
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 28 July 2011
Sec. Cellular Neurophysiology
volume 5 - 2011 | https://doi.org/10.3389/fncel.2011.00016
This article is part of the Research Topic GABA signaling in health and disease View all 18 articles
 Romain Nardou1,2,3 Sumii Yamamoto1,2,3 Asma Bhar1,2,3 Nail Burnashev1,2,3
Romain Nardou1,2,3 Sumii Yamamoto1,2,3 Asma Bhar1,2,3 Nail Burnashev1,2,3 Yehezkel Ben-Ari1,2,3* and Ilgam Khalilov1,2,3
Yehezkel Ben-Ari1,2,3* and Ilgam Khalilov1,2,3
Diazepam (DZP) and phenobarbital (PB) are extensively used as first and second line drugs to treat acute seizures in neonates and their actions are thought to be mediated by increasing the actions of GABAergic signals. Yet, their efficacy is variable with occasional failure or even aggravation of recurrent seizures questioning whether other mechanisms are not involved in their actions. We have now compared the effects of DZP and PB on ictal-like events (ILEs) in an in vitro model of mirror focus (MF). Using the three-compartment chamber with the two immature hippocampi and their commissural fibers placed in three different compartments, kainate was applied to one hippocampus and PB or DZP to the contralateral one, either after one ILE, or after many recurrent ILEs that produce an epileptogenic MF. We report that in contrast to PB, DZP aggravated propagating ILEs from the start, and did not prevent the formation of MF. PB reduced and DZP increased the network driven giant depolarizing potentials suggesting that PB may exert additional actions that are not mediated by GABA signaling. In keeping with this, PB but not DZP reduced field potentials recorded in the presence of GABA and NMDA receptor antagonists. These effects are mediated by a direct action on AMPA/kainate receptors since PB: (i) reduced AMPA/kainate receptor mediated currents induced by focal applications of glutamate; (ii) reduced the amplitude and the frequency of AMPA but not NMDA receptor mediated miniature excitatory postsynaptic currents (EPSCs); (iii) augmented the number of AMPA receptor mediated EPSCs failures evoked by minimal stimulation. These effects persisted in MF. Therefore, PB exerts its anticonvulsive actions partly by reducing AMPA/kainate receptors mediated EPSCs in addition to the pro-GABA effects. We suggest that PB may have advantage over DZP in the treatment of initial neonatal seizures since the additional reduction of glutamate receptors mediated signals may reduce the severity of neonatal seizures.
Neonatal seizures are inherently different from adult seizures and can be refractory to anti epileptic drugs (AEDs). Benzodiazepines – notably diazepam (DZP) – and phenobarbital (PB) are the AEDs of first choice to treat neonatal seizures (Wheless et al., 2007; Bassan et al., 2008). In general, PB is first used in neonatal seizures whereas diazepam or lorazepam are preferred in febrile seizures. They are thought to reduce seizures by reinforcing the efficacy of GABAergic inhibition through a direct action on GABA(A) receptors mediated currents. However, there are several complicating factors with their use. In many conditions they fail to block seizures, produce an electro-clinical disconnection of EEG and clinical signs (Connell et al., 1989; Painter et al., 1999; Boylan et al., 2002; Yanay et al., 2004; Guillet and Kwon, 2007; Kaindl et al., 2008) and even aggravate them (Loscher and Honack, 1989; Guerrini et al., 1998; Perucca et al., 1998; Painter et al., 1999; Goodkin et al., 2008; Goodkin and Kapur, 2009). These aggravating actions are at least partly mediated by a persistent increase of [Cl−]i leading to more seizures by enhancing the excitatory actions of GABA that heavily depend on this parameter (also see Cohen et al., 2002; Khalilov et al., 2003, 2005; Rivera et al., 2004; Glykys et al., 2009; Dzhala et al., 2010; Nardou et al., 2011). Experimental observations indicate that PB efficiently blocks early seizures but aggravates established ones when first applied after many recurrent seizures. This is due to a persistent accumulation of chloride and excitatory actions of GABA that will be further enhanced by PB (Glykys et al., 2009; Dzhala et al., 2010; Nardou et al., 2011). These effects are mediated by a persistent activation of the chloride importer NKCC1 and a down regulation and internalization of the chloride exporter KCC2 (Dzhala et al., 2010; Lee et al., 2010; Nardou et al., 2011). Clearly, the regulation of [Cl−]i constitutes a major source of problem to the use of these AEDs and stresses the importance of the history of seizures prior to treatment in determining the efficacy of the treatment.
Collectively however, these observations are based on the assumption that the actions of PB, DZP, and other members of this family are solely mediated by GABA signaling. Yet, there are several indications that this may not be the case. Barbiturate anesthetics have been reported to antagonize glutamate evoked currents in spinal neurons (MacDonald and Barker, 1978a,b, 1982; Macdonald and Kelly, 1994) and to reduce recombinant AMPA-type glutamate receptor channels expressed in cell lines (Jin et al., 2010). Also, barbiturates are more efficacious than benzodiazepines to treat various brain disorders including neonatal seizures (Cheng et al., 2002; Soderpalm, 2002; Carmo and Barr, 2005; Bartha et al., 2007) raising the possibility that additional non-GABA mediated signaling may underlie a better efficacy to treat various neonatal seizures. These observations stress the importance of both determining the possible non-GABA mediated actions of these AEDs and taking into account the differences between acute and recurrent more chronic seizures in order to determine possible aggravating deleterious actions.
Here, we compared the actions of PB and DZP on glutamate receptor mediated currents in naïve and “epileptic” immature hippocampal neurons. In that purpose, we used the triple chamber preparation that enables to apply to one chamber a convulsive agent (kainate) and an AED to the other and interrupt the connections after one or more propagated seizures (Khalilov et al., 1997, 1999, 2003). In this preparation, the contralateral hippocampus becomes epileptic as it generates spontaneous seizures when disconnected from the kainate treated side. This enables to compare the actions of AEDs on naïve and epileptic mirror foci. Using this preparation, we showed recently (Nardou et al., 2011) that PB efficiently blocks paroxysmal activity when applied with the initial seizures but aggravates them when applied late on a newly formed mirror focus (MF). This is due to an excessive increase of [Cl−]i leading to excitatory GABA that PB will further exacerbates (Nardou et al., 2011). We now report that in contrast to PB, DZP aggravates inaugurating, and established seizures. These differences are due to the selective actions of PB that reduces AMPA/kainate receptor mediated signals in addition to its pro-GABA actions. This dual action of PB on GABA and glutamate excitatory postsynaptic currents (EPSCs) provides an obvious important advantage of PB over DZP and may have as such important clinical implications.
Experiments were performed on neonatal Wistar rats (postnatal days P6–P8). All experiments have been carried out in accordance with the European Communities Council Directive of the 24 November 1986 (86/609/EEC).
The interconnected intact hippocampi were prepared according to the procedures described previously (Khalilov et al., 1997). In brief, neonatal rats and mice (P6–P8) were decapitated after hypothermic anesthesia, the brain rapidly removed to oxygenated (95% O2/5% CO2) ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 126, KCl 3.5, CaCl2 2.0, MgCl2 1.3, NaHCO3 25, NaH2PO4 1.2, and glucose 11 (pH 7.4). The two interconnected intact hippocampi were isolated and transferred into a beaker containing oxygenated ACSF and incubated at least 1 h before use. The hippocampi were placed into a fully submerged three-compartment chamber and superfused with oxygenated ACSF (30–32°C, 10–15 ml/min).
Slices (thickness 450 μm) were cut using a Leica VT-1000E vibratome (Leica Microsystems GmbH, Nussloch, Germany) or a McIIwain tissue chopper (slices 600 μm thick) and kept in oxygenated ACSF at room temperature for at least 1 h before use (Khalilov et al., 2005). Slices from epileptic hippocampi were prepared as described earlier (Nardou et al., 2009). Individual slices were then transferred to the recording chamber where they were fully submerged and superfused with oxygenated ACSF (30–32°C, 2–3 ml/min).
Hippocampal CA3 pyramidal neurons in the slices were visualized with infrared differential interference contrast microscopy (Nikon Eclipse FN).
Whole cell recordings were collected using an Axopatch 200B and MultiClamp 700B amplifiers (Axon Instruments, USA). Patch electrodes were made from borosilicate glass capillaries (G150F-15; Warner Instrument Corporation, Hamden, CT, USA) using the model PP-830 two-stage micropipette puller (Narishige, Tokyo, Japan). Patch pipette solution contained (in mM): CsCl 140, CaCl2 1, EGTA 10, HEPES 10, MgATP 2, and GTP 0.4 (pH 7.25), and an osmolarity of 280 mOsm. Patch pipettes had a resistance of 8–10 MΩ.
To evoke synaptic responses in pyramidal CA3 neurons, a concentric bipolar tungsten electrode (FHC, Bowdoinham, ME, USA) was placed in stratum lucidum. Single pulses were delivered using a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel) controlled by a PC, and stimulus intensity was controlled by a constant current stimulus isolator. AMPA/kainate receptor mediated EPSCs. EPSCs were evoked in the presence of NMDA and GABA receptor antagonists (40 μM D-APV, 10 μM bicuculline, and 2 μM CGP55845) and minimal afferent stimulation was used to activate one or few fibers. The minimal EPSCs were associated with occasional response failures. Their number was usually estimated by visual discrimination. The means of evoked EPSCs was determined by excluding the failures and making only means of the successful evoked responses. To confirm that responses were AMPA/kainate receptor mediated, an AMPA/kainate receptor antagonist (10 or 20 μM CNQX) was used to block responses.
Cells were identified by adding biocytin (0.4%) to the pipette solution for post hoc morphological analysis.
Nucleated patch clamp recordings were performed from CA3 pyramidal neurons in hippocampal slices using fast agonist application techniques in the standard external solution. After the whole cell configuration was made, nucleated (Sather et al., 1992) patches were pulled from the cell and placed in front of a piezo-controlled (P 245.70, Physik Instrumente, Waldbronn, Germany) fast application system with a double-barreled application pipette (Colquhoun et al., 1992). Glutamate (1 mM) pulses of 2 ms duration were applied every 10 s. Recordings were made using an EPC-9 amplifier (HEKA Electronics, Lambrecht, Germany) under visualization with a Leica microscope. The resistance of the pipettes was 4–5 MΩ. PB (100 μM) and diazepam (2 μM) were added to the extracellular solution. Experiments were performed at a holding potential of –60 mV. Current traces shown are averages of four to eight sweeps. Stimulus delivery and data acquisition was performed using Pulse software (Heka Elektronik, Lambrecht, Germany). Analyses were performed using IgorPro software (WaveMetrics, Lake Oswego, OR, USA).
Extracellular field potentials and multi-unit activities (MUAs) were recorded in the hippocampal slices and in the intact hippocampal preparations in vitro using tungsten wire electrodes (diameter: 50 μm, California Fine Wire, Grover Beach, CA, USA) and a low-noise multichannel DAM-8A amplifiers (WPI, GB; low filter: 0.1 Hz; high filter: 3 KHz; ×1000). Electrical stimulations were performed with a bipolar electrode (10–20 V, 40 μs).
The signals were digitized using an analog-to-digital converter (Digidata 1440A, Axon Instruments, USA). pCLAMP 10.1, Clampfit 10.1 (Axon Instruments, USA), MiniAnalysis 6.03 (Synaptosoft, Decatur, CA, USA), and Origin 7.5 (Microcal Software, USA) programs were used for the acquisition and analysis of the synaptic activities. Sampling interval per signal was 100 μs (10 kHz). Spikes (single unit activity) in extracellular recordings were detected by amplitude thresholding using MiniAnalysis 6.03 program. Power spectrum analysis was performed after applying a Hamming window function. Power was calculated by integrating the root mean square value of the signal in frequency bands from 1 to 1000 Hz. Time–frequency representations of ictal-like events were obtained using continuous Morlet wavelet transforms using AutoSignal v1.7 software (SeaSolve Software Inc.).
A picospritzer (General Valve Corporation, Fairfield, NJ, USA) was used to puff-apply (5–10 psi for 50–100 ms) of glutamate or GABA from a glass pipette at a distance of about 100–150 μm from the soma, adjusted such that postsynaptic current amplitudes did not exceed 300 pA.
Group measures are expressed as mean ± SEM; error bars also indicate SEM. The statistical significance of differences was assessed with the Student’s t-test. The level of significance was set at p < 0.05.
Antagonists bicuculline (10 μM), 6-cyano-7-nitroquinoxaline-2,3 dione (CNQX, 10 μM), D-2-Amino-5-phosphopentanoate (APV, 40 μM), tetrodotoxin (TTX, 1 μM), picrotoxin (PTX, 100 μM), CGP55845 (CGP, 2 μM); AEDs Diazepam (DZP, 2 μM) and (PB, 100 μM); and kainate (KA, 400 nM) were directly added to the perfusion solutions. The drugs used were purchased from Sigma (DZP, PB, TTX, PTX, CGP, glutamate, and kainic acid), Tocris (bicuculline, CNQX, APV), and Molecular Probes (biocytin).
We used the triple chamber preparation with the two intact hippocampi and the commissural connections in three different compartments (Khalilov et al., 1997, 1999). This enables to apply different agents on each compartment without diffusion and interference with the others (Khalilov et al., 2003). In a first series of experiments, we applied kainate (KA, 400 nM) to one hippocampus (referred to as the ipsilateral hippocampus, ipsi-) and recorded the activity from both the ipsi- and the contralateral (contra-) hippocampus that did not receive the convulsive agent (Figure 1A). In keeping with our earlier studies (Khalilov et al., 2003, 2005), KA generated an ictal-like event (ILE) with large amplitude (1–2 mV) and duration (1–2 min) ictal discharges (Figure 1A; see also Khalilov et al., 2003). These ILEs propagated to the contralateral hippocampus where they generated similar activities including high frequency (40–120 Hz) gamma oscillations (GOs; Figures 1A,B). In earlier experiments we have demonstrated that after circa 15 repeated unilateral KA applications, the contralateral hippocampus generated spontaneous ILEs when disconnected from the treated ipsilateral hippocampus (Khalilov et al., 2003, 2005). We referred to this as a newly formed secondary epileptogenic focus – or MF.
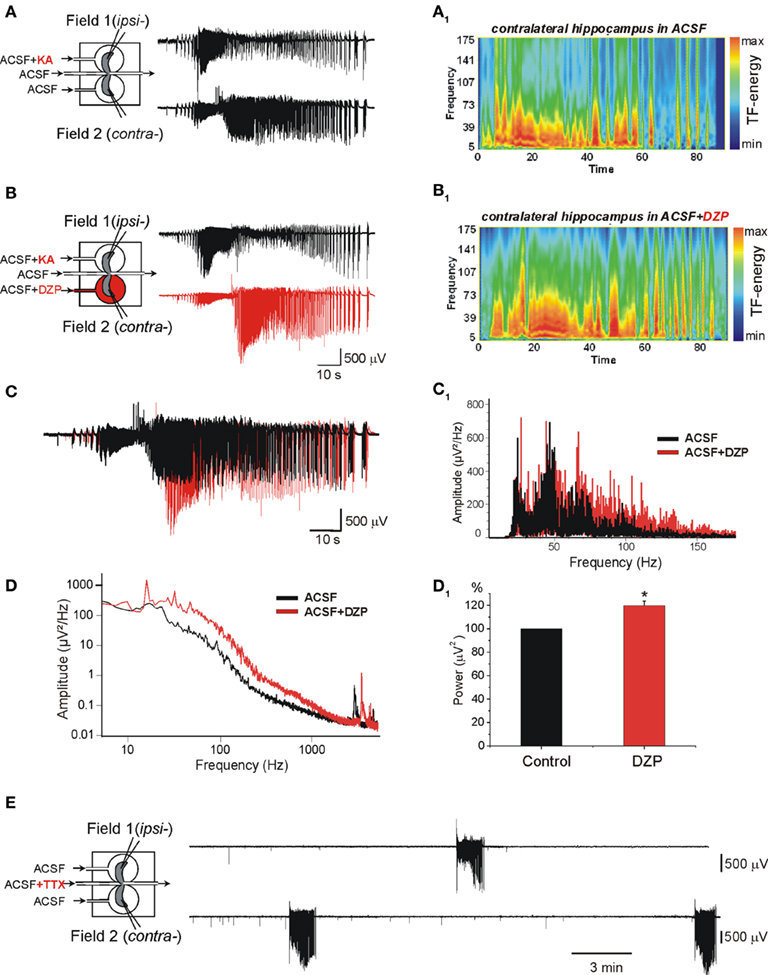
Figure 1. Diazepam (DZP) aggravates the propagating ictal-like events (ILEs) and does not prevent the formation of a mirror focus by repeated ILEs. (A,B) The scheme of the triple chamber preparation with the two intact hippocampi and their connecting inter-hemispheric commissure in independent chambers (left). Field recordings were made in the two hippocampi. (A) Kainate (KA) applied to one hippocampus (ipsilateral: ipsi-) generated seizure activity referred to as ILE that propagated to the contralateral hippocampus (contra-). The ILEs included high frequency gamma oscillations (GOs, 40–120 Hz) as shown in the time–frequency (TF) analysis (A1). (B) Application of DZP to contralateral hippocampus (2 μM, 15 min) before a second application of KA increased the ILE and GOs. TF analysis of ILE recorded in contra- is shown in (B1). (C) Superimposed ILEs [from (A,B)] recorded in contra- before (black) and after DZP application (red). DZP increased the amplitude and duration of ILE. (C1) Power spectra representation of ILEs recorded before (black) and after (red) DZP treatment [from (C)] after band-pass (40–150 Hz) filtering. (D) Example of power spectra of ILEs before and after DZP treatment [from (A,B)] used for calculation of power of ILEs. (D1) Normalized average power histogram of contralateral ILEs before and after DZP. Note that DZP application significantly increased average power of ILEs (by 19.2 ± 3.9%, n = 7 experiments, p < 0.05). (E) After 15 transient applications of KA in ipsi- with continuous application of DZP in contra-, disconnection of the two hippocampi by applications of TTX to the commissural chamber revealed that contralateral hippocampus generated spontaneous ILEs.
We have shown recently that early applications of (PB, 100 μM) to the contralateral hippocampus reduced dramatically the severity of propagating seizures and prevented the formation of a MF (Nardou et al., 2011). In contrast, in similar experiments, DZP (2 μM) augmented the severity of the propagating ILEs (Figures 1C,C1,D,D1).
The frequency of oscillations is an important component of the pathogenic actions of seizures. Indeed, in both human and experimental animals, high frequency oscillations (HFOs; >60 Hz) constitute a signature of the severity of recurrent seizures (Bragin et al., 1999; Khalilov et al., 2005; Le Van Quyen et al., 2006; Jacobs et al., 2008). In keeping with this, in the same preparation, the occurrence of GOs in propagating seizures is essential to induce the transformation of a naïve network to one that generates ILEs spontaneously (Khalilov et al., 2005). Whereas PB reduced the frequency of GOs and severity (power) of ILEs (Nardou et al., 2011), DZP augmented the range of GOs frequencies (Figures 1C1) and the power of ILEs (by 19.2 ± 3.9%, n = 7, p < 0.05) in the contralateral hippocampus already with the first application (Figures 1D1). DZP in contrast to PB did not prevent the formation of an epileptogenic MF (Figure 1E).
Therefore, PB and DZP exert opposite actions on severity of early ILEs in the intact hippocampus: PB reduces and DZP aggravates the propagating ILEs from the onset of their applications.
In earlier studies, we showed that once formed the MF generates spontaneous ILEs up to 1–2 days in an isolated hippocampus in vitro, and slices prepared from this hippocampus generate spontaneous and evoked interictal-like events (IILEs; Khalilov et al., 2003, 2005; Nardou et al., 2009, 2011). In whole hippocampus after the formation of MF, application of DZP significantly increased the severity of ILEs. The average power of spontaneous ILEs in MF hippocampus after bath application of DZP increased by 80 ± 19.8% (n = 4, p < 0.001; Figure 2A). In slices prepared from the hippocampus after MF formation, DZP increased both the frequency of IILEs and extracellular recorded spikes generated by the MF neurons (Figure 2B). In similar experiments, PB also aggravated the epileptiform activity generated by the MF by enhancing the strong excitatory actions of GABA (Nardou et al., 2011). Therefore, the difference between DZP and PB is valid for initial propagating seizures but not for spontaneous seizures generated by an epileptogenic MF suggesting that the persistent accumulation of [Cl−]i prevails in MF neurons rendering difficult the actions of GABA acting AEDs. Therefore, we next compared the actions of DZP and PB on ongoing network activity in slices obtained from control hippocampus.
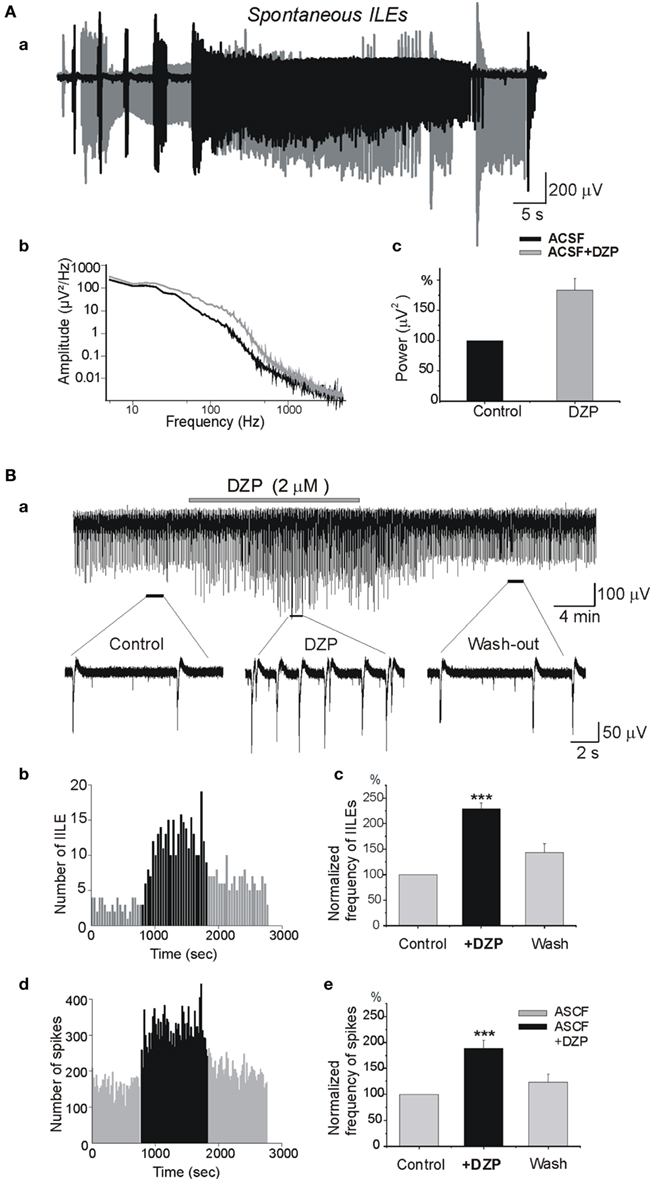
Figure 2. Diazepam aggravates the severity of epileptiform activities in the isolated mirror focus. (Aa) Superimposed ILEs generated spontaneously in the contralateral hippocampus after formation of a MF before (black) and after (gray) DZP application. (Ab) Power spectra of spontaneous ILEs before and after DZP treatment [from (Aa)]. (Ac) Average power histogram of spontaneous ILEs before and after DZP. DZP application significantly increased power of ILEs (by 80 ± 19.8%, n = 4, p < 0.001). (Ba) Extracellular recording in slices obtained from contralateral hippocampus after the formation of a MF. Slices generated spontaneous IILEs and multi-unit activities (MUAs) continuously that were reversibly aggravated by DZP (2 μM). (Bb) Quantification of IILEs from a single experiment [from (Ba)]. (Bc) Average quantification histogram of IILEs. (Bd) Quantification of spikes from single experiment [from (Ba)] and (Be) the average quantification histogram. Note that DZP increased the frequency of IILEs (by 128.7 ± 12.2%, n = 6, p < 0.001) and spikes (by 88.6 ± 16.2%, n = 6, p < 0.001).
In control conditions, the physiological pattern of the immature hippocampal network activity is characterized by spontaneous network driven giant depolarizing potentials (GDPs; Ben-Ari et al., 1989) that occur spontaneously and can be evoked by electrical stimulation. GDPs are generated by the converging actions of GABA and glutamatergic synapses (Ben Ari et al., 2007). We tested the effects of DZP on the frequency of occurrence of extracellular recorded GDPs and MUAs. As shown in Figure 3 DZP, applied at a concentration of 2 μM, increased the frequency of GDPs by 215.1 ± 13.6% (p < 0.001, n = 14) and the frequency of spikes by 170 ± 14.8% (p < 0.001, n = 14). In similar experiments, bath application of PB substantially reduced the amplitude of GDPs. PB reduced also the frequency of spontaneous spikes generated in CA3 pyramidal neurons by ∼50% (see Nardou et al., 2011). An additional non-exclusive mechanism is that GDP frequency being increased, there may also be a short-term depression of GABAergic inputs that would be superimposed on the effect of DZP.
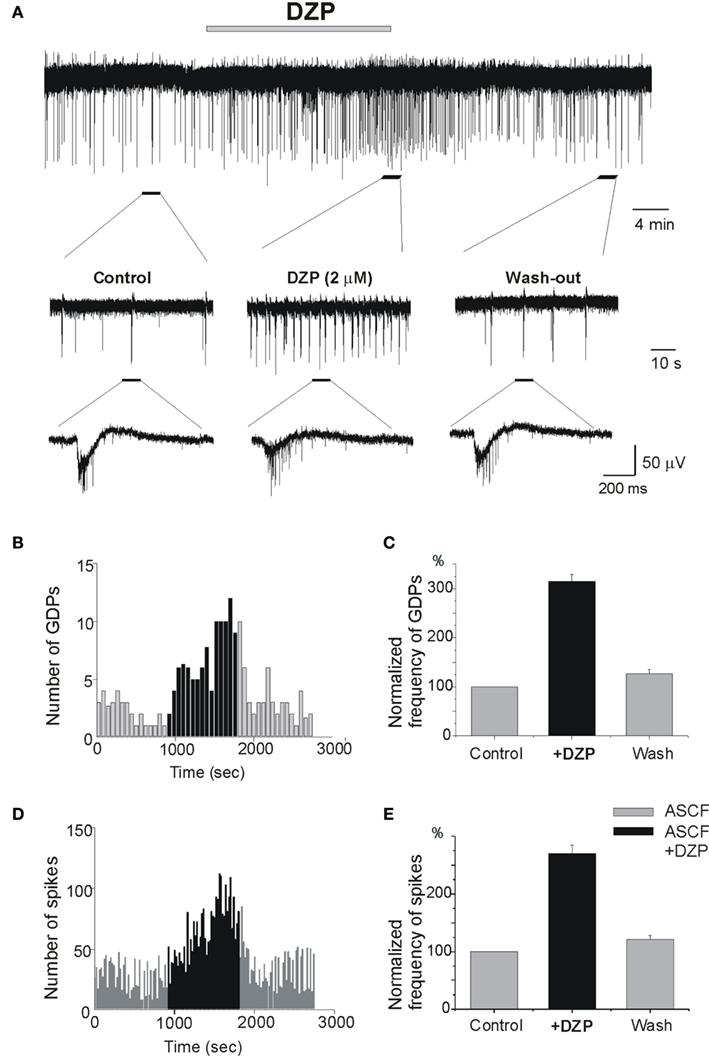
Figure 3. Diazepam increases ongoing neuronal activity and network driven GDPs. (A) Extracellular recording of the Giant Depolarizing Potentials (GDPs) and multi-unit activities (MUAs) in control slices to record network driven events and individual neuronal spikes, respectively. Non-filtered faster display traces are depicted below to show spikes. Note the increase of frequency of GDPs (B) and spikes (D) by DZP: (B–D) quantification of recorded GDPs and spikes from (A), Bin = 20 s. (C–E) Quantification of mean frequency of GDPs and spikes. DZP enhanced the frequency of GDPs (C) by 215.1 ± 13.6 (p < 0.001, n = 14) and spikes (E) by 170 ± 14.8% (p < 0.001, n = 14).
To determine the actions of DZP on GABA signaling, we tested the effects of DZP on responses generated by focal applications of GABA. As shown in Figure 4A, GABA was focally applied (100 μM, 100 ms) in the CA3c region while simultaneous CA3c field and whole cell recordings (from pyramidal neurons in CA3a region) were performed. Focal GABA application generated GDPs (Figure 4B). Application of DZP significantly increased the amplitude and duration of whole cell recorded responses (Figures 4B–E) accompanied by a 17.2 ± 2.7% (p < 0.05) as well as the area and decay time constant of the currents (Figures 4C–E) increase in the amplitude of extracellular recorded GDPs (not shown) and with a strong increase in the number of GDPs-associated spikes (98.1 ± 6.7%, p < 0.001, n = 5; Figure 4F). Similar focal application of GABA generated 2–3 action potentials (spikes) in cell attached recordings (see Nardou et al., 2011). In contrast to DZP, bath applications of PB (100 μM) significantly reduced the number of spikes (action potentials) from 2.7 ± 0.2 to 1.9 ± 0.2 (Nardou et al., 2011). Therefore, under control conditions PB reduces ongoing neuronal activity whereas DZP augments it. The observation that GABA acting PB reduces and DZP augments ongoing activity raises the possibility of non-GABA mediated actions of these AEDs. We therefore compared their effects on glutamatergic signals that are instrumental in the generation of early network activity.
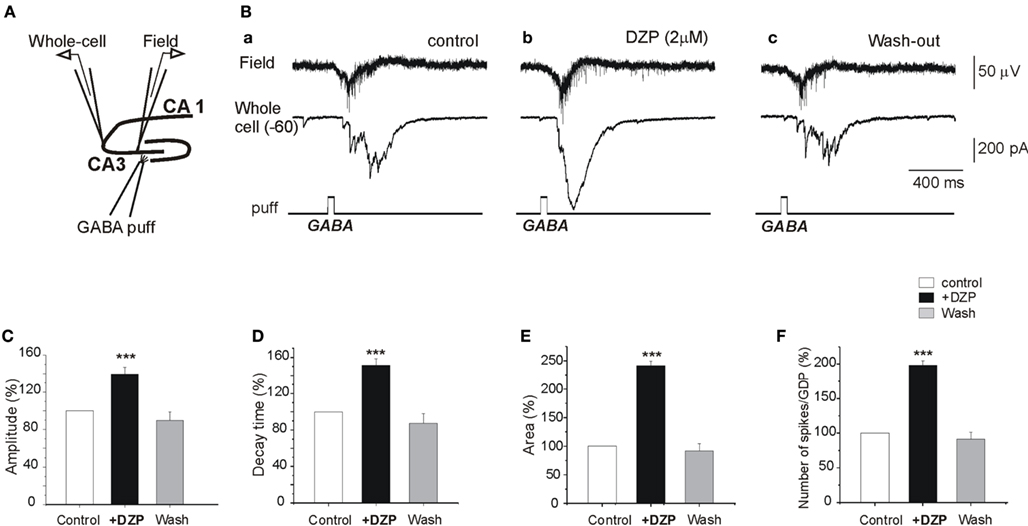
Figure 4. DZP enhances GABA excitation in naive neurons. (A) Experimental design. Simultaneous whole cell patch clamp recordings of a CA3a pyramidal neuron and field recordings in the CA3c pyramidal layer were made in control slices and GABA (100 μM) was focally applied (100 ms) by puff in the CA3c part of the pyramidal layer. (B) GABA puff generated a field and whole cell recorded GDPs (Ba) that was strongly augmented by DZP (Bb). The responses returned to control values after wash out of DZP (Bc). (C–E) Quantification of the effects of DZP on whole cell recorded postsynaptic currents: the amplitudes of the currents, the decay time and the area of whole cell recorded responses were significantly augmented by DZP. (F) Quantification histogram of mean number of spikes associated with puff GABA generated GDPs (a time window of 1 s from the GDP onset from GABA puff was analyzed).
To determine the effects of DZP and PB on glutamatergic activity, we first studied their effects on field potentials recorded in control slices in the presence of bicuculline (10 μM), CGP (2 μM) and APV (40 μM), respectively, to block GABA(A), GABA(B), and NMDA receptors. In these conditions, all or none IILEs are generated spontaneously and by local electrical stimuli (Figure 5). IILEs are mediated by AMPA/kainate receptors as they were fully blocked by the selective antagonist CNQX (10 μM, n = 13; Figure 5C). PB fully blocked the evoked and spontaneous IILEs (Figure 5A; n = 7). The frequency histograms (Figure 5Ad) reflect the powerful blockade of spikes and IILEs by PB. As 10 μm bicuculline do not always fully block GABAergic signaling (Strata and Cherubini, 1994) we also repeated these experiments with 100 μM picrotoxin instead of bicuculline and observed similar actions of PB (not shown, n = 3). In contrast, DZP (n = 6) failed to block or reduce these field potentials (Figure 5B). Thus, DZP did not reduce the evoked or the spontaneous IILEs. Therefore, PB but not DZP exerts a non-GABA mediated action on hippocampal neurons.
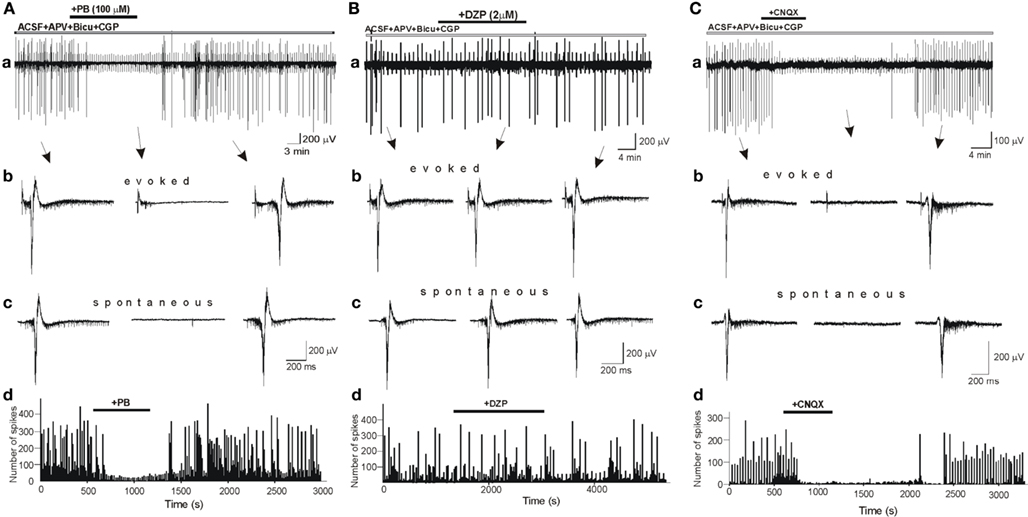
Figure 5. Phenobarbital but not diazepam blocks AMPA/kainate mediated inter ictal-like events (IILEs). (Aa,Ba) Extracellular recordings of spontaneous and evoked (electrical stimulation every 30 s) IILEs generated in control slices in the presence of GABA receptors antagonists – bicuculline (10 μM), CGP (2 μM), – and NMDA receptors antagonist – APV (40 μM). Non-filtered faster display traces of evoked (Ab,Bb,Cb) and spontaneous (Ac,Bc,Cc) IILEs. Generation of IILEs in these conditions is mediated by AMPA/kainate receptors as they were reversibly blocked by CNQX (C). Note that PB but not DZP completely blocked IILEs and reduced the frequency of spikes (Ad,Bd).
To examine the possible effects of PB and DZP on AMPA/kainate receptors, we next tested their effects on the currents evoked in whole cell recordings by focal pressure applications of glutamate. In the presence of NMDA and GABA receptor antagonists, focal applications of glutamate (100 μM, 100 ms) from patch pipettes generated large currents mediated by AMPA/kainate receptors as they are blocked by CNQX (n = 9; Figure 6C). PB reduced significantly the amplitude of these currents (to 67.2 ± 8.1%, p < 0.001, n = 4; Figure 6A). These effects fully recovered after wash out of PB (Figure 6Ad). In contrast, DZP had no effects on these currents (mean amplitude in DZP was 100.3 ± 4.9% of the control values, p > 0.05, n = 5; Figure 6B). Next, in similar experimental conditions using fast agonist application techniques (Colquhoun et al., 1992) glutamate (1 mM, 2 ms) was applied to the nucleated patch clamp recorded CA3 pyramidal neurons. As shown in Figure 7, PB significantly reduced the amplitude of glutamate induced currents (by 25.5 ± 0.5%, p < 0.001, n = 8). The decay time constant of these currents was not changed in the presence of PB (3.6 ± 0.5 ms in control and 3.4 ± 0.4 ms in the presence of PB, respectively, p > 0.5). In similar experiments DZP had no statistically significant effects on glutamate induced currents (mean current amplitude in DZP was 97.9 ± 3% of the control, p > 0.3, n = 4, not shown). In addition, we tested the effect of PB and DZP on NMDA receptor mediated currents. In the presence of AMPA/kainate and GABAreceptor antagonists focal puff applications of glutamate (100 μM, 100 ms) generated (at +40 mV holding potential) large amplitude outward currents. These currents are mediated by NMDA receptors as they were blocked by the NMDA receptor antagonist APV (40 μM, n = 11, not shown). As shown in Figure 8, neither PB nor DZP altered these currents. Therefore, PB selectively reduces AMPA/kainate receptor mediated currents.
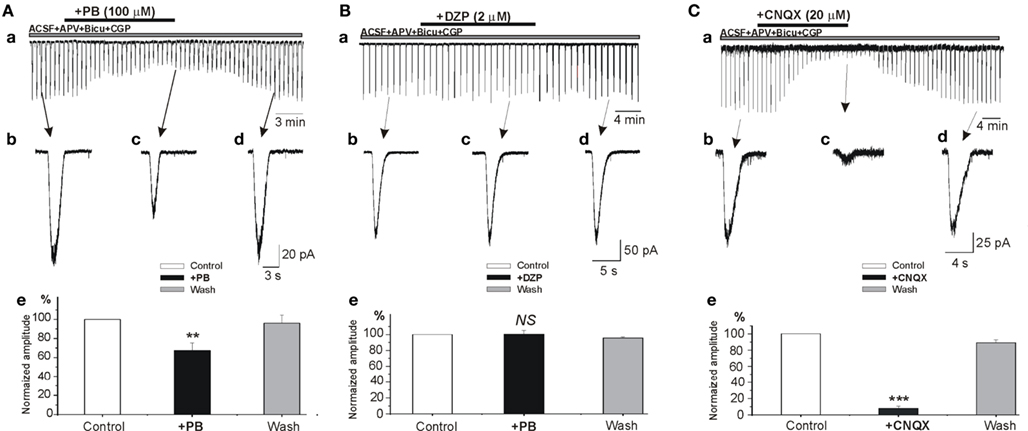
Figure 6. Phenobarbital but not diazepam reduces AMPA/kainate mediated currents. (A–C) Whole cell patch clamp recordings of currents induced by focal application of glutamate (100 μM, 100 ms) in the presence APV (40 μM). (Ae–Ce) Histograms of normalized averaged amplitudes of glutamate induced currents showing the effects of PB (Ae), DZP (Be), and CNQX (Ce). Note that phenobarbital but not diazepam significantly reduced the amplitude AMPA/kainate receptor mediated postsynaptic currents (67.2 ± 8.1%, p < 0.001, n = 4). NS, not statistically significant.
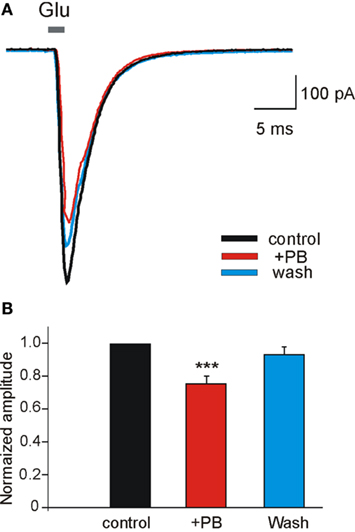
Figure 7. Phenobarbital reduces AMPA receptor mediated currents in nucleated patches. (A) Currents evoked in nucleated patches from CA3 pyramidal neurons from control slices. L-Glutamate (Glu, 1 mM) was applied in 2 ms pulses at a holding potential of −60 mV. Each trace is the average of 4–8 sweeps. Note that PB [(A) red trace] reduced the amplitude of AMPA/kainate mediated currents. (B) PB significantly reduced (by 25.5 ± 0.5%, p < 0.001, n = 8) the averaged amplitude of AMPA/kainate receptor mediated currents recorded in nucleated patches.
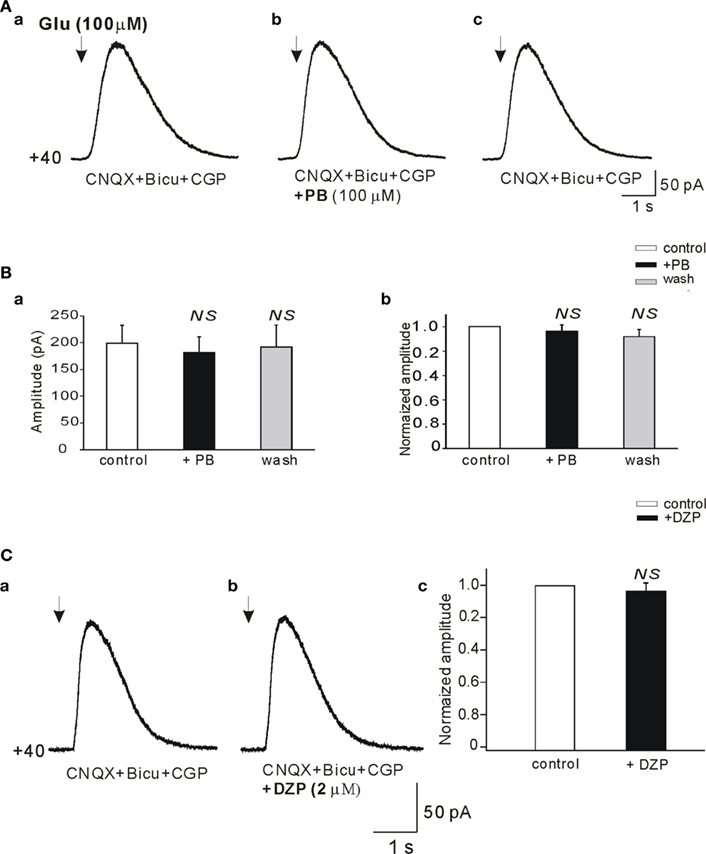
Figure 8. Phenobarbital does not reduce the NMDA receptor mediated currents. (A,Da,b) Whole cell patch clamp recordings (Vclamp = +40 mV) of currents induced by focal application of glutamate (100 μM, 100 ms, arrows) in the presence of bicuculline (10 μM), CGP (2 μM), and CNQX (20 μM; antagonist of AMPA/kainate receptors). Averaged NMDA receptor mediated currents before (Aa), in the presence (Ab), and after wash out of PB (Ac) recorded from the same pyramidal cell. (Ba) Histogram of averaged (n = 11) amplitudes of NMDA receptor mediated currents. (Bb) Histogram of averaged amplitudes normalized to control. Averaged NMDA receptor mediated currents before (Ca) and in the presence of DZP (Cb). (Cc) Histogram of averaged (n = 5) current amplitudes normalized to control. NS, not statistically significant.
To examine the pre- and postsynaptic mechanisms underlying the actions of PB, we used whole cell recordings to determined the effects of PB on the monosynaptic AMPA receptor-dependent EPSCs evoked by local electrical stimulations. We applied minimal electrical stimuli to generate all-or-none monosynaptic EPSCs (see Kullmann, 1994; Stevens and Wang, 1994). In the presence of NMDA and GABA receptor antagonists, an electrical stimulation evoked an all-or-none evoked AMPA receptor mediated EPSC (Figure 9). PB increased the number of failures of evoked AMPA receptor-dependent EPSCs (by 92.1 ± 30.1%, p < 0.001, n = 8; Figure 9Ae). In parallel experiments, miniature AMPA EPSCs (mEPSCs) were recorded in the presence of TTX to block ongoing network activity. PB reduced (Figure 9B) the amplitude (by 28.9%, from 25.9 ± 1.4 to 20.1 ± 0.9 pA, p < 0.01, n = 8 slices), and frequency of the mEPSCs (by 38.6 ± 8.0%, p < 0.01, n = 8 slices, not shown, 5 min recordings analyzed before, during and after wash out of PB). Therefore, PB exerts a selective action on ongoing and evoked glutamatergic exogenous and synaptic currents that is mediated by AMPA/kainate but not NMDA receptors.
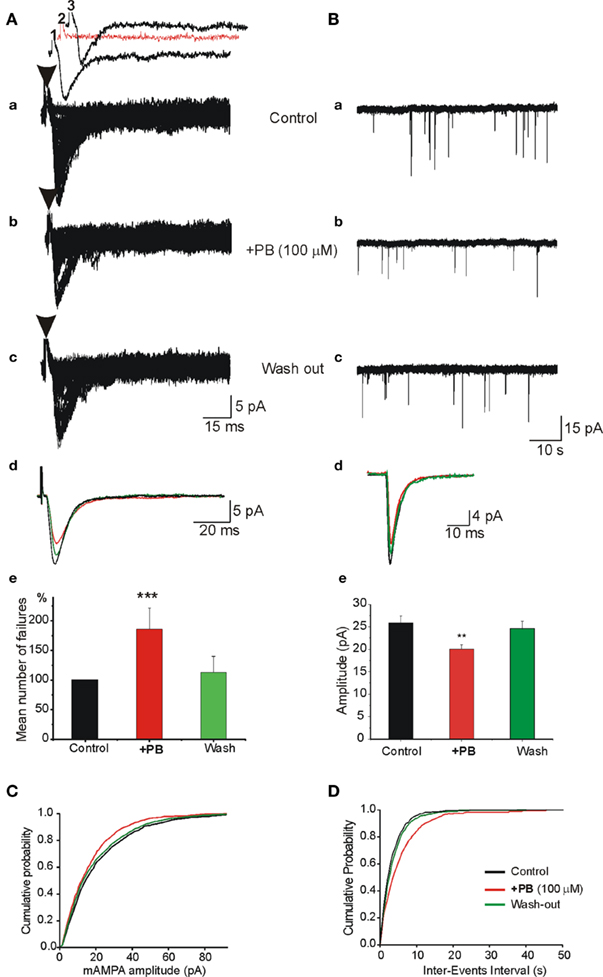
Figure 9. Pre- and postsynaptic actions of PB on AMPA/kainate receptor mediated excitatory postsynaptic synaptic currents (EPSCc). (A) In the presence of APV (40 μM) and bicuculline (10 μM), single fibers were electrically stimulated to evoke a mono synaptic AMPA receptor mediated EPSCs. The stimulation was adjusted to induce ∼20–50% failures [shown as red trace in (Aa2)]. (Aa–c) Single experiment obtained with repeated stimuli (30 stimuli every 10 s). (Aa) Control, (Ab) PB strongly enhanced the number of failures and reduced the average amplitude of EPSCs [see also (Ae)], (Ac) wash out of PB. (Ad) Superimposed averaged responses (the failures were discarded) from (Aa–c) to illustrate the effects of PB. Note the reduction of the averaged amplitude of EPSCs in the presence of PB (red trace). (Ae) Quantification of the mean number of normalized failures. Failure rate presented as “percentage of failure” (number of failure/total number of stimulation). Note the highly significant increase of the number of failures in the presence of PB (p < 0.001, n = 4 experiments). (B) PB reduces the amplitude of miniature AMPA receptor mediated EPSCs (mEPSCs). APV (40 μM) and bicuculline (10 μM) were applied throughout the recordings as well as TTX (1 μM) to generate mEPSCs. (Ba–c) Single traces to illustrate the actions of PB on AMPA receptor mediated mEPSCs. Note that PB (middle trace) reduced the amplitude of the minis. (Bd) Superimposed averaged traces of single mEPSCs to illustrate the effects of PB [from (Ba–c)]. Note the reduction of the mEPSCs amplitude (red trace). (Be) Quantification of the effects of PB. Note that PB reduced significantly (by 28.9%; from 25.9 ± 1.4 to 20.1 ± 0.9 pA, p < 0.01, n = 5 cells from different slices) the averaged amplitude of mEPSCs. PB also reduced the frequency of mini EPSCs (see text). (C) Cumulative probability distributions of mEPSCs amplitude in the presence of PB (red curve) demonstrate a leftward shift as compared with control and wash out. (D) Cumulative probability distribution of mEPSCs inter-event intervals in the presence of PB demonstrates a rightward shift as compared with control and wash out.
To examine whether PB reduces the AMPA/kainate receptor mediated currents in “epileptic” tissue we used slices obtained from contralateral hippocampus after the formation of MF. As shown in Figure 10Aa,b, in control conditions these MF slices generate spontaneous IILEs (see also Khalilov et al., 2003, 2005; Nardou et al., 2009, 2011). After blockade of GABA and NMDA receptors, the IILEs transformed into AMPA/kainate receptor mediated ILEs (Figure 10Ac) as they were reversibly blocked by CNQX (10 μM, not shown). Addition of PB (100 μM) in these conditions significantly reduced the amplitude (by 22.7 ± 4.2% to controls, n = 5, p < 0.001, not shown) and frequency of ILEs: PB increased the mean interval between ictal-like events by 318.1 ± 66.5% (p < 0.001, n = 5 slices; Figure 10C). PB significantly decreased also the power of AMPA/kainate receptor-dependent ictal-like events Figure 10B,C (by 26.9 ± 7.2%, n = 5, p < 0.001). Therefore, the inhibitory actions of PB on AMPA/kainate receptors are preserved in MF “epileptic” neurons.
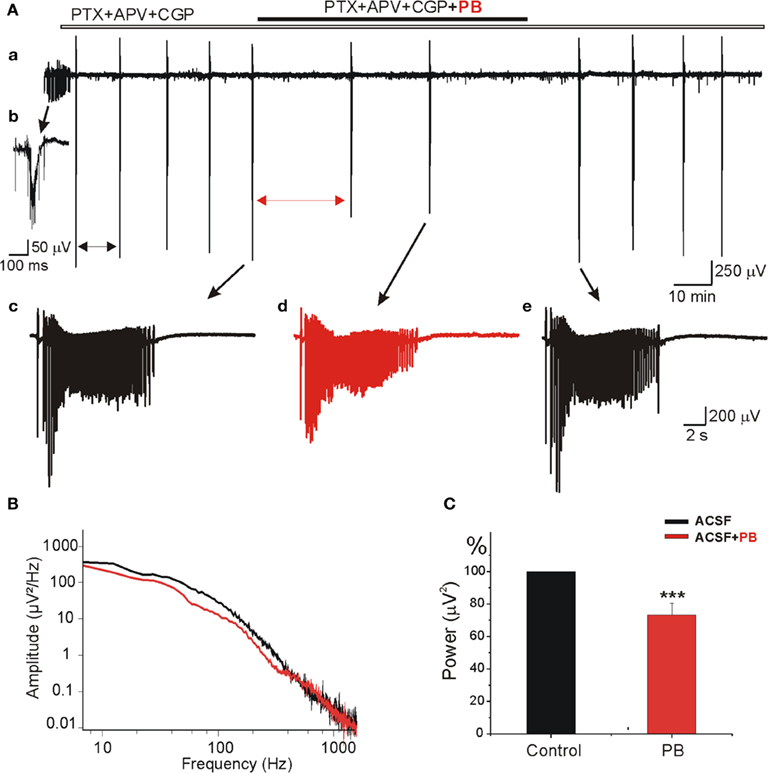
Figure 10. Phenobarbital reduces AMPA/kainate receptors field potentials in mirror focus neurons. (A) Extracellular field recording of spontaneous epileptiform activities in slices obtained from contralateral hippocampus after mirror focus formation. Recordings were performed in the presence of GABA(A) and GABA(C) receptors antagonist PTX (100 μM), GABA(B) receptor antagonist CGP (2 μM), and NMDA receptor antagonist APV (40 μM). (Ab) Example of epileptiform activities recorded in an “epileptic” slice from mirror focus before application of antagonists. (Ac–e) Examples of ILEs in the presence of GABA and NMDA receptors antagonists: before application (Ac), in the presence (Ad), and after PB wash out (Ae). Note the significant decrease of amplitude and frequency of ictal-like events. Mean interval between ictal-like events (double arrows) significantly increased (by 318.1 ± 66.5%, not shown). (B) Power spectrum [from (Ac–d)] showing the decrease of amplitude and frequency by PB. (C) Average power histogram of spontaneous ictal-like events before and after application of PB. PB significantly decreased the power of epileptiform events (by 26.9 ± 7.2%, n = 5, p < 0.001).
Our results first suggest that PB has a higher efficacy than DZP to reduce early seizures thanks to its dual action on GABA and AMPA/kainate receptor mediated currents. DZP aggravates inaugurating propagated seizures most likely because at that developmental stage many naïve neurons have elevated [Cl−]i. (Nardou et al., 2009). However, in ongoing epileptogenic mirror foci, where GABA strongly excites neurons, PB like DZP aggravates seizures in spite of the reduction of glutamatergic currents that it produces. Therefore, the efficacy of DZP like PB in reducing seizures is conditioned by the actions of GABA. When these are strongly excitatory due to a strong increase of [Cl−]i, the reduction of the excitation by glutamate will not suffice to block seizures. Our observations of direct actions of PB on AMPA/kainate but not NMDA currents suggest a specific not yet identified recognition site. In spite of the inherent limitations of in vitro experimental models, the present results suggest that for the treatment of early neonatal seizures PB may be more efficient than DZP. Furthermore, agents that exert a dual action on GABA and AMPA/kainate receptors may offer an interesting novel therapeutic perspective to treat neonatal seizures, provided that they are used at the earliest stages as the GABA polarity shift remains a fundamental point.
The effects of PB and DZP on GABAergic signals have been extensively investigated and will not be reviewed here (Barker and Gainer, 1973; Barker and Ransom, 1978; Barker and McBurney, 1979; Macdonald and Barker, 1979; Macdonald and McLean, 1982; Macdonald et al., 1986; Cheng et al., 2002; Soderpalm, 2002; Rogawski and Loscher, 2004; Czapinski et al., 2005; Macdonald and Rogawski, 2007; Stefan and Feuerstein, 2007). In short, both act directly on postsynaptic GABA receptors leading to a more powerful inhibition of target neurons. Classically, DZP potentiates GABA(A) receptor function by increasing channel opening frequency, whereas barbiturates increase channel open duration (Feng et al., 2004; Hanson and Czajkowski, 2008; Reid et al., 2009) by acting on different target sites on the GABA receptor channel complex (Feng et al., 2004; Hanson and Czajkowski, 2008; Mercado and Czajkowski, 2008). There are however several complicating factors. Benzodiazepine actions are highly dependent on the levels of GABA present (Mercik et al., 2007; Mozrzymas et al., 2007), the binding/unbinding of DZP from GABAA is highly complex (Jones-Davis et al., 2005; Bianchi et al., 2007a,b) and GABA receptor subunits and diazepam binding sites are internalized after seizures (Goodkin et al., 2008; Goodkin and Kapur, 2009). Systematic experimental comparisons of the actions of various GABA acting AEDs on neonatal network driven events have been seldom investigated. In the immature rat hippocampus, diazepam increases frequency yet slows down propagation of GDPs (Khalilov et al., 1999; Valeeva et al., 2010) whereas PB does not affect GDPs frequency (Dzhala et al., 2008) and reduces GDPs amplitude (Nardou et al., 2011). These observations are not readily compatible with an additional presynaptic action of PB on the release of GABA although this cannot be excluded at present. Collectively these observations are important considering the profound impact of ongoing network activity on transmitter release and the operation of developing cell assemblies. Thus, Cherubini and colleagues have elegantly shown that GDPs modulate GABA release via BDNF and other messengers (Sivakumaran et al., 2009). Since PB reduced and DZP augmented GDPs and early seizures we investigated the possibility that target sites other than GABA signaling could underline these differences.
Several studies have shown earlier that PB (and pentobarbital) reduced glutamatergic AMPA/kainate receptor mediated currents including: (i) a subunit selective action on kainate-induced currents in Xenopus oocytes expressing different GluR subunits (DildyMayfield et al., 1996; Minami et al., 1998); (ii) a reduction of glutamate currents in spinal cord neurons (Barker and Gainer, 1973; MacDonald and Barker, 1978b; Taverna et al., 1994; Joo et al., 1999). PB – like DZP (Rovira and Ben-Ari, 1994) – also reduced calcium currents (Werz and Macdonald, 1985). In contrast, zolpidem had no effects in young animals but decreased calcium and barium spikes in adults whereas midazolam increased barium spikes in both young and adult animals (Rovira and Ben-Ari, 1994). The contributions of these non-GABA mediated signaling remained however controversial (Kamiya et al., 1999; also see Barker and Gainer, 1973; Barker and Ransom, 1978).
Present results provide direct evidence that PB but not DZP tested in the same preparation exerts a strong modulation of glutamatergic receptors and synaptic glutamatergic currents. Thus, PB strongly reduced the amplitude and frequency of miniature AMPA/kainate receptor mediated EPSCs generated in the presence of TTX, indicating a direct pre- and postsynaptic action of PB. This was confirmed directly by showing that PB increased the frequency of failures evoked in an all-or-none manner by minimal electrical stimuli, and reduced the amplitude of the responses evoked by focal applications of glutamate in the presence of the same cocktail of receptor antagonists and directly the amplitude but not the kinetic of AMPA/kainate currents in nucleated patches. The molecular targets of these actions have not been determined and are outside the scope of the present study. Interestingly, presynaptic activation of kainate receptors in CA3 neurons alters GABA release raising the possibility of an indirect modulation of GABAergic signals by PB (Caiati et al., 2010).
The increase of the number of failures by PB as well as the reduction of the frequency of miniature EPSCs suggests also presynaptic effects in addition to the postsynaptic ones. Such effects have long been observed in a variety of preparations. Thus, PB decrease mono- and polysynaptic reflexes in the spinal cord (Nicholson et al., 1988) and the anesthetic actions of barbiturates have been ascribed in part by a reduction of presynaptic calcium entry and consequent reduction of neurotransmitter release in mouse spinal cord neurons (Heyer and Macdonald, 1982). Different concentration dependent actions of the pre- and postsynaptic actions of PB and barbiturates have also been reported in the frog neuromuscular junction (Proctor and Weakly, 1976). Clearly, these effects may also contribute to the anticonvulsive actions of PB but the importance of their contribution remains to be determined.
The effects of DZP on GDPs are quite striking and to some extent unexpected. They suggest that by enhancing the excitatory actions of GABA on immature neurons, agents that exert solely this action will produce unwarranted effects. The underlying mechanisms are not clear at present; they may include specific effects on interneurons that orchestrate GDPs (Bonifazi et al., 2009) and/or enhance possible depolarizing tonic GABA currents in pyramidal cells that is important at that early stage and provides a large current (Ge et al., 2006; Sebe et al., 2010). Benzodiazepines are extensively used to block status epilepticus. In adult animal models diazepam is systematically injected after administrations of kainate or pilocarpine to prevent animal death consequently to the recurrent seizures (Ben-Ari et al., 1978; Ben-Ari, 1985; Cavalheiro et al., 1996). In contrast, DZP – like PB – is not efficient to block seizures after recurrent seizures have taken place (see below). Here, we show that DZP aggravates seizures from their onset in neonatal hippocampi. The effects of DZP were highly reproducible, associated with a significant increase of the HFOs and consequently with the formation by seizures of an epileptogenic MF. Indeed, as shown earlier, there is a direct link between HFOs and the formation by seizures of an independent MF capable of generating seizures (Khalilov et al., 2005). These actions are also in keeping with our earlier report showing that functional GABAergic receptors are essential for the formation by seizures of a MF, since applications of a GABA receptor antagonist generated epileptiform events that however were neither associated with HFOs nor with the formation of an epileptogenic MF (Khalilov et al., 2005).
There is now direct evidence from humans and experimental animals that recurrent seizures produce an accumulation of [Cl−]i associated with a shift of the actions of GABA from inhibitory to excitatory (Cohen et al., 2002; Khalilov et al., 2003, 2005; Payne et al., 2003; Blaesse et al., 2009; Nardou et al., 2011). The underlying mechanisms are now better understood. It has been recently shown that GABA excites epileptic neurons because of high intracellular chloride mediated by a combined action of the NKCC1 chloride importer (Dzhala et al., 2010) and a down regulation of the chloride exporter KCC2 (Lee et al., 2010; Nardou et al., 2011), leading to strong excitatory actions of GABA that are exacerbated by PB. Therefore, although PB is more efficient than DZP to block early neonatal seizures, by shifting the actions of GABA to strongly excitatory, recurrent seizures abolish this advantage stressing the importance of an early aggressive treatment of seizures to avoid the loss of KCC2 activity and preserve the efficient actions of PB.
There are many obvious limitations to the in vitro preparations that restrict the relevance of these observations to a clinical situation including the lack of major cerebral connections, systemic events (vascular, hormonal, etc.) and the different species. Yet, the in vitro MF model used here has several advantages over conventional slices in that it enables to determine the activity of neuronal ensembles in relatively preserved conditions and to compare the actions of AEDs on acute and more chronic propagating seizures in neurons that have never been in contact with a convulsive agent. Our results suggest important advantages of PB over DZP since the former exerts a dual action and is not exclusively restricted to GABA signal. Although the magnitude of these effects is relatively small, they are stable and observed in naïve and “epileptic” neurons suggesting that AEDs with a dual action may be advantageous over agents that exert solely a pro-GABA action. This is particularly important in view of the vulnerability of intracellular chloride shifts with recurrent seizures that strongly limit the efficacy of GABA acting AEDs. There is now little doubt that in humans, like in experimental animals, GABA may excite epileptic neurons and this will seriously aggravate the situation. The history of seizures prior to AEDs is clearly an important element to take in consideration in order to preserve as much as possible the capacity of neurons to control their [Cl−]i.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Drs. Rustem Khazipov, Roman Tyzio and Diana Ferrari for their suggestions and criticism. Funding: This work was supported by the French Medical Research council (INSERM), the Université de la Méditerranée, the French agency of research ANR (L’Agence Nationale de la Recherche – to Ilgam Khalilov), Fondation pour la Recherche Médicale (FRM – to Romain Nardou), and the FP7 NEMO program: http://www.nemo-europe.com/(to Yehezkel Ben-Ari). Financial supported from University of Paris/INSERM “interface” programs to Yehezkel Ben-Ari.
Barker, J. L., and Gainer, H. (1973). Pentobarbital: selective depression of excitatory postsynaptic potentials. Science 182, 720–722.
Barker, J. L., and McBurney, R. N. (1979). Phenobarbitone modulation of postsynaptic GABA receptor function on cultured mammalian neurons. Proc. R. Soc. Lond. B Biol. Sci. 206, 319–327.
Barker, J. L., and Ransom, B. R. (1978). Pentobarbitone pharmacology of mammalian central neurones grown in tissue culture. J. Physiol. 280, 355–372.
Bartha, A. I., Shen, J., Katz, K. H., Mischel, R. E., Yap, K. R., Ivacko, J. A., Andrews, E. M., Ferriero, D. M., Ment, L. R., and Silverstein, F. S. (2007). Neonatal seizures: multicenter variability in current treatment practices. Pediatr. Neurol. 37, 85–90.
Bassan, H., Bental, Y., Shany, E., Berger, I., Froom, P., Levi, L., and Shiff, Y. (2008). Neonatal seizures: dilemmas in workup and management. Pediatr. Neurol. 38, 415–421.
Ben Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284.
Ben-Ari, Y. (1985). Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14, 375–403.
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaïarsa, J.-L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. (Lond.) 416, 303–325.
Ben-Ari, Y., Lagowska, Y., Le Gal La, S. G., Tremblay, E., Ottersen, O. P., and Naquet, R. (1978). Diazepam pretreatment reduces distant hippocampal damage induced by intra-amygdaloid injections of kainic acid. Eur. J. Pharmacol. 52, 419–420.
Bianchi, M. T., Botzolakis, E., and Macdonald, R. (2007a). Re-evaluating the benzodiazepine mechanism of action: from bench to bedside and back. Neurology 68, A269.
Bianchi, M. T., Botzolakis, E. J., Haas, K. F., Fisher, J. L., and Macdonald, R. L. (2007b). Microscopic kinetic determinants of macroscopic currents: insights from coupling and uncoupling of GABA(A) receptor desensitization and deactivation. J. Physiol. (Lond.) 584, 769–787.
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838.
Bonifazi, P., Goldin, M., Picardo, M. A., Jorquera, I., Cattani, A., Bianconi, G., Represa, A., Ben-Ari, Y., and Cossart, R. (2009). GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science 326, 1419–1424.
Boylan, G. B., Rennie, J. M., Pressler, R. M., Wilson, G., Morton, M., and Binnie, C. D. (2002). Phenobarbitone, neonatal seizures, and video-EEG. Arch. Dis. Child. Fetal Neonatal Ed. 86, 165–170.
Bragin, A., Engel, J. Jr., Wilson, C. L., Vizentin, E., and Mathern, G. W. (1999). Electrophysiologic analysis of a chronic seizure model after unilateral hippocampal KA injection. Epilepsia 40, 1210–1221.
Caiati, M. D., Sivakumaran, S., and Cherubini, E. (2010). In the developing rat hippocampus, endogenous activation of presynaptic kainate receptors reduces GABA release from mossy fiber terminals. J. Neurosci. 30, 1750–1759.
Carmo, K. B., and Barr, P. (2005). Drug treatment of neonatal seizures by neonatologists and paediatric neurologists. J. Paediatr. Child Health 41, 313–316.
Cavalheiro, E. A., Santos, N. F., and Priel, M. R. (1996). The pilocarpine model of epilepsy in mice. Epilepsia 37, 1015–1019.
Cheng, C., Roemer-Becuwe, C., and Pereira, J. (2002). When midazolam fails. J. Pain Symptom Manage. 23, 256–265.
Cohen, I., Navarro, V., Clemenceau, S., Baulac, M., and Miles, R. (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421.
Colquhoun, D., Jonas, P., and Sakmann, B. (1992). Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J. Physiol. (Lond.) 458, 261–287.
Connell, J., Oozeer, R., de, V. L., Dubowitz, L. M., and Dubowitz, V. (1989). Clinical and EEG response to anticonvulsants in neonatal seizures. Arch. Dis. Child. 64, 459–464.
Czapinski, P., Blaszczyk, B., and Czuczwar, S. J. (2005). Mechanisms of action of antiepileptic drugs. Curr. Top. Med. Chem. 5, 3–14.
DildyMayfield, J. E., Eger, E. I., and Harris, R. A. (1996). Anesthetics produce subunit-selective actions on glutamate receptors. J. Pharmacol. Exp. Ther. 276, 1058–1065.
Dzhala, V. I., Brumback, A. C., and Staley, K. J. (2008). Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann. Neurol. 63, 222–235.
Dzhala, V. I., Kuchibhotla, K. V., Glykys, J. C., Kahle, K. T., Swiercz, W. B., Feng, G., Kuner, T., Augustine, G. J., Bacskai, B. J., and Staley, K. J. (2010). Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J. Neurosci. 30, 11745–11761.
Feng, H. J., Bianchi, M. T., and Macdonald, R. L. (2004). Pentobarbital differentially modulates alpha 1 beta 3 delta and alpha 1 beta 3 gamma 2L GABA(A) receptor currents. Mol. Pharmacol. 66, 988–1003.
Ge, S., Goh, E. L., Sailor, K. A., Kitabatake, Y., Ming, G. L., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 2, 589–593.
Glykys, J., Dzhala, V. I., Kuchibhotla, K. V., Feng, G., Kuner, T., Augustine, G., Bacskai, B. J., and Staley, K. J. (2009). Differences in cortical vs. subcortical GABAergic signaling: a candidate mechanism of electroclinical dissociation of neonatal seizures. Neuron 10, 657–672.
Goodkin, H. P., Joshi, S., Mtchedlishvili, Z., Brar, J., and Kapur, J. (2008). Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 28, 2527–2538.
Goodkin, H. P., and Kapur, J. (2009). The impact of diazepam’s discovery on the treatment and understanding of status epilepticus. Epilepsia 50, 2011–2018.
Guerrini, R., Belmonte, A., and Genton, P. (1998). Antiepileptic drug-induced worsening of seizures in children. Epilepsia 39(Suppl. 3), S2–S10.
Guillet, R., and Kwon, J. (2007). Seizure recurrence and developmental disabilities after neonatal seizures: outcomes are unrelated to use of phenobarbital prophylaxis. J. Child Neurol. 22, 389–395.
Hanson, S. M., and Czajkowski, C. (2008). Structural mechanisms underlying benzodiazepine modulation of the GABA(A) receptor. J. Neurosci. 28, 3490–3499.
Heyer, E. J., and Macdonald, R. L. (1982). Barbiturate reduction of calcium-dependent action-potentials – correlation with anesthetic action. Brain Res. 236, 157–171.
Jacobs, J., LeVan, P., Chander, R., Hall, J., Dubeau, F., and Gotman, J. (2008). Interictal high-frequency oscillations (80-500 Hz) are an indicator of seizure onset areas independent of spikes in the human epileptic brain. Epilepsia 49, 1893–1907.
Jin, L. J., Schlesinger, F., Song, Y. P., Dengler, R., and Krampfl, K. (2010). The interaction of the neuroprotective compounds riluzole and phenobarbital with AMPA-type glutamate receptors: a patch-clamp study. Pharmacology 85, 54–62.
Jones-Davis, D. M., Song, L. Y., Gallagher, M. J., and Macdonald, R. L. (2005). Structural determinants of benzodiazepine allosteric regulation of GABA(A) receptor currents. J. Neurosci. 25, 8056–8065.
Joo, D. T., Xiong, Z. G., MacDonald, J. F., Jia, Z., Roder, J., Sonner, J., and Orser, B. A. (1999). Blockade of glutamate receptors and barbiturate anesthesia – increased sensitivity to pentobarbital-induced anesthesia despite reduced inhibition of AMPA receptors in GluR2 null mutant mice. Anesthesiology 91, 1329–1341.
Kaindl, A. M., Koppelstaetter, A., Nebrich, G., Stuwe, J., Sifringer, M., Zabel, C., Klose, J., and Ikonomidou, C. (2008). Brief alteration of NMDA or GABAA receptor-mediated neurotransmission has long term effects on the developing cerebral cortex. Mol. Cell. Proteomics 7, 2293–2310.
Kamiya, Y., Andoh, T., Furuya, R., Hattori, S., Watanabe, I., Sasaki, T., Ito, H., and Okumura, F. (1999). Comparison of the effects of convulsant and depressant barbiturate stereoisomers on AMPA-type glutamate receptors. Anesthesiology 90, 1704–1713.
Khalilov, I., Dzhala, V., Medina, I., Leinekugel, X., Melyan, Z., Lamsa, K., Khazipov, R., and Ben-Ari, Y. (1999). Maturation of kainate-induced epileptiform activities in interconnected intact neonatal limbic structures in vitro. Eur. J. Neurosci. 11, 3468–3480.
Khalilov, I., Esclapez, M., Medina, I., Aggoun, D., Lamsa, K., Leinekugel, X., Khazipov, R., and Ben-Ari, Y. (1997). A novel in vitro preparation: the intact hippocampal formation. Neuron 19, 743–749.
Khalilov, I., Holmes, G. L., and Ben Ari, Y. (2003). In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat. Neurosci. 6, 1079–1085.
Khalilov, I., Le Van, Q. M., Gozlan, H., and Ben Ari, Y. (2005). Epileptogenic actions of GABA and fast oscillations in the developing hippocampus. Neuron 48, 787–796.
Kullmann, D. M. (1994). Amplitude fluctuations of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron 12, 1111–1120.
Le Van Quyen, M., Khalilov, I., and Ben-Ari, Y. (2006). The dark side of high-frequency oscillations in the developing brain. Trends Neurosci. 29, 419–427.
Lee, H. H., Jurd, R., and Moss, S. J. (2010). Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 45, 173–179.
Loscher, W., and Honack, D. (1989). Comparison of the anticonvulsant efficacy of primidone and phenobarbital during chronic treatment of amygdala-kindled rats. Eur. J. Pharmacol. 162, 309–322.
MacDonald, J. F., and Barker, J. L. (1982). Multiple actions of picomolar concentrations of flurazepam on the excitability of cultured mouse spinal neurons. Brain Res. 246, 257–264.
MacDonald, R., and Barker, J. L. (1978a). Benzodiazepines specifically modulate GABA-mediated postsynaptic inhibition in cultured mammalian neurones. Nature 271, 563–564.
MacDonald, R. L., and Barker, J. L. (1978b). Different actions of anticonvulsant and anesthetic barbiturates demonstrated using mammalian neurons in cell-culture. Neurology 28, 367.
Macdonald, R. L., and Barker, J. L. (1979). Enhancement of GABA-mediated postsynaptic inhibition in cultured mammalian spinal-cord neurons – common-mode of anticonvulsant action. Brain Res. 167, 323–336.
Macdonald, R. L., and Kelly, K. M. (1994). Mechanisms of action of currently prescribed and newly developed antiepileptic drugs. Epilepsia 35(Suppl. 4), S41–S50.
Macdonald, R. L., and McLean, M. J. (1982). Cellular bases of barbiturate and phenytoin anticonvulsant drug action. Epilepsia 23(Suppl. 1), S7–S18.
Macdonald, R. L., and Rogawski, M. A. (2007). “Cellular effects of antiepileptic drugs,” in Epilepsy: A Comprehensive Textbook, 2nd Edn, eds J. Engel and T. A. Pedley (Philadelphia: Lippincott Williams and Wilkins), 1433–1445.
Macdonald, R. L., Weddle, M. G., and Gross, R. A. (1986). Benzodiazepine, beta-carboline, and barbiturate actions on GABA responses. Adv. Biochem. Psychopharmacol. 41, 67–78.
Mercado, J., and Czajkowski, C. (2008). Gamma-aminobutyric acid (GABA) and pentobarbital induce different conformational rearrangements in the GABA(A) receptor alpha 1 and beta(2) pre-M1 regions. J. Biol. Chem. 283, 15250–15257.
Mercik, K., Piast, M., and Mozrzymas, J. W. (2007). Benzodiazepine receptor agonists affect both binding and gating of recombinant alpha I beta 2 gamma 2 gamma-aminobutyric acid-A receptors. Neuroreport 18, 781–785.
Minami, K., Wick, M. J., Stern-Bach, Y., Dildy-Mayfield, J. E., Brozowski, S. J., Gonzales, E. L., Trudell, J. R., and Harris, R. A. (1998). Sites of volatile anesthetic action on kainate (glutamate receptor 6) receptors. J. Biol. Chem. 273, 8248–8255.
Mozrzymas, J. W., Wojtowicz, T., Piast, M., Lebida, K., Wyrembek, P., and Mercik, K. (2007). GABA transient sets the susceptibility of mIPSCs to modulation by benzodiazepine receptor agonists in rat hippocampal neurons. J. Physiol. (Lond.) 585, 29–46.
Nardou, R., Ben-Ari, Y., and Khalilov, I. (2009). Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J. Neurophysiol. 101, 2878–2888.
Nardou, R., Yamamoto, S., Chazal, G., Bhar, A., Ferrand, N., Dulac, O., Ben-Ari, Y., and Khalilov, I. (2011). Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain 134, 987–1002.
Nicholson, G. M., Spence, I., and Johnston, G. A. R. (1988). Differing actions of convulsant and nonconvulsant barbiturates – an electrophysiological study in the isolated spinal-cord of the rat. Neuropharmacology 27, 459–465.
Painter, M. J., Scher, M. S., Stein, A. D., Armatti, S., Wang, Z., Gardiner, J. C., Paneth, N., Minnigh, B., and Alvin, J. (1999). Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N. Engl. J. Med. 341, 485–489.
Payne, J. A., Rivera, C., Voipio, J., and Kaila, K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206.
Perucca, E., Gram, L., Avanzini, G., and Dulac, O. (1998). Antiepileptic drugs as a cause of worsening seizures. Epilepsia 39, 5–17.
Proctor, W. R., and Weakly, J. N. (1976). Comparison of presynaptic and post-synaptic actions of pentobarbital and phenobarbitone in neuromuscular-junction of frog. J. Physiol. (Lond.) 258, 257–268.
Reid, C. A., Taylor, N., Berkovic, S. F., and Petrou, S. (2009). Benzodiazepine efficacy in a mouse model of absence epilepsy based on a human mutation in the gamma2 GABA-A subunit. Epilepsia 50, 254–255.
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipilä, S., Payne, J. A., Minichiello, L., Saarma, M., and Kaila, K. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 24, 4683–4691.
Rogawski, M. A., and Loscher, W. (2004). The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 5, 553–564.
Rovira, C., and Ben-Ari, Y. (1994). Benzodiazepines modulate calcium spikes in young and adult hippocampal cells. Neuroreport 5, 2125–2129.
Sather, W., Dieudonné, S., MacDonald, J. F., and Ascher, P. (1992). Activation and desensitization of N-methyl-D-aspartate receptors in nucleated outside-out patches from mouse neurones. J. Physiol. 450, 643–672.
Sebe, J. Y., Looke-Stewart, E. C., Estrada, R. C., and Baraban, S. C. (2010). Robust tonic GABA currents can inhibit cell firing in mouse newborn neocortical pyramidal cells. Eur. J. Neurosci. 32, 1310–1318.
Sivakumaran, S., Mohajerani, M. H., and Cherubini, E. (2009). At immature mossy-fiber-CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J. Neurosci. 29, 2637–2647.
Soderpalm, B. (2002). Anticonvulsants: aspects of their mechanisms of action. Eur. J. Pain (Lond.) 6, 3–9.
Stefan, H., and Feuerstein, T. J. (2007). Novel anticonvulsant drugs. Pharmacol. Ther. 113, 165–183.
Stevens, C. F., and Wang, Y. (1994). Changes in reliability of synaptic function as a mechanism for plasticity. Nature 371, 704–707.
Strata, F., and Cherubini, E. (1994). Transient expression of a novel type of GABA response in rat CA3 hippocampal neurones during development. J. Physiol. (Lond.) 480, 493–503.
Taverna, F. A., Cameron, B. R., Hampson, D. L., Wang, L. Y., and MacDonald, J. F. (1994). Sensitivity of ampa receptors to pentobarbital. Eur. J. Pharmacol. Mol. Pharmacol. Sec. 267, R3–R5.
Valeeva, G., Abdullin, A., Tyzio, R., Skorinkin, A., Nikolski, E., Ben-Ari, Y., and Khazipov, R. (2010). Temporal coding at the immature depolarizing GABAergic synapse. Front. Cell. Neurosci. 4:17. doi: 10.3389/fncel.2010.00017
Werz, M. A., and Macdonald, R. L. (1985). Barbiturates decrease voltage-dependent calcium conductance of mouse neurons in dissociated cell-culture. Mol. Pharmacol. 28, 269–277.
Wheless, J. W., Clarke, D. F., Arzimanoglou, A., and Carpenter, D. (2007). Treatment of pediatric epilepsy: european expert opinion, 2007. Epileptic Disord. 9, 353–412.
Keywords: phenobarbital, diazepam, AMPA, immature hippocampus, seizures
Citation: Nardou R, Yamamoto S, Bhar A, Burnashev N, Ben-Ari Y and Khalilov I (2011) Phenobarbital but not diazepam reduces AMPA/kainate receptor mediated currents and exerts opposite actions on initial seizures in the neonatal rat hippocampus. Front. Cell. Neurosci. 5:16. doi: 10.3389/fncel.2011.00016
Received: 01 June 2011;
Accepted: 19 July 2011;
Published online: 28 July 2011.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyCopyright: © 2011 Nardou, Yamamoto, Bhar, Burnashev, Ben-Ari and Khalilov. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Yehezkel Ben-Ari, Campus Scientifique de Luminy, 163 route de Luminy, 13009 Marseille, France. e-mail:YmVuLWFyaUBpbm1lZC51bml2LW1ycy5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.