 Maria Rosito1
Maria Rosito1 Clotilde Lauro1
Clotilde Lauro1 Giuseppina Chece1
Giuseppina Chece1 Alessandra Porzia2
Alessandra Porzia2 Lucia Monaco1
Lucia Monaco1 Fabrizio Mainiero2
Fabrizio Mainiero2 Myriam Catalano1,3
Myriam Catalano1,3 Cristina Limatola1,3
Cristina Limatola1,3 Flavia Trettel1*
Flavia Trettel1*- 1Department of Physiology and Pharmacology, Istituto Pasteur Fondazione Cenci Bolognetti, Sapienza University of Rome, Rome, Italy
- 2Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy
- 3IRCSS NeuroMed, Pozzilli, Italy
Upon noxious insults, cells of the brain parenchyma activate endogenous self-protective mechanisms to counteract brain damage. Interplay between microglia and astrocytes can be determinant to build a physiological response to noxious stimuli arisen from injury or stress, thus understanding the cross talk between microglia and astrocytes would be helpful to elucidate the role of glial cells in endogenous protective mechanisms and might contribute to the development of new strategy to mobilize such program and reduce brain cell death. Here we demonstrate that chemokines CX3CL1 and CXCL16 are molecular players that synergistically drive cross-talk between neurons, microglia and astrocytes to promote physiological neuroprotective mechanisms that counteract neuronal cell death due to ischemic and excitotoxic insults. In an in vivo model of permanent middle cerebral artery occlusion (pMCAO) we found that exogenous administration of soluble CXCL16 reduces ischemic volume and that, upon pMCAO, endogenous CXCL16 signaling restrains brain damage, being ischemic volume reduced in mice that lack CXCL16 receptor. We demonstrated that CX3CL1, acting on microglia, elicits CXCL16 release from glia and this is important to induce neroprotection since lack of CXCL16 signaling impairs CX3CL1 neuroprotection against both in vitro Glu-excitotoxic insult and pMCAO. Moreover the activity of adenosine receptor A3R and the astrocytic release of CCL2 play also a role in trasmembrane chemokine neuroprotective effect, since their inactivation reduces CX3CL1- and CXCL16 induced neuroprotection.
Introduction
Glial cells, able to sense changes in brain environment, represent active players in various pathological conditions such as chronic neurodegenerative disease, trauma and stroke. It is now established that both microglia and astrocytes can play dual roles in the CNS having either detrimental or beneficial effects participating and enhancing inflammatory conditions, or limiting neuroinflammation, favoring repair and enhancing neuronal survival (Liu et al., 2011). Thus understanding the cross talk between microglia and astrocytes would be helpful to elucidate the role of glial cells in pathological conditions.
Microglia-astrocytes interplay is granted by different types of soluble mediators including ATP, adenosine, glutamate (Glu) (Boison et al., 2010; Burnstock et al., 2011; Franke et al., 2012; Pascual et al., 2012), growth factors and inflammatory cytokines (Hamby and Sofroniew, 2010). We have recently shown that the transmembrane chemokine CXCL16 and its receptor, CXCR6, are constitutively expressed in glia and neurons being able to drive neuroprotection against Glu excitotoxicity and oxygen glucose deprivation (OGD) insults in culture (Rosito et al., 2012). In particular we found that the neuroprotective activity of CXCL16 involves astrocytic release of CCL2 and the synergistic activity of adenosine and adenosine type 3 receptor (A3R) on astrocytes.
The other known trasmembrane chemokine CX3CL1 is constitutively expressed in the brain only by neurons, while its unique receptor CX3CR1 is exclusively present on microglial cells. Recently described as a neuronal “off signal” that keep microglia in resting state (Biber et al., 2007), in the last decade the role of CX3CL1-CX3CR1 signaling in modulating neuron viability has emerged in several studies on neurodegenerative and neuroinflammatory disease models (Soriano et al., 2002; Cardona et al., 2006; Huang et al., 2006; Dénes et al., 2008; Bhaskar et al., 2010; Fuhrmann et al., 2010; Lee et al., 2010; Cipriani et al., 2011). Moreover CX3CL1 ability to preserve neurons from excitotoxic insult has been shown both in vitro and in vivo: in particular CX3CL1 signaling in microglia determines the release of soluble factors, such as adenosine that, acting on the adenosine receptor type 1 (A1R), concur to neuroprotection against Glu excitotoxicity and cerebral ischemia (Limatola et al., 2005; Lauro et al., 2010; Cipriani et al., 2011; Catalano et al., 2013).
In the present paper we studied the interplay between trasmembrane chemokines and between glial cells in determining neuroprotection against excitotoxic insults. In particular we found that: (i) CXCL16 is able to reduce ischemic brain volume; (ii) following ischemic insults there is an overexpression of CXCL16; (iii) CXCL16 and CCL2 are released from glia upon CX3CL1 stimulation; and (iv) A3R and CXCR6 concur to CX3CL1 mediated neuroprotection.
Materials and Methods
Animals
Procedures using laboratory animals were in accordance with the international guidelines on the ethical use of animals from the European Communities Council Directive of 24 November 1986 (86/609/EEC). C57BL/6J (wt) and homozygous cxcr6gfp/gfp knock-in mice (Unutmaz et al., 2000) in which the coding region of the receptor has been substituted with the coding region of the Green Fluorescent Protein (GFP) were obtained from Jackson Laboratory (strain name B6.129P2-Cxcr6tm1Litt/J). A3R knockout mice (A3R–/–) (Salvatore et al., 2000) were also used. Animals of either sex were used.
Permanent Middle Cerebral Artery Occlusion (pMCAO)
Male mice (25–28 g, 10–12 weeks) were anesthetized with intraperitoneal Equitensine at 3.5 ml/kg (39 mM pentobarbital, 256 mM chloral hydrate, 86 mM MgSO4, 10% ethanol v/v, and 39.6% propyleneglycol v/v). The right MCA was permanently occluded by electrocoagulation as described previously (Storini et al., 2006). Mice were maintained at 37°C during surgery and sacrificed 24 h after pMCAO.
Intracerebroventricul (i.c.v.) Injection
Recombinant mouse CXCL16 or mouse CX3CL1 (Peprotech) was dissolved in saline solution and intracerebroventricularly injected 30 min before pMCAO. For dose-response experiments, mice were injected with 15, 70 and 150 pmol CXCL16/2 μl. Anesthetized animals were immobilized on a stereotaxic apparatus (David Kopf Instruments) and injected in the right cerebral ventricle (1 mm lateral and 3 mm deep, according to the atlas of Paxinos and Franklin, 2004). A constant rate of infusion (0.2 μl/min) was maintained with a pump (KD Scientific).
Brain Ischemic Volume Measurement
The extent of ischemic area was evaluated 24 h after ischemia. Mice were deeply anesthetized with Equitensine and transcardially perfused with ice-cold PBS (20 ml), pH 7.4, and paraformaldehyde (PFA; 4%, 50 ml) in PBS. The brains were carefully removed from the skull and transferred in 4% PFA at 4°C overnight, then to PBS/30% sucrose at 4°C overnight, frozen in isopentane at −45°C for 3 min, and then stored at −80°C until use. Twenty μm coronal brain cryosections were cut serially at 320 μm intervals and stained with cresyl violet. Infarct volumes were calculated by integration of the infarct areas on each brain slice, as described previously (Storini et al., 2006).
Primary Hippocampal Cultures
Primary hippocampal cultures were prepared from the brain of 0–2-day-old wild type (wt), cxcr6gfp/gfp and A3R–/– mice. In brief, after careful dissection from diencephalic structures, the meninges were removed and the hippocampi chopped and digested in 0.025% trypsin, in Hank’s balanced salt solution (HBSS) for 20 min at 37°C. Cells were mechanically dissociated and plated at a density of 2.5 × 105 in poly-L-lysine coated plastic 24-well dishes, in serum-free Neurobasal medium supplemented with B27, 0.5 mM L-glutamine and 100 μg/ml gentamicin. Successively, cells were kept at 37°C in 5% CO2 for 10–11 days in vitro (DIV) with a twice a week medium replacement (1:1 ratio). With this method we obtained 60–70% neurons, 30–35% astrocytes, 4–5% microglia, as determined with β-tubulin III, glial fibrillary acidic protein (GFAP), and isolectin IB4 staining (Lauro et al., 2010).
Oxygen Glucose Deprivation (OGD)
Primary hippocampal cultures (10–11 DIV) were exposed to OGD. Briefly, culture medium was replaced with modified Locke’s buffer (without glucose), bubbled with 95% N2/5% CO2, and transferred into an anaerobic chamber (Billups-Rothenberg MIC- 101) containing a mixture of 95% N2/5% CO2, and humidified at 37°C for 90 min. For the reperfusion conditions OGD was terminated by replacing the OGD medium with the original conditioned medium. For comparative purposes, control cultures were treated under normoxic conditions (95% O2/5% CO2) in complete Locke’s buffer supplemented with glucose (5.6 mM).
Glu Excitotoxicity
In primary hippocampal cultures (10–11 DIV) conditioned medium was removed and stored for later usage; neurons were washed and stimulated with Glu (100 μM, 30 min) in modified Locke’s buffer (without MgCl2 plus 1 μM glycine to stimulate all types of Glu receptors), in the presence or in the absence of recombinant mouse mCX3CL1 (100 nM, Peprotech). Under these experimental conditions, only neurons die (Chen et al., 2000; Rosito et al., 2012). After treatment, cells were re-incubated in the original conditioned medium for 18–20 h, treated with lysis buffer (0.5% ethylhexadecyldimethylammonium bromide, 0.28% acetic acid, 0.5% Triton X-100, 3 mM NaC1, 2 mM MgCl, in PBS pH 7.4) and counted in a hemocytometer for viability, as described (Volontè et al., 1994). Data were expressed as percentage of viable cells taking as 100% the number of viable cells in control cultures. Variability in the number of viable cells in control conditions never exceeded 10%.When necessary, cells were pre-treated with monoclonal mouse αCCL2 Ab (3 μg/ml, 30 min; R&D MAB479), rat IgG (3 μg/ml, 30 min; Santa Cruz Biotecnology sc-2032), 3-propyl-6-ethyl-5-[(ethylthio)carbonyl]-2phenyl-4-propyl-3-pyrinide carboxylate (MRS1523; 100 nM, Sigma) in culture medium; drugs were present also during and after Glu challenge.
RNA Extraction and Analysis
Total RNA from ipsilateral (ischemic core and penumbra) and controlateral (corresponding areas) brain emispheres of pMCAO mice, from primary hippocampal mixed cell cultures (5 × 105 cells), from primary astrocytes (2.5 × 105) and microglial cells (2.5 × 105), was extracted by the use of Trizol reagent (Invitrogen). Reverse transcription reaction was performed in a thermocycler (MJ Mini Personal Thermal Cycler; Biorad) using IScriptTM Reverse Transcription Supermix (Biorad) according to the manufacturer’s protocol. Real-time PCR (RT-PCR) was carried out in a I-Cycler IQ Multicolor RT-PCR Detection System (Biorad) using SsoFast EvaGreen Supermix (Biorad) according to the manufacturer’s instructions. The PCR protocol consisted of 40 cycles of denaturation at 95°C for 30 s and annealing/extension at 58°C for 30 s. For quantification analysis the comparative Threshold Cycle (Ct) method was used. The Ct values from each gene were normalized to the Ct value of β-actin or GAPDH in the same RNA samples. Relative quantification was performed using the 2−ΔΔCt method (Schmittgen and Livak, 2008) and expressed as fold change in arbitrary values. Primer sequences targeted against CXCL16 (BC019961.1, GenBank), mouse β-actin and GAPDH were as follows: CXCL16-forw. TCCTTTTCTTGTTGGCGCTG, CXCL16rev. CAGCGACACTGCCCCTGGT; β-actin-forw.AGAGGGAAATCGTGCGTGAC, β-actin-rev. CAATAGTGATGACCTGGCCGT; GAPDH- forw. TCGTCCCGTAGACAAAATGG, GAPDH-rev. CAAGGGGTTGAAGCTCAGAT.
Glial Primary Cultures
Primary cortical glial cells were prepared from 0–2-day-old wt mice. Cerebral cortices were chopped and digested in 30 U/ml papain for 40 min at 37°C followed by gentle trituration. The dissociated cells were washed, suspended in DMEM with 10% fetal bovine serum (FBS; Gibco) and 2 mM L-glutamine and plated at a density of 9–10 × 105 in 175 cm2 cell culture flasks. At confluence (10–14 DIV), glial cells were shaken for 2 h at 37°C to detach and collect microglial cells. Astrocytes which remained attached to the bottom of the flask were treated with trypsin and collected. These procedures gave almost pure (no more than 2% astrocyte contamination) microglial cell population, and astrocytes cell population (4–6% of microglia contamination), as verified by staining with GFAP and isolectin IB4.
Microglia-Astrocyte Co-Cultures
After 10–14 DIV, 8 × 105 microglial cells were re-plated and co-cultured for 48 h with astrocytes cells (8 × 105) seeded on 24 mm transwell cell-culture inserts (pore size 0.4 μm; Corning Life Sciences) which allows traffic of small diffusible substances, but prevents cell contact. After co-cultures, cells were treated with vehicle or soluble CX3CL1 (100 nM) for 18 h and upon stimulation proteins from cells and conditioned medium was collected and analyzed for Western blot. For CCL2 ELISA and mRNA analysis 2.5 × 105 astrocyte were re-plated on 12 mm transwell cell-culture inserts (pore size 0.4 μm; Corning Life Sciences) and co-cultured with 2.5 × 105 microglial cells. After 48 h cells were treated with vehicle or soluble CX3CL1 (100 nM) for 18 h. For ELISA conditioned medium (c.m.) was collected and analyzed according to the manufacturer (R&D Systems), while for mRNA extraction microglia and astrocytes were collected from the different transwell compartments.
Proteins Preparation
C.m. from microglia-astrocytes co-cultures were collected and concentrated by ultrafiltering on Ym-10 membrane (Centricon; Millipore). For cell membrane proteins preparation, cells were washed with phosphate-buffered saline and lysed for 15 min on ice in hypotonic buffer containing 10 mM HEPES pH 8, 1.5 mM MgCl2, 1 mM DDT, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride. After centrifugation at 1500 rpm for 5 min at 4°C, supernatant were ultra-centrifuged at 55000 rpm for 60 min, 4°C, and pellet were suspended in NaCl 10 mM.
Western blot Analysis
Protein samples were separated on 10% SDS-polyacrylamide gel and analyzed by Western immunoblot using a mouse CXCL16 antibody (0.2 μg/ml; R&D System, AF503) and HRP-tagged rabbit anti goat IgG secondary antibody (1:2000; Dako), and subsequently detected using a commercial chemiluminescent assay (Immun-Star WesternC Kit; Bio-Rad). Densitometric analysis was performed with Quantity One software (Biorad). Interpretation of western-blot bands for CXCL16 was according to Gough et al. (2004).
Statistical Analysis
The data are expressed as the means ± SEM. Where appropriate t-test, or analysis of variance (ANOVA) was used: we performed the parametric one-way ANOVA or two-way ANOVA followed by specific multiple comparison, as described in detail in figure legends. A value of p < 0.05 was considered significant. All statistical analysis was done using SigmaPlot 11.0 Software.
Results
Soluble CXCL16 Reduces the Ischemic Volume in Mouse Brain After pMCAO
Since we have recently demonstrated that CXCL16 is neuroprotective against Glu excitotoxicity in vitro (Rosito et al., 2012), we now investigated the ability of CXCL16 to induce neuroprotection in mice upon pMCAO. In wt mice, i.c.v. injection of soluble CXCL16, 30 min before induction of pMCAO, resulted in a decreased ischemic volume compared to wt mice injected with saline (n = 10) control solution: in particular, a significant reduction in ischemic volume was observed after injection of 70 and 150 pmol of CXCL16 (n = 4–8; p < 0.05), while no effect was observed upon injection of 15 pmol (n = 4; Figure 1A). Further experiments were performed at 70 pmol. The neuroprotective effect of CXCL16 was specific, being absent in mice that lack CXCR6 receptor (cxcr6gfp/gfp mice; Figure 1B). Two-way ANOVA analysis indicated a significant interaction between genotypes and treatments (p = 0.02) and post hoc evaluation revealed that CXCL16 was ineffective in reducing ischemic volume in cxcr6gfp/gfp mice (n = 6). In addition, in cxcr6gfp/gfp mice, pMCAO induced a significantly increased ischemic volume compared to wt animals suggesting that endogenous CXCL16-CXCR6 signaling contributes to restrain brain damage following ischemic insult.
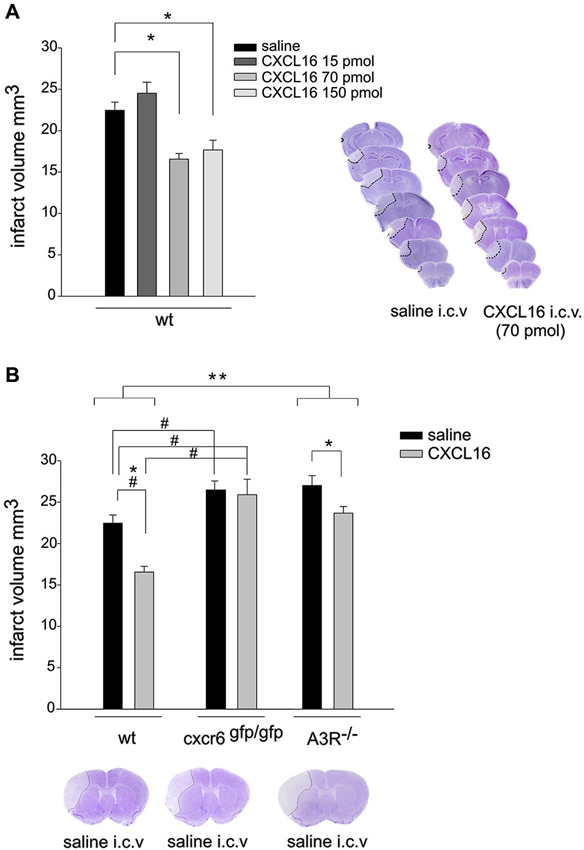
Figure 1. CXCL16 reduces ischemic volume in pMCAO. (A) CXCL16 dose-response effect. Left: Mice were i.c.v. injected with saline or CXCL16 (15–70-150 pmol) 30 min before pMCAO and analyzed for ischemic volume 24 h later (n = 4–10). Right: representative brain coronal sections from saline and CXCL16 i.c.v injected mice. Pale demarcated areas depict the ischemic lesions. (B) CXCL16 effect in wt, cxcr6 gfp/gfp, and A3R–/– mice. Mice of different genotypes (as indicated) were injected with saline or CXCL16 (70 pmol) before pMCAO and analyzed for ischemic volume (n = 6–10). Representative brain coronal section of pMCAO saline injected mice of different genotypes (Bottom). Results represent the mean ± SEM. Statistical analysis: one-way ANOVA followed by Holm-Sidak post-hoc test *p < 0.05 (A). Two-way ANOVA followed by Holm-Sidak post-hoc test * p < 0.05; ** p < 0.001; * refers to wt-A3R–/–comparison; #p < 0.05 refers to wt-cxcr6gfp/gfp comparison (B).
To investigate whether the protective effect of CXCL16 upon pMCAO requires the activity of A3R, we analyzed the effect of i.c.v. administration of CXCL16 in A3R–/– mice (n = 6–7; Figure 1B). Two-way ANOVA analysis reveals a significant differences between genotypes (p < 0.001), being the ischemic volume higher in A3R −/– vs. wt animals. CXCL16 administration was effective in reducing ischemic volume in both genotypes, (p < 0.05) but the reduction observed in A3R–/– mice was less pronounced (12.3% in A3R–/– vs. 26.3 % in wt).
Ischemic Insults Induce Up-Regulation of Endogenous CXCL16
Since CXCL16 signaling is determinant in reducing brain damage, we measured CXCL16 expression in the brain upon ischemia. RT-PCR analysis revealed that 24 h after pMCAO CXCL16 mRNA specifically increased in the ipsilateral hemisphere (n = 5; p < 0.001; Figure 2A), while no differences were observed in sham operated mice (not shown). Similar results were obtained in vitro, when hippocampal cultures were treated to induce OGD cell death (Rosito et al., 2012). After 90 min of OGD we observed a reduction of CXCL16 mRNA, followed by a significant increase after 2 h of recovery (n = 8–11; p < 0.05; Figure 2B).
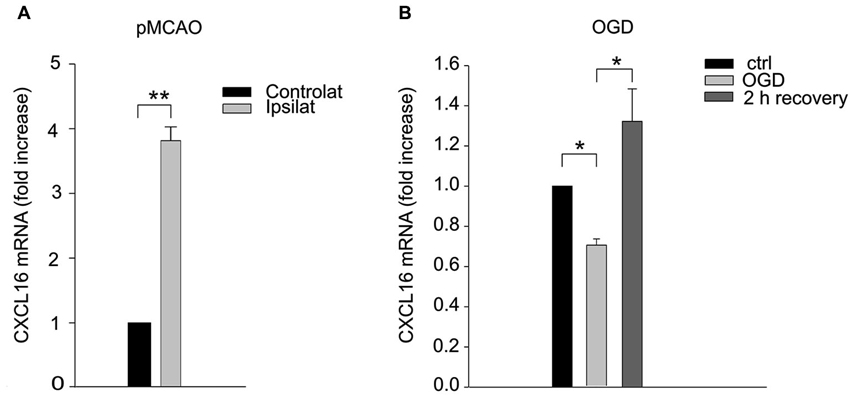
Figure 2. CXCL16 expression is increased upon ischemic insults. (A) CXCL16 mRNA quantification in pMCAO brain. qRT-PCR analysis in contralateral and ipsilateral emisphere 24 h after pMCAO (n = 5). (B) CXCL16 mRNA quantification in in vitro OGD. qRT-PCR analysis in primary hippocampal cultures after 90 min of OGD (n = 11) and after 2 h of recovery (n = 8). Data are expressed as mRNA fold increase normalized to β-actin. Results represent the mean ± SEM. Statistical analysis: Student’s t-test ** p < 0.001 (A); one-way ANOVA followed by Dunn’s post-hoc test * p < 0.05 (B).
CXCL16 is Released from Microglia and Astrocytes Upon CX3CL1 Stimulation
To investigate if CXCL16 could be released from glia upon treatment with the neuroprotective chemokine CX3CL1, conditioned media (c.m.) from microglia/astrocytes co-cultures (in transwell system, see Section Materials and Methods) treated or not with CX3CL1 (100 nM, 18 h), were analyzed for CXCL16 presence. Data shown in Figure 3A revealed a significant increase in soluble CXCL16 upon CX3CL1 treatment (n = 12; p < 0.001). Membrane fractions of both microglia and astrocytes were also analyzed and we found that after CX3CL1 treatment the mature form of CXCL16 was significantly increased (Figures 3B,C top panels; n = 6; p < 0.05). Interestingly, we also observed an increased expression of CXCL16 mRNA upon CX3CL1 stimulation both in microglia and astrocytes (Figures 3B,C bottom panels; n= 6–8; p < 0.05).
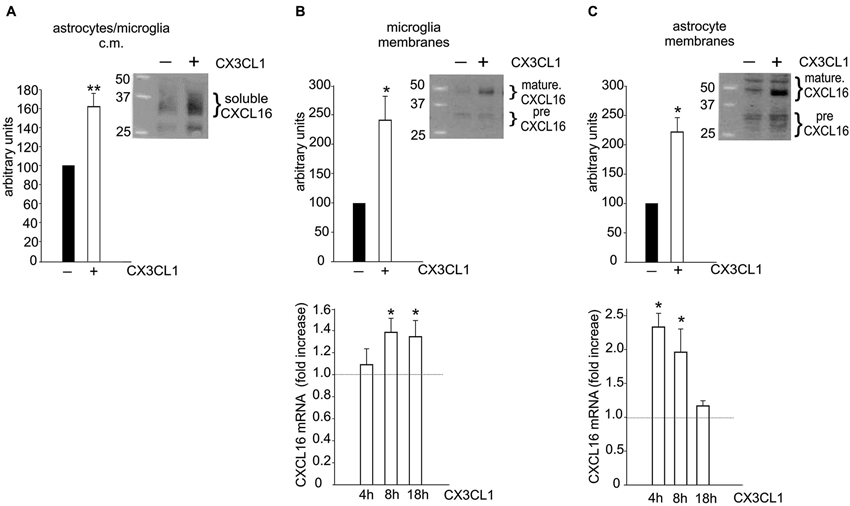
Figure 3. Glial cells increase CXCL16 expression upon CX3CL1 stimulation. (A) CX3CL1 induces the release of soluble CXCL16 from glial cells. Western-blot analysis for CXCL16 protein in the conditioned medium (c.m.) of astrocytes-microglia co-cultures stimulated or not with CX3CL1 (100 nM, 18 h). Representative blot is shown (n = 12). (B–C) CXCL16 up-regulation in microglia (B) and astrocytes (C) following CX3CL1 stimulation. Top: Western-blot analysis for mature and precursor (pre) CXCL16 species (Gough et al., 2004) in unstimulated and stimulated cells derived from co-cultures experiments. Representative blots are shown (n = 6). Bottom: qRT-PCR analysis for CXCL16 mRNA expression in microglia (B) and astrocytes (C) derived from co-cultures experiments treated or not with CX3CL1 for 4, 8, 18 h (n = 8). Data are expressed as mRNA fold increase normalized to GAPDH. Results represent the mean ± SEM. Statistical analysis: Student t-test * p < 0.05; ** p < 0.001 (A, B–C top panels); one-way ANOVA followed by Dunn’s post hoc test (B–C) bottom panels * p < 0.05.
CXCL16 is a Mediator of CX3CL1-Induced Neuroprotection Against Glu Excitotoxicity
Since CXCL16 acts on its unique receptor CXCR6, we performed experiments on the neuroprotective activity of CX3CL1 against Glu excitotoxicity in hippocampal cultures obtained from cxcr6gfp/gfp mice, to investigate the possible involvement of CXCL16 in CX3CL1-induced neuroprotection. As reported in Figure 4, CX3CL1 was less effective in preventing cell death in hippocampal cultures derived from mice that lack CXCL16 signaling, compared with wt cultures. In particular, statistical analysis (two-way ANOVA) indicated a significant interaction between genotypes and treatment, with a main effect of treatments (p = 0.004). In cxcr6gfp/gfp mice, a significant difference between Glu and both control and Glu/CX3CL1 treated cells was observed (n = 8–10; p < 0.05).
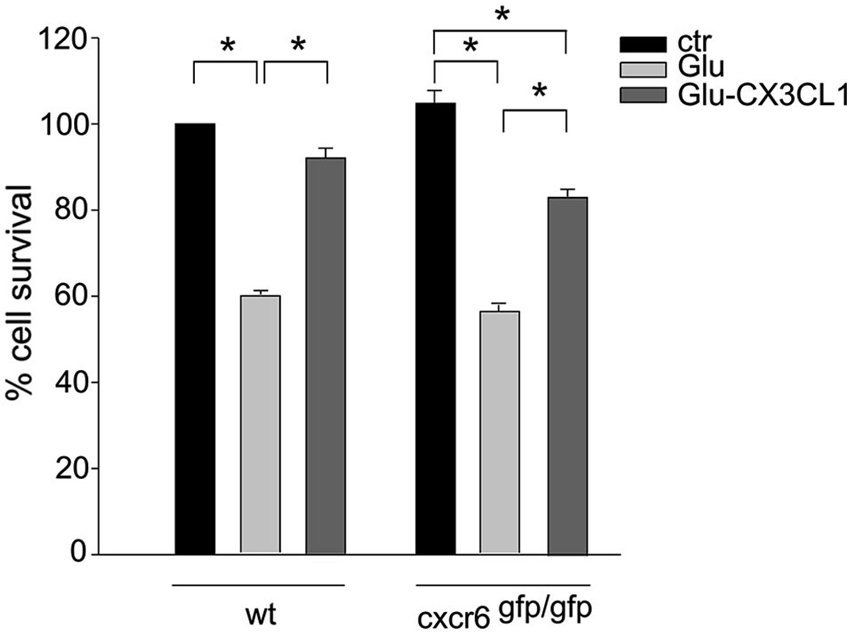
Figure 4. CXCL16 contributes to CX3CL1 neuroprotection against Glu toxic insult. Glu-excitotoxic experiments were performed in wt and cxcr6 gfp/gfp mice hippocampal cultures treated or not with CX3CL1 (n = 8–10). Data are expressed as percentage of viable cells in treated cultures taking as 100% the number of viable cells in wt control condition. Results represent the mean ± SEM. Statistical analysis: two-way ANOVA followed by Dunn’n post-hoc test * p < 0.05.
The Mediators of CXCL16 Neuroprotective Activity are Active Players in CX3CL1 Neuroprotection
The activity of CXCL16 and A3R on astrocytes and the consequential release of CCL2 are key events in CXCL16 induced neuroprotection (Rosito et al., 2012). To further corroborate the CX3CL1-CXCL16 connection in neuroprotection, we analyzed the contribution of A3R and CCL2 in this mechanism. We performed excitotoxic experiments in hippocampal cultures derived from A3R–/– mice: at difference with wt, in A3R–/– cultures CX3CL1 was less effective in preventing cell death (Figure 5A). Statistical analysis (two-way ANOVA) indicated a significant interaction between genotypes and treatment (p = 0.025), with a main effect of treatments. Both in wt and A3R–/– animals, a significant difference between Glu and both control and Glu/CX3CL1 treated cells was observed (post hoc analysis, n = 6; p < 0.05). According to this result, the A3R inhibitor MRS1523 reduced CX3CL1 neuroprotection (n = 5; p < 0.05; Figure 5B). Moreover, as reported in Figure 5C in the presence of neutralizing αCCL2 Ab (but not with control IgG, both used at 3 μg/ml), CX3CL1 was not able to induce neuroprotection (n = 4; p < 0.05).
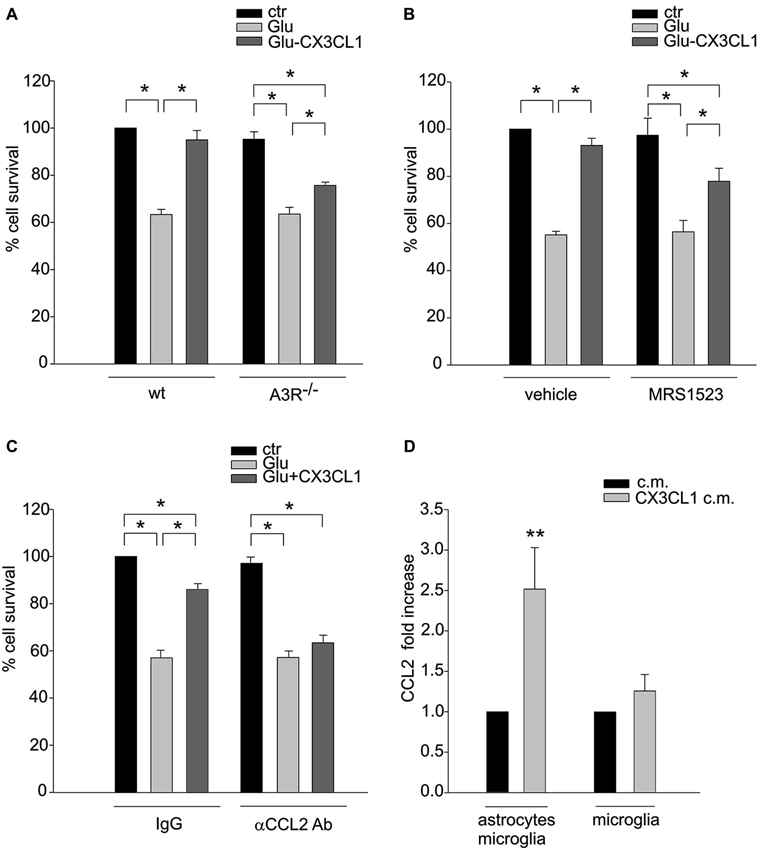
Figure 5. A3R activity and astrocytic CCL2 concur to CX3CL1 neuroprotection. (A) Genetic deletion of A3R reduces CX3CL1 neuroprotection. Glu-excitotoxic experiments were performed in hippocampal cells derived from wt or A3R–/– mice (n = 6). Data are expressed as percentage of viable cells in treated cultures taking as 100% the number of viable cells in wt control condition. (B) Pharmacological inhibition of A3R reduces CX3CL1 neuroprotection. Primary hippocampal cells were treated with A3R specific antagonists MRS1523 and used for Glu-excitotoxic experiments (n = 5). Data are expressed as percentage of viable cells in treated cultures taking as 100% the number of viable cells in vehicle control condition. (C) Neutralization of CCL2 activity prevents CX3CL1 neuroprotection. Glu-excitotoxic experiment were performed in hippocampal cultures in the presence of neutralizing αCCL2 Ab (3 μg/ml) or control IgG (3μg/ml) (n = 4–6). Data are expressed as percentage of viable cells in treated cultures taking as 100% the number of viable cells in IgG control condition. (D) CX3CL1 triggers CCL2 release from astrocytes. Microglia-astrocytes co-cultures or microglia alone were treated with CX3CL1or vehicle, and the c.m. were collected after 18 h. CCL2 levels in the media were measured by ELISA (n = 7–11). Results represent the mean ± SEM. Statistical analysis: two-way ANOVA followed by Holm-Sidak post-hoc test * p < 0.05 (A); one-way ANOVA followed by Holm-Sidak post-hoc test * p < 0.05 (B–C); Student’s t-test ** p < 0.001 (D).
To verify the hypothesis that CX3CL1 could also induce the release of CCL2, that concur to neuroprotection, we stimulated microglia-astrocytes co-cultures or microglia with CX3CL1 for 18 h and measured CCL2 level in the c.m. Figure 5D shows a basal release of CCL2 that increases upon CX3CL1 stimulation in microglia-astrocyte co-culture (n = 11; p < 0.001 Rank sum Test) but not in microglia alone (n = 7; p = 0.7 Rank sum Test), suggesting that CX3CL1 acting on microglia, induces the release of CCL2 from astrocytes.
CX3CL1 Neuroprotection Against pMCAO is Reduced in cxcr6gfp/gfp Mice
CX3CL1 is neuroprotective in pMCAO (Cipriani et al., 2011). To further confirm that CXCL16 contributes to CX3CL1 neuroprotection, wt and cxcr6gfp/gfp mice were i.c.v. injected with soluble CX3CL1, 30 min before induction of pMCAO: as reported in Figure 6, two-way ANOVA analysis reveals a significant difference between genotypes (p < 0.001), being the ischemic volume higher in cxcr6gfp/gfp mice vs. wt animals. CX3CL1 administration was effective in reducing ischemic volume in both genotypes (p < 0.05) but the reduction observed in cxcr6gfp/gfp mice was less pronounced being 10.9% (n = 6) vs. 25.6 % in wt (n = 4).
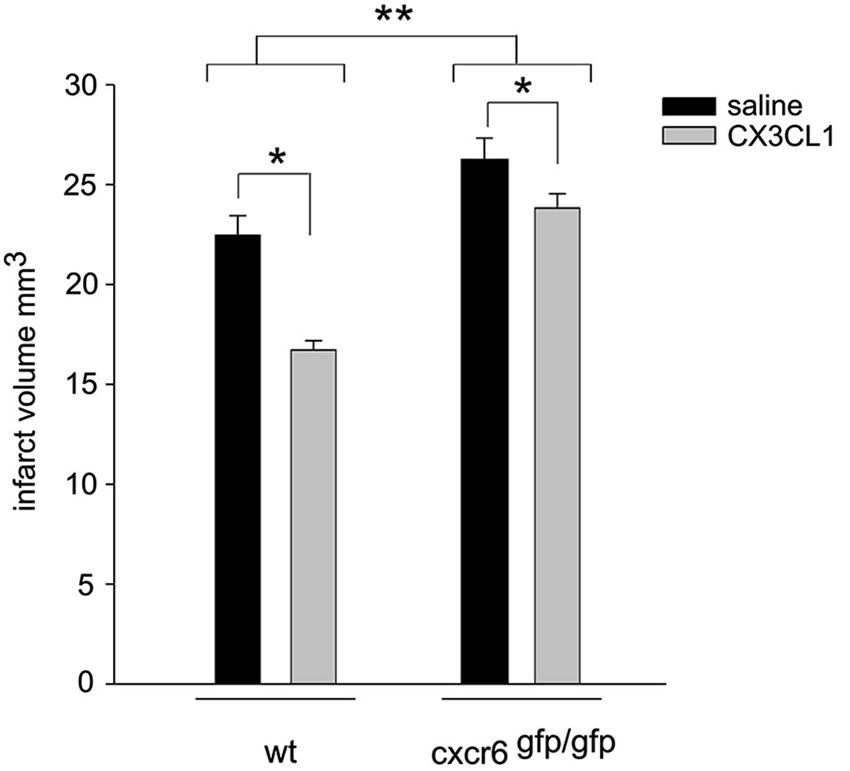
Figure 6. CX3CL1 neuroprotection against pMCAO is reduced in cxcr6 gfp/gfp mice. Mice were i.c.v. injected with saline or CX3CL1 (70 pmol) 30 min before pMCAO and analyzed for ischemic volume 24 h later (n = 4–10). Results represent the mean ± SEM. Statistical analysis: two-way ANOVA followed by Holm-Sidak post-hoc test * p < 0.05; ** p < 0.001.
Discussion
Glial cells, long thought to act as a mere “support” network, have been gaining increasing attention as crucial protagonists in a variety of neural functions including information processing but also cell viability. In the present paper we describe for the first time the ability of trasmembrane chemokines CX3CL1 and CXCL16 to drive molecular interplay between neurons, microglia and astrocytes in determining the neuroprotection against pMCAO and excitotoxic damage, showing that a concerted action of these cells is important to determine neuronal survival upon exposure to high level of Glu, a condition that normally occurs following ischemia (Castillo et al., 1996) but also in traumatic brain injuries (Zauner et al., 1996) or chronic neurodegenerative diseases (Shaw et al., 1995; Hallett and Standaert, 2004; Lipton, 2005).
In line with previous in vitro findings, we demonstrated that exogenous administration of soluble CXCL16 reduced brain ischemic volume following pMCAO; moreover we found that upon ischemic insult CXCL16 expression is increased in the ischemic hemisphere and that endogenous CXCL16 signaling is important per se to counteract brain damage, since in cxcr6gfp/gfp mice there is a significant increase in brain ischemic volume upon pMCAO. All together, these data indicate that CXCL16 represents a physiological mediator of self-protective mechanisms engaged by brain parenchyma to restrain cell damage following toxic insult. Upon brain ischemia, there is the simultaneous activation of destructive pathways leading to cell death but also of local protective mechanisms. Although the damaging effectors apparently prevail, evidences suggest that concomitant self-protective mechanisms might limit the resulting damage and set the stage for tissue repair and reorganization (Moskowitz et al., 2010; Iadecola and Anrather, 2011; Figure 7). Thus unveiling the molecular players that act in self-protective mechanism might provide new opportunity to treat brain pathologies.
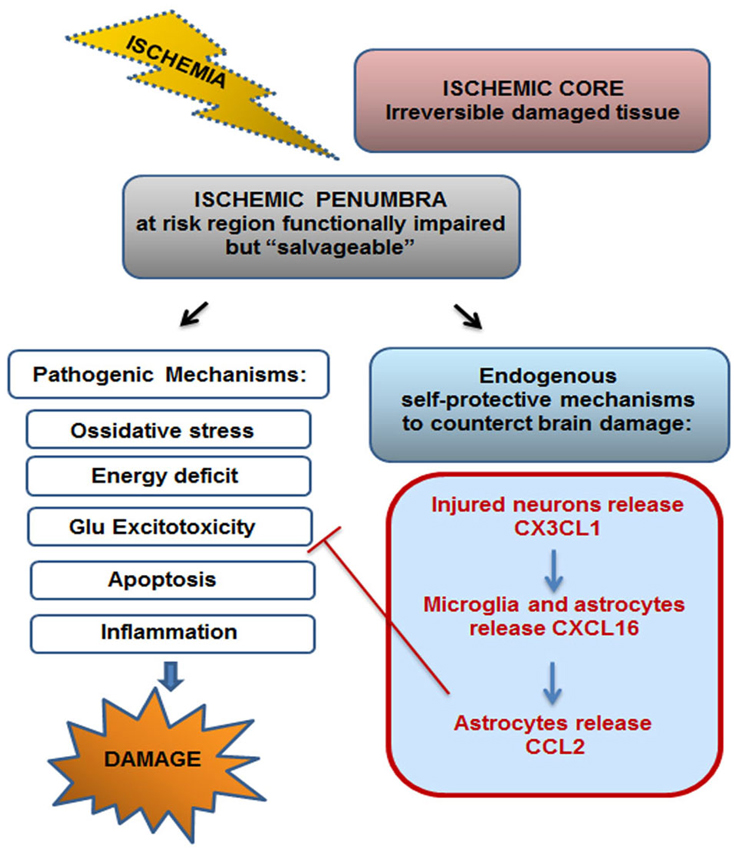
Figure 7. Endogenous self-protective mechanism drived by CX3CL1–CXCL16-CCL2 upon ischemia. Ischemic insult drives irreversible brain damage in ischemic core, while in the surrounding region (ischemic penumbra) brain tissue results to be functionally compromised but potentially “salvageable”. Upon ischemic insult both pathogenic and self-protective mechanisms occur in the penumbra. CX3CL1-CXCL16-CCL2 cascade, triggered by neurons-microglia-astrocytes cross-talk, represents one endogenous self-protective mechanism that contributes to the restriction of brain damage by counteracting Glu induced excitotoxicity.
Damaged neurons respond to neurotoxic insults releasing soluble factors that can be sensed by surrounding glia: CX3CL1, a chemokine selectively expressed by neurons in the nervous system, is one of such mediators being upregulated (Tarozzo et al., 2002; Zhu et al., 2009), cleaved and released upon ischemia and excitotoxic insult (Chapman et al., 2000; Limatola et al., 2005; Noda et al., 2011 ) and being able to drive neuroprotection (Limatola et al., 2005; Lauro et al., 2010; Cipriani et al., 2011). We reported here that, upon CX3CL1 stimulation, glial cells produce and release CXCL16, important for CX3CL1 neuroprotective effect. We speculate that CX3CL1 released from neurons upon ischemia might drive microglia-astrocytes cross-talk leading to CXCL16 increase. These data further corroborate the idea that, although the only direct targets of CX3CL1 are microglial cells, its neuroprotective effects are mediated by engagement of astrocytes that concur to limit excitotoxic cell death with the synergistic activity of adenosine (Catalano et al., 2013).
We do not know the mechanism that leads to the release of CXCL16 from glia, however it has been recently reported that activation of the purinergic receptor P2X7 induces CXCL16 shedding from RPMI8226 myeloma B cells (Pupovac et al., 2013). Hippocampal cells stimulation with CX3CL1 induces an increase in extracellular adenosine probably derived from released ATP, the effect being specifically blocked by the treatment with the ectonucleotidase inhibitor alpha-beta-methyleneadenosine 5′-diphosphate sodium salt (AOPCP) (Lauro et al., 2010). Since both astrocytes and microglia express P2X7 receptors, it could be hypothesized that CX3CL1 induces ATP release from microglia that, acting on P2X7 receptors, induces CXCL16 shedding from surrounding glial cells. A role for P2X7 in the release of neuroprotective mediators is in agreement with previous data showing that P2X7 activation reduces excitotoxic neuronal death, through TNF-α shedding from microglia (Suzuki et al., 2004).
Adenosine modulates neuron-glia communication (Boison et al., 2010) and can mediate neuroprotective effects through the activity of its own receptors: in this regards the activity of A1R is crucial to allow neuroprotection driven by CX3CL1, IL-6, oncostatin M (OSM), BDNF and erythropoietin (EPO) (Biber et al., 2008; Lauro et al., 2010; Moidunny et al., 2010). Also A3R activity can mediate neuroprotection since it has been shown that hypoxic conditions determine a wider neurodegeneration in A3R–/– mice (Fedorova et al., 2003) and i.c.v. injection of A3R selective agonist in mice reduces brain ischemic volume (Chen et al., 2006). In the present work, we confirmed an increased ischemic volume in A3R–/– mice compared to wt mice and found that the ability of CXCL16 to reduce ischemic volume is less pronounced in these mice. This is in line with our previous in vitro findings, where we have shown that soluble CXCL16 is able to promote neuronal survival against excitotoxic damage depending on A3R activity (Rosito et al., 2012), and in particular the synergistic activity of CXCL16 and A3R on astrocytes causes the release of CCL2 that act as a key mediator of neuroprotection.
We speculate that upon ischemic insult, CXCL16 released from glia concurs to endogenous neuroprotective mechanism elicited by neuronal CX3CL1 since we found that CX3CL1-induced neuroprotection was reduced in cxcr6gfp/gfp mice; both genetic and pharmacological inactivation of A3R reduces CX3CL1 neuroprotection against Glu excitotoxic insult; CX3CL1 is able to increase the release of CCL2 from astrocytes; CCL2 activity is important for CX3CL1 protective effect.
Nevertheless, our data showed that impairment of CXCL16 or A3R signaling in transgenic animals reduced, but did not totally prevented CX3CL1 neuroprotection, indicating that the mechanism we here proposed represent only a portion of the neuroprotective mechanisms driven by CX3CL1.
The involvement of A1R in CX3CL1 neuroprotection (Lauro et al., 2010) strongly suggests that there must be at least another mechanism, independent from CXCL16, important to protect cells from Glu excitoxicity: accordingly we have recently published that the activity of Glu transporter GLT1 on astrocytes is increased by CX3CL1, with mechanisms requiring A1R activation and this event is also crucial for CX3CL1 neuroprotection (Catalano et al., 2013).
In conclusion, the present work highlights the role played by chemokines as key endogenous modulators of the cross-talk between cells of brain parenchyma, that drive physiological neuroprotective mechanisms. In particular we demonstrated the existence of chemokine induced chemokine release (CX3CL1-CXCL16-CCL2) mechanism that involves neurons, microglia and astrocytes and that represents an endogenous self-protective mechanism that upon brain ischemia can limit cell damage in the ischemic penumbra, by counteracting neuronal cell death due to Glu excitotoxicity (Figure 7).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thanks Prof. Bertil B. Fredholm for critical reading and suggestions and for providing us A3R–/– mice.
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) Investigator Grant IG 12774.
References
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Biber, K., Neumann, H., Inoue, K., and Boddeke, H. W. (2007). Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 30, 596–602. doi: 10.1016/j.tins.2007.08.007
Biber, K., Pinto-Duarte, A., Wittendorp, M. C., Dolga, A. M., Fernandes, C. C., Von Frijtag Drabbe Künzel, J., et al. (2008). Interleukin-6 upregulates neuronal adenosine A1 receptors: implications for neuromodulation and neuroprotection. Neuropsychopharmacology 33, 2237–2250. doi: 10.1038/sj.npp.1301612
Boison, D., Chen, J. F., and Fredholm, B. B. (2010). Adenosine signaling and function in glial cells. Cell Death Differ. 17, 1071–1082. doi: 10.1038/cdd.2009.131
Burnstock, G., Fredholm, B. B., and Verkhratsky, A. (2011). Adenosine and ATP receptors in the brain. Curr. Top. Med. Chem. 11, 973–1011. doi: 10.2174/156802611795347627
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924. doi: 10.1038/nn1715
Castillo, J., Dávalos, A., Naveiro, J., and Noya, M. (1996). Neuroexcitatory amino acids and their relation to infarct size and neurological deficit in ischemic stroke. Stroke 27, 1060–1065. doi: 10.1161/01.str.27.6.1060
Catalano, M., Lauro, C., Cipriani, R., Chece, G., Ponzetta, A., Di Angelantonio, S., et al. (2013). CX3CL1 protects neurons against excitotoxicity enhancing GLT-1 activity on astrocytes. J. Neuroimmunol. 263, 75–82. doi: 10.1016/j.jneuroim.2013.07.020
Chapman, G. A., Moores, K., Harrison, D., Campbell, C. A., Stewart, B. R., and Strijbos, P. J. (2000). Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J. Neurosci. 20, 87–91.
Chen, G. J., Harvey, B. K., Shen, H., Chou, J., Victor, A., and Wang, Y. (2006). Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J. Neurosci. Res. 84, 1848–1855. doi: 10.1002/jnr.21071
Chen, C. J., Liao, S. L., and Kuo, J. S. (2000). Gliotoxic action of glutamate on cultured astrocytes. J. Neurochem. 75, 1557–1565. doi: 10.1046/j.1471-4159.2000.0751557.x
Cipriani, R., Villa, P., Chece, G., Lauro, C., Paladini, A., Micotti, E., et al. (2011). CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 31, 16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011
Dénes, A., Ferenczi, S., Halász, J., Környei, Z., and Kovács, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721. doi: 10.1038/jcbfm.2008.64
Fedorova, I. M., Jacobson, M. A., Basile, A., and Jacobson, K. A. (2003). Behavioral characterization of mice lacking the A3 adenosine receptor: sensitivity to hypoxic neurodegeneration. Cell. Mol. Neurobiol. 23, 431–447. doi: 10.1023/A:1023601007518
Franke, H., Verkhratsky, A., Burnstock, G., and Illes, P. (2012). Pathophysiology of astroglial purinergic signalling. Purinergic Signal. 8, 629–657. doi: 10.1007/s11302-012-9300-0
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Gough, P. J., Garton, K. J., Wille, P. T., Rychlewski, M., Dempsey, P. J., and Raines, E. W. (2004). A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand 16. J. Immunol. 172, 3678–3685. doi: 10.4049/jimmunol.172.6.3678
Hallett, P. J., and Standaert, D. G. (2004). Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol. Ther. 102, 155–174. doi: 10.1016/s0163-7258(04)00049-x
Hamby, M. E., and Sofroniew, M. V. (2010). Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7, 494–506. doi: 10.1016/j.nurt.2010.07.003
Huang, D., Shi, F. D., Jung, S., Pien, G. C., Wang, J., Salazar-Mather, T. P., et al. (2006). The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 20, 896–905. doi: 10.1096/fj.05-5465com
Iadecola, C., and Anrather, J. (2011). Stroke research at a crossroad: asking the brain for directions. Nat. Neurosci. 14, 1363–1368. doi: 10.1038/nn.2953
Lauro, C., Cipriani, R., Catalano, M., Trettel, F., Chece, G., Brusadin, V., et al. (2010). Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against Glu-induced death. Neuropsychopharmacology 35, 1550–1559. doi: 10.1038/npp.2010.26
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 177, 2549–2562. doi: 10.2353/ajpath.2010.100265
Limatola, C., Lauro, C., Catalano, M., Ciotti, M. T., Bertollini, C., Di Angelantonio, S., et al. (2005). Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J. Neuroimmunol. 166, 19–28. doi: 10.1016/j.jneuroim.2005.03.023
Lipton, S. A. (2005). The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr. Alzheimer Res. 2, 155–165. doi: 10.2174/1567205053585846
Liu, W., Tang, Y., and Feng, J. (2011). Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 89, 141–146. doi: 10.1016/j.lfs.2011.05.011
Moidunny, S., Dias, R. B., Wesseling, E., Sekino, Y., Boddeke, H. W., Sebastião, A. M., et al. (2010). Interleukin-6-type cytokines in neuroprotection and neuromodulation: oncostatin M, but not leukemia inhibitory factor, requires neuronal adenosine A1 receptor function. J. Neurochem. 114, 1667–1677. doi: 10.1111/j.1471-4159.2010.06881.x
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatment. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Noda, M., Doi, Y., Liang, J., Kawanokuchi, J., Sonobe, Y., Takeuchi, H., et al. (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 286, 2308–2319. doi: 10.1074/jbc.M110.169839
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A., and Bessis, A. (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U S A 109, E197–E205. doi: 10.1073/pnas.1111098109
Paxinos, G., and Franklin, K. B. J. (2004). The Mouse Brain in Stereotoxic Coordinates. Houston, Texas: Gulf Professional Publishing.
Pupovac, A., Foster, C. M., and Sluyter, R. (2013). Human P2X7 receptor activation induces the rapid shedding of CXCL16. Biochem. Biophys. Res. Commun. 432, 626–631. doi: 10.1016/j.bbrc.2013.01.134
Rosito, M., Deflorio, C., Limatola, C., and Trettel, F. (2012). CXCL16 orchestrates adenosine A3 receptor and MCP-1/CCL2 activity to protect neurons from excitotoxic cell death in the CNS. J. Neurosci. 32, 3154–3163. doi: 10.1523/JNEUROSCI.4046-11.2012
Salvatore, C. A., Tilley, S. L., Latour, A. M., Fletcher, D. S., Koller, B. H., and Jacobson, M. A. (2000). Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J. Biol. Chem. 275, 4429–4434. doi: 10.1074/jbc.275.6.4429
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Shaw, P. J., Forrest, V., Ince, P. G., Richardson, J. P., and Wastell, H. J. (1995). CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration 4, 209–216. doi: 10.1006/neur.1995.0026
Soriano, S. G., Amaravadi, L. S., Wang, Y. F., Zhou, H., Yu, G. X., Tonra, J. R., et al. (2002). Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 125, 59–65. doi: 10.1016/s0165-5728(02)00033-4
Storini, C., Bergamaschini, L., Gesuete, R., Rossi, E., Maiocchi, D., and De Simoni, M. G. (2006). Selective inhibition of plasma kallikrein protects brain from reperfusion injury. J. Pharmacol. Exp. Ther. 318, 849–854. doi: 10.1124/jpet.106.105064
Suzuki, T., Hide, I., Ido, K., Kohsaka, S., Inoue, K., and Nakata, Y. (2004). Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J. Neurosci. 24, 1–7. doi: 10.1523/jneurosci.3792-03.2004
Tarozzo, G., Campanella, M., Ghiani, M., Bulfone, A., and Beltramo, M. (2002). Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur. J. Neurosci. 15, 1663–1668. doi: 10.1046/j.1460-9568.2002.02007.x
Unutmaz, D., Xiang, W., Sunshine, M. J., Campbell, J., Butcher, E., and Littman, D. R. (2000). The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J. Immunol. 165, 3284–3292. doi: 10.4049/jimmunol.165.6.3284
Volontè, C., Ciotti, M. T., and Battistini, L. (1994). Development of a method for measuring cell number: application to CNS primary neuronal cultures. Cytometry 17, 274–276. doi: 10.1002/cyto.990170311
Zauner, A., Bullock, R., Kuta, A. J., Woodward, J., and Young, H. F. (1996). Glutamate release and cerebral blood flow after severe human head injury. Acta Neurochir. Suppl. 67, 40–44. doi: 10.1007/978-3-7091-6894-3_9
Keywords: CX3CL1, CXCL16, CCL2, A3R, glia cross-talk, neuroprotection, ischemia, excitotoxicity
Citation: Rosito M, Lauro C, Chece G, Porzia A, Monaco L, Mainiero F, Catalano M, Limatola C and Trettel F (2014) Trasmembrane chemokines CX3CL1 and CXCL16 drive interplay between neurons, microglia and astrocytes to counteract pMCAO and excitotoxic neuronal death. Front. Cell. Neurosci. 8:193. doi: 10.3389/fncel.2014.00193
Received: 06 May 2014; Accepted: 23 June 2014;
Published online: 10 July 2014.
Edited by:
Shawn Hayley, Carleton University, CanadaReviewed by:
Jiong Shi, Barrow Neurological Institute, USAShaohua Yang, University of North Texas Health Science Center, USA
Copyright © 2014 Rosito, Lauro, Chece, Porzia, Monaco, Mainiero, Catalano, Limatola and Trettel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flavia Trettel, Department of Physiology and Pharmacology, Istituto Pasteur Fondazione Cenci Bolognetti, Sapienza University of Rome, P.le Aldo Moro 5, Rome, Italy e-mail:ZmxhdmlhLnRyZXR0ZWxAdW5pcm9tYTEuaXQ=