
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 02 February 2015
Sec. Cellular Neurophysiology
Volume 9 - 2015 | https://doi.org/10.3389/fncel.2015.00012
This article is part of the Research Topic Neurodegeneration: from genetics to molecules View all 23 articles
 Marcio H. M. da Luz1,2Italo T. Peres1
Marcio H. M. da Luz1,2Italo T. Peres1 Tiago G. Santos3Vilma R. Martins3
Tiago G. Santos3Vilma R. Martins3 Marcelo Y. Icimoto4
Marcelo Y. Icimoto4 Kil S. Lee1*
Kil S. Lee1*Accumulation of protein aggregates is a histopathological hallmark of several neurodegenerative diseases, but in most cases the aggregation occurs without defined mutations or clinical histories, suggesting that certain endogenous metabolites can promote aggregation of specific proteins. One example that supports this hypothesis is dopamine and its metabolites. Dopamine metabolism generates several oxidative metabolites that induce aggregation of α-synuclein, and represents the main etiology of Parkinson's diseases. Because dopamine and its metabolites are unstable and can be highly reactive, we investigated whether these molecules can also affect other proteins that are prone to aggregate, such as cellular prion protein (PrPC). In this study, we showed that dopamine treatment of neuronal cells reduced the number of viable cells and increased the production of reactive oxygen species (ROS) as demonstrated in previous studies. Overall PrPC expression level was not altered by dopamine treatment, but its unglycosylated form was consistently reduced at 100 μM of dopamine. At the same concentration, the level of phosphorylated mTOR and 4EBP1 was also reduced. Moreover, dopamine treatment decreased the solubility of PrPC, and increased its accumulation in autophagosomal compartments with concomitant induction of LC3-II and p62/SQSTM1 levels. In vitro oxidation of dopamine promoted formation of high-order oligomers of recombinant prion protein. These results suggest that dopamine metabolites alter the conformation of PrPC, which in turn is sorted to degradation pathway, causing autophagosome overload and attenuation of protein synthesis. Accumulation of PrPC aggregates is an important feature of prion diseases. Thus, this study brings new insight into the dopamine metabolism as a source of endogenous metabolites capable of altering PrPC solubility and its subcellular localization.
Aberrant protein aggregation is a common hallmark of many neurodegenerative diseases, while a specific protein predominantly aggregates in each type of diseases (Brundin et al., 2010). Mutations that alter amino acid sequence or modifications of side chains by reactive molecules can disrupt the native fold (Tyedmers et al., 2010; Petrov and Zagrovic, 2011). Considering that proteins perform numerous biological activities, the accumulation of their aggregates can be toxic and effective clearance of structurally altered proteins can be essential for cellular survival (Tyedmers et al., 2010).
Most cases of neurodegenerative diseases are idiopathic and protein aggregation can occur without defined mutations or clinical histories that clearly justify the manifestation of the diseases (Alkhuja, 2013; Musiek and Schindler, 2013). This observation raises the possibility that certain endogenous metabolites can induce protein misfolding, and protein aggregates may accumulate upon excessive formation of such metabolites and/or failure of degradation pathways (Morris, 2013). For instance, reactive oxygen species (ROS) are produced as byproducts of several endogenous metabolic pathways, such as mitochondrial respiration and oxidase catalyzed reactions, which include the metabolism of catecholamine (Andreyev et al., 2005; Dikalov, 2011; Kodama et al., 2013). Among catecholamines, dopamine is the one that has the greatest propensity for oxidation (LaVoie et al., 2005). During dopamine metabolism, ROS are generated not only by enzymatic reactions but also by auto-oxidation of dopamine, producing highly reactive dopamine quinone (Asanuma et al., 2003). The accumulation of neuromelanin (which is synthesized from dopamine quinone) throughout the aging process is an indication of the production of dopamine quinone in physiological conditions (Herrero et al., 1993; Kim et al., 2006). Thus, enhanced dopamine metabolism can induce oxidative stress leading to mitocontrial dysfunction and disturbance in protein quality control system (Chen et al., 2008; Hastings, 2009). Although dopamine metabolism has been extensively linked to the aggregation of α-synuclein and Parkinson's disease (Cappai et al., 2005; Lee et al., 2011), dopamine oxidation can also induce the formation of adducts and aggregates of other proteins, such as cytoskeleton proteins and mitochondrial complexes (Van Laar et al., 2009). Moreover, dopamine can also oxidize prion protein in vitro in the presence of copper ions (Shiraishi and Nishikimi, 2002; Shiraishi et al., 2005).
Conformational alteration of cellular prion protein (PrPC) is an essential event for development of transmissible spongiform encephalopathies (TSE). However, in its normal conformation, PrPC plays important roles in cell growth, differentiation and survival (Linden et al., 2008). Previous studies have demonstrated that PrPC also possesses anti-oxidant properties, but the mechanisms related with this function are still under debate. The expression of PrPC appears to increase during the oxidative stress caused by ischemia and antioxidant defense mechanisms appear to require PrPC expression (McLennan et al., 2004; Beraldo et al., 2013). Direct reactions between PrPC and ROS have also been proposed (Bertuchi et al., 2012), suggesting that PrPC might play roles in scavenging ROS from the extracellular environment. PrPC is highly expressed in neurons and has flexible N-terminal domain enriched by amino acid residues that are more susceptible to oxidation (Linden et al., 2008; Abouelatta et al., 2009). Thus, it is plausible to hypothesize that PrPC can be a preferential target of ROS in the central nervous system with consequent inactivation of ROS activity. PrPC appears to participate in the regulation of dopamine metabolism (Lee et al., 1999; Adjou et al., 2008; Rial et al., 2014), but the role of PrPC in oxidative stress caused by dopamine oxidation has not yet been evaluated. In this study, we investigated the effects of dopamine toxicity on the expression levels, solubility and subcellular localization of PrPC, with concomitant analysis of cellular viability, protein synthesis and degradation pathways.
DMEM Media-Glutamax™-I and fetal bovine serum (FBS) were purchased from Life Technologies. Dopamine hydrochloride, thiazolyl blue tetrazolium bromide (MTT) and 2′, 7′-dichlorofluorescein diacetate (DCFDA) were purchased from Sigma-Aldrich. Complete Protease Inhibitor cocktail tablets were purchased from Roche and Pierce™ phosphatase inhibitor mini tablets were purchased from Thermo Scientific. All primary antibodies were purchased from Cell Signaling, except anti-prion SAF32, which was purchased from Cayman. Horseradish peroxidase conjugated secondary antibodies were purchased from Sigma-Aldrich and Alexa fluorophore conjugated antibodies and CellROX Green reagent were purchased from Life Technologies.
N2a cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in DMEM-Glutamax™-I (Dulbecco's Modified Eagle's Medium) supplemented with 10% FBS, Penicillin (100 U/mL) and Streptomycin (100 μg/mL). For dopamine treatment, 4 × 104 cells/cm2 were seeded. After 24 h, cells were treated with 50 or 100 μM dopamine diluted in DMEM containing 0.5% of FBS for 24 h.
Cells were treated with dopamine in 96-well plates for 24 h. After the treatment, cells were incubated with 100 μl of 1.2 mM MTT diluted in Krebs solution (NaCl 126 mM, KCl 2.5 mM, NaHCO3 25 mM, NaH2PO4 1.2 mM, MgCl2 1.2 mM, CaCl2 2.5 mM, D-glucose 10 mM) for 2 h. The MTT formazan were solubilized in 100 μl of DMSO. Insoluble materials were removed by centrifugation at 20,000 g for 2 min and absorbance of supernatant was measured at 540 nm.
Cells were treated with dopamine in 96-well plates for 24 h. After the treatment, cells were incubated with 10 μM of DCFDA or 5 μM CellRox Green in DMEM 0.5% FBS for 30 min. After washing twice with PBS or Krebs solution, the fluorescence was measured using plate reader (Bio-Tek). CellRox Green was also analyzed by Operetta high content screening system (Perkin Elmer).
Cells were lysed with lysis buffer (Tris 100 mM, pH 8.0, NaCl 130 mM, EDTA 10 mM, Triton X-100 1%, sodium deoxycholate 0.5%, complete protease inhibitor cocktail and Pierce phosphatase inhibitor mini tablet), and post-nuclear supernatants (PNS) were collected. The protein content in PNS was quantified using BCA protein assay kit (Thermo Scientific Pierce). Equal amounts of proteins were resolved by SDS-PAGE and transferred to PVDF membrane. For immunodetection, membranes were incubated with TBS-T (Tris 50 mM, NaCl 150 mM, Tween-20 0.1% pH 7.4) containing 5% BSA for 1 h. Then, the membranes were incubated with primary antibody for 1 h at room temperature or for 16 h at 4°C, then washed three times with TBS-T. Secondary antibody conjugated with peroxidase and SuperSignal™ West Pico Chemiluminescent Substrate were used to detect antigens labeled with specific antibodies. Images were digitalized and quantified using Alliance mini 4 m (UVITEC Cambridge). For each protein, at least three independent experiments were performed. In every experiment, the band intensity of the proteins of interest (PI) was normalized by the intensity of the GAPDH bands. Western blot is semi-quantitative assay and direct comparison of PI/GAPDH ratio between independent experiments is not possible. Thus, in order to enable this comparison, the average of PI/GAPDH ratio of three experimental groups (cells treated with 0, 50, and 100 μM of dopamine) was set as 100%, and the percentage of each group was calculated in each independent experiment. The mean percentage of all independent experiments was plotted with respective 95% confidence interval (CI95). An example of PrPC expression analysis was shown in supplementary data.
Post-nuclear supernatants were incubated with 1% sarkosyl for 10 min on ice, then ultracentrifuged for 2 h at 100,000 × g and at 4°C. Pellets were dissolved in sample buffer 4x (8% SDS, Tris HCl 250 mM, pH 6.8, 40% Glycerol, 0.08 mg/ml bromophenol blue and 1.4 M β-mercaptoetanol), and supernatants were incubated with 4 volumes of methanol for 2 h at −20°C for protein precipitation. After centrifugation for 20 min at 25,000× g and 4°C, methanol precipitated pellets were dissolved in Sample Buffer 4x. The level of PrPC in each fraction was assessed by western blot. To combine the results of multiple independent experiments, percentage of PrPC observed in each sample was calculated considering the average of six samples (Pellets and supernatants of 0, 50, and 100 μM) as 100%. This normalization was necessary because the absolute value of the band intensity is not comparable from one blot to another. The mean percentage of all independent experiments was plotted with respective CI95.
Cell surface proteins were biotinylated using Pierce Cell Surface Protein Isolation Kit (Thermo Scientific). The cells were harvested at time zero or after 1 h incubation in the presence or absence of 50 μM of dopamine. Biotinylated proteins were purified from the cell lysates using NeutrAvidin-agarose, and PrPC was detected by western blot using SAF32 antibody.
Cells were fixed with 3.7% paraformaldehyde diluted in PBS (pH 7) for 5 min and then with alkalinized 3.7% paraformaldehyde (pH 10) for 15 min. After washing twice with PBS, excess of paraformaldehyde was inactivated with glycine 100 mM and cells were permeabilized with Triton X-100 0.1% diluted in PBS for 10 min. To avoid unspecific bindings, cells were incubated with 1% BSA diluted in PBS for 1 h and then with primary antibodies diluted in PBS containing Triton X-100 0.1% for 1 h. After washing three times with PBS, cells were incubated with secondary antibody conjugated with Alexa 488 or Alexa 594 fluorophore for 1 h and then washed five times with PBS. Average images of six frames were acquired for each visual field using confocal microscope (Leica TCS SP8). Images were analyzed using Image J 1.48r. In each image, total area stained by anti-PrPC was defined as 100% and percentage of the area co-stained by anti-LC3-II was assessed using Colocalization Threshold plugin with default parameters (Bolte and Cordelieres, 2006). To estimate the distribution of PrPC in intracellular vesicles with distinct sizes, we counted the number of PrPC-positive particles and then categorized them by their size. The percentage of each category was calculated in relation to total number of particles counted in each group.
rPrP was prepared as described elsewhere (Zahn et al., 1997). The oligomerization was performed in 20 μl of Tris buffer (10 mM pH 7.4) containing 10 μg rPrP and dopamine at designated concentrations. This mixture was incubated for 24 h at 37°C protected from the light unless otherwise specified. Sodium metabisulfite (400 μM) was added whenever necessary. The product of oligomerization was separated in 4–20% gradient SDS polyacrylamide gel and revealed by silver staining.
All experiments, except ROS measurement and biotinylation of cell surface proteins, were repeated 3–5 times. For ROS measurement, one experiment was performed in triplicate for each dye and detection methods. Mean of independent experiments or replicates was plotted with respective CI95. The groups were considered different when their CI95 did not overlap. In this criterion, p-values are estimated to be lower than 0.01 (Cumming et al., 2007).
Treatment of neuronal cells with high concentration of dopamine for a prolonged period (typically 24 h) is known to evoke cytotoxic effects (Gomez-Santos et al., 2003; Yamakawa et al., 2010). To evaluate the effect of dopamine cytotoxicity on PrPC, we treated N2a cells with 50 and 100 μM of dopamine for 24 h. This treatment did not induce morphological alterations, but cell viability was gradually reduced as dopamine concentration was increased (Figures 1A,B). Dopamine treatment also induced ROS production as assessed by CellROX Green reagent and 2′,7′-dichlorofluorescein diacetate (DCFDA), compounds that show enhanced fluorescence upon oxidation. Both reagents showed similar results in high-throughput plate reader (Figure 1C), but the increase of CellRox fluorescence induced by 50 μM of dopamine was not statistically significant. This subtle difference between two dyes can be explained by their different reactivity. CellROX Green was also analyzed by high content cell analyzer. As shown in Figure 1D, brighter fluorescent spots were observed in cells treated with 50 and 100 μM of dopamine. According to the manufacturer, this pattern is due to the primary location of CellROX in the nucleus and mitochondria upon oxidation.
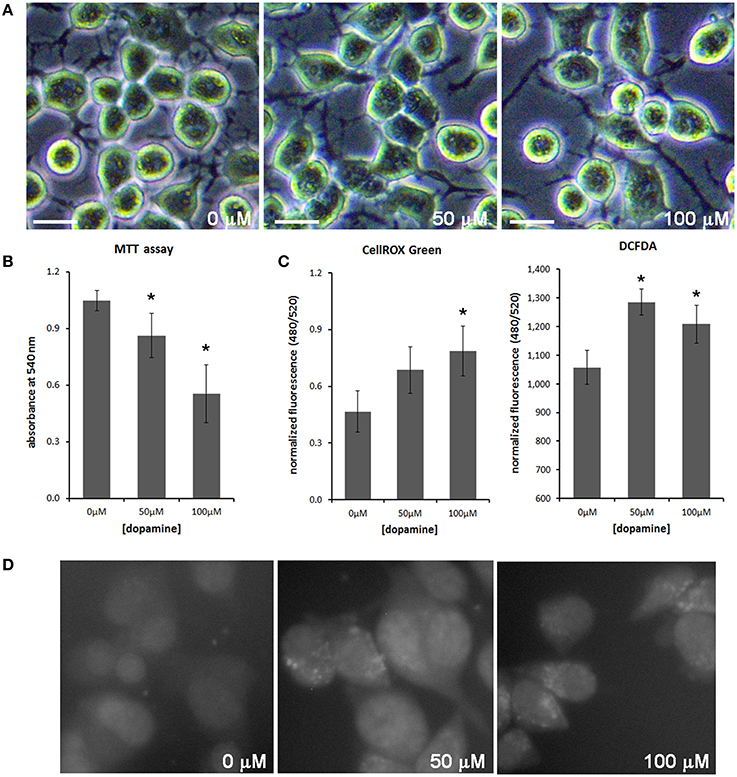
Figure 1. Dopamine reduced cell viability and increased ROS production. The morphology of N2a cells was not altered with dopamine treatment for 24 h (A), but the viability assessed by the formation of MTT formazan was reduced after dopamine treatment (B). Mean of four independent experiments was represented with respective CI95 (B). ROS production was evaluated using CellROX Green reagent and DCFDA. Fluorescence was measured by high-throughput plate reader and normalized by total protein contents/well or by cell viability. Mean of triplicate was plotted with respective CI95 (C). CellROX Green was also analyzed by high content cell analyzer. Representative field images are shown (D). *no overlap of CI95 between indicated group and control group (0 μM). scale bar = 20 μM.
The treatment with 50 and 100 μM of dopamine did not alter the total amount of PrPC (Figures 2A,B). However, the fast-migrating band (#) was decreased with 100 μM of dopamine (Figures 2A,C). This band probably represents newly synthesized unglycosylated form rather than N-terminally truncated PrPC because this band migrated close to 26 kDa marker and was recognized by SAF32, an antibody that binds to octa-repeat region located in N-terminal PrPC. The specific reduction of this immature form can indicate that dopamine toxicity might have affected protein synthesis. Thus, we verified the level of mTOR (a master regulator of growth and protein synthesis) together with 4EBP1 (a repressor of cap-dependent translation upon dephosphorylation). As shown in Figures 3A–D, phosphorylation of mTOR and 4EBP1 was significantly decreased by 100 μM of dopamine. Also, faster migration of total 4EBP1 bands indicated lower degree of phosphorylation. These data indicate that protein synthesis might be compromised in this condition. In addition, the expression level of ER chaperone BiP was increased with 50 μM of dopamine (Figures 3E,F), suggesting augment of misfolded proteins in ER. However, at 100 μM, BiP expression returned to control level (Figures 3E,F), presumably due to the attenuated protein synthesis. On the other hand, eIF2-α phosphorylation was not altered by dopamine treatment (Figures 3E,F).
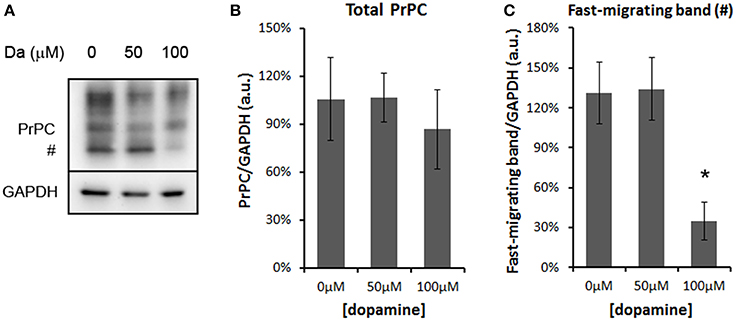
Figure 2. Dopamine reduced the expression level of unglycosylated PrPC. Treatment of N2a cells with dopamine for 24 h did not alter overall expression of PrPC, but reduced the level of fast-migrating unglycosylated band of PrPC (#, A). The intensity of total PrPC (B) or fast-migrating band (C) was normalized by respective GAPDH band. Means of 5 independent experiments were plotted with respective CI95. *no overlap of CI95 between indicated group and control group (0 μM).
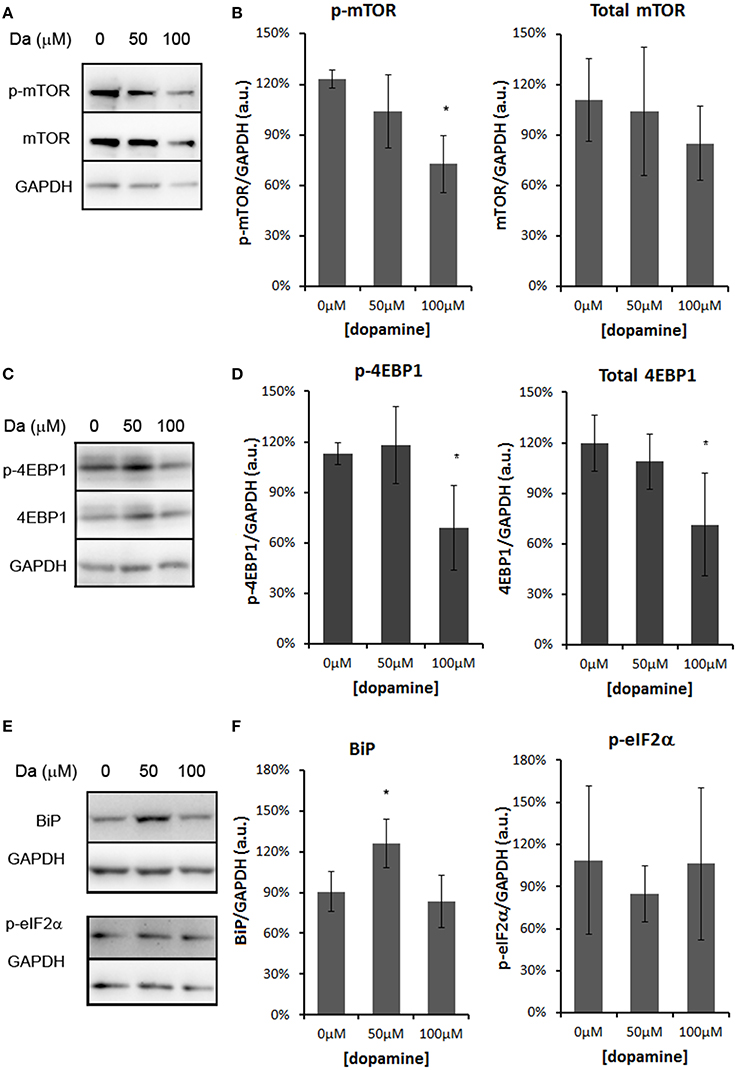
Figure 3. Dopamine treatment compromised protein synthesis. The level of phosphorylated mTOR (p-mTOR) was reduced by 100 μM of dopamine, while total mTOR was not altered (A,B). The level of phosphorylated 4EBP-1 (p-4EBP-1) and total 4EBP1 was reduced by 100 μM of dopamine (C,D). The level of BiP was induced by 50 μM of dopamine, while the phosphorylated eIF2α (p-eIF2α) was not altered (E,F). Band intensity of p-mTOR, mTOR, p-4EBP1, total 4EBP1, BiP and p-eIF2α was normalized by the respective GAPDH band and mean of three independent experiments was plotted with respective CI95 (B, D, and F). *no overlap of CI95 between indicated group and control group (0 μM).
All protein fold changes were calculated based on the loading control GAPDH. Thus, to ensure the constitutive expression of GAPDH in our experimental conditions, we compared the GAPDH expression to α-tubulin, another frequently used loading control. As shown in Figure 4, GAPDH expression was not significantly changed by dopamine treatment.
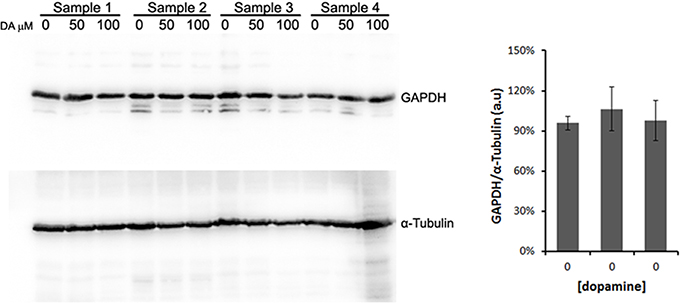
Figure 4. Validation of GAPDH as loading control. N2a cells were treated with dopamine at designated concentrations and cell lysates were used to evaluate the expression levels of GAPDH and α-tubulin in four independent samples. The band intensity of GAPDH was normalized by the intensity of α-tubulin. Mean normalized intensity was plotted with respective CI95.
Altogether, these data indicate that 100 μM of dopamine compromises cellular viability, redox balance and protein synthesis, which may affect the synthesis of PrPC.
An intriguing fact is that even with reduced protein synthesis, we did not observe a significant reduction of the mature form of PrPC. One possibility is that dopamine treatment may reduce the turnover of PrPC. To test this hypothesis, we biotinylated cell surface protein and observed a turnover of PrPC. After 1 h of incubation, a substantial amount of biotinylated PrPC was degraded in both dopamine-treated (50 μM) and untreated cells compared to input (Figure 5A, comparing second and third lane with first lane). However, a higher amount of biotinylated PrPC remained in dopamine-treated cells compared to untreated cells, indicating that dopamine treatment reduced PrPC turnover (Figure 5A, comparing second lane with third lane). To verify whether this reduced turnover is due to the altered biochemical characteristics, we evaluated the solubility of PrPC in 1% sarkosyl. As shown in Figure 5B, an increased amount of PrPC was recovered in insoluble pellet fraction after treatment with 50 and 100 μM of dopamine (Figure 5B). To further evaluate the effects of dopamine on PrPC aggregation, in vitro experiments were conducted using recombinant mouse PrPC (rPrP). Dopamine induced the formation of SDS-resistant higher-order oligomers of rPrP in a time and concentration-dependent manner (Figure 5C). Oligomerization was prevented in the presence of antioxidant (sodium metabisulfite 400 μM) (Figure 5C, last two lanes). These results indicate that oxidative metabolites of dopamine can induce the accumulation of insoluble PrPC. In the presence of metabisulfite, monomer rPrP migrated slowly. Even after reduction of proteins with DTT, sulfhydryl groups can be readily reoxidized and form disulfide bond (Wall, 1971), and reduced recombinant PrP migrates more slowly than oxidized form (Lee and Eisenberg, 2003). Likely, metabisulfite prevented the reoxidation of sulfydryl groups, resulting in slower migration (Figure 5C).
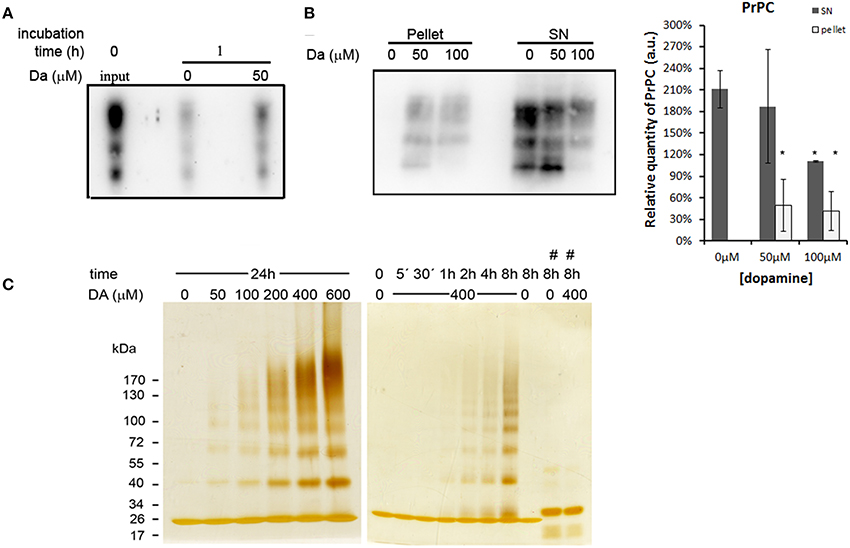
Figure 5. Dopamine reduced turnover and solubility of PrPC. Cell surface proteins was biotinylated and chased at time zero (input) or after 1 h incubation in the presence or absence of 50 μM of dopamine. After 1 h, dopamine treated cells showed higher amount of remaining PrPC compared to untreated cells (A). After dopamine treatment, insoluble proteins in sarkosyl 1% were recovered by ultracentrifugation. Higher amounts of PrPC were recovered in pellet fractions and lower amount in supernatants (SN), indicating lower solubility (B). Mean band intensity of three independent experiments was plotted with respective CI95. Dark gray bars represent SN and light gray bars represent pellet quantification (B). *no overlap of CI95 between indicated group and control group (0 μM). rPrP (10 μg) was incubated with designated concentration of dopamine (DA) for 24 h or with 400 μM of dopamine for indicated times (C). When 400 μM of metabisulfite (#) was added, the oligomerization was prevented (C). rPrP monomer migrated close to 26 kDa marker. Three independent experiments were performed and representative images are shown.
Autophagy is one of the pathways that degrade prion protein (Homma et al., 2014). Since dopamine treatment induced accumulation of insoluble PrPC, we evaluated whether these aggregates were sorted to autophagosomes, labeled by LC3-II (an autophagosome membrane associated protein). As shown in Figure 6, the degree of colocalization between PrPC and LC3-II was increased by dopamine treatment (Figures 6, 7A). Of note, the intracellular organelles that contained PrPC were frequently enlarged in cells treated with dopamine (Figures 6, 7B) and they were mostly positive for LC3-II staining (Figure 6).
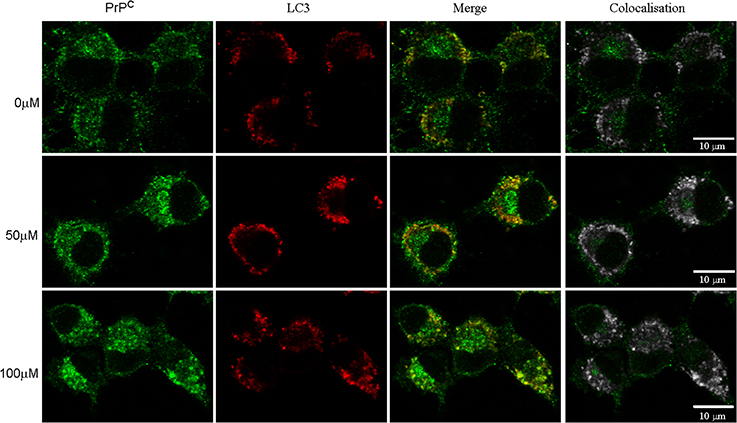
Figure 6. Dopamine induced PrPC sorting to autophagosomes. After dopamine treatment, endogenous PrPC (green) and LC3-I/LC3-II (red) was detected using specific antibodies. Third column shows merged images. Right column shows colocalized pixels in gray scale. Scale bar = 10 μm.
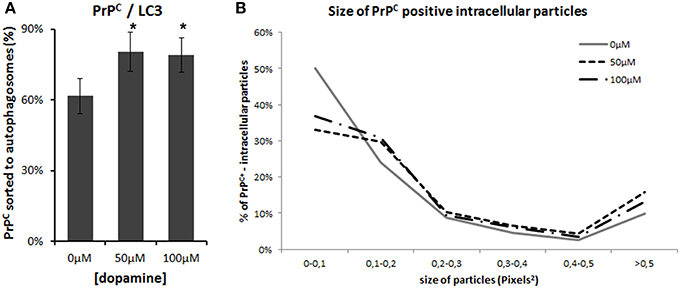
Figure 7. Image analysis. Percentage of PrPC-positive area that colocaize with LC3-II was calculated using colocalization threshold plugins of Image J. The graphs represent mean of 5 to 6 images with respective CI95 (A). * no overlap of CI95 between indicated group and control group. The area of PrPC-positive intracellular vesicles was measured in pixel2 using Image J and was categorized by their area. Percentage of each category was calculated. (B). Dopamine treated cells (dotted lines) showed lower percentage of smaller vesicles and higher percentage of vesicles that measured more than 0.5 pixel2.
In addition, treatment of the N2a cells with 100 μM of dopamine increased the levels of p62/SQSTM1 and LC3-II (Figures 8A,B). The former transports ubiquitinated proteins to the autophagosome and remains associated to LC3-II, which is produced by lipidation of LC3-I (Ravikumar et al., 2010). The accumulation of both proteins occurred without the induction of Beclin-1, a component of the complex that initiates the formation of autophagosome (Figures 8A,B) (Ravikumar et al., 2010). Thus, these data suggest that the accumulation of autophagosomes was not due to the activation of autophagy, but rather the failure in further processing of autophagosomal cargo. The immunofluorescence data also showed the accumulation and enlargement of LC3-II positive-vesicles (Figure 6).
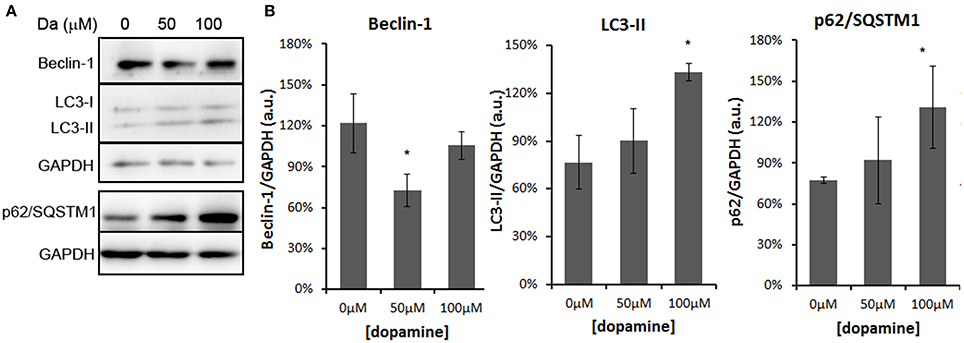
Figure 8. Dopamine treatment induced accumulation of autophagosomes. The level of Beclin-1 was reduced by 50 μM of dopamine and restored to control level at 100 μM, while p62/SQSTM1 and LC3-II was increased by 100 μM of dopamine (A). Beclin-1 and LC3-II were analyzed in the same gradient gel. Band intensity of Beclin-1, p62/SQSTM1, and LC3-II was normalized by the respective GAPDH band and mean of three independent experiments was plotted with respective CI95 (B). *no overlap of CI95 between indicated group and control group (0 μM).
Overall, these results indicate that toxic concentration of dopamine altered the solubility of PrPC and promoted its accumulation in autophagosomes, affecting autophagic flux and attenuating protein synthesis.
Dopamine is an important neurotransmitter that controls several functions such as physical movement, emotional process and alertness (Kauer and Malenka, 2007). In spite of its importance in physiology, dopamine metabolism has been pointed out as the main etiology of Parkinson's disease due to the inherent instability and reactivity of dopamine and its metabolites, capable to induce oxidative damage in biomolecules (Hastings, 2009). In this study, we demonstrated that dopamine treatment altered the solubility of PrPC and promoted its accumulation in autophagosomes in neuronal cells. Auto-oxidation of dopamine also induced the formation of SDS-resistant oligomers of unglycosylated recombinant prion protein. Previous studies have demonstrated that inhibition of complex glycosylation during synthetic pathway or expression of unglycosylated PrP facilitated its conversion into PrPSc (Korth et al., 2000; Winklhofer et al., 2003). In our study, dopamine induced aggregation of both glycosylated neuronal PrPC and unglycosylated rPrP, but with different biochemical characteristics regarding to the resistance to SDS.
Although the conversion of PrPC into abnormal prion protein (PrPSc) is an essential event for the development of TSE, molecular mechanisms of conformational transition between PrPC and PrPSc are poorly understood. Nevertheless, previous studies have demonstrated that PrPSc-like aggregates were found in normal brains at low levels (Yuan et al., 2006). Glycosylation pattern, retrograde transport from ER to cytosol and inhibition of proteosomal activity might contribute to this spontaneous production of PrPSc-like aggregates (Korth et al., 2000; Yedidia et al., 2001; Ma and Lindquist, 2002; Winklhofer et al., 2003). The aggregation of PrPC in response to redox alteration has also been proposed as a cytoprotective mechanism (Das et al., 2010). However, if the aggregates are not effectively degraded by autophagy, other proteins might co-aggregate causing cytotoxicity (Das et al., 2010). In this study, we observed that dopamine treatment increased the amount of LC3-II and p62/SQSTM1, both involved in autophagosomal cargo recruitment. Concomitantly, PrPC was also accumulated in autophagosomes. These data indicate that the degradation of autophagosomal cargo by lysosome was not effective. Therefore, it is plausible to speculate that the reduced cell viability observed after dopamine treatment is at least partially due to the accumulation of PrPC aggregates in autophagosomes. Similarly, PrPC aggregation might have negatively affected protein synthesis since a previous study has demonstrated that protein synthesis was corrupted in N2a cells infected with prions (Roffe et al., 2010).
Endocytosis of PrPC can be stimulated by several compounds such as Cu2+, nucleic acids and heme (Lee et al., 2001, 2007; Kocisko et al., 2006). An intriguing fact is that these compounds can alter the structure of PrPC and confer cytotoxicity at high concentration (Thakur et al., 2011; Macedo et al., 2012). These findings suggest that PrPC might function as a scavenging receptor for toxic molecules. Dopamine can induce toxicity by rapid auto-oxidation producing reactive dopamine-quinone (Asanuma et al., 2003; Chen et al., 2008). Binding of toxic molecules to PrPC, including dopamine metabolites, might induce structural alterations, which in turn, may trigger endocytosis for further degradation. Frequent binding of toxic molecules that cause conformational alterations of PrPC can eventually overload the degradation pathways, instigating the accumulation of protein aggregates.
Certain conditions, such as stress, drug addiction, iron deficiency, L-DOPA treatment or schizophrenia are known to increase dopamine metabolism (Nelson et al., 1997; de la Fuente-Fernandez et al., 2004; Elliott and Beveridge, 2005; Kim et al., 2005). Our findings raise a possibility that chronic exposure to these conditions may facilitate PrPC aggregation. Possibly, dopamine and its oxidative metabolites can have more general roles in other protein aggregation, but the specificity of the target can be ruled by subcellular compartmentalization, brain region specificity and susceptibility of aggregation. Identification of endogenous metabolites that induce aggregation of a specific protein can contribute to better understanding of idiopathic neurodegenerative diseases and provide new molecular targets for treatment.
Marcio H. M. da Luz conducted all experiments except rPrP oligomerization and analyzed the results. Italo T. Peres performed rPrP oligomerization assay. Tiago G. Santos acquired confocal images. Vilma R. Martins participated in study design and interpretation of data and revised the manuscript. Marcelo Y. Icimoto purified rPrP. Kil S. Lee participated in study design, data analysis and interpretation, and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by grants from FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo: 2008/06152-9, 2009/14027-2, 2013/22413-5, 2012/18093-2), CAPES (Coordenação de Aperfeiçoamento de pessoal de Nível Superior), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and EMU (programa de equipamentos multiusuários).
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fncel.2015.00012/abstract
ROS, Reactive oxygen species; PrPC, Cellular prion protein; PrPSc, abnormal prion protein; SDS, Sodium dodecyl sulfate; mTOR, Mammalian target of rapamycin; 4EBP1, 4E binding protein 1; ER, Endoplasmic reticulum; eIF2-α, Eukariotic initiation factor 2-α; p62/SQSTM1, Sequestosome 1; LC3, Microtubule-associated protein 1 light chain 3; TSE, Transmissible spongiform encephalopathies.
Abouelatta, A. I., Campanali, A. A., Ekkati, A. R., Shamoun, M., Kalapugama, S., and Kodanko, J. J. (2009). Oxidation of the natural amino acids by a ferryl complex: kinetic and mechanistic studies with peptide model compounds. Inorg. Chem. 48, 7729–7739. doi: 10.1021/ic900527c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adjou, K. T., Dilda, P., Aumond, P., Gueddari, S., Deslys, J. P., Dormont, D., et al. (2008). Increase of monoamine oxidase-B activity in the brain of scrapie-infected hamsters. Neurochem. Int. 52, 1416–1421. doi: 10.1016/j.neuint.2008.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alkhuja, S. (2013). Parkinson disease: research update and clinical management. South Med. J. 106, 334. doi: 10.1097/SMJ.0b013e318290f72a
Andreyev, A. Y., Kushnareva, Y. E., and Starkov, A. A. (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc.) 70, 200–214. doi: 10.1007/s10541-005-0102-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asanuma, M., Miyazaki, I., and Ogawa, N. (2003). Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson's disease. Neurotox. Res. 5, 165–176. doi: 10.1007/BF03033137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beraldo, F. H., Soares, I. N., Goncalves, D. F., Fan, J., Thomas, A. A., Santos, T. G., et al. (2013). Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. 27, 3594–3607. doi: 10.1096/fj.13-232280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertuchi, F. R., Bourgeon, D. M., Landemberger, M. C., Martins, V. R., and Cerchiaro, G. (2012). PrPC displays an essential protective role from oxidative stress in an astrocyte cell line derived from PrPC knockout mice. Biochem. Biophys. Res. Commun. 418, 27–32. doi: 10.1016/j.bbrc.2011.12.098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bolte, S., and Cordelieres, F. P. (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232. doi: 10.1111/j.1365-2818.2006.01706.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brundin, P., Melki, R., and Kopito, R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307. doi: 10.1038/nrm2873
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cappai, R., Leck, S. L., Tew, D. J., Williamson, N. A., Smith, D. P., Galatis, D., et al. (2005). Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 19, 1377–1379. doi: 10.1096/fj.04-3437fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L., Ding, Y., Cagniard, B., Van Laar, A. D., Mortimer, A., Chi, W., et al. (2008). Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 28, 425–433. doi: 10.1523/JNEUROSCI.3602-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cumming, G., Fidler, F., and Vaux, D. L. (2007). Error bars in experimental biology. J. Cell Biol. 177, 7–11. doi: 10.1083/jcb.200611141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Das, D., Luo, X., Singh, A., Gu, Y., Ghosh, S., Mukhopadhyay, C. K., et al. (2010). Paradoxical role of prion protein aggregates in redox-iron induced toxicity. PLoS ONE 5:e11420. doi: 10.1371/journal.pone.0011420
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de la Fuente-Fernandez, R., Sossi, V., Huang, Z., Furtado, S., Lu, J. Q., Calne, D. B., et al. (2004). Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson's disease: implications for dyskinesias. Brain 127, 2747–2754. doi: 10.1093/brain/awh290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elliott, J. M., and Beveridge, T. J. (2005). Psychostimulants and monoamine transporters: upsetting the balance. Curr. Opin. Pharmacol. 5, 94–100. doi: 10.1016/j.coph.2004.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gomez-Santos, C., Ferrer, I., Santidrian, A. F., Barrachina, M., Gil, J., and Ambrosio, S. (2003). Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J. Neurosci. Res. 73, 341–350. doi: 10.1002/jnr.10663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hastings, T. G. (2009). The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson's disease. J. Bioenerg. Biomembr. 41, 469–472. doi: 10.1007/s10863-009-9257-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herrero, M. T., Hirsch, E. C., Kastner, A., Luquin, M. R., Javoy-Agid, F., Gonzalo, L. M., et al. (1993). Neuromelanin accumulation with age in catecholaminergic neurons from Macaca fascicularis brainstem. Dev. Neurosci. 15, 37–48. doi: 10.1159/000111315
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Homma, T., Ishibashi, D., Nakagaki, T., Satoh, K., Sano, K., Atarashi, R., et al. (2014). Increased expression of p62/SQSTM1 in prion diseases and its association with pathogenic prion protein. Sci. Rep. 4, 4504. doi: 10.1038/srep04504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kauer, J. A., and Malenka, R. C. (2007). Synaptic plasticity and addiction. Nat. Rev. Neurosci. 8, 844–858. doi: 10.1038/nrn2234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, S. T., Choi, J. H., Chang, J. W., Kim, S. W., and Hwang, O. (2005). Immobilization stress causes increases in tetrahydrobiopterin, dopamine, and neuromelanin and oxidative damage in the nigrostriatal system. J. Neurochem. 95, 89–98. doi: 10.1111/j.1471-4159.2005.03342.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, S. T., Choi, J. H., Kim, D., and Hwang, O. (2006). Increases in TH immunoreactivity, neuromelanin and degeneration in the substantia nigra of middleaged mice. Neurosci. Lett. 396, 263–268. doi: 10.1016/j.neulet.2005.11.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kocisko, D. A., Vaillant, A., Lee, K. S., Arnold, K. M., Bertholet, N., Race, R. E., et al. (2006). Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 50, 1034–1044. doi: 10.1128/AAC.50.3.1034-1044.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kodama, R., Kato, M., Furuta, S., Ueno, S., Zhang, Y., Matsuno, K., et al. (2013). ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 18, 32–41. doi: 10.1111/gtc.12015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Korth, C., Kaneko, K., and Prusiner, S. B. (2000). Expression of unglycosylated mutated prion protein facilitates PrP(Sc) formation in neuroblastoma cells infected with different prion strains. J. Gen. Virol. 81, 2555–2563.
LaVoie, M. J., Ostaszewski, B. L., Weihofen, A., Schlossmacher, M. G., and Selkoe, D. J. (2005). Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 11, 1214–1221. doi: 10.1038/nm1314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. G., Park, S. J., Choi, E. K., Carp, R. I., and Kim, Y. S. (1999). Increased expression of prion protein is associated with changes in dopamine metabolism and MAO activity in PC12 cells. J Mol. Neurosci. 13, 121–126. doi: 10.1385/JMN:13:1-2:121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. J., Baek, S. M., Ho, D. H., Suk, J. E., Cho, E. D., and Lee, S. J. (2011). Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp. Mol. Med. 43, 216–222. doi: 10.3858/emm.2011.43.4.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, K. S., Magalhaes, A. C., Zanata, S. M., Brentani, R. R., Martins, V. R., and Prado, M. A. (2001). Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J. Neurochem. 79, 79–87. doi: 10.1046/j.1471-4159.2001.00529.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, K. S., Raymond, L. D., Schoen, B., Raymond, G. J., Kett, L., Moore, R. A., et al. (2007). Hemin interactions and alterations of the subcellular localization of prion protein. J. Biol. Chem. 282, 36525–36533. doi: 10.1074/jbc.M705620200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S., and Eisenberg, D. (2003). Seeded conversion of recombinant prion protein to a disulfide-bonded oligomer by a reduction-oxidation process. Nat. Struct. Biol. 10, 725–730. doi: 10.1038/nsb961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, J., and Lindquist, S. (2002). Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. science 298, 1785–1788. doi: 10.1126/science.1073619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Macedo, B., Millen, T. A., Braga, C. A., Gomes, M. P., Ferreira, P. S., Kraineva, J., et al. (2012). Nonspecific prion protein-nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry 51, 5402–5413. doi: 10.1021/bi300440e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McLennan, N. F., Brennan, P. M., McNeill, A., Davies, I., Fotheringham, A., Rennison, K. A., et al. (2004). Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 165, 227–235. doi: 10.1016/S0002-9440(10)63291-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morris, J. C. (2013). Neurodegenerative disorders of aging: the down side of rising longevity. Mo. Med. 110, 393–394.
Musiek, E. S., and Schindler, S. E. (2013). Alzheimer disease: current concepts & future directions. Mo. Med. 110, 395–400.
Nelson, C., Erikson, K., Pinero, D. J., and Beard, J. L. (1997). In vivo dopamine metabolism is altered in iron-deficient anemic rats. J. Nutr. 127, 2282–2288.
Petrov, D., and Zagrovic, B. (2011). Microscopic analysis of protein oxidative damage: effect of carbonylation on structure, dynamics, and aggregability of villin headpiece. J. Am. Chem. Soc. 133, 7016–7024. doi: 10.1021/ja110577e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ravikumar, B., Sarkar, S., Davies, J. E., Futter, M., Garcia-Arencibia, M., Green-Thompson, Z. W., et al. (2010). Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435. doi: 10.1152/physrev.00030.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rial, D., Pamplona, F. A., Moreira, E. L., Moreira, K. M., Hipolide, D., Rodrigues, D. I., et al. (2014). Cellular prion protein is present in dopaminergic neurons and modulates the dopaminergic system. Eur. J. Neurosci. doi: 10.1111/ejn.12600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roffe, M., Beraldo, F. H., Bester, R., Nunziante, M., Bach, C., Mancini, G., et al. (2010). Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. U.S.A. 107, 13147–13152. doi: 10.1073/pnas.1000784107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shiraishi, N., Inai, Y., Bi, W., and Nishikimi, M. (2005). Fragmentation and dimerization of copper-loaded prion protein by copper-catalysed oxidation. Biochem. J. 387, 247–255. doi: 10.1042/BJ20041561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shiraishi, N., and Nishikimi, M. (2002). Carbonyl formation on a copper-bound prion protein fragment, PrP23-98, associated with its dopamine oxidase activity. FEBS Lett. 511, 118–122. doi: 10.1016/S0014-5793(01)03324-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thakur, A. K., Srivastava, A. K., Srinivas, V., Chary, K. V., and Rao, C. M. (2011). Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J. Biol. Chem. 286, 38533–38545. doi: 10.1074/jbc.M111.265645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tyedmers, J., Mogk, A., and Bukau, B. (2010). Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788. doi: 10.1038/nrm2993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Laar, V. S., Mishizen, A. J., Cascio, M., and Hastings, T. G. (2009). Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol. Dis. 34, 487–500. doi: 10.1016/j.nbd.2009.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wall, J. S. (1971). Disulfide bonds: determination, location and influence on molecular properties of proteins. J. Agr. Food Chem. 19, 619–625 doi: 10.1021/jf60176a003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Winklhofer, K. F., Heller, U., Reintjes, A., and Tatzelt, J. (2003). Inhibition of complex glycosylation increases the formation of PrPsc. Traffic 4, 313–322. doi: 10.1034/j.1600-0854.2003.00088.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamakawa, K., Izumi, Y., Takeuchi, H., Yamamoto, N., Kume, T., Akaike, A., et al. (2010). Dopamine facilitates alpha-synuclein oligomerization in human neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 391, 129–134. doi: 10.1016/j.bbrc.2009.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yedidia, Y., Horonchik, L., Tzaban, S., Yanai, A., and Taraboulos, A. (2001). Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 20, 5383–5391. doi: 10.1093/emboj/20.19.5383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuan, J., Xiao, X., McGeehan, J., Dong, Z., Cali, I., Fujioka, H., et al. (2006). Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J. Biol. Chem. 281, 34848–34858. doi: 10.1074/jbc.M602238200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zahn, R., von Schroetter, C., and Wuthrich, K. (1997). Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 417, 400–404. doi: 10.1016/S0014-5793(97)01330-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: dopamine, protein aggregation, prion, protein synthesis, autophagy, neurodegeneration
Citation: da Luz MHM, Peres IT, Santos TG, Martins VR, Icimoto MY and Lee KS (2015) Dopamine induces the accumulation of insoluble prion protein and affects autophagic flux. Front. Cell. Neurosci. 9:12. doi: 10.3389/fncel.2015.00012
Received: 21 October 2014; Accepted: 09 January 2015;
Published online: 02 February 2015.
Edited by:
Victoria Campos, Instituto Nacional de Neurologia y Neurocirugia, MexicoReviewed by:
Yong-Sun Kim, Hallym University, South KoreaCopyright © 2015 da Luz, Peres, Santos, Martins, Icimoto and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kil S. Lee, Department of Biochemistry, Universidade Federal de São Paulo, Rua Pedro de Toledo 669, 8° andar, Vila Clementino, São Paulo, SP 04039-032, Brazil e-mail:a2lsX3N1bl9sZWVAeWFob28uY29tLmJy;a3NsZWVAdW5pZmVzcC5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.