1
Institute of Neuroscience Consiglio Nazionale delle Ricerche, Pisa, Italy
2
Department of Psychology, Florence University, Florence, Italy
3
Laboratory of Neurobiology, Scuola Normale Superiore, Pisa, Italy
Experience is required for the shaping and refinement of developing neural circuits during well defined periods of early postnatal development called critical periods. Many studies in the visual cortex have shown that intracortical GABAergic circuitry plays a crucial role in defining the time course of the critical period for ocular dominance plasticity. With the end of the critical period, neural plasticity wanes and recovery from the effects of visual defects on visual acuity (amblyopia) or binocularity is much reduced or absent. Recent results pointed out that intracortical inhibition is a fundamental limiting factor for adult cortical plasticity and that its reduction by means of different pharmacological and environmental strategies makes it possible to greatly enhance plasticity in the adult visual cortex, promoting ocular dominance plasticity and recovery from amblyopia. Here we focus on the role of intracortical GABAergic circuitry in controlling both developmental and adult cortical plasticity. We shall also discuss the potential clinical application of these findings to neurological disorders in which synaptic plasticity is compromised because of excessive intracortical inhibition.
What we are, how we behave, how we talk, how we perceive, crucially depends upon how our brain has developed. Maturation of neural circuits is a protracted phase of brain development which, though initially guided by genetically coded cues, requires in most cases to be shaped and refined by the interaction of the individual with the environment. To allow this shaping action, developing neural circuits exhibit a great degree of plasticity so that they can be modified, functionally and morphologically, in response to experience. Neural plasticity is high during well defined periods of early postnatal development called critical periods (CPs). As experience drives the remodeling of neural connections, determining the maturation of neural circuits and of the corresponding functions, both circuits and functions become progressively less modifiable by experience and CPs draw towards their closure. As a result of this decline in plasticity, recovery from the effects of defective neural circuit development is significantly reduced or lost after the end of the CP. Recent studies, however, reveal that it is possible to enhance neural plasticity in the adult cortex. The visual cortex has long been a paradigmatic model to study the influence of experience on the development and plasticity of neural connections and its molecular basis. Studies in the visual cortex have shown that intracortical GABAergic circuitry is at the core both of developmental plasticity and CP control and of adult cortical plasticity limitations.
Intracortical GABAergic inhibition (IGI) plays a crucial role in shaping visual cortical receptive fields and their development. Maturation of visual acuity is well correlated with the decrease of mean receptive field size of neurons in the primary visual cortex (Fagiolini et al., 1994
) and with the postnatal development of IGI (Huang et al., 1999
). Both visual acuity maturation and IGI development are dependent from visual experience: dark rearing, which prevents visual acuity development (Fagiolini et al., 1994
), also affects the developmental increase of IGI (Benevento et al., 1995
; Gianfranceschi et al., 2003
). IGI development has been therefore suggested to contribute to visual acuity development and a direct evidence for this comes from BDNF overexpressing mice (Huang et al., 1999
). In these mice BDNF expression in the visual cortex is higher than in wt mice starting from very early postnatal ages and this correlates with a precocious development of IGI, as shown by the faster developmental increase in inhibitory postsynaptic currents and GABA biosynthetic enzyme GAD65 in the perisomatic region of pyramidal cells. This precocious IGI development is accompanied by an accelerated development of visual acuity.
Recently, the use of the environmental enrichment (EE) paradigm has contributed to strengthen the importance of IGI development for the functional development of the visual cortex. EE is a combination of complex sensory-motor stimulation obtained by rearing large groups of animals in wide stimulating environments where a variety of objects are used to promote exploration, cognitive activity, social interaction and physical exercise (for a review, see Sale et al., 2009
). EE accelerates (Cancedda et al., 2004
; Sale et al., 2004
), and prevents dark rearing deleterious effects on, visual acuity development (Bartoletti et al., 2004
), promoting the maturation of IGI. In addition, Insuline-like Growth Factor-1 (IGF-1), a possible mediator of EE effects on visual cortex development, is also likely to act targeting IGI development (Ciucci et al., 2007
).
The best characterized contribution of IGI to developmental plasticity is that to the opening and closure of the CP for ocular dominance (OD) plasticity (Huang et al., 1999
; Hensch, 2005
; Morishita and Hensch, 2008
). If vision in one eye is defective in man or experimentally impaired by monocular deprivation (MD) during infancy in mammals, visual acuity of that eye does not develop normally (a condition called amblyopia), binocular vision is impaired, visual cortical neurons become strongly dominated by the non deprived eye (OD shift) and the proportion of binocular neurons greatly decreases (Hubel and Wiesel, 1970
; Fagiolini et al., 1994
). If the visual deprivation is removed early in development, the ensuing recovery is very good; on the contrary, recovery is very limited if defect removal is delayed to adulthood. Also MD effectiveness in modifying the OD of visual cortical neurons declines with age (Hubel and Wiesel, 1970
; Hensch, 2005
). Hensch et al. (1998)
have shown that inhibitory neurotransmission is necessary for the manifestation of OD plasticity in response to MD. In transgenic mice lacking the 65-kD isoform of the GABA-synthesizing enzyme GAD (GAD65), OD plasticity in response to MD is deficient. OD plasticity in these animals can be rescued if GABAergic transmission is enhanced in the visual cortex by means of benzodiazepines. This suggests that the development of GABAergic intracortical inhibition is an enabling factor for CP opening. Indeed, Fagiolini and Hensch (2000)
have shown that precocious enhancement of IGI by the early administration of diazepam to the visual cortex accelerates the opening of the CP (Figure 1
).
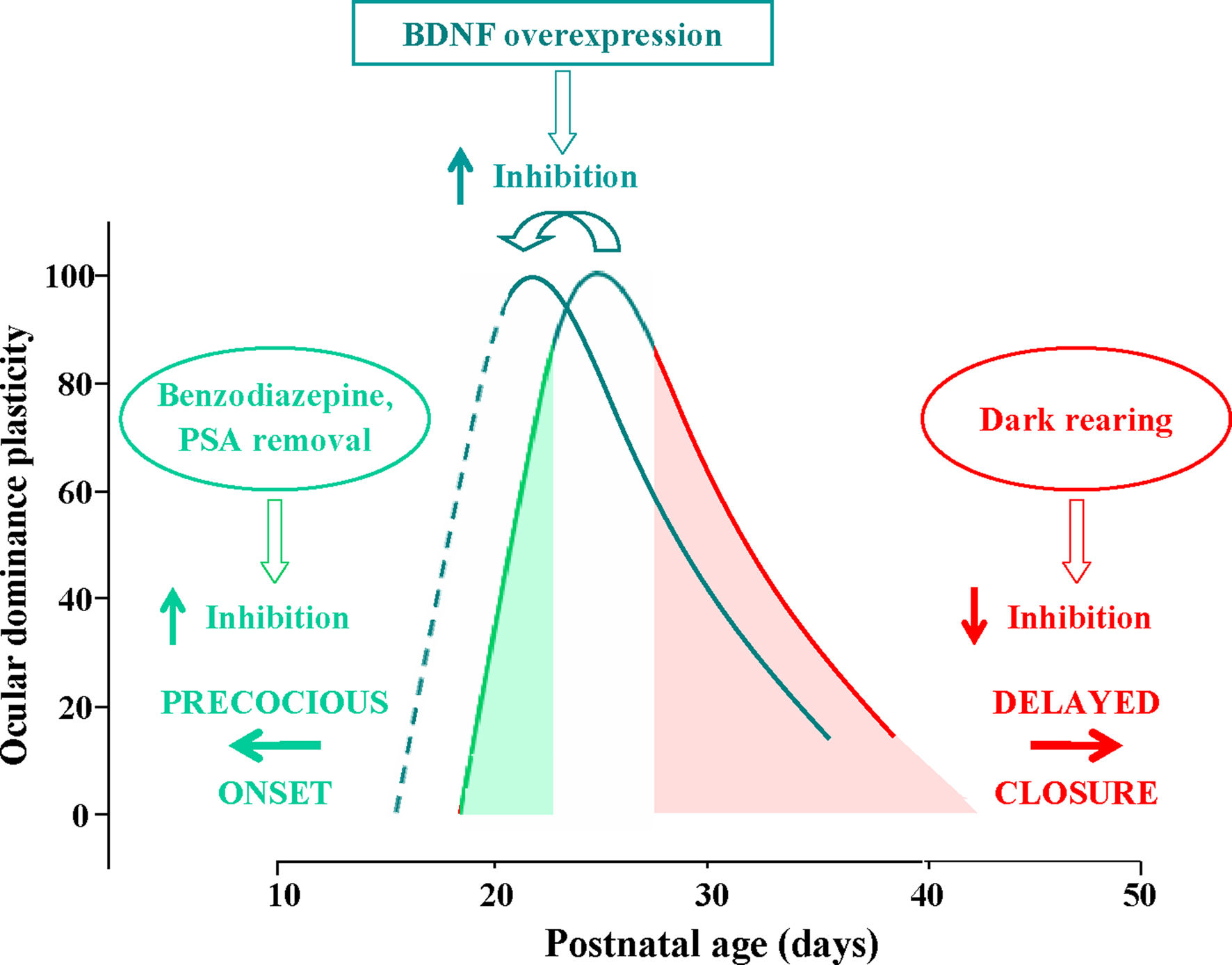
Figure 1. Time course of the critical period (CP) for ocular dominance (OD) plasticity in response to monocular deprivation in rodents; OD plasticity is normalized to the CP peak’s level. CP onset can be anticipated by increasing intracortical inhibition through benzodiazepine treatment or PSA (see text) removal (green arrow pointing left). Conversely, the end of the CP can be delayed by preventing the maturation of GABAergic inhibition through dark rearing from birth (red arrow pointing right). The effects of dark rearing can be counteracted by concomitant EE (see text). BDNF over-expression promotes a faster maturation of GABAergic interneurons, acting both on the onset and on the closure of the CP, thus shifting leftward the entire developmental plasticity curve.
A specific subset of inhibitory interneurons, the parvalbumin-positive large basket cells, seems to be crucial for the control of CP opening (Fagiolini et al., 2004
). Moreover, Di Cristo et al. (2007)
have shown that premature removal in the visual cortex of polysialic acid (PSA) presented by the neural cell adhesion molecule, results in a precocious maturation of perisomatic innervation by basket interneurons, enhanced inhibitory synaptic transmission, and an earlier onset of OD plasticity (Figure 1
).
Sugiyama et al. (2008)
have demonstrated that another factor might impinge on IGI development to promote CP onset. Otx2 homeoprotein is transported from the retina to the visual cortex in an activity-dependent way and it contributes to the regulation of the onset of visual cortex CP plasticity. Otx2controls maturation of cortical parvalbumin-positive GABAergic interneurons. In addition, it promotes the developmental organization of specific components of the extracellular matrix, the Chondroitin Sulphate Proteoglycans (CSPGs), into tightly woven nets surrounding cortical neurons called perineuronal nets (PNNs). These nets are preferentially found around parvalbumin-positive GABAergic interneurons and might contribute to the development of their functional properties and to the onset of CP, possibly also making Otx-2 available to them.
Development of inhibition seems also to determine the decline of visual cortex plasticity as development proceeds and CPs draw to their end. The results obtained in mice with precocious BDNF expression clearly show that an accelerated development of IGI results in an early closure of the CP for the effects of monocular deprivation (Hanover et al., 1999
; Huang et al., 1999
) (Figure 1
). Similarly, animals exposed to EE exhibit a precocious decline of synaptic plasticity in the visual cortex (Cancedda et al., 2004
) and EE counteracts the effects of dark rearing on the closure of the CP for OD plasticity, in good correlation with the preserved developmental formation of PNNs around GABAergic interneurons and of GAD65 puncta rings around the cell body of pyramidal cells (Bartoletti et al., 2004
). This is in accordance with the well known finding that IGI matures in correspondence with CP progression (see Hensch, 2005
) and that dark rearing, which delays CP closure, delays inhibition development (Figure 1
). They are also in agreement with the data by Fagiolini and Hensch (2000)
who observed that, employing long-term monocular deprivation as a probe for OD plasticity, the CP is prolonged into adulthood in GAD65 knock-out mice.
These findings suggest that IGI levels cross two thresholds during development, the first setting the point after which inhibition levels are enough to allow OD plasticity to be expressed (beginning of CP); as development proceeds, the inhibitory tone further increases and crosses a second threshold, after which inhibition drastically reduces the potential for plasticity (closure of CP) (Feldman, 2000
).
Recent studies revealed a previously unsuspected plasticity potential for the adult visual cortex, challenging the classic notion that OD plasticity or amblyopia recovery are possible only early in development during the CP. In this new work, the capability to reduce intracortical inhibition levels has emerged as critical for the restoration of plasticity.
The most direct demonstration that GABAergic inhibition actively restricts plasticity in the adult visual cortex derives from the very recent report that a pharmacological reduction of inhibition obtained by intracortical infusion of either mercaptopropionic acid (MPA, an inhibitor of GABA synthesis) or picrotoxin (PTX, a GABAA receptor antagonist), at doses which do not alter visual cortical neuron responsiveness, reactivates OD plasticity in response to MD in adult rats (Harauzov et al., 2010
) (Figure 2
). This is accompanied, in visual cortical slices of MD animals with reduced inhibition, by the reinstatement of white matter-induced LTP in the II-III layer, a form of plasticity normally restricted to the CP due to the maturation of IGI. These results indicate that in normal adult animals, where inhibitory processes have completed their development, a brief reduction of GABAergic inhibition is sufficient to reopen a window of plasticity in the visual cortex well after the normal closure of the CP. This supports the hypothesis that the development of inhibition contributes to CP closure. In the visual cortex of MD animals with reduced inhibition there is a decrease in the expression of CSPGs, which follows the reactivation of OD plasticity. The reduction in PNN density observed in MPA or PTX-treated animals after MD could be an indication that the reactivation of OD plasticity caused by reducing the inhibitory tone sets in motion endogenous mechanisms of extracellular matrix remodelling (Mataga et al., 2002
, 2004
; Oray et al., 2004
; Hensch, 2005
). It has to be underlined, however, that the OD shift induced by MD in adult animals with reduced intracortical inhibition (Harauzov et al., 2010
) is smaller than that induced during the CP. This suggests that other mechanisms which restrain the expression of OD plasticity are still in place. One of these factors could be the density of PNNs, which are known to develop in correlation with CP closure and have been shown to be non permissive for OD plasticity (Pizzorusso et al., 2002
, 2006
). The reduction in PNN density in MPA or PTX-treated animals is partial, therefore PNN density remains higher than during the peak of the critical period (Pizzorusso et al., 2002
). In addition, the presence of factors limiting chromatin remodelling (Putignano et al., 2007
), or signalling from extracellular factors, for example from myelin proteins on Nogo receptors (McGee et al., 2005
) as well as the characteristics of adhesion molecules (Hensch, 2005
), might contribute to partially restrain the full expression of long-term OD changes in response to MD in animals with reduced intracortical inhibition.
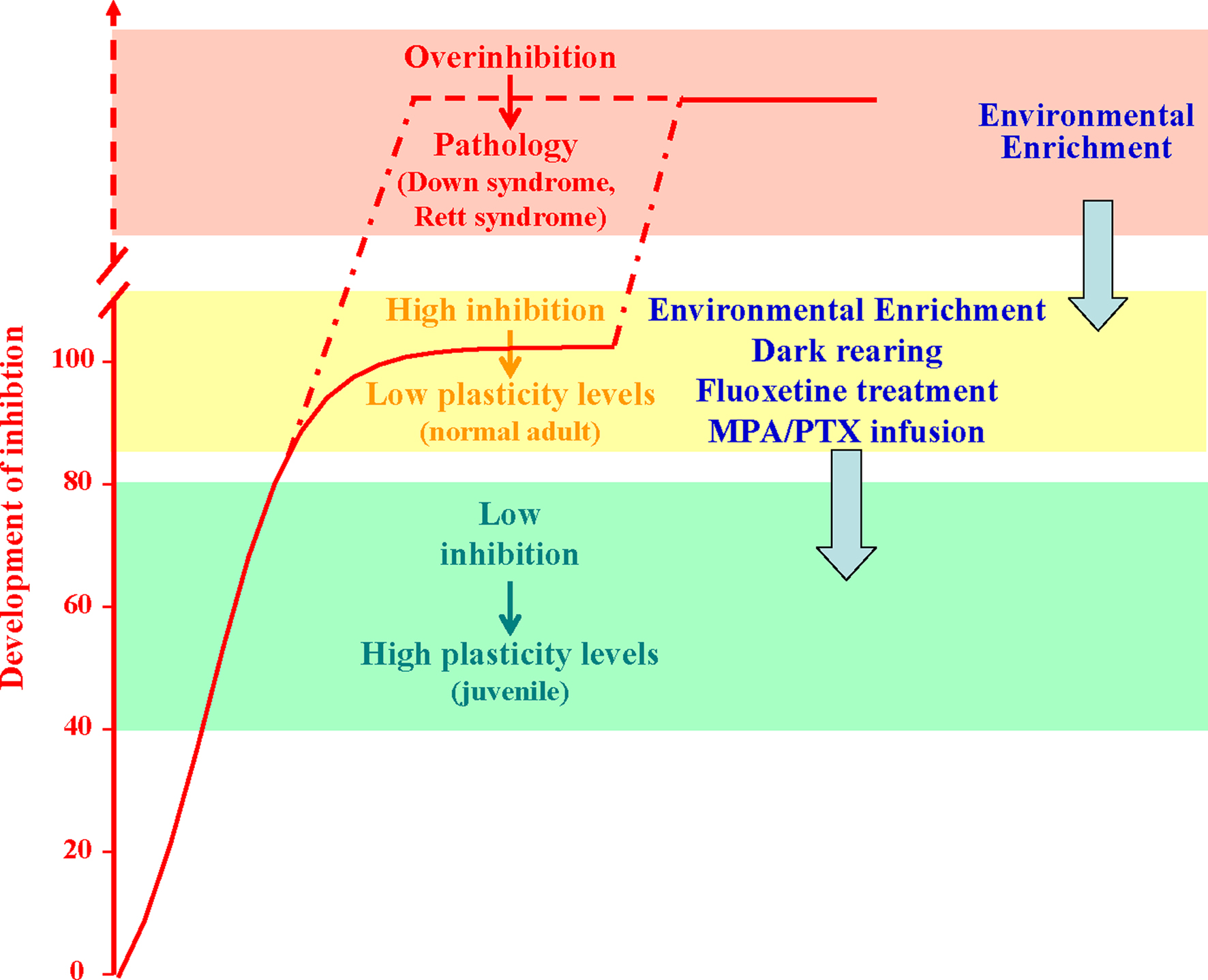
Figure 2. Developmental increase of brain GABAergic inhibition levels (normalized to the normal adult values; red curve) is paralleled by a progressive reduction of experience-dependent plasticity. Plasticity is high during early development (green block) and very low in the adult brain (yellow block). Anomalous increases in the strength of inhibitory neural circuits may lead to over-inhibition linked to permanent deficits in synaptic plasticity and neural development, like in the Rett syndrome and in the Down’s syndrome. Reducing GABAergic inhibition with pharmacological (blockers of GABA synthesis or GABA receptor antagonists, fluoxetine) or environmental (EE, and dark exposure) treatments can increase plasticity in the adult brain, enabling OD plasticity and favoring recovery from amblyopia. The capability of EE to reduce GABAergic inhibition makes this paradigm eligible for therapeutic applications also in the treatment of developmental intellectual disabilities.
In the attempt to manipulate the balance between excitation and inhibition using non invasive strategies eligible to be transferred to humans, we exploited the paradigm of EE. We found that a 3-weeks exposure of adult amblyopic rats to EE promotes a complete recovery of both visual acuity and OD (Figure 2
). This striking effect was associated with a threefold reduction in the basal levels of GABA detected by in vivo brain microdialysis in the visual cortex contralateral to the previously amblyopic eye (Sale et al., 2007
). A reduced cortical inhibition was also detectable at the synaptic plasticity level, since the visual cortical slices of EE rats displayed a full reinstatement of white matter-induced LTP. IGI reduction is crucial for the plasticity enhancement induced by EE, because the recovery of visual functions is completely prevented by cortical infusion of diazepam directly into the primary visual cortex during the EE period (Sale et al., 2007
). Consistent with the results of Harauzov et al. (2010)
, we also found a reduction in PNN density in the animals exposed to EE, a result that strengthens the notion of a possible link between the functional state of the extracellular matrix and the intracortical inhibitory tone. Interestingly, repetitive transcranial magnetic stimulation in humans, which increases cortical excitability, transiently improves contrast sensitivity in adult amblyopes, likely acting on the excitation/inhibition balance (Thompson et al., 2008
).
Recent studies reported that exposing adult rats to complete darkness, a treatment qualitatively very different from EE, can favor plasticity in the adult brain (He et al., 2006
, 2007
) (Figure 2
). There is indirect evidence that also in this case the enhanced visual cortical plasticity may be related to a reduced expression of GABAA receptors relative to AMPA receptors. This could cause a shift in the balance between inhibition and excitation towards levels more similar to those found in the immature cortex.
With the ambitious goal to search for possible enviromimetics, molecular factors that might be exploited to reproduce the beneficial effects elicited by EE, we came back to the classic observation that the neurotransmitter systems characterized by diffuse projections throughout the entire brain, i.e. the serotoninergic, noradrenergic and dopaminergic systems, are all set in motion by EE (see van Praag et al., 2000
). These neuromodulators have a great influence on plasticity processes in the adult brain, with a fundamental role in regulating the arousal state and attentional processes (see Gu, 2002
), which are important components of the animal response to the enriched experience. Therefore, it should be possible to mimic the EE effects on adult visual cortex plasticity by an artificial modulation of one or more of these transmitters. Widely prescribed drugs for the treatment of depression, the so-called selective serotonin reuptake inhibitors (SSRIs), act by enhancing extracellular serotonin and noradrenalin levels, even if the relationship between acute increases in these neurotrasmitters and the clinical antidepressant effect has remained unclear. We recently showed that chronic treatment with fluoxetine (Prozac), a SSRI used to treat depression, obsessive-compulsive disorder and panic attacks, induces a reinstatement of OD plasticity in response to MD and a full recovery from amblyopia in adult animals (Maya Vetencourt et al., 2008
) (Figure 2
). As found for EE rats, these functional effects are associated with a marked reduction of GABAergic inhibition and are completely prevented by cortical diazepam administration.
The crucial involvement of IGI in limiting adult cortical plasticity could have implications also for other forms of brain repair, emerging as a possible target for behavioural or pharmacological interventions following brain lesions. A change in inhibitory tone has indeed been found in perilesional regions in patients with stroke in the motor cortex; treatment-associated cortical reorganization, which was correlated to the recovery of motor function, was influenced by the distribution of inhibitory properties within the representation area prior to therapy (Liepert et al., 2006
).
There is increasing consensus on the concept that not only does brain inhibition control the closure of CPs and adult cortical plasticity, but that it may also be linked to the pathogenesis of developmental intellectual disabilities (see Fernandez and Garner, 2007
for a recent review). Anomalous increases in the strength of inhibitory neural circuits during brain maturation may lead to permanent deficits in synaptic plasticity and neural development (Figure 2
). This over-inhibition may be the consequence of environmental disturbances, such as prenatal protein malnutrition (see Morgane et al., 2002
) but its etiology is more frequently linked to genetic alterations, with the two paradigmatic examples of Rett syndrome (RS) and Down’s syndrome (DS).
RS is an X-linked, severe progressive disorder of CNS development which causes a complex neurological and neurobehavioural phenotype characterized by mild-to-moderate mental retardation and highly disabling dysfunctions in motor coordination skills (Hagberg et al., 1983
). The main mutations are found in the gene encoding the methyl-CpG-binding protein (MeCP2) (Amir et al., 1999
), a factor critically involved in the epigenetic regulation of the expression of genes essential for plasticity, such as BDNF (Chen et al., 2003
; Chang et al., 2006
). Electrophysiological studies from MeCP2 transgenic mice reported a reduced neuronal activity in cortical and hippocampal neurons, pointing to a shift in the balance between inhibition and excitation as a likely mechanism underlying the motor and cognitive phenotype (Dani et al., 2005
; Asaka et al., 2006
; Fernandez and Garner, 2007
). Given that persons with RS have an increased sensitivity to sedative drugs which act potentiating GABAergic inhibition (Tofil et al., 2006
), it is tempting to speculate that overinhibition may also be present in the brain of human RS patients. Both BDNF over-expression and EE are successful in rescuing or ameliorating the main deficits detectable at the behavioural and electrophysiological level inMeCP2mutant mice (Chang et al., 2006
; Kondo et al., 2008
; Lonetti et al., 2010
). At least for EE, this may directly result from a decrease in the GABAergic tone prompted by exposure to EE stimulation.
DS is caused by triplication of chromosome 21 (Chr21) as result of a misdivision in the sperm or the egg prior to conception, and it is the most diffused genetic cause of mental retardation. The most intensively studied mouse model of DS is the Ts65Dn line, that carries a triplication of a critical segment of Chr16, syntenic with human Chr21 (Gardiner et al., 2003
). Ts65dn mice recapitulate all main hallmarks of the DS phenotype (Richtsmeier et al., 2002
; Hunter et al., 2003
; Driscoll et al., 2004
; Wenger et al., 2004
). There is increasing evidence that the cognitive deficits displayed by Ts65Dn mice can be attributable to excessive hippocampal inhibition. Accordingly, Fernandez et al. (2007)
have shown that the spatial learning disabilities and the failure to induce LTP in the hippocampus observed in Ts65Dn mice can be rescued by administration of non-competitive antagonists of GABAA receptors. It remains unclear whether it is possible to induce a reduction of GABAergic inhibition in Ts65Dn mice through a non- pharmacological approach. The capability of EE to reduce GABAergic inhibition makes this paradigm eligible for therapeutic applications in the treatment of DS (e.g., Martinez-Cué et al., 2002
; Dierssen et al., 2003
) (Figure 2
).
In conclusion, intracortical inhibition is a crucial determinant of cortical plasticity, both during development and in the adult. In typical development, it contributes to the maturation of the cortex and of cortex-dependent functions; moreover it might play a role in the intellectual disabilities found in atypical development. In the adult, it limits remodeling of neural connections, tightly controlling neural plasticity and exhibiting spontaneous changes under pathological conditions. Harnessing these changes might favor plasticity and promote functional recovery.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Italian Ministry of the University and Scientific and Technological Research COFIN.