
- 1NeuroBiology and Genetics Group, Genetic Medicine Research Centre, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, UPM Serdang, Malaysia
- 2Clinical Genetics Unit, Department of Obstetrics and Gynaecology, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, UPM Serdang, Malaysia
- 3Department of Human Anatomy, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, UPM Serdang, Malaysia
Intellectual disability (ID) is one of the many features manifested in various genetic syndromes leading to deficits in cognitive function among affected individuals. ID is a feature affected by polygenes and multiple environmental factors. It leads to a broad spectrum of affected clinical and behavioral characteristics among patients. Until now, the causative mechanism of ID is unknown and the progression of the condition is poorly understood. Advancement in technology and research had identified various genetic abnormalities and defects as the potential cause of ID. However, the link between these abnormalities with ID is remained inconclusive and the roles of many newly discovered genetic components such as non-coding RNAs have not been thoroughly investigated. In this review, we aim to consolidate and assimilate the latest development and findings on a class of small non-coding RNAs known as microRNAs (miRNAs) involvement in ID development and progression with special focus on Down syndrome (DS) and X-linked ID (XLID) [including Fragile X syndrome (FXS)].
Introduction
Intellectual disability (ID), also known as mental retardation, is the most common developmental disability and affects about 1–3% of the world population. It is a condition defined as having a significantly impaired cognitive ability and adaptive behaviors before the age of 18 (Daily et al., 2000). In clinical practice, diagnosis of ID in a patient is based on the intelligence quotient (IQ) below 70 at the age of 5 or older and deficits in two or more adaptive behaviors, such as communication, self-care, social skills, community access skills, self-direction, health, and safety (Kaufmann et al., 2007; Van Bokhoven, 2011). Conventionally, ID is divided into two distinctive groups; syndromic ID and non-syndromic ID. In syndromic ID, patients are presented with additional clinical features such as physical deformities or metabolic defects whereas intellectual impairment is the sole clinical features for non-syndromic ID (Kaufman et al., 2010; Van Bokhoven, 2011). However, there are considerable challenges in distinguishing both groups as some clinical features are very subtle and difficult to be diagnosed. Therefore, multiple comparisons have to be made between patients with common etiology to identify the genetic defects associated with the clinical features presented.
In addition to the classification based on clinical features, ID can be categorized according to the level of severity based on IQ of the patient. Patients with IQ below 20 is categorized as profound ID, IQ of 20–34 as severe ID, IQ of 35–49 as moderate ID, and IQ of 50–70 as mild ID (American Psychiatric Association, 2000). However, most studies often use a simpler classification where the patients are categorized as mild ID when the IQ lies between 50 and 70 or severe ID when the IQ is below 50. Under this simpler classification, only 20% of mild ID cases have any conclusive genetic causes whereas other cases are largely attributed by environmental factors such as malnutrition during pregnancy, infections, fetal alcohol syndrome, premature birth or exposure to neurotoxic compounds (Van Bokhoven, 2011). By contrast, about 50–65% of severe ID cases are commonly associated with genetic disorders resulted from chromosomal abnormalities, dysregulation of genetic imprinting, or monogenic disorders (Van Bokhoven, 2011).
The most prevalent genetic abnormalities associated with ID is chromosomal aberrations, such as trisomy 21 in Down syndrome (DS), trisomy 18 in Edward syndrome, the presence of extra chromosome X in males with Klinefelter syndrome or monosomy X in females with Turner syndrome, and chromosomal deletions at 22q11 in DiGeorge syndrome. Angelman syndrome and Prader–Willi syndrome are examples of genetic disorders caused by dysregulation of genetic imprinting, with ID as one of the common clinical features. On the other hand, monogenic disorder for ID is rare and commonly manifested as non-syndromic ID. It is also commonly known as X-linked ID (XLID) because most of the associated genes are located on the X chromosome. Coffin–Lowry syndrome is an example of XLID that arises as a result of X-linked dominant inheritance of mutated RPS6KA3 (ribosomal protein S6 kinase, 90 kDa polypeptide 3) on the X chromosome (Kleefstra et al., 2005).
Understanding the affected molecular pathways and cellular processes due to genetic abnormalities will help to unravel mechanisms underlying ID. However, the association between the genetic abnormalities and ID has remained unclear. In the past two decades, the discoveries of microRNAs (miRNAs), a class of small non-coding RNA about 18–24 nucleotides (nt), had brought new perspective to the studies of ID (Bartel, 2004). The discovery of a small RNA transcript lin-4 in 1993 through a genetic screening in the nematodes Caenorhabditis elegans and its roles in regulation of lin-14 translation via antisense RNA–RNA interaction have brought forth the beginning of the era of miRNAs and non-coding RNAs (Lee et al., 1993; Wightman et al., 1993). The era of miRNAs truly took off after the second miRNA, let-7, was discovered in 2000 in C. elegans. Let-7 functions similarly as lin-4 and was found to be conserved throughout metazoans (Reinhart et al., 2000). This finding sparks off a large scale searches for other miRNAs and their roles in regulating cellular and molecular processes (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001).
Since the discovery of lin-4 and let-7, a total of 21,264 miRNA entries were indexed in miRBase (www.mirbase.org; miRBase v19 accessed on 8/11/2012) for various organisms ranging from mammals (e.g., human, chimpanzee, mouse, cattle) to zebrafish, plants, arthropods, and viruses. Figure 1A shows the number of miRNAs indexed in miRBase v19 for selected organisms and the human genome harbors the greatest number of miRNAs that are reported to date (approximately 10% of all known miRNAs). In addition, the miRNA genes are not randomly distributed across the mammalian genome because some chromosomes in human (e.g., HSA19) and mouse (e.g., MMU2 and MMU12) genomes have exceptionally high density of miRNA genes as compared to the global average (Figure 2). The non-random genomic distribution of miRNA genes across the human genome or within a chromosome has been associated with dysregulated expression levels of miRNAs that may lead to diseases (Calin et al., 2004; Albano et al., 2010). Besides, changes of the copy number for miRNA genes due to aneuploidy may serve as one of the many causes of ID. Somatic mosaicisms in neurons due to aneuploidy have been identified in the mammalian brain and constitute only a small proportion of all neurons. These aneuploid neurons are resulted from errors in chromosomal segregation during normal progenitor cells proliferation and are integrated within circuitries of the normal brain (Rehen et al., 2001, 2005; Yang et al., 2003; Kingsbury et al., 2005). Taken together, the effect of non-random genomic distribution and imbalance dosage of miRNA genes in ID development may serve as an important piece of missing puzzle in understanding various genetic syndromes of ID.
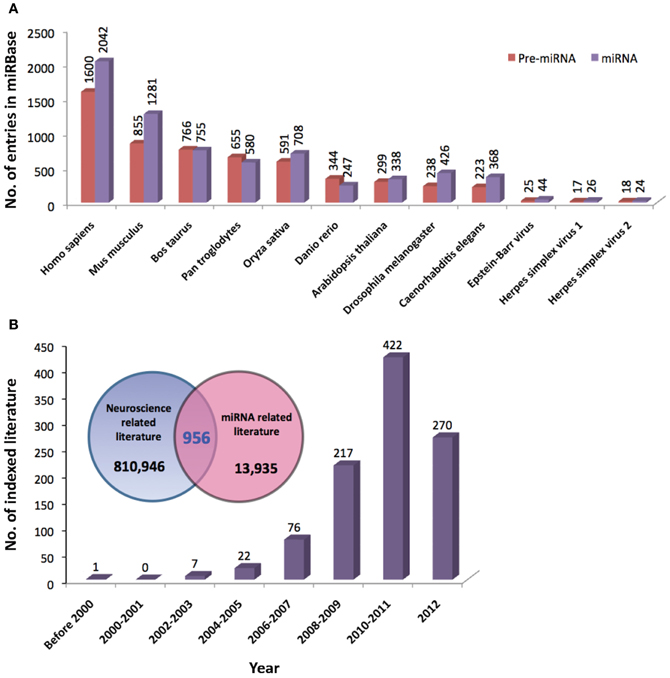
Figure 1. (A) Total number of precursor and mature miRNAs from various species of organisms indexed by miRBase (www.miRBase.org; miRBase v19 accessed on 8/11/2012). (B) The Venn diagram shows the total number of neuroscience- and miRNA-related literature indexed in PubMed. Neuroscience-related literature were searched based on the following terms/functions used in the title/abstract of the literature; (1) neuroscience or (2) brain or (3) neuron or (4) neuron or (5) glia or (6) nervous system. MiRNA-related literature were searched based on (1) miRNA or (2) microRNA or (3) lin-4 or (4) let-7 terms/functions. Total numbers of overlapping literature between the two groups of literature were searched based on the combined terms/functions: neuroscience-related search terms AND miRNA-related search terms. The graph shows the number of neuroscience-miRNA-related literature indexed in PubMed since 2000.
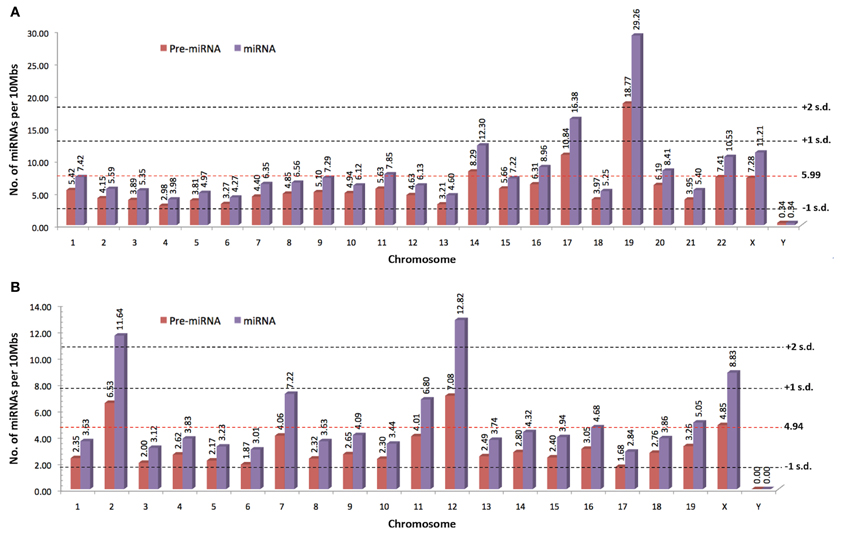
Figure 2. The normalized number of precursor and mature miRNAs per 10 Mbs indexed by miRBase for (A) human and (B) mouse (www.miRBase.org; miRBase v19 accessed on 8/11/2012). Red-dotted horizontal lines denote the average mature miRNAs per 10Mbs across the genome. Gray-dotted horizontal lines denote the number of standard deviation (SD) from the global average value.
To date, a great number of miRNAs have been shown to be associated with the development and function of nervous system (Christensen and Schratt, 2009; Fiore et al., 2011; Im and Kenny, 2012; McNeill and Van Vactor, 2012). The role of miRNAs in molecular regulation of gene expression in neurons (Siegel et al., 2011), learning and memory (Konopka et al., 2011), and the development of neurological disorders (Forero et al., 2010; Satoh, 2010) has been extensively reviewed. Since 2000, the total number of indexed literature in PubMed that are related to the field of neuroscience and miRNA has increased tremendously (Figure 1B). However, the role of miRNAs in ID has not gained much attention to date, thus in this review, we will provide a brief overview on miRNA biogenesis and function and highlight the involvement of miRNAs in molecular mechanisms related to ID with special focus on DS, XLID, and Fragile X syndrome (FXS).
MiRNA Biogenesis and Function
MiRNAs are transcribed from their genes by either RNA polymerase II or III into a long primary transcript (pri-miRNAs) that folded itself into a hairpin structure (Figure 3) (Lee et al., 2004; Borchert et al., 2006). These miRNAs genes are located in both coding and non-coding genes across different regions in the genome. Frequently, miRNAs are transcribed from the introns of protein coding genes (Rodriguez et al., 2004; Saini et al., 2007). In addition, miRNAs are also transcribed from the non-coding region as part of long non-coding RNAs and are often arranged in clusters in the genome, leading to the formation of several miRNAs from the same pri-miRNA transcript (Rodriguez et al., 2004).
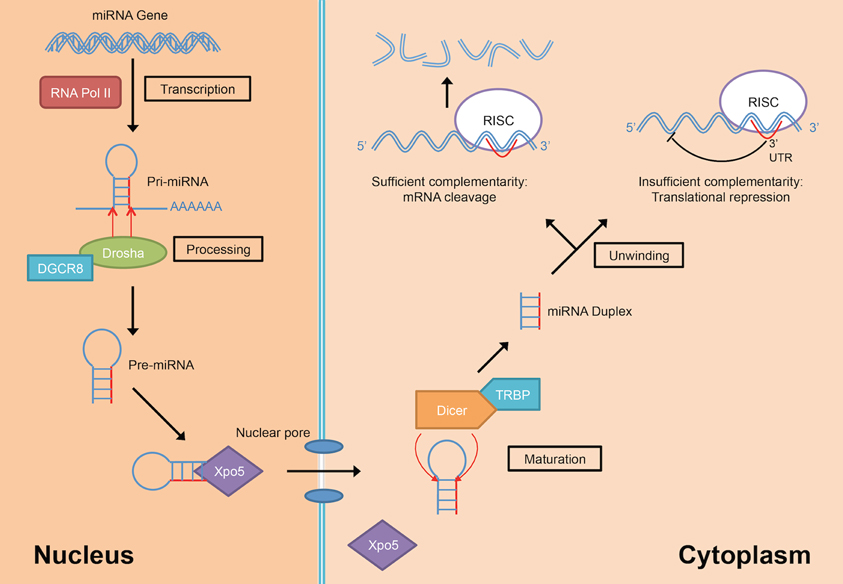
Figure 3. MiRNA biogenesis and mode of action (DGCR8, DiGeorge Critical Region 8; RISC, RNA-induced silencing complex; RNA Pol II, RNA polymerase II; TRBP, Tar-RNA binding protein; Xpo5, Exportin-5).
Following the transcription into pri-miRNA, the primary transcripts are then further processed in two stages by two different enzymes from the RNase III family; Drosha in the nucleus and Dicer in the cytoplasm. Drosha, in concert with DiGeorge Critical Region 8 (DGCR8) protein, facilitate the nuclear cleavage of pri-miRNAs into a ~60–90 nt stem loop intermediate resembles hairpin and termed as precursor miRNAs (pre-miRNAs) (Lee et al., 2003). The cleavage happened at the 5′ and 3′ arms of the pri-miRNAs hairpin by the action of two RNase domains on Drosha (Han et al., 2004). During the cleavage, DGCR8 is needed to stabilize the Drosha at its middle domain as well as to determine the exact cleavage site in the pri-miRNA. Subsequently, the resulted pre-miRNA hairpin is exported into the cytoplasm with the aid from a nuclear transport receptor, Exportin-5 (Xpo5) (Yi et al., 2003; Lund et al., 2004).
In the cytoplasm, pre-miRNAs are further processed into RNA duplexes of miRNA:miRNA* by Dicer with the help of its cofactor, Tar-RNA binding protein (TRBP) (Chendrimada et al., 2005; Haase et al., 2005). There is an additional cleavage step for miRNAs that have high degree of complementarity along the hairpin stem by the action of Argonaute-2 (Ago2) which generates a nick in the middle of the prospective passenger strand (complementary strand to the mature miRNA that usually exists in low level), resulting in Ago2-cleaved pre-miRNA (ac-pre-miRNA) (Diederichs and Haber, 2007). The loop on both pre-miRNA and ac-pre-miRNA are then cleaved by Dicer to generate a miRNA duplex of about 22 nt in length.
The miRNA duplex generated from Dicer-mediated cleavage need to be separated into a functional guide strand that is complementary to the target and the passenger strand is usually degraded. The generation of the single stranded guide strand as the mature miRNA requires the unwinding activity of helicase. However, a universal helicase responsible for this has not been identified to date. The single stranded guide miRNA will be incorporated into a ribonucleoprotein effector complex, termed as RNA-induced silencing complex (RISC). Then, the miRNA directs the RISC to the target mRNA by binding to the 3′ UTR of the targeted mRNA. Nucleotides at positions 2–8 at 5′ region of the miRNA is known as the seed sequence and is involved in recognizing the target mRNA (Bartel, 2004). The choice of mRNA posttranscriptional regulation mechanisms depends on the complementarity of the seed sequence as well as supplementary binding of 3′ region of the miRNA with the 3′ UTR of the target mRNA. If the miRNA has perfect or sufficient complementarity to the mRNA, RISC will cleave off the target mRNA leading to mRNA degradation. Translational repression will happen instead if the miRNA:mRNA binding is imperfect (Bartel, 2004) leading to increased rate of degradation due to deadenylation of the mRNA (Eulalio et al., 2009). The deadenylation of mRNAs by miRNAs initiated the hypothesis that complete deadenylation leads to disruption of the interaction of poly(A)-binding protein and the cap, thus reducing the translation of the mRNAs. However, a recent publication demonstrated that in the case of miR-430, the translational repression occurred independent of the deadenylation (Bazzini et al., 2012). The author suggested that the miRNAs with partial complementarity to its target may trigger both translational repression and deadenylation to ensure maximum target mRNA repression and degradation.
MiRNA and Down Syndrome
DS is a genetic disorder of trisomy human chromosome 21 (HSA21) with live birth prevalence of 11.2 per 10,000 births (Loane et al., 2012). All DS patients exhibit ID. They also develop other clinical features such as typical craniofacial appearance (e.g., brachycephaly, epicanthic fold and protruding tongue), hypotonia, congenital heart defects, early onset of Alzheimer's disease (AD), dementia as well as cognitive impairment (Van Cleve and Cohen, 2006; Van Cleve et al., 2006). Mild to severe intellectual disabilities were observed in DS patients with reported average value of 50 in IQ and learning disabilities involving both long-term and short-term memory formation (Brown et al., 2003; Vicari et al., 2005). The extra HSA21 caused various neuroanatomical abnormalities in DS patients such as reduction in brain size, brain weight as well as neuronal density, neuronal distribution, and dendritic abnormalities at cellular level (Wisniewski, 1990; Takashima et al., 1994; Kaufmann and Moser, 2000). There have been many hypotheses proposed to correlate the extra HSA21 to DS phenotypes such as gene dosage imbalance hypothesis, amplified developmental instability hypothesis, and critical region hypothesis (Delabar et al., 1993; Pritchard and Kola, 1999; Antonarakis et al., 2004). There is growing interest in epigenetic modifications, allelic differences, gene dosage compensation and also additive, subtractive, and epistatic patterns of genetic contribution to DS phenotypes (Dey, 2011).
Latest statistic from miRBase (www.mirbase.org; miRBase v19 accessed on 21/12/2012) indicates that there are 19 precursors and 26 mature miRNAs located on HSA21 but only five of them have been implicated in DS so far such as miR-155, miR-802, miR-125b-2, let-7c, and miR-99a (as summarized in Table 1) (Kuhn et al., 2008). These miRNAs are overexpressed in DS fetal hippocampus and heart samples and are exclusively expressed in neurons but not in microglial cells or astrocytes. Interestingly, neurons expressing these miRNAs were found increased by 10–15 fold in DS fetal hippocampus samples as compared to age- and sex-matched controls (Kuhn et al., 2008).
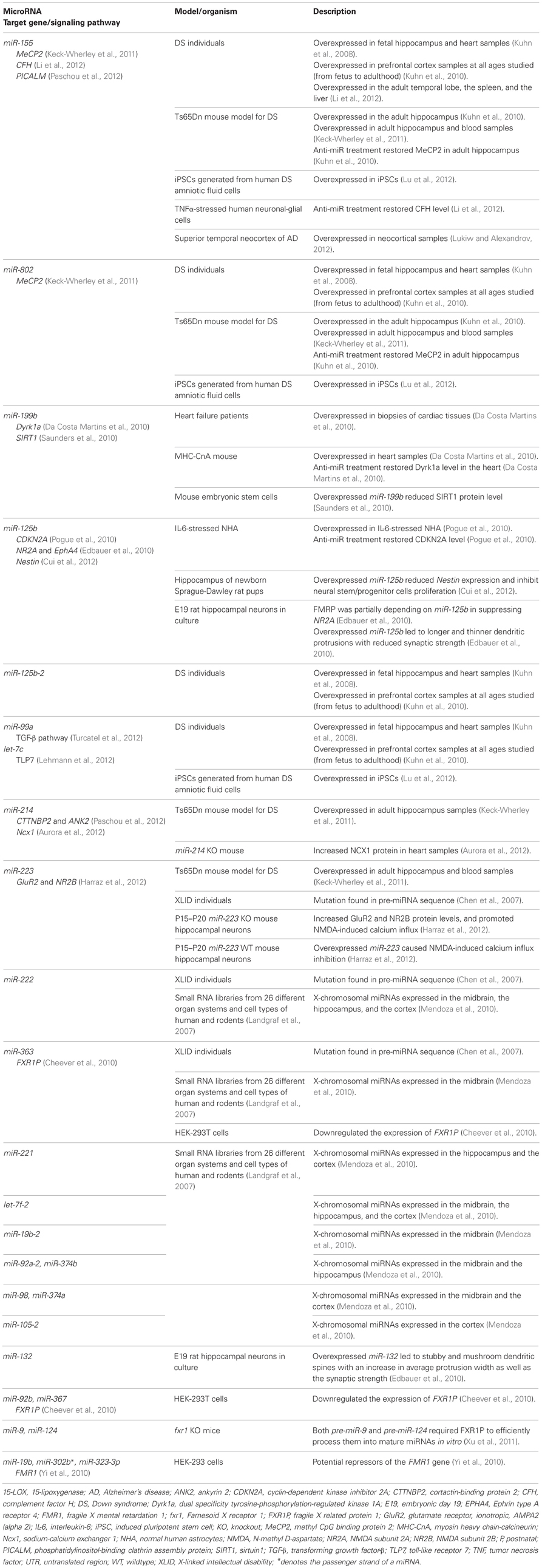
Table 1. Summary of miRNAs that are associated with intellectual disabilities.
Trisomic overexpression of HSA21-derived miRNA is believed to contribute, in part, to the defective neurodevelopment in DS individuals. Overexpression of miR-155 and miR-802 (Table 1) in Ts65Dn (a genetic mouse model of DS) hippocampus may contribute to impaired hippocampal synaptic plasticity and neurogenesis (Keck-Wherley et al., 2011). Both miR-155 and miR-802 target the methyl CpG binding protein 2 (MeCP2) (Kuhn et al., 2010), which is associated with deleterious effects on development as seen in Rett Syndrome (Cohen et al., 2003). Silencing of these two miRNAs in Ts65Dn is able to restore the MeCP2 expression and its target genes (Kuhn et al., 2010). Overexpression of miR-155 and miR-802 and low expression of MeCP2 were observed in induced pluripotent stem cells (iPSC) from human DS amniotic fluids cells (Lu et al., 2012). Interestingly, a study in mouse model of Rett Syndrome showed that MeCP2 repressed miR-199b expression in the mouse brain (Urdinguio et al., 2010) and miR-199b has been shown to suppress sirtuin 1 (SIRT1) (Saunders et al., 2010), which is involved in dendritic development (Codocedo et al., 2012). Reduction of SIRT1 promotes neurogenic potential of adult subventricular zone and hippocampal neural precursors (Saharan et al., 2013). As aforementioned, overexpression of miR-155 and miR-802 have been found to repress MeCP2 expression (Kuhn et al., 2010), therefore these HSA21-miRNAs in DS brain are expected to repress MeCP2 and subsequently relieve the suppression on miR-199b and decreased SIRT1 expression. This interconnected cascade of regulations suggests a series of complex events that may be disrupted in DS brain development. Selective inactivation on these HSA21-derived miRNAs can be employed as a novel therapeutic tool in the future.
DS is also characterized by brain deficits and systemic immune pathology such as activated microglia and increased inflammatory signaling. Overexpression of miR-155 and repression of complement factor H (CFH), an essential repressor of the innate immune response were observed in brains and peripheral tissues of DS individuals (Li et al., 2012). Interestingly, miR-155 was overexpressed in AD and was found to regulate CFH expression (Lukiw and Alexandrov, 2012). It was predicted to interact with presynaptic mRNAs such as phosphatidylinositol-binding clathrin assembly protein (PICALM) (Paschou et al., 2012), which has been implicated in AD (Melville et al., 2012). As DS patients are prone to develop early onset AD, association between miR-155 and brain pathology should be further investigated to unravel its role in regulating innate immune and inflammatory responses.
There have been efforts made to unravel the role of miR-125b (summarized in Table 1) in the neuropathology of DS subjects. MiR-125b was significantly upregulated in interleukin-6-stressed normal human astrocytes (Pogue et al., 2010). Anti-miR-125b treatment attenuated astrogliosis and increases cyclin-dependent kinase inhibitor 2A (CDKN2A), a negative regulator of cell growth. Astrogliosis in DS brains could be attributed to the glial proliferative effect of miR-125b via suppression of CDKN2A (Pogue et al., 2010). In addition, miR-125b also targeted glutamate receptor, ionotropic, N-methyl d-aspartate subunit 2A (NR2A), Ephrin type A receptor 4 (EPHA4), and Nestin (Edbauer et al., 2010; Cui et al., 2012). Mir-125b is crucial for spine morphology development and neuronal survival (Schaefer et al., 2007). It stimulates neurite outgrowth and dendritic branching (Le et al., 2009) which produced longer and thinner dendritic spine with reduced spine width by targeting NR2A (Edbauer et al., 2010). Suppression of NR2A and EphA4 have been implicated in long-term potentiation (LTP) and/or long-term depression (LTD) processes (Zhao and Constantine-Paton, 2007; Filosa et al., 2009; Xu et al., 2009). In a Luciferase reporter assay, miR-125b was shown to target 3′UTR of Nestin and reduced both Nestin mRNA and protein levels. When overexpressed, miR-125b inhibited neural stem/progenitor cells proliferation via suppression of Nestin (Cui et al., 2012). Taken together, it is evident that the overexpression of miR-125b may lead to significant changes in neuronal development. The role of miR-125b in targeting various transcripts leading to impaired synaptogenesis and LTP in DS subjects should be investigated.
Mir-99a regulates transforming growth factor-β (TGF-β) pathway (Turcatel et al., 2012) which directly involved in retrograde synaptic signaling (Sanyal et al., 2004) and regulates Wnt signaling in stem cell differentiation (Cai et al., 2013). On the other hand, let-7c is highly expressed in the hippocampal region of adult mice (Bak et al., 2008). It has been shown to cause neuronal loss in a dose- and time-dependent manner by acting on toll-like receptor 7 (TLR7) (Lehmann et al., 2012). Role of mir-99a and let-7c in regulating neuronal function in DS individuals warrant further investigations.
Children with DS often experience auditory deficits (Cunningham and McArthur, 2006), language difficulties which include reading impairment (Lemons and Fuchs, 2010; Nash and Heath, 2011) and limited verbal short-term memory capacity (Jarrold et al., 2002; Brock and Jarrold, 2005). These DS-associated impairments will generally lead to learning disabilities in DS children. MiR-125b-2, let-7c, and miR-99a are the three HSA21-derived miRNAs which orthologs were found expressed in the mouse inner ear (Weston et al., 2006). An increased in the size of the large conductance calcium activated potassium (BK) current (which is known to determine electrical tuning) was observed in Tc1 (another mouse model for DS that carries a significant portion of HSA21) inner hair cells as compared to control (Kuhn et al., 2012). Thus, the roles of miR-125b-2, let-7c, and miR-99a should be validated in human samples to better understand their roles in regulating auditory development or function in DS individuals.
Other non HSA21-derived miRNAs such as miR-214 and miR-223 were found overexpressed in the adult Ts65Dn hippocampus (Keck-Wherley et al., 2011) but their expression profiles have not been extensively characterized in DS individuals. Mir-214 was predicted to regulate cortactin-binding protein 2 (CTTNBP2) and ankyrin 2 (ANK2) that code for synaptic protein (Paschou et al., 2012). It also repressed sodium-calcium exchanger 1 (Ncx1) (Aurora et al., 2012), which is crucial for spine-dendrites compartmentalization (Lörincz et al., 2007) and presynaptic calcium recovery mechanism in cerebellar parallel fiber (Roome et al., 2013). MiR-223 has been reported to function as a regulator on glutamate receptor subunits NR2B and GluR2 and its overexpression led to NMDA-induced calcium influx inhibition on hippocampal neurons (Harraz et al., 2012). Downstream gene targets of both miR-214 and miR-223 have important roles in regulating neuronal development and function, however, the direct association between these miRNAs and their targets with neuronal dysfunction in DS are yet to be determined.
MiRNAs derived from other chromosomes but targeted genes located on HSA21 may contribute to the phenotypes of DS. For instance, both mouse chromosome 9-derived miR-199b and human chromosome 2-derived miR-1246 are known to downregulate DS associated dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A gene (Dyrk1a—encoded in MMU16; DYRK1A—encoded in HSA21) and subsequently induce apoptosis through nuclear factor of activated T-cells (NFAT) pathway (Da Costa Martins et al., 2010). Dyrk1a with dysregulated NFAT signaling has been reported in DS individuals (Arron et al., 2006). NFAT signaling pathway has been implicated in AD (Abdul et al., 2009; Hudry et al., 2012). Based on the AD model, further investigation of both miR-199b:Dyrk1a and miR-1246:DYRK1A interactions may provide an insight on mechanisms mediated by these miRNAs in ID progression among DS individuals.
In brief, HSA21-harbored miRNAs are directly involved in neuronal differentiation, neuronal development, synaptic plasticity and more. Their roles in DS may shed light in understanding the underlying mechanisms in the development of ID More excitingly, miRNA-based therapies have been shown to restore expression of targeted genes as well as their downstream activities. Restoration of defective neurogenesis, gliogenesis and cell cycles events have not been fully explored and these warrant further investigation to better understand ID development in DS patients.
MiRNA and X-Linked Intellectual Disabilities
X chromosome plays a major role in the development of sexual characteristics and it harbors many genes that are involved in cognitive functions. XLID broadly refer to different forms of ID that are inherited as X-linked traits. First reported in 1938, Lionel Penrose observed higher number of males suffered from ID than female from a survey and classification of those in institutional care and their relatives (Penrose, 1938). He also found that the ratio between male to female ID patients was 1.25:1, which was eventually confirmed by many other studies in USA, Canada, Australia as well as Europe with all the studies came into agreement that about 30% excess of males as compared to female were affected by ID (Raymond, 2006). From various family studies, it was then discovered that many of the ID was inherited as X-linked trait. Hence, XLID typically affects the male population with mild to severe ID. Females are less likely to be affected by XLID. If they are affected, they developed milder ID symptoms than the male. This is due to X chromosome inactivation where one of the two X chromosomes in female is inactivated randomly early in development. The X-inactivation is a dosage correction mechanism in female to prevent the double dosage effect of the genes on both X chromosomes. For female with XLID, the mechanism could reduce the effect of mutated or deleted genes by inactivating the affected X chromosome.
XLID is manifested in more than 150 syndromes, including FXS, Klinefelter syndrome, Rett syndrome, Coffin–Lowry syndrome, and X-linked alpha thalassemia. To date, 102 X-linked genes have been associated to 82 syndromes with mutations in these genes described as the causes (Stevenson et al., 2012). However, the identification of genes that are responsible for other XLID syndromes had been unsuccessful due to the rarity of the syndromes or the difficulty in identifying the genes involved. This has impeded the progression in XLID related research.
In 2007, a large-scale mutation screening of miRNAs in patients with ID was reported (Chen et al., 2007). A cohort of 464 patients with XLID has been screened for mutations in 13 known, brain expressed X-chromosomal miRNAs. Four nucleotide changes in three different pre-miRNAs (miR-222, miR-223, and miR-363; Table 1) have been observed in the study though the changes appeared to be functionally neutral and did not affect the function of the respective miRNAs. The authors suggested that the functionally neutral mutations on the miRNAs indicate a strong selection on the pre-miRNA and thus reflecting on the general importance of miRNA system.
Mendoza and colleagues are interested in the potential roles of brain expressed X-chromosomal miRNAs especially in the context of human intelligence and they attempted such investigation by using informatics tools (Mendoza et al., 2010). By mining the Mammalian miRNA Expression Atlas based on Small RNA Library Sequencing (Landgraf et al., 2007), 77 human X-chromosomal miRNAs was identified and ten were further selected for target genes prediction as well as gene ontologies (GO) analysis by virtue of their differential levels in the cortex, hippocampus and midbrain. The ten miRNAs selected were let-7f-2, miR-19b-2, miR-92a-2, miR-98, miR-105-2, miR-221, miR-222, miR-363, miR-374a, and miR-374b (Table 1). Out of the 10 miRNAs, only let-7f-2 and miR-222 were expressed in all the three brain regions studied indicating that they may play an important role in regulating brain function. Target genes for all the 10 selected miRNAs were predicted using in silico tools (MiRanda, TargetScan, and MirTarget2) and results from all analyses were pooled for GO annotation enrichment analysis. The enrichment analysis showed that many biological processes were associated with brain development including the development of the forebrain, midbrain as well as hippocampus. These results show that X chromosome is a potential repository for genes highly expressed in the brain and may have roles in regulating the development or function of nervous system.
MiRNA and Fragile X Syndrome
FXS, also known as Martin Bell syndrome, is one of the most common forms of XLID with an estimated prevalence of 1 in 4000 males and 1 in 8000 females (Warren and Sherman, 2001). The syndrome is transmitted as an X-linked dominant trait and individuals with the syndrome are presented with mild to severe ID (IQs in the range of 20–70), mild abnormal facial features (mainly in males), as well as macroorchidism in postpubescent males (Warren and Nelson, 1994). The loss-of-function mutation on fragile mental retardation 1 (FMR1) gene, due to the trinucleotide expansion of CGG repeat in 5′ untranslated region (UTR), has been identified as the cause for FXS. The large trinucleotide expansion has been associated with hypermethylation of both the CGG repeat and upstream CpG islands, resulting in transcriptional silencing of FMR1, which leads to the loss of its product, fragile X mental retardation protein (FMRP) (Jin et al., 2004a). As a translation repressor, miRNAs may play a role in downregulating the production of FMRP. For example, miR-19b, miR-302b*, and miR-323-3p (Table 1) were shown to target 3′ UTR of FMR1 in a luciferase reporter gene assay (Yi et al., 2010). This suggests that the expression of FMRP is susceptible to miRNA regulation by targeting FMR1 transcripts. Such incident, however, have not been demonstrated or validated in FXS subjects to date.
FMRP is an RNA-binding protein which plays a role in mRNA transport and translational regulation especially in the synaptic region of neurons. Loss of functional FMRP impairs normal synaptic plasticity, which is believed to be the molecular basis for ID in FXS patients (Bassell and Warren, 2008). Comparing miRNAs targets predicted by miRanda with datasets of FMRP-bound mRNAs, 74% of the mRNAs were found to be matching with miRNAs target genes (John et al., 2004). This suggests that some of the translation may be regulated through actions of both FMRP and miRNAs, indicating a close association between both. Indeed, it was shown that FMRP interacts with miRNAs as well as its machinery including Dicer and mammalian ortholog of Argonaute 1 (AGO1) in mammalian cell cultures (Jin et al., 2004b). FMRP also facilitate the assembly of miRNAs on specific target mRNAs by acting as a miRNA acceptor protein for Dicer (Plante et al., 2006). RNAs targeted by FMRP were involved in synaptic signaling pathways such as synaptic LTP, glutamate receptor signaling, neuropathic pain signaling, GABA receptor signaling, synaptic LTD and CREB signaling in neurons. FMRP bound to its RNA targets mainly at the coding sequences and stalled ribosomes (Darnell et al., 2011). Loss of FMRP function relieved the ribosomal stalling, thus suggesting that FMRP may regulate translation at the level of elongation (Darnell et al., 2011). Since FMRP could act as a miRNA acceptor from Dicer, any defect in the function of FMRP will not only cause abnormalities in miRNA function but translational regulation of its targeted mRNAs as well.
FMRP-associated miR-125b and miR-132 have a crucial role in regulating the structure and function of the synapses (Edbauer et al., 2010). By overexpressing miR-125b in mouse hippocampal neurons in culture, longer and thinner dendritic protrusions with reduced synaptic strength were observed. Overexpression of miR-132, however, resulted in stubby and mushroom spines with an increased average protrusion width as well as the synaptic strength (Table 1). When FMRP was downregulated in vitro, overexpression of both miR-125b and miR-132 did not cause any phenotypic changes to the dendritic spine morphology suggesting both FMRP and its associated miRNAs were needed for translation regulation. Together with FMRP, miR-125b targeted and suppressed NR2A, a subunit of NMDA (N-methyl-D-aspartate) receptor (Edbauer et al., 2010), which has an important role in LTP in cortex and amygdala, spike-timing-dependent plasticity and ocular dominance plasticity (Li et al., 2002; Zhao et al., 2005; Desai et al., 2006; Dölen, 2007; Meredith et al., 2007). All these findings further solidified the potential role of miR-125b and FMRP in the synaptic plasticity and function.
In addition to FMRP, another paralogs of FMRP known as Fragile X-related protein 1 (FXR1P) was targeted and regulated by miR-92b, miR-363, and miR-367 (Table 1). Overexpression of miR-367 downregulated the expression of FXR1P in human HEK-293 and HeLa cell lines (Cheever et al., 2010). Interestingly, FXR1P was required to efficiently process pre-miR-9 and pre-miR-124 into mature miRNAs in vitro. Loss of FXR1P led to the downregulation of the brain specific miRNAs, that were crucial to several aspects of neuronal development and function (Xu et al., 2011). Mutations in either miRNA recognition sites for miR-92b, miR-363, or miR-367 on FXR1P will result in increased levels of FXR1P that leads to a lower level of miR-9 and miR-124. Collectively these findings imply that FXR1P are far more important than previously thought due to the facts that FXR1P functions in processing pre-miRNAs and yet FXR1P itself are regulated by miRNAs. This central role of FXR1P leads us to postulate that mutations on FXR1P may contribute to FXS essentially due to disrupted miRNA-mediated translation regulation.
To date, there are no conclusive evidences that showed direct link between miRNAs and FXS. However, the prospect of miRNAs in answering much of the question about FXS continues to encourage researchers in pursuing miRNA as a potential breakthrough to understand FXS. With its role as a regulator in the expression of multitude of genes, miRNAs definitely will help in filling much of the gaps in our understanding of XLID.
Conclusion
As much as we have progressed in the discovery of genes or mechanisms responsible for ID, many other syndromes where ID was implicated remained elusive and under-investigated. These pose a great challenge to researchers in their effort to better understand the “universal” mechanism underlying the development and progression of broad-spectrum and heterogenous ID conditions. In present day, miRNAs have the potential to bridge the missing link between genetic abnormalities with ID especially in cases where the causative genes could not be identified. In addition, miRNAs' unique characteristics of being able to target multiple genes or that a single gene could be targeted by multiple miRNAs may explain the reason behind some severe ID conditions where multiple genes are affected. The one-to-one, one-to-many, and many-to-one mode of miRNA:mRNA interactions provide us with a great versatility in designing experiments to study the development and progression of ID as well as develop therapeutic strategies aiming at pharmacologic inhibition of ID-associated miRNAs. Besides drug based approach, various molecular therapies based on small interfering RNA (siRNA) and locked nucleic acid (LNA) have been tested and hold promises in treating genetic disorders (Frieden and Ørum, 2008; Fluiter et al., 2009; Bernardo et al., 2012; Chabot et al., 2012). In addition, the advent of nanotechnology-based delivery system as well as advancement of stem cell researches would make the goal of treating and improving the cognitive function of individuals with ID a reality in near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Research University Grant Scheme/RUGS, Universiti Putra Malaysia/UPM (04-01-11-1163RU) awarded to Pike-See Cheah; Exploratory Research Grant Scheme, Ministry of Higher Education/MOHE, Malaysia (ERGS/1/11/SKK/UPM/03/1) awarded to Pike-See Cheah; Fundamental Research Grant Scheme, MOHE, Malaysia (04-01-12-1126FR) awarded to King-Hwa Ling; RUGS, UPM (04-02-12-2120RU) awarded to King-Hwa Ling. Wei-Hong Siew was a recipient of MyMaster scholarship from Ministry of Higher Education, Malaysia and Graduate Research Fellowship from Universiti Putra Malaysia. Kai-Leng Tan was a recipient of MyPhD scholarship from Ministry of Higher Education, Malaysia.
References
Abdul, H. M., Sama, M. A., Furman, J. L., Mathis, D. M., Beckett, T. L., Weidner, A. M., et al. (2009). Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 29, 12957–12969.
Albano, F., Anelli, L., Zagaria, A., Coccaro, N., Casieri, P., Rossi, A. R., et al. (2010). Non random distribution of genomic features in breakpoint regions involved in chronic myeloid leukemia cases with variant t(9;22) or additional chromosomal rearrangements. Mol. Cancer 9:120. doi: 10.1186/1476-4598-9-120
American Psychiatric Association. (2000). Diagnostical and Statistical Manual of Mental Disorders: DSM-IV-TR: 4th Edition Text Revision. Washington, DC: American Psychiatric Publishing.
Antonarakis, S. E., Lyle, R., Dermitzakis, E. T., Reymond, A., and Deutsch, S. (2004). Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat. Rev. Genet. 5, 725–738.
Arron, J. R., Winslow, M. M., Polleri, A., Chang, C.-P., Wu, H., Gao, X., et al. (2006). NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441, 595–600.
Aurora, A. B., Mahmoud, A. I., Luo, X., Johnson, B. A., Van Rooij, E., Matsuzaki, S., et al. (2012). MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Invest. 122, 1222–1232.
Bak, M., Silahtaroglu, A., MØller, M., Christensen, M., Rath, M. F., Skryabin, B., et al. (2008). MicroRNA expression in the adult mouse central nervous system. RNA 14, 432–444.
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214.
Bazzini, A. A., Lee, M. T., and Giraldez, A. J. (2012). Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237.
Bernardo, B. C., Gao, X.-M., Winbanks, C. E., Boey, E. J. H., Tham, Y. K., Kiriazis, H., et al. (2012). Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. U.S.A. 109, 17615–17620.
Borchert, G. M., Lanier, W., and Davidson, B. L. (2006). RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 13, 1097–1101.
Brock, J., and Jarrold, C. (2005). Serial order reconstruction in Down syndrome: evidence for a selective deficit in verbal short-term memory. J. Child Psychol. Psychiatry 46, 304–316.
Brown, J. H., Johnson, M. H., Paterson, S. J., Gilmore, R., Longhi, E., and Karmiloff-Smith, A. (2003). Spatial representation and attention in toddlers with Williams syndrome and Down syndrome. Neuropsychologia 41, 1037–1046.
Cai, J., Schleidt, S., Pelta-heller, J., Hutchings, D., Cannarsa, G., and Iacovitti, L. (2013). BMP and TGF- b pathway mediators are critical upstream regulators of Wnt signaling during midbrain dopamine differentiation in human pluripotent stem cells. Dev. Biol. 376, 62–73.
Calin, G. A., Sevignani, C., Dumitru, C. D., Hyslop, T., Noch, E., Yendamuri, S., et al. (2004). Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. U.S.A. 101, 2999–3004.
Chabot, S., Orio, J., Castanier, R., Bellard, E., Nielsen, S. J., Golzio, M., et al. (2012). LNA-based oligonucleotide electrotransfer for miRNA inhibition. Mol. Ther. 20, 1590–1598.
Cheever, A., Blackwell, E., and Ceman, S. (2010). Fragile X protein family member FXR1P is regulated by microRNAs. RNA 16, 1530–1539.
Chen, W., Jensen, L. R., Gecz, J., Fryns, J.-P., Moraine, C., De Brouwer, A., et al. (2007). Mutation screening of brain-expressed X-chromosomal miRNA genes in 464 patients with nonsyndromic X-linked mental retardation. Eur. J. Hum. Genet. 15, 375–378.
Chendrimada, T. P., Gregory, R. I., Kumaraswamy, E., Norman, J., Cooch, N., Nishikura, K., et al. (2005). TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740–744.
Christensen, M., and Schratt, G. M. (2009). microRNA involvement in developmental and functional aspects of the nervous system and in neurological diseases. Neurosci. Lett. 466, 55–62.
Codocedo, J. F., Allard, C., Godoy, J. A., Varela-Nallar, L., and Inestrosa, N. C. (2012). SIRT1 regulates dendritic development in hippocampal neurons. PLoS ONE 7:e47073. doi: 10.1371/journal.pone.0047073
Cohen, D. R., Matarazzo, V., Palmer, A. M., Tu, Y., Jeon, O.-H., Pevsner, J., et al. (2003). Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol. Cell. Neurosci. 22, 417–429.
Cui, Y., Xiao, Z., Han, J., Sun, J., Ding, W., Zhao, Y., et al. (2012). MiR-125b orchestrates cell proliferation, differentiation and migration in neural stem/progenitor cells by targeting Nestin. BMC Neurosci. 13:116. doi: 10.1186/1471-2202-13-116
Cunningham, C., and McArthur, K. (2006). Hearing loss and treatment in young Down's syndrome children. Child Care Health Dev. 7, 357–374.
Da Costa Martins, P. A., Salic, K., Gladka, M. M., Armand, A.-S., Leptidis, S., El Azzouzi, H., et al. (2010). MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol. 12, 1220–1227.
Daily, D. K., Ardinger, H. H., and Holmes, G. E. (2000). Identification and evaluation of mental retardation. Am. Fam. Physician 61, 1059–1067, 1070.
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y. S., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261.
Delabar, J. M., Theophile, D., Rahmani, Z., Chettouh, Z., Blouin, J. L., Prieur, M., et al. (1993). Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur. J. Hum. Genet. 1, 114–124.
Desai, N. S., Casimiro, T. M., Gruber, S. M., and Vanderklish, P. W. (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 96, 1734–1745.
Diederichs, S., and Haber, D. A. (2007). Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131, 1097–1108.
Dölen, G., Osterweil, E., Rao, B. S. S., Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962.
Edbauer, D., Neilson, J. R., Foster, K. A., Wang, C.-F., Seeburg, D. P., Batterton, M. N., et al. (2010). Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65, 373–384.
Eulalio, A., Huntzinger, E., Nishihara, T., Rehwinkel, J., Fauser, M., and Izaurralde, E. (2009). Deadenylation is a widespread effect of miRNA regulation. RNA 15, 21–32.
Filosa, A., Paixão, S., Honsek, S. D., Carmona, M. A., Becker, L., Feddersen, B., et al. (2009). Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat. Neurosci. 12, 1285–1292.
Fiore, R., Khudayberdiev, S., Saba, R., and Schratt, G. (2011). MicroRNA function in the nervous system. Prog. Mol. Biol. Transl. Sci. 102, 47–100.
Fluiter, K., Mook, O. R. F., and Baas, F. (2009). The therapeutic potential of LNA-modified siRNAs: reduction of off-target effects by chemical modification of the siRNA sequence. Methods Mol. Biol. 487, 189–203.
Forero, D. A., Van der Ven, K., Callaerts, P., and Del-Favero, J. (2010). miRNA genes and the brain: implications for psychiatric disorders. Hum. Mutat. 31, 1195–1204.
Frieden, M., and Ørum, H. (2008). Locked nucleic acid holds promise in the treatment of cancer. Curr. Pharm. Des. 14, 1138–1142.
Haase, A. D., Jaskiewicz, L., Zhang, H., Lainé, S., Sack, R., Gatignol, A., et al. (2005). TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 6, 961–967.
Han, J., Lee, Y., Yeom, K.-H., Kim, Y.-K., Jin, H., and Kim, V. N. (2004). The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18, 3016–3027.
Harraz, M. M., Eacker, S. M., Wang, X., Dawson, T. M., and Dawson, V. L. (2012). MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 18962–18967.
Hudry, E., Wu, H.-Y., Arbel-Ornath, M., Hashimoto, T., Matsouaka, R., Fan, Z., et al. (2012). Inhibition of the NFAT pathway alleviates amyloid β neurotoxicity in a mouse model of Alzheimer's disease. J. Neurosci. 32, 3176–3192.
Im, H.-I., and Kenny, P. J. (2012). MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 35, 325–334.
Jarrold, C., Baddeley, A. D., and Phillips, C. E. (2002). Verbal short-term memory in Down syndrome: a problem of memory, audition, or speech? J. Speech Lang. Hear. Res. 45, 531–544.
Jin, P., Alisch, R. S., and Warren, S. T. (2004a). RNA and microRNAs in fragile X mental retardation. Nat. Cell Biol. 6, 1048–1053.
Jin, P., Zarnescu, D. C., Ceman, S., Nakamoto, M., Mowrey, J., Jongens, T. A., et al. (2004b). Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat. Neurosci. 7, 113–117.
John, B., Enright, A. J., Aravin, A., Tuschl, T., Sander, C., and Marks, D. S. (2004). Human MicroRNA targets. PLoS Biol. 2:e363. doi: 10.1371/journal.pbio.0020363
Kaufman, L., Ayub, M., and Vincent, J. B. (2010). The genetic basis of non-syndromic intellectual disability: a review. J. Neurodev. Disord. 2, 182–209.
Kaufmann, W. E., Carter, J. C., Bukelis, I., and Lieberman, D. N. (2007). “Neurobiology of Genetic Mental Retardation,” in Neurobiology of Disease, ed S. Gilman (San Diego, CA: Elsevier Academic Press), 563–573.
Kaufmann, W. E., and Moser, H. W. (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 10, 981–991.
Keck-Wherley, J., Grover, D., Bhattacharyya, S., Xu, X., Holman, D., Lombardini, E. D., et al. (2011). Abnormal microRNA expression in Ts65Dn hippocampus and whole blood: contributions to Down syndrome phenotypes. Dev. Neurosci. 33, 451–467.
Kingsbury, M. A., Friedman, B., McConnell, M. J., Rehen, S. K., Yang, A. H., Kaushal, D., et al. (2005). Aneuploid neurons are functionally active and integrated into brain circuitry. Proc. Natl. Acad. Sci. U.S.A. 102, 6143–6147.
Kleefstra, T., Smidt, M., Banning, M. J. G., Oudakker, A. R., Van Esch, H., De Brouwer, A. P. M., et al. (2005). Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J. Med. Genet. 42, 299–306.
Konopka, W., Schütz, G., and Kaczmarek, L. (2011). The microRNA contribution to learning and memory. Neuroscientist 17, 468–474.
Kuhn, D. E., Nuovo, G. J., Martin, M. M., Malana, G. E., Pleister, A. P., Jiang, J., et al. (2008). Human chromosome 21-derived miRNAs are overexpressed in down syndrome brains and hearts. Biochem. Biophys. Res. Commun. 370, 473–477.
Kuhn, D. E., Nuovo, G. J., Terry, A. V., Martin, M. M., Malana, G. E., Sansom, S. E., et al. (2010). Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human Down syndrome brains. J. Biol. Chem. 285, 1529–1543.
Kuhn, S., Ingham, N., Pearson, S., Gribble, S. M., Clayton, S., Steel, K. P., et al. (2012). Auditory function in the Tc1 mouse model of down syndrome suggests a limited region of human chromosome 21 involved in otitis media. PLoS ONE 7:e31433. doi: 10.1371/journal.pone.0031433
Lagos-Quintana, M., Rauhut, R., Lendeckel, W., and Tuschl, T. (2001). Identification of novel genes coding for small expressed RNAs. Science 294, 853–858.
Landgraf, P., Rusu, M., Sheridan, R., Sewer, A., Iovino, N., Aravin, A., et al. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414.
Lau, N. C., Lim, L. P., Weinstein, E. G., and Bartel, D. P. (2001). An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862.
Le, M. T. N., Xie, H., Zhou, B., Chia, P. H., Rizk, P., Um, M., et al. (2009). MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol. Cell. Biol. 29, 5290–5305.
Lee, R. C., and Ambros, V. (2001). An extensive class of small RNAs in Caenorhabditis elegans. Science 294, 862–864.
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The, C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854.
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., et al. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419.
Lee, Y., Kim, M., Han, J., Yeom, K.-H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060.
Lehmann, S. M., Krüger, C., Park, B., Derkow, K., Rosenberger, K., Baumgart, J., et al. (2012). An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 15, 827–835.
Lemons, C. J., and Fuchs, D. (2010). Phonological awareness of children with Down syndrome: its role in learning to read and the effectiveness of related interventions. Res. Dev. Disabil. 31, 316–330.
Li, J., Pelletier, M. R., Perez Velazquez, J.-L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151.
Li, Y. Y., Alexandrov, P. N., Pogue, A. I., Zhao, Y., Bhattacharjee, S., and Lukiw, W. J. (2012). miRNA-155 upregulation and complement factor H deficits in Down's syndrome. Neuroreport 23, 168–173.
Loane, M., Morris, J. K., Addor, M.-C., Arriola, L., Budd, J., Doray, B., et al. (2012). Twenty-year trends in the prevalence of Down syndrome and other trisomies in Europe: impact of maternal age and prenatal screening. Eur. J. Hum. Genet. 21, 27–33.
Lörincz, A., Rózsa, B., Katona, G., Vizi, E. S., and Tamás, G. (2007). Differential distribution of NCX1 contributes to spine-dendrite compartmentalization in CA1 pyramidal cells. Proc. Natl. Acad. Sci. U.S.A. 104, 1033–1038.
Lu, H.-E., Yang, Y.-C., Chen, S.-M., Su, H.-L., Huang, P.-C., Tsai, M.-S., et al. (2012). Modeling neurogenesis impairment in Down syndrome with induced pluripotent stem cells from Trisomy 21 amniotic fluid cells. Exp. Cell Res. 319, 498–505.
Lukiw, W. J., and Alexandrov, P. N. (2012). Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer's disease (AD) brain. Mol. Neurobiol. 46, 11–19.
Lund, E., Güttinger, S., Calado, A., Dahlberg, J. E., and Kutay, U. (2004). Nuclear export of microRNA precursors. Science 303, 95–98.
Melville, S. A., Buros, J., Parrado, A. R., Vardarajan, B., Logue, M. W., Shen, L., et al. (2012). Multiple loci influencing hippocampal degeneration identified by genome scan. Ann. Neurol. 72, 65–75.
Mendoza, R. B., Labastilla, E. M., Endriga, M. A., Deocaris, C. C., and Deocaris, C. C. (2010). Systems analysis of X-chromosomal miRNAs and their target genes using informatics tools. J. Trends Chem. 1, 18–23.
Meredith, R. M., Holmgren, C. D., Weidum, M., Burnashev, N., and Mansvelder, H. D. (2007). Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron 54, 627–638.
Nash, H., and Heath, J. (2011). The role of vocabulary, working memory and inference making ability in reading comprehension in Down syndrome. Res. Dev. Disabil. 32, 1782–1791.
Paschou, M., Paraskevopoulou, M. D., Vlachos, I. S., Koukouraki, P., Hatzigeorgiou, A. G., and Doxakis, E. (2012). miRNA regulons associated with synaptic function. PLoS ONE 7:e46189. doi: 10.1371/journal.pone.0046189
Penrose, L. S. (1938). A Clinical and Genetic Study of 1280 Cases of Mental Defect. London: H. M. Stationery Office.
Plante, I., Davidovic, L., Ouellet, D. L., Gobeil, L.-A., Tremblay, S., Khandjian, E. W., et al. (2006). Dicer-derived microRNAs are utilized by the fragile X mental retardation protein for assembly on target RNAs. J. Biomed. Biotechnol. 2006, 1–12.
Pogue, A. I., Cui, J. G., Li, Y. Y., Zhao, Y., Culicchia, F., and Lukiw, W. J. (2010). Micro RNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci. Lett. 476, 18–22.
Pritchard, M. A., and Kola, I. (1999). The “gene dosage effect” hypothesis versus the “amplified developmental instability” hypothesis in Down syndrome. J. Neural Transm. Suppl. 57, 293–303.
Rehen, S. K., McConnell, M. J., Kaushal, D., Kingsbury, M. A., Yang, A. H., and Chun, J. (2001). Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl. Acad. Sci. U.S.A. 98, 13361–13366.
Rehen, S. K., Yung, Y. C., McCreight, M. P., Kaushal, D., Yang, A. H., Almeida, B. S. V., et al. (2005). Constitutional aneuploidy in the normal human brain. J. Neurosci. 25, 2176–2180.
Reinhart, B. J., Slack, F. J., Basson, M., Pasquinelli, A. E., Bettinger, J. C., Rougvie, A. E., et al. (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906.
Rodriguez, A., Griffiths-Jones, S., Ashurst, J. L., and Bradley, A. (2004). Identification of mammalian microRNA host genes and transcription units. Genome Res. 14, 1902–1910.
Roome, C. J., Knöpfel, T., and Empson, R. M. (2013). Functional contributions of the plasma membrane calcium ATPase and the sodium-calcium exchanger at mouse parallel fibre to Purkinje neuron synapses. Pflügers Arch. 465, 319–331.
Saharan, S., Jhaveri, D. J., and Bartlett, P. F. (2013). SIRT1 regulates the neurogenic potential of neural precursors in the adult subventricular zone and hippocampus. J. Neurosci. Res. 91, 642–659.
Saini, H. K., Griffiths-Jones, S., and Enright, A. J. (2007). Genomic analysis of human microRNA transcripts. Proc. Natl. Acad. Sci. U.S.A. 104, 17719–17724.
Sanyal, S., Kim, S. M., and Ramaswami, M. (2004). Retrograde regulation in the CNS; neuron-specific interpretations of TGF-beta signaling. Neuron 41, 845–848.
Satoh, J. (2010). MicroRNAs and their therapeutic potential for human diseases: aberrant microRNA expression in Alzheimer's disease brains. J. Pharmacol. Sci. 114, 269–275.
Saunders, L. R., Sharma, A. D., Tawney, J., Nakagawa, M., Okita, K., Yamanaka, S., et al. (2010). miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging 2, 415–431.
Schaefer, A., O'Carroll, D., Tan, C. L., Hillman, D., Sugimori, M., Llinas, R., et al. (2007). Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 204, 1553–1558.
Siegel, G., Saba, R., and Schratt, G. (2011). microRNAs in neurons: manifold regulatory roles at the synapse. Curr. Opin. Genet. Dev. 21, 491–497.
Stevenson, R. E., Schwartz, C. E., and Rogers, R. C. (2012). Atlas of X-Linked Intellectual Disability Syndromes. New York: NY, Oxford University Press.
Takashima, S., Iida, K., Mito, T., and Arima, M. (1994). Dendritic and histochemical development and ageing in patients with Down's syndrome. J. Intellect. Disabil. Res. 38(Pt 3), 265–273.
Turcatel, G., Rubin, N., El-Hashash, A., and Warburton, D. (2012). MIR-99a and MIR-99b modulate TGF-β induced epithelial to mesenchymal plasticity in normal murine mammary gland cells. PLoS ONE 7:e31032. doi: 10.1371/journal.pone.0031032
Urdinguio, R. G., Fernandez, A. F., Lopez-Nieva, P., Rossi, S., Huertas, D., Kulis, M., et al. (2010). Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 5, 656–663.
Van Bokhoven, H. (2011). Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 45, 81–104.
Van Cleve, S. N., Cannon, S., and Cohen, W. I. (2006). Part II: Clinical Practice Guidelines for adolescents and young adults with Down Syndrome: 12 to 21 Years. J. Pediatr. Health Care 20, 198–205.
Van Cleve, S. N., and Cohen, W. I. (2006). Part I: clinical practice guidelines for children with Down syndrome from birth to 12 years. J. Pediatr. Health Care 20, 47–54.
Vicari, S., Bellucci, S., and Carlesimo, G. A. (2005). Visual and spatial long-term memory: differential pattern of impairments in Williams and Down syndromes. Dev. Med. Child Neurol. 47, 305–311.
Warren, S. T., and Nelson, D. L. (1994). Advances in molecular analysis of fragile X syndrome. JAMA 271, 536–542.
Warren, S. T., and Sherman, S. L. (2001). “The fragile X syndrome,” in The Metabolic and Molecular Basis of Inherited Disease, Vol. 1, eds C. R. Scriver, A. L. Beaudet, D. Valle, B. Childs, K. W. Kinzler, and B. Vogelstein (New York: NY, McGraw-Hill), 1257–1290.
Weston, M. D., Pierce, M. L., Rocha-Sanchez, S., Beisel, K. W., and Soukup, G. A. (2006). MicroRNA gene expression in the mouse inner ear. Brain Res. 1111, 95–104.
Wightman, B., Ha, I., and Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862.
Wisniewski, K. E. (1990). Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. Am. J. Med. Genet. Suppl. 7, 274–281.
Xu, X.-L., Zong, R., Li, Z., Biswas, M. H. U., Fang, Z., Nelson, D. L., et al. (2011). FXR1P but not FMRP regulates the levels of mammalian brain-specific microRNA-9 and microRNA-124. J. Neurosci. 31, 13705–13709.
Xu, Z., Chen, R.-Q., Gu, Q.-H., Yan, J.-Z., Wang, S.-H., Liu, S.-Y., et al. (2009). Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci. 29, 8764–8773.
Yang, A. H., Kaushal, D., Rehen, S. K., Kriedt, K., Kingsbury, M. A., McConnell, M. J., et al. (2003). Chromosome segregation defects contribute to aneuploidy in normal neural progenitor cells. J. Neurosci. 23, 10454–10462.
Yi, R., Qin, Y., Macara, I. G., and Cullen, B. R. (2003). Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 17, 3011–3016.
Yi, Y. H., Sun, X. S., Qin, J. M., Zhao, Q. H., Liao, W. P., and Long, Y. S. (2010). Experimental identification of microRNA targets on the 3′ untranslated region of human FMR1 gene. J. Neurosci. Methods 190, 34–38.
Zhao, J.-P., and Constantine-Paton, M. (2007). NR2A-/- mice lack long-term potentiation but retain NMDA receptor and L-type Ca2+ channel-dependent long-term depression in the juvenile superior colliculus. J. Neurosci. 27, 13649–13654.
Keywords: Down syndrome, brain development, cognitive function, Fragile X syndrome, X-linked genetic disease, non-coding RNA, neuronal development, mental retardation
Citation: Siew W-H, Tan K-L, Abbaspour Babaei M, Cheah P-S and Ling K-H (2013) MicroRNAs and intellectual disability (ID) in Down syndrome, X-linked ID, and Fragile X syndrome. Front. Cell. Neurosci. 7:41. doi: 10.3389/fncel.2013.00041
Received: 04 January 2013; Accepted: 27 March 2013;
Published online: 15 April 2013.
Edited by:
Eran Meshorer, The Hebrew University of Jerusalem, IsraelReviewed by:
Nobuhiro Harada, Fujita Health University School of Medicine, JapanSebastian Kadener, The Hebrew University, Israel
Hermona Soreq, The Hebrew University of Jerusalem, Israel
Copyright © 2013 Siew, Tan, Abbaspour Babaei, Cheah and Ling. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: King-Hwa Ling, Clinical Genetics Unit, Department of Obstetrics and Gynaecology, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Level 6, Block B, 43400 UPM Serdang, Selangor, Malaysia. e-mail: khling@medic.upm.edu.my