 Gabriele Deidda
Gabriele Deidda Ignacio F. Bozarth
Ignacio F. Bozarth Laura Cancedda
Laura Cancedda- Department of Neuroscience and Brain Technologies, Istituto Italiano di Tecnologia, Genova, Italy
During mammalian ontogenesis, the neurotransmitter GABA is a fundamental regulator of neuronal networks. In neuronal development, GABAergic signaling regulates neural proliferation, migration, differentiation, and neuronal-network wiring. In the adult, GABA orchestrates the activity of different neuronal cell-types largely interconnected, by powerfully modulating synaptic activity. GABA exerts these functions by binding to chloride-permeable ionotropic GABAA receptors and metabotropic GABAB receptors. According to its functional importance during development, GABA is implicated in a number of neurodevelopmental disorders such as autism, Fragile X, Rett syndrome, Down syndrome, schizophrenia, Tourette's syndrome and neurofibromatosis. The strength and polarity of GABAergic transmission is continuously modulated during physiological, but also pathological conditions. For GABAergic transmission through GABAA receptors, strength regulation is achieved by different mechanisms such as modulation of GABAA receptors themselves, variation of intracellular chloride concentration, and alteration in GABA metabolism. In the never-ending effort to find possible treatments for GABA-related neurological diseases, of great importance would be modulating GABAergic transmission in a safe and possibly physiological way, without the dangers of either silencing network activity or causing epileptic seizures. In this review, we will discuss the different ways to modulate GABAergic transmission normally at work both during physiological and pathological conditions. Our aim is to highlight new research perspectives for therapeutic treatments that reinstate natural and physiological brain functions in neuro-pathological conditions.
Introduction
Gamma-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the mature brain by mainly binding to chloride (Cl−) and bicarbonate (HCO3−)-permeable GABAA receptors (GABAAR; Mohler, 2007; Hubner and Holthoff, 2013) and metabotropic GABAB receptors. The regulation of neuronal activity by GABA depends on its metabolism, expression and activity of the pre- and postsynaptic receptors, and on the electrochemical gradient of Cl− across the cellular membrane.
GABA is synthesized from glutamate as a substrate by two glutamic acid decarboxylase enzymes (GAD) of different molecular weight: GAD67 and GAD65 (encoded by the Gad1 and Gad2 genes, respectively; Pinal and Tobin, 1998). The two isoforms show striking differences in their developmental expression (Kiser et al., 1998; Buddhala et al., 2009), subcellular localization (Dupuy and Houser, 1996; Buddhala et al., 2009), enzymatic activity (Battaglioli et al., 2003; Fenalti et al., 2007), and gene regulation (Feldblum et al., 1993; Pinal and Tobin, 1998; Buddhala et al., 2009). In particular, GAD67 has a cytosolic localization mostly in the neuronal soma, and it provides basal level of GABA synthesis. Conversely, GAD65 is preferentially located in the axonal terminal and it provides additional supply of GABA in condition of metabolic demand (Asada et al., 1997; Kash et al., 1997; Namchuk et al., 1997; Buddhala et al., 2009).
Once synthesized, vesicular GABA transporters (VGATs), which are embedded in presynaptic vesicular membranes, use the electrochemical gradient for H+ to shuffle and pack GABA into small synaptic vesicles (Roth et al., 2012). Upon fusion of the synaptic vesicles to the cell membrane due to incoming action potentials, GABA is released in the synaptic cleft where it acts on ionotropic GABAA and GABAC, as well as metabotropic GABAB receptors. The magnitude and direction of the ionic current through GABAARs exquisitely depends on its driving force, defined as the difference between the electrochemical equilibrium potential of Cl− anions (reversal potential, ECl) and the resting membrane potential of the neuron (Vm). If this difference is positive or negative, there will be a net flux of Cl− anions through the plasma membrane following GABAAR opening, and this will result in a change in the membrane potential of the neuron. In particular, the net flux of Cl− anions through GABAAR (i.e., toward inside or outside the cell) critically hinges on its intracellular concentration ([Cl−]i). In neurons, two main chloride cotransporters are responsible for setting [Cl−]i. The Na+/K+/Cl− cotransporter NKCC1 (Blaesse et al., 2009), which imports Cl− into the neuron, and the K+/Cl− cotransporter KCC2, which exports Cl− out of the neuron (Rivera et al., 1999; Sernagor et al., 2010; Kahle et al., 2013). When ECl is close to Vm, GABA will exert its inhibitory action by a shunting inhibitory mechanism. Indeed, the local increase in membrane GABAAR conductance will “hold” the neuron at the ECl, reducing the amplitude of subsequent excitatory postsynaptic potentials (following Ohm's law) and thus shunting any excitatory input (Gonzalez-Burgos et al., 2011).
The termination of GABA action at synapses depends on GABA reuptake into nerve terminals and astrocytes by GABA transporters located at the cell membrane (GATs; Lee et al., 2006b). Finally, the catabolism of GABA depends on the action of GABA transaminase enzyme (GABA-T) and succinate semialdehyde dehydrogenase enzyme (SSADH), which convert GABA into intermediates of the Krebs cycle and substrates for new production of glutamate.
During early development, GABA is depolarizing and mostly excitatory due to high [Cl−]i, and it plays a key role by regulating a number of processes including migration, morphological maturation and differentiation of neurons (Ben-Ari et al., 2007; Wang and Kriegstein, 2009; Ben-Ari et al., 2012). Accordingly, GABAergic signaling has been implicated in a number of neurodevelopmental disorders, such as autism (Tabuchi et al., 2007; Coghlan et al., 2012), Fragile X (Curia et al., 2009; Coghlan et al., 2012), Rett Syndrome (Medrihan et al., 2008; Coghlan et al., 2012), Down Syndrome (Chakrabarti et al., 2010; Costa and Scott-McKean, 2013), Schizophrenia (Lewis et al., 2012), Tourette Syndrome (Kalanithi et al., 2005; Di Cristo, 2007), and Neurofibromatosis type I (Costa et al., 2002; Diggs-Andrews and Gutmann, 2013). Unfortunately, in most cases GABAergic drugs are dangerous because either they silence the neuronal network activity in a developing brain or, more often, they trigger epilepsy in patients with neurodevelopmental disorders, who -similarly to infants- (Connell et al., 1989; Sankar and Painter, 2005; Briggs and Galanopoulou, 2011; Loscher et al., 2013) are at higher risk of seizures per se (Rissman and Mobley, 2011; Coghlan et al., 2012; Ostendorf et al., 2013; Williams et al., 2013). Finding viable therapeutic treatments targeting GABAergic transmission in neurodevelopmental disorders is thus both of great importance and a big challenge. Addressing this issue implies the study of the fine modulation of GABAergic transmission during physiological conditions as a starting point to tackle dysfunctional transmission in neurodevelopmental disorders. The overall goal of such a challenging approach is to find a physiologically subtler way to safely modulate GABAergic signaling without hurting the delicate equilibrium of the neuronal-network activity in a developing brain.
In the first part of this review, we will discuss different ways to modulate GABAergic transmission naturally at work during physiological conditions. In the second part, we will discuss how GABAergic transmission is affected in disease, and we will give examples of therapeutic approaches that are currently utilized to reinstate brain functions in neuro-pathological conditions.
Modulation of GABAergic Transmission Under Normal Physiological Conditions
Modulation of GABAergic transmission is achieved under different physiological conditions in the brain in diverse ways, ranging from regulation of GABAAR expression/activity and GABA metabolism to regulation of intracellular chloride concentration.
Modulation of GABAA Receptor Function
GABAARs are composed of 5 protein subunits belonging to 19 different classes (α1–6, β1–3, γ1–3, δ, ϵ, θ, π, ρ1–3; Simon et al., 2004; Luscher et al., 2011; Hubner and Holthoff, 2013; Fritschy and Panzanelli, 2014). The assembly of GABAARs as heteropentamers produces complex heterogeneity in their structure, which is the major determinant of their pharmacological profile and kinetics (Barnard et al., 1998; Luscher et al., 2011; Fritschy and Panzanelli, 2014). In adult mammals, the most common configuration at the synapse comprises two α1, two β2, and one γ2 subunits. This composition guarantees fast and short-lasting (phasic) inhibition mediated by fast desensitizing postsynaptic receptors. Interestingly, GABAAR composition changes during ontogenesis, in order to meet the needs of continuously developing neuronal networks. During early development, when synaptogenesis is not fully established, GABA signaling through GABAARs is mainly achieved at extra synapticsites. Extrasynaptic GABAARs contain α5 or δ, as well as β2/3 and γ subunits, allowing “ambient” GABA to elicit long-lasting (tonic) currents that result in persistent depolarization in young neurons (Cellot and Cherubini, 2013). This tonic activation of extrasynaptic GABAARs is critical at that time, as it regulates cell migration, proliferation, neurite growth and synapse formation (Ben-Ari et al., 2007; Wang and Kriegstein, 2009). Interestingly, when synaptogenesis begins, α5 subunits are downregulated and its neuronal localization switches from the soma to the dendritic compartment in the hippocampus (Ramos et al., 2004).
Upon the establishment of structural synapses, functional communication between neurons requires GABAARs localized at the synapses. At that time, the first formation of primitive functional neuronal circuits follows the hebbian rule “neurons that fire together wire together” (Ben-Ari et al., 2007). Consequently, GABAAR kinetics are determined by the widespread distribution of α2/3 subunits, which ensures long-lasting currents well-suited for increasing probability of concurrent spiking from different cells (Fritschy et al., 1994; Uusi-Oukari and Korpi, 2010). As development progresses, α2 and α3 subunits are downregulated, whereas α1 and α6 subunits are up-regulated (Laurie et al., 1992; Poulter et al., 1992; Uusi-Oukari and Korpi, 2010). This results in a faster decay time of GABAergic events, which allows precise setting of the temporal window for synaptic integration (Pouille and Scanziani, 2001) and synchronization of neuronal networks (Cobb et al., 1995; Whittington et al., 2011). Of note, intracellular chloride itself has been recently shown to influence the subunit composition, possibly acting as an intracellular messenger (Succol et al., 2012).
Once perinatal development is completed, production of new neurons in the adult (adult neurogenesis) remains restricted only to certain brain niches in the dentate gyrus of the hippocampus and in the olfactory bulb. Interestingly, adult neurogenesis is also under the control of GABAergic transmission (Fritschy and Panzanelli, 2014). Not surprisingly, neurogenesis in early development and in the adult share many similarities. For instance, availability of different GABAAR subunits provides a molecular mechanism to precisely control neuronal maturation in the dentate gyrus at spatio-temporal level. Indeed, neuronal progenitors cells (NPCs) express functional GABAARs composed of α5β3γ2 subunits mediating a tonic response to GABA that maintains NPCs in a quiescence state (Song et al., 2012). Interestingly, mice lacking α4 subunits mediating GABAA tonic currents, show increased proliferation and impaired dendritic maturation at early stages. Remarkably, this effect is not present in the mouse line lacking δ subunit, also mediating GABAA tonic currents (Duveau et al., 2011). Conversely, α2 subunit, mediating GABAA phasic currents, controls positioning of newborn neurons and dendritic maturation at later stages (Duveau et al., 2011). In the olfactory bulb, specific GABAAR subunits also regulate adult neurogenesis, as for example α2 subunit regulates neuronal maturation (Pallotto et al., 2012).
Once neuronal circuits are established, and generation of new neurons continues only in discrete niches, the developing brain still retains the potential to learn and respond to experience with plastic changes in neuronal connections. Again, brain plasticity is finely regulated by GABAergic transmission (Fritschy and Panzanelli, 2014). Long-term potentiation (LTP) is a long-lasting increase in the synaptic efficacy elicited by reliable experimental protocols of short, high-frequency stimulation of presynaptic fibers, and it is thus considered as an established model of learning and brain plasticity (Bliss and Collingridge, 1993; Feldman, 2009, 2012). LTP can be easily induced in different regions of the brain (Feldman, 2009, 2012), and in all these structures, GABAAR activity prevents or reduces LTP magnitude in vitro (Wigstrom and Gustafsson, 1983; Grover and Yan, 1999; Kleschevnikov et al., 2004; Harauzov et al., 2010; Arima-Yoshida et al., 2011; Contestabile et al., 2013) and in vivo (Hirai and Okada, 1993; Kaibara and Leung, 1993; Matsuyama et al., 2008; Feldman, 2012). In particular, in the visual cortex, the peculiar contribution of GABAAR signaling on LTP depends on the so-called “critical period,” a process of experience-dependent maturation during a specific temporal window after birth. Only during the critical period, but not in the adult, it is possible to induce LTP and another paradigm of plasticity in vivo called monocular deprivation (MD), consisting of a functional and an anatomical plastic rearrangement of visual neuronal circuits after a brief period of sight deprivation in one eye (Hensch, 2004; Levelt and Hubener, 2012). Interestingly, GABA controls both the opening and the closure of critical period for plasticity paradigms LTP and MD (Heimel et al., 2011; Levelt and Hubener, 2012). Importantly, critical-period plasticity depends on specific GABAARs subunits, as demonstrated by the finding that only neuronal circuits containing α1 GABAAR subunits drive cortical plasticity (Fagiolini et al., 2004; Fritschy and Panzanelli, 2014).
One important mechanism regulating trafficking of GABAARs as well as their kinetics is phosphorylation of specific α, β, and γ subunits by diverse serine/threonine or tyrosine kinases (Kittler and Moss, 2003; Vithlani et al., 2011). In particular, α4 phosphorylation by protein kinase C (PKC) increases insertion and stability of α4/β3-containing GABAAR (Abramian et al., 2010), but also decreases surface expression and activity of α4/β2/δ containing receptors, possibly suggesting that their differential regulation could be due to the expression of different β subunits (Bright and Smart, 2013). Moreover, phosphorylation of β1 subunit by cAMP-dependent kinase (PKA) decreases GABA currents, whereas β2 subunit does not respond to PKA (Brandon et al., 2002; Kittler and Moss, 2003). On the other hand, β2 subunit can be phosphorylated by Akt increasing postsynaptic GABAAR expression (Wang et al., 2003c) and by calcium/calmodulin-dependent protein kinase II (CaMKII) increasing the amplitude of spontaneous inhibitory postsynaptic currents (IPSC; Houston et al., 2008). Conversely, when phosphorylating the β3 subunit, CaMKII increases the decay time of sIPSCs (Houston et al., 2008), but also increases surface expression of α5/β3-containing receptors and tonic currents in different experimental systems (Saliba et al., 2012). Moreover, phosphorylation of the β3 subunit by PKA increases GABA currents (McDonald et al., 1998; Brandon et al., 2002). Lastly, phosphorylation of the γ2 subunit decreases the internalization rate of GABAARs and therefore increases their surface expression (Kittler et al., 2008).
Some GABAAR subunits are basally phosphorylated (Brandon et al., 2000, 2001), and some phosphatases dephosphorylate specific subunits (Jones and Westbrook, 1997; Huang and Dillon, 1998; Lu et al., 2000; Wang et al., 2003b; Jovanovic et al., 2004; Terunuma et al., 2004; Muir et al., 2010). This suggests that physiological modulation of the phosphorylation state of GABAARs is finely tuned and may regulate their function. Specifically, protein phosphatases 1α and 2A (but not 2B, i.e., calcineurin) dephosphorylate the β3 subunit of the GABAAR, leading to decrease of mIPSC amplitude in hippocampal neurons (Jovanovic et al., 2004; Terunuma et al., 2004). On the other hand, NMDA-dependent activation of calcineurin mediates long-term depression (LTD) at inhibitory synapses through γ2 subunit dephosphorylation and consequent reduction of GABAAR responses (Lu et al., 2000; Wang et al., 2003b). Possibly, GABAAR cluster dispersal is responsible for the reduction of GABAAR responses, given that NMDA-dependent activation of calcineurin and consequent GABAAR dephosphorylation increases lateral diffusion of the receptor itself (Muir et al., 2010).
The efficacy of GABAAergic transmission is also determined by the number of GABAARs at the synaptic site. As this depends on the rates of insertion and removal of the receptor themselves, endocytosis and rapid recycling (Kittler et al., 2000) or ubiquitin-dependent degradation of GABAARs (Saliba et al., 2007) can also modify the efficacy of postsynaptic inhibition. Accordingly, changes in lateral diffusion of GABAARs inside and outside of the synapse, may play a major role in the regulation of GABAAergic synaptic transmission (Triller and Choquet, 2005). Indeed, as described for glutamate receptors (Heine et al., 2008; Petrini et al., 2009), rapid exchange of desensitized receptors between synaptic and extrasynaptic site could allow a higher current to flow at the postsynaptic site.
Modulation of GABA Metabolism
Modulation of GABAergic signaling can also occur at metabolic level, including both anabolism and catabolism of GABA.
GAD enzymes synthetize GABA and they are differentially expressed during ontogenesis, probably reflecting diverse contributions to GABAergic regulation of brain activity. GAD65 and GAD67 are mainly expressed during postnatal development and in the mature brain, whereas two truncated forms of GAD67 (GAD25 and GAD44) are mainly expressed during embryonic development (Buddhala et al., 2009). GAD25 represents the regulatory amino-terminal domain of GAD67, whereas GAD44 represents the enzymatic carboxy-terminal. Although the role of GAD25 and GAD44 during embryonic development has not been understood yet, their expression -at a time when cell proliferation, migration and synaptogenesis occur- has suggested that they could play a role in these processes (Popp et al., 2009). As development progresses, GAD25 and GAD44 expression diminishes accompanied by an increased expression of the active forms of GAD -GAD65 and GAD67- and a concomitant increase of GABA synthesis (Varju et al., 2002).
In particular, GAD67 is expressed earlier than GAD65 (Kiser et al., 1998), it is ubiquitously present in the neuron (Esclapez et al., 1994), and it is expressed as a permanently active enzyme (Lindefors, 1993). Conversely, GAD65 is expressed later than GAD67 (Kiser et al., 1998), it preferentially localizes at synaptic terminals (Esclapez et al., 1994), and it is largely present as an inactive apoenzyme, which can be activated by neuronal activity (Lindefors, 1993; Buddhala et al., 2009). Thus, GAD65 and GAD67 seem to provide a dual system for the control of GABAergic transmission: at earlier stages, GAD67 contributes to tonic GABA transmission necessary for proper development of neuronal circuits (Kaufman et al., 1991; Esclapez et al., 1994). In line with this, GAD67 knockout (KO) mice, which exhibit a 93% reduction of GABA levels, die perinatally (Asada et al., 1997). Once circuits are established (but still immature), GAD65 is expressed and implicated in sustained phasic synaptic transmission in response to neuronal activity (Hensch et al., 1998; Tian et al., 1999; Buddhala et al., 2009). This phasic synaptic transmission supports GABA in reaching a threshold level necessary to render neuronal circuit responsive to experience by functional and anatomical plastic shaping (Fagiolini and Hensch, 2000; Heimel et al., 2011; Levelt and Hubener, 2012). Indeed, GAD65 KO mice are profoundly affected in their ability to respond to monocular deprivation (Hensch et al., 1998).
GABA vesicular transporters (VGAT) mediate transport of GABA into small presynaptic vesicles. Interestingly, absence of VGAT causes embryonic lethality in VGAT KO mice, indicating a fundamental role for GABA release from synaptic vesicles in brain development (Wojcik et al., 2006). Based on the increased expression of VGAT during development, which parallels the developmental switch of GABAergic responses from excitatory to inhibitory, it was proposed that VGAT may also play a role in regulating the latter switch. Nevertheless, the lack of a difference between the expression of KCC2 in VGAT KO in comparison to wild type mice, seems to rule out this hypothesis (Wojcik et al., 2006; Boulland and Chaudhry, 2012). On the other hand, VGAT could still regulate the strength of presynaptic GABAAergic transmission acutely (Kullmann et al., 2005), as in vitro experiments show that VGAT co-transports Cl− anions together with GABA (Juge et al., 2009), probably rendering [Cl−]i lower in the presynaptic terminal. Finally, VGAT promotes GABAergic transmission also indirectly by promoting GABA synthesis. Indeed, VGAT forms a complex with GAD65 in the nerve terminal, and its absence in the VGAT KO mice results in a loss of GAD65 itself from the presynaptic terminal (Jin et al., 2003; Wojcik et al., 2006).
The control of GABA release represents another key step in the regulation of GABAergic transmission. In this respect, an interesting phenomenon is represented by kainate receptors (KARs), which are expressed at presynaptic terminals of GABAergic interneurons. Activation of these receptors results in a decrease in the amplitude of GABAergic currents and a disinhibition of the postsynaptic neuron (Sihra and Rodriguez-Moreno, 2011). Also extracellular-signal-regulated kinases (ERK) can modulate GABA release during neuronal activity (Cui et al., 2008). Moreover, non-vesicular GABA release (i.e., via reversal of GAT-1), contributing to GABA phasic transmission (Wu et al., 2007), can be stimulated by nitric oxide (Tarasenko et al., 2014).
Once GABA is released, GABA transporters at the cell membrane of axon terminals are responsible for GABA removal from the synaptic cleft. GATs modulate GABAergic transmission by regulating the time course of both phasic and tonic GABAergic responses. Even if diffusion of GABA out of the synaptic cleft is the primary determinant for the termination of the response, GABA-uptake inhibitors greatly prolong the phasic conductance associated with application of GABA, pointing to the role of GATs in the termination of GABA-evoked postsynaptic potentials (Dingledine and Korn, 1985; Zhou and Danbolt, 2013). Moreover, GABA uptake becomes of primary importance in conditions of sustained phasic activity of GABAergic neurons (Kersante et al., 2013). On the other hand, GAT regulation is fundamental also for tonic inhibition, as this requires continuous receptor activity elicited by ambient GABA. Indeed, blockade of GABA uptake by GABA transporter blocker NO711 unmasks tonic inhibition in pyramidal neurons in the hippocampus (Semyanov et al., 2003; Walker and Semyanov, 2008). Thus, far, four GATs (GAT-1, GAT-2, GAT-3, BGT-1) have been isolated (Roth and Draguhn, 2012; Zhou and Danbolt, 2013). As for the different GAD isoforms, GATs are differently expressed during development, possibly reflecting diverse needs of dynamically developing neuronal networks. For example, during development, GAT-3 is the mostly expressed in neurons, GAT-2 is poorly expressed, and GAT-1 expression increases gradually (Minelli et al., 1995; Evans et al., 1996; Zhou and Danbolt, 2013). As a result, GAT-1 is the most abundant GAT isoform expressed in neuronal and in astrocytic terminals and GAT-3 is expressed only in astrocytes in the adult cortex (Evans et al., 1996). How the expression of different GATs at diverse time of development and in different cell types may influence GABAergic transmission still needs to be elucidated.
Regulation of Cation/Chloride Cotransporters
Modulation of GABAergic transmission by variation in [Cl−]i is a mechanism naturally at work under a number or circumstances. Interestingly, as Cl− reversal potential (ECl) rests very close to the resting membrane potential of the neuron (Vm), even small changes in [Cl−]i can result in significant shifts of the ECl, for instance from hyperpolarizing to depolarizing. The most characterized example of modulation of GABAergic transmission by variations in [Cl−]i occurs during ontogenesis. Indeed, a progressive decrease of [Cl−]i is responsible for the shift of the polarity of GABAergic transmission from depolarizing and mostly excitatory during early development to hyperpolarizing and inhibitory in the adult. This developmental shift of GABAergic signaling has been evolutionarily conserved from reptiles to humans, and it possibly provides excitation in the developing brain without the risk of glutamatergic excitotoxicity (Ben-Ari, 2002). Interestingly, this switch also occurs during adult neurogenesis in the dentate gyrus of the hippocampus and in the olfactory bulb (Wang et al., 2003a; Ge et al., 2006, 2008; Sernagor et al., 2010; Young et al., 2010; Ming and Song, 2011; Young et al., 2012).
The higher [Cl−]i in young neurons depends on high expression of NKCC1 (Dzhala et al., 2005), and it results in depolarizing GABAA transmission. As development proceeds, KCC2 becomes expressed at higher levels than NKCC1 (Rivera et al., 1999; Dzhala et al., 2005), and hyperpolarizing GABAergic transmission takes over (Ben-Ari, 2002; Ben-Ari et al., 2007; Cancedda et al., 2007; Wang and Kriegstein, 2009; Sernagor et al., 2010). Of note, the functional maturation of GABAAR signaling from depolarizing to hyperpolarizing occurs earlier in females than in males (Galanopoulou, 2008), mostly due to the different sex-related expression of NKCC1 and KCC2 (Nunez and McCarthy, 2007).
Another example of how plasticity of ECl controls GABAergic transmission and brain function under particular physiological circumstances is in the regulation of the circadian rhythm. Mammals, as many other species, entrain their behavior and hormonal daily oscillations to the internal biological clock, which is hierarchically mastered by neurons in the suprachiasmatic nucleus (SCN) of the hypothalamus (Lehman et al., 1987). Interestingly, ECl undergoes changes during the circadian rhythm in the SCN, although the effective polarity of GABAAergic transmission is still controversial. Indeed, while first studies suggested GABA to be excitatory at night and inhibitory during the day (Liou and Albers, 1990; Bos and Mirmiran, 1993), a later study revealed opposite actions (Wagner et al., 1997). Possibly, the discrepancy between the two sets of results originated from the use of diverse experimental conditions during the extracellular-spike recordings (i.e., standard ACSF extracellular solution vs. solutions modified in chloride concentration) to assess the polarity of GABAA responses. To resolve this controversy, De Jeu and coworkers directly measured ECl by means of gramicidin-perforated patch-clamp recordings, which allow preservation of [Cl−]i and, consequently, a correct assessment of the polarity of GABAA responses (De Jeu and Pennartz, 2002). They found a significant day vs. night difference in ECl with more depolarized values during the night than during the day. In line with this, successive studies showed that NKCC1 expression was higher at night (Choi et al., 2008). Importantly, the excitatory effect of GABA was reduced by application of NKCC1-inhibitor and loop diuretic bumetanide in brain slices, and absent in Nkcc1 knock-out mice. This points to a regulation of NKCC1 expression to achieve plasticity of ECl during the circadian rhythm. This mechanism was proposed to counteract propagation of excitatory signals during the day (Choi et al., 2008).
Another example of modulation of GABAAergic transmission through cation/chloride cotransporters during a particular physiological condition is the shift in ECl during labor. Birth is a stressful event associated with high risk of injury for the fetal brain, and hypoxic-ischemic brain damage is one of the principal causes of death and neurological impairment in infants (Volpe, 2008). Neuronal activity requires a high metabolic demand since action-potential generation and propagation consumes 47% of available ATP in neurons, while postsynaptic potentials consume another 34% (Attwell and Laughlin, 2001). Consequently, as neuronal activity cannot be easily sustained in condition of hypoxia, the presence of a depolarizing action of GABAergic transmission during delivery could further limit availability of energy supply caused by delivery-induced hypoxia, as experiments on high neuronal activity during seizures would suggest (Dzhala et al., 2000). In a very interesting study, Tyzio and co-workers investigated whether activity of cation/chloride cotransporters during delivery may be altered (Tyzio et al., 2006). They found a shift of ECl to hyperpolarizing GABA responses 1–2 h before delivery. Interestingly, authors also found that bath application of oxytocin, which is massively released before delivery to induce parturition (Gimpl and Fahrenholz, 2001), suppressed GABA-mediated excitation and induced a negative shift in ECl in slices from animals 2 days before delivery. Finally, authors demonstrated that oxytocin-mediated effect occurred via inhibition of NKCC1 chloride cotransporter by treatment with bumetanide (Tyzio et al., 2006).
Delivery is also potentially painful for the newborn (Fitzgerald, 2005), possibly leading to long-term pathological consequences (Volpe, 2008, 2009). In a follow up study, Mazzuca and coworkers demonstrated that oxytocin exerts an analgesic action against pain in newborn rat pups, assessed in terms of thermal tail-flick test and pain-induced vocalization. Also in this case, the effect of oxytocin was achieved by a reduction of GABA depolarization via inhibition of NKCC1, as bumetanide recapitulated the analgesic effects of oxytocin (Mazzuca et al., 2011). These findings are in line with studies in humans indicating that infants born by elective caesarean (c-) section show a lower threshold for pain in comparison to infants born by natural vaginal delivery (Bergqvist et al., 2009), possibly due to the lack of release of maternal oxytocin during delivery by c-section (Gimpl and Fahrenholz, 2001).
The subtle modulation of GABAergic transmission through cation/chloride cotransporters in condition of limiting energy supply finds another example in the animal kingdom (Tyzio et al., 2006). As mentioned above, neuronal activity requires a high metabolic demand and it cannot be easily sustained in condition of hypoxia (Attwell and Laughlin, 2001). The pond snail Lymnae stagnalis is a bimodal breather that can use cutaneous gas exchange under well-oxygenated water, and lung exchange in the air atmosphere in hypoxic condition, which naturally encounters during summer time when the pond water where it lives dries out (Jones, 1961). Remarkably, the pond snail is able to survive under harsh hypoxic experimental conditions such as 40 h in a N2-bubbled environment (Inoue et al., 2001). This is due to a decrease in neuronal activity mediated by a switch in GABA polarity under hypoxic conditions, and it promotes survival by reducing the energetic demand of the brain (Cheung et al., 2006). Indeed, in normal conditions, GABA exerts an excitatory action in the dorsal ganglia of the pond snail, which helps in maintaining basal levels of spontaneous neuronal activity. Conversely, under hypoxic condition, neurons become less active, an effect mimicked by inhibition of NKCC1 activity with bath application of bumetanide.
Modulation of GABAergic transmission by regulation of cation/chloride cotransporters also occurs at synaptic level. For example, an activity-induced depolarizing shift in ECl has been described after repetitive postsynaptic spiking (Fiumelli et al., 2005), tetanic stimulation (Kaila et al., 1997), correlated synaptic activity (Woodin et al., 2003), and rebound burst activity (Wang et al., 2006a). Moreover, some evidence also suggests a role for cation/chloride cotransporters in LTP, although with contrasting results. In particular, while some authors report that following high-frequency stimulation and LTP induction there is a downregulation of KCC2, pointing to a more depolarized ECl (Wang et al., 2006b), other studies indicate that LTP induction results in a shift in ECl toward more hyperpolarized values (Xu and Sastry, 2007). Of note, in one study, authors found that chloride channel CLC-3 contributes to reducing LTP magnitude, as CLC-3 KO mice show an excessive induction of LTP (Farmer et al., 2013).
In agreement with the studies described above, in the presence of increased neuronal activity driven by physiological sensory stimulation, the brain responds also modulating the plasticity of ECl by changes in cation/chloride cotransporters. For example, environmental enrichment (EE), which is a combination of inanimate and social stimuli providing a complex sensory-motor stimulation (Rosenzweig et al., 1978; Sale et al., 2014), promotes a precocious maturation of GABAergic transmission in the visual system (Cancedda et al., 2004; Heimel et al., 2011; Levelt and Hubener, 2012). Interestingly, this precocious maturation is accompanied by an accelerated transition of GABA action from depolarizing to hyperpolarizing, possibly due to a concomitant increased expression of KCC2 (He et al., 2010). Finally, recent studies reveal that also maternal deprivation can affect ECl, determining a negative shift of GABA responses. This effect seems to be mediated by a higher expression of KCC2 cotransporter (Galanopoulou, 2008). Thus, the plasticity of ECl plays a major role in regulating naturally relevant behaviors.
Modulation of GABAergic Transmission in Pathology and Therapeutic Perspectives
Given the pivotal role of GABA in the regulation of brain development and functioning, it is not surprising that alterations in the GABAergic system have been implicated in a number of neurodevelopmental disorders (Di Cristo et al., 2011). As we described above for the modulation of GABAergic signaling in physiology, the alterations of the GABAergic system in neurodevelopmental disorders can occur at the level of GABA receptors, metabolism and/or cation/chloride cotransporters (Table 1).
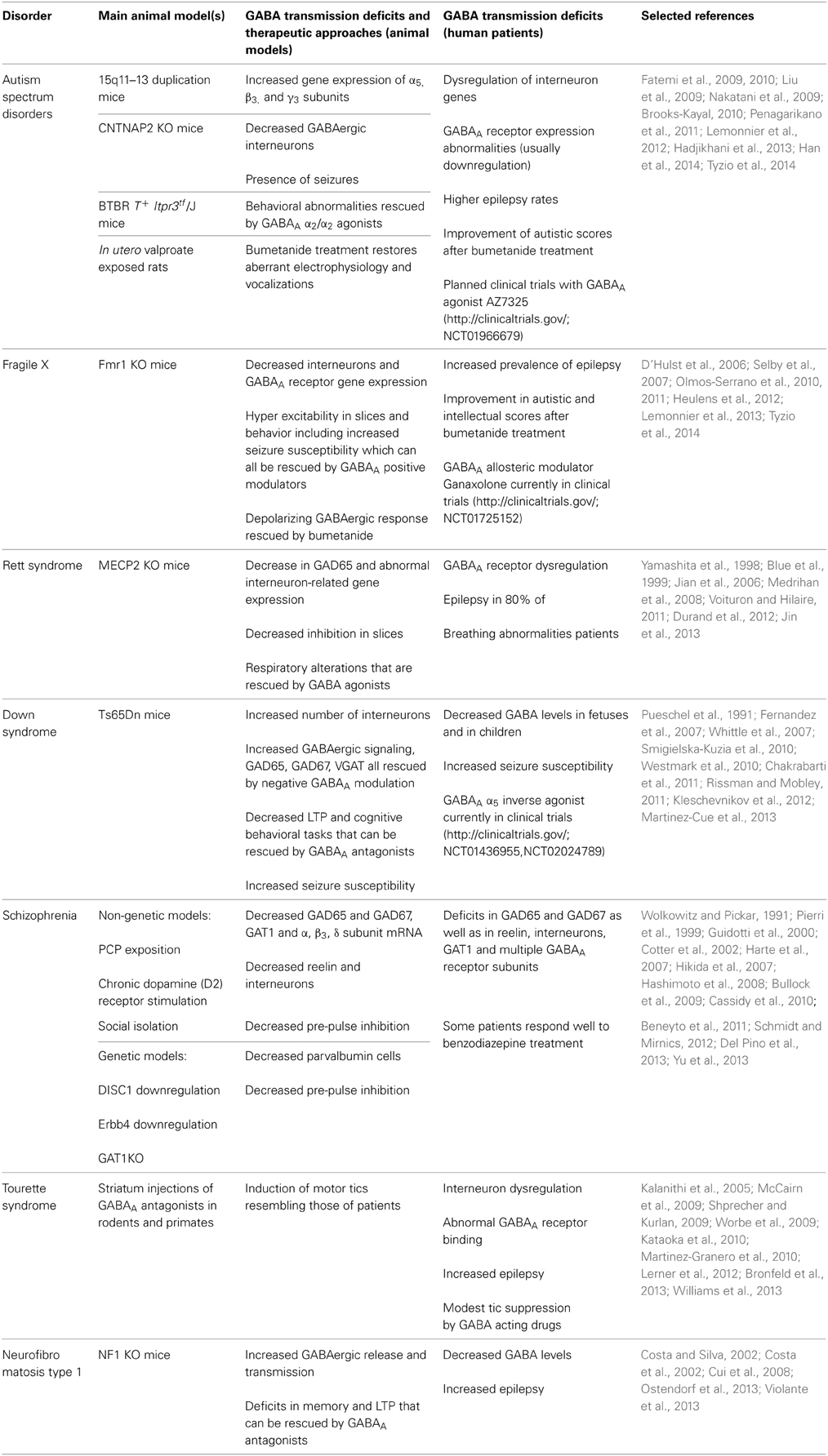
Table 1. Summary of GABAA signaling alterations in neurodevelopmental disorders.
Autism Spectrum Disorders
Autism spectrum disorders (ASDs) are a group of syndromes of diverse etiology with a common set of core symptoms that include compromised social interaction, impaired communication, and repetitive behaviors (Pizzarelli and Cherubini, 2011). ASDs can be idiopathic or co-morbid with other syndromes such as Fragile X or Rett syndrome. Notwithstanding the wide variation in the etiology, the consistency in symptomatology suggests that the mechanisms underlying the pathology of ASDs are common. Mounting evidence indicates that one such mechanism is an impairment in the neuronal excitatory/inhibitory balance (Pizzarelli and Cherubini, 2011), as also suggested by the high prevalence of epilepsy in patients with ASDs (Brooks-Kayal, 2010). Accordingly, several studies have identified mutations in different GABAAR subunits as risk factors for the development of ASDs (Coghlan et al., 2012) and autism-related genes have been found to be mostly expressed in interneurons (Xu et al., 2014). In particular, GABRB3, GABRA5, and GABRG3 genes were described as risk factors (Cook et al., 1998; Menold et al., 2001; Buxbaum et al., 2002; Shao et al., 2003; McCauley et al., 2004), and GABRA4 was found to be involved in ASD on its own, but also through interaction with GABRB1 (Ma et al., 2005). Moreover, GABAAR subunits α1–5, β1 and β3 and GAD65 and 67 are downregulated in autistic brains (Fatemi et al., 2002, 2009, 2010; Mendez et al., 2013). Accordingly, GABA levels are decreased in the frontal lobe and in the auditory cortex of autistic patients (Harada et al., 2011; Rojas et al., 2013). In addition, mutations in interneuron-associated genes DLX1 and DLX2 have been described as susceptibility factors for ASD. Finally, benzodiazepine binding to GABAARs is decreased in the hippocampus and frontal cortex of ASD patients (Blatt et al., 2001; Guptill et al., 2007; Liu et al., 2009; Mori et al., 2012).
Benzodiazepines have been used to treat autistic patients along with other antiepileptic drugs (AEDs), mostly because of the comorbidity of ASD and epilepsy. Interestingly, it was proposed that antiepileptics might have an effect on modulating behavioral aspects, as in some studies antiepileptic drugs improved socialization and communication skills (Di Martino and Tuchman, 2001). Nevertheless, consensus on this issue has not been reached yet. In fact, a study by Marrosu and coworkers showed that the use of diazepam had a paradoxical effect by increasing anxiety and aggression in autistic children (Marrosu et al., 1987). This observation led to the hypothesis that GABA could be having excitatory actions in ASD, resulting in a groundbreaking pilot study to test the therapeutical actions of chronic bumetanide treatment in ASD children. By inhibition of NKCC1, the treatment was designed to revert the polarity of GABAergic response from possibly depolarizing to hyperpolarizing in autistic children (Lemonnier and Ben-Ari, 2010). The reduction in autistic scores and the absence of side effects over 3 months of treatment in 5 autistic children led the research group to perform a larger and double-blind clinical trial with 54 patients (Lemonnier et al., 2012). After 3 months of treatment, patients showed a significant amelioration of their symptoms. In addition, this effect was still noticeable (although to a lesser degree) after a 1 month wash-out period. Finally, the same group has recently showed that adolescents and young adults show improvement in emotion recognition after 10 months of treatment with bumetanide (Hadjikhani et al., 2013). Interestingly, an abolished GABA excitatory-inhibitory shift during delivery leading to a chronic deficient chloride regulation and autistic-like behaviors has been recently shown in the valproate and fragile X rodent models of autism (Tyzio et al., 2014). Of note, a planned clinical trial with a selective α2/3 selective agonist (AZ7325) will recruit patients soon (http://clinicaltrials.gov/; NCT01966679).
Reinforcing the hypothesis of an involvement of faulty GABAergic transmission in autism, mice with 15q11-13 duplication and contactin-associated protein-like 2 (CNTNAP2) KO mice -two mutations present in some autistic patients- present autistic-like behaviors associated with increased α5, β3, γ3 subunit mRNAs as well as decreased number of parvalbumin, calretinin and neuropeptid Y interneurons, respectively (Nakatani et al., 2009; Penagarikano et al., 2011). Further correlation between compromised GABAergic transmission and ASD was found in a mouse mutant of voltage-gated ion channels where the mutation was restricted to forebrain interneurons. These animals exhibited autistic behaviors (i.e., decreased social interaction) that were rescued by benzodiazepine treatment (Han et al., 2012). Moreover, in the BTBR T+ Itpr3tf/J (a mouse strain that is used as a model of idiopathic autism given its behavioral profile including poor social interaction) low doses of a drug acting on α2/3 subunits (i.e., L-838,417) ameliorate short- and long-term memory as well as social interaction deficits (Han et al., 2014). Finally, the NHE9 gene which encodes a (Na+, K+)/H+ exchanger, is a genetic factor that increases risk for ASD and could potentially affect GABA activity by altering ionic gradients and consequent [Cl−]i (Morrow et al., 2008).
Fragile X Syndrome
Fragile X is a monogenic disorder caused by mutations in the FMR1 gene (Bagni and Greenough, 2005). This gene encodes for the Fragile X mental retardation protein (FMRP), which regulates the translation of a wide array of mRNAs. As a considerable amount of Fragile X patients show autistic symptoms and/or comorbidity with epilepsy, neuronal excitatory/inhibitory imbalance has been hypothesized also in these patients (Coghlan et al., 2012). Accordingly, hyper excitability in slices has been described in the Fmr1 KO mouse model (Gibson et al., 2008; Olmos-Serrano et al., 2010; Paluszkiewicz et al., 2011; Goncalves et al., 2013). In particular, recordings in these mice showed decreased sIPSCs and mIPSCs as well as decreased tonic inhibition in the amygdala, and increased firing rate and synchrony in the cortex (Olmos-Serrano et al., 2010; Goncalves et al., 2013 but also see Centonze et al., 2008). Interestingly, this hyperexcitability at synaptic level was accompanied by hyperexcitability at behavioral level, as demonstrated by increased activity in the open field. This could be rescued by GABAA agonist THIP -which increases tonic currents by mainly acting on the δ subunit- at doses that did not have an effect on wild-type mice (Olmos-Serrano et al., 2011).
Among possible causes for the neuronal excitatory/inhibitory imbalance in Fragile X, deficits at different levels of the GABAergic signaling have been described. In particular, decreased density of PV-expressing interneurons was described in Fmr1 KO (Selby et al., 2007). Moreover, deficits in GABAAR mRNA and protein levels for different subunits (i.e., α1, α3, α4, β1, β2, δ, γ1, and γ2) were reported in Fmr1 KO, although some authors found these defects only during development (El Idrissi et al., 2005; D'Hulst et al., 2006; Gantois et al., 2006; Adusei et al., 2010). Besides downregulation of GABAAR subunits, also defects in GABA metabolism have been described, as mRNA for GAD1 and protein expression of GAD65/67 was decreased in Fmr1 KO mice (D'Hulst et al., 2009; Olmos-Serrano et al., 2010), although these studies contrast with others that found GAD65/67 to be increased in these mice (El Idrissi et al., 2005; Adusei et al., 2010). Moreover, mRNA for GAT1 and 4, succinate semialdehyde dehydrogenase (SSADH) and GABA-T are decreased whereas GABA release probability is increased in Fmr1 KO mice (Centonze et al., 2008). Finally, gephyrin (a GABAergic postsynaptic marker) expression is decreased in the cortex of these mice (D'Hulst et al., 2009).
In line with the above findings hypothesizing a decrease in GABAergic transmission in Fragile X, a number of in vivo studies attempted to rescue behavioral deficits in mouse models by increasing GABAergic signaling. For example, administration of GABAA agonist taurine rescued performance in the passive avoidance task in Fmr1 KO mice (El Idrissi et al., 2009). Moreover, hyperexcitability in slices and behavior in mice were rescued by the agonist THIP (Olmos-Serrano et al., 2010, 2011). Recently, treatment with diazepam or ganaxolone (a neurostreroid that can positively modulate GABAARs) rescued audiogenic-induced seizures in Fmr1 KO animals (Heulens et al., 2012). The fact that the δ subunit is downregulated in Fmr1 KO mice and the encouraging results with THIP and ganaxolone -both of which act through the δ subunit- suggest that the δ subunit of the GABAAR could be a good therapeutic target in Fragile X. Indeed, ganaxolone is being tested in a Phase 2 clinical trial for children with Fragile X (NCT01725152, http://clinicaltrials.gov/).
Finally, given their earlier success in treating autistic children with bumetanide (Lemonnier et al., 2012), as well as the high incidence of autism in Fragile X patients, Lemonnier and colleagues used bumetanide to treat a child with Fragile X and concomitant autistic diagnosis (Lemonnier et al., 2013). Interestingly, after 3 months of treatment, the child achieved an improvement in several clinical tests addressing autistic behavior and intellectual disability. As this is a promising single case report, further studies will have to evaluate the therapeutic potential of bumetanide treatment for Fragile X on a larger cohort of patients. This is in line with recent articles reporting that switch of GABAergic transmission from depolarizing to hyperpolarizing is delayed in Fmr1KO mice (He et al., 2014). Nevertheless, future treatments of Fragile X adults with bumetanide, may require further pre-clinical investigation because excitatory GABAergic transmission seems to persist in the adult hippocampus whereas inhibitory GABAergic transmission takes eventually over in the cortex of Fmr1KO mice (He et al., 2014; Tyzio et al., 2014).
Rett Syndrome
Rett syndrome is a chromosome X-linked neurodevelopmental disorder caused by mutations in the Methyl-CpG-binding protein 2 (MECP2) gene. Rett syndrome patients develop normally until the age of 6–18 months, but then they undergo a developmental regression that courses with loss of speech, motor coordination, breathing abnormalities and cognitive impairment (Amir et al., 1999; Leblanc and Fagiolini, 2011). Also, epilepsy is present in 80% of patients (Jian et al., 2006). As for the other neurodevelopmental disorders described above, a number of evidence in Rett syndrome patients point to alteration of GABAergic transmission. Indeed, density of GABA receptors is increased in young patients (Blue et al., 1999), and benzodiazepine binding to GABAAR is decreased in adult patients (Yamashita et al., 1998). Interestingly, a recent study found KCC2 levels decreased in cerebrospinal fluid (CSF) of Rett syndrome patients (Duarte et al., 2013). Although these studies in patients point to a general alteration in GABAergic signaling in Rett syndrome, whether this alteration entails decreased or increased transmission is not clear yet.
Experiments in MECP2 deficient mice also point to a general impairment in the GABAergic system, with contrasting results on whether GABAergic transmission is increased or decreased depending on the brain area analyzed. In particular, in postnatal ventro-lateral medulla decreased levels of α2 GABAAR subunit-expression and a decrease in amplitude and frequency of sIPSCs were described (Medrihan et al., 2008). Conversely, recordings from layer V pyramidal neurons of older MECP2 KO mice showed no change in mIPSCs (Dani et al., 2005). Moreover, MECP2 KO mice present altered expression of VGAT in the thalamus with decreased expression in the ventro-basal complex and increased expression in the reticular thalamic nucleus accompanied by frequencies of mIPSCs changing in the same direction (Zhang et al., 2010). Finally, a decrease in GABA release has been also described in MECP2 deficient mice (Medrihan et al., 2008; Jin et al., 2013).
A demonstration for a pivotal role for GABAergic system in Rett syndrome was provided by a study that deleted MECP2 exclusively in GABAergic neurons (Chao et al., 2010). In this mouse model, many of the Rett syndrome symptoms such as increased stereotypical behaviors, progressive motor and respiratory dysfunctions, alterations in social behavior and premature death were reproduced and the epileptic phenotype clearly emerged. Accordingly, MECP2 is a regulator of gene transcription highly expressed in GABAergic neurons (Akbarian et al., 2001), and its deletion causes a decrease in mRNA levels of GAD65, as well as in calbindin and calretinin, although an increase in parvalbumin (Durand et al., 2012). Moreover, one of the targets of MECP2 is GABRB3 (Leblanc and Fagiolini, 2011). Finally, also indirect findings in MECP2 deficient mice suggest a role for GABA in the pathology of this disorder. Indeed, dark rearing, an intervention which strongly affects GABAergic transmission (Benevento et al., 1995; Lee et al., 2006a), was able to rescue parvalbumin interneuron connectivity in the visual cortex and visual acuity of MECP2 KO mice (Durand et al., 2012). Of note, the respiratory alterations found in these mice were alleviated by treatment with benzodiazepine Midazolam, further suggesting the involvement of the GABAergic system in the general pathology of Rett syndrome disorder (Voituron and Hilaire, 2011). Yet, deletion of MECP2 exclusively in excitatory neurons has been recently shown to lead to epileptic phenotype and reduction of GABAergic synapses, indicating that MECP2 is also important in these cells (Zhang et al., 2014).
Down Syndrome
Down syndrome (DS) is a genetic condition caused by the trisomy of the twenty first chromosome and it is the most common genetic cause of intellectual disability accompanied in adulthood by Alzheimer-like pathology (Costa and Scott-McKean, 2013). Despite its relative frequency (i.e., 1 in 700 live births), the molecular mechanisms that mediate it are largely unknown. Nevertheless, several data from both patients and mouse models of DS support the notion that alterations in GABAergic signaling may underlie the cognitive component of this pathology (Kleschevnikov et al., 2004; Fernandez et al., 2007; Chakrabarti et al., 2010; Martinez-Cue et al., 2013). Indeed, GABA levels are reduced in the fetal frontal cortex (Whittle et al., 2007) and in the temporal lobes of children with DS (Smigielska-Kuzia et al., 2010). Conversely, results from postmortem adult brains are mostly unclear describing a decrease in the number of calbindin and parvalbumin interneurons in cortex, but a non-significant trend to reduced GABA in different brain regions, possibly because experiments were performed on brains from patients who had developed Alzheimer-like pathology (Reynolds and Warner, 1988; Kobayashi et al., 1990; Seidl et al., 2001). Additionally, a study with human neural progenitor cells found increased α2 and decreased α5 and β3 GABAAR-subunit expression (Bhattacharyya et al., 2009). Thus, as for Rett syndrome, studies on DS patients point to a general alteration in GABAergic signaling, although it is not clear yet whether this alteration is due to a decreased or an increased transmission.
To aid experimental investigation on DS, a number of mouse models have been generated, and among those, the Ts65Dn is the best characterized (Costa and Scott-McKean, 2013). Notably, a large body of work has pointed to a role for increased GABAergic transmission in these mice. First, a reorganization of inhibitory synapses of granule cells in the dentate gyrus and an increase in the number of calretinin, parvalbumin, somatostatin and calbindin interneurons were observed in the cortex (Belichenko et al., 2004; Chakrabarti et al., 2010; Perez-Cremades et al., 2010). Accordingly, GABAergic markers such as GAD65, GAD67, VGAT and gephyrin were elevated (Martinez-Cue et al., 2013). Moreover, GABA release (Begenisic et al., 2014) and evoked IPSCs of both GABAA and GABAB receptors were also increased (Kleschevnikov et al., 2012), with some authors finding increased GABAergic transmission particularly during the second postnatal week (Mitra et al., 2012). As Belichenko and coworkers found a decrease in β2/3 receptor at 3, but not at 8 months in the Ts65Dn model, this could indicate that not only the receptor distribution, but also its expression during certain developmental windows could be relevant to the disease (Belichenko et al., 2009b). Finally, Ts65Dn and Ts1Cje models of DS, showed decreased LTP and increased LTD in the hippocampus, which could be rescued by the use of GABAA antagonists (Siarey et al., 1999; Kleschevnikov et al., 2004; Costa and Grybko, 2005; Belichenko et al., 2007, 2009a; Fernandez et al., 2007). At the behavioral level, a number of treatments aimed at decreasing GABAergic transmission have been attempted in mouse models of DS with the aim of rescuing cognitive deficits. In particular, Fernandez and colleagues treated Ts65Dn animals with several GABAA antagonists (i.e., picrotoxin, bilobalide and pentylenetetrazole) and observed an improvement in declarative memory in the novel-object recognition and the spontaneous-alternation tasks (Fernandez et al., 2007). Interestingly, 2-week treatment with pentylenetetrazole maintained improved memory in the novel object recognition task up to 2 months after treatment discontinuation. Furthermore, an independent study showed that pentylenetetrazole was also able to rescue spatial memory in the Morris water maze in Ts65Dn mice (Rueda et al., 2008).
Unfortunately, chronic administration of GABAA antagonists carries the risk of increasing seizure susceptibility. Thus, this pharmacological approach may not be readily translated into clinical practice because of the higher incidence of seizures that characterizes DS patients (and mouse models; Westmark et al., 2010; Rissman and Mobley, 2011). In the search of a better pharmacological approach that would avoid the risk of further increasing seizure susceptibility in DS, two independent groups specifically targeted the α5 subunit of the GABAAR. α5 subunit is highly expressed in the hippocampus (Sur et al., 1998) and authors predicted that its antagonization would provide enough disinhibition to rescue hippocampal-dependent memory. On the other hand, since α5 is not widely expressed in other brain regions, authors also predicted that risk of seizure induction would be less by a α5 subunit antagonist than by general GABAAR antagonists. In particular, Braudeau and coworkers found that treatment with an α5 inverse agonist rescued spatial memory as well as object-recognition memory in Ts65Dn mice (El Idrissi et al., 2005; Braudeau et al., 2011a,b; Potier et al., 2014). Moreover, recent findings describe that treatment with a negative allosteric modulator of α5 subunit-containing GABAARs rescues spatial memory, Schaffer collateral LTP, GAD65, GAD67, VGAT and gephyrin levels in Ts65Dn mice (Martinez-Cue et al., 2013). Interestingly, this treatment also increases cellular proliferation in the dentate gyrus (Martinez-Cue et al., 2013). This could indicate restored adult neurogenesis, which is also affected in DS models and partly responsible for the cognitive impairment (Rueda et al., 2005; Clark et al., 2006; Ishihara et al., 2010; Contestabile et al., 2013). Based on all of these studies, Hoffmann-La Roche has recently completed a Phase 1 and started a Phase 2 clinical trial (NCT01436955, NCT02024789, http://clinicaltrials.gov/) with an α5 inverse agonist aimed at measuring safety and cognitive improvement in DS patients, but no results have been posted yet.
Finally, also indirect evidence suggests that GABAergic treatments may ameliorate DS symptoms. For example, treating Ts65Dn pups with fluoxetine results in the normalization of a number of anatomical defects in the dentate gyrus-CA3 region of the hippocampus (Stagni et al., 2013), and treatment during adulthood rescues hippocampal adult neurogenesis (Clark et al., 2006). Since fluoxetine strongly and directly alters GABAergic hippocampal neurotransmission independently of its effect on amine reuptake systems (Mendez et al., 2012), it is tempting to speculate that fluoxetine effect on DS may be mediated by GABAergic transmission. Accordingly, a chronic treatment with fluoxetine normalizes GABA release and promotes recovery of cognitive impairment and hippocampal synaptic plasticity in adult Ts65Dn mice (Begenisic et al., 2014).
Thus, based on studies on animal models, treatment of DS with drugs targeting GABAergic transmission seems a promising venue. Nevertheless, in the context of finding GABAergic treatment for DS with reduced side effects, it will be essential to investigate the mechanisms involved in increased seizure prevalence in both humans and mouse models (Pueschel et al., 1991; Westmark et al., 2010).
Schizophrenia
Schizophrenia is a neurodevelopmental disorder characterized by psychosis and cognitive deficits (Lewis et al., 2012). Multiple evidence shows that GABA plays a role in the development of schizophrenia by affecting mostly (but not only) cognitive aspects of the disorder (Ahn et al., 2011; Gonzalez-Burgos et al., 2011). Indeed, alterations in most domains of GABA-signaling have been described in schizophrenic patients. For example, variations in the GAD1 gene can increase risk of schizophrenia, and schizophrenic patients who carry a single nucleotide polymorphism in this gene have increased GAD25/GAD67 ratios in the hippocampus, suggesting that an immature GABAergic system is retained (Hyde et al., 2011). Moreover, GAD67, GAD65, and reelin (a protein mainly secreted by GABAergic interneurons) mRNAs and proteins are decreased in a number of brain regions, including the prefrontal cortex and cerebellum of schizophrenic patients (Guidotti et al., 2000; Fatemi et al., 2005; Hashimoto et al., 2008). These reductions are also observed in rodent models of schizophrenia. For example, chronic stimulation of D2 dopaminergic receptors reduces GAD67 expression (Schmidt and Mirnics, 2012), NMDA-receptor antagonism by phencyclidine (PCP) reduces mRNA levels of GAD65 and GAD67 (Bullock et al., 2009), isolation of rats leads to a decrease in reelin protein as well as number of reelin-positive cells in the dentate gyrus (Cassidy et al., 2010), and neonatal ventral hippocampus lesions decrease GAD67 mRNA in adult rats, an effect that can be counteracted by antipsychotic treatments (Lipska et al., 2003).
Specific alterations to GABAergic interneurons such as a decrease in the density of calbindin interneurons of the anterior cingulate cortex (Cotter et al., 2002) and a decrease in mRNA for markers of different interneuron subpopulations (i.e., somatostatin, parvalbumin and cholecystokinin) across different regions have been also described in schizophrenic patients (Hashimoto et al., 2008; Fung et al., 2010). Interestingly, parvalbumin downregulation is also common to genetic (Erbb4, DISC1) and non-genetic (PCP treatment, methylazoxymethanol administration during gestation, isolation) rodent and primate models (PCP-treatment; Cochran et al., 2003; Harte et al., 2007; Hikida et al., 2007; Morrow et al., 2007; Fisahn et al., 2009). In particular, deletion of Erbb4 from fast-spiking interneurons in mice leads to a decrease in GAD67 and parvalbumin protein levels, aberrant neuronal excitability and behavioral abnormalities (impairment in working memory, sociability and pre-pulse inhibition), consistent with a schizophrenic phenotype (Del Pino et al., 2013). Moreover, chandelier interneuron terminals in the prefrontal cortex of patients show a decreased GAT1 expression (Woo et al., 1998; Pierri et al., 1999), and GAT1 mRNA is decreased in cerebellum of PCP-treated rats with GAT1 positive chandelier interneuron cartridges decreased in the cortex of isolated rats (Bloomfield et al., 2008; Bullock et al., 2009). Taking advantage of this knowledge, Yu and colleagues explored the behavioral phenotype of GAT1 KO mice, and concluded it could be used as a model of schizophrenia (Yu et al., 2013). Although the above described results on GAD and on the different subpopulations of interneurons in schizophrenia would predict a net decrease in GABA production, measurements of total GABA in brains of patients by magnetic resonance spectroscopy (MRS) are conflicting, describing both a decrease and an increase (Goto et al., 2009; Ongur et al., 2010; Yoon et al., 2010).
Alterations in different GABAAR subunits were also reported in schizophrenic patients, including decrease in mRNA for α1, α5, β2, δ, and θ subunits and increase in α2 and ρ2, subunits (Volk et al., 2002; Hashimoto et al., 2008; Beneyto et al., 2011; Fatemi et al., 2013). On the other hand, increased mRNA levels of α1, α6, β2, β3, and δ, as well as GABAAR binding have been described in animal models (Endo et al., 2007; Bullock et al., 2009). A postmortem study showing increased GABAAR binding in the prefrontal cortex of schizophrenic patients further adds to the theory of disrupted GABAergic activity in this disorder (Benes et al., 1996). Thus, from this experimental evidence GABAergic transmission appears either increased or decreased in schizophrenic patients and animal models, depending on the brain area.
Interestingly, benzodiazepine treatment (alone or in combination with antipsychotics) has been used with mixed results for the treatment of anxiety and management of psychotic symptoms in schizophrenia. Nevertheless, 30–50% of patients respond favorably, possibly suggesting that GABA alterations are present in some patients, but not in others (Wolkowitz and Pickar, 1991). Data showing that antagonism of GABAARs by iomazenil treatment increases psychosis in only 50% of schizophrenics (and not in healthy controls) further strengthen this hypothesis (Ahn et al., 2011). To take advantage of this seeming therapeutic opportunity, Lewis and colleagues performed a first clinical trial using a benzodiazepine-like drug with specificity for receptors containing α2 and α3 subunits, thus avoiding α1 and α5, which are mostly involved in the side effects of conventional benzodiazepines. This study showed improved delayed memory in the in schizophrenic patients (Lewis et al., 2008). Nevertheless, a latter study did not find any improvement using the same compound in a larger sample of patients (Buchanan et al., 2011). As discussed above, the difference may rest in the fact that the impairment of the GABAergic signaling may be present only in a subpopulation of patients.
Finally, also factors that influence [Cl−]i concentration have been implicated in schizophrenia. For instance, the SLC12A2 gene, which encodes for NKCC1, has been reported to be a susceptibility gene for schizophrenia (Potkin et al., 2009). Notably, Kim and colleagues found that an interaction between a single nucleotide polymorphism in the SLC12A2 gene and a single nucleotide polymorphism in the Disrupted in schizophrenia 1 (DISC1) gene increased risk for the development of schizophrenia (Kim et al., 2012). Moreover, KCC2 mRNA was decreased together with increased NKCC1/KCC2 ratios in hippocampus of patients (Hyde et al., 2011). Interestingly, the same group has recently reported differential expression of NKCC1 splice variants and an increase of a new transcript variant of the KCC2 gene in schizophrenia patients (Tao et al., 2012; Morita et al., 2014). Finally, although expression of NKCC1 and KCC2 mRNA was found unaltered in a study on prefrontal cortex of schizophrenic patients, the same study found increased expression of OXSR1 and WNK3 transcripts, which act posttranscriptionally to up-regulate NKCC1 and downregulate KCC2 activity (Arion and Lewis, 2011).
Tourette Syndrome
Tourette Syndrome (TS) is a neurodevelopmental disorder characterized by the presence of movement and vocalization tics. It is often comorbid with obsessive compulsive behavior, anxiety, attention deficit hyperactivity disorder and sleep disorders (Jankovic, 2001; McNaught and Mink, 2011). It also presents with a higher incidence of epilepsy (Williams et al., 2013). As for the other neurodevelopmental disorders described above, these symptoms suggest an altered neuronal excitatory/inhibitory ratio, with a possible implication of GABAergic signaling. Accordingly, postmortem staining of basal ganglia of TS patients revealed a decrease in the number of parvalbumin (PV) interneurons in the caudate and putamen as well as an increase in the same population in the globus pallidus pars interna (Kalanithi et al., 2005; Kataoka et al., 2010). Furthermore, a recent study on GABAAR binding in TS patients has shown a decreased in several regions (i.e., ventral caudate, putamen, nucleus acumbens and globus pallidus) and increase in others (i.e., substancia nigra, cerebellum and left periaductal gray; Lerner et al., 2012). Moreover, a recent genetic study found a correlation between severity of symptoms and increased blood expression of GABAAR subunit α2–4, β1, ρ1, and GABAAR-associated protein in patients (Tian et al., 2011). The same study also showed that genes encoding for the α4 and γ1 GABAAR subunits presented alternative splicing (Tian et al., 2011). Further strengthening the idea of an implication of the GABAergic system in TS is the fact that microinjections of GABAA antagonists into the striatum of both mice and primates induce motor tics resembling those of Tourette patients (McCairn et al., 2009; Worbe et al., 2009; Bronfeld et al., 2013). Furthermore, pharmacological blockade of fast-spiking interneurons in the striatum is sufficient alone to elicit movement abnormalities also resembling those of TS (Gittis et al., 2011). Finally, there is some clinical evidence of GABAergic drugs being useful to control tics. In particular, clonazepam and levetiracetam have shown a modest profile in tic suppression, although more double-blind trials are needed to draw definitive conclusions (Shprecher and Kurlan, 2009; Martinez-Granero et al., 2010).
Neurofibromatosis Type 1
Neurofibromatosis type 1 is a genetic disorder caused by mutations in the NF1 gene. The phenotype is not homogeneous, and it includes presence of neurofibromas, cognitive impairment and increased epilepsy (Costa and Silva, 2002; Diggs-Andrews and Gutmann, 2013; Ostendorf et al., 2013). Increased hyperactivity, sleep disturbance, and emotional issues are also present (Johnson et al., 2005; Leschziner et al., 2013).
Both evidence in patients and studies in the NF1 KO mouse model of neurofibromatosis type 1 point to defects in GABAergic transmission, although with contrasting results. A recent study used MRS to measure total GABA in the visual cortex of NF1 patients and found that its level was decreased in comparison to controls (Violante et al., 2013). Conversely, work with NF1 mutant mice showed an increased GABA release as well as GABAergic transmission, and hypothesized them to be a cause for cognitive impairment (Costa et al., 2002; Cui et al., 2008; Shilyansky et al., 2010). In particular, NF1 KO mice showed an increase in the amplitude of evoked IPSPs, in the frequency of spontaneous IPSCs, and in the frequency of miniature IPSCs when measured in high potassium medium (Costa et al., 2002; Cui et al., 2008; Shilyansky et al., 2010). Cui and colleagues also observed that the increase in frequency of the miniature IPSCs was still present when the NF1 mutation was induced exclusively in interneurons, but not when it was only present in pyramidal neurons, again pointing to a key role of GABAergic transmission in the disease (Cui et al., 2008). Interestingly, NF1 KO mice also show deficits in CA1hippocampal LTP, spatial learning and working memory that can be reverted by the GABAA antagonist picrotoxin (Costa et al., 2002; Cui et al., 2008; Shilyansky et al., 2010).
Succinic Semialdehyde Dehydrohenase Deficiency, Tuberous Sclerosis Complex, Angelman Syndrome, Prader-Willi Syndrome
Although less frequent and less studied, other neurodevelopmental disorders have also been related to defective GABAergic transmission.
Succinic semialdehyde dehydrohenase deficiency (SSDHD) is caused by a deficit of the GABA catabolitic enzyme succinic semialdehyde dehydrohenase and it thus results in an increase of GABA and γ-hydroxybutiryc acid (GHB, a by-product of GABA catabolism). The disorder courses with mental retardation, autistic-like behavior, hypotonia and seizures (Pearl et al., 2011; Vogel et al., 2013).
Tuberous sclerosis complex (TSC) is caused by loss of function of TSC1 and TSC2 genes, and courses with presence of cortical tubers, mental retardation, autistic-like behavior and epilepsy. Interestingly, studies of the tubers have shown decreased expression of GABAAR α1 subunit, decreased benzodiazepine-receptor binding and decreased expression of KCC2, as well as increased GABA levels and increased expression of NKCC1 (Talos et al., 2012). Interestingly, treatment with vigabatrin (an inhibitor of GABA-T) improves cognitive and behavioral issues and it is effective for controlling seizures in TSC (Jambaque et al., 2000).
Angelman and the closely related Prader-Willi Syndromes, are the result of deletion/methylation of chromosome region15q11–13. If the mutation is of maternal origin, it will give rise to Angelman Syndrome, whereas if the mutation is of paternal origin, the result will be Prader-Willi Syndrome (Zafeiriou et al., 2013). Interestingly, although considered distinct pathologies, both disorders share symptoms such as mental retardation and autism-like behaviors, and Angelman Syndrome patients are at higher risk of developing seizures (Buiting, 2010; Zafeiriou et al., 2013). Since the chromosomal region involved in these two disorders contains the genes of three GABAAR subunits (i.e., α5, β3, and γ3; Fiumara et al., 2010), this may indicate an involvement of the GABAergic system, as confirmed by some studies suggesting abnormal GABA metabolism in these disorders (Dhossche et al., 2005).
GABAergic Therapies: Toward Innovation and Beyond
Neurodevelopmental disorders are a number of heterogeneous conditions that share a strong social and emotional impact on the lives of patients and their families. Although their etiology is very different and they are characterized by a variety of clinical pictures, they surprisingly share a large number of symptoms such as cognitive impairment, increased seizure susceptibility and sleep disturbance. Interestingly, an excitatory/inhibitory imbalance in neuronal activity -at least partly due to a disruption in the GABAergic system- also appears as a common feature to most of these pathologies.
From a historical point of view, research aimed to develop therapeutic strategies mostly addressing either GABAAR function or GABA metabolism, possibly because it simply represented the more intuitive approach. For example, in a number of studies researchers directly targeted GABA signaling by treatment of animal models with GABAA agonists (i.e., autism, Fragile X, Rett syndrome; Olmos-Serrano et al., 2011; Voituron and Hilaire, 2011; Han et al., 2012) or antagonists (i.e., Down syndrome and Neurofibromatosis type I; Fernandez et al., 2007; Cui et al., 2008). Although encouraging results indicate improvement of both cognitive and non-cognitive symptoms in these models, it is important to be cautious when translating data from animal models to humans. For example, certain apparent discrepancies between animal models of the diseases and patients such as those described for Down syndrome, Rett syndrome, schizophrenia, and Neurofibromatosis type 1 have to be properly addressed, possibly through parallel studies in animals and humans by the same researchers with similar experimental approaches, when achievable.
Since most discussed developmental disorders present increased risk of epileptic seizures, adequate controls for seizure activity should be performed. Furthermore, since GABA is a very widespread neurotransmitter acting both at the CNS and PNS levels and in a number of periphery organs (Roberts and Krause, 1982; Watanabe et al., 2002), treatments with GABAA antagonists could lead to excessive disinhibition through the blocking of both phasic and shunting inhibition and treatment with GABAA agonists can lead to undesired side effects such as drowsiness and muscle relaxation (Rudolph et al., 1999; Griebel and Holmes, 2013). Thus, issues on the potentials of translating experimental findings into the clinical practice, forced researchers to find more subtle ways to modulate GABAergic transmission. The discovery that different GABAAR subunits finely modulate GABAergic responses in physiology (e.g., tonic vs. phasic inhibition) inspired researchers to target specific subunits in neurodevelopmental disorders to reduce side effects of GABAAergic pharmacological treatments (e.g., the specific antagonists of the α5 subunit in the Ts65Dn animal model of Down syndrome to recover cognitive deficits without pro-epileptic side effects; Braudeau et al., 2011b; Martinez-Cue et al., 2013). In addition, GABAA agonists selective for the α2,3 subunits are being researched as potential anxiolytic/analgesic drugs that lack the sedative effects, which are mainly mediated by α1 (Mohler, 2011), and these drugs are also being tested for schizophrenia (Lewis et al., 2008; Buchanan et al., 2011; Mohler, 2011). Moreover, it has been recently argued that drugs acting on the GABAA δ subunit can aid the treatment of Fragile X (Hagerman et al., 2009; Olmos-Serrano et al., 2011; Heulens et al., 2012). Given the latter encouraging results, it is possible that future therapies -if aimed at targeting GABAAR subunits specifically affected in the diverse diseases- will present even further reduced side effects by restoring proper brain function with no effect on other “non-diseased” receptors. However, further research focusing on alteration of specific GABAA subunits in each different neurodevelopmental disorder and further research focusing on developing drugs that selectively act on these specific subunits are a prerequisite before treatment implementation (Mohler, 2011).
The discovery that action of NKCC1/KCC2 could indirectly modulate GABAergic transmission opened new avenue for therapeutic research readably translatable into clinical trials. First, high expression of NKCC1 in the neonatal brain was suggested to be a concomitant cause aggravating epilepsy in infants (Dzhala et al., 2005). Then, modulation of GABAergic transmission trough variation in the activity of cation/Cl− cotransporters was described under particular physiological conditions (e.g., delivery and adult neurogenesis) and in neurodevelopmental disorders (e.g., Fragile X, Rett syndrome, schizophrenia). As NKCC1 inhibitor bumetanide is an FDA-approved loop diuretic extensively used in the past with very mild side effects (Kahle and Staley, 2008), clinicians were readily allowed to perform clinical trials on the use of bumetanide to treat infant and adult epilepsy, and more recently autism in young patients with encouraging results (Kahle et al., 2009; Lemonnier and Ben-Ari, 2010; Lemonnier et al., 2012; Eftekhari et al., 2013; Hadjikhani et al., 2013). As neurodevelopmental disorders are characterized by increased susceptibility to seizures, diuretic treatment seems a very promising approach also because loop diuretics have been clinically tested in the past as antiepileptics by a mechanism proposed to be independent from GABAergic transmission (Maa et al., 2011). Indeed, bumetanide decreases cell volume and increases the extracellular space, both of which contribute to decrease neuronal excitability and synchrony of neuronal network by means of the so called “ephaptic effect,” namely a synapse-independent form of propagation of electrical signals (Haglund and Hochman, 2005; Maa et al., 2011; Hochman, 2012). Moreover, action on cation chloride cotransporters is predicted to be a subtler approach with milder side effects than direct action on GABAARs by agonists/antagonists as shunting inhibition (which critically depends on the number of open GABAARs) results only slightly affected by small changes in [Cl−]i. Nevertheless, as bumetanide does not pass well the blood-brain barrier (BBB) in mice, more lipophylic bumetanide prodrugs are being developed to address this issue, while avoiding the diuretic effect due to bumetanide action on NKCC2, an isoform present in the kidney. Some of these drugs have recently been tested in rodents and seem to have better brain penetration (Loscher et al., 2013; Tollner et al., 2014). On the other hand, a completely new compound (CLP257) was very recently described to restore Cl− balance by increasing KCC2 expression at the plasma membrane and it was able to rescue nociceptive responses in a rat model of neuropathic pain, providing a new and efficient tool to tackle hyperexcitability in the SNC (Gagnon et al., 2013).
Finally, also environmental enrichment (EE), which exerts a profound influence over GABAergic transmission (Cancedda et al., 2004; Sale et al., 2010) results in behavioral, synaptic and connectivity rescues in models of neurodevelopmental disorders (Restivo et al., 2005; Nithianantharajah and Hannan, 2006; Lonetti et al., 2010; Begenisic et al., 2011; Durand et al., 2012; Hannan, 2014), and could thus be considered a possible therapeutic approach. In particular, EE rescues spatial memory and dentate gyrus LTP as well as neuronal proliferation in Ts65Dn animals (Begenisic et al., 2011; Chakrabarti et al., 2011). Moreover, cortical LTP, anxiety behavior, motor coordination and spatial learning could be rescued in MECP2 null mice by EE (Kondo et al., 2008; Nag et al., 2009; Kerr et al., 2010; Lonetti et al., 2010) with an indication that the inhibitory GABAergic system could be preferentially responsible for the effect (Boggio et al., 2010; Lonetti et al., 2010). Furthermore, EE restored environment exploration and dendritic branching/length in Fmr1KO mice (Restivo et al., 2005), and it was able to ameliorate some symptoms in a genetic model of schizophrenia (McOmish et al., 2008). Finally, behavioral impairments such as nociception, motor coordination, pre-pulse inhibition, stereotyped behavior, grooming, memory deficits, anxiety, social deficits were rescued in a number of mice models of ASD reared in an EE (Schneider et al., 2006; Lacaria et al., 2012; Reynolds et al., 2013). Although EE does seem ideal as a non-invasive treatment, there is however controversy as to how well it will translate to the human setting, given that some of the effects that are sometimes reported in environmentally enriched animals could be just considered as the rescue of the environmental deprivation of the standard housing conditions of laboratory animals (Nithianantharajah and Hannan, 2006; Sale et al., 2014). Nevertheless, the first recent attempts to translate EE into the humans setting have proven useful for the treatment of autism (Woo and Leon, 2013).
Concluding Remarks
In conclusion, we reviewed that modulation of GABAergic transmission plays a pivotal role in regulating brain activity under physiological conditions during development, and disruption of GABAergic signaling is a hallmark of many neurodevelopmental disorders. Thus, correcting GABA signaling in pathological conditions could reinstate physiological level of brain function in a pathological background. On the other hand, because of the complex role for GABA in regulating physiological neuronal activity during development and in adulthood, careful consideration should be paid in the treatment of neurodevelopmental disorders by GABAergic therapies to decrease side effects. We propose that further dissection of the molecular mechanisms in place for the regulation of brain development under physiological conditions may inspire researchers to address GABAergic transmission with a subtle and physiological approach also in neurodevelopmental disorders. Moreover, a detailed study of the specific defects in GABAergic transmission in the different neurodevelopmental disorders must be achieved before being able to design the best therapeutic strategies. Also, it will be compulsory to perform thorough research on humans as data in animal models are at times not paralleled by findings in humans. In the near future, studies with human-derived induced pluripotent stem (iPS) cells will greatly aid to resolve this issue, by unraveling the fine molecular pathways affected in each disorder in humans. Indeed, cell lines derived from human patients are already available for mutations related to ASD such as SHANK3, as well as for Fragile X, Rett syndrome, Down syndrome, and schizophrenia (Marchetto et al., 2010; Urbach et al., 2010; Brennand et al., 2011; Li et al., 2012). Thus, basic research on the role of GABAergic transmission during physiological and pathological brain development is a fundamental prerequisite in the design of innovative pharmacological treatments with reduced side effects.
Conflict of Interest Statement
Laura Cancedda is on the Provisional Application: US 61/919,195, 2013. Modulators of Intracellular Chloride Concentration For Treating An Intellectual Disability.
Acknowledgments
The work was supported by Compagnia di San Paolo # 2008.1267 and Telethon # GGP10135 and GGP13187.
References
Abramian, A. M., Comenencia-Ortiz, E., Vithlani, M., Tretter, E. V., Sieghart, W., Davies, P. A., et al. (2010). Protein kinase C phosphorylation regulates membrane insertion of GABAA receptor subtypes that mediate tonic inhibition. J. Biol. Chem. 285, 41795–41805. doi: 10.1074/jbc.M110.149229
Adusei, D. C., Pacey, L. K., Chen, D., and Hampson, D. R. (2010). Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology 59, 167–171. doi: 10.1016/j.neuropharm.2010.05.002
Ahn, K., Gil, R., Seibyl, J., Sewell, R. A., and D'Souza, D. C. (2011). Probing GABA receptor function in schizophrenia with iomazenil. Neuropsychopharmacology 36, 677–683. doi: 10.1038/npp.2010.198
Akbarian, S., Chen, R. Z., Gribnau, J., Rasmussen, T. P., Fong, H., Jaenisch, R., et al. (2001). Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol. Dis. 8, 784–791. doi: 10.1006/nbdi.2001.0420
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Arima-Yoshida, F., Watabe, A. M., and Manabe, T. (2011). The mechanisms of the strong inhibitory modulation of long-term potentiation in the rat dentate gyrus. Eur. J. Neurosci. 33, 1637–1646. doi: 10.1111/j.1460-9568.2011.07657.x
Arion, D., and Lewis, D. A. (2011). Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch. Gen. Psychiatry 68, 21–31. doi: 10.1001/archgenpsychiatry.2010.114
Asada, H., Kawamura, Y., Maruyama, K., Kume, H., Ding, R. G., Kanbara, N., et al. (1997). Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 94, 6496–6499. doi: 10.1073/pnas.94.12.6496
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Bagni, C., and Greenough, W. T. (2005). From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci. 6, 376–387. doi: 10.1038/nrn1667
Barnard, E. A., Skolnick, P., Olsen, R. W., Mohler, H., Sieghart, W., Biggio, G., et al. (1998). International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 50, 291–313.
Battaglioli, G., Liu, H., and Martin, D. L. (2003). Kinetic differences between the isoforms of glutamate decarboxylase: implications for the regulation of GABA synthesis. J. Neurochem. 86, 879–887. doi: 10.1046/j.1471-4159.2003.01910.x
Begenisic, T., Baroncelli, L., Sansevero, G., Milanese, M., Bonifacino, T., Bonanno, G., et al. (2014). Fluoxetine in adulthood normalizes GABA release and rescues hippocampal synaptic plasticity and spatial memory in a mouse model of Down syndrome. Neurobiol. Dis. 63, 12–19. doi: 10.1016/j.nbd.2013.11.010
Begenisic, T., Spolidoro, M., Braschi, C., Baroncelli, L., Milanese, M., Pietra, G.,et al. (2011). Environmental enrichment decreases GABAergic inhibition and improves cognitive abilities, synaptic plasticity, and visual functions in a mouse model of Down syndrome. Front. Cell Neurosci. 5:29. doi: 10.3389/fncel.2011.00029
Belichenko, N. P., Belichenko, P. V., Kleschevnikov, A. M., Salehi, A., Reeves, R. H., and Mobley, W. C. (2009a). The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J. Neurosci. 29, 5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009
Belichenko, P. V., Kleschevnikov, A. M., Masliah, E., Wu, C., Takimoto-Kimura, R., Salehi, A., et al. (2009b). Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of Down syndrome. J. Comp. Neurol. 512, 453–466. doi: 10.1002/cne.21895
Belichenko, P. V., Kleschevnikov, A. M., Salehi, A., Epstein, C. J., and Mobley, W. C. (2007). Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. J. Comp. Neurol. 504, 329–345. doi: 10.1002/cne.21433
Belichenko, P. V., Masliah, E., Kleschevnikov, A. M., Villar, A. J., Epstein, C. J., Salehi, A., et al. (2004). Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J. Comp. Neurol. 480, 281–298. doi: 10.1002/cne.20337
Ben-Ari, Y. (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Ben-Ari, Y., Woodin, M. A., Sernagor, E., Cancedda, L., Vinay, L., Rivera, C., et al. (2012). Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever! Front. Cell Neurosci. 6:35. doi: 10.3389/fncel.2012.00035
Benes, F. M., Vincent, S. L., Marie, A., and Khan, Y. (1996). Up-regulation of GABAA receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience 75, 1021–1031. doi: 10.1016/0306-4522(96)00328-4
Benevento, L. A., Bakkum, B. W., and Cohen, R. S. (1995). gamma-Aminobutyric acid and somatostatin immunoreactivity in the visual cortex of normal and dark-reared rats. Brain Res. 689, 172–182. doi: 10.1016/0006-8993(95)00553-3
Beneyto, M., Abbott, A., Hashimoto, T., and Lewis, D. A. (2011). Lamina-specific alterations in cortical GABA(A) receptor subunit expression in schizophrenia. Cereb. Cortex 21, 999–1011. doi: 10.1093/cercor/bhq169
Bergqvist, L. L., Katz-Salamon, M., Hertegard, S., Anand, K. J., and Lagercrantz, H. (2009). Mode of delivery modulates physiological and behavioral responses to neonatal pain. J. Perinatol. 29, 44–50. doi: 10.1038/jp.2008.129
Bhattacharyya, A., McMillan, E., Chen, S. I., Wallace, K., and Svendsen, C. N. (2009). A critical period in cortical interneuron neurogenesis in down syndrome revealed by human neural progenitor cells. Dev. Neurosci. 31, 497–510. doi: 10.1159/000236899
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. doi: 10.1016/j.neuron.2009.03.003
Blatt, G. J., Fitzgerald, C. M., Guptill, J. T., Booker, A. B., Kemper, T. L., and Bauman, M. L. (2001). Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J. Autism. Dev. Disord. 31, 537–543. doi: 10.1023/A:1013238809666
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Bloomfield, C., French, S. J., Jones, D. N., Reavill, C., Southam, E., Cilia, J., et al. (2008). Chandelier cartridges in the prefrontal cortex are reduced in isolation reared rats. Synapse 62, 628–631. doi: 10.1002/syn.20521
Blue, M. E., Naidu, S., and Johnston, M. V. (1999). Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp. Neurol. 156, 345–352. doi: 10.1006/exnr.1999.7030
Boggio, E. M., Lonetti, G., Pizzorusso, T., and Giustetto, M. (2010). Synaptic determinants of rett syndrome. Front. Synaptic Neurosci. 2:28. doi: 10.3389/fnsyn.2010.00028
Bos, N. P., and Mirmiran, M. (1993). Effects of excitatory and inhibitory amino acids on neuronal discharges in the cultured suprachiasmatic nucleus. Brain Res. Bull. 31, 67–72. doi: 10.1016/0361-9230(93)90012-Z
Boulland, J. L., and Chaudhry, F. A. (2012). Ontogenetic changes in the distribution of the vesicular GABA transporter VGAT correlate with the excitation/inhibition shift of GABA action. Neurochem. Int. 61, 506–516. doi: 10.1016/j.neuint.2012.03.018
Brandon, N., Jovanovic, J., and Moss, S. (2002). Multiple roles of protein kinases in the modulation of gamma-aminobutyric acid(A) receptor function and cell surface expression. Pharmacol. Ther. 94, 113–122. doi: 10.1016/S0163-7258(02)00175-4
Brandon, N. J., Delmas, P., Hill, J., Smart, T. G., and Moss, S. J. (2001). Constitutive tyrosine phosphorylation of the GABA(A) receptor gamma 2 subunit in rat brain. Neuropharmacology 41, 745–752. doi: 10.1016/S0028-3908(01)00121-6
Brandon, N. J., Delmas, P., Kittler, J. T., McDonald, B. J., Sieghart, W., Brown, D. A., et al. (2000). GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. J. Biol. Chem. 275, 38856–38862. doi: 10.1074/jbc.M004910200
Braudeau, J., Dauphinot, L., Duchon, A., Loistron, A., Dodd, R. H., Herault, Y., et al. (2011a). Chronic treatment with a promnesiant GABA-A alpha5-selective inverse agonist increases immediate early genes expression during memory processing in mice and rectifies their expression levels in a down syndrome mouse model. Adv. Pharmacol. Sci. 2011, 153218. doi: 10.1155/2011/153218
Braudeau, J., Delatour, B., Duchon, A., Pereira, P. L., Dauphinot, L., De Chaumont, F., et al. (2011b). Specific targeting of the GABA-A receptor alpha5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J. Psychopharmacol. 25, 1030–1042. doi: 10.1177/0269881111405366
Brennand, K. J., Simone, A., Jou, J., Gelboin-Burkhart, C., Tran, N., Sangar, S., et al. (2011). Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225. doi: 10.1038/nature09915
Briggs, S. W., and Galanopoulou, A. S. (2011). Altered GABA signaling in early life epilepsies. Neural Plast. 2011:527605. doi: 10.1155/2011/527605
Bright, D. P., and Smart, T. G. (2013). Protein kinase C regulates tonic GABA(A) receptor-mediated inhibition in the hippocampus and thalamus. Eur. J. Neurosci. 38, 3408–3423. doi: 10.1111/ejn.12352
Bronfeld, M., Yael, D., Belelovsky, K., and Bar-Gad, I. (2013). Motor tics evoked by striatal disinhibition in the rat. Front. Syst. Neurosci. 7:50. doi: 10.3389/fnsys.2013.00050
Brooks-Kayal, A. (2010). Epilepsy and autism spectrum disorders: are there common developmental mechanisms? Brain Dev. 32, 731–738. doi: 10.1016/j.braindev.2010.04.010
Buchanan, R. W., Keefe, R. S., Lieberman, J. A., Barch, D. M., Csernansky, J. G., Goff, D. C., et al. (2011). A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol. Psychiatry 69, 442–449. doi: 10.1016/j.biopsych.2010.09.052
Buddhala, C., Hsu, C. C., and Wu, J. Y. (2009). A novel mechanism for GABA synthesis and packaging into synaptic vesicles. Neurochem. Int. 55, 9–12. doi: 10.1016/j.neuint.2009.01.020
Buiting, K. (2010). Prader-willi syndrome and angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 365–376. doi: 10.1002/ajmg.c.30273
Bullock, W. M., Bolognani, F., Botta, P., Valenzuela, C. F., and Perrone-Bizzozero, N. I. (2009). Schizophrenia-like GABAergic gene expression deficits in cerebellar Golgi cells from rats chronically exposed to low-dose phencyclidine. Neurochem. Int. 55, 775–782. doi: 10.1016/j.neuint.2009.07.010
Buxbaum, J. D., Silverman, J. M., Smith, C. J., Greenberg, D. A., Kilifarski, M., Reichert, J., et al. (2002). Association between a GABRB3 polymorphism and autism. Mol. Psychiatry 7, 311–316. doi: 10.1038/sj.mp.4001011
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M. M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007
Cancedda, L., Putignano, E., Sale, A., Viegi, A., Berardi, N., and Maffei, L. (2004). Acceleration of visual system development by environmental enrichment. J. Neurosci. 24, 4840–4848. doi: 10.1523/JNEUROSCI.0845-04.2004
Cassidy, A. W., Mulvany, S. K., Pangalos, M. N., Murphy, K. J., and Regan, C. M. (2010). Reduced reelin protein synthesis in ventral hippocampus of isolation reared Wistar rats accompanies impaired avoidance conditioning. Behav. Brain Res. 213, 130–134. doi: 10.1016/j.bbr.2010.04.040
Cellot, G., and Cherubini, E. (2013). Functional role of ambient GABA in refining neuronal circuits early in postnatal development. Front. Neural Circuits 7:136. doi: 10.3389/fncir.2013.00136
Centonze, D., Rossi, S., Mercaldo, V., Napoli, I., Ciotti, M. T., De Chiara, V., et al. (2008). Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol. Psychiatry 63, 963–973. doi: 10.1016/j.biopsych.2007.09.008
Chakrabarti, L., Best, T. K., Cramer, N. P., Carney, R. S., Isaac, J. T., Galdzicki, Z., et al. (2010). Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat. Neurosci. 13, 927–934. doi: 10.1038/nn.2600
Chakrabarti, L., Scafidi, J., Gallo, V., and Haydar, T. F. (2011). Environmental enrichment rescues postnatal neurogenesis defect in the male and female Ts65Dn mouse model of Down syndrome. Dev. Neurosci. 33, 428–441. doi: 10.1159/000329423
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Cheung, U., Moghaddasi, M., Hall, H. L., Smith, J. J., Buck, L. T., and Woodin, M. A. (2006). Excitatory actions of GABA mediate severe-hypoxia-induced depression of neuronal activity in the pond snail (Lymnaea stagnalis). J. Exp. Biol. 209, 4429–4435. doi: 10.1242/jeb.02553
Choi, H. J., Lee, C. J., Schroeder, A., Kim, Y. S., Jung, S. H., Kim, J. S., et al. (2008). Excitatory actions of GABA in the suprachiasmatic nucleus. J. Neurosci. 28, 5450–5459. doi: 10.1523/JNEUROSCI.5750-07.2008
Clark, S., Schwalbe, J., Stasko, M. R., Yarowsky, P. J., and Costa, A. C. (2006). Fluoxetine rescues deficient neurogenesis in hippocampus of the Ts65Dn mouse model for Down syndrome. Exp. Neurol. 200, 256–261. doi: 10.1016/j.expneurol.2006.02.005
Cobb, S. R., Buhl, E. H., Halasy, K., Paulsen, O., and Somogyi, P. (1995). Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378, 75–78. doi: 10.1038/378075a0
Cochran, S. M., Kennedy, M., McKerchar, C. E., Steward, L. J., Pratt, J. A., and Morris, B. J. (2003). Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 28, 265–275. doi: 10.1038/sj.npp.1300031
Coghlan, S., Horder, J., Inkster, B., Mendez, M. A., Murphy, D. G., and Nutt, D. J. (2012). GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci. Biobehav. Rev. 36, 2044–2055. doi: 10.1016/j.neubiorev.2012.07.005
Connell, J., Oozeer, R., De Vries, L., Dubowitz, L. M., and Dubowitz, V. (1989). Clinical and EEG response to anticonvulsants in neonatal seizures. Arch. Dis. Child 64, 459–464. doi: 10.1136/adc.64.4_Spec_No.459
Contestabile, A., Greco, B., Ghezzi, D., Tucci, V., Benfenati, F., and Gasparini, L. (2013). Lithium rescues synaptic plasticity and memory in Down syndrome mice. J. Clin. Invest. 123, 348–361. doi: 10.1172/JCI64650
Cook, E. H. Jr. Courchesne, R. Y., Cox, N. J., Lord, C., Gonen, D., Guter, S. J., et al. (1998). Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. Am. J. Hum. Genet. 62, 1077–1083. doi: 10.1086/301832
Costa, A. C., and Grybko, M. J. (2005). Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci. Lett. 382, 317–322. doi: 10.1016/j.neulet.2005.03.031
Costa, A. C., and Scott-McKean, J. J. (2013). Prospects for improving brain function in individuals with Down syndrome. CNS Drugs. 27, 679–702. doi: 10.1007/s40263-013-0089-3
Costa, R. M., Federov, N. B., Kogan, J. H., Murphy, G. G., Stern, J., Ohno, M., et al. (2002). Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415, 526–530. doi: 10.1038/nature711
Costa, R. M., and Silva, A. J. (2002). Molecular and cellular mechanisms underlying the cognitive deficits associated with neurofibromatosis 1. J. Child Neurol. 17, 622–626. discussion: 627–629, 646–651.
Cotter, D., Landau, S., Beasley, C., Stevenson, R., Chana, G., Macmillan, L., et al. (2002). The density and spatial distribution of GABAergic neurons, labelled using calcium binding proteins, in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia. Biol. Psychiatry 51, 377–386. doi: 10.1016/S0006-3223(01)01243-4
Cui, Y., Costa, R. M., Murphy, G. G., Elgersma, Y., Zhu, Y., Gutmann, D. H., et al. (2008). Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 135, 549–560. doi: 10.1016/j.cell.2008.09.060
Curia, G., Papouin, T., Seguela, P., and Avoli, M. (2009). Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 19, 1515–1520. doi: 10.1093/cercor/bhn159
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565. doi: 10.1073/pnas.0506071102
De Jeu, M., and Pennartz, C. (2002). Circadian modulation of GABA function in the rat suprachiasmatic nucleus: excitatory effects during the night phase. J. Neurophysiol. 87, 834–844.
Del Pino, I., Garcia-Frigola, C., Dehorter, N., Brotons-Mas, J. R., Alvarez-Salvado, E., Martinez De Lagran, M., et al. (2013). Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168. doi: 10.1016/j.neuron.2013.07.010
Dhossche, D. M., Song, Y., and Liu, Y. (2005). Is there a connection between autism, Prader-Willi syndrome, catatonia, and GABA? Int. Rev. Neurobiol. 71, 189–216. doi: 10.1016/S0074-7742(05)71009-6
D'Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., et al. (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 1121, 238–245. doi: 10.1016/j.brainres.2006.08.115
D'Hulst, C., Heulens, I., Brouwer, J. R., Willemsen, R., De Geest, N., Reeve, S. P., et al. (2009). Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 1253, 176–183. doi: 10.1016/j.brainres.2008.11.075
Di Cristo, G. (2007). Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin. Genet. 72, 1–8. doi: 10.1111/j.1399-0004.2007.00822.x
Di Cristo, G., Pizzorusso, T., Cancedda, L., and Sernagor, E. (2011). GABAergic circuit development and its implication for CNS disorders. Neural Plast. 2011:623705. doi: 10.1155/2011/623705
Diggs-Andrews, K. A., and Gutmann, D. H. (2013). Modeling cognitive dysfunction in neurofibromatosis-1. Trends Neurosci. 36, 237–247. doi: 10.1016/j.tins.2012.12.002
Di Martino, A., and Tuchman, R. F. (2001). Antiepileptic drugs: affective use in autism spectrum disorders. Pediatr. Neurol. 25, 199–207. doi: 10.1016/S0887-8994(01)00276-4
Dingledine, R., and Korn, S. J. (1985). Gamma-aminobutyric acid uptake and the termination of inhibitory synaptic potentials in the rat hippocampal slice. J. Physiol. 366, 387–409.
Duarte, S. T., Armstrong, J., Roche, A., Ortez, C., Perez, A., O'Callaghan Mdel, M., et al. (2013). Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with rett syndrome. PLoS ONE 8:e68851. doi: 10.1371/journal.pone.0068851
Dupuy, S. T., and Houser, C. R. (1996). Prominent expression of two forms of glutamate decarboxylase in the embryonic and early postnatal rat hippocampal formation. J. Neurosci. 16, 6919–6932.
Durand, S., Patrizi, A., Quast, K. B., Hachigian, L., Pavlyuk, R., Saxena, A., et al. (2012). NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron 76, 1078–1090. doi: 10.1016/j.neuron.2012.12.004
Duveau, V., Laustela, S., Barth, L., Gianolini, F., Vogt, K. E., Keist, R., et al. (2011). Spatiotemporal specificity of GABAA receptor-mediated regulation of adult hippocampal neurogenesis. Eur. J. Neurosci. 34, 362–373. doi: 10.1111/j.1460-9568.2011.07782.x
Dzhala, V., Ben-Ari, Y., and Khazipov, R. (2000). Seizures accelerate anoxia-induced neuronal death in the neonatal rat hippocampus. Ann. Neurol. 48, 632–640. doi: 10.1002/1531-8249(200010)48:4<632::AID-ANA10>3.0.CO;2-3
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., et al. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213. doi: 10.1038/nm1301
Eftekhari, S., Mehvari Habibabadi, J., Najafi Ziarani, M., Hashemi Fesharaki, S. S., Gharakhani, M., Mostafavi, H., et al. (2013). Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia 54, e9–e12. doi: 10.1111/j.1528-1167.2012.03654.x
El Idrissi, A., Boukarrou, L., Dokin, C., and Brown, W. T. (2009). Taurine improves congestive functions in a mouse model of fragile X syndrome. Adv. Exp. Med. Biol. 643, 191–198. doi: 10.1007/978-0-387-75681-3_19
El Idrissi, A., Ding, X. H., Scalia, J., Trenkner, E., Brown, W. T., and Dobkin, C. (2005). Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 377, 141–146. doi: 10.1016/j.neulet.2004.11.087
Endo, K., Hori, T., Abe, S., and Asada, T. (2007). Alterations in GABA(A) receptor expression in neonatal ventral hippocampal lesioned rats: comparison of prepubertal and postpubertal periods. Synapse 61, 357–366. doi: 10.1002/syn.20393
Esclapez, M., Tillakaratne, N. J., Kaufman, D. L., Tobin, A. J., and Houser, C. R. (1994). Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J. Neurosci. 14, 1834–1855.
Evans, J. E., Frostholm, A., and Rotter, A. (1996). Embryonic and postnatal expression of four gamma-aminobutyric acid transporter mRNAs in the mouse brain and leptomeninges. J. Comp. Neurol. 376, 431–446. doi: 10.1002/(SICI)1096-9861(19961216)376:3<431::AID-CNE6>3.0.CO;2-3
Fagiolini, M., Fritschy, J. M., Low, K., Mohler, H., Rudolph, U., and Hensch, T. K. (2004). Specific GABAA circuits for visual cortical plasticity. Science 303, 1681–1683. doi: 10.1126/science.1091032
Fagiolini, M., and Hensch, T. K. (2000). Inhibitory threshold for critical-period activation in primary visual cortex. Nature 404, 183–186. doi: 10.1038/35004582
Farmer, L. M., Le, B. N., and Nelson, D. J. (2013). CLC-3 chloride channels moderate long-term potentiation at Schaffer collateral-CA1 synapses. J. Physiol. 591, 1001–1015. doi: 10.1113/jphysiol.2012.243485
Fatemi, S. H., Folsom, T. D., Rooney, R. J., and Thuras, P. D. (2013). mRNA and protein expression for novel GABAA receptors theta and rho2 are altered in schizophrenia and mood disorders; relevance to FMRP-mGluR5 signaling pathway. Transl. Psychiatry 3, e271. doi: 10.1038/tp.2013.46
Fatemi, S. H., Halt, A. R., Stary, J. M., Kanodia, R., Schulz, S. C., and Realmuto, G. R. (2002). Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 52, 805–810. doi: 10.1016/S0006-3223(02)01430-0
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rooney, R. J., Patel, D. H., and Thuras, P. D. (2010). mRNA and protein levels for GABAAalpha4, alpha5, beta1 and GABABR1 receptors are altered in brains from subjects with autism. J. Autism. Dev. Disord. 40, 743–750. doi: 10.1007/s10803-009-0924-z
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., and Thuras, P. D. (2009). GABA(A) receptor downregulation in brains of subjects with autism. J. Autism Dev. Disord. 39, 223–230. doi: 10.1007/s10803-008-0646-7
Fatemi, S. H., Stary, J. M., Earle, J. A., Araghi-Niknam, M., and Eagan, E. (2005). GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr. Res. 72, 109–122. doi: 10.1016/j.schres.2004.02.017
Feldblum, S., Erlander, M. G., and Tobin, A. J. (1993). Different distributions of GAD65 and GAD67 mRNAs suggest that the two glutamate decarboxylases play distinctive functional roles. J. Neurosci. Res. 34, 689–706. doi: 10.1002/jnr.490340612
Feldman, D. E. (2009). Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55. doi: 10.1146/annurev.neuro.051508.135516
Feldman, D. E. (2012). The spike-timing dependence of plasticity. Neuron 75, 556–571. doi: 10.1016/j.neuron.2012.08.001
Fenalti, G., Law, R. H., Buckle, A. M., Langendorf, C., Tuck, K., Rosado, C. J., et al. (2007). GABA production by glutamic acid decarboxylase is regulated by a dynamic catalytic loop. Nat. Struct. Mol. Biol. 14, 280–286. doi: 10.1038/nsmb1228
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 10, 411–413. doi: 10.1038/nn1860
Fisahn, A., Neddens, J., Yan, L., and Buonanno, A. (2009). Neuregulin-1 modulates hippocampal gamma oscillations: implications for schizophrenia. Cereb. Cortex 19, 612–618. doi: 10.1093/cercor/bhn107
Fitzgerald, M. (2005). The development of nociceptive circuits. Nat. Rev. Neurosci. 6, 507–520. doi: 10.1038/nrn1701
Fiumara, A., Pittala, A., Cocuzza, M., and Sorge, G. (2010). Epilepsy in patients with Angelman syndrome. Ital. J. Pediatr. 36:31. doi: 10.1186/1824-7288-36-31
Fiumelli, H., Cancedda, L., and Poo, M. M. (2005). Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron 48, 773–786. doi: 10.1016/j.neuron.2005.10.025
Fritschy, J. M., and Panzanelli, P. (2014). GABA receptors and plasticity of inhibitory neurotransmission in the central nervous system. Eur. J. Neurosci. doi: 10.1111/ejn.12534. [Epub ahead of print].
Fritschy, J. M., Paysan, J., Enna, A., and Mohler, H. (1994). Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J. Neurosci. 14, 5302–5324.
Fung, S. J., Webster, M. J., Sivagnanasundaram, S., Duncan, C., Elashoff, M., and Weickert, C. S. (2010). Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 167, 1479–1488. doi: 10.1176/appi.ajp.2010.09060784
Gagnon, M., Bergeron, M. J., Lavertu, G., Castonguay, A., Tripathy, S., Bonin, R. P., et al. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19, 1524–1528. doi: 10.1038/nm.3356
Galanopoulou, A. S. (2008). Dissociated gender-specific effects of recurrent seizures on GABA signaling in CA1 pyramidal neurons: role of GABA(A) receptors. J. Neurosci. 28, 1557–1567. doi: 10.1523/JNEUROSCI.5180-07.2008
Gantois, I., Vandesompele, J., Speleman, F., Reyniers, E., D'Hooge, R., Severijnen, L. A., et al. (2006). Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol. Dis. 21, 346–357. doi: 10.1016/j.nbd.2005.07.017
Ge, S., Goh, E. L., Sailor, K. A., Kitabatake, Y., Ming, G. L., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593. doi: 10.1038/nature04404
Ge, S., Sailor, K. A., Ming, G. L., and Song, H. (2008). Synaptic integration and plasticity of new neurons in the adult hippocampus. J. Physiol. 586, 3759–3765. doi: 10.1113/jphysiol.2008.155655
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Gimpl, G., and Fahrenholz, F. (2001). The oxytocin receptor system: structure, function, and regulation. Physiol. Rev. 81, 629–683.
Gittis, A. H., Leventhal, D. K., Fensterheim, B. A., Pettibone, J. R., Berke, J. D., and Kreitzer, A. C. (2011). Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. J. Neurosci. 31, 15727–15731. doi: 10.1523/JNEUROSCI.3875-11.2011
Goncalves, J. T., Anstey, J. E., Golshani, P., and Portera-Cailliau, C. (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 16, 903–909. doi: 10.1038/nn.3415
Gonzalez-Burgos, G., Fish, K. N., and Lewis, D. A. (2011). GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. 2011, 723184. doi: 10.1155/2011/723184
Goto, N., Yoshimura, R., Moriya, J., Kakeda, S., Ueda, N., Ikenouchi-Sugita, A., et al. (2009). Reduction of brain gamma-aminobutyric acid (GABA) concentrations in early-stage schizophrenia patients: 3T Proton MRS study. Schizophr. Res. 112, 192–193. doi: 10.1016/j.schres.2009.04.026
Griebel, G., and Holmes, A. (2013). 50 years of hurdles and hope in anxiolytic drug discovery. Nat. Rev. Drug Discov. 12, 667–687. doi: 10.1038/nrd4075
Grover, L. M., and Yan, C. (1999). Blockade of GABAA receptors facilitates induction of NMDA receptor-independent long-term potentiation. J. Neurophysiol. 81, 2814–2822.
Guidotti, A., Auta, J., Davis, J. M., Di-Giorgi-Gerevini, V., Dwivedi, Y., Grayson, D. R., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 57, 1061–1069. doi: 10.1001/archpsyc.57.11.1061
Guptill, J. T., Booker, A. B., Gibbs, T. T., Kemper, T. L., Bauman, M. L., and Blatt, G. J. (2007). [3H]-flunitrazepam-labeled benzodiazepine binding sites in the hippocampal formation in autism: a multiple concentration autoradiographic study. J. Autism Dev. Disord. 37, 911–920. doi: 10.1007/s10803-006-0226-7
Hadjikhani, N., Zurcher, N. R., Rogier, O., Ruest, T., Hippolyte, L., Ben-Ari, Y., et al. (2013). Improving emotional face perception in autism with diuretic bumetanide: a proof-of-concept behavioral and functional brain imaging pilot study. Autism. doi: 10.1177/1362361313514141. [Epub ahead of print].
Hagerman, R. J., Berry-Kravis, E., Kaufmann, W. E., Ono, M. Y., Tartaglia, N., Lachiewicz, A., et al. (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123, 378–390. doi: 10.1542/peds.2008-0317
Haglund, M. M., and Hochman, D. W. (2005). Furosemide and mannitol suppression of epileptic activity in the human brain. J. Neurophysiol. 94, 907–918. doi: 10.1152/jn.00944.2004
Han, S., Tai, C., Jones, C. J., Scheuer, T., and Catterall, W. A. (2014). Enhancement of Inhibitory Neurotransmission by GABAA Receptors Having alpha2,3-Subunits Ameliorates Behavioral Deficits in a Mouse Model of Autism. Neuron 81, 1282–1289. doi: 10.1016/j.neuron.2014.01.016
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Hannan, A. J. (2014). Review: environmental enrichment and brain repair: harnessing the therapeutic effects of cognitive stimulation and physical activity to enhance experience-dependent plasticity. Neuropathol. Appl. Neurobiol. 40, 13–25. doi: 10.1111/nan.12102
Harada, M., Taki, M. M., Nose, A., Kubo, H., Mori, K., Nishitani, H., et al. (2011). Non-invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-editing proton MR spectroscopy using a clinical 3 tesla instrument. J. Autism Dev. Disord. 41, 447–454. doi: 10.1007/s10803-010-1065-0
Harauzov, A., Spolidoro, M., Dicristo, G., De Pasquale, R., Cancedda, L., Pizzorusso, T., et al. (2010). Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J. Neurosci. 30, 361–371. doi: 10.1523/JNEUROSCI.2233-09.2010
Harte, M. K., Powell, S. B., Swerdlow, N. R., Geyer, M. A., and Reynolds, G. P. (2007). Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J. Neural Transm. 114, 893–898. doi: 10.1007/s00702-007-0627-6
Hashimoto, T., Bazmi, H. H., Mirnics, K., Wu, Q., Sampson, A. R., and Lewis, D. A. (2008). Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 165, 479–489. doi: 10.1176/appi.ajp.2007.07081223
He, Q., Nomura, T., Xu, J., and Contractor, A. (2014). The developmental switch in GABA polarity is delayed in Fragile X mice. J. Neurosci. 34, 446–450. doi: 10.1523/JNEUROSCI.4447-13.2014
He, S., Ma, J., Liu, N., and Yu, X. (2010). Early enriched environment promotes neonatal GABAergic neurotransmission and accelerates synapse maturation. J. Neurosci. 30, 7910–7916. doi: 10.1523/JNEUROSCI.6375-09.2010
Heimel, J. A., Van Versendaal, D., and Levelt, C. N. (2011). The role of GABAergic inhibition in ocular dominance plasticity. Neural Plast. 2011, 391763. doi: 10.1155/2011/391763
Heine, M., Groc, L., Frischknecht, R., Beique, J. C., Lounis, B., Rumbaugh, G., et al. (2008). Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science 320, 201–205. doi: 10.1126/science.1152089
Hensch, T. K. (2004). Critical period regulation. Annu. Rev. Neurosci. 27, 549–579. doi: 10.1146/annurev.neuro.27.070203.144327
Hensch, T. K., Fagiolini, M., Mataga, N., Stryker, M. P., Baekkeskov, S., and Kash, S. F. (1998). Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 282, 1504–1508. doi: 10.1126/science.282.5393.1504
Heulens, I., D'Hulst, C., Van Dam, D., De Deyn, P. P., and Kooy, R. F. (2012). Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav. Brain Res. 229, 244–249. doi: 10.1016/j.bbr.2012.01.031
Hikida, T., Jaaro-Peled, H., Seshadri, S., Oishi, K., Hookway, C., Kong, S., et al. (2007). Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. U.S.A. 104, 14501–14506. doi: 10.1073/pnas.0704774104
Hirai, H., and Okada, Y. (1993). Ipsilateral corticotectal pathway inhibits the formation of long-term potentiation (LTP) in the rat superior colliculus through GABAergic mechanism. Brain Res. 629, 23–30. doi: 10.1016/0006-8993(93)90476-4
Hochman, D. W. (2012). The extracellular space and epileptic activity in the adult brain: explaining the antiepileptic effects of furosemide and bumetanide. Epilepsia 53(Suppl. 1), 18–25. doi: 10.1111/j.1528-1167.2012.03471.x
Houston, C. M., Hosie, A. M., and Smart, T. G. (2008). Distinct regulation of beta2 and beta3 subunit-containing cerebellar synaptic GABAA receptors by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 28, 7574–7584. doi: 10.1523/JNEUROSCI.5531-07.2008
Huang, R. Q., and Dillon, G. H. (1998). Maintenance of recombinant type A gamma-aminobutyric acid receptor function: role of protein tyrosine phosphorylation and calcineurin. J. Pharmacol. Exp. Ther. 286, 243–255.
Hubner, C. A., and Holthoff, K. (2013). Anion transport and GABA signaling. Front. Cell Neurosci. 7:177. doi: 10.3389/fncel.2013.00177
Hyde, T. M., Lipska, B. K., Ali, T., Mathew, S. V., Law, A. J., Metitiri, O. E., et al. (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011
Inoue, T., Haque, Z., Lukowiak, K., and Syed, N. I. (2001). Hypoxia-induced respiratory patterned activity in Lymnaea originates at the periphery. J. Neurophysiol. 86, 156–163.
Ishihara, K., Amano, K., Takaki, E., Shimohata, A., Sago, H., Epstein, C. J., et al. (2010). Enlarged brain ventricles and impaired neurogenesis in the Ts1Cje and Ts2Cje mouse models of Down syndrome. Cereb. Cortex 20, 1131–1143. doi: 10.1093/cercor/bhp176
Jambaque, I., Chiron, C., Dumas, C., Mumford, J., and Dulac, O. (2000). Mental and behavioural outcome of infantile epilepsy treated by vigabatrin in tuberous sclerosis patients. Epilepsy Res. 38, 151–160. doi: 10.1016/S0920-1211(99)00082-0
Jankovic, J. (2001). Tourette's syndrome. N. Engl. J. Med. 345, 1184–1192. doi: 10.1056/NEJMra010032
Jian, L., Nagarajan, L., De Klerk, N., Ravine, D., Bower, C., Anderson, A., et al. (2006). Predictors of seizure onset in Rett syndrome. J. Pediatr. 149, 542–547. doi: 10.1016/j.jpeds.2006.06.015
Jin, H., Wu, H., Osterhaus, G., Wei, J., Davis, K., Sha, D., et al. (2003). Demonstration of functional coupling between gamma -aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 100, 4293–4298. doi: 10.1073/pnas.0730698100
Jin, X., Cui, N., Zhong, W., Jin, X. T., and Jiang, C. (2013). GABAergic synaptic inputs of locus coeruleus neurons in wild-type and Mecp2-null mice. Am. J. Physiol. Cell Physiol. 304, C844–C857. doi: 10.1152/ajpcell.00399.2012
Johnson, H., Wiggs, L., Stores, G., and Huson, S. M. (2005). Psychological disturbance and sleep disorders in children with neurofibromatosis type 1. Dev. Med. Child Neurol. 47, 237–242. doi: 10.1017/S0012162205000460
Jones, J. D. (1961). Aspects of respiration in Planorbis corneus L. and Lymnaea stagnalis L. (Gastropoda: Pulmonata). Comp. Biochem. Physiol. 4, 1–29. doi: 10.1016/0010-406X(61)90042-1
Jones, M. V., and Westbrook, G. L. (1997). Shaping of IPSCs by endogenous calcineurin activity. J. Neurosci. 17, 7626–7633.
Jovanovic, J. N., Thomas, P., Kittler, J. T., Smart, T. G., and Moss, S. J. (2004). Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J. Neurosci. 24, 522–530. doi: 10.1523/JNEUROSCI.3606-03.2004
Juge, N., Muroyama, A., Hiasa, M., Omote, H., and Moriyama, Y. (2009). Vesicular inhibitory amino acid transporter is a Cl-/gamma-aminobutyrate Co-transporter. J. Biol. Chem. 284, 35073–35078. doi: 10.1074/jbc.M109.062414
Kahle, K. T., Barnett, S. M., Sassower, K. C., and Staley, K. J. (2009). Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na(+)-K(+)-2Cl(-) cotransporter NKCC1. J. Child. Neurol. 24, 572–576. doi: 10.1177/0883073809333526
Kahle, K. T., Deeb, T. Z., Puskarjov, M., Silayeva, L., Liang, B., Kaila, K., et al. (2013). Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 36, 726–737. doi: 10.1016/j.tins.2013.08.006
Kahle, K. T., and Staley, K. J. (2008). The bumetanide-sensitive Na-K-2Cl cotransporter NKCC1 as a potential target of a novel mechanism-based treatment strategy for neonatal seizures. Neurosurg. Focus. 25, E22. doi: 10.3171/FOC/2008/25/9/E22
Kaibara, T., and Leung, L. S. (1993). Basal versus apical dendritic long-term potentiation of commissural afferents to hippocampal CA1: a current-source density study. J. Neurosci. 13, 2391–2404.
Kaila, K., Lamsa, K., Smirnov, S., Taira, T., and Voipio, J. (1997). Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J. Neurosci. 17, 7662–7672.
Kalanithi, P. S., Zheng, W., Kataoka, Y., Difiglia, M., Grantz, H., Saper, C. B., et al. (2005). Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 13307–13312. doi: 10.1073/pnas.0502624102
Kash, S. F., Johnson, R. S., Tecott, L. H., Noebels, J. L., Mayfield, R. D., Hanahan, D., et al. (1997). Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 94, 14060–14065. doi: 10.1073/pnas.94.25.14060
Kataoka, Y., Kalanithi, P. S., Grantz, H., Schwartz, M. L., Saper, C., Leckman, J. F., et al. (2010). Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J. Comp. Neurol. 518, 277–291. doi: 10.1002/cne.22206
Kaufman, D. L., Houser, C. R., and Tobin, A. J. (1991). Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J. Neurochem. 56, 720–723. doi: 10.1111/j.1471-4159.1991.tb08211.x
Kerr, B., Silva, P. A., Walz, K., and Young, J. I. (2010). Unconventional transcriptional response to environmental enrichment in a mouse model of Rett syndrome. PLoS ONE 5:e11534. doi: 10.1371/journal.pone.0011534
Kersante, F., Rowley, S. C., Pavlov, I., Gutierrez-Mecinas, M., Semyanov, A., Reul, J. M., et al. (2013). A functional role for both -aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J. Physiol. 591, 2429–2441. doi: 10.1113/jphysiol.2012.246298
Kim, J. Y., Liu, C. Y., Zhang, F., Duan, X., Wen, Z., Song, J., et al. (2012). Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 148, 1051–1064. doi: 10.1016/j.cell.2011.12.037
Kiser, P. J., Cooper, N. G., and Mower, G. D. (1998). Expression of two forms of glutamic acid decarboxylase (GAD67 and GAD65) during postnatal development of rat somatosensory barrel cortex. J. Comp. Neurol. 402, 62–74. doi: 10.1002/(SICI)1096-9861(19981207)402:1<62::AID-CNE5>3.0.CO;2-M
Kittler, J. T., Chen, G., Kukhtina, V., Vahedi-Faridi, A., Gu, Z., Tretter, V., et al. (2008). Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor gamma2 subunit. Proc. Natl. Acad. Sci. U.S.A. 105, 3616–3621. doi: 10.1073/pnas.0707920105
Kittler, J. T., Delmas, P., Jovanovic, J. N., Brown, D. A., Smart, T. G., and Moss, S. J. (2000). Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J. Neurosci. 20, 7972–7977.
Kittler, J. T., and Moss, S. J. (2003). Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol. 13, 341–347. doi: 10.1016/S0959-4388(03)00064-3
Kleschevnikov, A. M., Belichenko, P. V., Gall, J., George, L., Nosheny, R., Maloney, M. T., et al. (2012). Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 45, 683–691. doi: 10.1016/j.nbd.2011.10.009
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 24, 8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004
Kobayashi, K., Emson, P. C., Mountjoy, C. Q., Thornton, S. N., Lawson, D. E., and Mann, D. M. (1990). Cerebral cortical calbindin D28K and parvalbumin neurones in Down's syndrome. Neurosci. Lett. 113, 17–22. doi: 10.1016/0304-3940(90)90487-T
Kondo, M., Gray, L. J., Pelka, G. J., Christodoulou, J., Tam, P. P., and Hannan, A. J. (2008). Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome–Mecp2 gene dosage effects and BDNF expression. Eur. J. Neurosci. 27, 3342–3350. doi: 10.1111/j.1460-9568.2008.06305.x
Kullmann, D. M., Ruiz, A., Rusakov, D. M., Scott, R., Semyanov, A., and Walker, M. C. (2005). Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog. Biophys. Mol. Biol. 87, 33–46. doi: 10.1016/j.pbiomolbio.2004.06.003
Lacaria, M., Spencer, C., Gu, W., Paylor, R., and Lupski, J. R. (2012). Enriched rearing improves behavioral responses of an animal model for CNV-based autistic-like traits. Hum. Mol. Genet. 21, 3083–3096. doi: 10.1093/hmg/dds124
Laurie, D. J., Wisden, W., and Seeburg, P. H. (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 12, 4151–4172.
Leblanc, J. J., and Fagiolini, M. (2011). Autism: a “critical period” disorder? Neural Plast. 2011, 921680. doi: 10.1155/2011/921680
Lee, E. J., Gibo, T. L., and Grzywacz, N. M. (2006a). Dark-rearing-induced reduction of GABA and GAD and prevention of the effect by BDNF in the mouse retina. Eur. J. Neurosci. 24, 2118–2134. doi: 10.1111/j.1460-9568.2006.05078.x
Lee, T. S., Bjornsen, L. P., Paz, C., Kim, J. H., Spencer, S. S., Spencer, D. D., et al. (2006b). GAT1 and GAT3 expression are differently localized in the human epileptogenic hippocampus. Acta Neuropathol. 111, 351–363. doi: 10.1007/s00401-005-0017-9
Lehman, M. N., Silver, R., Gladstone, W. R., Kahn, R. M., Gibson, M., and Bittman, E. L. (1987). Circadian rhythmicity restored by neural transplant. Immunocytochemical characterization of the graft and its integration with the host brain. J. Neurosci. 7, 1626–1638.
Lemonnier, E., and Ben-Ari, Y. (2010). The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 99, 1885–1888. doi: 10.1111/j.1651-2227.2010.01933.x
Lemonnier, E., Degrez, C., Phelep, M., Tyzio, R., Josse, F., Grandgeorge, M., et al. (2012). A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2, e202. doi: 10.1038/tp.2012.124
Lemonnier, E., Robin, G., Degrez, C., Tyzio, R., Grandgeorge, M., and Ben-Ari, Y. (2013). Treating Fragile X syndrome with the diuretic bumetanide: a case report. Acta Paediatr. 102, e288–e290. doi: 10.1111/apa.12235
Lerner, A., Bagic, A., Simmons, J. M., Mari, Z., Bonne, O., Xu, B., et al. (2012). Widespread abnormality of the gamma-aminobutyric acid-ergic system in Tourette syndrome. Brain 135, 1926–1936. doi: 10.1093/brain/aws104
Leschziner, G. D., Golding, J. F., and Ferner, R. E. (2013). Sleep disturbance as part of the neurofibromatosis type 1 phenotype in adults. Am. J. Med. Genet. A 161, 1319–1322. doi: 10.1002/ajmg.a.35915
Levelt, C. N., and Hubener, M. (2012). Critical-period plasticity in the visual cortex. Annu. Rev. Neurosci. 35, 309–330. doi: 10.1146/annurev-neuro-061010-113813
Lewis, D. A., Cho, R. Y., Carter, C. S., Eklund, K., Forster, S., Kelly, M. A., et al. (2008). Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am. J. Psychiatry 165, 1585–1593. doi: 10.1176/appi.ajp.2008.08030395
Lewis, D. A., Curley, A. A., Glausier, J. R., and Volk, D. W. (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35, 57–67. doi: 10.1016/j.tins.2011.10.004
Li, L. B., Chang, K. H., Wang, P. R., Hirata, R. K., Papayannopoulou, T., and Russell, D. W. (2012). Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem. Cell 11, 615–619. doi: 10.1016/j.stem.2012.08.004
Lindefors, N. (1993). Dopaminergic regulation of glutamic acid decarboxylase mRNA expression and GABA release in the striatum: a review. Prog. Neuropsychopharmacol. Biol. Psychiatry 17, 887–903. doi: 10.1016/0278-5846(93)90018-N
Liou, S. Y., and Albers, H. E. (1990). Single unit response of neurons within the hamster suprachiasmatic nucleus to GABA and low chloride perfusate during the day and night. Brain Res. Bull. 25, 93–98. doi: 10.1016/0361-9230(90)90257-Z
Lipska, B. K., Lerman, D. N., Khaing, Z. Z., Weickert, C. S., and Weinberger, D. R. (2003). Gene expression in dopamine and GABA systems in an animal model of schizophrenia: effects of antipsychotic drugs. Eur. J. Neurosci. 18, 391–402. doi: 10.1046/j.1460-9568.2003.02738.x
Liu, X., Novosedlik, N., Wang, A., Hudson, M. L., Cohen, I. L., Chudley, A. E., et al. (2009). The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur. J. Hum. Genet. 17, 228–235. doi: 10.1038/ejhg.2008.148
Lonetti, G., Angelucci, A., Morando, L., Boggio, E. M., Giustetto, M., and Pizzorusso, T. (2010). Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol. Psychiatry 67, 657–665. doi: 10.1016/j.biopsych.2009.12.022
Loscher, W., Puskarjov, M., and Kaila, K. (2013). Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69, 62–74. doi: 10.1016/j.neuropharm.2012.05.045
Lu, Y. M., Mansuy, I. M., Kandel, E. R., and Roder, J. (2000). Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 26, 197–205. doi: 10.1016/S0896-6273(00)81150-2
Luscher, B., Fuchs, T., and Kilpatrick, C. L. (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409. doi: 10.1016/j.neuron.2011.03.024
Ma, D. Q., Whitehead, P. L., Menold, M. M., Martin, E. R., Ashley-Koch, A. E., Mei, H., et al. (2005). Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am. J. Hum. Genet. 77, 377–388. doi: 10.1086/433195
Maa, E. H., Kahle, K. T., Walcott, B. P., Spitz, M. C., and Staley, K. J. (2011). Diuretics and epilepsy: will the past and present meet? Epilepsia 52, 1559–1569. doi: 10.1111/j.1528-1167.2011.03203.x
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Marrosu, F., Marrosu, G., Rachel, M. G., and Biggio, G. (1987). Paradoxical reactions elicited by diazepam in children with classic autism. Funct. Neurol. 2, 355–361.
Martinez-Cue, C., Martinez, P., Rueda, N., Vidal, R., Garcia, S., Vidal, V., et al. (2013). Reducing GABAA alpha5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome. J. Neurosci. 33, 3953–3966. doi: 10.1523/JNEUROSCI.1203-12.2013
Martinez-Granero, M. A., Garcia-Perez, A., and Montanes, F. (2010). Levetiracetam as an alternative therapy for Tourette syndrome. Neuropsychiatr. Dis. Treat. 6, 309–316. doi: 10.2147/NDT.S6371
Matsuyama, S., Taniguchi, T., Kadoyama, K., and Matsumoto, A. (2008). Long-term potentiation-like facilitation through GABAA receptor blockade in the mouse dentate gyrus in vivo. Neuroreport 19, 1809–1813. doi: 10.1097/WNR.0b013e328319ab94
Mazzuca, M., Minlebaev, M., Shakirzyanova, A., Tyzio, R., Taccola, G., Janackova, S., et al. (2011). Newborn Analgesia Mediated by Oxytocin during Delivery. Front. Cell Neurosci. 5:3. doi: 10.3389/fncel.2011.00003
McCairn, K. W., Bronfeld, M., Belelovsky, K., and Bar-Gad, I. (2009). The neurophysiological correlates of motor tics following focal striatal disinhibition. Brain 132, 2125–2138. doi: 10.1093/brain/awp142
McCauley, J. L., Olson, L. M., Delahanty, R., Amin, T., Nurmi, E. L., Organ, E. L.,et al. (2004). A linkage disequilibrium map of the 1-Mb 15q12 GABA(A) receptor subunit cluster and association to autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 131B, 51–59. doi: 10.1002/ajmg.b.30038
McDonald, B. J., Amato, A., Connolly, C. N., Benke, D., Moss, S. J., and Smart, T. G. (1998). Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat. Neurosci. 1, 23–28. doi: 10.1038/223
McNaught, K. S., and Mink, J. W. (2011). Advances in understanding and treatment of Tourette syndrome. Nat. Rev. Neurol. 7, 667–676. doi: 10.1038/nrneurol.2011.167
McOmish, C. E., Burrows, E., Howard, M., Scarr, E., Kim, D., Shin, H. S., et al. (2008). Phospholipase C-beta1 knockout mice exhibit endophenotypes modeling schizophrenia which are rescued by environmental enrichment and clozapine administration. Mol. Psychiatry 13, 661–672. doi: 10.1038/sj.mp.4002046
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., et al. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121. doi: 10.1152/jn.00826.2007
Mendez, M. A., Horder, J., Myers, J., Coghlan, S., Stokes, P., Erritzoe, D., et al. (2013). The brain GABA-benzodiazepine receptor alpha-5 subtype in autism spectrum disorder: a pilot [(11)C]Ro15-4513 positron emission tomography study. Neuropharmacology 68, 195–201. doi: 10.1016/j.neuropharm.2012.04.008
Mendez, P., Pazienti, A., Szabo, G., and Bacci, A. (2012). Direct alteration of a specific inhibitory circuit of the hippocampus by antidepressants. J. Neurosci. 32, 16616–16628. doi: 10.1523/JNEUROSCI.1720-12.2012
Menold, M. M., Shao, Y., Wolpert, C. M., Donnelly, S. L., Raiford, K. L., Martin, E. R., et al. (2001). Association analysis of chromosome 15 gabaa receptor subunit genes in autistic disorder. J. Neurogenet. 15, 245–259. doi: 10.3109/01677060109167380
Minelli, A., Brecha, N. C., Karschin, C., Debiasi, S., and Conti, F. (1995). GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J. Neurosci. 15, 7734–7746.
Ming, G. L., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702. doi: 10.1016/j.neuron.2011.05.001
Mitra, A., Blank, M., and Madison, D. V. (2012). Developmentally altered inhibition in Ts65Dn, a mouse model of Down syndrome. Brain Res. 1440, 1–8. doi: 10.1016/j.brainres.2011.12.034
Mohler, H. (2007). Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J. Neurochem. 102, 1–12. doi: 10.1111/j.1471-4159.2007.04454.x
Mohler, H. (2011). The rise of a new GABA pharmacology. Neuropharmacology 60, 1042–1049. doi: 10.1016/j.neuropharm.2010.10.020
Mori, T., Mori, K., Fujii, E., Toda, Y., Miyazaki, M., Harada, M., et al. (2012). Evaluation of the GABAergic nervous system in autistic brain: (123)I-iomazenil SPECT study. Brain Dev. 34, 648–654. doi: 10.1016/j.braindev.2011.10.007
Morita, Y., Callicott, J. H., Testa, L. R., Mighdoll, M. I., Dickinson, D., Chen, Q., et al. (2014). Characteristics of the cation cotransporter NKCC1 in human brain: alternate transcripts, expression in development, and potential relationships to brain function and schizophrenia. J. Neurosci. 34, 4929–4940. doi: 10.1523/JNEUROSCI.1423-13.2014
Morrow, B. A., Elsworth, J. D., and Roth, R. H. (2007). Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacology (Berl.) 192, 283–290. doi: 10.1007/s00213-007-0708-0
Morrow, E. M., Yoo, S. Y., Flavell, S. W., Kim, T. K., Lin, Y., Hill, R. S., et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. doi: 10.1126/science.1157657
Muir, J., Arancibia-Carcamo, I. L., Macaskill, A. F., Smith, K. R., Griffin, L. D., and Kittler, J. T. (2010). NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the gamma2 subunit. Proc. Natl. Acad. Sci. U.S.A. 107, 16679–16684. doi: 10.1073/pnas.1000589107
Nag, N., Moriuchi, J. M., Peitzman, C. G., Ward, B. C., Kolodny, N. H., and Berger-Sweeney, J. E. (2009). Environmental enrichment alters locomotor behaviour and ventricular volume in Mecp2 1lox mice. Behav. Brain Res. 196, 44–48. doi: 10.1016/j.bbr.2008.07.008
Nakatani, J., Tamada, K., Hatanaka, F., Ise, S., Ohta, H., Inoue, K., et al. (2009). Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137, 1235–1246. doi: 10.1016/j.cell.2009.04.024
Namchuk, M., Lindsay, L., Turck, C. W., Kanaani, J., and Baekkeskov, S. (1997). Phosphorylation of serine residues 3, 6, 10, and 13 distinguishes membrane anchored from soluble glutamic acid decarboxylase 65 and is restricted to glutamic acid decarboxylase 65alpha. J. Biol. Chem. 272, 1548–1557. doi: 10.1074/jbc.272.3.1548
Nithianantharajah, J., and Hannan, A. J. (2006). Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat. Rev. Neurosci. 7, 697–709. doi: 10.1038/nrn1970
Nunez, J. L., and McCarthy, M. M. (2007). Evidence for an extended duration of GABA-mediated excitation in the developing male versus female hippocampus. Dev. Neurobiol. 67, 1879–1890. doi: 10.1002/dneu.20567
Olmos-Serrano, J. L., Corbin, J. G., and Burns, M. P. (2011). The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev. Neurosci. 33, 395–403. doi: 10.1159/000332884
Olmos-Serrano, J. L., Paluszkiewicz, S. M., Martin, B. S., Kaufmann, W. E., Corbin, J. G., and Huntsman, M. M. (2010). Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 30, 9929–9938. doi: 10.1523/JNEUROSCI.1714-10.2010
Ongur, D., Prescot, A. P., McCarthy, J., Cohen, B. M., and Renshaw, P. F. (2010). Elevated gamma-aminobutyric acid levels in chronic schizophrenia. Biol. Psychiatry 68, 667–670. doi: 10.1016/j.biopsych.2010.05.016
Ostendorf, A. P., Gutmann, D. H., and Weisenberg, J. L. (2013). Epilepsy in individuals with neurofibromatosis type 1. Epilepsia 54, 1810–1814. doi: 10.1111/epi.12348
Pallotto, M., Nissant, A., Fritschy, J. M., Rudolph, U., Sassoe-Pognetto, M., Panzanelli, P., et al. (2012). Early formation of GABAergic synapses governs the development of adult-born neurons in the olfactory bulb. J. Neurosci. 32, 9103–9115. doi: 10.1523/JNEUROSCI.0214-12.2012
Paluszkiewicz, S. M., Olmos-Serrano, J. L., Corbin, J. G., and Huntsman, M. M. (2011). Impaired inhibitory control of cortical synchronization in fragile X syndrome. J. Neurophysiol. 106, 2264–2272. doi: 10.1152/jn.00421.2011
Pearl, P. L., Shukla, L., Theodore, W. H., Jakobs, C., and Michael Gibson, K. (2011). Epilepsy in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. Brain Dev. 33, 796–805. doi: 10.1016/j.braindev.2011.04.013
Penagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Perez-Cremades, D., Hernandez, S., Blasco-Ibanez, J. M., Crespo, C., Nacher, J., and Varea, E. (2010). Alteration of inhibitory circuits in the somatosensory cortex of Ts65Dn mice, a model for Down's syndrome. J. Neural Transm. 117, 445–455. doi: 10.1007/s00702-010-0376-9
Petrini, E. M., Lu, J., Cognet, L., Lounis, B., Ehlers, M. D., and Choquet, D. (2009). Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron 63, 92–105. doi: 10.1016/j.neuron.2009.05.025
Pierri, J. N., Chaudry, A. S., Woo, T. U., and Lewis, D. A. (1999). Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am. J. Psychiatry 156, 1709–1719.
Pinal, C. S., and Tobin, A. J. (1998). Uniqueness and redundancy in GABA production. Perspect. Dev. Neurobiol. 5, 109–118.
Pizzarelli, R., and Cherubini, E. (2011). Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011, 297153. doi: 10.1155/2011/297153
Popp, A., Urbach, A., Witte, O. W., and Frahm, C. (2009). Adult and embryonic GAD transcripts are spatiotemporally regulated during postnatal development in the rat brain. PLoS ONE 4:e4371. doi: 10.1371/journal.pone.0004371
Potier, M. C., Braudeau, J., Dauphinot, L., and Delatour, B. (2014). Reducing GABAergic inhibition restores cognitive functions in a mouse model of Down syndrome. CNS Neurol. Disord. Drug Targets 13, 8–15. doi: 10.2174/18715273113126660185
Potkin, S. G., Turner, J. A., Guffanti, G., Lakatos, A., Fallon, J. H., Nguyen, D. D., et al (2009). A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr. Bull. 35, 96–108. doi: 10.1093/schbul/sbn155
Pouille, F., and Scanziani, M. (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293, 1159–1163. doi: 10.1126/science.1060342
Poulter, M. O., Barker, J. L., O'Carroll, A. M., Lolait, S. J., and Mahan, L. C. (1992). Differential and transient expression of GABAA receptor alpha-subunit mRNAs in the developing rat CNS. J. Neurosci. 12, 2888–2900.
Pueschel, S. M., Louis, S., and McKnight, P. (1991). Seizure disorders in Down syndrome. Arch. Neurol. 48, 318–320. doi: 10.1001/archneur.1991.00530150088024
Ramos, B., Lopez-Tellez, J. F., Vela, J., Baglietto-Vargas, D., Del Rio, J. C., Ruano, D., et al. (2004). Expression of alpha 5 GABAA receptor subunit in developing rat hippocampus. Brain Res. Dev. Brain Res. 151, 87–98. doi: 10.1016/j.devbrainres.2004.04.003
Restivo, L., Ferrari, F., Passino, E., Sgobio, C., Bock, J., Oostra, B. A., et al. (2005). Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 11557–11562. doi: 10.1073/pnas.0504984102
Reynolds, G. P., and Warner, C. E. (1988). Amino acid neurotransmitter deficits in adult Down's syndrome brain tissue. Neurosci. Lett. 94, 224–227. doi: 10.1016/0304-3940(88)90299-6
Reynolds, S., Urruela, M., and Devine, D. P. (2013). Effects of environmental enrichment on repetitive behaviors in the BTBR T+tf/J mouse model of autism. Autism Res. 6, 337–343. doi: 10.1002/aur.1298
Rissman, R. A., and Mobley, W. C. (2011). Implications for treatment: GABAA receptors in aging, Down syndrome and Alzheimer's disease. J. Neurochem. 117, 613–622. doi: 10.1111/j.1471-4159.2011.07237.x
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Roberts, E., and Krause, D. N. (1982). Gamma-Aminobutyric acid system in cardiovascular and cerebrovascular function. Isr. J. Med. Sci. 18, 75–81.
Rojas, D. C., Singel, D., Steinmetz, S., Hepburn, S., and Brown, M. S. (2013). Decreased left perisylvian GABA concentration in children with autism and unaffected siblings. Neuroimage 86, 28–34. doi: 10.1016/j.neuroimage.2013.01.045
Rosenzweig, M. R., Bennett, E. L., Hebert, M., and Morimoto, H. (1978). Social grouping cannot account for cerebral effects of enriched environments. Brain Res. 153, 563–576. doi: 10.1016/0006-8993(78)90340-2
Roth, F. C., and Draguhn, A. (2012). GABA metabolism and transport: effects on synaptic efficacy. Neural Plast. 2012:805830. doi: 10.1155/2012/805830
Roth, T. C. 2nd. Ladage, L. D., Freas, C. A., and Pravosudov, V. V. (2012). Variation in memory and the hippocampus across populations from different climates: a common garden approach. Proc. Biol. Sci. 279, 402–410. doi: 10.1098/rspb.2011.1020
Rudolph, U., Crestani, F., Benke, D., Brunig, I., Benson, J. A., Fritschy, J. M., et al. (1999). Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 401, 796–800. doi: 10.1038/44579
Rueda, N., Florez, J., and Martinez-Cue, C. (2008). Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neurosci. Lett. 433, 22–27. doi: 10.1016/j.neulet.2007.12.039
Rueda, N., Mostany, R., Pazos, A., Florez, J., and Martinez-Cue, C. (2005). Cell proliferation is reduced in the dentate gyrus of aged but not young Ts65Dn mice, a model of Down syndrome. Neurosci. Lett. 380, 197–201. doi: 10.1016/j.neulet.2005.01.039
Sale, A., Berardi, N., and Maffei, L. (2014). Environment and brain plasticity: towards an endogenous pharmacotherapy. Physiol. Rev. 94, 189–234. doi: 10.1152/physrev.00036.2012
Sale, A., Berardi, N., Spolidoro, M., Baroncelli, L., and Maffei, L. (2010). GABAergic inhibition in visual cortical plasticity. Front. Cell Neurosci. 4:10. doi: 10.3389/fncel.2010.00010
Saliba, R. S., Kretschmannova, K., and Moss, S. J. (2012). Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 31, 2937–2951. doi: 10.1038/emboj.2012.109
Saliba, R. S., Michels, G., Jacob, T. C., Pangalos, M. N., and Moss, S. J. (2007). Activity-dependent ubiquitination of GABA(A) receptors regulates their accumulation at synaptic sites. J. Neurosci. 27, 13341–13351. doi: 10.1523/JNEUROSCI.3277-07.2007
Sankar, R., and Painter, M. J. (2005). Neonatal seizures: after all these years we still love what doesn't work. Neurology 64, 776–777. doi: 10.1212/01.WNL.0000157320.78071.6D
Schmidt, M. J., and Mirnics, K. (2012). Modeling interneuron dysfunction in schizophrenia. Dev. Neurosci. 34, 152–158. doi: 10.1159/000336731
Schneider, T., Turczak, J., and Przewlocki, R. (2006). Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology 31, 36–46. doi: 10.1038/sj.npp.1300767
Seidl, R., Cairns, N., Singewald, N., Kaehler, S. T., and Lubec, G. (2001). Differences between GABA levels in Alzheimer's disease and Down syndrome with Alzheimer-like neuropathology. Naunyn. Schmiedebergs Arch. Pharmacol. 363, 139–145. doi: 10.1007/s002100000346
Selby, L., Zhang, C., and Sun, Q. Q. (2007). Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci. Lett. 412, 227–232. doi: 10.1016/j.neulet.2006.11.062
Semyanov, A., Walker, M. C., and Kullmann, D. M. (2003). GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat. Neurosci. 6, 484–490. doi: 10.1038/nn1043
Sernagor, E., Chabrol, F., Bony, G., and Cancedda, L. (2010). GABAergic control of neurite outgrowth and remodeling during development and adult neurogenesis: general rules and differences in diverse systems. Front. Cell Neurosci. 4:11. doi: 10.3389/fncel.2010.00011
Shao, Y., Cuccaro, M. L., Hauser, E. R., Raiford, K. L., Menold, M. M., Wolpert, C. M., et al. (2003). Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am. J. Hum. Genet. 72, 539–548. doi: 10.1086/367846
Shilyansky, C., Karlsgodt, K. H., Cummings, D. M., Sidiropoulou, K., Hardt, M., James, A. S., et al. (2010). Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proc. Natl. Acad. Sci. U.S.A. 107, 13141–13146. doi: 10.1073/pnas.1004829107
Shprecher, D., and Kurlan, R. (2009). The management of tics. Mov. Disord. 24, 15–24. doi: 10.1002/mds.22378
Siarey, R. J., Carlson, E. J., Epstein, C. J., Balbo, A., Rapoport, S. I., and Galdzicki, Z. (1999). Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 38, 1917–1920. doi: 10.1016/S0028-3908(99)00083-0
Sihra, T. S., and Rodriguez-Moreno, A. (2011). Metabotropic actions of kainate receptors in the control of GABA release. Adv. Exp. Med. Biol. 717, 1–10. doi: 10.1007/978-1-4419-9557-5_1
Simon, J., Wakimoto, H., Fujita, N., Lalande, M., and Barnard, E. A. (2004). Analysis of the set of GABA(A) receptor genes in the human genome. J. Biol. Chem. 279, 41422–41435. doi: 10.1074/jbc.M401354200
Smigielska-Kuzia, J., Bockowski, L., Sobaniec, W., Kulak, W., and Sendrowski, K. (2010). Amino acid metabolic processes in the temporal lobes assessed by proton magnetic resonance spectroscopy (1H MRS) in children with Down syndrome. Pharmacol. Rep. 62, 1070–1077. doi: 10.1016/S1734-1140(10)70369-8
Song, J., Zhong, C., Bonaguidi, M. A., Sun, G. J., Hsu, D., Gu, Y., et al. (2012). Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489, 150–154. doi: 10.1038/nature11306
Stagni, F., Magistretti, J., Guidi, S., Ciani, E., Mangano, C., Calza, L., et al. (2013). Pharmacotherapy with fluoxetine restores functional connectivity from the dentate gyrus to field CA3 in the Ts65Dn mouse model of down syndrome. PLoS ONE 8:e61689. doi: 10.1371/journal.pone.0061689
Succol, F., Fiumelli, H., Benfenati, F., Cancedda, L., and Barberis, A. (2012). Intracellular chloride concentration influences the GABAA receptor subunit composition. Nat. Commun. 3, 738. doi: 10.1038/ncomms1744
Sur, C., Quirk, K., Dewar, D., Atack, J., and McKernan, R. (1998). Rat and human hippocampal alpha5 subunit-containing gamma-aminobutyric AcidA receptors have alpha5 beta3 gamma2 pharmacological characteristics. Mol. Pharmacol. 54, 928–933.
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Talos, D. M., Sun, H., Kosaras, B., Joseph, A., Folkerth, R. D., Poduri, A., et al. (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 71, 539–551. doi: 10.1002/ana.22696
Tao, R., Li, C., Newburn, E. N., Ye, T., Lipska, B. K., Herman, M. M., et al. (2012). Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J. Neurosci. 32, 5216–5222. doi: 10.1523/JNEUROSCI.4626-11.2012
Tarasenko, A., Krupko, O., and Himmelreich, N. (2014). New insights into molecular mechanism(s) underlying the presynaptic action of nitric oxide on GABA release. Biochim. Biophys. Acta. 1840, 1923–1932. doi: 10.1016/j.bbagen.2014.01.030
Terunuma, M., Jang, I. S., Ha, S. H., Kittler, J. T., Kanematsu, T., Jovanovic, J. N., et al. (2004). GABAA receptor phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. J. Neurosci. 24, 7074–7084. doi: 10.1523/JNEUROSCI.1323-04.2004
Tian, N., Petersen, C., Kash, S., Baekkeskov, S., Copenhagen, D., and Nicoll, R. (1999). The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc. Natl. Acad. Sci. U.S.A. 96, 12911–12916. doi: 10.1073/pnas.96.22.12911
Tian, Y., Gunther, J. R., Liao, I. H., Liu, D., Ander, B. P., Stamova, B. S., et al. (2011). GABA- and acetylcholine-related gene expression in blood correlate with tic severity and microarray evidence for alternative splicing in Tourette syndrome: a pilot study. Brain Res. 1381, 228–236. doi: 10.1016/j.brainres.2011.01.026
Tollner, K., Brandt, C., Topfer, M., Brunhofer, G., Erker, T., Gabriel, M., et al. (2014). A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann Neurol. 75, 550–562. doi: 10.1002/ana.24124
Triller, A., and Choquet, D. (2005). Surface trafficking of receptors between synaptic and extrasynaptic membranes: and yet they do move! Trends Neurosci. 28, 133–139. doi: 10.1016/j.tins.2005.01.001
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hubner, C. A., Represa, A., et al. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792. doi: 10.1126/science.1133212
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Urbach, A., Bar-Nur, O., Daley, G. Q., and Benvenisty, N. (2010). Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem. Cell 6, 407–411. doi: 10.1016/j.stem.2010.04.005
Uusi-Oukari, M., and Korpi, E. R. (2010). Regulation of GABA(A) receptor subunit expression by pharmacological agents. Pharmacol. Rev. 62, 97–135. doi: 10.1124/pr.109.002063
Varju, P., Katarova, Z., Madarasz, E., and Szabo, G. (2002). Sequential induction of embryonic and adult forms of glutamic acid decarboxylase during in vitro-induced neurogenesis in cloned neuroectodermal cell-line, NE-7C2. J. Neurochem. 80, 605–615. doi: 10.1046/j.0022-3042.2001.00733.x
Violante, I. R., Ribeiro, M. J., Edden, R. A., Guimaraes, P., Bernardino, I., Rebola, J., et al. (2013). GABA deficit in the visual cortex of patients with neurofibromatosis type 1: genotype-phenotype correlations and functional impact. Brain 136, 918–925. doi: 10.1093/brain/aws368
Vithlani, M., Terunuma, M., and Moss, S. J. (2011). The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol. Rev. 91, 1009–1022. doi: 10.1152/physrev.00015.2010
Vogel, K. R., Pearl, P. L., Theodore, W. H., McCarter, R. C., Jakobs, C., and Gibson, K. M. (2013). Thirty years beyond discovery—clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J. Inherit. Metab. Dis. 36, 401–410. doi: 10.1007/s10545-012-9499-5
Voituron, N., and Hilaire, G. (2011). The benzodiazepine Midazolam mitigates the breathing defects of Mecp2-deficient mice. Respir. Physiol. Neurobiol. 177, 56–60. doi: 10.1016/j.resp.2011.02.002
Volk, D. W., Pierri, J. N., Fritschy, J. M., Auh, S., Sampson, A. R., and Lewis, D. A. (2002). Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb. Cortex 12, 1063–1070. doi: 10.1093/cercor/12.10.1063
Volpe, J. J. (2009). Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 8, 110–124. doi: 10.1016/S1474-4422(08)70294-1
Wagner, S., Castel, M., Gainer, H., and Yarom, Y. (1997). GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature 387, 598–603. doi: 10.1038/42468
Walker, M. C., and Semyanov, A. (2008). Regulation of excitability by extrasynaptic GABA(A) receptors. Results Probl. Cell Differ. 44, 29–48. doi: 10.1007/400_2007_030
Wang, D. D., and Kriegstein, A. R. (2009). Defining the role of GABA in cortical development. J. Physiol. 587, 1873–1879. doi: 10.1113/jphysiol.2008.167635
Wang, D. D., Krueger, D. D., and Bordey, A. (2003a). GABA depolarizes neuronal progenitors of the postnatal subventricular zone via GABAA receptor activation. J. Physiol. 550, 785–800. doi: 10.1113/jphysiol.2003.042572
Wang, J., Liu, S., Haditsch, U., Tu, W., Cochrane, K., Ahmadian, G., et al. (2003b). Interaction of calcineurin and type-A GABA receptor gamma 2 subunits produces long-term depression at CA1 inhibitory synapses. J. Neurosci. 23, 826–836.
Wang, L., Kitai, S. T., and Xiang, Z. (2006a). Activity-dependent bidirectional modification of inhibitory synaptic transmission in rat subthalamic neurons. J. Neurosci. 26, 7321–7327. doi: 10.1523/JNEUROSCI.4656-05.2006
Wang, Q., Liu, L., Pei, L., Ju, W., Ahmadian, G., Lu, J., et al. (2003c). Control of synaptic strength, a novel function of Akt. Neuron 38, 915–928. doi: 10.1016/S0896-6273(03)00356-8
Wang, W., Gong, N., and Xu, T. L. (2006b). Downregulation of KCC2 following LTP contributes to EPSP-spike potentiation in rat hippocampus. Biochem. Biophys. Res. Commun. 343, 1209–1215. doi: 10.1016/j.bbrc.2006.03.038
Watanabe, M., Maemura, K., Kanbara, K., Tamayama, T., and Hayasaki, H. (2002). GABA and GABA receptors in the central nervous system and other organs. Int. Rev. Cytol. 213, 1–47. doi: 10.1016/S0074-7696(02)13011-7
Westmark, C. J., Westmark, P. R., and Malter, J. S. (2010). Alzheimer's disease and Down syndrome rodent models exhibit audiogenic seizures. J. Alzheimers Dis. 20, 1009–1013. doi: 10.3233/JAD-2010-100087
Whittington, M. A., Cunningham, M. O., Lebeau, F. E., Racca, C., and Traub, R. D. (2011). Multiple origins of the cortical gamma rhythm. Dev. Neurobiol. 71, 92–106. doi: 10.1002/dneu.20814
Whittle, N., Sartori, S. B., Dierssen, M., Lubec, G., and Singewald, N. (2007). Fetal Down syndrome brains exhibit aberrant levels of neurotransmitters critical for normal brain development. Pediatrics 120, e1465–e1471. doi: 10.1542/peds.2006-3448
Wigstrom, H., and Gustafsson, B. (1983). Large long-lasting potentiation in the dentate gyrus in vitro during blockade of inhibition. Brain Res. 275, 153–158. doi: 10.1016/0006-8993(83)90428-6
Williams, D., Stern, J. S., Grabecki, K., Simmons, H., and Robertson, M. M. (2013). Epilepsy in Tourette Syndrome. J. Neurol. Neurosurg. Psychiatry 84:e1. doi: 10.1136/jnnp-2013-306103.31
Wojcik, S. M., Katsurabayashi, S., Guillemin, I., Friauf, E., Rosenmund, C., Brose, N., et al. (2006). A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron 50, 575–587. doi: 10.1016/j.neuron.2006.04.016
Wolkowitz, O. M., and Pickar, D. (1991). Benzodiazepines in the treatment of schizophrenia: a review and reappraisal. Am. J. Psychiatry 148, 714–726.
Woo, C. C., and Leon, M. (2013). Environmental enrichment as an effective treatment for autism: a randomized controlled trial. Behav. Neurosci. 127, 487–497. doi: 10.1037/a0033010
Woo, T. U., Whitehead, R. E., Melchitzky, D. S., and Lewis, D. A. (1998). A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 95, 5341–5346. doi: 10.1073/pnas.95.9.5341
Woodin, M. A., Ganguly, K., and Poo, M. M. (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transporter activity. Neuron 39, 807–820. doi: 10.1016/S0896-6273(03)00507-5
Worbe, Y., Baup, N., Grabli, D., Chaigneau, M., Mounayar, S., McCairn, K., et al. (2009). Behavioral and movement disorders induced by local inhibitory dysfunction in primate striatum. Cereb. Cortex 19, 1844–1856. doi: 10.1093/cercor/bhn214
Wu, Y., Wang, W., Diez-Sampedro, A., and Richerson, G. B. (2007). Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 56, 851–865. doi: 10.1016/j.neuron.2007.10.021
Xu, J. Y., and Sastry, B. R. (2007). Theta-bursts induce a shift in reversal potentials for GABA-A receptor-mediated postsynaptic currents in rat hippocampal CA1 neurons. Exp. Neurol. 204, 836–839. doi: 10.1016/j.expneurol.2007.01.004
Xu, X., Wells, A. B., O'Brien, D. R., Nehorai, A., and Dougherty, J. D. (2014). Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci. 34, 1420–1431. doi: 10.1523/JNEUROSCI.4488-13.2014
Yamashita, Y., Matsuishi, T., Ishibashi, M., Kimura, A., Onishi, Y., Yonekura, Y., et al. (1998). Decrease in benzodiazepine receptor binding in the brains of adult patients with Rett syndrome. J. Neurol. Sci. 154, 146–150. doi: 10.1016/S0022-510X(97)00223-2
Yoon, J. H., Maddock, R. J., Rokem, A., Silver, M. A., Minzenberg, M. J., Ragland, J. D., et al. S. (2010). GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J. Neurosci. 30, 3777–3781. doi: 10.1523/JNEUROSCI.6158-09.2010
Young, S. Z., Platel, J. C., Nielsen, J. V., Jensen, N. A., and Bordey, A. (2010). GABA(A) increases calcium in subventricular zone astrocyte-like cells through L- and T-type voltage-gated calcium channels. Front. Cell Neurosci. 4:8. doi: 10.3389/fncel.2010.00008
Young, S. Z., Taylor, M. M., Wu, S., Ikeda-Matsuo, Y., Kubera, C., and Bordey, A. (2012). NKCC1 knockdown decreases neuron production through GABA(A)-regulated neural progenitor proliferation and delays dendrite development. J. Neurosci. 32, 13630–13638. doi: 10.1523/JNEUROSCI.2864-12.2012
Yu, Z., Fang, Q., Xiao, X., Wang, Y. Z., Cai, Y. Q., Cao, H., et al. (2013). GABA transporter-1 deficiency confers schizophrenia-like behavioral phenotypes. PLoS ONE 8:e69883. doi: 10.1371/journal.pone.0069883
Zafeiriou, D. I., Ververi, A., Dafoulis, V., Kalyva, E., and Vargiami, E. (2013). Autism spectrum disorders: the quest for genetic syndromes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162B, 327–366. doi: 10.1002/ajmg.b.32152
Zhang, W., Peterson, M., Beyer, B., Frankel, W. N., and Zhang, Z. W. (2014). Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J. Neurosci. 34, 2754–2763. doi: 10.1523/JNEUROSCI.4900-12.2014
Zhang, Z. W., Zak, J. D., and Liu, H. (2010). MeCP2 is required for normal development of GABAergic circuits in the thalamus. J. Neurophysiol. 103, 2470–2481. doi: 10.1152/jn.00601.2009
Keywords: GABA, neurodevelopmental disorders, cation chloride cotransporters, GABAA receptor, GABA metabolism
Citation: Deidda G, Bozarth IF and Cancedda L (2014) Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front. Cell. Neurosci. 8:119. doi: 10.3389/fncel.2014.00119
Received: 31 January 2014; Accepted: 14 April 2014;
Published online: 22 May 2014.
Edited by:
Graziella DiCristo, University of Montreal, CanadaReviewed by:
Enrico Cherubini, International School for Advanced Studies, ItalyJasmina N Jovanovic, University College London, UK
Copyright © 2014 Deidda, Bozarth and Cancedda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Cancedda, Department of Neuroscience and Brain Technologies, Istituto Italiano di Tecnologia, via Morego, 30, 16163 Genova, Italy e-mail:bGF1cmEuY2FuY2VkZGFAaWl0Lml0
†These authors have contributed equally to this work.