 Dania Vecchia
Dania Vecchia Angelita Tottene
Angelita Tottene Arn M.J.M. van den Maagdenberg
Arn M.J.M. van den Maagdenberg Daniela Pietrobon
Daniela Pietrobon- 1Department of Biomedical Sciences, University of Padova, and Consiglio Nazionale delle Ricerche (CNR) Institute of Neuroscience, Padova, Italy
- 2Departments of Human Genetics and Neurology, Leiden University Medical Centre, Leiden, Netherlands
Familial hemiplegic migraine type 1 (FHM1) is caused by gain-of-function mutations in CaV2.1 (P/Q-type) Ca2+ channels. Knockin (KI) mice carrying the FHM1 R192Q missense mutation show enhanced cortical excitatory synaptic transmission at pyramidal cell synapses but unaltered cortical inhibitory neurotransmission at fast-spiking interneuron synapses. Enhanced cortical glutamate release was shown to cause the facilitation of cortical spreading depression (CSD) in R192Q KI mice. It, however, remains unknown how other FHM1 mutations affect cortical synaptic transmission. Here, we studied neurotransmission in cortical neurons in microculture from KI mice carrying the S218L mutation, which causes a severe FHM syndrome in humans and an allele-dosage dependent facilitation of experimental CSD in KI mice, which is larger than that caused by the R192Q mutation. We show gain-of-function of excitatory neurotransmission, due to increased action-potential evoked Ca2+ influx and increased probability of glutamate release at pyramidal cell synapses, but unaltered inhibitory neurotransmission at multipolar interneuron synapses in S218L KI mice. In contrast with the larger gain-of-function of neuronal CaV2.1 current in homozygous than heterozygous S218L KI mice, the gain-of-function of evoked glutamate release, the paired-pulse ratio and the Ca2+ dependence of the excitatory postsynaptic current were similar in homozygous and heterozygous S218L KI mice, suggesting compensatory changes in the homozygous mice. Furthermore, we reveal a unique feature of S218L KI cortical synapses which is the presence of a fraction of mutant CaV2.1 channels being open at resting potential. Our data suggest that, while the gain-of-function of evoked glutamate release may explain the facilitation of CSD in heterozygous S218L KI mice, the further facilitation of CSD in homozygous S218L KI mice is due to other CaV2.1-dependent mechanisms, that likely include Ca2+ influx at voltages sub-threshold for action potential generation.
Introduction
CaV2.1 (P/Q-type) Ca2+ channels play a prominent role in initiating action potential (AP)-evoked neurotransmitter release at brain excitatory and inhibitory synapses (Pietrobon, 2005, 2010). Several neurological disorders including familial hemiplegic migraine type 1 (FHM1), a rare monogenic form of migraine with aura, are caused by mutations in the CACNA1A gene, which encodes for the α1A pore-forming subunit of CaV2.1 channels (Ophoff et al., 1996; de Vries et al., 2009; Pietrobon, 2010). Migraine is a common brain disorder whose key manifestation is the recurrence of attacks of unilateral headache due to activation of the trigeminovascular pain pathway (Pietrobon and Moskowitz, 2013). There is compelling evidence that headache mechanisms can be triggered by cortical spreading depression (CSD), the phenomenon that underlies migraine aura (Noseda and Burstein, 2013; Pietrobon and Moskowitz, 2013, 2014). The mechanisms of primary brain dysfunction that lead to the onset of a migraine attack and to increased susceptibility to CSD, however, remain a major open issue. Insights into these mechanisms can be obtained from the functional analysis of transgenic knockin (KI) mice carrying FHM1 missense mutations that were introduced into the orthologous Cacna1a gene. FHM1 KI mice were shown to exhibit a lower threshold for induction of experimental CSD and a higher velocity of CSD propagation compared with wild-type (WT) mice (van den Maagdenberg et al., 2004, 2010).
FHM1 mutations produce gain-of-function of single human recombinant CaV2.1 channels, mainly due to channel activation at lower voltages and increased channel open probability (Tottene et al., 2002, 2005; Pietrobon, 2013). Accordingly, a larger P/Q-type Ca2+ current density in a broad range of mild depolarizations was measured in several types of neurons (including cortical pyramidal cells) from KI mice that carry either the FHM1 R192Q or the S218L mutation (van den Maagdenberg et al., 2004, 2010; Tottene et al., 2009; Inchauspe et al., 2010; Fioretti et al., 2011; Gao et al., 2012; Di Guilmi et al., 2014). Whereas mutation R192Q in humans causes pure FHM (Ophoff et al., 1996), mutation S218L is associated with a severe clinical syndrome, that may consist of cerebellar ataxia, seizures, coma and sometimes fatal cerebral edema triggered by a trivial head trauma, in addition to FHM (Kors et al., 2001; Stam et al., 2009). In accordance with the more severe clinical phenotype, the S218L mutation produces: (i) a larger shift to lower voltages of activation of human CaV2.1 channels than the R192Q mutation (Hans et al., 1999; Tottene et al., 2005; Pietrobon, 2010); (ii) a larger gain-of-function of the P/Q-type Ca2+ current at low voltages in neurons of homozygous S218L (SL/SL) compared with homozygous R192Q (RQ/RQ) KI mice (van den Maagdenberg et al., 2004, 2010; Inchauspe et al., 2010; Di Guilmi et al., 2014); and (iii) a larger facilitation of induction and propagation of experimental CSD in SL/SL compared with RQ/RQ KI mice in vivo (Eikermann-Haerter et al., 2009, 2011; van den Maagdenberg et al., 2010). Both the gain-of-function of the neuronal P/Q-type Ca2+ current and the facilitation of experimental CSD in heterozygous S218L (SL/WT) KI mice were similar to those in RQ/RQ KI mice (van den Maagdenberg et al., 2010).
Investigation of cortical synaptic transmission in RQ/RQ KI mice revealed an increased strength of excitatory neurotrans-mission at cortical pyramidal cell synapses, due to increased probability of glutamate release consequent to increased AP-evoked Ca2+ influx through presynaptic P/Q-type Ca2+ channels compared with those at WT synapses (Tottene et al., 2009). In striking contrast, inhibitory synaptic transmission at fast-spiking and other multipolar interneuron synapses was unaltered, due to unaltered AP-evoked Ca2+ influx and GABA release (Tottene et al., 2009; Vecchia et al., 2014). Restoration of AP-evoked glutamate release at cortical pyramidal cell synapses of RQ/RQ KI mice to the WT value completely eliminated the facilitation of experimental CSD, thus supporting a causative link between enhanced glutamate release and facilitation of CSD in FHM1 KI mice (Tottene et al., 2009). The differential effect of the R192Q mutation at cortical excitatory and inhibitory synapses suggests altered regulation of the cortical excitatory-inhibitory balance in FHM1 and points to a disruption of this balance and neuronal network hyperactivity as the basis for an episodic vulnerability to CSD ignition in migraine (Tottene et al., 2009; Vecchia and Pietrobon, 2012).
The effect of other FHM1 mutations, including the more severe S218L mutation, on cortical synaptic transmission remains unknown. Here, we studied excitatory and inhibitory neurotransmission in microcultures of cortical neurons from neonatal S218L KI and WT mice to investigate: (i) whether the S218L mutation, like the R192Q mutation, enhances excitatory neurotransmission by increasing AP-evoked Ca2+ influx and glutamate release at cortical pyramidal cell synapses, but does not affect inhibitory neurotransmission at multipolar interneuron synapses; (ii) whether the gain-of-function of evoked glutamate release in SL/WT and SL/SL KI mice correlates with the similar gain-of-function of the P/Q-type Ca2+ current and facilitation of CSD in SL/WT and RQ/RQ KI mice and with the larger gain-of-function of the P/Q-type Ca2+ current and facilitation of CSD in SL/SL than SL/WT and RQ/RQ KI mice; and (iii) whether, as a consequence of the particularly low threshold of activation of mutant S218L channels, a fraction of these channels are open at resting membrane potential in cortical glutamatergic synaptic terminals.
Materials and Methods
Preparation of Cortical Neurons in Microculture
Knockin mice carrying the S218L mutation in the Cacna1a gene were generated as previously described (van den Maagdenberg et al., 2010). Cortical neurons were isolated from P0-2 homozygous SL/SL or heterozygous SL/WT KI mice and WT mice with the same genetic background following the procedure of Levi et al. (1984). The neurons were cultured on glial microislands (at the density of 6,000–25,000 cells/mL) essentially as reported in Brody and Yue (2000) for hippocampal neurons, except for the following details: astrocytes culture medium was Basal Eagle’s Medium (BME) plus 10% fetal bovine serum, 25 mM KCl, 2 mM glutamine and 50 µg/mL gentamicin; neuronal medium was Neurobasal A plus 2% B27 Supplement, 0.5 mM glutamine and 1% PSN Antibiotic mix (all from Gibco); only half of the volume of the astrocytes medium was replaced with neuronal medium to allow the astrocytes to condition the medium before neuron plating. Every 4 days half of the volume of neuronal medium was refreshed.
Single cortical neurons grown on glial microislands form synaptic connections onto themselves (autapses) with properties very similar to those of synapses between neurons; these autaptic connections are by definition monosynaptic, offer an unusually homogeneous population of synapses producing large synaptic responses and solution exchanges can be fast and complete (Bekkers and Stevens, 1991).
All experimental procedures were carried out in accordance with the Italian Animal Welfare Act and with the Use Committee guidelines of the University of Padova and were approved by the local authority veterinary service.
Electrophysiological Recordings and Data Analysis
Whole-cell patch-clamp recordings were made at room temperature following standard techniques. Electrical signals were recorded through an Axopatch-200B or Multiclamp 700B patch-clamp amplifier and digitized using a Digidata 1440A or Digidata 1322A interface and pClamp software (Axon Instruments).
Microislands containing several glial cells and a single neuron were selected for recording of evoked postsynaptic currents (PSCs) in voltage-clamp mode (sampling 5 KHz; filter 1 KHz) after 8–14 days (DIVs) in culture. APs in the unclamped processes were induced by a 2 ms voltage pulse to +20 mV every 10 s from a holding potential of −80 mV. The evoked PSCs were measured at −80 mV and −90 mV for excitatory and inhibitory PSC, respectively. Single neurons with a pyramidal cell morphology characterized by a typical prominent process emanating from a triangular/pyramidal soma were selected for recording glutamate receptor-mediated excitatory postsynaptic currents (EPSCs), as in Tottene et al. (2009). The currents recorded in the presence of 5 µM NBQX were subtracted to all records to obtain the evoked EPSCs (displayed in the figures after blanking 1–2 ms around each stimulus artifact for clarity). Single neurons with irregular soma morphology and multiple asymmetrical processes emanating from it (multipolar interneurons) were selected for recording inhibitory postsynaptic currents (IPSCs), as described in Vecchia et al. (2014). Although the GABAA-mediated IPSCs recorded at −90 mV are inward currents (given the predicted and measured Erev of −69 and −63 mV, respectively), they were easily distinguished from glutamate receptor-mediated EPSCs on the basis of their slower time course and their complete inhibition by 20 µM bicuculline (Ascent Scientific-Abcam). The currents recorded in the presence of bicuculline were subtracted to all records to obtain the evoked IPSCs (displayed in the figures after blanking 1–2 ms around each stimulus artifact for clarity). After PSC stabilization (typically 3 min after break-in), 5–10 sweeps were averaged to obtain the PSC amplitude.
The pipette solution contained (in mM): 110 K-methane-sulfonate, 5 MgCl2, 30 HEPES, 3 EGTA, 4 ATP, 0.5 GTP and 1 cAMP (pH 7.4 with KOH). The extracellular solution contained (in mM): 145 NaCl, 3 KCl, 10 HEPES, 10 glucose, 1 MgCl2, 2 CaCl2, 0.05 D-AP5 (pH 7.4 with NaOH). In the experiments testing the effect of the peptide toxin ω-AgaIVA (Aga) (Peptide Institute Inc.), cytochrome C (0.1 mg/mL) was added to the solution.
A liquid junction potential (LJP) of −8 mV should be added to all voltages to obtain the correct membrane potentials (Neher, 1992). Patch-clamp pipettes had resistances of 1.8–2.5 MΩ. Compensation (typically 60–80%) for series resistance was used (2.5–6 MΩ after compensation).
The relative number of neurons recorded from WT and KI mice at different DIVs was closely matched.
Acute coronal slices of the barrel cortex were prepared from P16-18 mice as described in Tottene et al. (2009). Miniature EPSCs (mEPSCs) were recorded from layer 2/3 pyramidal cells that were identified based on their morphology and firing pattern in response to current injection as in Tottene et al. (2009). The pipette solution contained (in mM): 6 KCl, 114 K-gluconate, 10 HEPES, 10 phosphocreatine, 4 MgATP, 0.3 NaGTP (pH 7.25 with KOH, osmolarity 300 mOsm with sucrose. Slice were superfused with a modified ACSF containing (in mM): 125 NaCl, 3.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 0.5 MgCl2, 1 CaCl2, 25 glucose saturated with 95% O2 5% CO2. In order to record mEPSCs, after the break-in and the recording of the firing pattern, slices were superfused with the same ACSF solution containing (in µM): TTX 0.2, D-AP5 50, bicuculline 20 and cytochrome C (0.1 mg/mL). mEPSCs were acquired at −69 mV for at least 3 min (sampling 10 KHz; filter 2 KHz) before and after application of 400 nM Aga and analyzed using Clampfit 10.0 (Axon Instruments), as in Tottene et al. (2009). Series resistance was not compensated (Rs was below 25 MΩ with less than 20% variation); LJP −12 mV.
Data are given as mean ± SEM; stars indicate a statistically significant difference from control assessed by the unpaired or paired Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Results
To assess whether the FHM1 S218L mutation leads to increased AP-evoked Ca2+ influx and increased glutamate release at cortical pyramidal cell synapses, we studied excitatory neurotransmission in cortical neurons from neonatal SL/WT KI and WT mice grown in microculture. EPSCs were evoked in single cortical pyramidal cells by brief depolarizing voltage steps eliciting an AP in the unclamped axonal processes (Figure 1A, left inset). Tottene et al. (2009) have previously shown that P/Q-, N- and R-type Ca2+ channels cooperate in controlling glutamate release at cortical pyramidal cell autapses from WT mice (cf. superadditivity of the fractional EPSC inhibition by specific blockers of P/Q-, N- and R-type Ca2+ channels).
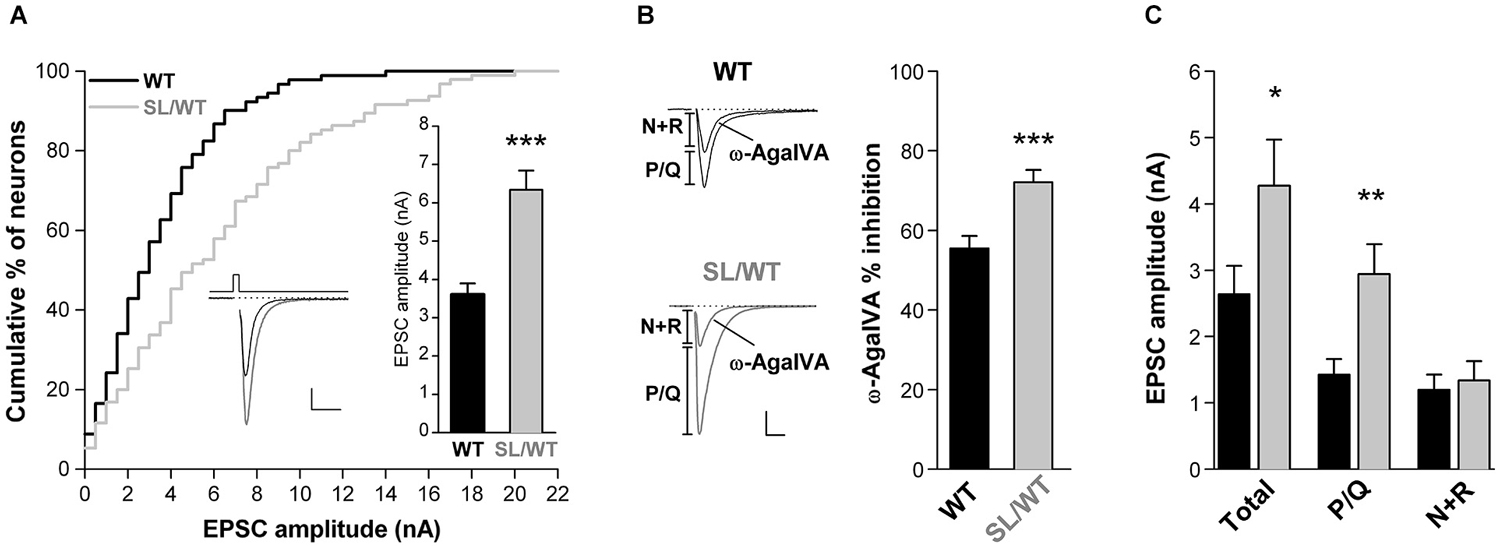
Figure 1. Increased EPSC amplitude and increased contribution of P/Q-type Ca2+ channels to excitatory neurotransmission in cortical pyramidal cell autapses of SL/WT KI mice. (A) Cumulative distribution and average value (right inset) of EPSC amplitudes evoked in single WT and SL/WT cortical pyramidal cells in microculture for 10 to 14 days (DIV). EPSC amplitude: 6.3 ± 0.5 nA (n = 95) in SL/WT and 3.6 ± 0.3 nA (n = 91) in WT neurons (t-test: p = 5.0 × 10−6). Left inset: representative EPSC traces from a WT (black) and a SL/WT (gray) neuron; scale bars 10 ms, 1 nA. The relative number of neurons recorded from WT and KI mice at different DIV were closely matched, because EPSC amplitude continued to increase from 10 to 14 DIV in both control and SL/WT neurons (Tottene et al., 2009). (B) Contribution of P/Q-type Ca2+ channels to excitatory neurotransmission evaluated from the fraction of the EPSC inhibited by Aga (200 nM, a saturating concentration since the EPSC was not further inhibited by 400 nM Aga (Tottene et al., 2009)) in cortical pyramidal cells in microculture (DIV 8–12): 72 ± 3% (n = 19) in SL/WT and 56 ± 3% (n = 21) in WT neurons (t-test: p = 6.1 × 10−4). Representative EPSC traces before and after Aga are shown on the left; scale bars: 10 ms, 1 nA. The relative number of neurons recorded from WT and KI mice at different DIV were closely matched. (C) Average values of the amplitudes of the EPSC (total) and its Aga-sensitive (P/Q) and Aga-insensitive (N+R) components (same neurons as in B). The P/Q and N+R components for each cell were obtained (as shown in the representative traces in B) as difference of EPSC amplitude before and after Aga (P/Q) and the EPSC amplitude remaining after Aga (N+R). Values of EPSC amplitudes: 4.3 ± 0.7 in SL/WT vs. 2.6 ± 0.4 in WT (Total; t-test: p = 0.048); 2.9 ± 0.5 in SL/WT vs. 1.4 ± 0.2 in WT (P/Q; t-test: p = 0.0038); 1.3 ± 0.3 in SL/WT vs. 1.2 ± 0.2 in WT (N+R; t-test: p = 0.70).
If the S218L mutation leads to increased AP-evoked Ca2+ influx through P/Q-type Ca2+ channels located at the active zones and, as a consequence, to increased probability of vesicle release, one predicts a larger AP-evoked EPSC amplitude and a larger contribution of P/Q-type Ca2+ channels to neurotransmission in SL/WT KI relative to WT mice, as previously found in RQ/RQ KI mice (Tottene et al., 2009). Indeed, the average EPSC amplitude was 1.8 times larger in SL/WT KI than in WT mice (Figure 1A), and a larger fraction of the EPSC was inhibited by the specific P/Q-type Ca2+ channel inhibitor ω-AgaIVA (Aga, 200 nM) in KI compared with WT neurons (Figure 1B), indicating a larger contribution of P/Q-type Ca2+ channels to synaptic transmission in SL/WT KI mice. The analysis of the absolute size of the component of the EPSC inhibited by this P/Q-type channel blocker (Tottene et al., 2009) reveals that the increased probability of release consequent to increased Ca2+ influx through P/Q-type Ca2+ channels completely accounts for the increased EPSC amplitude in SL/WT KI mice. In fact, the amplitude of the Aga-sensitive component was 2.1 times larger in KI than in WT mice, and the difference in size of this component in the two genotypes (1.5 nA) was similar to the difference in size of the corresponding total EPSC amplitude (1.6 nA; Figure 1C). On the other hand, the amplitude of the Aga-insensitive component was similar in KI and WT mice (Figure 1C). These findings are inconsistent with significant changes in the size of the readily releasable pool (RRP) of vesicles, at each synapse and/or in the number of synapses in SL/WT neurons, because change in the RRP or in the number of synapses would equally affect the Aga-sensitive and Aga-insensitive components of the EPSC (Tottene et al., 2009).
Further supporting the conclusion of an increased AP-evoked Ca2+ influx through presynaptic mutant CaV2.1 channels is the finding that in SL/WT KI mice the dependence of the EPSC on the extracellular concentration of Ca2+ ions [Ca2+]out was shifted to lower [Ca2+]out with unaltered cooperativity coefficient relative to WT (EC50: 0.90 ± 0.07 mM (n = 11) in SL/WT vs. 1.65 ± 0.10 mM (n = 7) in WT; p = 1.0 × 10−5) (Figure 2A), as previously found in RQ/RQ KI mice (Tottene et al., 2009).
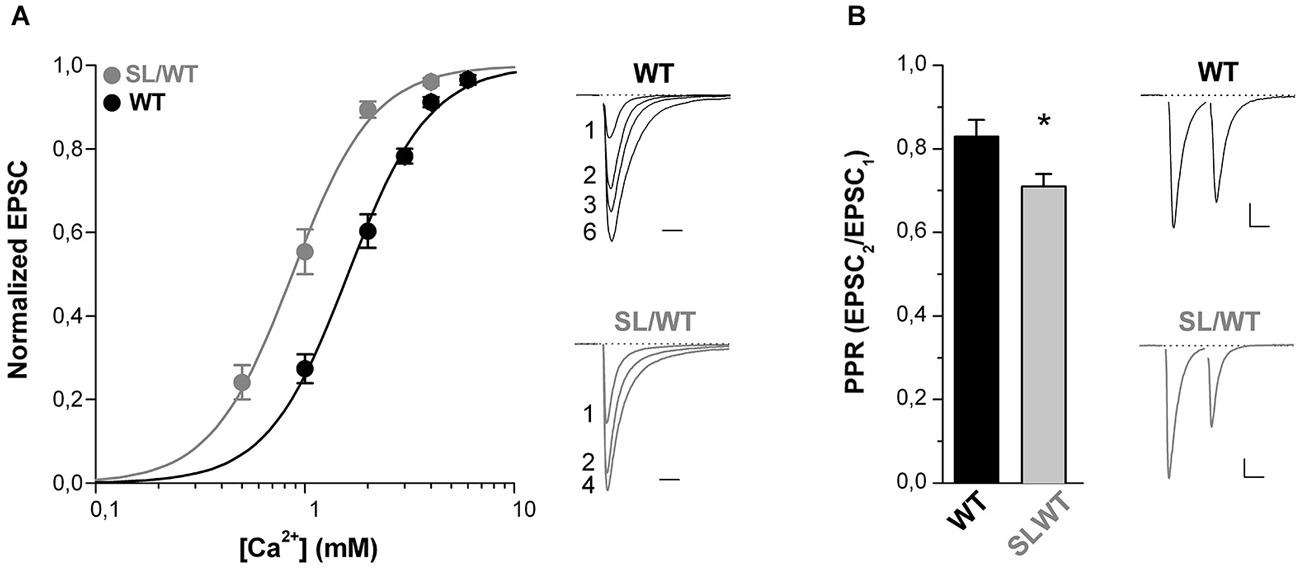
Figure 2. The dependence of the EPSC on extracellular [Ca2+] is shifted to lower [Ca2+] and the paired-pulse ratio is decreased at cortical pyramidal cell autapses of SL/WT KI mice. (A) Left panel: normalized EPSC amplitudes as a function of extracellular [Ca2+] evoked in cortical pyramidal cells in microculture (DIV 9–14) from WT (n = 7) and SL/WT KI (n = 11) mice. In each cell the EPSC data points were fitted according to the Hill equation: EPSC = EPSCmax([Ca2+]n/(EC50)n+[Ca2+]n) and the EPSC amplitudes were then normalized to the EPSCmax obtained from the fit. The average normalized EPSC data points were fitted with EC50 = 0.87 mM, n = 2.2 for SL/WT KI mice and EC50 = 1.6 mM, n = 2.2 for WT mice. Right panel: representative normalized EPSC traces recorded at the indicated [Ca2+] (in mM) from a WT and a SL/WT neuron. Scale bar: 10 ms. (B) Paired-pulse ratios (PPR = EPSC2/EPSC1) evoked by two depolarizing stimuli at 50 Hz in cortical pyramidal cells in microculture (DIV 10–14). PPR values: 0.71 ± 0.03 (n = 43) in SL/WT and 0.83 ± 0.04 (n = 51) in WT neurons (t-test: p = 0.032). Right: representative EPSC traces evoked by two depolarizing stimuli at 50 Hz in a WT and a SL/WT pyramidal cell; scale bars 10 ms, 0.25 nA and 10 ms, 1 nA for WT and SL/WT, respectively.
Moreover, the finding that the paired-pulse ratio (PPR, the ratio between the amplitudes of the second and first EPSC elicited by two AP stimuli at 50 Hz) was lower in SL/WT KI than WT mice (Figure 2B) is consistent with and further supports the conclusion that the S218L mutation leads to an increased probability of vesicle release at individual cortical synapses.
The gain-of-function of both the evoked EPSC and the contribution of P/Q-type Ca2+ channels to neurotransmission as well as the Ca2+ dependence of the EPSC and the PPR are quantitatively similar in cortical pyramidal cell autapses of heterozygous SL/WT KI and homozygous RQ/RQ KI mice. In fact, according to Tottene et al. (2009), the evoked EPSC at cortical RQ/RQ KI pyramidal cell autapses is 1.7 times larger relative to WT, the fractional inhibition by Aga is 78 ± 3% and the EC50 of the Ca2+ dependence is 0.94 mM. Moreover, we measured a PPR of 0.63 ± 0.03 in response to 50 Hz paired-pulses at RQ/RQ cortical pyramidal cell autapses (n = 43). The similar increase in AP-evoked Ca2+ influx and probability of release in SL/WT and RQ/RQ KI mice correlates with the relatively small difference in gain-of-function of the CaV2.1 current at low voltages in neurons from these mice (van den Maagdenberg et al., 2004, 2010).
To assess whether, as previously shown for the R192Q mutation (Tottene et al., 2009; Vecchia et al., 2014), the S218L mutation also does not affect inhibitory synaptic transmission at fast-spiking and other multipolar interneuron synapses, we studied inhibitory neurotransmission in cortical neurons from neonatal SL/WT and WT neurons grown in microculture. The recordings were performed on neurons characterized by irregular soma morphology with multiple asymmetrical processes emanating from it (cf. Figure 1 in Vecchia et al. (2014)), most of which (62–70%), but not all, had firing properties typical of fast-spiking interneurons (Vecchia et al., 2014). IPSCs were evoked in single cortical multipolar interneurons by brief depolarizing voltage steps (Figure 3A, left inset). Both the average amplitude and the fractional inhibition by Aga of the evoked IPSCs were similar in SL/WT and WT interneurons (Figure 3). From this we conclude that the S218L mutation, similar to the R192Q mutation, enhances excitatory neurotransmission at cortical pyramidal cell autapses but does not affect inhibitory neurotransmission at multipolar interneuron autapses, despite the large contribution of P/Q-type Ca2+channels in controlling GABA release at these synapses.
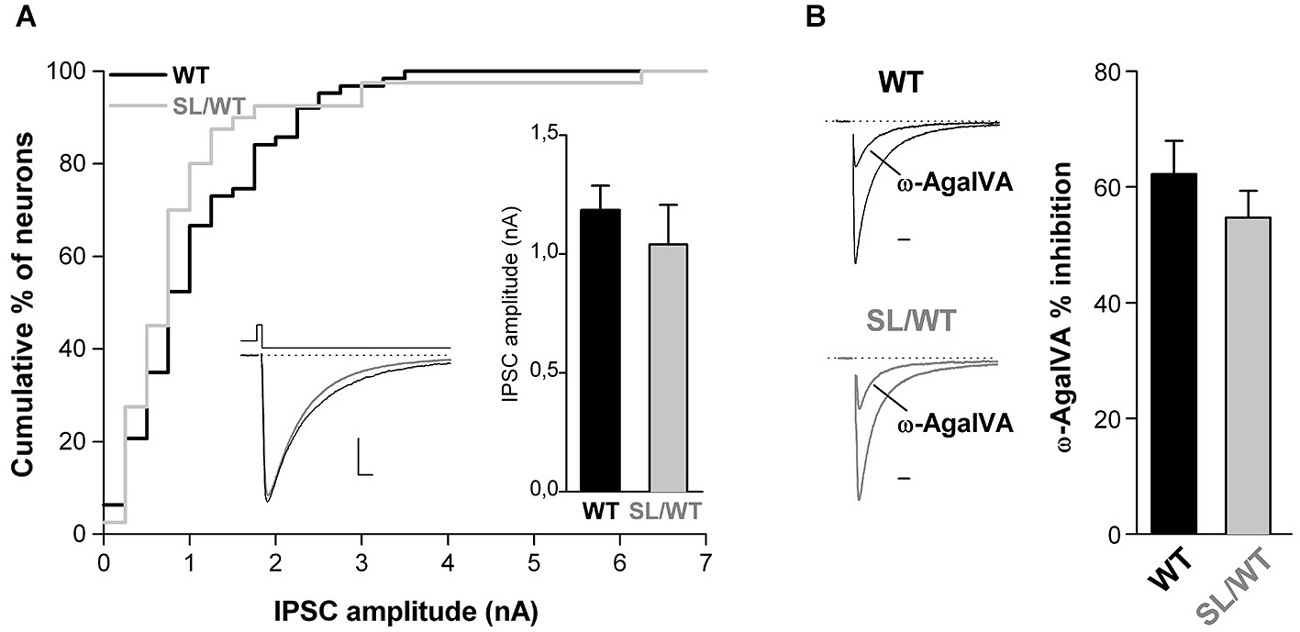
Figure 3. Unaltered evoked IPSC amplitude and unaltered contribution of P/Q-type Ca2+ channels to inhibitory neurotransmission at cortical multipolar interneuron autapses of SL/WT KI mice. (A) Cumulative distribution and average value (right inset) of IPSC amplitudes evoked in single WT and SL/WT cortical multipolar interneurons in microculture (DIV 10–14). IPSC amplitude: 1.0 ± 0.2 nA (n = 40) in SL/WT and 1.2 ± 0.1 nA (n = 63) in WT interneurons (t-test: p = 0.44). Left inset: representative IPSC traces from a WT (black) and SL/WT (gray) interneuron; scale bars: 10 ms, 250 pA. (B) Contribution of P/Q-type Ca2+ channels to inhibitory autaptic neurotransmission evaluated from the fraction of the evoked IPSC inhibited by Aga (200 nM, a saturating concentration since the IPSC was not further inhibited by 400 nM Aga, n = 4 not shown) in WT and SL/WT cortical multipolar interneurons in microculture (DIV 10–14): 55 ± 5% (n = 13) in SL/WT and 62 ± 6% (n = 17) in WT interneurons (t-test: p = 0.34). Representative IPSC traces before and after Aga (normalized to the control value) are shown on the left (scale bar: 10 ms).
The unaltered inhibitory synaptic transmission and the similar increase in the strength of excitatory synaptic transmission in SL/WT and RQ/RQ KI mice, which is in good correlation with the similar facilitation of experimental CSD in vivo (van den Maagdenberg et al., 2010), are consistent with and support the conclusion that enhanced glutamate release at cortical pyramidal cell synapses may explain the facilitation of CSD in SL/WT KI mice, as directly shown in RQ/RQ KI mice (Tottene et al., 2009). Given the larger facilitation of CSD in SL/SL compared with SL/WT KI mice (cf. 67% vs. 38% reduction in CSD threshold and 140% vs. 42% increase in CSD velocity in SL/SL vs. SL/WT KI), that correlates with the larger gain-of-function of the CaV2.1 current in SL/SL compared with SL/WT neurons (van den Maagdenberg et al., 2010), one would expect a larger increase in the strength of excitatory synaptic transmission at cortical pyramidal cell synapses in SL/SL compared with SL/WT KI mice.
In contrast to this expectation, however, similar average EPSC amplitudes were evoked in SL/SL and SL/WT cortical pyramidal cells in microculture (Figure 4A). Moreover, the PPR was also similar (Figure 4B), suggesting a similar probability of release in neurons of the two genotypes. This is not due to near saturation of the presynaptic Ca2+ sensor at SL/WT autapses (cf. maximal value of evoked EPSC only 1.1 times larger than the EPSC at 2 mM Ca2+ in Figure 2A) because, even decreasing the [Ca2+]out down to 0.5 mM did not reveal a larger EPSC in SL/SL compared to SL/WT KI mice (Figure 4C). Indeed, the evoked EPSC at SL/SL and SL/WT autapses showed a similar dependence on [Ca2+]out (EC50 = 1.08 ± 0.14 mM for SL/SL KI (n = 10) and EC50 = 0.90 ± 0.07 mM for SL/WT KI (n = 11); p = 0.26) and, in a larger set of experiments, the ratio of the EPSCs evoked at 1 mM and 2 mM [Ca2+]out was also similar in SL/SL and SL/WT KI mice (0.55 ± 0.04 in SL/SL (n = 21) and 0.61 ± 0.04 in SL/WT (n = 17); p = 0.23). If, as expected from the larger gain-of-function of the neuronal CaV2.1 current in SL/SL relative to SL/WT KI mice (van den Maagdenberg et al., 2010), AP-evoked Ca2+ influx at the active zones is larger in SL/SL KI mice, and if no compensatory changes occur at SL/SL synapses (whereby the affinity of the presynaptic Ca2+ sensor is decreased and/or the capacity to rapidly buffer presynaptic Ca2+ is increased at the active zones of SL/SL compared with SL/WT KI mice), then the Ca2+ dependence of the EPSC at SL/SL autapses should be shifted to lower [Ca2+]out relative to that at SL/WT KI autapses. The finding of a similar Ca2+ dependence of the EPSC in SL/SL and SL/WT KI mice is consistent with the existence of compensatory changes like those mentioned already and/or with a compensatory decrease in the number of functional CaV2.1 channels at the active zones in SL/SL KI mice (see Section Discussion). Whatever the exact underlying compensatory mechanism, the similar gain-of-function of excitatory neurotransmission at cortical pyramidal cell synapses of SL/SL and SL/WT (and RQ/RQ) KI mice contrasts with the larger facilitation of CSD in SL/SL compared to SL/WT (and RQ/RQ) KI mice (van den Maagdenberg et al., 2010), and suggests that the further facilitation of CSD in SL/SL KI mice is due to gain-of-function of CaV2.1-dependent mechanisms that are different from evoked glutamate release at cortical pyramidal cell synapses.
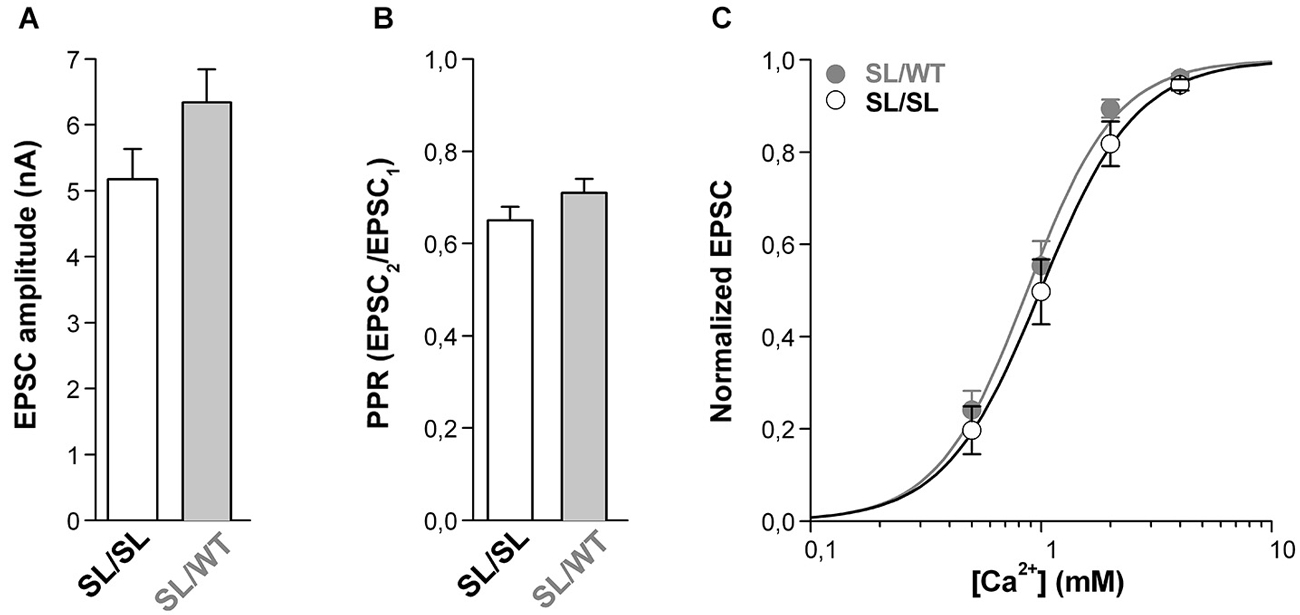
Figure 4. Similar gain-of-function of excitatory neurotransmission at cortical pyramidal cell autapses of homozygous SL/SL and heterozygous SL/WT KI mice. (A) Average value of EPSC amplitudes evoked in single SL/SL and SL/WT cortical pyramidal cells in microculture (DIV 10–14). EPSC amplitudes: 5.2 ± 0.5 nA in SL/SL (n = 46) and 6.3 ± 0.5 nA in SL/WT (n = 95) neurons (t-test: p = 0.14). (B) PPR evoked by two depolarizing stimuli at 50 Hz in cortical pyramidal cells in microculture (DIV 10–14) from SL/SL (n = 36) and SL/WT KI mice (n = 43). PPR: 0.65 ± 0.03 and 0.71 ± 0.03 in SL/SL and SL/WT neurons, respectively (t-test: p = 0.15). (C) Normalized EPSC amplitudes as a function of extracellular [Ca2+] evoked in cortical pyramidal cells in microculture (DIV 10–14) from SL/SL (n = 10) and SL/WT KI (n = 11) mice. The average normalized EPSC data points were fitted with EC50 = 0.99 mM, n = 2.1 for SL/SL KI mice and EC50 = 0.87 mM, n = 2.2 for SL/WT KI mice.
Notably, it has been recently shown that at Calyx of Held terminals a fraction of mutant CaV2.1 channels is open at resting membrane potential in SL/SL KI but not RQ/RQ KI and WT mice; as a consequence, the intraterminal [Ca2+] and the frequency of mEPSCs were larger in SL/SL than WT calyces (Di Guilmi et al., 2014).
To investigate whether a fraction of mutant CaV2.1 channels open at resting membrane potential at glutamatergic synaptic terminals of cortical neurons from SL/SL and SL/WT KI mice, we recorded the frequency of mEPSCs in layer 2/3 pyramidal cells in acute cortical slices and measured its sensitivity to the specific P/Q-type Ca2+ channel inhibitor Aga. The frequency of mEPSCs was significantly reduced after application of Aga (400 nM) in both SL/SL (30 ± 4%) and SL/WT (21 ± 4%) cortical slices (Figure 5). In contrast, Aga did not affect mEPSC frequency in RQ/RQ slices (data not shown), in agreement with the similar frequency of mEPSCs reported in RQ/RQ KI and WT mice (Tottene et al., 2009). These data support the conclusion that a fraction of mutant CaV2.1 channels open at resting membrane potential in cortical excitatory presynaptic terminals in both SL/SL and SL/WT KI mice, but not in RQ/RQ KI mice. Although there is a trend of a larger reduction of mEPSC frequency in SL/SL compared with SL/WT KI cortical pyramidal cells, the difference is not statistically significant.
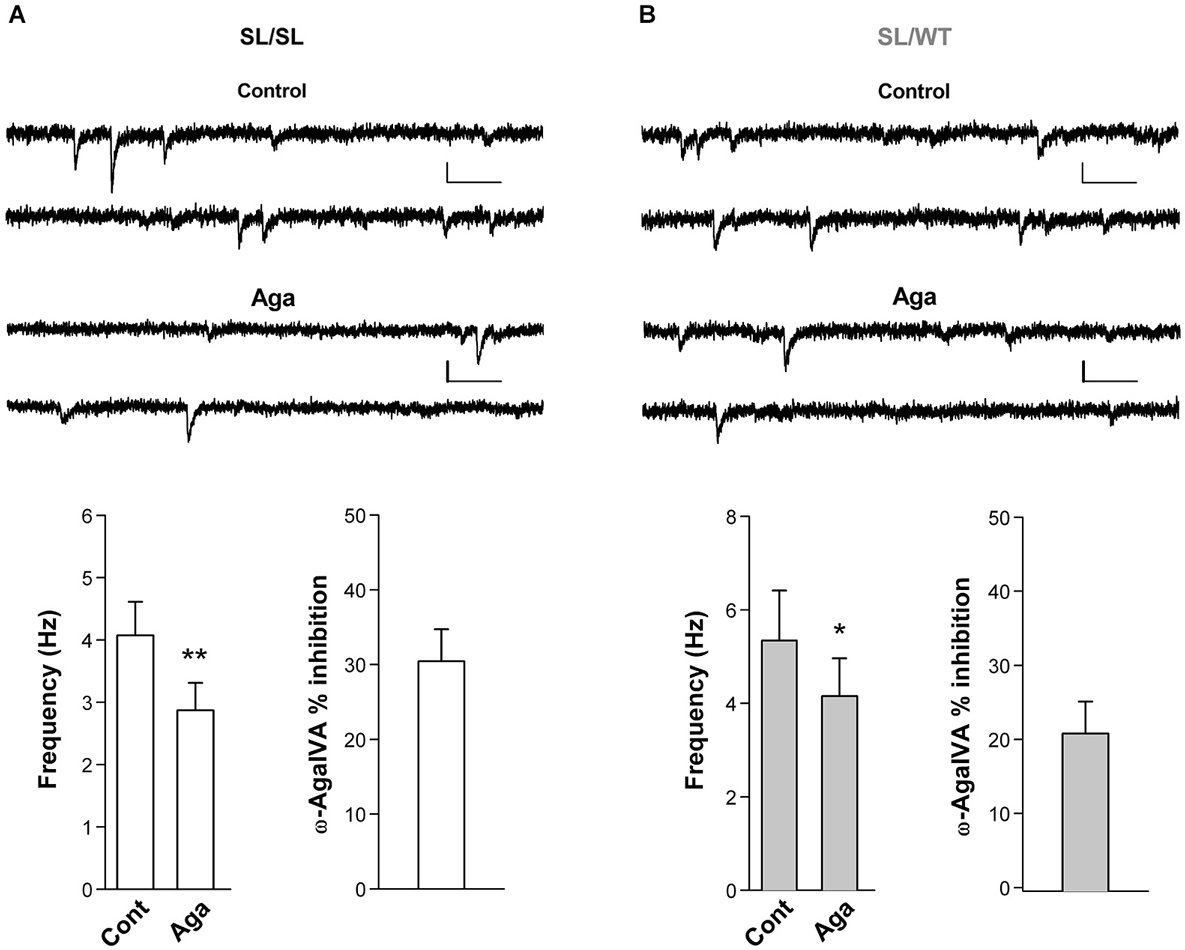
Figure 5. Inhibition of P/Q-type Ca2+ channels reduces the frequency of miniature EPSCs in layer 2/3 pyramidal cells in acute cortical slices from SL/SL and SL/WT KI mice. (A) Representative traces of mEPSCs recorded at −69 mV in a layer 2/3 pyramidal cell and average values of mEPSC frequency (n = 7) in control and after application of Aga (400 nM) in acute cortical slices from P16-18 SL/SL KI mice (Paired t-test: p = 0.0026); also shown is the average value of the percent inhibition of mEPSC frequency by the drug in each cell: 30 ± 4% (n = 7). Scale bars: 50 ms and 10 pA. (B) As in (A), but recordings are from P16-17 SL/WT KI mice (n = 7) (Paired t-test: p = 0.012). Average value of the percent inhibition of mEPSC frequency by the drug in each cell: 21 ± 4%, n = 7. The difference in percent inhibition of mEPSC frequency by Aga in SL/SL and SL/WT neurons is not statistically significant (p = 0.14). Both amplitudes and frequencies of mEPSCs were similar in SL/SL and SL/WT KI mice: 8.3 ± 0.5 pA and 4.1 ± 0.5 Hz in SL/SL vs. 9.1 ± 0.7 pA and 5.3 ± 1.1 Hz in SL/WT; (t-test: p = 0.38 and 0.31, respectively).
Discussion
Our analysis of excitatory and inhibitory synaptic transmission in microcultures of cortical neurons from S218L KI and WT mice revealed that the severer FHM1 S218L mutation, like previously found for the milder FHM1 R192Q mutation (Tottene et al., 2009; Vecchia et al., 2014), enhances excitatory neurotransmission at cortical pyramidal cell synapses without affecting inhibitory neurotransmission at multipolar interneuron synapses, despite the large contribution of P/Q-type Ca2+ channels in controlling GABA release at these synapses. This finding is consistent with the conclusion that the differential effect on cortical excitatory and inhibitory synaptic transmission is likely a common feature of FHM1 mutations, and further supports the idea that the neuronal circuits that dynamically maintain a tight balance between excitation and inhibition are likely altered in FHM1. A plausible working hypothesis is that dysregulation of the cortical excitatory-inhibitory balance in FHM1 may, under certain conditions (cf. migraine triggers), lead to hyperactivity of cortical circuits, mainly due to excessive recurrent excitation, that may create the conditions for the initiation of spontaneous CSDs (e.g., by increasing the extracellular K+ concentration, [K+]e, above a critical value; Vecchia and Pietrobon, 2012; Pietrobon and Moskowitz, 2014).
The increased strength of excitatory neurotransmission at cortical pyramidal cell autapses of S218L KI mice is due to increased probability of glutamate release consequent to increased AP-evoked Ca2+ influx through presynaptic P/Q-type Ca2+ channels. In fact, as previously shown for R192Q KI mice (Tottene et al., 2009): (i) the larger AP-evoked EPSC amplitude in S218L KI compared with WT mice was accompanied by a larger contribution of P/Q-type Ca2+ channels to synaptic transmission; (ii) the dependence of the EPSC on [Ca2+]out was shifted to lower values; and (iii) the PPR was lower in S218L KI than in WT mice. The gain-of-function of both the evoked EPSC and the contribution of P/Q-type Ca2+ channels to neurotransmission as well as the Ca2+ dependence of the EPSC and the PPR at cortical pyramidal cell autapses of heterozygous SL/WT KI mice were quantitatively similar to those reported for homozygous RQ/RQ KI mice (Tottene et al., 2009), in good correlation with the similar gain-of-function of the neuronal P/Q-type Ca2+ current at low voltages and the similar facilitation of experimental CSD in vivo in SL/WT and RQ/RQ KI mice (van den Maagdenberg et al., 2010). This good correlation is consistent with and supports the conclusion that enhanced glutamate release at cortical pyramidal cell synapses may explain the facilitation of induction and propagation of CSD in SL/WT KI mice, as directly shown in RQ/RQ KI mice (Tottene et al., 2009).
However, in contrast with the larger neuronal P/Q-type Ca2+ current at low voltages and the larger facilitation of CSD in homozygous SL/SL compared with heterozygous SL/WT KI mice (van den Maagdenberg et al., 2010), the strength of excitatory transmission at cortical pyramidal cell autapses, the PPR and the Ca2+ dependence of the EPSC were all similar in SL/SL and SL/WT KI mice. These findings strongly suggest the existence of compensatory changes in SL/SL KI mice that prevent excessive glutamate release, and are consistent with e.g., a decreased affinity of the presynaptic Ca2+ sensor and/or a decrease in the number of functional CaV2.1 channels at the active zones in SL/SL compared with SL/WT (and RQ/RQ) KI mice to compensate for the larger Ca2+ influx through open mutant CaV2.1 channels. Evidence for the latter compensatory mechanism at Calyx of Held terminals of SL/SL KI mice is provided by the finding that, while the gain-of function of the Ca2+ current at low voltages is larger in Calyx terminals from SL/SL than RQ/RQ KI mice, the Ca2+ current at high voltages is reduced in SL/SL but unchanged in RQ/RQ KI compared with WT terminals (Inchauspe et al., 2010; Di Guilmi et al., 2014). This compensatory decrease in the number of functional Ca2+ channels may explain a decreased AP-evoked Ca2+ current in SL/SL KI compared with WT Calyx terminals (Di Guilmi et al., 2014). In contrast, AP-evoked Ca2+ influx is larger relative to WT at cortical pyramidal cell synapses (cf. left-shifted EPSC Ca2+ dependence) and cerebellar granule cell synapses (Adams et al., 2010) of SL/SL KI mice. The different alterations of AP-evoked Ca2+ influx at these different synapses may reflect the different durations of the AP (much shorter at Calyx terminals: Inchauspe et al., 2010; Di Guilmi et al., 2014) and/or different compensatory mechanisms.
Whatever the exact underlying compensatory mechanism, the similar gain-of-function of excitatory neurotransmission at cortical pyramidal cell synapses of SL/SL and SL/WT (and RQ/RQ) KI mice contrasts with the larger facilitation of experimental CSD in SL/SL compared with SL/WT (and RQ/RQ) KI mice (van den Maagdenberg et al., 2010), and suggests that the further facilitation of CSD in SL/SL KI mice is due to gain-of-function of CaV2.1-dependent mechanisms different from evoked glutamate release at cortical pyramidal cell synapses. Gain-of-function of CaV2.1-dependent mechanisms different from evoked-glutamate release should also underlie the unique cortical susceptibility to repetitive CSD events and the unique propensity of CSD to spread into subcortical structures (e.g., the hippocampus) observed in S218L (both larger in SL/SL than SL/WT) but not R192Q KI mice (van den Maagdenberg et al., 2010; Eikermann-Haerter et al., 2011).
A feature of cortical synaptic transmission in S218L KI mice not present in R192Q KI mice is the presence of a fraction of mutant CaV2.1 channels that is open at resting potential in cortical excitatory synaptic terminals, as shown by the reduction in mEPSCs frequency produced by the specific P/Q-type Ca2+ channel blocker in S218L cortical slices (but not R192Q slices). As a trend, the fractional reduction was larger in SL/SL than SL/WT KI mice, but the difference did not reach statistical significance, possibly because even in SL/SL pyramidal cells only 30% of mEPSCs were Aga-sensitive. The compensatory changes that prevent a larger gain-of-function of evoked glutamate release in SL/SL compared with SL/WT KI mice may also reduce the difference in the fraction of Aga-sensitive mEPSCs between the two genotypes, and, in the case of a decreased affinity of presynaptic Ca2+ sensor(s), may account for the similar mEPSCs frequency in SL/SL and SL/WT pyramidal cells.
The fraction of mutant CaV2.1 channels that are open at rest in cortical excitatory synaptic terminals is much smaller than that at Calyx terminals (cf. much larger, 76%, reduction of mEPSCs frequency by Aga at Calyx of Held synapses: Di Guilmi et al., 2014), probably as a consequence of expression of CaV2.1 channels with a higher activation threshold (cf. half-voltage of activation of the WT P/Q-type Ca2+ current of −8 mV in cortical pyramidal cells (Tottene et al., 2009) and of −30 mV in calyces (Di Guilmi et al., 2014)), and a smaller shift to lower voltages produced by the S218L mutation (7 mV in pyramidal cells vs. 11 mV in calyces). As a consequence, one predicts a smaller rise of the intraterminal basal [Ca]in in cortical excitatory terminals than measured in SL/SL Calyx terminals (Di Guilmi et al., 2014); this probably explains why the mechanism of facilitation of release (independent of Ca2+ influx and dependent on increased basal [Ca]in) that has been proposed to underlie the gain-of-function of AP-evoked glutamate release in Calyx (Di Guilmi et al., 2014), does not appear to contribute to the gain-of-function of excitatory transmission at SL/SL cortical pyramidal cell synapses. Nonetheless, since V resting of cortical pyramidal cells in vivo in awake animals and also in sleeping or anesthetized animals during the up-states is 10–20 mV more depolarized than that in acute cortical slices (Gentet et al., 2010; Mateo et al., 2011), the fraction of open mutant CaV2.1 channels and the rise of basal [Ca]in might be larger in S218L KI mice in vivo.
One can envisage several mechanisms by which Ca2+ influx at negative voltages sub-threshold for AP generation through presynaptic and postsynaptic mutant CaV2.1 channels (Westenbroek et al., 1995) and increased basal [Ca]in in cortical neurons may contribute to lower the threshold for CSD induction, increase the rate and extent of CSD spread and make the brain susceptible to repetitive CSD events. Available data suggest that a local regenerative [K+]e increase, [K+]e-induced synaptic glutamate release following opening of CaV2.1 channels and activation of NMDARs (and possibly postsynaptic CaV2.1 channels) are key elements in the positive feedback cycle that ignites CSD (Pietrobon and Moskowitz, 2014). This model predicts a lower threshold for CSD initiation and a higher rate of CSD propagation (mediated by diffusion of K+: Pietrobon and Moskowitz, 2014) in SL/SL than in SL/WT KI mice, even with similar AP-evoked glutamate release in the two genotypes, if a larger fraction of mutant CaV2.1 channels opens at sub-threshold depolarizations induced by local [K+]e rise in SL/SL KI mice. Moreover, CaV2.1-dependent release of other neurotransmitters or neuromodulators (e.g., CGRP: Tozzi et al., 2012) may be enhanced in SL/SL KI mice relative to SL/WT and RQ/RQ KI mice and might contribute to the further facilitation of CSD in homozygous S218L KI mice. It is also possible that only in SL/SL neurons [Ca]in-activated cationic channels contribute to the net self-sustaining inward current necessary to initiate the positive feedback cycle that ignites CSD (Somjen et al., 2009; Pietrobon and Moskowitz, 2014). Finally, a constant basal Ca2+ influx would put a metabolic burden on cortical neurons of S218L (but not R192Q) KI mice that may prolong the transient hypoxia and slow the recovery of cerebral flow after CSD (Pietrobon and Moskowitz, 2014) and in general make the recovery from CSD more difficult. Since another peculiarity of mutant S218L Ca2+ channels, besides the particularly low threshold of activation, is the incomplete inactivation during prolonged depolarizations (Tottene et al., 2005), a larger increase of [Ca]in during CSD in cortical neurons may also contribute to a slower recovery from CSD in S218L KI mice. A slower recovery from CSD could provide a plausible explanation for the unique susceptibility to recurrent CSDs (van den Maagdenberg et al., 2010) and the unique propensity of CSD to spread into subcortical structures such as hippocampus and thalamus as well as the more severe motor deficits after CSD (Eikermann-Haerter et al., 2009, 2011) as observed in S218L KI mice. It has also been proposed that these unique features conferred by the S218L mutation may underlie the severe and clinically broad phenotype that is seen in patients carrying the S218L but not in patients carrying the R192Q mutation (Eikermann-Haerter et al., 2009, 2011; van den Maagdenberg et al., 2010).
Author and Contributions
Dania Vecchia performed the experiments, analysed the data, discussed findings and interpretation of the data and contributed to drafting the manuscript.
Angelita Tottene contributed to the acquisition, analysis and discussion of the data and revised critically the manuscript.
Arn M.J.M. van den Maagdenberg contributed to the discussion and interpretation of the data, provided reagents and analytic tools, and revised critically the manuscript.
Daniela Pietrobon conceived and designed the study, discussed findings and interpretation of the data and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Telethon Italy grant GGP06234 (to Daniela Pietrobon), Fondazione Cariparo Excellence Project: Calcium Signaling in Health and Disease (to Daniela Pietrobon), the Italian Ministry of University and Research PRIN2007, 2010 (to Daniela Pietrobon), the University of Padova Strategic Project: Physiopathology of Signaling in Neuronal Tissue and Progetto Ateneo 2012 (to Daniela Pietrobon), the Centre for Medical Systems Biology (CMSB) in the framework of the Netherlands Genomics Initiative (NGI) (to Arn M.J.M. van den Maagdenberg). Thanks to L. Broos for technical assistance.
References
Adams, P. J., Rungta, R. L., Garcia, E., van den Maagdenberg, A. M., MacVicar, B. A., and Snutch, T. P. (2010). Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc. Natl. Acad. Sci. U S A 107, 18694–18699. doi: 10.1073/pnas.1009500107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bekkers, J. M., and Stevens, C. F. (1991). Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc. Natl. Acad. Sci. U S A 88, 7834–7838. doi: 10.1073/pnas.88.17.7834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brody, D. L., and Yue, D. T. (2000). Release-independent short-term synaptic depression in cultured hippocampal neurons. J. Neurosci. 20, 2480–2494.
de Vries, B., Frants, R. R., Ferrari, M. D., and van den Maagdenberg, A. M. (2009). Molecular genetics of migraine. Hum. Genet. 126, 115–132. doi: 10.1007/s00439-009-0684-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Guilmi, M. N., Wang, T., Inchauspe, C. G., Forsythe, I. D., Ferrari, M. D., van den Maagdenberg, A. M., et al. (2014). Synaptic gain-of-function effects of mutant Cav2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. J. Neurosci. 34, 7047–7058. doi: 10.1523/JNEUROSCI.2526-13.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eikermann-Haerter, K., Dileköz, E., Kudo, C., Savitz, S. I., Waeber, C., Baum, M. J., et al. (2009). Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J. Clin. Invest. 119, 99–109. doi: 10.1172/JCI36059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eikermann-Haerter, K., Yuzawa, I., Qin, T., Wang, Y., Baek, K., Kim, Y. R., et al. (2011). Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J. Neurosci. 31, 5755–5763. doi: 10.1523/JNEUROSCI.5346-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fioretti, B., Catacuzzeno, L., Sforna, L., Gerke-Duncan, M. B., van den Maagdenberg, A. M., Franciolini, F., et al. (2011). Trigeminal ganglion neuron subtype-specific alterations of Ca(V)2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J. Physiol. 589, 5879–5895. doi: 10.1113/jphysiol.2011.220533
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, Z., Todorov, B., Barrett, C. F., van Dorp, S., Ferrari, M. D., van den Maagdenberg, A. M., et al. (2012). Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L) mutant mice. J. Neurosci. 32, 15533–15546. doi: 10.1523/JNEUROSCI.2454-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gentet, L. J., Avermann, M., Matyas, F., Staiger, J. F., and Petersen, C. C. (2010). Membrane potential dynamics of GABAergic neurons in the barrel cortex of behaving mice. Neuron 65, 422–435. doi: 10.1016/j.neuron.2010.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hans, M., Luvisetto, S., Williams, M. E., Spagnolo, M., Urrutia, A., Tottene, A., et al. (1999). Functional consequences of mutations in the human a1A calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 19, 1610–1619.
Inchauspe, C. G., Urbano, F. J., Di Guilmi, M. N., Forsythe, I. D., Ferrari, M. D., van den Maagdenberg, A. M., et al. (2010). Gain of function in FHM-1 Ca(V)2.1 knock-in mice is related to the shape of the action potential. J. Neurophysiol. 104, 291–299. doi: 10.1152/jn.00034.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kors, E. E., Terwindt, G. M., Vermeulen, F. L., Fitzsimons, R. B., Jardine, P. E., Heywood, P., et al. (2001). Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 49, 753–760. doi: 10.1002/ana.1031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levi, G., Aloisi, M., Ciotti, M., and Gallo, V. (1984). Autoradiographic localization and depolarization-induced release of amino acids in differentiating granule cells cultures. Brain Res. 290, 77–86. doi: 10.1016/0006-8993(84)90737-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mateo, C., Avermann, M., Gentet, L. J., Zhang, F., Deisseroth, K., and Petersen, C. C. (2011). In vivo optogenetic stimulation of neocortical excitatory neurons drives brain-state-dependent inhibition. Curr. Biol. 21, 1593–1602. doi: 10.1016/j.cub.2011.08.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neher, E. (1992). Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 207, 123–131. doi: 10.1016/0076-6879(92)07008-c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Noseda, R., and Burstein, R. (2013). Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization and modulation of pain. Pain 154(Suppl. 1), S44–53. doi: 10.1016/j.pain.2013.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N., van Eijk, R., Oefner, P. J., Hoffman, S. M. G., et al. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552. doi: 10.1016/s0092-8674(00)81373-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pietrobon, D. (2005). Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr. Opin. Neurobiol. 15, 257–265. doi: 10.1016/j.conb.2005.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pietrobon, D. (2010). CaV2.1 channelopathies. Pflugers Arch. 460, 375–393. doi: 10.1007/s00424-010-0802-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pietrobon, D. (2013). Calcium channels and migraine. Biochim. Biophys. Acta 1828, 1655–1665. doi: 10.1016/j.bbamem.2012.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pietrobon, D., and Moskowitz, M. A. (2013). Pathophysiology of migraine. Annu. Rev. Physiol. 75, 365–391. doi: 10.1146/annurev-physiol-030212-183717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pietrobon, D., and Moskowitz, M. A. (2014). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 15, 379–393. doi: 10.1038/nrn3770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Somjen, G. G., Kager, H., and Wadman, W. J. (2009). Calcium sensitive non-selective cation current promotes seizure-like discharges and spreading depression in a model neuron. J. Comput. Neurosci. 26, 139–147. doi: 10.1007/s10827-008-0103-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stam, A. H., Luijckx, G. J., Poll-Thé, B. T., Ginjaar, I. B., Frants, R. R., Haan, J., et al. (2009). Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J. Neurol. Neurosurg. Psychiatry 80, 1125–1129. doi: 10.1136/jnnp.2009.177279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tottene, A., Conti, R., Fabbro, A., Vecchia, D., Shapovalova, M., Santello, M., et al. (2009). Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 61, 762–773. doi: 10.1016/j.neuron.2009.01.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tottene, A., Fellin, T., Pagnutti, S., Luvisetto, S., Striessnig, J., Fletcher, C., et al. (2002). Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. U S A 99, 13284–13289. doi: 10.1073/pnas.192242399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tottene, A., Pivotto, F., Fellin, T., Cesetti, T., van den Maagdenberg, A. M., and Pietrobon, D. (2005). Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 280, 17678–17686. doi: 10.1074/jbc.m501110200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tozzi, A., de Iure, A., Di Filippo, M., Costa, C., Caproni, S., Pisani, A., et al. (2012). Critical role of calcitonin gene-related peptide receptors in cortical spreading depression. Proc. Natl. Acad. Sci. U S A 109, 18985–18990. doi: 10.1073/pnas.1215435109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van den Maagdenberg, A. M., Pietrobon, D., Pizzorusso, T., Kaja, S., Broos, L. A., Cesetti, T., et al. (2004). A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41, 701–710. doi: 10.1016/s0896-6273(04)00085-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van den Maagdenberg, A. M., Pizzorusso, T., Kaja, S., Terpolilli, N., Shapovalova, M., Hoebeek, F. E., et al. (2010). High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann. Neurol. 67, 85–98. doi: 10.1002/ana.21815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vecchia, D., and Pietrobon, D. (2012). Migraine: a disorder of brain excitatory-inhibitory balance? Trends Neurosci. 35, 507–520. doi: 10.1016/j.tins.2012.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vecchia, D., Tottene, A., van den Maagdenberg, A. M., and Pietrobon, D. (2014). Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in CaV2.1 knockin migraine mice. Neurobiol. Dis. 69, 225–234. doi: 10.1016/j.nbd.2014.05.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: calcium channel, synaptic transmission, cortex, migraine, spreading depression, excitatory, inhibitory, knockin mouse
Citation: Vecchia D, Tottene A, van den Maagdenberg AMJM and Pietrobon D (2015) Abnormal cortical synaptic transmission in CaV2.1 knockin mice with the S218L missense mutation which causes a severe familial hemiplegic migraine syndrome in humans. Front. Cell. Neurosci. 9:8. doi: 10.3389/fncel.2015.00008
Received: 22 December 2014; Paper pending published: 29 December 2014;
Accepted: 08 January 2015; Published online: 17 February 2015.
Edited by:
Maria Cristina D’Adamo, University of Perugia, ItalyReviewed by:
Paolo Calabresi, Santa Maria della Misericordia Hospital, ItalySalvatore Salomone, Catania University, Italy
Copyright © 2015 Vecchia, Tottene, van den Maagdenberg and Pietrobon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Pietrobon, Department of Biomedical Sciences, University of Padova, and Consiglio Nazionale delle Ricerche (CNR) Institute of Neuroscience, Via U. Bassi 58, 35121 Padova, Italy e-mail: daniela.pietrobon@unipd.it