 Carolina A. Oliva1,2
Carolina A. Oliva1,2 Jessica Y. Vargas
Jessica Y. Vargas Nibaldo C. Inestrosa
Nibaldo C. Inestrosa
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 03 December 2013
Sec. Cellular Neurophysiology
Volume 7 - 2013 | https://doi.org/10.3389/fncel.2013.00224
This article is part of the Research Topic Developmental molecules in the mature nervous system awaken by experience. View all 7 articles
During development of the central nervous system the Wnt signaling pathway has been implicated in a wide spectrum of physiological processes, including neuronal connectivity and synapse formation. Wnt proteins and components of the Wnt pathway are expressed in the brain since early development to the adult life, however, little is known about its role in mature synapses. Here, we review evidences indicating that Wnt proteins participate in the remodeling of pre- and post-synaptic regions, thus modulating synaptic function. We include the most recent data in the literature showing that Wnts are constantly released in the brain to maintain the basal neural activity. Also, we review the evidences that involve components of the Wnt pathway in the development of neurological and mental disorders, including a special emphasis on in vivo studies that relate behavioral abnormalities to deficiencies in Wnt signaling. Finally, we include the evidences that support a neuroprotective role of Wnt proteins in Alzheimer’s disease. We postulate that deregulation in Wnt signaling might have a fundamental role in the origin of neurological diseases, by altering the synaptic function at stages where the phenotype is not yet established but when the cognitive decline starts.
Among all developmental processes that involve Wnt signaling, some of the more prominent are the primary embryo axis formation, segmentation processes, and appendage patterning in Drosophila; organogenesis from worms to mice, as well as stem cell proliferation (Niehrs, 2010; Jackson and Eisenmann, 2012; Wang et al., 2012; Wray and Hartmann, 2012). Wnt signaling has been implicated in various diseases, including colon cancer and melanoma (Polakis, 2012), and also in neurodegenerative diseases (Inestrosa and Arenas, 2010; Berwick and Harvey, 2012), reflecting its relevance in fundamental biological processes across species (Clevers and Nusse, 2012). Because Wnt gene is fundamental to determine a normal neural phenotype (McMahon and Moon, 1989b), in this review we focus on its function related to the central nervous system (CNS), from embryonic neural development to higher brain function in the adult brain.
The Wnt signaling comprises a complex cascade of components that are under many regulatory steps. The Wnt proteins family includes 19 members present in mammals. The prototypical Wnt receptor is the seven transmembrane-receptor Frizzled (Fz; Bhanot et al., 1996; Rulifson et al., 2000; van Amerongen et al., 2008). There are also co-receptors been described, such as the members of the low-density lipoprotein receptor-related protein 5/6 (LRP5/6; Tamai et al., 2000; Wehrli et al., 2000; Mao et al., 2001; MacDonald et al., 2009), the single-pass transmembrane receptors Tyr kinase-like orphan receptor (ROR), protein Tyr kinase 7 (PTK7), receptor Tyr kinase (RYK), and muscle skeletal receptor Tyr kinase (MUSK) (Oishi et al., 2003; Lu et al., 2004; Cadigan and Liu, 2006; Gordon and Nusse, 2006; Green et al., 2008; Jing et al., 2009; Fradkin et al., 2010; Minami et al., 2010; Peradziryi et al., 2012), and the co-receptors from the proteoglycan families (Kikuchi et al., 2011). There are several regulatory steps for the activity of these receptors. Not only they can be intracellularly phosphorylated, but also there are several secreted antagonists that can act extracellularly to modify their activity, like Cerberus and Dickkopf-related protein 1 (Dkk-1) that bind LRP blocking the interaction to Wnt/Frizzled (Mao et al., 2001); the secreted Frizzled-related protein (sFRP) that binds directly to Wnt because of the similarity they have with Fz (Finch et al., 1997; Rattner et al., 1997); the Wnt inhibitory factor (WIF); Sclerostin (and its homolog Wise; Cruciat and Niehrs, 2013), and two classes of Wnt agonists, the R-spondin family and Norrin (Cruciat and Niehrs, 2013).
To date there is an enormous amount of information about Wnt signaling components and how they are compromised in different phenotypes (Figure 1). Historically, Wnt proteins has been classified as either canonical or non-canonical (Gordon and Nusse, 2006). For instance, Wnt1, Wnt3a, Wnt7a/b, and Wnt8 are common activators of the canonical pathway, while Wnt4, Wnt5a, and Wnt11 are mainly activators of the non-canonical pathway (Gordon and Nusse, 2006; MacDonald et al., 2009; Kikuchi et al., 2011). However, this traditional differentiation seems to be not as simple as was initially defined and more aspects should be considered. The fact that there are many different Wnt ligands and more than 15 different receptors and co-receptors allows enormous possibilities of interaction. Moreover, as result of these interactions, different intracellular cascades could be activated which makes that the cellular response turns difficult to be predicted (van Amerongen and Nusse, 2009). Furthermore, some evidences in the literature suggest that the activation of canonical or non-canonical pathway in a cell by a particular Wnt ligand may depend on the cellular context and the specificity by which Wnt binds to the receptor and co-receptor, and is not a property of the ligand itself (Mikels and Nusse, 2006b; Grumolato et al., 2010). It has been shown that Wnt ligands can also compete for the binding to specific receptors, causing the inhibition of the reciprocal signaling pathway (Grumolato et al., 2010). Despite this, some combinations of Wnt ligand-Fz receptor allow to predict the activation of a specific pathway. For example, the binding of Wnt3a ligand to Fz1 receptor activates the canonical pathway in PC12 cells (Chacon et al., 2008). Furthermore, if the Fz receptor binds to the co-receptor LRP5/6, it determines the activation of the canonical pathway, but if Fz binds to the co-receptor ROR1/2 the non-canonical pathway is activated instead.
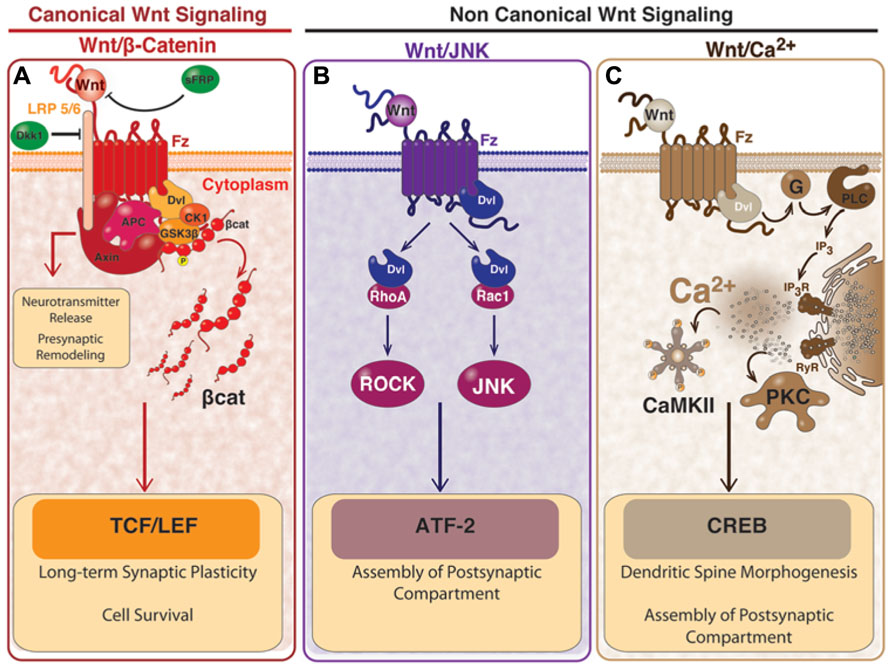
FIGURE 1. Wnt signaling pathways and the effects in mature synapses. (A) activation of canonical Wnt/β-catenin dependent pathway starts with the binding of Wnt ligand to Fz receptor and to the co-receptor LRP5/6, which induces the recruitment of Dvl and causes the inhibition of the “β-catenin destruction complex” formed by Axin, APC, GSK3β, and CK1. The inhibition of the “β-catenin destruction complex” causes the accumulation of β-catenin, which is no longer phosphorylated and it translocates to the nucleus where it activates transcription of Wnt target genes mediated by TCF/LEF. Canonical Wnt pathway participates in synaptic plasticity and cell survival. A divergent pathway involving proteins of the “β-catenin destruction complex” participates in the clustering of pre-synaptic proteins and the neurotransmitter release. The Wnt/β-catenin pathway can be blocked by Dkk-1 which binds to LRP, or through the sFRP which binds directly to Wnt ligand. (B) In the non-canonical Wnt/JNK pathway, activation of Dvl by the binding of Wnt to Fz, induces the activation of Rho and Rac small GTPases. Activation of Rho signals through ROCK and the activation of Rac signals through the JNK. This pathway is involved in the clustering of post-synaptic proteins and can also lead gene transcription mediated by ATF2. (C) In the non-canonical Wnt/Ca2+ pathway, the binding of Wnt to Fz, activates the heterotrimeric G-proteins which in turn signal through PLC and IP3 to induce the release of the intracellular Ca2+ and the activation of PKC and CaMKII. In this pathway, the transcription factor CREB is activated. Wnt/Ca2+ pathway is involved in the clustering of post-synaptic proteins and the dendritic spine morphogenesis. This figure is a courtesy of Felipe Serrano, Ph.D. student.
The first Wnt signaling pathway identified was the “canonical pathway” (Figure 1). This pathway is also called Wnt/β-catenin pathway since the induction of some embryonic structures in Xenopus (McMahon and Moon, 1989a) and the transformation of mouse mammary cells (Wong et al., 1994) are strongly correlated with the increment in β-catenin/TCF levels (Shimizu et al., 1997). The activation of the Wnt/β-catenin pathway has been prominently involved in regulating cell differentiation and proliferation (Toledo et al., 2008; Niehrs, 2010; Wray and Hartmann, 2012) controlling growth and cell fate specification during development (Cadigan and Nusse, 1997). Since deregulation of β-catenin/TCF levels is a relevant feature of a variety of cancers (Reya and Clevers, 2005), this aspect of Wnt signaling is, to date, the most investigated (Kim et al., 2013).
In steady-state or in the absence of Wnt stimulation, the cytoplasmic cellular levels of β-catenin are low since casein kinase-1α (CK-1α) and glycogen synthase kinase-3β (GSK-3β) sequentially phosphorylates the protein, targeting β-catenin for ubiquitination and proteasome degradation (Aberle et al., 1997; Liu et al., 2002; Nusse and Varmus, 2012). Instead, in the presence of Wnt ligand, Wnt binds both Fz and LRP5/6 forming a Wnt-receptor complex that recruits the protein disheveled (Dvl), which oligomerizes in the plasma membrane making a platform for the recruitment and allocation of the “β-catenin destruction complex” (Bilic et al., 2007). This complex is formed by the scaffold protein Axin, GSK-3β, CK-1α, and the tumor suppressor adenomatous polyposis coli (APC; Cliffe et al., 2003; Schwarz-Romond et al., 2007; Gao and Chen, 2010). Once the complex is recruited, CK-1α phosphorylates LRP5/6 which causes the inhibition of the “β-catenin destruction complex” (Figure 1). As consequence of this inhibition, β-catenin is stabilized and accumulated in the cytoplasm and can enter to the nucleus to activate the transcription of Wnt target genes (Logan and Nusse, 2004) under the control of the TCF/LEF transcription factors (T-cell factor, TCF /lymphoid enhancer factor, LEF; Clevers and Nusse, 2012). There are several Wnt target genes that are activated in this process, including c-Myc, cyclin D1, Axin2, and Ca2+-calmodulin-dependent protein kinase type IV (CamKIV; Toledo et al., 2008; Arrazola et al., 2009; Hodar et al., 2010; Nusse and Varmus, 2012).
There are at least two other pathways activated by Wnt ligands which effects are independent of β-catenin. These pathways, known as the “non-canonical pathways,” are grouped according the Wnt receptor and co-receptors involved, and also by the intracellular signal triggered: the planar cell polarity (PCP) pathway and the Ca2+ pathway (Figure 1). The Wnt/PCP pathway was first identified in Drosophila where it regulates tissue polarity and cell migration during development (Adler, 2002; Veeman et al., 2003). The Wnt/PCP pathway signals through c-Jun N-terminal kinase (JNK) to control cell polarity, hence it is also known as the Wnt/JNK pathway (Boutros et al., 1998; Yamanaka et al., 2002). In this pathway, the binding of Wnt to the Fz receptor on the membrane surface is followed by the activation of Rho and Rac small GTPases which in turn stimulate ROCK and JNK, respectively (Figure 1). The downstream effect of this pathway is the regulation of the cytoskeletal organization, cell motility, and gene expression by JNK-dependent transcription factors, for example the activating transcription factor 2 (ATF2; Figure 1), with concomitant activation of its target genes (Simons and Mlodzik, 2008; Kikuchi et al., 2011).
The Wnt/Ca2+ pathway is mostly a G-protein dependent signaling pathway (Slusarski et al., 1997a; Kohn and Moon, 2005). The activation of the Wnt/Ca2+ pathway requires the binding of Wnt to Fz on the membrane surface to stimulate heterotrimeric G-proteins (Slusarski et al., 1997a,b), which activates, in turn, the phospholipase-C (PLC). As a result, PLC in turn stimulates the generation of diacylglycerol (DG) and inositol-1,4,5-thriphosphate [Ins(1,4,5)P3]. This last one triggers the increase of the intracellular Ca2+-release, decreases cyclic guanosine monophosphate (cGMP), and activates the Ca2+-calmodulin-dependent protein kinase II (CaMKII), calcineurin, and protein kinase-C (PKC; Veeman et al., 2003; Kohn and Moon, 2005; Montcouquiol et al., 2006). In this process several transcription factors are activated, including the cAMP Response Element-Binding Protein-1 (CREB; Figure 1).
The classification of individual Wnt proteins in canonical and non-canonical is used now to indicate the activation of a signaling pathway, either β-catenin dependent or independent (Mikels and Nusse, 2006a, b; van Amerongen and Nusse, 2009). It has been shown that the induction of canonical or non-canonical pathway depends on the particular co-receptor activated (Grumolato et al., 2010). Moreover, the activation of a specific co-receptor by a Wnt ligand involves a common mechanism that requires the recruitment of Dvl, Axin, and GSK-3β in order to trigger either canonical or non-canonical pathways (Grumolato et al., 2010). Strikingly, Wnt ligands can compete for the Fz receptor at the cell surface and cause inhibition of the reciprocal pathway (Grumolato et al., 2010), therefore providing another control point to determine the specificity of signaling.
During the establishment of neuronal connectivity at early and late postnatal development, molecules such as the brain-derived neurotrophic factor (BDNF), bone morphogenetic protein (BMP), and Wnts act in the postnatal nervous system to help the recently formed synapses to establish and consolidate the new cellular interactions, and later to lead synaptic remodeling and plasticity throughout life (Lu, 2003; Marques, 2005; Speese and Budnik, 2007). BDNF is not only required during early development for neuronal survival and function (Duman and Voleti, 2012), but also in the adult brain to improve synaptic transmission, long-term potentiation (LTP), and ocular dominance plasticity (Magby et al., 2006; Maya Vetencourt et al., 2008; Duman and Voleti, 2012). Recently, it has been demonstrated that BDNF is a direct target of the canonical Wnt pathway in glial cells (Yi et al., 2012). Moreover, an activator of Wnt induces BDNF in retina glial cells and Wnt3a induces BDNF in a retina ganglion cell line (Seitz et al., 2010; Fragoso et al., 2011), linking both BDNF and Wnt signaling pathways in common effectors actions (Yi et al., 2012). Like BDNF, Wnt proteins remain expressed in the postnatal brain where they play a fundamental role modulating the connectivity between pre- and post-synaptic regions, participating in the remodeling of synaptic structures and regulating synaptic function (Inestrosa and Arenas, 2010; Wisniewska, 2013). Next we briefly discuss the histological evidence that shows the expression of the Wnt pathway in the postnatal brain (for more detail review (Oliva et al., 2013)).
The Wnt ligands and several components of the Wnt pathway have been identified in the adult brain in the major subdivisions of the cerebral cortex, in the olfactory bulb (OB), and olfactory related areas, in the hippocampal formation, neocortex, and in the thalamus (Shimogori et al., 2004; Wayman et al., 2006; Cerpa et al., 2008; Davis et al., 2008; Gogolla et al., 2009). In situ hybridization shows that expression of different Wnts is particularly high in those areas where the neurons are continuously renewed, such as OB and DG (Shimogori et al., 2004) and during the period of dendrite development in the hippocampus (Wayman et al., 2006).
The presence of some components of the Wnt signaling in the thalamus during postnatal life is also relevant (Shimogori et al., 2004; Wisniewska, 2013), since the thalamus is the relay for most of the sensory information to the cortex. A recent work shows that β-catenin regulates the expression of a group of genes in the thalamus (Wisniewska et al., 2012). These highly expressed genes encode for proteins involved in neuronal excitability, including voltage-gated ion channels, neurotransmitter receptors, synaptic vesicle proteins, and synaptic structural proteins. As such, they underlie cell membrane conductances of Ca2+, K+, and Cl- ions (in the case of the GABA receptor) and directly propagate, inhibit, or modify electric signals. Hence, β-catenin could be relevant to determine the degree of excitability in the thalamic neurons by regulating the expression of those genes and influencing the thalamocortical circuit activity (Wisniewska et al., 2012; Wisniewska, 2013). Since sensory information is continuously processed during the lifespan of an animal it is likely that Wnt signaling plays an important role in thalamocortical connectivity based on experience. However, further studies are required to address this important issue.
Whether Wnt ligands expression is crucial to determine the final structural and functional pattern in a mature circuit of the adult brain is matter of speculation. However, the presence of Wnt components in many brain areas, including those that are actively participating in neurogenesis, or those that are implicated in sensory processing and superior cognitive processes, suggest that Wnt pathway is involved in more than only structural functions. In the next sections we review the evidences that relate Wnt to active structural and functional processing in mature synapses, and we discuss the importance that this can have to modulate active depending processes, such as synaptic plasticity.
Wnt ligands have been linked to the assembly of structural components in the pre-synaptic compartments. For example, Wnt7a can act as a retrograde signal from granular cells (GC) in the cerebellum to induce pre-synaptic differentiation in the mossy fiber (MF), working as a synaptogenic factor (Hall et al., 2000; Ahmad-Annuar et al., 2006). Wnt7a induces axonal spreading, increases the size of the growth cone and its branching (Lucas and Salinas, 1997; Hall et al., 2000) and probably contributes to the formation of the active zones because it increases the clustering of synapsin I, a synaptic vesicle component (Hall et al., 2000). Wnt7a also induce clustering of synaptophysin, synaptotagmin, and SV-2 without affecting the clustering of post-synaptic proteins like the post-synaptic density protein-95 (PSD-95) (Cerpa et al., 2008). Moreover, a mutant mouse deficient in Wnt7a shows a delayed synaptic maturation (Hall et al., 2000) supporting the notion of the ligand Wnt7a modulating the pre-synaptic compartment. On the other hand, lithium, a pharmacological inhibitor of GSK-3β, mimics the effect of Wnt7a on axonal spreading and branching in cerebellar granular cells in vitro, suggesting that GSK-3β also participates in synaptic maturation (Lucas and Salinas, 1997). However, GSK-3β is not required for the clustering of pre-synaptic vesicle proteins induced by Wnt7a (Cerpa et al., 2008). Instead, clustering induction correlates with β-catenin stabilization, does not involve gene expression, and is also mimicked by Wnt3a. It has been suggested that Wnt7a requires Dvl1 to regulate the normal recycling rate of synaptic vesicles, and the deficiency of both proteins (Wnt7a/Dvl1 double null mutant mice) significantly reduces the miniature excitatory post-synaptic current (mEPSC) frequency, an indication of a defect in the neurotransmitter release (Hall et al., 2000; Ahmad-Annuar et al., 2006). Additionally, Wnt7a increases the frequency of mEPSC and decreases the rate of paired-pulse facilitation (PPF) (Cerpa et al., 2008), a protocol used to distinguish the involvement of the pre-synaptic from the post-synaptic terminal (Foster and McNaughton, 1991). Also, using FM dye it has been proved that Wnt7a stimulates the recycling and accelerates the exocytosis of synaptic vesicles (Hall et al., 2000; Ahmad-Annuar et al., 2006; Cerpa et al., 2008). Wnt7b and Wnt3a also increase the number of pre-synaptic puncta suggesting a role for these ligands in the pre-synaptic assembly (Ahmad-Annuar et al., 2006; Cerpa et al., 2008; Davis et al., 2008). Altogether these evidences support the role of Wnt in the maturation and function of pre-synaptic terminals.
Wnt7a has been also involved in receptors trafficking since it increases the number and size of co-clusters of the pre-synaptic α7-nicotinic acetylcholine receptors (α7-nAChR) and APC in hippocampal neurons, as well as it modulates the α7-nAChR trafficking to the nerve terminal (Farias et al., 2007). This evidence indicates that Wnt pathway is actively involved in the receptors functional availability in the pre-synaptic terminal.
Most of the ligands that are able to modulate pre-synaptic differentiation have shown to activate the Wnt/β-catenin signaling pathway. Despite that, nanomolar concentrations of Wnt3a which also modulates the recycling and exocytosis of synaptic vesicles in the hippocampal synapses, increases the frequency of mEPSC through a mechanism that involves Ca2+ entrance (Cerpa et al., 2008; Avila et al., 2010), suggesting a cross-talk between canonical Wnt pathway and the Wnt/Ca2+ signaling (Avila et al., 2010). This evidence implies that some of the components associated with the non-canonical pathway may also be involved in the functionality of the pre-synaptic nerve terminal. Altogether these evidences suggest that Wnt ligands that activate canonical Wnt pathway are involved in the pre-synaptic structure assembly and participate during consolidation of synaptic connectivity.
Wnt signaling can actively modulate the structural and functional assembly of the post-synaptic terminal. For instance, Wnt5a modulates excitatory synaptic transmission (Cerpa et al., 2010, 2011), specifically up-regulating excitatory synaptic currents through NMDA receptors (NMDARs), but not AMPA receptors (AMPARs), through activation of PKC and JNK, and facilitating the induction of excitatory LTP (Cerpa et al., 2011). Moreover, Wnt5a actively modulates inhibitory synapses in the hippocampus, increasing the turnover of GABAA receptor (GABAARs) in the surface and increasing GABA currents by a post-synaptic mechanisms that involved Wnt/Ca2+/CaMKII (Cuitino et al., 2010). Wnt5a also participates in the dendritic spine morphogenesis, inducing de novo dendritic spines and causing an increase in the volume and density of pre-existent spines, enhancing the efficacy of hippocampal glutamatergic synapses (Varela-Nallar et al., 2010). Wnt5a also induces the increment of calcium in the synaptic puncta of cultured hippocampal neurons, suggesting the activation of Wnt/Ca2+ signaling pathway (Varela-Nallar et al., 2010) through a mechanism that involved fast phosphorylation of CaMKII (Farias et al., 2009). Wnt5a also modulates post-synaptic protein assembly by increasing the clustering of PSD-95, without affecting the total levels of this protein, via activation of Wnt/JNK signaling pathway (Farias et al., 2009).
Among the non-typical functions attributable to the ligands, we consider some examples. It has been shown that Wnt3a induces clustering of AChRs in motoneurons during the neuromuscular innervation (Henriquez et al., 2008). This effect requires Dvl1 and agrin, a protoglycan released by motoneurons, but does not involve Wnt/β-catenin pathway. Instead, Wnt3a induces AChRs aggregation through activation of Rac1 (Henriquez et al., 2008). However, Wnt3a through the activation of Wnt/β-catenin pathway inhibits agrin-induced AChR clusters, suggesting that Wnt signaling dynamically regulates the post-synaptic assembly during the establishment of the neuromuscular junction (Wang et al., 2008).
In another example, Wnt7a has also been shown to regulate the post-synaptic compartment by stimulating the morphogenesis and function of excitatory dendritic spines, without affecting the inhibitory connectivity in the hippocampus (Ciani et al., 2011). It has been shown that Wnt7a requires Dvl expression to induce spine growth in pre-existed post-synaptic spines, as well as the activation of CaMKII to induce spine maturation (Ciani et al., 2011). On the other hand, it has been shown that the ligand Wnt7b and Dvl, independently of Wnt/GSK-3β, can activate Rac and downstream JNK to increase dendritic length and branching in immature hippocampal neurons (Rosso et al., 2005).
Interestingly, while the activation of canonical pathway by Wnt3a, Wnt7a, or Wnt7b induces synaptogenesis through β-catenin stability, activation of non-canonical pathway through Wnt5a negatively regulates synaptogenesis (Davis et al., 2008), which could constitute a competitive mechanism to balance the synapse formation in the brain. Altogether these evidences support the idea of a new role for traditional canonical Wnt ligands, which associated with the non-canonical pathway may also be involved in the functionality of the post-synaptic region.
In the previous sections we mainly reviewed the role of Wnt signaling pathways in synaptic structure and in particular functional changes through development. In this section we address how Wnt signaling may be relevant for physiological functions during activity and non-activity dependent processes.
Several evidences in the literature show that Wnt signaling is constitutively released during basal synaptic activities. Using sFRP1, a physiological Wnt signaling inhibitor, and using a reporter cell line that expresses GFP, it has been shown that hippocampal cultures are spontaneously secreting measurable amounts of endogenous Wnt (Rosso et al., 2005). Wnt is also constitutively released in slices and specifically regulates the function of NMDAR (Cerpa et al., 2011). Moreover, it has been shown that using PKC or JNK inhibitors to block the downstream signaling, the basal NMDAR synaptic neurotransmission is reduced, suggesting that Wnt is using non-canonical components of Wnt pathway to maintain glutamatergic neurotransmission (Cerpa et al., 2011). sFRP1 also reduces the amplitude of evoked fEPSP in hippocampal slices, confirming that Wnt is being released to maintain the basal tone of synaptic activity (Varela-Nallar et al., 2010). These evidences strongly support the notion that during basal activity Wnt molecules are being spontaneously released to keep functional connectivity. Indeed, it has been observed that local inhibition of mossy fibers with sFRP decreases the levels of Wnt7a/b protein, suppressing the increased effect that an enriched ambient has on the synapses number (Gogolla et al., 2009). Interestingly, in animals housed in control conditions, sFRP also reduces the synapses number, suggesting that Wnt signaling is required to generate structural plasticity (Gogolla et al., 2009). Recently, we identify a very dynamic effect of Wnt ligands, modulating the spike rate in brain slices in vitro. Using a slice model in mice that can generate spontaneous oscillations we recently demonstrated that Wnt3a perfused into the recording solution is able to modulate the spike frequency, while antibodies against Wnt ligands significantly affect the oscillation (Oliva et al., 2013). The possibility that Wnt is being constantly released to exert functional effects on the cells in a network is intriguing, as well as exciting possibility. Nevertheless, the function of the released Wnts in neuronal network remains to be resolved.
Several studies have shown that the neocortex and the hippocampus are electrically active even from embryonic stages, and this early neuronal activity has a potent trophic role, mediating neuronal survival (Golbs et al., 2011) and maturation of molecular, cellular, and structural features of cortical circuits (Owens and Kriegstein, 2002). This spontaneous activity in the form of spontaneous action potentials plays a fundamental role in the fine tuning of connectivity in several regions of the brain (Masland, 1977; Ben-Ari et al., 1989; Yuste et al., 1992; Feller et al., 1996; Garaschuk et al., 1998; Komuro and Rakic, 1998; Moody and Bosma, 2005; Mire et al., 2012). The establishment of early network patterns during the first weeks of life has different functions and the underlying mechanisms are different of those of mature network. The presence of molecules such as Wnt and BDNF during particular early developmental moments may help in the establishment and consolidation of the recently formed synapses (Lu, 2003; Marques, 2005; Speese and Budnik, 2007), contributing to the circuitry maturation and consolidating the rhythmic activity that can sustain adult behavior (Egorov and Draguhn, 2012). How Wnt signaling is contributing to the establishment of mature neural circuits, is still matter of speculation, but based on the histological and functional evidences, it is expected to be important.
Wnt ligands and receptors are expressed in areas of the brain that undergo plasticity, suggesting that during an activity-dependent process Wnt is released to regulate synaptic transmission and ulterior structural modifications. Dendritic spines, direct targets of the Wnt action, have been considered as one of the first visible effectors of plasticity (experience dependent)-induced changes (Alvarez and Sabatini, 2007; Holtmaat and Svoboda, 2009; Bosch and Hayashi, 2012). During development, synaptogenesis contributes to establish the connectivity within neuronal circuits (Holtmaat and Svoboda, 2009). Later, during postnatal development and adolescence, activity-dependent spine maintenance or elimination contributes to the remodeling of neuronal circuits (Alvarez and Sabatini, 2007). In this section we review the evidence of Wnt signaling as one of the signaling pathways responsible for structural dendritic changes during activity-dependent plasticity.
Using high K+ solution, a solution used to mimic the depolarization caused by persistent neuronal activity (Ryan et al., 1993), Yu and Malenka found that the increment in the number and length of dendritic branches in culture hippocampal neurons depends on β-catenin availability (Yu and Malenka, 2003) and showed that this effect is reduced by Dkk-1. Using a GFP-reporter cell line to detect Wnt they demonstrated that neuronal depolarization with K+, increases Wnt secretion from hippocampal neurons but not from astrocytes, contributing to the idea that changes in dendritic arborization depend on activity-induced Wnt release (Yu and Malenka, 2003). Moreover, CaMKIV that has been also shown to regulate depolarization-dependent dendritic growth requires β-catenin (Redmond et al., 2002; Yu and Malenka, 2003).
Other evidences show that Wnt released in an activity-dependent manner modifies the structure and function of dendritic branches. Neuronal activity induced by high K+ sequentially activates NMDARs, CaMKK, CaMKI, Ras, MEK/ERK, and CREB-dependent transcription of Wnt2 (Wayman et al., 2006), indicating that Wnt2 could be a CREB-responsive gene. Indeed, Wnt2 can stimulate dendrite development by itself (Wayman et al., 2006). Along these lines, another report showed that the Wnt inhibitor WIF, which prevents that Wnt binds its receptor Fz (Han and Lin, 2005), suppresses dendritic arborization induced by high K+. Based on a previous report indicating that CaMKIV initiates the cascade culminating in CREB activation and dendritic maturation (Redmond et al., 2002), it is plausible that this effect is mediated through CREB-dependent Wnt (Wayman et al., 2006). This evidence strongly suggests that sustained activity can induce synthesis of more Wnt to potentiate the cellular response. Altogether, these evidences suggest an active role of Wnt in determine the strength and remodeling of the synapse in response to an activity-dependent process.
Using a more physiological paradigm to mimic neuronal depolarization, such as theta burst stimulation or high frequency stimulation (HFS) to induce LTP (Larson et al., 1986), it has been also shown an increases in the expression of Wnt signaling components and the Wnt release in an activity-dependent manner. LTP induces changes in the mRNA levels and the immunostaining pattern of several Wnt proteins, such as Wnt3a, Fz4, β-catenin, and Dvl3 (Chen et al., 2006). LTP also induces accumulation of β-catenin in the nucleus and the increment of several Wnt-target genes, confirming the activation of Wnt/β-catenin/TCF pathway by tetanic stimulation (Chen et al., 2006). In fact, the size and number of Wnt3a puncta diminishes after tetanic stimulation, suggesting that Wnt3a is being released in a process that depends of NMDAR activation (Chen et al., 2006). Moreover, the treatment with a specific antibody against Wnt3a shows a significant reduction on LTP, while incubations with lithium enhances LTP, also showing that Wnt3a is endogenously released to activate Wnt/β-catenin pathway (Chen et al., 2006). These studies strongly suggest that the presence of Wnt ligand and/or the activation of the canonical pathway are required to induce LTP.
On the other hand, a recent report has shown that neuronal activity decreases the expression of sFRP3, releasing the Wnt pathway from its natural inhibitor (Jang et al., 2013). The reduction in sFRP3 is essential for the activity-dependent neuronal maturation process in the hippocampus, increasing neurogenesis (Jang et al., 2013), a mechanism that can be used to overcome neuronal death. This evidence suggests an important effect of neural activity on the neurogenesis induction, process that appears to be under tight control of Wnt signaling. More studies are required to confirm this hypothesis.
The evidences discussed here reveal that sustained depolarization activates Wnt signaling, increases synaptic transmission and facilitates LTP in hippocampal brain slices and in cultured neurons. Wnts also have a significant effect controlling the basal activity on brain circuits, suggesting an important role for Wnt signaling in the regulation of spontaneous and activity-dependent plasticity (Inestrosa and Arenas, 2010; Budnik and Salinas, 2011). However, there is still a lack of in vivo evidence about how Wnt signaling may affect physiological synaptic processes, for example during active behavior.
Recent studies have suggested a role for the Wnt/β-catenin pathway in the memory formation of adults (Maguschak and Ressler, 2008, 2011), while, deregulation of the Wnt/β-catenin pathway has been implicated in the Alzheimer’s diseases (ADs) pathogenesis (Moon et al., 2004), which is associated with memory loss. However, it remains unknown how Wnt signaling is involved in learning and memory processes in the adult. Recently, it has been reported that stereotaxic injection of the Wnt antagonist Dkk-1 in the basolateral amygdala interferes with the long-term memory consolidation without affecting short-term memory (Maguschak and Ressler, 2011). Remarkably, administration of Wnt1 during fear memory formation also interferes with the long-term memory consolidation in the amygdala (Maguschak and Ressler, 2011). The Wnt1 administration avoids the transient decrease in the Wnt1 mRNA that occurs immediately after fear conditioning, suggesting that the rapid decrease in the expression of Wnt1 might be critical for the fear memory formation (Maguschak and Ressler, 2011). However, not only the Wnt1 mRNA but also several genes related to Wnt signaling are rapidly downregulated during fear learning and normalized during memory consolidation (Maguschak and Ressler, 2011). Also, during fear memory consolidation in the amygdala there is a transient increment of β-catenin mRNA levels, while the β-catenin deletion specifically affects fear memory consolidation of adult mice (Maguschak and Ressler, 2008).
On the other hand, a selective increment in the hippocampal levels of Wnt7 and Wnt5a, but not Wnt3 isoform, during consolidation and recall of spatial memory has been reported (Tabatadze et al., 2012). The levels of Wnt7 in the hippocampus of animals trained in a water maze hidden platform, are enhanced in comparison to the animals trained with a visible platform (Tabatadze et al., 2012). Despite in the visible platform the memory acquisition is better, animals trained in the hidden platform have better retention memory after 30 days of training, that correlates with elevated levels of Wnt7 (Tabatadze et al., 2012). The cells that concentrate the expression of Wnt7 are the granular cells in the DG, showing a correlation between the expression of Wnt7 in this region with the establishment of the long-lasting memory (Tabatadze et al., 2012).
Recently, it has been reported that the activation of Wnt/β-catenin pathway is also required for memory consolidation of a hippocampal-dependent object recognition task (Fortress et al., 2013). Intrahippocampal administration of Dkk-1 avoids familiar object recognition and generates a strong reduction in Wnt-related proteins such as β-catenin, Cyclin D1, c-myc, and Wnt7, evidencing the role of canonical Wnt signaling in hippocampal consolidation memory (Fortress et al., 2013). In another study it has been shown that an aged APC heterozygous knockout (APC+/-) mice exhibit a severe insufficiency in the working memory performance suggesting that a correct regulation of Wnt signaling is determinant for the appropriate functioning of the CNS (Koshimizu et al., 2011). More evidence has recently involved Wnt signaling in olfactory memory in Drosophila, showing that armadillo (homolog to β-catenin), wingless (Wnt ligand), and arrow (co-receptor) are necessary for long term memory formation (Tan et al., 2013), indicating that these mechanisms are evolutionary conserved among spacies.
Altogether, these evidences support the idea that Wnt/β-catenin pathway is involved in long-term memory consolidation and that the activation of Wnt signaling in response to a sensory stimulus induces functional long-lasting changes improving the animal’s performance. In the next section, we review the evidences in the literature showing that deregulation of the Wnt pathway might be related to major neurological disorders.
During the last decades, increasing data has suggested that the progression of a neurological disease is the result of severe neuroanatomical abnormalities. However, more evidences shows that several changes in the synaptic connectivity such as the imbalance between excitation/inhibition may result into a cascade of multiple effects at the neural network level which in many cases precedes cell degeneration or death (Hickey et al., 2008; Hermann et al., 2009; Palop and Mucke, 2010; Penzes et al., 2013). Excitatory/inhibitory balance critically regulates cortical network function, neural network activity, and behaviors associated with psychiatric disorders (Penzes et al., 2013). Multiple lines of evidence implicate excitatory and inhibitory synaptic circuits in the cortex, striatum, and hippocampus as key cellular substrates for pathogenesis in these disorders. In fact, major neurological diseases such as autism, schizophrenia (SZ), bipolar disorder, and major neurodegenerative disorders including AD, Huntington’s disease (HD), and Parkinson’s disease (PD) share these characteristics, that the neurodegenerative diseases show progressive neuronal loss. In this section we review the most recent studies related to the progression of the most common neurological diseases in which Wnt signaling pathway and its components has been suggested to be relevant (Figure 2).
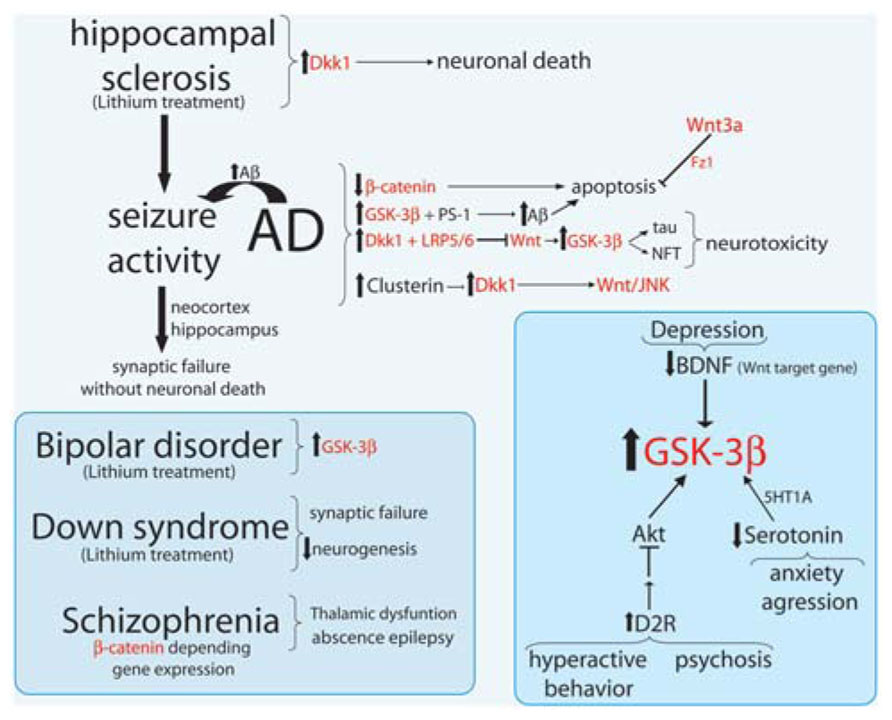
FIGURE 2. Major neurological disorders and their relationship with Wnt signaling components. Summary scheme of Wnt signaling components (in red) involved in Alzheimer disease (AD), seizure activity (epilepsy), hippocampal sclerosis, bipolar disorder, down syndrome, and Schizophrenia.
Alzheimer’s disease is a neurodegenerative disorder characterized by progressive deterioration of the individual cognitive functions, mainly caused by synaptic damage and neuronal death in specific regions of the brain (Selkoe, 2001; Mattson, 2004; Walsh and Selkoe, 2004). Distinctive features of the AD brains are the presence of senile plaques, composed by extracellular deposits of amyloid-β (Aβ) peptides and the neurofibrillary tangles (NFT), composed by intracellular aggregates of hyper-phosphorylated tau protein (Mayeux and Stern, 2012). Despite there is not clarity yet about the triggering factors for AD, there are some candidates which are also common factors to several other neuronal diseases (Jope and Johnson, 2004), among which are components of the Wnt signaling machinery (Figure 2). One of them is the GSK-3β (Hooper et al., 2008), a constitutively active protein and whose expression is increased in the hippocampus of AD patients (Blalock et al., 2004). Over-expression of GSK-3β in mice prevents the induction of LTP and reduces the spatial learning (Hernandez et al., 2002; Hooper et al., 2007), linking the characteristic memory failure in AD to the increment in GSK-3β. The same transgenic mouse exhibits tau hyperphosphorylation, low β-catenin in the nucleus, and neurodegeneration by NFT (Lucas et al., 2001), features that can be prevented by chronic lithium exposure (Engel et al., 2006a, b). In fact, GSK-3 activity mediates Aβ production from its precursor amyloid precursor protein (APP; Phiel et al., 2003; Ryder et al., 2003). It has been shown that GSK-3β binds presenilin-1 (PS-1), a protein involved in the proteolytic cleavage of the APP, to produce the Aβ peptide. The expression of a mutant PS-1 shows an increment in GSK-3 activity and in the hyperphosphorylated tau protein in cells (Takashima et al., 1998; Zhang et al., 1998). In cultured neurons the toxicity mediated by Aβ depends on increased GSK-3β activity, but it reverses when GSK-3β expression or its activity is blocked (Takashima et al., 1993, 1996; Busciglio et al., 1995; Alvarez et al., 1999).
We previously demonstrated that activation of Wnt signaling inhibits GSK-3β, and leads to neuroprotection in both hippocampal cultured neurons and in vivo transgenic model of AD (Garrido et al., 2002; Inestrosa et al., 2002; De Ferrari et al., 2003; Alvarez et al., 2004; Quintanilla et al., 2005). A marked decrease in the levels of β-catenin has been found in the brain of early onset familial AD patients with PS-1 mutations (Zhang et al., 1998). It has been shown that PS-1 form a complex with β-catenin, similarly to how β-catenin is associated to GSK-3 and to APC (Zhang et al., 1998). The inhibition of the β-catenin/TCF pathway enhances the vulnerability to apoptosis (Zhang et al., 1998), suggesting that when β-catenin is stabilized and TCF is activated, it could lead to neuronal survival. Moreover, Li and colleagues found that tau hyperphosphorylation, which inhibits competitively phosphorylation of β-catenin by GSK-3β, protects the cells from apoptosis (Li et al., 2007). Wnt3a can also protect from Aβ-induced apoptosis through binding Fz1 but no Fz2 (Chacon et al., 2008) and the genetic variations in LRP-6 have been associated with late-onset AD (De Ferrari et al., 2007), supporting the evidences that alterations in Wnt/β-catenin signaling could be involved in AD.
Another molecule of the Wnt pathway involved in the AD phenotype is Dkk-1, that is the cross-link between the pathway to survival/apoptosis that relate Aβ and Wnt signaling (Figure 2). While canonical Wnt/β-catenin endorses cell survival, Aβ induces the expression of the natural antagonist Dkk-1. In AD brains there are an up-regulated expression of Dkk-1 compared to healthy individuals (Caricasole et al., 2004), while the chronic overexpression of Dkk-1 in transgenic mice causes an age-related tau phosphorylation and cognitive deficits (Killick et al., 2012). During Aβ exposure there is an induction of Dkk-1 expression that depends on p53 (Wang et al., 2000; Zhang et al., 2002; Killick et al., 2012). Since Dkk-1 binds LRP5/6 to inhibit Wnt signaling, the lack of inhibition of GSK-3β facilitates tau hyperphosphorylation and the NFT formations, leading neurotoxicity and apoptosis caused by Aβ peptides (Caricasole et al., 2004). Dkk-1 knock-down almost completely avoid neuronal death and tau phosphorylation, blocking neurotoxicity induced by Aβ (Caricasole et al., 2004). Dkk-1 can also reversibly reduce the amount of synaptic proteins and the number of active pre-synaptic sites, by inducing synaptic disassembly of both pre- and post-synaptic sites (Purro et al., 2012). However, using an antibody anti-Dkk-1 the synaptic loss induced by Aβ can be blocked (Purro et al., 2012). Recently it has been reported that Dkk-1 expression increases in an age-dependent manner and causes the reduction of hippocampal neurogenesis. In neural progenitors with inducible loss of Dkk1 not only enhance neurogenesis and the TCF/LEF activity, but reverses the cognitive decline related to deleterious acquisition and memory consolidation (Seib et al., 2013). These evidences might have several implications for future treatments of dementias and neurodegenerative diseases, yet more studies are required.
New evidences also support a link between Aβ toxicity and Wnt pathway. Besides APOE locus, classically associated with susceptibility for late-onset AD, it has been identified a new susceptibility factor in late-onset AD, a loci at CLU (Lambert et al., 2009). Both APOE and CLU, which are considered the most abundant apolipoproteins in the CNS (Roheim et al., 1979), have been involved in Aβ clearance from the brain through the blood-brain barrier (DeMattos et al., 2004; Lambert et al., 2009). Clusterin, a product of CLU promotes cell survival, is increased in AD models (Killick et al., 2012) and is regulated by Wnt signaling via TCF1 (Schepeler et al., 2007). Suppression of the Wnt signaling pathway in colon carcinoma cells lead to up-regulation of CLU (Schepeler et al., 2007). The knock-down of clusterin protects against Aβ neurotoxicity and prevents the induction of Dkk-1 by Aβ (Killick et al., 2012). In contrast, Aβ targets clusterin allowing its intracellular accumulation and initiating the neurotoxicity process (Killick et al., 2012).
The C-terminal of Dkk-1 protein, which is the domain that antagonizes the canonical Wnt pathway through binding LRP5/6, is responsible for the activation of gene transcription of genes involved in AD-like pathology (Killick et al., 2012). Interestingly, Dkk-1 blocks Wnt/β-catenin signaling and activates the non-canonical Wnt/JNK pathway, leading the increment in c-Jun activity (Killick et al., 2012). Thus, activation of gene transcription in the presence of Aβ occurs due to that Dkk-1 activates Wnt/JNK pathway, which leads to tau phosphorylation. Moreover, clusterin and several genes from the Wnt/JNK signaling have been found in AD human-brain (Killick et al., 2012), supporting the idea that Aβ induces clusterin/Dkk-1/JNK pathway to produces neurotoxicity.
On the other hand, lithium an activator of Wnt/β-catenin signaling (Klein and Melton, 1996; Gould et al., 2004) indirectly induces protection against Aβ neurotoxicity in mice models of AD (Rockenstein et al., 2007). In a study from our laboratory we reported that in vivo activation of Wnt pathway by lithium reduces memory loss and Aβ aggregates in APP/PS1 double transgenic mice, an AD animal model (Toledo and Inestrosa, 2010). Lithium also reduces the level of tau aggregates and axonal degeneration in transgenic mice over-expressing human tau, suggesting that Wnt pathway can also control tautopathy progression (Noble et al., 2005). Moreover, lithium improves spatial memory after being decreased by Aβ fibrils injections (De Ferrari et al., 2003; Toledo and Inestrosa, 2010) and interferes with the cleavage of APP causing a reduction in Aβ levels (Phiel et al., 2003).
Interestingly, we observed that in vivo activation of Wnt signaling also improves episodic memory of APP/PS1 mice (personal observations). These results agree with recent findings showing that the activation of Wnt/β-catenin signaling through the inhibition of GSK-3β, can reverses the hippocampus-dependent learning deficits in a mouse model of fragile-X syndrome (Guo et al., 2012). More studies are required, at present, to determine the nature of this effect.
It has been described that transgenic mouse with familial mutation associated to AD presents high levels of altered neuronal activity, indicated by the electroencephalographic studies and for the susceptibility to develop seizure activity in several brain regions, including neocortex and hippocampus (Palop et al., 2007). The same incidence has been found in AD patients (Menendez, 2005). This epileptiform activity in vivo occurs in absence of neuronal loss, supporting the hypothesis that an aberrant neuronal activity is a primary effect of the high Aβ levels and not a consequence of the neurodegeneration (Palop et al., 2007; Palop and Mucke, 2009). In a recent work it has been shown that the overexpression of the APP intracellular domain (AICD) increases the susceptibility to have spontaneous seizures, deficits in LTP and seizure-induced drugs generation (Vogt et al., 2011). Moreover, in these models the seizure susceptibility and severity depends on the levels of AICD; at higher AICD levels, most severe seizures (Vogt et al., 2011). These effects are independent of the levels of Aβ, because a mice overexpressing hAPP but with a mutation in AICD does not generates deficits in LTP or memory, despite the levels of Aβ (Galvan et al., 2006; Saganich et al., 2006). Both data are not contradictory and suggest that not only high levels of Aβ but also overexpressed AICD are required to cause seizure susceptibility. Interestingly, the overexpression of AICD increases the levels of active GSK-3β (Ryan and Pimplikar, 2005), something that also occur in AD patients (Hooper et al., 2008). Whether this evidence relates Wnt signaling with seizure susceptibility has not been demonstrated yet. However, cells with NFT inclusions and positives to active GSK-3β are allocated in brain regions where Wnt components have been well described (Pei et al., 1999).
On the other hand, an early expression of Dkk-1 has been found in patients with hippocampal sclerosis, characterized by temporal lobe epilepsy, severe neuronal loss and gliosis (Busceti et al., 2007). Like in hypoxia/ischemia in vivo and in excitotoxicity-related neuronal death in vitro (Cappuccio et al., 2005), the expression of Dkk-1 in hippocampal sclerosis correlates with the neuronal death (Busceti et al., 2007). In fact, the increase in Dkk-1 precedes neuronal death in both the olfactory neurons as in the hippocampus of rats that developed seizures induced by kainate (Busceti et al., 2007). Animals previously injected with lithium are protected against neuronal loss, but not against the kainate-induced seizures (Busceti et al., 2007). Intracerebral administration of Dkk-1 antisense almost completely prevents seizure-induced neuronal death (Caricasole et al., 2004; Busceti et al., 2007), but not ictal activity seizures (Busceti et al., 2007). These evidences suggest that the mechanisms for neuronal loss and seizure activity are separated, raising the possibility to use therapies targeting Wnt signaling components to protect against the neuronal loss induced by epilepsy.
In the last years an increased amount of evidence has shown that components of Wnt pathways are involved in major psychiatric disorders. Deregulation of Wnt signaling has been implicated in bipolar disorder (Klein and Melton, 1996; Grimes and Jope, 2001; Gould and Manji, 2002), in autism (Moon et al., 2004; De Ferrari and Moon, 2006), and schizophrenia (Miyaoka et al., 1999; Yang et al., 2003; Ftouh et al., 2005; De Ferrari and Moon, 2006; Inestrosa et al., 2012), among others. Bipolar disorder is a mental disorder characterized by episodes of depression interweaved by mania (Gould and Manji, 2002) but also for other misbalances such as sleep and circadian rhythm disturbances (Kaladchibachi et al., 2007; Ahnaou and Drinkenburg, 2011). At the molecular level, several evidences have converged to show that a dysregulation of GSK-3β cascade is involved in bipolar disorder (Klein and Melton, 1996; Grimes and Jope, 2001; Gould and Manji, 2002; Valvezan and Klein, 2012). It has been shown that mice with constitutively activated GSK-3β exhibit the same disturbances in sleep-wake cycle, locomotor activity, and body temperature, all major features that also can be found in bipolar patients (Tilleman et al., 2002; Prickaerts et al., 2006; Ahnaou and Drinkenburg, 2011). Lithium has been widely used as a treatment for mood disorders. Although GSK-3 seems to be the most plausible target for lithium (Valvezan and Klein, 2012), it has not been confirmed yet that GSK-3 is necessary for the action of lithium, since GSK-3 may activate several downstream mechanisms by which exerts its effects, such as Wnt and Akt signaling pathways (Valvezan and Klein, 2012). An evidence that link lithium effect with Wnt signaling is the fact that lithium stimulates neurogenesis (Samuels and Hen, 2011). Lithium, through GSK-3β inhibition, activates the canonical Wnt pathway to induce neurogenesis in the adult hippocampus (Lie et al., 2005). Moreover, transgenic mice overexpressing β-catenin shows the same behavior than lithium-treated animals suggesting that behavioral effects of lithium may be mediated through the increment of β-catenin (Gould et al., 2007). Additional studies are required to investigate the involvement of Wnt in lithium-mediated neurogenesis and behavior.
Recently, lithium has been also used to treat Ts65Dn mice, a model of Down syndrome. It has been shown that these animals exhibit several dysfunctions, such as synaptic failures and a significant reduced neurogenesis (Contestabile et al., 2013). Lithium, through the activation of canonical Wnt pathway, recovered the adult neurogenesis in these mice by stimulating the proliferation of neural precursor cells and increasing the number of newborn cells turning into mature neurons in the DG (Chen et al., 2000; Wexler et al., 2008; Contestabile et al., 2013). More importantly, the consequence of this treatment is the improvement of synaptic plasticity and cognitive functions dependent on hippocampal plasticity (Contestabile et al., 2013). It has been shown that Down syndrome has an excessive GABA inhibition that could exert a negative effect on the neurogenesis during embryonic and postnatal development, and on the excitatory neurotransmission, altogether leading to performance impairment (Kleschevnikov et al., 2004; Fernandez et al., 2007). Whether activation of Wnt pathway by lithium helps to partially restore the imbalance in the neurotransmission in this model, is a matter of further investigations. However, if so it suggests a possible therapeutic treatment to improve cognitive deficits in this disorder.
Wnt components such as GSK-3β have been also involved in psychotic and hyperactivity behavior, also referred as dopamine/serotonin-associated behaviors (Beaulieu et al., 2009). Several studies have converged evidence for an involvement of Akt and GSK-3 pathway in dopamine-related behavior and in the mechanism of action of some psychoactive drugs (Beaulieu et al., 2004; Beaulieu et al., 2005). It has been shown that overactivation of dopamine receptor-2 (D2R) causes hyperactivity through up-regulation of GSK-3β. The fact that the activation of Wnt pathway causes the inactivation of GSK3β could provide a window where to explore the potential usage of Wnt analogs as a treatment to improve the physical manifestation of psychosis and hyperactivity disorders.
Similar approaches directed to regulate GSK-3β can be found in the serotonin-related behavior. One of the consequences of the reduction of brain serotonin levels is the pronounced increment of GSK-3β and several emotional abnormalities such as anxiety and aggression in rodents (Beaulieu et al., 2008). Treating this animals with a specific GSK-3β inhibitor or breeding this animal with an animal deficient in GSK-3β, completely rescues the behavioral abnormalities associated with the lack of serotonin (Lucki et al., 2001; Weisstaub et al., 2006). This strongly suggests that GSK-3β signaling mediate behavioral effects of serotonin. Moreover, it has been shown that the typical antipsychotic drug haloperidol and the two atypicals risperidone and clozapine increase the protein level of β-catenin, Dvl-3, Axin, GSK-3α/β, and Wnt5a in prefrontal cortex, and β-catenin, GSK-3α/β, and Dvl-3 in the dopamine centers of the ventral midbrain (Alimohamad et al., 2005a,b; Sutton et al., 2007). The antipsychotics drugs do not have any effect on Wnt1 and Wnt3a, on Fz, or JNK or c-JNK (Sutton et al., 2007). These treatments cause an increment of β-catenin in the cytoplasm and nucleus and involve TCF-mediated gene transcription, suggesting that an abnormal expression of these proteins could underlie the serotonin-mediated behavior.
Recently, important evidences have related depression to Wnt signaling cascades (Duman and Voleti, 2012). It has been shown that a decreased BDNF expression underlies the stress and depression and, conversely, the use of antidepressants causes enhanced BDNF expression that could mediate the beneficial effects in the hippocampus and prefrontal cortex (Duman and Monteggia, 2006; Krishnan and Nestler, 2008). In fact, BDNF is sufficient to produce antidepressant behavioral effect; however deletion of BDNF is not enough to induce depression (Duman and Monteggia, 2006; Schmidt and Duman, 2007). BDNF has been recently described as a target of Wnt signaling (Yi et al., 2012). On the other hand, it remains unresolved whether the increase in BDNF levels is the result of Wnt signaling activation in these brain regions. However, other evidences could sustain this possibility. Also, it has been reported that GSK3 is required for the rapid antidepressant actions of ketamine (Beurel et al., 2011). Ketamine increases GSK3 phosphorylation in the mouse hippocampus and cerebral cortex (Beurel et al., 2011), while microarray studies show that the use of antidepressants differentially regulates the expression of Wnts, Fz receptors and Dvl in hippocampus, as well as Tcf/Lef, demonstrating the relevancy of the Wnt signaling components in the antidepressant effects (Okamoto et al., 2010; Duman and Voleti, 2012). Still more studies are required to elucidate the causes and potential beneficial effects of Wnt signaling on depression and related mood disorders.
As we previously mentioned, it has been shown that β-catenin regulates the expression of a novel group of genes in the thalamus (Wisniewska et al., 2012). However, it is still not clear whether variations in the level of β-catenin affect the expression of the genes that encode voltage gated Ca2+ channels (VGCCs) and neurotransmitter receptors, shaping neuronal excitability in vivo. If so, then inappropriate activity of β-catenin might affect the proper signal transmission in thalamocortical circuits. Thalamocortical desynchronization underlies absence epilepsy (Hughes, 2009). Specifically, the T-type voltage-gated channel Cav3.1 has been proposed to be implicated in absence seizures (Barclay et al., 2001; Liu et al., 2007; Ernst et al., 2009). Moreover, anticonvulsant drugs target voltage-gated channels (Errington et al., 2005). In the opposite, schizophrenia has also been associated with thalamic dysfunction (Harms et al., 2007; Smith et al., 2011; Marenco et al., 2012), while a group of synaptic genes involved in excitability have been found to be associated with the risk of schizophrenia (Lips et al., 2012). These results suggest a possible role for β-catenin-dependent gene expression in thalamic pathologies.
Literature has been recently populated with evidences indicating that changes during development could account for neurological disease vulnerability in the adulthood (Penzes et al., 2011; Penzes et al., 2013). Genetic mutations or environmental insults that perturb a normal developmental progression could constitute an early signature of developmental disorders. Nevertheless, disturbances that affect synapse morphogenesis and/or function during a specific developmental time or in a particular brain region, also may determine the consequent neuropathological symptom, such as neuronal circuit alterations and ultimately cognitive and behavioral symptoms. Mutations associated to initial cognitive disabilities may occur in molecules that play an essential role in regulating brain synapse formation and plasticity. Given the relevancy that Wnt signaling pathways have in these processes, we hypothesized that failures in establishing spines dendritic network could be caused by failures in the Wnt signaling pathways during development.
Some recent findings have provided important evidences to support this idea. Individuals with autism, SZ and AD show alterations in the density of dendritic spines in cortical pyramidal cells (Penzes et al., 2011) and also exhibit functional and structural changes in the inhibitory circuits (Oblak et al., 2010, 2011; Chattopadhyaya and Cristo, 2012). SZ patients show a profound reduction in spines density in prefrontal cortex and in primary auditory cortex, and also a reduction in the spines density and size in the hippocampal CA3 region In fact, the disrupted in schizophrenia (DISC1) gene is highly abundant in spines and it has also been associated to other psychiatric disorders (Schumacher et al., 2009). Indeed, DISC1 is known to interact with several well-established regulators of dendritic spine morphogenesis (Millar et al., 2003). Kalirin-7 via activation of a common Wnt pathway downstream effector Rac1 was found to directly regulate the effects of DISC1 on the spines morphology (Hayashi-Takagi et al., 2010). Kalirin- knockout mice remarkably show low spines density and behavioral schizophrenia-related behaviors at an age in mice that match to human adolescence, when the symptoms in humans emerges.
Emerging evidences suggest a role for Wnt pathway in synaptic maintenance and function of the adult brain. Activation of Wnt pathway has an effect in both basal and evoked activity. As we have discussed in this review, Wnt leads to rapid modification of the pre-synaptic compartment, regulating the clustering of synaptic proteins and the recycling of synaptic vesicles, while at the post-synaptic compartment Wnt modulates the assembly of the post-synaptic apparatus and the dendritic spine morphogenesis. Recent evidences indicate that activation of Wnt pathway modulates the efficacy of the excitatory and inhibitory synaptic transmission through divergent mechanisms. Moreover, activation of Wnt pathway enhances synaptic plasticity and the cognitive function of the adult brain. In vivo studies have shown that Wnt pathway is involved in both hippocampal- and amygdala-dependent memory. Interestingly, these studies have evidenced that endogenous Wnt activity is required for the normal functioning of neuronal circuits and that the inhibition of Wnt pathway can cause synaptic plasticity impairments and memory deficits. Moreover, the inhibition of Wnt pathway also causes a reduction in the basal spontaneous activity, which suggests that Wnt participates in the establishment of mature neuronal networks. Deregulation of Wnt pathway has been implicated in neurological pathologies, including AD, SZ, and mood disorders. Particularly, Wnt activator may be therapeutically relevant in AD since it has been shown activation of Wnt displays a neuroprotective effect and resolves the synaptic and cognitive impairment in mouse model of AD. In summary, the evidences discussed here show that Wnt pathway plays a key role in the synaptic function of mature nervous system, constituting a promising therapeutic target for the treatment of certain neurological diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Basal Center of Excellence in Science and Technology CONICYT-CARE (PFB 12/2007) and Fondecyt N° 1120156 to Nibaldo C. Inestrosa. A postdoctoral fellowship from MECESUP to Carolina A. Oliva and a predoctoral fellowship from Fundación Gran Mariscal de Ayacucho to Jessica Y. Vargas are also thanked.
Aberle, H., Bauer, A., Stappert, J., Kispert, A., and Kemler, R. (1997). Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804. doi: 10.1093/emboj/16.13.3797
Adler, P. N. (2002). Planar signaling and morphogenesis in Drosophila. Dev. Cell 2, 525–535. doi: 10.1016/S1534-5807(02)00176-4
Ahmad-Annuar, A., Ciani, L., Simeonidis, I., Herreros, J., Fredj, N. B., Rosso, S. B., et al. (2006). Signaling across the synapse: a role for Wnt and dishevelled in presynaptic assembly and neurotransmitter release. J. Cell Biol. 174, 127–139. doi: 10.1083/jcb.200511054
Ahnaou, A., and Drinkenburg, W. H. (2011). Disruption of glycogen synthase kinase-3-beta activity leads to abnormalities in physiological measures in mice. Behav. Brain Res. 221, 246–252. doi: 10.1016/j.bbr.2011.03.004
Alimohamad, H., Rajakumar, N., Seah, Y. H., and Rushlow, W. (2005a). Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol. Psychiatry 57, 533–542. doi: 10.1016/j.biopsych.2004.11.036
Alimohamad, H., Sutton, L., Mouyal, J., Rajakumar, N., and Rushlow, W. J. (2005b). The effects of antipsychotics on beta-catenin, glycogen synthase kinase-3 and dishevelled in the ventral midbrain of rats. J. Neurochem. 95, 513–525. doi: 10.1111/j.1471-4159.2005.03388.x
Alvarez, A. R., Godoy, J. A., Mullendorff, K., Olivares, G. H., Bronfman, M., and Inestrosa, N. C. (2004). Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp. Cell Res. 297, 186–196. doi: 10.1016/j.yexcr.2004.02.028
Alvarez, G., Munoz-Montano, J. R., Satrustegui, J., Avila, J., Bogonez, E., and Diaz-Nido, J. (1999). Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 453, 260–264. doi: 10.1016/S0014-5793(99)00685-7
Alvarez, V. A., and Sabatini, B. L. (2007). Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 30, 79–97. doi: 10.1146/annurev.neuro.30.051606.094222
Arrazola, M. S., Varela-Nallar, L., Colombres, M., Toledo, E. M., Cruzat, F., Pavez, L., et al. (2009). Calcium/calmodulin-dependent protein kinase type IV is a target gene of the Wnt/beta-catenin signaling pathway. J. Cell. Physiol. 221, 658–667. doi: 10.1002/jcp.21902
Avila, M. E., Sepulveda, F. J., Burgos, C. F., Moraga-Cid, G., Parodi, J., Moon, R. T., et al. (2010). Canonical Wnt3a modulates intracellular calcium and enhances excitatory neurotransmission in hippocampal neurons. J. Biol. Chem. 285, 18939–18947. doi: 10.1074/jbc.M110.103028
Barclay, J., Balaguero, N., Mione, M., Ackerman, S. L., Letts, V. A., Brodbeck, J., et al. (2001). Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J. Neurosci. 21, 6095–6104.
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2009). Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 49, 327–347. doi: 10.1146/annurev.pharmtox.011008.145634
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273. doi: 10.1016/j.cell.2005.05.012
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., et al. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104. doi: 10.1073/pnas.0307921101
Beaulieu, J. M., Zhang, X., Rodriguiz, R. M., Sotnikova, T. D., Cools, M. J., Wetsel, W. C., et al. (2008). Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 1333–1338. doi: 10.1073/pnas.0711496105
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325.
Berwick, D. C., and Harvey, K. (2012). The importance of Wnt signalling for neurodegeneration in Parkinson’s disease. Biochem. Soc. Trans. 40, 1123–1128. doi: 10.1042/BST20120122
Beurel, E., Song, L., and Jope, R. S. (2011). Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol. Psychiatry 16, 1068–1070. doi: 10.1038/mp.2011.47
Bhanot, P., Brink, M., Samos, C. H., Hsieh, J. C., Wang, Y., Macke, J. P., et al. (1996). A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 382, 225–230. doi: 10.1038/382225a0
Bilic, J., Huang, Y. L., Davidson, G., Zimmermann, T., Cruciat, C. M., Bienz, M., et al. (2007). Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619–1622. doi: 10.1126/science.1137065
Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R., and Landfield, P. W. (2004). Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A. 101, 2173–2178. doi: 10.1073/pnas.0308512100
Bosch, M., and Hayashi, Y. (2012). Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 22, 383–388. doi: 10.1016/j.conb.2011.09.002
Boutros, M., Paricio, N., Strutt, D. I., and Mlodzik, M. (1998). Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell 94, 109–118. doi: 10.1016/S0092-8674(00)81226-X
Budnik, V., and Salinas, P. C. (2011). Wnt signaling during synaptic development and plasticity. Curr. Opin. Neurobiol. 21, 151–159. doi: 10.1016/j.conb.2010.12.002
Busceti, C. L., Biagioni, F., Aronica, E., Riozzi, B., Storto, M., Battaglia, G., et al. (2007). Induction of the Wnt inhibitor, Dickkopf-1, is associated with neurodegeneration related to temporal lobe epilepsy. Epilepsia 48, 694–705. doi: 10.1111/j.1528-1167.2007.01055.x
Busciglio, J., Lorenzo, A., Yeh, J., and Yankner, B. A. (1995). beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14, 879–888. doi: 10.1016/0896-6273(95)90232-5
Cadigan, K. M., and Liu, Y. I. (2006). Wnt signaling: complexity at the surface. J. Cell Sci. 119, 395–402. doi: 10.1242/jcs.02826
Cadigan, K. M., and Nusse, R. (1997). Wnt signaling: a common theme in animal development. Genes Dev. 11, 3286–3305. doi: 10.1101/gad.11.24.3286
Cappuccio, I., Calderone, A., Busceti, C. L., Biagioni, F., Pontarelli, F., Bruno, V., et al. (2005). Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J. Neurosci. 25, 2647–2657. doi: 10.1523/JNEUROSCI.5230-04.2005
Caricasole, A., Copani, A., Caraci, F., Aronica, E., Rozemuller, A. J., Caruso, A., et al. (2004). Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J. Neurosci. 24, 6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004
Cerpa, W., Farias, G. G., Godoy, J. A., Fuenzalida, M., Bonansco, C., and Inestrosa, N. C. (2010). Wnt-5a occludes Abeta oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol. Neurodegener. 5, 3. doi: 10.1186/1750-1326-5-3
Cerpa, W., Gambrill, A., Inestrosa, N. C., and Barria, A. (2011). Regulation of NMDA-receptor synaptic transmission by Wnt signaling. J. Neurosci. 31, 9466–9471. doi: 10.1523/JNEUROSCI.6311-10.2011
Cerpa, W., Godoy, J. A., Alfaro, I., Farias, G. G., Metcalfe, M. J., Fuentealba, R., et al. (2008). Wnt-7a modulates the synaptic vesicle cycle and synaptic transmission in hippocampal neurons. J. Biol. Chem. 283, 5918–5927. doi: 10.1074/jbc.M705943200
Chacon, M. A., Varela-Nallar, L., and Inestrosa, N. C. (2008). Frizzled-1 is involved in the neuroprotective effect of Wnt3a against Abeta oligomers. J. Cell. Physiol. 217, 215–227. doi: 10.1002/jcp.21497
Chattopadhyaya, B., and Cristo, G. D. (2012). GABAergic circuit dysfunctions in neurodevelopmental disorders. Front. Psychiatry 3:51. doi: 10.3389/fpsyt.2012.00051
Chen, J., Park, C. S., and Tang, S. J. (2006). Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J. Biol. Chem. 281, 11910–11916. doi: 10.1074/jbc.M511920200
Chen, G., Rajkowska, G., Du, F., Seraji-Bozorgzad, N., and Manji, H. K. (2000). Enhancement of hippocampal neurogenesis by lithium. J. Neurochem. 75, 1729–1734. doi: 10.1046/j.1471-4159.2000.0751729.x
Ciani, L., Boyle, K. A., Dickins, E., Sahores, M., Anane, D., Lopes, D. M., et al. (2011). Wnt7a signaling promotes dendritic spine growth and synaptic strength through Ca(2)(+)/Calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. U.S.A. 108, 10732–10737. doi: 10.1073/pnas.1018132108
Clevers, H., and Nusse, R. (2012). Wnt/beta-catenin signaling and disease. Cell 149, 1192–1205. doi: 10.1016/j.cell.2012.05.012
Cliffe, A., Hamada, F., and Bienz, M. (2003). A role of dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr. Biol. 13, 960–966. doi: 10.1016/S0960-9822(03)00370-1
Contestabile, A., Greco, B., Ghezzi, D., Tucci, V., Benfenati, F., and Gasparini, L. (2013). Lithium rescues synaptic plasticity and memory in Down syndrome mice. J. Clin. Invest. 123, 348–361. doi: 10.1172/JCI64650
Cruciat, C. M., and Niehrs, C. (2013). Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb. Perspect. Med. 3, a015081. doi: 10.1101/cshperspect.a015081
Cuitino, L., Godoy, J. A., Farias, G. G., Couve, A., Bonansco, C., Fuenzalida, M., et al. (2010). Wnt-5a modulates recycling of functional GABAA receptors on hippocampal neurons. J. Neurosci. 30, 8411–8420. doi: 10.1523/JNEUROSCI.5736-09.2010
Davis, E. K., Zou, Y., and Ghosh, A. (2008). Wnts acting through canonical and noncanonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev. 3, 32. doi: 10.1186/1749-8104-3-32
De Ferrari, G. V., Chacon, M. A., Barria, M. I., Garrido, J. L., Godoy, J. A., Olivares, G., et al. (2003). Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol. Psychiatry 8, 195–208. doi: 10.1038/sj.mp.4001208
De Ferrari, G. V., and Moon, R. T. (2006). The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 25, 7545–7553. doi: 10.1038/sj.onc.1210064
De Ferrari, G. V., Papassotiropoulos, A., Biechele, T., Wavrant De-Vrieze, F., Avila, M. E., Major, M. B., et al. (2007). Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 104, 9434–9439. doi: 10.1073/pnas.0603523104
DeMattos, R. B., Cirrito, J. R., Parsadanian, M., May, P. C., O’Dell, M. A., Taylor, J. W., et al. (2004). ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41, 193–202. doi: 10.1016/S0896-6273(03)00850-X
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127. doi: 10.1016/j.biopsych.2006.02.013
Duman, R. S., and Voleti, B. (2012). Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 35, 47–56. doi: 10.1016/j.tins.2011.11.004
Egorov, A. V., and Draguhn, A. (2012). Development of coherent neuronal activity patterns in mammalian cortical networks: common principles and local hetereogeneity. Mech. Dev. 130, 412–423. doi: 10.1016/j.mod.2012.09.006
Engel, T., Goni-Oliver, P., Lucas, J. J., Avila, J., and Hernandez, F. (2006a). Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 99, 1445–1455. doi: 10.1111/j.1471-4159.2006.04139.x
Engel, T., Hernandez, F., Avila, J., and Lucas, J. J. (2006b). Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 26, 5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006
Ernst, W. L., Zhang, Y., Yoo, J. W., Ernst, S. J., and Noebels, J. L. (2009). Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J. Neurosci. 29, 1615–1625. doi: 10.1523/JNEUROSCI.2081-08.2009
Errington, A. C., Stohr, T., and Lees, G. (2005). Voltage gated ion channels: targets for anticonvulsant drugs. Curr. Top. Med. Chem. 5, 15–30. doi: 10.2174/1568026053386872
Farias, G. G., Alfaro, I. E., Cerpa, W., Grabowski, C. P., Godoy, J. A., Bonansco, C., et al. (2009). Wnt-5a/JNK signaling promotes the clustering of PSD-95 in hippocampal neurons. J. Biol. Chem. 284, 15857–15866. doi: 10.1074/jbc.M808986200
Farias, G. G., Valles, A. S., Colombres, M., Godoy, J. A., Toledo, E. M., Lukas, R. J., et al. (2007). Wnt-7a induces presynaptic colocalization of alpha 7-nicotinic acetylcholine receptors and adenomatous polyposis coli in hippocampal neurons. J. Neurosci. 27, 5313–5325. doi: 10.1523/JNEUROSCI.3934-06.2007
Feller, M. B., Wellis, D. P., Stellwagen, D., Werblin, F. S., and Shatz, C. J. (1996). Requirement for cholinergic synaptic transmission in the propagation of spontaneous retinal waves. Science 272, 1182–1187. doi: 10.1126/science.272.5265.1182
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of down syndrome. Nat. Neurosci. 10, 411–413. doi:10.1038/nn1860
Finch, P. W., He, X., Kelley, M. J., Uren, A., Schaudies, R. P., Popescu, N. C., et al. (1997). Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc. Natl. Acad. Sci. U.S.A. 94, 6770–6775. doi: 10.1073/pnas.94.13.6770
Fortress, A. M., Schram, S. L., Tuscher, J. J., and Frick, K. M. (2013). Canonical Wnt signaling is necessary for object recognition memory consolidation. J. Neurosci. 33, 12619–12626. doi: 10.1523/JNEUROSCI.0659-13.2013
Foster, T. C., and McNaughton, B. L. (1991). Long-term enhancement of CA1 synaptic transmission is due to increased quantal size, not quantal content. Hippocampus 1, 79–91. doi: 10.1002/hipo.450010108
Fradkin, L. G., Dura, J. M., and Noordermeer, J. N. (2010). Ryks: new partners for Wnts in the developing and regenerating nervous system. Trends Neurosci. 33, 84–92. doi: 10.1016/j.tins.2009.11.005
Fragoso, M. A., Yi, H., Nakamura, R. E., and Hackam, A. S. (2011). The Wnt signaling pathway protects retinal ganglion cell 5 (RGC-5) cells from elevated pressure. Cell Mol. Neurobiol. 31, 163–173. doi: 10.1007/s10571-010-9603-z
Ftouh, S., Akbar, M. T., Hirsch, S. R., and de Belleroche, J. S. (2005). Down-regulation of Dickkopf 3, a regulator of the Wnt signalling pathway, in elderly schizophrenic subjects. J. Neurochem. 94, 520–530. doi: 10.1111/j.1471-4159.2005.03239.x
Galvan, V., Gorostiza, O. F., Banwait, S., Ataie, M., Logvinova, A. V., Sitaraman, S., et al. (2006). Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl. Acad. Sci. U.S.A. 103, 7130–7135. doi: 10.1073/pnas.0509695103
Gao, C., and Chen, Y. G. (2010). Dishevelled: the hub of Wnt signaling. Cell. Signal. 22, 717–727. doi: 10.1016/j.cellsig.2009.11.021
Garaschuk, O., Hanse, E., and Konnerth, A. (1998). Developmental profile and synaptic origin of early network oscillations in the CA1 region of rat neonatal hippocampus. J. Physiol. 507(Pt 1), 219–236. doi: 10.1111/j.1469-7793.1998.219bu.x
Garrido, J. L., Godoy, J. A., Alvarez, A., Bronfman, M., and Inestrosa, N. C. (2002). Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 16, 1982–1984. doi:10.1096/fj.02-0327fje
Gogolla, N., Galimberti, I., Deguchi, Y., and Caroni, P. (2009). Wnt signaling mediates experience-related regulation of synapse numbers and mossy fiber connectivities in the adult hippocampus. Neuron 62, 510–525. doi: 10.1016/j.neuron.2009.04.022
Golbs, A., Nimmervoll, B., Sun, J. J., Sava, I. E., and Luhmann, H. J. (2011). Control of programmed cell death by distinct electrical activity patterns. Cereb. Cortex 21, 1192–1202. doi: 10.1093/cercor/bhq200
Gordon, M. D., and Nusse, R. (2006). Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22429–22433. doi: 10.1074/jbc.R600015200
Gould, T. D., Chen, G., and Manji, H. K. (2004). In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology 29, 32–38. doi: 10.1038/sj.npp.1300283
Gould, T. D., Einat, H., O’Donnell, K. C., Picchini, A. M., Schloesser, R. J., and Manji, H. K. (2007). Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology 32, 2173–2183. doi: 10.1038/sj.npp.1301338
Gould, T. D., and Manji, H. K. (2002). The Wnt signaling pathway in bipolar disorder. Neuroscientist 8, 497–511. doi: 10.1177/107385802237176
Green, J. L., Kuntz, S. G., and Sternberg, P. W. (2008). Ror receptor tyrosine kinases: orphans no more. Trends Cell Biol. 18, 536–544. doi: 10.1016/j.tcb.2008.08.006
Grimes, C. A., and Jope, R. S. (2001). The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65, 391–426. doi: 10.1016/S0301-0082(01)00011-9
Grumolato, L., Liu, G., Mong, P., Mudbhary, R., Biswas, R., Arroyave, R., et al. (2010). Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 24, 2517–2530. doi: 10.1101/gad.1957710
Guo, W., Murthy, A. C., Zhang, L., Johnson, E. B., Schaller, E. G., Allan, A. M., et al. (2012). Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 21, 681–691. doi: 10.1093/hmg/ddr501
Hall, A. C., Lucas, F. R., and Salinas, P. C. (2000). Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell 100, 525–535. doi: 10.1016/S0092-8674(00)80689-3
Han, C., and Lin, X. (2005). Shifted from Wnt to Hedgehog signaling pathways. Mol. Cell 17, 321–322. doi: 10.1016/j.molcel.2005.01.009
Harms, M. P., Wang, L., Mamah, D., Barch, D. M., Thompson, P. A., and Csernansky, J. G. (2007). Thalamic shape abnormalities in individuals with schizophrenia and their nonpsychotic siblings. J. Neurosci. 27, 13835–13842. doi: 10.1523/JNEUROSCI.2571-07.2007
Hayashi-Takagi, A., Takaki, M., Graziane, N., Seshadri, S., Murdoch, H., Dunlop, A. J., et al. (2010). Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 13, 327–332. doi: 10.1038/nn.2487
Henriquez, J. P., Webb, A., Bence, M., Bildsoe, H., Sahores, M., Hughes, S. M., et al. (2008). Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. U.S.A. 105, 18812–18817. doi: 10.1073/pnas.0806300105
Hermann, D., Both, M., Ebert, U., Gross, G., Schoemaker, H., Draguhn, A., et al. (2009). Synaptic transmission is impaired prior to plaque formation in amyloid precursor protein-overexpressing mice without altering behaviorally-correlated sharp wave-ripple complexes. Neuroscience 162, 1081–1090. doi: 10.1016/j.neuroscience.2009.05.044
Hernandez, F., Borrell, J., Guaza, C., Avila, J., and Lucas, J. J. (2002). Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 83, 1529–1533. doi: 10.1046/j.1471-4159.2002.01269.x
Hickey, M. A., Kosmalska, A., Enayati, J., Cohen, R., Zeitlin, S., Levine, M. S., et al. (2008). Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience 157, 280–295. doi: 10.1016/j.neuroscience.2008.08.041
Hodar, C., Assar, R., Colombres, M., Aravena, A., Pavez, L., Gonzalez, M., et al. (2010). Genome-wide identification of new Wnt/beta-catenin target genes in the human genome using CART method. BMC Genomics 11:348. doi: 10.1186/1471-2164-11-348
Holtmaat, A., and Svoboda, K. (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658. doi: 10.1038/nrn2699
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104, 1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., et al. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86. doi: 10.1111/j.1460-9568.2006.05245.x
Hughes, J. R. (2009). Absence seizures: a review of recent reports with new concepts. Epilepsy Behav. 15, 404–412. doi: 10.1016/j.yebeh.2009.06.007
Inestrosa, N. C., and Arenas, E. (2010). Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 11, 77–86. doi: 10.1038/nrn2755
Inestrosa, N., De Ferrari, G. V., Garrido, J. L., Alvarez, A., Olivares, G. H., Barria, M. I., et al. (2002). Wnt signaling involvement in beta-amyloid-dependent neurodegeneration. Neurochem. Int. 41, 341–344. doi: 10.1016/S0197-0186(02)00056-6
Inestrosa, N. C., Montecinos-Oliva, C., and Fuenzalida, M. (2012). Wnt signaling: role in Alzheimer disease and schizophrenia. J. Neuroimmune Pharmacol. 7, 788–807. doi: 10.1007/s11481-012-9417-5
Jackson, B. M., and Eisenmann, D. M. (2012). Beta-catenin-dependent Wnt signaling in C. elegans: teaching an old dog a new trick. Cold Spring Harb. Perspect. Biol. 4, a007948. doi: 10.1101/cshperspect.a007948
Jang, M. H., Bonaguidi, M. A., Kitabatake, Y., Sun, J., Song, J., Kang, E., et al. (2013). Secreted frizzled-related protein 3 regulates activity-dependent adult hippocampal neurogenesis. Cell Stem Cell 12, 215–223. doi: 10.1016/j.stem.2012.11.021
Jing, L., Lefebvre, J. L., Gordon, L. R., and Granato, M. (2009). Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron 61, 721–733. doi: 10.1016/j.neuron.2008.break12.025
Jope, R. S., and Johnson, G. V. (2004). The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29, 95–102. doi: 10.1016/j.tibs.2003.12.004
Kaladchibachi, S. A., Doble, B., Anthopoulos, N., Woodgett, J. R., and Manoukian, A. S. (2007). Glycogen synthase kinase 3, circadian rhythms, and bipolar disorder: a molecular link in the therapeutic action of lithium. J. Circadian Rhythms 5, 3. doi: 10.1186/1740-3391-5-3
Kikuchi, A., Yamamoto, H., Sato, A., and Matsumoto, S. (2011). New insights into the mechanism of Wnt signaling pathway activation. Int. Rev. Cell Mol. Biol. 291, 21–71. doi: 10.1016/B978-0-12-386035-4.00002-1
Killick, R., Ribe, E. M., Al-Shawi, R., Malik, B., Hooper, C., Fernandes, C., et al. (2012). Clusterin regulates beta-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry. doi: 10.1038/mp.2012.163 [Epub ahead of print].
Kim, W., Kim, M., and Jho, E. H. (2013). Wnt/beta-catenin signalling: from plasma membrane to nucleus. Biochem. J. 450, 9–21. doi: 10.1042/BJ20121284
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459. doi: 10.1073/pnas.93.16.8455
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of down syndrome. J. Neurosci. 24, 8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004
Kohn, A. D., and Moon, R. T. (2005). Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium 38, 439–446. doi: 10.1016/j.ceca.2005.06.022
Komuro, H., and Rakic, P. (1998). Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca2+ fluctuations. J. Neurobiol. 37, 110–130. doi: 10.1002/(SICI)1097-4695(199810)37:1<110::AID-NEU9>3.0.CO;2-C
Koshimizu, H., Fukui, Y., Takao, K., Ohira, K., Tanda, K., Nakanishi, K., et al. (2011). Adenomatous polyposis coli heterozygous knockout mice display hypoactivity and age-dependent working memory deficits. Front. Behav. Neurosci. 5:85. doi: 10.3389/fnbeh.2011.00085
Krishnan, V., and Nestler, E. J. (2008). The molecular neurobiology of depression. Nature 455, 894–902. doi: 10.1038/nature07455
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Larson, J., Wong, D., and Lynch, G. (1986). Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 368, 347–350. doi: 10.1016/0006-8993(86)90579-2
Li, H. L., Wang, H. H., Liu, S. J., Deng, Y. Q., Zhang, Y. J., Tian, Q., et al. (2007). Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 104, 3591–3596. doi: 10.1073/pnas.0609303104
Lie, D. C., Colamarino, S. A., Song, H. J., Desire, L., Mira, H., Consiglio, A., et al. (2005). Wnt signalling regulates adult hippocampal neurogenesis. Nature 437, 1370–1375. doi: 10.1038/nature04108
Lips, E. S., Cornelisse, L. N., Toonen, R. F., Min, J. L., Hultman, C. M., Holmans, P. A., et al. (2012). Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol. Psychiatry 17, 996–1006. doi: 10.1038/mp.2011.117
Liu, X. B., Coble, J., van Luijtelaar, G., and Jones, E. G. (2007). Reticular nucleus-specific changes in alpha3 subunit protein at GABA synapses in genetically epilepsy-prone rats. Proc. Natl. Acad. Sci. U.S.A. 104, 12512–12517. doi: 10.1073/pnas.0705320104
Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G. H., Tan, Y., et al. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847. doi: 10.1016/S0092-8674(02)00685-2
Logan, C. Y., and Nusse, R. (2004). The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810. doi: 10.1146/annurev.cellbio.20.010403.113126
Lu, B. (2003). BDNF and activity-dependent synaptic modulation. Learn. Mem. 10, 86–98. doi: 10.1101/lm.54603
Lu, W., Yamamoto, V., Ortega, B., and Baltimore, D. (2004). Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 119, 97–108. doi: 10.1016/j.cell.2004.09.019
Lucas, F. R., and Salinas, P. C. (1997). WNT-7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Dev. Biol. 192, 31–44. doi: 10.1006/dbio.1997.8734
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39. doi: 10.1093/emboj/20.1.27
Lucki, I., Dalvi, A., and Mayorga, A. J. (2001). Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology 155, 315–322. doi: 10.1007/s002130100694
MacDonald, B. T., Tamai, K., and He, X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. doi: 10.1016/j.devcel.2009.06.016
Magby, J. P., Bi, C., Chen, Z. Y., Lee, F. S., and Plummer, M. R. (2006). Single-cell characterization of retrograde signaling by brain-derived neurotrophic factor. J. Neurosci. 26, 13531–13536. doi: 10.1523/JNEUROSCI.4576-06.2006
Maguschak, K. A., and Ressler, K. J. (2008). Beta-catenin is required for memory consolidation. Nat. Neurosci. 11, 1319–1326. doi: 10.1038/nn.2198
Maguschak, K. A., and Ressler, K. J. (2011). Wnt signaling in amygdala-dependent learning and memory. J. Neurosci. 31, 13057–13067. doi: 10.1523/JNEUROSCI.3248-11.2011
Mao, B., Wu, W., Li, Y., Hoppe, D., Stannek, P., Glinka, A., et al. (2001). LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 411, 321–325. doi: 10.1038/35077108
Marenco, S., Stein, J. L., Savostyanova, A. A., Sambataro, F., Tan, H. Y., Goldman, A. L., et al. (2012). Investigation of anatomical thalamo-cortical connectivity and FMRI activation in schizophrenia. Neuropsychopharmacology 37, 499–507. doi: 10.1038/npp.2011.215
Marques, G. (2005). Morphogens and synaptogenesis in Drosophila. J. Neurobiol. 64, 417–434. doi: 10.1002/neu.20165
Masland, R. H. (1977). Maturation of function in the developing rabbit retina. J. Comp. Neurol. 175, 275–286. doi: 10.1002/cne.901750303
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/nature02621
Maya Vetencourt, J. F., Sale, A., Viegi, A., Baroncelli, L., De Pasquale, R., O’Leary, O. F., et al. (2008). The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 320, 385–388. doi: 10.1126/science.1150516
Mayeux, R., and Stern, Y. (2012). Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, pii: a006239. doi: 10.1038/nrneurol.2011.2. Epub 2011 Feb 8.
McMahon, A. P., and Moon, R. T. (1989a). Ectopic expression of the proto-oncogene int-1 in Xenopus embryos leads to duplication of the embryonic axis. Cell 58, 1075–1084. doi: 10.1016/0092-8674(89)90506-0
McMahon, A. P., and Moon, R. T. (1989b). Int-1 – a proto-oncogene involved in cell signalling. Development 107(Suppl), 161–167.
Menendez, M. (2005). Down syndrome, Alzheimer’s disease and seizures. Brain Dev. 27, 246–252. doi: 10.1016/j.braindev.2004.07.008
Mikels, A. J., and Nusse, R. (2006a). Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 4:e115. doi: 10.1371/journal.pbio.0040115
Mikels, A. J., and Nusse, R. (2006b). Wnts as ligands: processing, secretion and reception. Oncogene 25, 7461–7468. doi: 10.1038/sj.onc.1210053
Millar, J. K., Christie, S., and Porteous, D. J. (2003). Yeast two-hybrid screens implicate DISC1 in brain development and function. Biochem. Biophys. Res. Commun. 311, 1019–1025. doi: 10.1016/j.bbrc.2003.10.101
Minami, Y., Oishi, I., Endo, M., and Nishita, M. (2010). Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: their implications in developmental morphogenesis and human diseases. Dev. Dyn. 239, 1–15. doi: 10.1002/dvdy.21991.
Mire, E., Mezzera, C., Leyva-Diaz, E., Paternain, A. V., Squarzoni, P., Bluy, L., et al. (2012). Spontaneous activity regulates Robo1 transcription to mediate a switch in thalamocortical axon growth. Nat. Neurosci. 15, 1134–1143. doi: 10.1038/nn.3160
Miyaoka, T., Seno, H., and Ishino, H. (1999). Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 38, 1–6. doi: 10.1016/S0920-9964(98)00179-0
Montcouquiol, M., Crenshaw, E. B. III., and Kelley, M. W. (2006). Noncanonical Wnt signaling and neural polarity. Annu. Rev. Neurosci. 29, 363–386. doi: 10.1146/annurev.neuro.29.051605.112933
Moody, W. J., and Bosma, M. M. (2005). Ion channel development, spontaneous activity, and activity-dependent development in nerve and muscle cells. Physiol. Rev. 85, 883–941. doi: 10.1152/physrev.00017.2004
Moon, R. T., Kohn, A. D., De Ferrari, G. V., and Kaykas, A. (2004). WNT and beta-catenin signalling: diseases and therapies. Nat. Rev. Genet. 5, 691–701. doi: 10.1038/nrg1427
Niehrs, C. (2010). On growth and form: a Cartesian coordinate system of Wnt and BMP signaling specifies bilaterian body axes. Development 137, 845–857. doi: 10.1242/dev.039651
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., et al. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995. doi: 10.1073/pnas.0500466102
Nusse, R., and Varmus, H. (2012). Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J. 31, 2670–2684. doi: 10.1038/emboj.2012.146
Oblak, A. L., Gibbs, T. T., and Blatt, G. J. (2010). Decreased GABA(B) receptors in the cingulate cortex and fusiform gyrus in autism. J. Neurochem. 114, 1414–1423. doi: 10.1111/j.1471-4159.2010.06858.x. Epub 2010 Jun 12.
Oblak, A. L., Gibbs, T. T., and Blatt, G. J. (2011). Reduced GABAA receptors and benzodiazepine binding sites in the posterior cingulate cortex and fusiform gyrus in autism. Brain Res. 1380, 218–228. doi: 10.1016/j.brainres.2010.09.021
Oishi, I., Suzuki, H., Onishi, N., Takada, R., Kani, S., Ohkawara, B., et al. (2003). The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 8, 645–654. doi: 10.1046/j.1365-2443.2003.00662.x
Okamoto, H., Voleti, B., Banasr, M., Sarhan, M., Duric, V., Girgenti, M. J., et al. (2010). Wnt2 expression and signaling is increased by different classes of antidepressant treatments. Biol. Psychiatry 68, 521–527. doi: 10.1016/j.biopsych.2010.04.023
Oliva, C. A., Vargas, J. Y., and Inestrosa, N. C. (2013). Wnt signaling: role in LTP, neural networks and memory. Ageing Res. Rev. 12, 786–800. doi: 10.1016/j.arr.2013.03.006
Owens, D. F., and Kriegstein, A. R. (2002). Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 3, 715–727. doi: 10.1038/nrn919
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Palop, J. J., and Mucke, L. (2009). Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 66, 435–440. doi: 10.1001/archneurol.2009.15
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Pei, J. J., Braak, E., Braak, H., Grundke-Iqbal, I., Iqbal, K., Winblad, B., et al. (1999). Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 58, 1010–1019. doi: 10.1097/00005072-199909000-00011
Penzes, P., Buonanno, A., Passafarro, M., Sala, C., and Sweet, R. A. (2013). Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J. Neurochem. 126, 165–182. doi: 10.1111/jnc.12261
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Peradziryi, H., Tolwinski, N. S., and Borchers, A. (2012). The many roles of PTK7: a versatile regulator of cell-cell communication. Arch. Biochem. Biophys. 524, 71–76. doi: 10.1016/j.abb.2011.12.019
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439. doi: 10.1038/nature01640
Polakis, P. (2012). Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 4, pii: a008052. doi: 10.1101/cshperspect.a008052
Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., van Craenendonck, H., Goris, I., et al. (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J. Neurosci. 26, 9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006
Purro, S. A., Dickins, E. M., and Salinas, P. C. (2012). The secreted Wnt antagonist Dickkopf-1 is required for amyloid beta-mediated synaptic loss. J. Neurosci. 32, 3492–3498. doi: 10.1523/JNEUROSCI.4562-11.2012
Quintanilla, R. A., Munoz, F. J., Metcalfe, M. J., Hitschfeld, M., Olivares, G., Godoy, J. A., et al. (2005). Trolox and 17beta-estradiol protect against amyloid beta-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J. Biol. Chem. 280, 11615–11625. doi: 10.1074/jbc.M411936200
Rattner, A., Hsieh, J. C., Smallwood, P. M., Gilbert, D. J., Copeland, N. G., Jenkins, N. A., et al. (1997). A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors. Proc. Natl. Acad. Sci. U.S.A. 94, 2859–2863. doi: 10.1073/pnas.94.7.2859
Redmond, L., Kashani, A. H., and Ghosh, A. (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34, 999–1010. doi: 10.1016/S0896-6273(02)00737-7
Reya, T., and Clevers, H. (2005). Wnt signalling in stem cells and cancer. Nature 434, 843–850. doi: 10.1038/nature03319
Rockenstein, E., Torrance, M., Adame, A., Mante, M., Bar-on, P., Rose, J. B., et al. (2007). Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 27, 1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007
Roheim, P. S., Carey, M., Forte, T., and Vega, G. L. (1979). Apolipoproteins in human cerebrospinal fluid. Proc. Natl. Acad. Sci. U.S.A. 76, 4646–4649. doi: 10.1073/pnas.76.9.4646
Rosso, S. B., Sussman, D., Wynshaw-Boris, A., and Salinas, P. C. (2005). Wnt signaling through dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8, 34–42. doi: 10.1038/nn1374
Rulifson, E. J., Wu, C. H., and Nusse, R. (2000). Pathway specificity by the bifunctional receptor frizzled is determined by affinity for wingless. Mol. Cell 6, 117–126. doi:10.1016/S1097-2765(05)00018-3
Ryan, K. A., and Pimplikar, S. W. (2005). Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J. Cell Biol. 171, 327–335. doi: 10.1083/jcb.200505078
Ryan, T. A., Reuter, H., Wendland, B., Schweizer, F. E., Tsien, R. W., and Smith, S. J. (1993). The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron 11, 713–724. doi: 10.1016/0896-6273(93)90081-2
Ryder, J., Su, Y., Liu, F., Li, B., Zhou, Y., and Ni, B. (2003). Divergent roles of GSK3 and CDK5 in APP processing. Biochem. Biophys. Res. Commun. 312, 922–929. doi: 10.1016/j.bbrc.2003.11.014
Saganich, M. J., Schroeder, B. E., Galvan, V., Bredesen, D. E., Koo, E. H., and Heinemann, S. F. (2006). Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J. Neurosci. 26, 13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006
Samuels, B. A., and Hen, R. (2011). Neurogenesis and affective disorders. Eur. J. Neurosci. 33, 1152–1159. doi: 10.1111/j.1460-9568.2011.07614.x
Schepeler, T., Mansilla, F., Christensen, L. L., Orntoft, T. F., and Andersen, C. L. (2007). Clusterin expression can be modulated by changes in TCF1-mediated Wnt signaling. J. Mol. Signal. 2, 6. doi: 10.1186/1750-2187-2-6
Schmidt, H. D., and Duman, R. S. (2007). The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav. Pharmacol. 18, 391–418. doi: 10.1097/FBP.0b013e3282ee2aa8
Schumacher, J., Laje, G., Abou Jamra, R., Becker, T., Muhleisen, T. W., Vasilescu, C., et al. (2009). The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum. Mol. Genet. 18, 2719–2727. doi: 10.1093/hmg/ddp204
Schwarz-Romond, T., Fiedler, M., Shibata, N., Butler, P. J., Kikuchi, A., Higuchi, Y., et al. (2007). The DIX domain of dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 14, 484–492. doi: 10.1038/nsmb1247
Seib, D. R., Corsini, N. S., Ellwanger, K., Plaas, C., Mateos, A., Pitzer, C., et al. (2013). Loss of Dickkopf-1 restores neurogenesis in old age and counteracts cognitive decline. Cell Stem Cell 12, 204–214. doi: 10.1016/j.stem.2012.11.010
Seitz, R., Hackl, S., Seibuchner, T., Tamm, E. R., and Ohlmann, A. (2010). Norrin mediates neuroprotective effects on retinal ganglion cells via activation of the Wnt/beta-catenin signaling pathway and the induction of neuroprotective growth factors in Muller cells. J. Neurosci. 30, 5998–6010. doi: 10.1523/JNEUROSCI.0730-10.2010
Selkoe, D. J. (2001). Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. 3, 75–80.
Shimizu, H., Julius, M. A., Giarre, M., Zheng, Z., Brown, A. M., and Kitajewski, J. (1997). Transformation by Wnt family proteins correlates with regulation of beta-catenin. Cell Growth Differ. 8, 1349–1358.
Shimogori, T., VanSant, J., Paik, E., and Grove, E. A. (2004). Members of the Wnt, Fz, and Frp gene families expressed in postnatal mouse cerebral cortex. J. Comp. Neurol. 473, 496–510. doi: 10.1002/cne.20135
Simons, M., and Mlodzik, M. (2008). Planar cell polarity signaling: from fly development to human disease. Annu. Rev. Genet. 42, 517–540. doi: 10.1146/annurev.genet.42.110807.091432
Slusarski, D. C., Corces, V. G., and Moon, R. T. (1997a). Interaction of Wnt and a frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 390, 410–413. doi: 10.1038/37138
Slusarski, D. C., Yang-Snyder, J., Busa, W. B., and Moon, R. T. (1997b). Modulation of embryonic intracellular Ca2+ signaling by Wnt-5A. Dev. Biol. 182, 114–120. doi: 10.1006/dbio.1996.8463
Smith, M. J., Wang, L., Cronenwett, W., Mamah, D., Barch, D. M., and Csernansky, J. G. (2011). Thalamic morphology in schizophrenia and schizoaffective disorder. J. Psychiatr. Res. 45, 378–385. doi: 10.1016/j.jpsychires.2010.08.003
Speese, S. D., and Budnik, V. (2007). Wnts: up-and-coming at the synapse. Trends Neurosci. 30, 268–275. doi: 10.1016/j.tins.2007.04.003
Sutton, L. P., Honardoust, D., Mouyal, J., Rajakumar, N., and Rushlow, W. J. (2007). Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J. Neurochem. 102, 153–169. doi: 10.1111/j.1471-4159.2007.04527.x
Tabatadze, N., Tomas, C., McGonigal, R., Lin, B., Schook, A., and Routtenberg, A. (2012). Wnt transmembrane signaling and long-term spatial memory. Hippocampus 22, 1228–1241. doi: 10.1002/hipo.20991
Takashima, A., Murayama, M., Murayama, O., Kohno, T., Honda, T., Yasutake, K., et al. (1998). Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc. Natl. Acad. Sci. U.S.A. 95, 9637–9641. doi: 10.1073/pnas.95.16.9637
Takashima, A., Noguchi, K., Michel, G., Mercken, M., Hoshi, M., Ishiguro, K., et al. (1996). Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci. Lett. 203, 33–36. doi: 10.1016/0304-3940(95)12257-5
Takashima, A., Noguchi, K., Sato, K., Hoshino, T., and Imahori, K. (1993). Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 90, 7789–7793. doi: 10.1073/pnas.90.16.7789
Tamai, K., Semenov, M., Kato, Y., Spokony, R., Liu, C., Katsuyama, Y., et al. (2000). LDL-receptor-related proteins in Wnt signal transduction. Nature 407, 530–535. doi: 10.1038/35035117
Tan, Y., Yu, D., Busto, G. U., Wilson, C., and Davis, R. L. (2013). Wnt signaling is required for long-term memory formation. Cell Rep. 4, 1082–1089. doi: 10.1016/j.celrep.2013.08.007
Tilleman, K., Stevens, I., Spittaels, K., Haute, C. V., Clerens, S., Van Den Bergh, G., et al. (2002). Differential expression of brain proteins in glycogen synthase kinase-3 transgenic mice: a proteomics point of view. Proteomics 2, 94–104. doi: 10.1002/1615-9861(200201)2:1<94::AID-PROT94>3.0.CO;2-W
Toledo, E. M., Colombres, M., and Inestrosa, N. C. (2008). Wnt signaling in neuroprotection and stem cell differentiation. Prog. Neurobiol. 86, 281–296. doi: 10.1016/j.pneurobio.2008.08.001
Toledo, E. M., and Inestrosa, N. C. (2010). Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol. Psychiatry 15, 272–285, 228. doi: 10.1038/mp.2009.72
Valvezan, A. J., and Klein, P. S. (2012). GSK-3 and Wnt signaling in neurogenesis and bipolar disorder. Front. Mol. Neurosci. 5:1. doi: 10.3389/fnmol.2012.00001
van Amerongen, R., Mikels, A., and Nusse, R. (2008). Alternative wnt signaling is initiated by distinct receptors. Sci. Signal. 1, re9. doi: 10.1126/scisignal.135re9
van Amerongen, R., and Nusse, R. (2009). Towards an integrated view of Wnt signaling in development. Development 136, 3205–3214. doi: 10.1242/dev.033910
Varela-Nallar, L., Alfaro, I. E., Serrano, F. G., Parodi, J., and Inestrosa, N. C. (2010). Wingless-type family member 5A (Wnt-5a) stimulates synaptic differentiation and function of glutamatergic synapses. Proc. Natl. Acad. Sci. U.S.A. 107, 21164–21169. doi: 10.1073/pnas.1010011107
Veeman, M. T., Axelrod, J. D., and Moon, R. T. (2003). A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell 5, 367–377. doi: 10.1016/S1534-5807(03)00266-1
Vogt, D. L., Thomas, D., Galvan, V., Bredesen, D. E., Lamb, B. T., and Pimplikar, S. W. (2011). Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol. Aging 32, 1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002
Walsh, D. M., and Selkoe, D. J. (2004). Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44, 181–193. doi: 10.1016/j.neuron.2004.09.010
Wang, J., Ruan, N. J., Qian, L., Lei, W. L., Chen, F., and Luo, Z. G. (2008). Wnt/beta-catenin signaling suppresses Rapsyn expression and inhibits acetylcholine receptor clustering at the neuromuscular junction. J. Biol. Chem. 283, 21668–21675. doi: 10.1074/jbc.M709939200
Wang, J., Shou, J., and Chen, X. (2000). Dickkopf-1, an inhibitor of the Wnt signaling pathway, is induced by p53. Oncogene 19, 1843–1848. doi: 10.1038/sj.onc.1203503
Wang, J., Sinha, T., and Wynshaw-Boris, A. (2012). Wnt signaling in mammalian development: lessons from mouse genetics. Cold Spring Harb. Perspect. Biol. 4, pii: a007963. doi: 10.1101/cshperspect.a007963
Wayman, G. A., Impey, S., Marks, D., Saneyoshi, T., Grant, W. F., Derkach, V., et al. (2006). Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 50, 897–909. doi: 10.1016/j.neuron.2006.05.008
Wehrli, M., Dougan, S. T., Caldwell, K., O’Keefe, L., Schwartz, S., Vaizel-Ohayon, D., et al. (2000). arrow encodes an LDL-receptor-related protein essential for wingless signalling. Nature 407, 527–530. doi: 10.1038/35035110
Weisstaub, N. V., Zhou, M., Lira, A., Lambe, E., Gonzalez-Maeso, J., Hornung, J. P., et al. (2006). Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science 313, 536–540. doi: 10.1126/science.1123432
Wexler, E. M., Geschwind, D. H., and Palmer, T. D. (2008). Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol. Psychiatry 13, 285–292. doi: 10.1038/sj.mp.4002093
Wisniewska, M. B. (2013). Physiological role of beta-catenin/TCF signaling in neurons of the adult brain. Neurochem. Res. 38, 1144–1155. doi: 10.1007/s11064-013-0980-9
Wisniewska, M. B., Nagalski, A., Dabrowski, M., Misztal, K., and Kuznicki, J. (2012). Novel beta-catenin target genes identified in thalamic neurons encode modulators of neuronal excitability. BMC Genomics 13:635. doi: 10.1186/1471-2164-13-635
Wong, G. T., Gavin, B. J., and McMahon, A. P. (1994). Differential transformation of mammary epithelial cells by Wnt genes. Mol. Cell. Biol. 14, 6278–6286. doi: 10.1128/MCB.14.9.6278
Wray, J., and Hartmann, C. (2012). WNTing embryonic stem cells. Trends Cell Biol. 22, 159–168. doi: 10.1016/j.tcb.2011.11.004
Yamanaka, H., Moriguchi, T., Masuyama, N., Kusakabe, M., Hanafusa, H., Takada, R., et al. (2002). JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 3, 69–75. doi: 10.1093/embo-reports/kvf008
Yang, J., Si, T., Ling, Y., Ruan, Y., Han, Y., Wang, X., et al. (2003). Association study of the human FZD3 locus with schizophrenia. Biol. Psychiatry 54, 1298–1301. doi: 10.1016/S0006-3223(03)00291-9
Yi, H., Hu, J., Qian, J., and Hackam, A. S. (2012). Expression of brain-derived neurotrophic factor is regulated by the Wnt signaling pathway. Neuroreport 23, 189–194. doi: 10.1097/WNR.0b013e32834fab06
Yu, X., and Malenka, R. C. (2003). Beta-catenin is critical for dendritic morphogenesis. Nat. Neurosci. 6, 1169–1177. doi: 10.1038/nn1132
Yuste, R., Peinado, A., and Katz, L. C. (1992). Neuronal domains in developing neocortex. Science 257, 665–669. doi: 10.1126/science.1496379
Zhang, Y., McLaughlin, R., Goodyer, C., and LeBlanc, A. (2002). Selective cytotoxicity of intracellular amyloid beta peptide1-42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 156, 519–529. doi: 10.1083/jcb.200110119
Keywords: Wnt in adult brain, neurodegenerative diseases, Wnt in circuit development, Wnt signaling pathways, spontaneous activity, activity-dependent processes
Citation: Oliva CA, Vargas JY and Inestrosa NC (2013) Wnts in adult brain: from synaptic plasticity to cognitive deficiencies. Front. Cell. Neurosci. 7:224. doi: 10.3389/fncel.2013.00224
Received: 27 May 2013; Accepted: 03 November 2013;
Published online: 03 December 2013.
Edited by:
Lavinia Alberi, University of Fribourg, SwitzerlandReviewed by:
Urs Albrecht, University of Fribourg, SwitzerlandCopyright © 2013 Oliva, Vargas and Inestrosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nibaldo C. Inestrosa, Centro de Envejecimiento y Regeneración, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Alameda 340, P.O. Box 114-D, Santiago, Chile e-mail:bmluZXN0cm9zYUBiaW8ucHVjLmNs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.