocytic and microglial physiology, eliciting toxic pro-inflammatory responses, including TNF-α release, contributing to brain damage in AD. In addition, it is possible that exposure to increased AβO levels leads to impairments in mechanisms essential for proper brain function, such as neuromodulator release and phagocytosis. Glial loss-of-function instigated by AβOs could impact neurotransmission and synapse homeostasis, a possibility supported by recent reports that glial gene expression is essential for memory and may be disrupted in AD (Matsuno et al., 2015; Sekar et al., 2015). Thus, glial-mediated effects initiated by AβOs in the brain may occur by increased release of toxic molecules, impairment of essential glial functions in neuromodulation, or both.
Several questions pertaining to the mechanisms of AD pathology have been resolved after three decades of research centered on the Aβ peptide. Yet, many major questions still demand answers, particularly regarding the specific synaptotoxic/neurotoxic outcomes of the accumulation of low and high molecular weight AβOs in the AD brain. Further, the precise mechanisms by which oligomers trigger neurotoxicity are not fully understood (Benilova and De Strooper, 2011). One possibility is that AβOs bind to a multi-protein receptor complex at the neuronal surface, and this triggers parallel aberrant activation of several signaling pathways that result in synapse failure and neuronal damage (Krafft and Klein, 2010; Ferreira and Klein, 2011). Another possibility is that different-sized oligomers bind to different receptors, triggering selective neurotoxic pathways. In this regard, we recently obtained evidence that high and low molecular weight oligomers do not compete for the same receptor at neuronal surfaces (Achurra and Ferreira, unpublished data). It may well be, therefore, that the neuronal impact of low molecular weight oligomers (which specifically results in persistent structural damage to synapses revealed by synaptophysin loss; Figueiredo et al., 2013) differs substantially in terms of the receptors and activated pathways from the impact of high molecular weight oligomers, which appears to be centrally mediated by NMDARs and can be blocked by the NMDAR channel blocker memantine (Figueiredo et al., 2013).
Another major gap in our current knowledge comprises the interaction of oligomers with glial cells (notably, astrocytes and microglia), how such interaction may lead to potentially damaging aberrant activation of glial cells, and the potential role of glial cells in oligomer clearance. In particular, nothing is known about the potentially different interactions of LMW and HMW oligomers with glial cells.
Lastly, we feel that major efforts should be directed at generating improved AD models. Most of the currently available animal models are based on introduction of familial AD-linked mutations in rodents, resulting in abnormally elevated Aβ levels and deposition, and AD-like pathology. While such transgenic models have revealed many important aspects of AD progression, they can at best be considered models for familial (early onset) cases of AD, which represent no more than 5% of the total disease burden (LaFerla and Oddo, 2005). Moreover, as previously pointed out (Selkoe, 2011), transgenic mouse models of AD do not recapitulate the full spectrum of AD pathology. For example, transgenic mouse models of AD lack the NFTs that are a hallmark of human pathology, and induction of tangle formation in mice depends on expression of a mutant form of tau that is not found in AD patients (LaFerla and Green, 2012). Most transgenic mouse models also fail to exhibit neuronal death as observed in later states of AD. It is noteworthy that a recently developed AD rat model engineered to express mutant human APP and PS1 genes was reported to develop cognitive impairment accompanied by AβO production, tau pathology and neuronal loss (Cohen et al., 2013), and a mouse model developed by LaFerla and co-workers expressing Arctic APP at low levels presented enhanced AβO formation and wild-type tau pathology (Chabrier et al., 2012).
Also noteworthy is that APP transgenic mice express different Aβ assemblies (from monomers to plaques), making it difficult to assign specific pathogenic roles and mechanisms to each type of Aβ aggregate. An attempt to overcome this hurdle was the development of a mouse model for AD that markedly accumulates AβOs and exhibits synapse and memory loss in the absence of amyloid fibrils and plaque deposition (Tomiyama et al., 2010). Similar cognitive impairment was found in another transgenic model at pre-plaque ages (Ferretti et al., 2011).
The inability to recapitulate cardinal features of the neuropathology of AD, on top of the essential differences between rodent and human brains in terms of the neural circuitry and architecture, may explain why a large number of therapeutic approaches that were validated in transgenic mouse models of AD have disappointingly failed in human trials.
Recent approaches have been undertaken to develop alternative experimental models that might be relevant to sporadic AD and to human pathology. We have shown that human adult cortical tissue slices can be maintained in culture for several days, allowing investigation of the effects of AβOs in ex-vivo human brain tissue (Sebollela et al., 2012). This approach has allowed an initial investigation of oligomer-induced changes in gene expression in human brain tissue, and, among other findings, showed that synaptophysin is markedly down-regulated, at both mRNA and protein levels, in human brain tissue exposed to AβOs (Sebollela et al., 2012). Clearly, access to surgical human brain tissue represents a bottleneck for wider dissemination of this experimental model. Nonetheless, the relevance of results obtained in intact human neural tissue should not be underestimated in considering the translational potential of this model for development of novel therapeutic approaches in AD.
In terms of in vivo modeling of sporadic AD, two recent initiatives are worth mentioning. First, i.c.v. injection of AβOs in mice triggers aberrant signaling pathways (Bomfim et al., 2012; Lourenco et al., 2013) that culminate with memory loss and behavioral alterations that recapitulate clinical observations in AD patients (Figueiredo et al., 2013; Ledo et al., 2013; Lourenco et al., 2013). The relative ease of implementation and robustness of this approach make it an attractive alternative for pathway elucidation as well as for screening of novel treatments or preventative schemes. It should be kept in mind, however, that this represents an acute model, as neuropathology and cognitive deficits are detected between one day and a few weeks following a single i.c.v. injection of AβOs, much faster than the course of several months or years for disease progression in transgenic mice or humans, respectively. It is possible, therefore, that the acute impact of AβOs in mice brains triggers signaling pathways that might not play significant roles in the chronic nature of human disease. Moreover, mechanistic studies in rodents might not fully recapitulate human disease, as rodents do not spontaneously develop AD features such as Aβ deposition and accelerated cognitive decline. Therefore, the creation of novel animal models that better approximate human AD seems warranted.
Development of a novel monkey model of AD was recently reported (Forny-Germano et al., 2014). I.c.v. infusion of AβOs in cynomolgus monkeys triggered brain pathology that was quite reminiscent of neuropathological alterations that are characteristic of AD, including tau hyperphosphorylation, formation of neurofibrillary inclusions, and synapse loss. The similarity in pathological features triggered by oligomers and the overall close proximity between macaque and human brains suggests that this novel model may be very relevant for studies aimed to understand in greater detail the mechanisms of pathogenesis instigated by AβOs, and to improve translation of experimental results into therapeutic approaches. Such therapeutic interventions could be useful not only in AD but also, as recently proposed (Morrison and Baxter, 2014), to maintain synapse health in age-related cognitive impairment and to prevent the onset of dementia in the elderly.
Finally, induced pluripotent stem cells (iPSCs) derived from AD patient fibroblasts (Yagi et al., 2011; Israel et al., 2012; Kondo et al., 2013) have been generated in the past 4 years. AD-derived cells present increased tau phosphorylation (Israel et al., 2012) and Aβ oligomer accumulation (Kondo et al., 2013), and similar findings were described in a 3D culture of neural stem cells derived from familial AD cases (Choi et al., 2014). Generation of sporadic AD-derived iPSCs is of special interest as it might provide insight into how non-familial AD cases initiate and develop. As technology for iPSC generation develops and costs are reduced, it is anticipated that such cells will become a valuable resource for high-content screening of potential neuroprotective or disease-modifying approaches.
In conclusion, compelling evidence supports the notion that AβOs are the proximal toxins that cause synapse failure and, ultimately, memory loss in AD. Although questions remain to be answered, e.g., concerning the identity (or identities) of the neuronal receptor(s) to which oligomers bind and the size/nature of the most synaptotoxic species, we now know a great deal about aberrant signaling pathways and mechanisms by which oligomers affect neuronal physiology and, specifically, synapse function (Figure 1). Recent efforts have also aimed to develop experimental models that more closely recapitulate the human disease and allow for detailed testing of novel potentially disease-modifying therapeutics. Together, such developments offer reasons for optimism in the quest towards development of effective approaches to prevent or rescue synapse damage in AD.
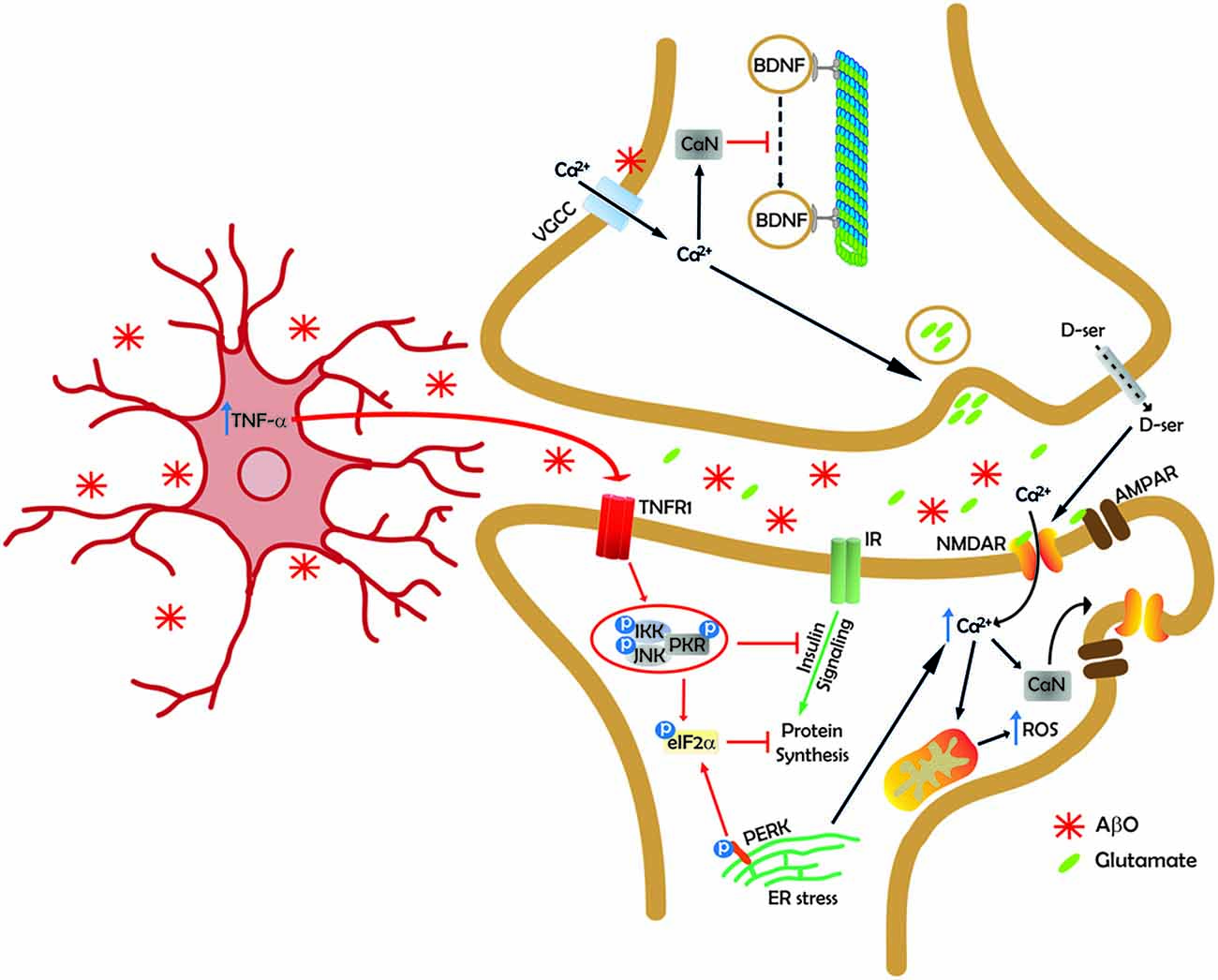
Figure 1. Synapse under siege. A simplified scheme illustrating some of the mechanisms by which AβOs (AβOs; represented as red asterisks) impact synapse plasticity and function. At the pre-synaptic terminal, AβOs inhibit microtubule-based fast axonal transport (FAT) in a tau-independent manner (Decker et al., 2010b; Ramser et al., 2013; Gan and Silverman, 2015; Takach et al., 2015), resulting in reduced or interrupted transport of various cargoes, including brain-derived neurotrophic factor (BDNF), to synapses. FAT inhibition may involve physical or functional interaction of AβOs with axonal voltage-gated calcium channels (VGCC), calcium influx into the axon and activation of calcineurin (CaN). Deregulated calcium levels also triggers increased pre-synaptic glutamate release (Brito-Moreira et al., 2011). Release of the NMDA receptor (NMDAR) co-agonist, D-serine, is also increased in neurons exposed to AβOs (Brito-Moreira et al., 2011; Madeira et al., 2015), resulting in elevated basal excitatory tonus. At the dendritic spine, AβOs aberrantly activate NMDARs, triggering increased calcium influx and increased production of reactive oxygen species (ROS; De Felice et al., 2007; Decker et al., 2010a; Saraiva et al., 2010; Paula-Lima et al., 2011), likely mediated by mitochondrial dysfunction. NMDAR-mediated calcium influx causes ryanodyne receptor-mediated calcium release from endoplasmic reticulum (ER) stores (Paula-Lima et al., 2011). Elevated calcium levels at the spine activate CaN to dephosphorylate NMDARs and AMPARs at specific phosphoepitopes, causing their removal from synapses and internalization (Snyder et al., 2005; Hsieh et al., 2006; Jürgensen et al., 2011). CaN activation has also been implicated in Aβ-induced spine loss (Wu et al., 2010; not represented in the scheme for simplicity). Calcium-dependent activation of calcium/calmodulin-dependent kinase II (CaMKII) and casein kinase II (CKII) also appears to mediate removal of NMDARs from synapses (De Felice et al., 2009; not represented in the current scheme). AβOs block insulin signaling by instigating removal of insulin receptors (IRs) from the neuronal plasma membrane (De Felice et al., 2009; not represented here), inhibiting IR autophosphorylation (Zhao et al., 2008; not represented), and by inhibiting IR-mediated tyrosine phosphorylation of the insulin receptor substrate-1 (IRS-1; Bomfim et al., 2012; not represented). AβOs instigate brain microgliosis and increased microglial production/secretion of TNF-α (Ledo et al., 2013; unpublished results). Aberrant TNF-α signaling activates cell stress kinases (c-Jun N-terminal kinase, JNK; IκB kinase, IKK; double-stranded DNA-dependent protein kinase, PKR; Bomfim et al., 2012; Lourenco et al., 2013), resulting in serine phosphorylation and further inhibition of IRS-1. PKR also phosphorylates eukaryotic initiation factor 2 α (eIF2α; Lourenco et al., 2013). eIF2α is further phosphorylated by PKR-related ER resident kinase (PERK), a kinase that executes one of the three branches of the unfolded protein response (UPR) that is activated by ER stress in neurons exposed to AβOs (Lourenco et al., 2013; Ma et al., 2013). Insulin signaling inhibition and eIF2α-P lead to blockade of protein synthesis, essential for synaptic plasticity (Buffington et al., 2014). Additional mechanisms (not included here) thought to further contribute to synapse failure include deregulation of the actin cytoskeleton at spines (Lacor et al., 2004; Davis et al., 2011), loss of pre- and post-synaptic proteins (e.g., synaptophysin, PSD-95; Roselli et al., 2005; Sebollela et al., 2012; Figueiredo et al., 2013), Fyn kinase-mediated surface removal of NMDARs and spine loss (Um et al., 2012), among others.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Work in the authors’ laboratory has been supported by grants from National Institute for Translational Neuroscience/Brazil (to STF), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (to STF and FGF), and Human Frontier Science Program (HFSP) (to FGF). MVL and MMO are pre-doctoral fellows supported by CNPq and Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES), respectively.
Abisambra, J. F., Jinwal, U. K., Blair, L. J., O’Leary, J. C. 3rd, Li, Q., Brady, S., et al. (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 33, 9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013
Alzheimer, A. (1907). Uber eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. Phychish Gerichtl. Med. Berlin 64, 146–148.
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An english translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Alzheimer’s Association. (2015). 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 11, 332–384. doi: 10.1016/j.jalz.2015.02.003
An, K., Klyubin, I., Kim, Y., Jung, J. H., Mably, A. J., O’Dowd, S. T., et al. (2013). Exosomes neutralize synaptic-plasticity-disrupting activity of Abeta assemblies in vivo. Mol. Brain 6:47. doi: 10.1186/1756-6606-6-47
Anker, J. N., Hall, W. P., Lambert, M. P., Velasco, P. T., Mrksich, M., Klein, W. L., et al. (2009). Detection and identification of bioanalytes with high resolution LSPR spectroscopy and MALDI mass spectrometry. J. Phys. Chem. C Nanomater. Interfaces 113, 5891–5894. doi: 10.1021/jp900266k
Arispe, N., Pollard, H. B., and Rojas, E. (1993). Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. U S A 90, 10573–10577. doi: 10.1073/pnas.90.22.10573
Arispe, N., Pollard, H. B., and Rojas, E. (1996). Zn2+ interaction with Alzheimer amyloid beta protein calcium channels. Proc. Natl. Acad. Sci. U S A 93, 1710–1715. doi: 10.1073/pnas.93.4.1710
Baker-Nigh, A., Vahedi, S., Davis, E. G., Weintraub, S., Bigio, E. H., Klein, W. L., et al. (2015). Neuronal amyloid-beta accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain doi: 10.1093/brain/awv024. [Epub ahead of print].
Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., et al. (2010). Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U S A 107, 2295–2300. doi: 10.1073/pnas.0911829107
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Bao, F., Wicklund, L., Lacor, P. N., Klein, W. L., Nordberg, A., and Marutle, A. (2012). Different beta-amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiol. Aging 33, 825.e1–825.e13. doi: 10.1016/j.neurobiolaging.2011.05.003
Benilova, I., and De Strooper, B. (2011). An overlooked neurotoxic species in Alzheimer’s disease. Nat. Neurosci. 14, 949–950. doi: 10.1038/nn.2871
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Beraldo, F. H., Arantes, C. P., Santos, T. G., Machado, C. F., Roffe, M., Hajj, G. N., et al. (2011). Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 25, 265–279. doi: 10.1096/fj.10-161653
Bernstein, S. L., Dupuis, N. F., Lazo, N. D., Wyttenbach, T., Condron, M. M., Bitan, G., et al. (2009). Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 1, 326–331. doi: 10.1038/nchem.247
Bie, B., Wu, J., Yang, H., Xu, J. J., Brown, D. L., and Naguib, M. (2014). Epigenetic suppression of neuroligin 1 underlies amyloid-induced memory deficiency. Nat. Neurosci. 17, 223–231. doi: 10.1038/nn.3618
Bitan, G., Kirkitadze, M. D., Lomakin, A., Vollers, S. S., Benedek, G. B., and Teplow, D. B. (2003). Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U S A 100, 330–335. doi: 10.1073/pnas.222681699
Bjorklund, N. L., Reese, L. C., Sadagoparamanujam, V. M., Ghirardi, V., Woltjer, R. L., and Taglialatela, G. (2012). Absence of amyloid beta oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol. Neurodegener. 7:23. doi: 10.1186/1750-1326-7-23
Bomfim, T. R., Forny-Germano, L., Sathler, L. B., Brito-Moreira, J., Houzel, J. C., Decker, H., et al. (2012). An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Invest. 122, 1339–1353. doi: 10.1172/jci57256
Brito-Moreira, J., Paula-Lima, A. C., Bomfim, T. R., Oliveira, F. B., Sepúlveda, F. J., De Mello, F. G., et al. (2011). Aβ oligomers induce glutamate release from hippocampal neurons. Curr. Alzheimer Res. 8, 552–562. doi: 10.2174/156720511796391917
Brodaty, H., Breteler, M. M., Dekosky, S. T., Dorenlot, P., Fratiglioni, L., Hock, C., et al. (2011). The world of dementia beyond 2020. J. Am. Geriatr. Soc. 59, 923–927. doi: 10.1111/j.1532-5415.2011.03365.x
Brouillette, J., Caillierez, R., Zommer, N., Alves-Pires, C., Benilova, I., Blum, D., et al. (2012). Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-β1–42 oligomers are revealed in vivo by using a novel animal model. J. Neurosci. 32, 7852–7861. doi: 10.1523/jneurosci.5901-11.2012
Buffington, S. A., Huang, W., and Costa-Mattioli, M. (2014). Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci. 37, 17–38. doi: 10.1146/annurev-neuro-071013-014100
Caetano, F. A., Beraldo, F. H., Hajj, G. N., Guimaraes, A. L., Jurgensen, S., Wasilewska-Sampaio, A. P., et al. (2011). Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J. Neurochem. 117, 538–553. doi: 10.1111/j.1471-4159.2011.07225.x
Calamai, M., and Pavone, F. S. (2013). Partitioning and confinement of GM1 ganglioside induced by amyloid aggregates. FEBS Lett. 587, 1385–1391. doi: 10.1016/j.febslet.2013.03.014
Cameron, B., Tse, W., Lamb, R., Li, X., Lamb, B. T., and Landreth, G. E. (2012). Loss of interleukin receptor-associated kinase 4 signaling suppresses amyloid pathology and alters microglial phenotype in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 15112–15123. doi: 10.1523/jneurosci.1729-12.2012
Canale, C., Seghezza, S., Vilasi, S., Carrotta, R., Bulone, D., Diaspro, A., et al. (2013). Different effects of Alzheimer’s peptide Abeta(1–40) oligomers and fibrils on supported lipid membranes. Biophys. Chem. 182, 23–29. doi: 10.1016/j.bpc.2013.07.010
Carrero, I., Gonzalo, M. R., Martin, B., Sanz-Anquela, J. M., Arévalo-Serrano, J., and Gonzalo-Ruiz, A. (2012). Oligomers of β-amyloid protein (Aβ1–42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1β, tumour necrosis factor-α and a nuclear factor κ-B mechanism in the rat brain. Exp. Neurol. 236, 215–227. doi: 10.1016/j.expneurol.2012.05.004
Chabrier, M. A., Blurton-Jones, M., Agazaryan, A. A., Nerhus, J. L., Martinez-Coria, H., and LaFerla, F. M. (2012). Soluble aβ promotes wild-type tau pathology in vivo. J. Neurosci. 32, 17345–17350. doi: 10.1523/JNEUROSCI.0172-12.2012
Chacón, M. A., Varela-Nallar, L., and Inestrosa, N. C. (2008). Frizzled-1 is involved in the neuroprotective effect of Wnt3a against Abeta oligomers. J. Cell. Physiol. 217, 215–227. doi: 10.1002/jcp.21497
Chafekar, S. M., Hoozemans, J. J., Zwart, R., Baas, F., and Scheper, W. (2007). Abeta 1–42 induces mild endoplasmic reticulum stress in an aggregation state-dependent manner. Antioxid. Redox Signal. 9, 2245–2254. doi: 10.1089/ars.2007.1797
Chang, R. C., Wong, A. K., Ng, H. K., and Hugon, J. (2002). Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13, 2429–2432. doi: 10.1097/00001756-200212200-00011
Chen, Y. R., and Glabe, C. G. (2006). Distinct early folding and aggregation properties of Alzheimer amyloid-beta peptides Abeta40 and Abeta42: stable trimer or tetramer formation by Abeta42. J. Biol. Chem. 281, 24414–24422. doi: 10.1074/jbc.m602363200
Chiu, S. L., Chen, C. M., and Cline, H. T. (2008). Insulin receptor signaling regulates synapse number, dendritic plasticity and circuit function in vivo. Neuron 58, 708–719. doi: 10.1016/j.neuron.2008.04.01
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’Avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278. doi: 10.1038/nature13800
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
Cohen, R. M., Rezai-Zadeh, K., Weitz, T. M., Rentsendorj, A., Gate, D., Spivak, I., et al. (2013). A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta and frank neuronal loss. J. Neurosci. 33, 6245–6256. doi: 10.1523/JNEUROSCI.3672-12.2013
Costa-Mattioli, M., Gobert, D., Harding, H., Herdy, B., Azzi, M., Bruno, M., et al. (2005). Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436, 1166–1173. doi: 10.1038/nature03897
Costa-Mattioli, M., Gobert, D., Stern, E., Gamache, K., Colina, R., Cuello, C., et al. (2007). eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129, 195–206. doi: 10.1016/j.cell.2007.01.050
Davis, R. C., Marsden, I. T., Maloney, M. T., Minamide, L. S., Podlisny, M., Selkoe, D. J., et al. (2011). Amyloid beta dimers/trimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin-phosphorylation. Mol. Neurodegener. 6:10. doi: 10.1186/1750-1326-6-10
Decker, H., Jürgensen, S., Adrover, M. F., Brito-Moreira, J., Bomfim, T. R., Klein, W. L., et al. (2010a). N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-beta peptide oligomers. J. Neurochem. 115, 1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x
Decker, H., Lo, K. Y., Unger, S. M., Ferreira, S. T., and Silverman, M. A. (2010b). Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J. Neurosci. 30, 9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010
De Felice, F. G. (2013). Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J. Clin. Invest. 123, 531–539. doi: 10.1172/jci64595
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.m607483200
De Felice, F. G., Vieira, M. N., Bomfim, T. R., Decker, H., Velasco, P. T., Lambert, M. P., et al. (2009). Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. U S A 106, 1971–1976. doi: 10.1073/pnas.0809158106
De Felice, F. G., Vieira, M. N., Saraiva, L. M., Figueroa-Villar, J. D., Garcia-Abreu, J., Liu, R., et al. (2004). Targeting the neurotoxic species in Alzheimer’s disease: inhibitors of Abeta oligomerization. FASEB J. 18, 1366–1372. doi: 10.1096/fj.04-1764com
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 29, 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
De Ferrari, G. V., Chacón, M. A., Barría, M. I., Garrido, J. L., Godoy, J. A., Olivares, G., et al. (2003). Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol. Psychiatry 8, 195–208. doi: 10.1038/sj.mp.4001208
Desikan, R. S., McEvoy, L. K., Thompson, W. K., Holland, D., Roddey, J. C., Blennow, K., et al. (2011). Amyloid-beta associated volume loss occurs only in the presence of phospho-tau. Ann. Neurol. 70, 657–661. doi: 10.1002/ana.22509
Dias-Santagata, D., Fulga, T. A., Duttaroy, A., and Feany, M. B. (2007). Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Invest. 117, 236–245. doi: 10.1172/jci28769
Dickson, D. W., Crystal, H. A., Bevona, C., Honer, W., Vincent, I., and Davies, P. (1995). Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 16, 285–298; discussion 298–304. doi: 10.1016/0197-4580(95)00013-5
Dinamarca, M. C., Weinstein, D., Monasterio, O., and Inestrosa, N. C. (2011). The synaptic protein neuroligin-1 interacts with the amyloid β-peptide. Is there a role in Alzheimer’s disease? Biochemistry 50, 8127–8137. doi: 10.1021/bi201246t
Di Prisco, G. V., Huang, W., Buffington, S. A., Hsu, C. C., Bonnen, P. E., Placzek, A. N., et al. (2014). Translational control of mGluR-dependent long-term depression and object-place learning by eIF2α. Nat. Neurosci. 17, 1073–1082. doi: 10.1038/nn.3754
Di Scala, C., Chahinian, H., Yahi, N., Garmy, N., and Fantini, J. (2014). Interaction of Alzheimer’s beta-amyloid peptides with cholesterol: mechanistic insights into amyloid pore formation. Biochemistry 53, 4489–4502. doi: 10.1021/bi500373k
Dohler, F., Sepulveda-Falla, D., Krasemann, S., Altmeppen, H., Schlüter, H., Hildebrand, D., et al. (2014). High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer’s disease. Brain 137, 873–886. doi: 10.1093/brain/awt375
Fernandez, A. M., and Torres-Alemán, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Ferreira, S. T., and Klein, W. L. (2011). The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 96, 529–543. doi: 10.1016/j.nlm.2011.08.003
Ferreira, S. T., Vieira, M. N., and De Felice, F. G. (2007). Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life 59, 332–345. doi: 10.1080/15216540701283882
Ferretti, M. T., Partridge, V., Leon, W. C., Canneva, F., Allard, S., Arvanitis, D. N., et al. (2011). Transgenic mice as a model of pre-clinical Alzheimer’s disease. Curr. Alzheimer Res. 8, 4–23. doi: 10.2174/156720511794604561
Figueiredo, C. P., Clarke, J. R., Ledo, J. H., Ribeiro, F. C., Costa, C. V., Melo, H. M., et al. (2013). Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 33, 9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013
Finder, V. H., Vodopivec, I., Nitsch, R. M., and Glockshuber, R. (2010). The recombinant amyloid-β peptide Aβ1–42 aggregates faster and is more neurotoxic than synthetic Aβ1–42. J. Mol. Biol. 396, 9–18. doi: 10.1016/j.jmb.2009.12.016
Fluharty, B. R., Biasini, E., Stravalaci, M., Sclip, A., Diomede, L., Balducci, C., et al. (2013). An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866. doi: 10.1074/jbc.M112.423954
Forloni, G., and Balducci, C. (2011). β-amyloid oligomers and prion protein: fatal attraction? Prion 5, 10–15. doi: 10.4161/pri.5.1.14367
Forny-Germano, L., Lyra e Silva, N. M., Batista, A. F., Brito-Moreira, J., Gralle, M., Boehnke, S. E., et al. (2014). Alzheimer’s disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J. Neurosci. 34, 13629–13643. doi: 10.1523/JNEUROSCI.1353-14.2014
Fowler, S. W., Chiang, A. C., Savjani, R. R., Larson, M. E., Sherman, M. A., Schuler, D. R., et al. (2014). Genetic modulation of soluble Abeta rescues cognitive and synaptic impairment in a mouse model of Alzheimer’s disease. J. Neurosci. 34, 7871–7885. doi: 10.1523/JNEUROSCI.0572-14.2014
Frandemiche, M. L., De Seranno, S., Rush, T., Borel, E., Elie, A., Arnal, I., et al. (2014). Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 34, 6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014
Frost, B., Hemberg, M., Lewis, J., and Feany, M. B. (2014). Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 17, 357–366. doi: 10.1038/nn.3639
Fu, A. K., Hung, K. W., Huang, H., Gu, S., Shen, Y., Cheng, E. Y., et al. (2014). Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 111, 9959–9964. doi: 10.1073/pnas.1405803111
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Fukumoto, H., Tokuda, T., Kasai, T., Ishigami, N., Hidaka, H., Kondo, M., et al. (2010). High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 24, 2716–2726. doi: 10.1096/fj.09-150359
Gan, K. J., and Silverman, M. A. (2015). Dendritic and axonal mechanisms of Ca2+ elevation impair BDNF transport in Aβ oligomer-treated hippocampal neurons. Mol. Biol. Cell 26, 1058–1071. doi: 10.1091/mbc.E14-12-1612
Garai, K., and Frieden, C. (2013). Quantitative analysis of the time course of Abeta oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Abeta. Proc. Natl. Acad. Sci. U S A 110, 3321–3326. doi: 10.1073/pnas.1222478110
Garcia-Osta, A., and Alberini, C. M. (2009). Amyloid beta mediates memory formation. Learn. Mem. 16, 267–272. doi: 10.1101/lm.1310209
Georganopoulou, D. G., Chang, L., Nam, J. M., Thaxton, C. S., Mufson, E. J., Klein, W. L., et al. (2005). Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 102, 2273–2276. doi: 10.1073/pnas.0409336102
Ghosh, S., Wu, M. D., Shaftel, S. S., Kyrkanides, S., LaFerla, F. M., Olschowka, J. A., et al. (2013). Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 33, 5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013
Gimbel, D. A., Nygaard, H. B., Coffey, E. E., Gunther, E. C., Laurén, J., Gimbel, Z. A., et al. (2010). Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/s0006-291x(84)80190-4
Gong, B., Cao, Z., Zheng, P., Vitolo, O. V., Liu, S., Staniszewski, A., et al. (2006). Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 126, 775–788. doi: 10.1016/j.cell.2006.06.046
Gong, Y., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., et al. (2003). Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U S A 100, 10417–10422. doi: 10.1073/pnas.1834302100
Gralle, M., and Ferreira, S. T. (2007). Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog. Neurobiol. 82, 11–32. doi: 10.1016/j.pneurobio.2007.02.001
Gregor, M. F., and Hotamisligil, G. S. (2011). Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445. doi: 10.1146/annurev-immunol-031210-101322
Guillot-Sestier, M. V., Doty, K. R., Gate, D., Rodriguez, J. Jr., Leung, B. P., Rezai-Zadeh, K., et al. (2015). Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 85, 534–548. doi: 10.1016/j.neuron.2014.12.068
Haes, A. J., Chang, L., Klein, W. L., and Van Duyne, R. P. (2005). Detection of a biomarker for Alzheimer’s disease from synthetic and clinical samples using a nanoscale optical biosensor. J. Am. Chem. Soc. 127, 2264–2271. doi: 10.1021/ja044087q
Hamilton, A., Esseltine, J. L., DeVries, R. A., Cregan, S. P., and Ferguson, S. S. (2014). Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 7:40. doi: 10.1186/1756-6606-7-40
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
He, P., Zhong, Z., Lindholm, K., Berning, L., Lee, W., Lemere, C., et al. (2007). Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 178, 829–841. doi: 10.1083/jcb.200705042
Hellstrand, E., Boland, B., Walsh, D. M., and Linse, S. (2010). Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 1, 13–18. doi: 10.1021/cn900015v
Herrup, K. (2010). Reimagining Alzheimer’s disease–an age-based hypothesis. J. Neurosci. 30, 16755–16762. doi: 10.1523/JNEUROSCI.4521-10.2010
Herskovits, A. Z., Locascio, J. J., Peskind, E. R., Li, G., and Hyman, B. T. (2013). A Luminex assay detects amyloid β oligomers in Alzheimer’s disease cerebrospinal fluid. PLoS One 8:e67898. doi: 10.1371/journal.pone.0067898
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Hong, S., Ostaszewski, B. L., Yang, T., O’Malley, T. T., Jin, M., Yanagisawa, K., et al. (2014). Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82, 308–319. doi: 10.1016/j.neuron.2014.02.027
Hoozemans, J. J., van Haastert, E. S., Nijholt, D. A., Rozemuller, A. J., Eikelenboom, P., and Scheper, W. (2009). The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 174, 1241–1251. doi: 10.2353/ajpath.2009.080814
Hoozemans, J. J., Veerhuis, R., Van Haastert, E. S., Rozemuller, J. M., Baas, F., Eikelenboom, P., et al. (2005). The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 110, 165–172. doi: 10.1007/s00401-005-1038-0
Hotamisligil, G. S., Peraldi, P., Budavari, A., Ellis, R., White, M. F., and Spiegelman, B. M. (1996). IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 271, 665–668. doi: 10.1126/science.271.5249.665
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. U S A 96, 3228–3233. doi: 10.1073/pnas.96.6.3228
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Hu, N. W., Nicoll, A. J., Zhang, D., Mably, A. J., O’Malley, T., Purro, S. A., et al. (2014). mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 5:3374. doi: 10.1038/ncomms4374
Hu, N. W., Smith, I. M., Walsh, D. M., and Rowan, M. J. (2008). Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain 131, 2414–2424. doi: 10.1093/brain/awn174
Hung, L. W., Ciccotosto, G. D., Giannakis, E., Tew, D. J., Perez, K., Masters, C. L., et al. (2008). Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J. Neurosci. 28, 11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Izzo, N. J., Staniszewski, A., To, L., Fa, M., Teich, A. F., Saeed, F., et al. (2014a). Alzheimer’s therapeutics targeting amyloid beta 1–42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One 9:e111898. doi: 10.1371/journal.pone.0111898
Izzo, N. J., Xu, J., Zeng, C., Kirk, M. J., Mozzoni, K., Silky, C., et al. (2014b). Alzheimer’s therapeutics targeting amyloid beta 1–42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One 9:e111899. doi: 10.1371/journal.pone.0111899
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U S A 108, 5819–5824. doi: 10.1073/pnas.1017033108
Jürgensen, S., Antonio, L. L., Mussi, G. E., Brito-Moreira, J., Bomfim, T. R., De Felice, F. G., et al. (2011). Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-beta oligomers. J. Biol. Chem. 286, 3270–3276. doi: 10.1074/jbc.M110.177790
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Kaufman, A. C., Salazar, S. V., Haas, L. T., Yang, J., Kostylev, M. A., Jeng, A. T., et al. (2015). Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. doi: 10.1002/ana.24394. [Epub ahead of print].
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kessels, H. W., Nguyen, L. N., Nabavi, S., and Malinow, R. (2010). The prion protein as a receptor for amyloid-beta. Nature 466, E3–E4; discussion E4–E5. doi: 10.1038/nature09217
Khosravani, H., Zhang, Y., Tsutsui, S., Hameed, S., Altier, C., Hamid, J., et al. (2008). Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 181, 551–565. doi: 10.1083/jcb.200711002
Klein, W. L., Krafft, G. A., and Finch, G. E. (2001). Targeting small Aβ oligomers: the solution for Alzheimer’s disease conundrum? Trends Neurosci. 24, 219–224. doi: 10.1016/s0166-2236(00)01749-5
Klyubin, I., Betts, V., Welzel, A. T., Blennow, K., Zetterberg, H., Wallin, A., et al. (2008). Amyloid β protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J. Neurosci. 28, 4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008
Klyubin, I., Cullen, W. K., Hu, N. W., and Rowan, M. J. (2012). Alzheimer’s disease Aβ assemblies mediating rapid disruption of synaptic plasticity and memory. Mol. Brain 5:25. doi: 10.1186/1756-6606-5-25
Klyubin, I., Walsh, D. M., Lemere, C. A., Cullen, W. K., Shankar, G. M., Betts, V., et al. (2005). Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 11, 556–561. doi: 10.1038/nm1234
Koffie, R. M., Meyer-Luehmann, M., Hashimoto, T., Adams, K. W., Mielke, M. L., Garcia-Alloza, M., et al. (2009). Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U S A 106, 4012–4017. doi: 10.1073/pnas.0811698106
Kokubo, H., Kayed, R., Glabe, C. G., and Yamaguchi, H. (2005). Soluble Aβ oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer’s disease brain. Brain Res. 1031, 222–228. doi: 10.1016/j.brainres.2004.10.041
Kondo, T., Asai, M., Tsukita, K., Kutoku, Y., Ohsawa, Y., Sunada, Y., et al. (2013). Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12, 487–496. doi: 10.1016/j.stem.2013.01.009
Kowall, N. W., Beal, M. F., Busciglio, J., Duffy, L. K., and Yankner, B. A. (1991). An in vivo model for the neurodegenerative effects of beta amyloid and protection by substance P. Proc. Natl. Acad. Sci. U S A 88, 7247–7251. doi: 10.1073/pnas.88.16.7247
Krafft, G. A., and Klein, W. L. (2010). ADDLs and the signaling web that leads to Alzheimer’s disease. Neuropharmacology 59, 230–242. doi: 10.1016/j.neuropharm.2010.07.012
Kuo, Y. M., Emmerling, M. R., Vigo-Pelfrey, C., Kasunic, T. C., Kirkpatrick, J. B., Murdoch, G. H., et al. (1996). Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 271, 4077–4081. doi: 10.1074/jbc.271.8.4077
Kurup, P., Zhang, Y., Xu, J., Venkitaramani, D. V., Haroutunian, V., Greengard, P., et al. (2010). Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J. Neurosci. 30, 5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010
Lacor, P. N., Buniel, M. C., Chang, L., Fernandez, S. J., Gong, Y., Viola, K. L., et al. (2004). Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 24, 10191–10200. doi: 10.1523/jneurosci.3432-04.2004
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., et al. (2007). Abeta oligomer-induced aberrations in synapse composition, shape and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807. doi: 10.1523/jneurosci.3501-06.2007
LaFerla, F. M., and Green, K. N. (2012). Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006320. doi: 10.1101/cshperspect.a006320
LaFerla, F. M., and Oddo, S. (2005). Alzheimer’s disease: Aβ, tau and synaptic dysfunction. Trends Mol. Med. 11, 170–176. doi: 10.1016/j.molmed.2005.02.009
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U S A 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lambert, M. P., Velasco, P. T., Chang, L., Viola, K. L., Fernandez, S., Lacor, P. N., et al. (2007). Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem. 100, 23–35. doi: 10.1111/j.1471-4159.2006.04157.x
Lambert, M. P., Viola, K. L., Chromy, B. A., Chang, L., Morgan, T. E., Yu, J., et al. (2001). Vaccination with soluble Aβ oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 79, 595–605. doi: 10.1046/j.1471-4159.2001.00592.x
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Ledo, J. H., Azevedo, E. P., Clarke, J. R., Ribeiro, F. C., Figueiredo, C. P., Foguel, D., et al. (2013). Amyloid-β oligomers link depressive-like behavior and cognitive deficits in mice. Mol. Psychiatry 18, 1053–1054. doi: 10.1038/mp.2012.168
Lee, J., Gillman, A. L., Jang, H., Ramachandran, S., Kagan, B. L., Nussinov, R., et al. (2014a). Role of the fast kinetics of pyroglutamate-modified amyloid-beta oligomers in membrane binding and membrane permeability. Biochemistry 53, 4704–4714. doi: 10.1021/bi500587p
Lee, L., Kosuri, P., and Arancio, O. (2014b). Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients. J. Alzheimers Dis. 38, 49–62. doi: 10.3233/JAD-130740
Lesné, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Lesné, S. E., Sherman, M. A., Grant, M., Kuskowski, M., Schneider, J. A., Bennett, D. A., et al. (2013). Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 136, 1383–1398. doi: 10.1093/brain/awt062
Li, S., Hong, S., Shepardson, N. E., Walsh, D. M., Shankar, G. M., and Selkoe, D. (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. doi: 10.1016/j.neuron.2009.05.012
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Lorenzo, A., and Yankner, B. A. (1994). Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. U S A 91, 12243–12247. doi: 10.1073/pnas.91.25.12243
Lourenco, M. V., Clarke, J. R., Frozza, R. L., Bomfim, T. R., Forny-Germano, L., Batista, A. F., et al. (2013). TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. doi: 10.1016/j.cmet.2013.11.002
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Ma, Q. L., Yang, F., Rosario, E. R., Ubeda, O. J., Beech, W., Gant, D. J., et al. (2009). β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J. Neurosci. 29, 9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009
Madeira, C., Lourenco, M. V., Vargas-Lopes, C., Suemoto, C. K., Brandao, C. O., Reis, T., et al. (2015). d-serine levels in Alzheimer’s disease: implications for novel biomarker development. Transl. Psychiatry 5:e561. doi: 10.1038/tp.2015.52
Magdesian, M. H., Carvalho, M. M., Mendes, F. A., Saraiva, L. M., Juliano, M. A., Juliano, L., et al. (2008). Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling. J. Biol. Chem. 283, 9359–9368. doi: 10.1074/jbc.M707108200
Mandelkow, E. M., and Mandelkow, E. (2012). Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
Masliah, E., Mallory, M., Hansen, L., DeTeresa, R., and Terry, R. D. (1993). Quantitative synaptic alterations in the human neocortex during normal aging. Neurology 43, 192–197. doi: 10.1212/WNL.43.1_Part_1.192
Masliah, E., Terry, R. D., Mallory, M., Alford, M., and Hansen, L. A. (1990). Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am. J. Pathol. 137, 1293–1297.
Masters, C. L., Multhaup, G., Simms, G., Pottgiesser, J., Martins, R. N., and Beyreuther, K. (1985). Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 4, 2757–2763.
Matsumura, S., Shinoda, K., Yamada, M., Yokojima, S., Inoue, M., Ohnishi, T., et al. (2011). Two distinct amyloid beta-protein (Abeta) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology and toxicity analyses. J. Biol. Chem. 286, 11555–11562. doi: 10.1074/jbc.M110.181313
Matsuno, M., Horiuchi, J., Yuasa, Y., Ofusa, K., Miyashita, T., Masuda, T., et al. (2015). Long-term memory formation in Drosophila requires training-dependent glial transcription. J. Neurosci. 35, 5557–5565. doi: 10.1523/JNEUROSCI.3865-14.2015
McAlpine, F. E., Lee, J. K., Harms, A. S., Ruhn, K. A., Blurton-Jones, M., Hong, J., et al. (2009). Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 34, 163–177. doi: 10.1016/j.nbd.2009.01.006
McDonald, J. M., Cairns, N. J., Taylor-Reinwald, L., Holtzman, D., and Walsh, D. M. (2012). The levels of water-soluble and triton-soluble Aβ are increased in Alzheimer’s disease brain. Brain Res. 1450, 138–147. doi: 10.1016/j.brainres.2012.02.041
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Beyreuther, K., et al. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m
Miñano-Molina, A. J., España, J., Martín, E., Barneda-Zahonero, B., Fadó, R., Solé, M., et al. (2011). Soluble oligomers of amyloid-beta peptide disrupt membrane trafficking of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J. Biol. Chem. 286, 27311–27321. doi: 10.1074/jbc.M111.227504
Morrison, J. H., and Baxter, M. G. (2014). Synaptic health. JAMA Psychiatry 71, 835–837. doi: 10.1001/jamapsychiatry.2014.380
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058.
Narayan, P., Holmstrom, K. M., Kim, D. H., Whitcomb, D. J., Wilson, M. R., St George-Hyslop, P., et al. (2014). Rare individual amyloid-β oligomers act on astrocytes to initiate neuronal damage. Biochemistry 53, 2442–2453. doi: 10.1021/bi401606f
Nielsen, H. M., Mulder, S. D., Belien, J. A., Musters, R. J., Eikelenboom, P., and Veerhuis, R. (2010). Astrocytic A beta 1–42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins. Glia 58, 1235–1246. doi: 10.1002/glia.21004
Ogawa, M., Tsukuda, M., Yamaguchi, T., Ikeda, K., Okada, T., Yano, Y., et al. (2011). Ganglioside-mediated aggregation of amyloid beta-proteins (Aβ): comparison between Aβ-(1–42) and Aβ- (1–40). J. Neurochem. 116, 851–857. doi: 10.1111/j.1471-4159.2010.06997.x
O’Malley, T. T., Oktaviani, N. A., Zhang, D., Lomakin, A., O’Nuallain, B., Linse, S., et al. (2014). Aβ dimers differ from monomers in structural propensity, aggregation paths and population of synaptotoxic assemblies. Biochem. J. 461, 413–426. doi: 10.1042/bj20140219
O’Nuallain, B., Freir, D. B., Nicoll, A. J., Risse, E., Ferguson, N., Herron, C. E., et al. (2010). Amyloid β-protein dimers rapidly form stable synaptotoxic protofibrils. J. Neurosci. 30, 14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010
O’Nuallain, B., Klyubin, I., Mc Donald, J. M., Foster, J. S., Welzel, A., Barry, A., et al. (2011). A monoclonal antibody against synthetic Aβ dimer assemblies neutralizes brain-derived synaptic plasticity-disrupting Abeta. J. Neurochem. 119, 189–201. doi: 10.1111/j.1471-4159.2011.07389.x
Overk, C. R., and Masliah, E. (2014). Toward a unified therapeutics approach targeting putative amyloid-beta oligomer receptors. Proc. Natl. Acad. Sci. U S A 111, 13680–13681. doi: 10.1073/pnas.1414554111
Patel, A. N., and Jhamandas, J. H. (2012). Neuronal receptors as targets for the action of amyloid-beta protein (Aβ) in the brain. Expert Rev. Mol. Med. 14:e2. doi: 10.1017/s1462399411002134
Paula-Lima, A. C., Adasme, T., Sanmartin, C., Sebollela, A., Hetz, C., Carrasco, M. A., et al. (2011). Amyloid β-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid. Redox Signal. 14, 1209–1223. doi: 10.1089/ars.2010.3287
Perez-Nievas, B. G., Stein, T. D., Tai, H. C., Dols-Icardo, O., Scotton, T. C., Barroeta-Espar, I., et al. (2013). Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526. doi: 10.1093/brain/awt171
Peters, C., Espinoza, M. P., Gallegos, S., Opazo, C., and Aguayo, L. G. (2015). Alzheimer’s Aβ interacts with cellular prion protein inducing neuronal membrane damage and synaptotoxicity. Neurobiol. Aging 36, 1369–1377. doi: 10.1016/j.neurobiolaging.2014.11.019
Pike, C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G., and Cotman, C. W. (1993). Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 13, 1676–1687.
Podlisny, M. B., Ostaszewski, B. L., Squazzo, S. L., Koo, E. H., Rydell, R. E., Teplow, D. B., et al. (1995). Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J. Biol. Chem. 270, 9564–9570.
Poling, A., Morgan-Paisley, K., Panos, J. J., Kim, E. M., O’Hare, E., Cleary, J. P., et al. (2008). Oligomers of the amyloid-beta protein disrupt working memory: confirmation with two behavioral procedures. Behav. Brain Res. 193, 230–234. doi: 10.1016/j.bbr.2008.06.001
Pooler, A. M., Polydoro, M., Maury, E. A., Nicholls, S. B., Reddy, S. M., Wegmann, S., et al. (2015). Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol. Commun. 3:14. doi: 10.1186/s40478-015-0199-x
Puzzo, D., Privitera, L., Fà, M., Staniszewski, A., Hashimoto, G., Aziz, F., et al. (2011). Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 69, 819–830. doi: 10.1002/ana.22313
Puzzo, D., Privitera, L., Leznik, E., Fà, M., Staniszewski, A., Palmeri, A., et al. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008
Ramser, E. M., Gan, K. J., Decker, H., Fan, E. Y., Suzuki, M. M., Ferreira, S. T., et al. (2013). Amyloid-β oligomers induce tau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol. Biol. Cell 24, 2494–2505. doi: 10.1091/mbc.E12-12-0858
Rangachari, V., Moore, B. D., Reed, D. K., Sonoda, L. K., Bridges, A. W., Conboy, E., et al. (2007). Amyloid-β(1–42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochemistry 46, 12451–12462. doi: 10.1021/bi701213s
Rasool, S., Martinez-Coria, H., Wu, J. W., LaFerla, F., and Glabe, C. G. (2013). Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Aβ deposition and tau pathology in 3xTg-AD mice. J. Neurochem. 126, 473–482. doi: 10.1111/jnc.12305
Reed, M. N., Hofmeister, J. J., Jungbauer, L., Welzel, A. T., Yu, C., Sherman, M. A., et al. (2011). Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol. Aging 32, 1784–1794. doi: 10.1016/j.neurobiolaging.2009.11.007
Renner, M., Lacor, P. N., Velasco, P. T., Xu, J., Contractor, A., Klein, W. L., et al. (2010). Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754. doi: 10.1016/j.neuron.2010.04.029
Ricciarelli, R., Puzzo, D., Bruno, O., Canepa, E., Gardella, E., Rivera, D., et al. (2014). A novel mechanism for cyclic adenosine monophosphate-mediated memory formation: role of amyloid beta. Ann. Neurol. 75, 602–607. doi: 10.1002/ana.24130
Ricobaraza, A., Cuadrado-Tejedor, M., Marco, S., Pérez-Otaño, I., and García-Osta, A. (2012). Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22, 1040–1050. doi: 10.1002/hipo.20883
Ricobaraza, A., Cuadrado-Tejedor, M., Perez-Mediavilla, A., Frechilla, D., Del Río, J., and García-Osta, A. (2009). Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 34, 1721–1732. doi: 10.1038/npp.2008.229
Ríos, J. A., Cisternas, P., Arrese, M., Barja, S., and Inestrosa, N. C. (2014). Is Alzheimer’s disease related to metabolic syndrome? A Wnt signaling conundrum. Prog. Neurobiol. 121, 125–146. doi: 10.1016/j.pneurobio.2014.07.004
Roher, A. E., Chaney, M. O., Kuo, Y. M., Webster, S. D., Stine, W. B., Haverkamp, L. J., et al. (1996). Morphology and toxicity of Aβ-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J. Biol. Chem. 271, 20631–20635. doi: 10.1074/jbc.271.34.20631
Roselli, F., Tirard, M., Lu, J., Hutzler, P., Lamberti, P., Livrea, P., et al. (2005). Soluble β-amyloid1–40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 25, 11061–11070. doi: 10.1523/jneurosci.3034-05.2005
Rowan, M. J., Klyubin, I., Wang, Q., and Anwyl, R. (2005). Synaptic plasticity disruption by amyloid beta protein: modulation by potential Alzheimer’s disease modifying therapies. Biochem. Soc. Trans. 33, 563–567. doi: 10.1042/bst0330563
Saijo, K., and Glass, C. K. (2011). Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 11, 775–787. doi: 10.1038/nri3086
Saitoh, T., Sundsmo, M., Roch, J. M., Kimura, N., Cole, G., Schubert, D., et al. (1989). Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell 58, 615–622. doi: 10.1016/0092-8674(89)90096-2
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. U S A 109, E2895–E2903. doi: 10.1073/pnas.1121081109
Saraiva, L. M., Seixas Da Silva, G. S., Galina, A., Da-Silva, W. S., Klein, W. L., Ferreira, S. T., et al. (2010). Amyloid-beta triggers the release of neuronal hexokinase 1 from mitochondria. PLoS One 5:e15230. doi: 10.1371/journal.pone.0015230
Sebollela, A., Freitas-Correa, L., Oliveira, F. F., Paula-Lima, A. C., Saraiva, L. M., Martins, S. M., et al. (2012). Amyloid-β oligomers induce differential gene expression in adult human brain slices. J. Biol. Chem. 287, 7436–7445. doi: 10.1074/jbc.m111.298471
Sekar, S., McDonald, J., Cuyugan, L., Aldrich, J., Kurdoglu, A., Adkins, J., et al. (2015). Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 36, 583–591. doi: 10.1016/j.neurobiolaging.2014.09.027
Selkoe, D. J. (2002a). Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J. Clin. Invest. 110, 1375–1381. doi: 10.1172/jci16783
Selkoe, D. J. (2002b). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Selkoe, D. J. (2011). Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 17, 1060–1065. doi: 10.1038/nm.2460
Serrano-Pozo, A., William, C. M., Ferrer, I., Uro-Coste, E., Delisle, M. B., Maurage, C. A., et al. (2010). Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain 133, 1312–1327. doi: 10.1093/brain/awq056
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/jneurosci.4970-06.2007
Shankar, G. M., Leissring, M. A., Adame, A., Sun, X., Spooner, E., Masliah, E., et al. (2009). Biochemical and immunohistochemical analysis of an Alzheimer’s disease mouse model reveals the presence of multiple cerebral Aβ assembly forms throughout life. Neurobiol. Dis. 36, 293–302. doi: 10.1016/j.nbd.2009.07.021
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Silva-Alvarez, C., Arrázola, M. S., Godoy, J. A., Ordenes, D., and Inestrosa, N. C. (2013). Canonical Wnt signaling protects hippocampal neurons from Aβ oligomers: role of non-canonical Wnt-5a/Ca(2+) in mitochondrial dynamics. Front. Cell. Neurosci. 7:97. doi: 10.3389/fncel.2013.00097
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., et al. (2005). Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 8, 1051–1058. doi: 10.1038/nn1503
Suzuki, K., Hayashi, Y., Nakahara, S., Kumazaki, H., Prox, J., Horiuchi, K., et al. (2012). Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 76, 410–422. doi: 10.1016/j.neuron.2012.10.003
Takach, O., Gill, T. B., and Silverman, M. A. (2015). Modulation of insulin signaling rescues BDNF transport defects independent of tau in amyloid-β oligomer-treated hippocampal neurons. Neurobiol. Aging 36, 1378–1382. doi: 10.1016/j.neurobiolaging.2014.11.018
Takahashi, R. H., Almeida, C. G., Kearney, P. F., Yu, F., Lin, M. T., Milner, T. A., et al. (2004). Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 24, 3592–3599. doi: 10.1523/jneurosci.5167-03.2004
Takamura, A., Sato, Y., Watabe, D., Okamoto, Y., Nakata, T., Kawarabayashi, T., et al. (2012). Sortilin is required for toxic action of Aα oligomers (AαOs): extracellular AαOs trigger apoptosis and intraneuronal AαOs impair degradation pathways. Life Sci. 91, 1177–1186. doi: 10.1016/j.lfs.2012.04.038
Takeda, S., Hashimoto, T., Roe, A. D., Hori, Y., Spires-Jones, T. L., and Hyman, B. T. (2013). Brain interstitial oligomeric amyloid beta increases with age and is resistant to clearance from brain in a mouse model of Alzheimer’s disease. FASEB J. 27, 3239–3248. doi: 10.1096/fj.13-229666
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/JCI59903
Tanzi, R. E., and Bertram, L. (2001). New frontiers in Alzheimer’s disease genetics. Neuron 32, 181–184. doi: 10.1016/s0896-6273(01)00476-7
Tarczyluk, M. A., Nagel, D. A., Rhein Parri, H., Tse, E. H., Brown, J. E., Coleman, M. D., et al. (2015). Amyloid γ 1–42 induces hypometabolism in human stem cell-derived neuron and astrocyte networks. J. Cereb. Blood Flow Metab. doi: 10.1038/jcbfm.2015.58. [Epub ahead of print].
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., Deteresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Tofoleanu, F., and Buchete, N. V. (2012). Alzheimer Aβ peptide interactions with lipid membranes: fibrils, oligomers and polymorphic amyloid channels. Prion 6, 339–345. doi: 10.4161/pri.21022
Toledo, E. M., and Inestrosa, N. C. (2010). Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol. Psychiatry 15, 272–285, 228. doi: 10.1038/mp.2009.72
Tomiyama, T., Matsuyama, S., Iso, H., Umeda, T., Takuma, H., Ohnishi, K., et al. (2010). A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation and neuronal loss in vivo. J. Neurosci. 30, 4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010
Townsend, M., Mehta, T., and Selkoe, D. J. (2007). Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J. Biol. Chem. 282, 33305–33312. doi: 10.1074/jbc.m610390200
Townsend, M., Shankar, G. M., Mehta, T., Walsh, D. M., and Selkoe, D. J. (2006). Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J. Physiol. 572, 477–492. doi: 10.1113/jphysiol.2005.103754
Tsigelny, I. F., Sharikov, Y., Kouznetsova, V. L., Greenberg, J. P., Wrasidlo, W., Gonzalez, T., et al. (2014). Structural diversity of Alzheimer’s disease amyloid-beta dimers and their role in oligomerization and fibril formation. J. Alzheimers Dis. 39, 583–600. doi: 10.3233/JAD-131589
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Varela-Nallar, L., and Inestrosa, N. C. (2013). Wnt signaling in the regulation of adult hippocampal neurogenesis. Front. Cell. Neurosci. 7:100. doi: 10.3389/fncel.2013.00100
Vargas, J. Y., Fuenzalida, M., and Inestrosa, N. C. (2014). In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J. Neurosci. 34, 2191–2202. doi: 10.1523/JNEUROSCI.0862-13.2014
Velasco, P. T., Heffern, M. C., Sebollela, A., Popova, I. A., Lacor, P. N., Lee, K. B., et al. (2012). Synapse-binding subpopulations of Abeta oligomers sensitive to peptide assembly blockers and scFv antibodies. ACS Chem. Neurosci. 3, 972–981. doi: 10.1021/cn300122k
Viola, K. L., and Klein, W. L. (2015). Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment and diagnosis. Acta Neuropathol. 129, 183–206. doi: 10.1007/s00401-015-1386-3
Viola, K. L., Sbarboro, J., Sureka, R., De, M., Bicca, M. A., Wang, J., et al. (2015). Towards non-invasive diagnostic imaging of early-stage Alzheimer’s disease. Nat. Nanotechnol. 10, 91–98. doi: 10.1038/nnano.2014.254
Vossel, K. A., Beagle, A. J., Rabinovici, G. D., Shu, H., Lee, S. E., Naasan, G., et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. doi: 10.1001/jamaneurol.2013.136
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, H. W., Pasternak, J. F., Kuo, H., Ristic, H., Lambert, M. P., Chromy, B., et al. (2002). Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 924, 133–140. doi: 10.1016/s0006-8993(01)03058-x
Westerman, M. A., Cooper-Blacketer, D., Mariash, A., Kotilinek, L., Kawarabayashi, T., Younkin, L. H., et al. (2002). The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 22, 1858–1867. doi: 10.3410/f.1009109.145810
Williams, T. L., Johnson, B. R., Urbanc, B., Jenkins, A. T., Connell, S. D., and Serpell, L. C. (2011). Aβ42 oligomers, but not fibrils, simultaneously bind to and cause damage to ganglioside-containing lipid membranes. Biochem. J. 439, 67–77. doi: 10.1042/BJ20110750
Wu, H. Y., Hudry, E., Hashimoto, T., Kuchibhotla, K., Rozkalne, A., Fan, Z., et al. (2010). Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification and neuritic dystrophies through calcineurin activation. J. Neurosci. 30, 2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010
Xia, W., Yang, T., Shankar, G., Smith, I. M., Shen, Y., Walsh, D. M., et al. (2009). A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 66, 190–199. doi: 10.1001/archneurol.2008.565
Yagi, T., Ito, D., Okada, Y., Akamatsu, W., Nihei, Y., Yoshizaki, T., et al. (2011). Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 20, 4530–4539. doi: 10.1093/hmg/ddr394
Yamamoto, N., Matsubara, T., Sobue, K., Tanida, M., Kasahara, R., Naruse, K., et al. (2012). Brain insulin resistance accelerates Abeta fibrillogenesis by inducing GM1 ganglioside clustering in the presynaptic membranes. J. Neurochem. 121, 619–628. doi: 10.1111/j.1471-4159.2012.07668.x
Yamamoto, N., Tanida, M., Kasahara, R., Sobue, K., and Suzuki, K. (2014). Leptin inhibits amyloid beta-protein fibrillogenesis by decreasing GM1 gangliosides on the neuronal cell surface through PI3K/Akt/mTOR pathway. J. Neurochem. 131, 323–332. doi: 10.1111/jnc.12828
Yamanaka, M., Ishikawa, T., Griep, A., Axt, D., Kummer, M. P., and Heneka, M. T. (2012). PPARgamma/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 32, 17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012
Yates, E. A., Owens, S. L., Lynch, M. F., Cucco, E. M., Umbaugh, C. S., and Legleiter, J. (2013). Specific domains of Aβ facilitate aggregation on and association with lipid bilayers. J. Mol. Biol. 425, 1915–1933. doi: 10.1016/j.jmb.2013.03.022
Yoon, S. O., Park, D. J., Ryu, J. C., Ozer, H. G., Tep, C., Shin, Y. J., et al. (2012). JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 75, 824–837. doi: 10.1016/j.neuron.2012.06.024
Zempel, H., Luedtke, J., Kumar, Y., Biernat, J., Dawson, H., Mandelkow, E., et al. (2013). Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 32, 2920–2937. doi: 10.1038/emboj.2013.207
Zempel, H., and Mandelkow, E. M. (2012). Linking amyloid-β and tau: amyloid-β induced synaptic dysfunction via local wreckage of the neuronal cytoskeleton. Neurodegener. Dis. 10, 64–72. doi: 10.1159/000332816
Zempel, H., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2010). Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010
Zhang, Q., Guo, S., Zhang, X., Tang, S., Wang, L., Han, X., et al. (2015). Amyloid β oligomer-induced ERK1/2-dependent serine 636/639 phosphorylation of insulin receptor substrate-1 impairs insulin signaling and glycogen storage in human astrocytes. Gene 561, 76–81. doi: 10.1016/j.gene.2015.02.011
Zhang, Y., Kurup, P., Xu, J., Anderson, G. M., Greengard, P., Nairn, A. C., et al. (2011). Reduced levels of the tyrosine phosphatase STEP block beta amyloid-mediated GluA1/GluA2 receptor internalization. J. Neurochem. 119, 664–672. doi: 10.1111/j.1471-4159.2011.07450.x
Zhang, Y. J., Shi, J. M., Bai, C. J., Wang, H., Li, H. Y., Wu, Y., et al. (2012). Intra-membrane oligomerization and extra-membrane oligomerization of amyloid-beta peptide are competing processes as a result of distinct patterns of motif interplay. J. Biol. Chem. 287, 748–756. doi: 10.1074/jbc.m111.281295
Zhao, W. Q., and Alkon, D. L. (2001). Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 177, 125–134. doi: 10.1016/s0303-7207(01)00455-5
Zhao, W. Q., Chen, H., Quon, M. J., and Alkon, D. L. (2004). Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 490, 71–81. doi: 10.1016/j.ejphar.2004.02.045
Zhao, W. Q., De Felice, F. G., Fernandez, S., Chen, H., Lambert, M. P., Quon, M. J., et al. (2008). Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 22, 246–260. doi: 10.1096/fj.06-7703com
Zhao, W. Q., Santini, F., Breese, R., Ross, D., Zhang, X. D., Stone, D. J., et al. (2010). Inhibition of calcineurin-mediated endocytosis and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J. Biol. Chem. 285, 7619–7632. doi: 10.1074/jbc.m109.057182
Keywords: Alzheimer’s disease, amyloid-β oligomers, synapse failure, neuronal dysfunction, memory loss
Citation: Ferreira ST, Lourenco MV, Oliveira MM and De Felice FG (2015) Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 9:191. doi: 10.3389/fncel.2015.00191
Received: 12 March 2015; Accepted: 30 April 2015;
Published online: 26 May 2015.
Edited by:
Rakez Kayed, University of Texas Medical Branch, USAReviewed by:
Ottavio Arancio, Columbia University, USACopyright © 2015 Ferreira, Lourenco, Oliveira and De Felice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio T. Ferreira, Institute of Biophysics Carlos Chagas Filho, Federal University of Rio de Janeiro, Av. Carlos Chagas 373, Bloco C, Sala C1-031, Rio de Janeiro, RJ 21944-590, Brazil,ZmVycmVpcmFAYmlvcW1lZC51ZnJqLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.