 Songfen Wu1,2,3
Songfen Wu1,2,3 Kuan Li
Kuan Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Behav. Neurosci. , 12 February 2025
Sec. Learning and Memory
Volume 19 - 2025 | https://doi.org/10.3389/fnbeh.2025.1529522
Introduction: NRBF2, a component of autophagy-associated PIK3C3/VPS34-containing phosphatidylinositol 3-kinase complex, plays a crucial role in learning and memory processes, yet its specific impact on memory and the underlying molecular mechanisms remains unclear.
Methods: Here, we utilized NRBF2 knockout mice to examine its influence on the time course of fear memory. Employing quantitative PCR, Western blot analysis, behavioral tests, and electrophysiology, we investigated the mechanisms through which NRBF2 affects memory processing.
Results: We observed an increase in Nrbf2 mRNA levels at 6 and 12 h, and protein levels at 6 h post fear conditioning. Depletion of NRBF2 impaired memory acquisition, short-term, and long-term memory without causing any anxiety-like behavior. Interestingly, inhibition of Vps34 and autophagy by SAR405 disrupted fear memory consolidation, while leaving memory acquisition, short-term memory, and long-term potentiation (LTP) unaffected. Our results suggested that NRBF2 deletion impaired memory acquisition through an autophagy-independent pathway and provided novel insights into the role of NRBF2 in the central nervous system.
Discussion: This study offer new insights into the role of NRBF2 and highlight the potential of targeting NRBF2 as a therapeutic strategy for addressing cognitive deficits associated with various disorders.
Autophagy is an evolutionarily conserved catabolic process that encompasses the initiation, nucleation, elongation, maturation, fusion, and degradation stages (Li et al., 2020). This process plays a pivotal role in neurodevelopment and neurotransmitter release (Kuijpers and Haucke, 2021), contributing to the maintenance of neuronal integrity and synaptic plasticity (Nikoletopoulou et al., 2017). Impairments in autophagy are associated with memory deficits in aging and Alzheimer’s disease (AD) (He and Klionsky, 2009; Nixon, 2013; Yang et al., 2017). Nuclear Receptor Binding Factor 2 (NRBF2) is initially identified as a coregulator that interacts with nuclear receptors to modulate transcriptional activity (Yasumo et al., 2000; Flores et al., 2004). Recent studies have revealed NRBF2 as the fifth subunit of the active PIK3C3/VPS34-containing class III phosphatidylinositol 3-kinase (PtdIns3K) complex, a critical component of autophagy (Young et al., 2016; Ohashi, 2021). Accumulating evidence has suggested that NRBF2 is crucial for autophagosome formation and maturation. Through its MIT domain, NRBF2 directly interacts with Atg14L, enhancing Vps34 kinase activity and facilitating autophagy initiation (Lu et al., 2014). Moreover, NRBF2 acts as a RAB7 effector, supporting autophagosome maturation (Cai et al., 2021). Both NRBF2 and autophagy have been implicated in the regulation of learning and memory processes (Lachance et al., 2019; Hylin et al., 2018; Li et al., 2019).
Unique molecular mechanisms are known to underlie different stages of memory formation, specifically the acquisition, consolidation, and reconsolidation (Johansen et al., 2011). Learning and acquisition are the basis of memory, while consolidation is a process of the conversion of labile short-term memory (STM) into stable long-term memory (LTM) (McGaugh, 2000; Nader and Hardt, 2009; Bush et al., 2010). Synaptic plasticity is fundamental to learning and memory, and the abnormality of its two different forms, long-term potentiation (LTP) and long-term depression (LTD), is regarded as cellular mechanisms underlying memory deficits (Bin Ibrahim et al., 2022). Our previous study found that inhibiting autophagy in basolateral amygdala, either by targeting the Vps34 or Atg5, resulted in increased inhibitory synaptic transmission and impaired LTM, while preserving memory acquisition and STM integrity (Li et al., 2019). Two other studies have also demonstrated that autophagy inhibition is essential for LTD induction (Shehata et al., 2012; Pan et al., 2021). The involvement of NRBF2 in memory has also been previously documented. Nrbf2 knockout (NRBF2-KO) mice exhibited memory deficits and decreased autophagy in hippocampus, accompanied by decreased LTP (Lachance et al., 2019), whereas Nrbf2 conditional knockout mice in the nervous system showed impaired spatial memory with minimal autophagy deficits, indicating an autophagy-independent pathway (Ouyang et al., 2020). These findings have led to the intriguing hypothesis that NRBF2 may influence memory formation and LTP through non-autophagy pathways. However, the precise role of NRBF2 in each memory stage and its association with autophagy in memory regulation remains to be fully elucidated.
To investigate whether NRBF2 affects the acquisition and consolidation of fear memories, particularly through the autophagy pathway, we employed fear conditioning tests and field potential recording techniques to explore the involvement of NRBF2 in the memory process. Additionally, SAR405, a Vps34 inhibitor, was used to elucidate the underlying association with autophagy. Our results showed that depletion of NRBF2 impaired memory acquisition and subsequent STM and LTM, while inhibition of autophagy by SAR405 disrupted fear memory consolidation, without altering memory acquisition, short-term memory, and LTP. These results suggest that NRBF2 deletion impairs memory acquisition through an autophagy-independent pathway and provide novel insights into the role of NRBF2 in the central nervous system.
NRBF2–/– mice utilized in this study were acquired from Prof. Jia-Hong Lu (University of Macau, Taipa, Macau SAR China). NRBF2 heterozygous mutant mice were bred with C57BL/6 J mice and interbred to generate the NRBF2–/– mice. All animals were housed under a 12/12 h light/dark cycle with lights on at 7:00 AM, and maintained at a constant temperature (22 ± 2 °C) and humidity of 50 ± 10%, with ad libitum access to food and water. Five mice were acclimated per cage, and male mice aged 8–11 weeks were used for the experiment. All experimental procedures were approved by the Animal Welfare Committee of Huazhong University of Science and Technology and the Ethics Committee at the Shenzhen People’s Hospital, in accordance with the ARRIVE guidelines 2.0 (Percie du Sert et al., 2020). All methods were carried out following with the relevant guidelines and regulations. At the end of the experiments, the mice used for behavioral testing were euthanized in a CO2 chamber.
Total RNA was extracted from hippocampal homogenates of 8–10-week-old wild-type littermate mice using TRIzol® Reagent (Invitrogen, Cat# 15596018CN) following the manufacturer’s protocol. A quantity of 1 μg of RNA was then reverse transcription to generate cDNA libraries was performed using the cDNA synthesis kit (Vazyme, Cat# R212-02) according to the provided protocol. Quantitative PCR (qPCR) was conducted with the Universal SYBR qPCR Master Mix (Vazyme, Cat# Q511-02). Gene expression analysis was carried out using the 2–Δ Δ CT method. The primers used for transcript analysis were as follows: Nrbf2 - F: AAG GAC CCC TCA ACC TTG CT, Nrbf2 - R: CAG TTC CAG TGA TAA GTG AGC C; GAPDH - F: AAC GAC CCC TTC ATT GAC, GAPDH - R: TCC ACG ACA TAC TCA GCA C.
The procedures were processed according to our previous protocol with minor modifications (Li et al., 2019; Shi et al., 2018). In brief, hippocampal tissue from each mouse was homogenized in RIPA buffer (MCE, HY-K1001), supplemented with 1 mM PMSF, 1 × protease inhibitor cocktail and 1 × phosphatase inhibitor cocktail (Sigma). Samples were centrifuged at 12,000 g for 20 min at 4°C. Lysates were heated at 100°C with 6× loading buffer (Beyotime) for 10 min and then stored at −20°C until next step. A total of 20 μg protein per sample was loaded on 10% SDS-PAGE gel, and then transferred onto PVDF membranes (Millipore) and blocked in 5% bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 2 h at room temperature. The transferred membranes were incubated overnight at 4°C with primary antibodies: NRBF2 rabbit mAb (1:1000, Proteintech, 24858-1-AP) and GAPDH mouse mAb (1:60000, Invitrogen, AM4300). After three washes with TBST, the membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2000) in TBST with 1% BSA for 2 h at room temperature. Following additional washes, the membranes were reacted with an enhanced chemiluminescence reagent (Epizyme). Images were scanned and captured with Micro Chemi (Bio-rad, ChemiDoc XRS+) and the optical densities of the detected bands were quantified using ImageJ software (NIH).
The open field test (OFT) is commonly used to assess spontaneous locomotor activity and exploratory behavior in response to novel environments in rodents (Sun et al., 2024). The OFT was conducted as described in a previous study with minor modifications (Shi et al., 2018; Shoji and Miyakawa, 2021). Before the experiment, the mice were habituated to the investigator’s handling for 180 s on three consecutive days in the laboratory where the mice were subjected to the OFT. The mice were placed in a square open arena (40 cm × 40 cm × 40 cm), and allowed to explore the area freely for 20 min. Their activity was recorded and analyzed using the Tru Scan Activity System (Coulbourn Instruments). The central area was illuminated to 100 lx using LED lights mounted on the ceiling of each apparatus. The arena surface was cleaned with 70% ethanol after each trial.
The elevated plus maze (EPM) apparatus consists of a central area (5 cm × 5 cm), two open arms (25 cm × 5 cm), and two enclosed arms (25 cm × 5 cm × 15 cm). The maze is positioned 50 cm above the ground in a room. The EPM procedure was performed as previously described in a study, with minor modifications (Painsipp et al., 2008). The light intensity at the central quadrangle was 70 lux, on the open arms 80 lux and in the closed arms 40 lux. The mice were placed in the central square, facing one of the open arms, and were allowed 5 min to explore the maze. The time spent by each mouse in each arm and the number of arm entries during the 5-min exploration were recorded using the DigBehv Animal Behavior Analysis System.
The light-dark (LD) box apparatus (45 cm × 27 cm × 27 cm) consisted of two compartments: a light compartment and a dark compartment, separated by an opaque plexiglass partition with a 5 cm diameter hole. This hole allowed the mouse to access the surrounding arena. The LD test was performed following our previously established protocol, with minor adjustments (Shi et al., 2018). The light compartment was illuminated with a strong light source (400 lux). The mice were individually placed in the center of the light compartment, facing away from the hole, and were allowed to explore the apparatus freely for 10 min. The time spent by the mice in the light and dark compartments was recorded using the DigBehv Animal Behavior Analysis System.
The fear conditioning test was performed according to our previous protocol with minor adjustments (Li et al., 2019; Wilensky et al., 2006). All the experiments were conducted in two contexts: context A and context B. The conditioning chamber (context A, 32 cm × 26 cm × 30 cm) was sound-attenuating, with foot shocks administered through a stainless grid floor. The test chamber (context B, 32 cm × 26 cm × 30 cm) was brightly illuminated and featured a flat black plastic floor. The apparatus was illuminated by a light source with an intensity of 20 lux. The chambers were cleaned with 70% ethanol before each session. Before the experiment, the mice were habituated to the investigator’s handling for 180s on three consecutive days in the laboratory, where they would later undergo the fear conditioning test. On day 1 (habituation), the mice were introduced to context A and allowed to freely explore for 5 min. On the training day (day 2), a tone conditioned stimulus (CS, 80 dB) was presented for 29 s that co-terminated with a single electric foot shock as an unconditioned stimulus (US, 0.7 mA, 1 s). This pairing was repeated five times, with a 60-s interval between CS-US presentations. The mice were returned to their home cages 30 s after the last foot shock. Fear memory test was performed at 3 h and 24 h after fear conditioning. During the memory test in context B, the mice were exposed to five tones (30 s each) with a 30-s interval. Fear memory was assessed by quantifying the percentage of freezing time during the test periods.
For SAR405 treatment, SAR405 (1 μM, ApexBio) was intracranial injected to the hippocampus. The mice were then subjected to the habituation procedure in conditioning chamber, and fear conditioning training was performed 24 hours after the injection, as described above.
C57BL/6 J mice were anesthetized with pentobarbital sodium (60 mg/kg, i.p.) and then mounted in a stereotaxic apparatus (RWD Life Science, China). 22-gauge stainless steel guide cannulas were bilaterally implanted dorsal to the CA1 region (AP: −1.7, ML: ± 1.5, DV: −1.8). They were then given 1 week to recover from surgery. When intracranial injection was performed before fear conditioning, a 33-gauge injection cannula was used to replace the inner sealing wire and protruded 1 mm beyond the guide cannula. Drugs were infused into the hippocampus at a rate of 0.5 μL/min with a total volume of 1.0 μL/side. SAR405 (1 μM, ApexBio) was used in the intra-hippocampus injections.
Field excitatory postsynaptic potentials (fEPSPs) were recorded using the field potential recording technique, following our previous protocol with slight modifications (Wang et al., 2015; Wu et al., 2022). In brief, the brains of C57BL/6J mice, which did not undergo any behavioral experiments, were sectioned into 400 μm-thick coronal slices containing the hippocampus using a microslicer (Leica VT1000 S; Leica Biosystems, Germany). The slices were incubated in artificial cerebrospinal fluid (ACSF) for at least 1.5 h at 27 °C. Subsequently, individual slice was transferred to a perfusion-type recording chamber and continuously superfused with ACSF pre-gassed with 95% O2/5% CO2 using a constant-current pump (HL-2, Shanghai JingDa Biochemical Instrument). A bipolar electrode was positioned in the Schaffer collaterals, and fEPSPs were recorded in the CA1 stratum radiatum layer using a glass micropipette filled with 3 M NaCl (2–5 MΩ, pulled from borosilicate capillaries). The recordings were collected and analyzed using a multi-channel physiological signal acquisition and processing system (RM6240BD, Chengyi, China). The stimulation intensity was gradually increased, and the corresponding fEPSP amplitudes were recorded. The stimulation intensity was then adjusted to produce a basal fEPSP amplitude corresponding to 1/3 to 1/2 of the maximum fEPSP amplitude, with a stimulation frequency of 0.033 Hz and an inter-stimulus interval of 30 s. After recording stable fEPSPs for at least 15 min, three trains of high-frequency stimulation (HFS) were applied to induce long-term potentiation (LTP). Each train consisted of 100 pulses delivered at 100 Hz, with a 30-s interval between trains. The stimulation intensity remained constant throughout the LTP recording. The initial slopes of fEPSPs recorded prior to HFS were measured and used as the baseline, with subsequent responses expressed as a percentage of this baseline level. For paired-pulse ratio (PPR) recordings, a second stimulus was delivered following the first with intervals of 50 ms and 90 ms. The PPR was calculated as the ratio of the second fEPSP amplitude to the first. For pharmacological experiments, SAR405 (1 μM) or a vehicle was added to the perfusing ACSF to assess its effects on LTP and PPR.
All data in this study were analyzed using GraphPad Prism version 8.0 (GraphPad Software). All experiments were biologically replicated at least three times. Dots in the figure represented single mice or independent experiments. The sample sizes were described in the relevant figure legends. Comparisons between two groups were evaluated using an unpaired Student’s t-test. For multiple group comparisons, one-way analysis of variance (ANOVA) followed by Tukey’s test was applied. Fear conditioning training results were assessed using repeated two-way ANOVA with Bonferroni’s post-hoc test. Statistical tests were shown in each figure’s legend. Data were considered as statistically significant when P-values were <0.05 (*), <0.01(**), <0.001 (***) or <0.0001 (****). All data were shown as mean ± SEM.
To map the time window of Nrbf2 mRNA and protein levels after fear conditioning, qPCR and WB were employed to assess NRBF2 levels in whole hippocampal tissue from wild-type mice at 1 h, 3 h, 6 h, 12 h and 24 h after fear conditioning, as well as the naïve group, which did not undergo fear conditioning (Figure 1A). The results showed that compared to the naïve group, the level of Nrbf2 mRNA was significantly elevated at 6 h and 12 h after training (Figure 1B) (F5,30 = 3.721, p = 0.009), while the NRBF2 protein level showed a significant increase at 6 h after training (Figures 1C, D) (F5,54 = 2.451, p = 0.045), indicating the involvement of NRBF2 in fear memory formation.
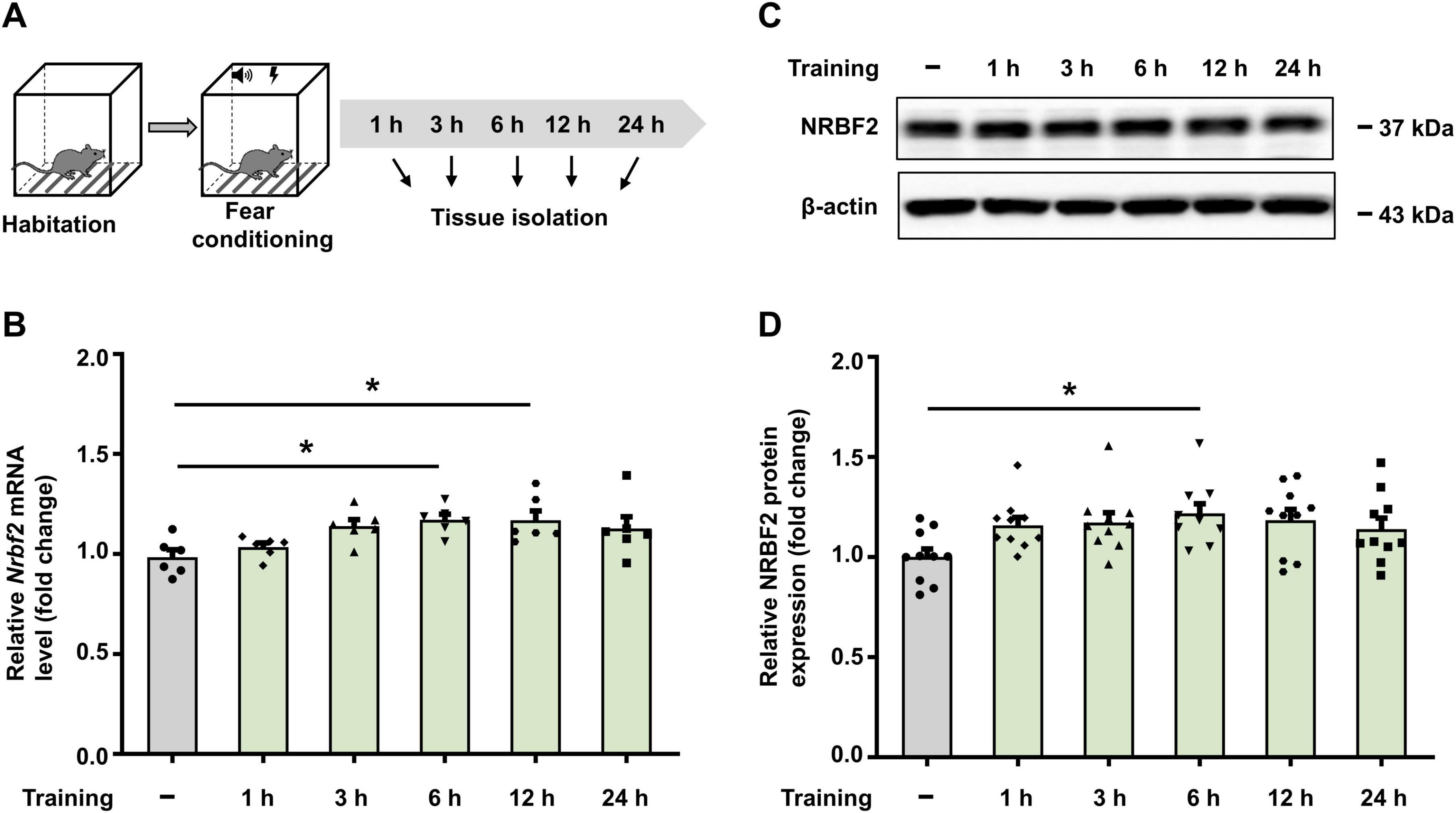
Figure 1. Fear conditioning increases Nrbf2 mRNA and protein levels. (A) Schematic of experimental design for hippocampus tissue extraction. (B) Quantitative PCR results showing the Nrbf2 mRNA level in the hippocampus at 1 h, 3 h, 6 h, 12 h and 24 h post-fear conditioning (n = 6 mice/group, one-way ANOVA with Tukey’s test, F5,30 = 3.721, p = 0.009; post hoc.
To substantiate the role of NRBF2 in learning and memory, we utilized NRBF2-KO mice (Yang et al., 2017), which were confirmed by complete elimination of NRBF2 protein in the hippocampus through WB analysis (Figures 2A, B) (t10 = 11.07, ****p < 0.0001). Subsequently, we investigated the potential influence of NRBF2 depletion on locomotor and anxiety-like behaviors by assessing behavioral changes in NRBF2-KO and wild-type (WT) mice using an open field test (OFT), elevated plus maze (EPM) test, and light-dark (LD) box test. Our results showed no significant alterations in exploratory behaviors, a key indicator of anxiety-like behaviors, as evidenced by the comparable the percentage of time spent in central area of the OFT (Figures 2C, D) (t17 = 0.685, p = 0.503), the time spent in the open arms (Figure 2F) (t17 = 0.088, p = 0.931) and entries into the open arms (Figure 2G) (t17 = 1.005, p = 0.329) of the EPM, as well as the percentage of time spent in the light compartment of the LD box (Figure 2H) (t17 = 0.708, p = 0.488) between NRBF2-KO and WT mice. Furthermore, the total distance traveled by the NRBF2-KO mice in the OFT was not significantly different from that of their control counterparts (Figure 2E) (t17 = 0.210, p = 0.836). These data show that NRBF2 depletion does not affect motor function and anxiety-like behavior.
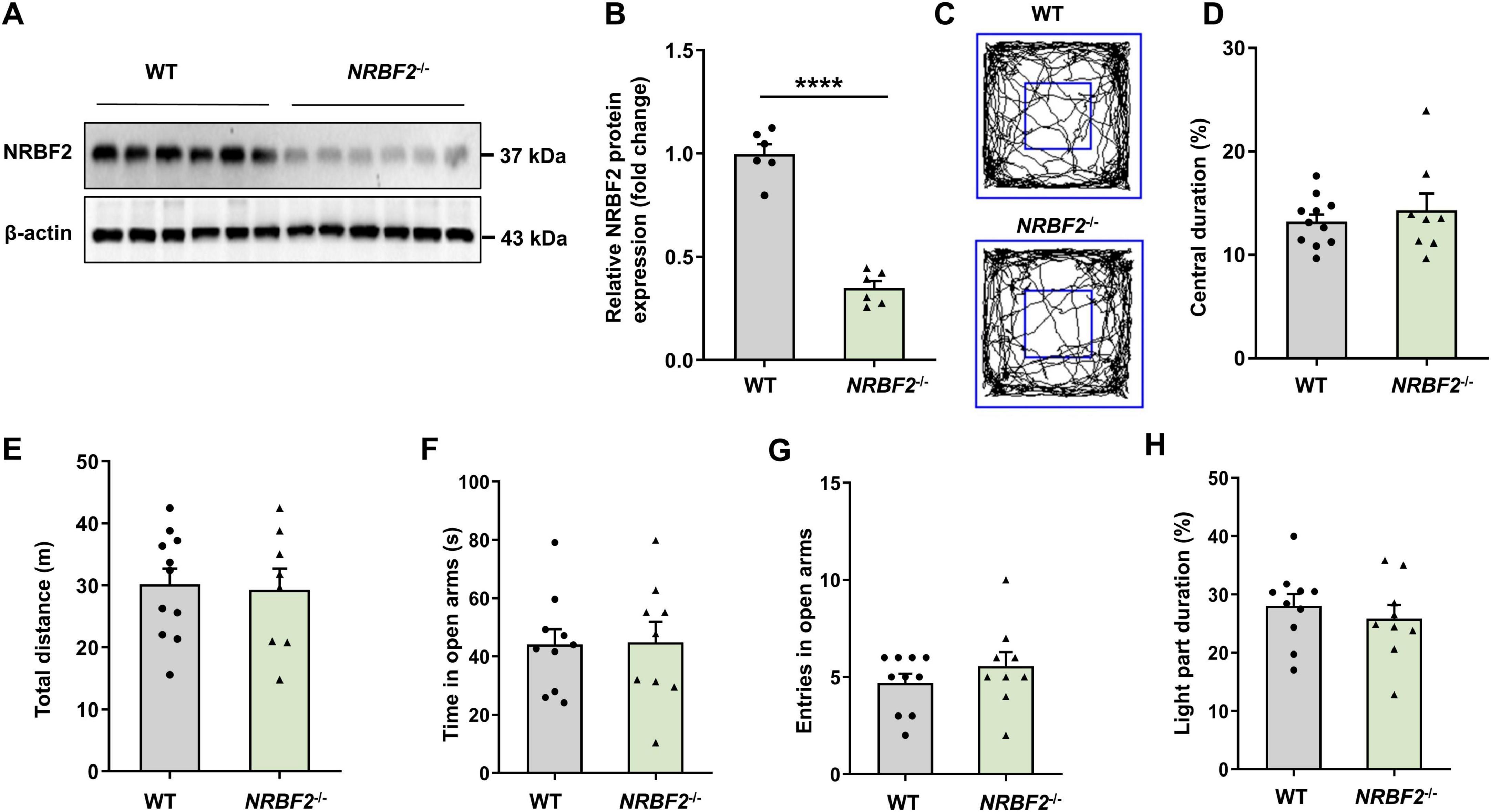
Figure 2. NRBF2-KO has no effect on locomotion and anxiety-like behavior. (A,B) Representative WB images (A) and quantification data (B) assess expression of NRBF2 in the hippocampus of mice (n = 6 mice/group, unpaired t-test, t10 = 11.07, ****p < 0.0001). (C) Representative traces showing the movement of NRBF2-KO and WT mice in the open field test. (D,E) Time spent in center zone (D) and total distance traveled (E) in the open field test (n = 11 mice in WT and 8 in NRBF2-KO group; (D) unpaired t-test, t17 = 0.685, p = 0.503; (E) unpaired t-test, t17 = 0.210, p = 0.836). (F,G) Time spent (F) and entries (G) in the open arms of the elevated plus maze test (n = 10 mice in WT and 9 in NRBF2-KO group; (F) unpaired t-test, t17 = 0.088, p = 0.931; (G) unpaired t-test, t17 = 1.005, p = 0.329). (H) The percentage of time spent in the light compartment of the light-dark box test (n = 10 mice in WT and 9 in NRBF2-KO group; unpaired t-test, t17 = 0.708, p = 0.488). Data were shown as mean ± SEM.
In light of the observed elevation in NRBF2 levels following fear conditioning, we conducted fear conditioning tests to assess the influence of NRBF2 depletion on fear memory, quantifying freezing behavior percentage (Figure 3A). Notably, during fear training, NRBF2-KO mice displayed a significantly lower freezing percentage compared to WT mice, indicating impaired learning (Figure 3B) (F1,19 = 11.54, p = 0.003). Furthermore, NRBF2-KO mice exhibited reduced freezing behavior at both short-term memory (STM, 3 h) (Figure 3C) (t19 = 2.550, *p = 0.02) and long-term memory (LTM, 24 h) intervals (Figure 3D) (t19 = 3.276, **p = 0.004), suggesting a deficit in memory acquisition affecting both STM and LTM (Bush et al., 2010; Makkar et al., 2010). Our findings underscored that NRBF2 deletion impaired memory acquisition, along with subsequent STM and LTM. Comparable performance in the OFT, EPM and LD tests excluded the possibility that alterations in freezing behaviors stemmed from variations in locomotor activity or anxiety-related behaviors.
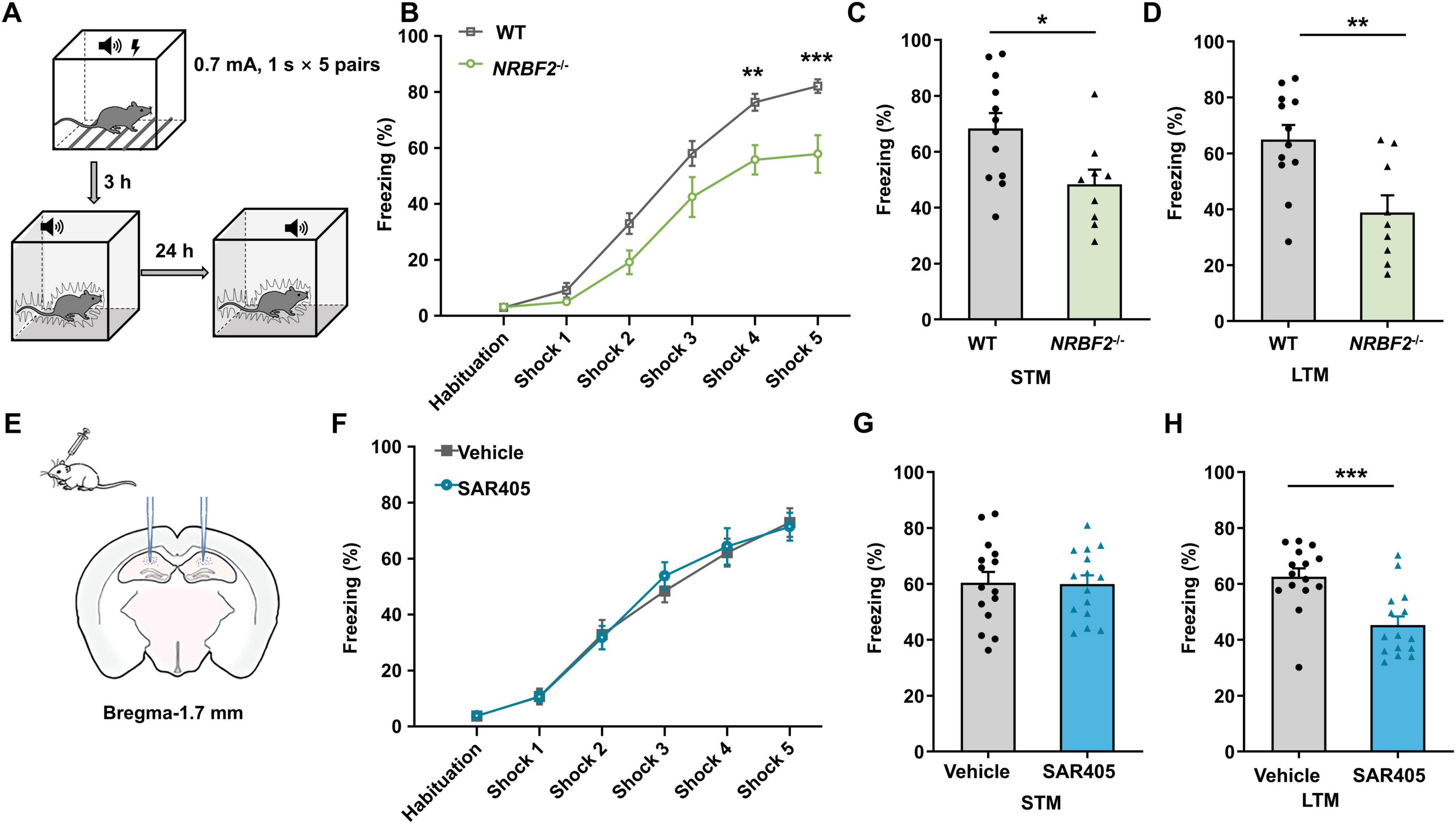
Figure 3. Effects of NRBF2 Deletion and SAR405 treatment on each of the fear memory stages. (A) Schematics of the procedure of fear conditioning. (B) The freezing curves during the training session showed the memory acquisition process in NRBF2-KO mice (n = 12 mice in WT and 9 in NRBF2-KO group, repeated two-way ANOVA, F1,19 = 11.05, p = 0.004; post hoc.
It’s well-recognized that NRBF2 plays a pivotal role as a component of the Vps34 complex and regulates complex kinase activity in autophagy induction (Ohashi, 2021). Our previous study (Li et al., 2019) demonstrated that Vps34 inhibition by SAR405 in the amygdala impairs memory consolidation, so we further investigated the impact of Vps34 inhibition in the hippocampus on learning and memory. This investigation aimed to elucidate whether NRBF2 deletion yields similar effects to Vps34 inhibition. SAR405, a selective inhibitor of Vps34 commonly used for autophagy inhibition (Schlütermann et al., 2018; An and Harper, 2018; Ronan et al., 2014), was locally delivered to the bilateral hippocampus before fear conditioning (Figure 3E). Our findings revealed that SAR405 treatment impaired freezing behaviors at 24 h after training (t28 = 4.027, ***p < 0.001), with no differences observed during fear training (F1,28 = 0.044, p = 0.835) and in STM (t28 = 0.097, p = 0.932) (Figures 3F–H), Thus, our results suggested that SAR405 pretreatment specifically attenuated fear memory consolidation while leaving memory acquisition unaffected.
Enhanced synaptic plasticity in the hippocampus constitutes the fundamental cellular mechanism for spatial learning and memory, and limiting LTP expression has been shown to result in a deficit in fear acquisition, which subsequently translates into disrupted fear memory (Nabavi et al., 2014; Takeuchi et al., 2014). A recent study has reported a reduced maintenance of LTP in NRBF2-KO animals compared to WT (Lachance et al., 2019), consistent with our findings that NRBF2 deletion impaired memory acquisition. However, it remains unclear whether these deficits in memory acquisition and LTP are mediated through the Vps34 complex-induced autophagy mechanism. To address this question, we investigated the effects of SAR405 on synaptic transmission and LTP. Electrophysiological recordings were conducted to measure the field excitatory postsynaptic potential (fEPSP) in the CA3-CA1 region, and high-frequency stimulation (HFS) was employed to induce LTP. Our results, as depicted in Figures 4A–C, revealed that SAR405 treatment of the slices did not affect baseline fEPSP or HFS-induced LTP compared to the vehicle group. Furthermore, to investigate whether SAR405 affects presynaptic mechanisms, we measured the paired-pulse ratio (PPR), which is an indicator of presynaptic neurotransmitter release. We observed no significant change in the PPR ratio (Figures 4D, E) (F1,6 = 0.453, p = 0.526), indicating that SAR405 did not affect the probability of releasing vesicles at the presynaptic terminal. Taken together, these data suggest that the inhibition of Vps34 with SAR405 is not necessary for excitatory synaptic transmission and LTP.
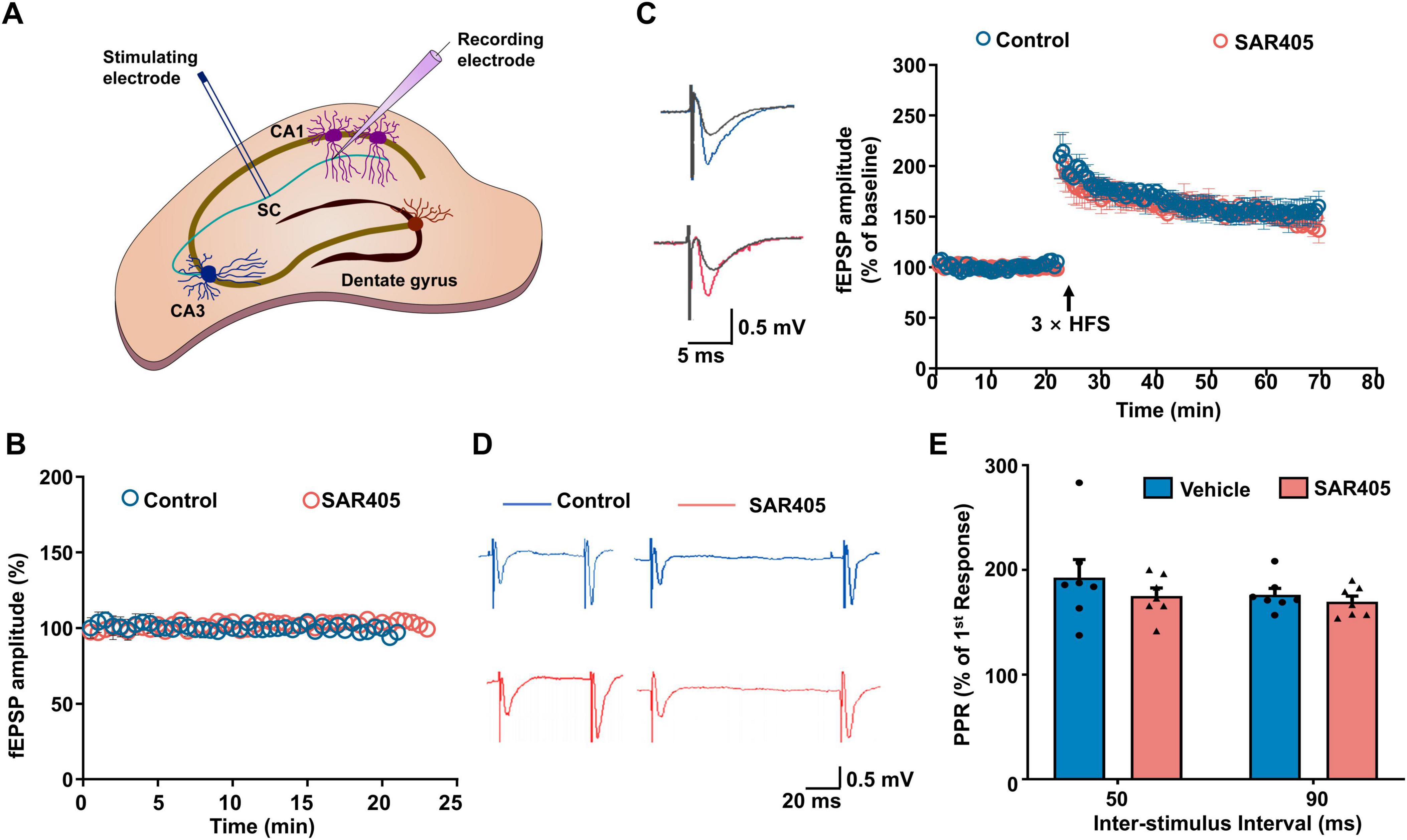
Figure 4. SAR405 does not affect synaptic plasticity in the hippocampus. (A) Schematic diagram of the field excitatory postsynaptic potential (fEPSP) in Schaffer collateral-CA1 pyramidal cells. (B) Baseline fEPSP slope was recorded after slices were treated with SAR405 or vehicle (nvehicle = 4 slices, 4 mice, nSAR405 = 4 slices, 4 mice). (C) The baseline and LTP recording (nvehicle = 6 slices, 5 mice, nSAR405 = 6 slices, 4 mice). (D) Representative traces of paired-pulse stimulation, which were evoked with inter-stimulus intervals of 50 ms and 90 ms in SAR405 treated and vehicle mice. (E) PPR was not significantly different in SAR405 treated compared with vehicle mice (nvehicle = 7 slices, 4 mice, nSAR405 = 7 slices, 4 mice, repeat two-way ANOVA, F1,6 = 0.453, p = 0.526). Result is presented as mean ± SEM.
The present study demonstrates that the levels of Nrbf2 mRNA and protein increase following fear conditioning, and depletion of NRBF2 impairs fear memory acquisition as well as subsequent STM and LTM. Although NRBF2 is a key component of the Vps34 complex involved in autophagy induction, pharmacological inhibition of Vps34 by SAR405 selectively impairs memory consolidation without affecting memory acquisition or LTP (Figure 5). Our study provides evidence that NRBF2 influences memory acquisition in a non-autophagy dependent manner, while the induction of autophagy by the Vps34 complex is necessary for memory consolidation.
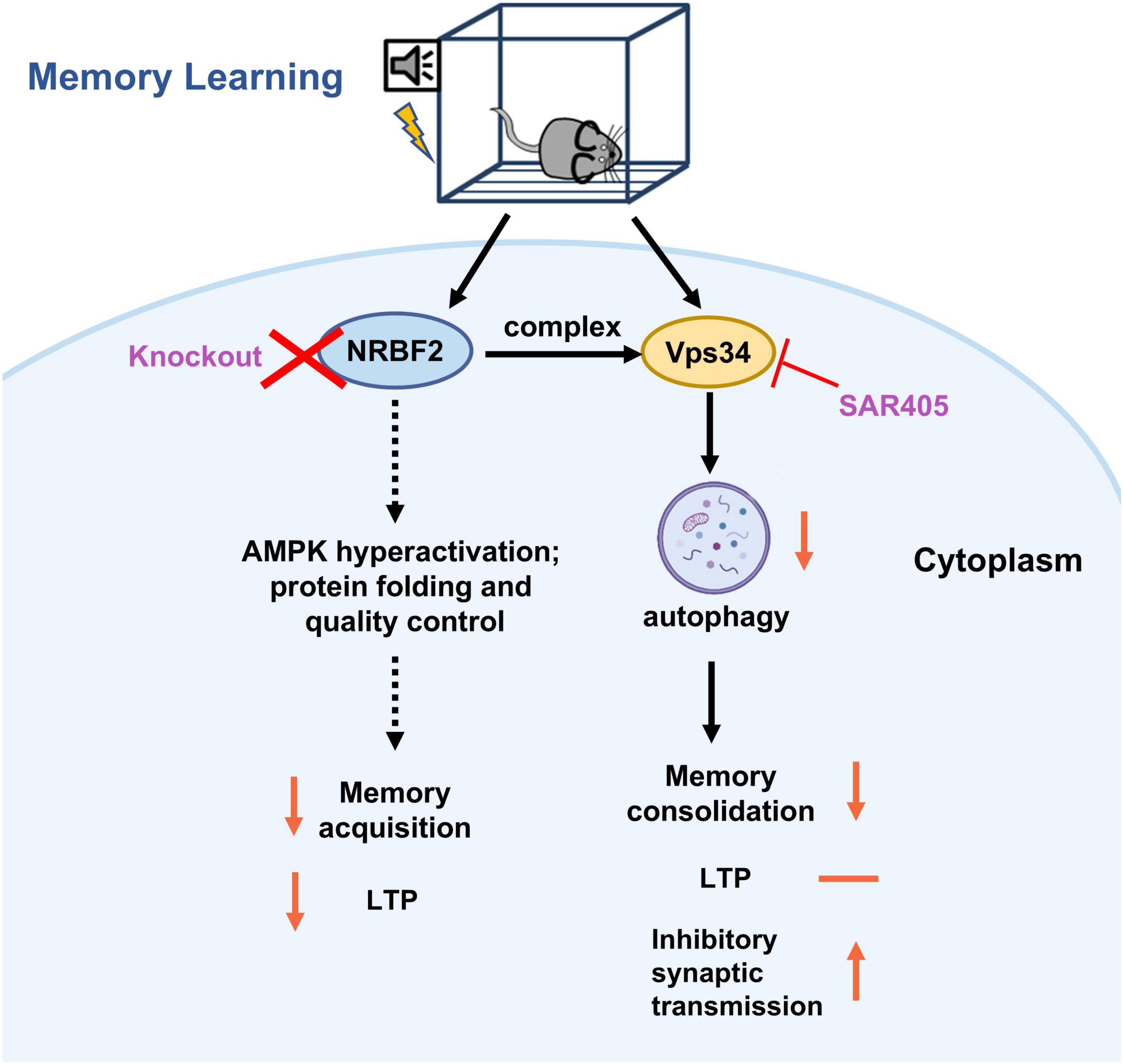
Figure 5. The schematic representation illustrates the effects and underlying mechanisms of NRBF2 on the memory acquisition process. During the learning and memory process, knockout of NRBF2, a component of the Vps34 complex, disrupts fear memory acquisition and LTP, potentially through the hyperactivation of AMPK (Lachance et al., 2019) and its involvement in protein folding and quality control (Ouyang et al., 2020). In contrast, inhibition of Vps34 by SAR405 selectively impairs memory consolidation by increasing inhibitory synaptic transmission (Li et al., 2019), without affecting memory acquisition and LTP.
NRBF2 has been reported to be necessary for learning and memory (Ouyang et al., 2020), as well as cognitive impairment in Alzheimer’s disease (AD) patients (Yang et al., 2017; Lachance et al., 2019). Previous researches have demonstrated contextual memory deficits in NRBF2-KO animals through various cognitive tests, including radial-arm maze, fear conditioning, object location task and water maze (Lachance et al., 2019; Ouyang et al., 2020). Consistent with these findings, our study observed increased NRBF2 levels in the hippocampus following fear conditioning and demonstrated memory deficits upon NRBF2 deletion. Moreover, loss of NRBF2 does not affect motor abilities or anxiety-like behaviors in mice, but has been shown to induce a depression-like phenotype (Zhang et al., 2023). Additionally, we delineated the temporal dynamics of fear memory upon NRBF2 deletion and unequivocally showed that NRBF2 contributes to the acquisition stage of learning and memory.
NRBF2 has been identified as a protein that interacts with the Vps34 complex to induce autophagy (Behrends et al., 2010; Cao et al., 2014). Our previous research demonstrated that the infusion of the Vps34 inhibitor SAR405 into the amygdala disrupted memory consolidation by interfering with autophagy and inhibitory neurotransmission, while leaving memory acquisition unaffected (Li et al., 2019). Several other studies have also showed that autophagy inhibition, whether pharmacologically (3-MA and Spautin-1) or genetically (knockdown of Atg7, BECN1, or LC3B), impairs LTM while leaving STM intact (Pandey et al., 2021; Hylin et al., 2018). Consistent with these studies, our results demonstrated that SAR405 infusion into the hippocampus impaired memory consolidation. Collectively, these data confirm that inhibiting autophagy does not affect learning acquisition and STM.
In addition to the disparities observed in memory acquisition and consolidation between NRBF2 knockdown and autophagy inhibition, their impacts on synaptic plasticity also differ in cellular mechanisms. Autophagy has been shown to mediate the degradation of PSD-95 in synapses, which is required for NMDA receptor-dependent LTD (Compans et al., 2021). Pharmacological inhibition of autophagy using rapamycin and trehalose blocked LTD but had no effect on LTP and paired-pulse ratio (Kallergi et al., 2022). Similarly, genetic inhibition of autophagy through Atg5 knockout and Beclin-1 siRNAs showed that autophagy is necessary for LTD induction but not for LTP (Shen et al., 2020). Consistent with these findings, we found that SAR405 did not affect basal synaptic transmission and LTP in the hippocampus. However, deletion of NRBF2 led to impaired LTP maintenance (Lachance et al., 2019). These observations support the hypothesis that NRBF2 may regulate learning and memory through non-autophagy pathways (Ouyang et al., 2020; Ohashi, 2021).
Several potential mechanisms have been proposed to explain the regulation of memory acquisition by NRBF2. Hyperactivation of AMP-activated protein kinase (AMPK) and reduced activity of the mammalian target of rapamycin (mTOR) observed in NRBF2-KO mice may contribute to impaired memory and LTP (Lachance et al., 2019). Notably, AMPK has been shown to modulate LTP, with its activation partially inhibits LTP maintenance by suppressing the mTOR pathway (Herzig and Shaw, 2018; Kim et al., 2011). Moreover, mTOR is involved in the establishment of long-lasting LTP by mediating LTP-related protein synthesis (Tsokas et al., 2007). However, further direct evidence is needed to validate this hypothesis, particularly regarding the involvement of the AMPK-mTOR signaling pathway in autophagy regulation (Alers et al., 2012). Another inference from RNA-seq analysis suggested that NRBF2 regulated learning and memory by modulating networks associated with the retinoic acid receptors, protein folding, and quality control (Ouyang et al., 2020). Given that memory formation requires extensive mRNA transcription and protein translation, it is conceivable that NRBF2 may contribute to these processes. However, elucidating the specific mechanisms underlying these hypotheses will require further exploration through multi-omics and behavioral experiments.
Collectively, our study has provided convincing evidence that NRBF2 deletion impairs memory acquisition through an autophagy-independent pathway. These findings offer new insights into the role of NRBF2 and highlight the potential of targeting NRBF2 as a therapeutic strategy for addressing cognitive deficits associated with various disorders.
The original contributions presented in this study are included in this article/supplementary material, further inquiries can be directed to the corresponding author.
The animal studies were approved by the Animal Welfare Committee of Huazhong University of Science & Technology and the Ethics Committee at the Shenzhen People’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
SW: Formal analysis, Investigation, Methodology, Writing – original draft. HZ: Investigation, Methodology, Validation, Writing – original draft. KL: Funding acquisition, Investigation, Supervision, Writing – review and editing. XZ: Investigation, Funding acquisition, Writing – review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Shenzhen Science and Technology Innovation Program, grant number: JCYJ20190807150005699 and JCYJ20220530152216036, Shenzhen Key Medical Discipline Construction Fund (grant no. SZXK059), and Basic and Applied Basic Research Foundation of Guangdong Province (2020A1515110993).
We thank Dr. Jiahong Lu for providing NRBF2 KO mice.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alers, S., Löffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11. doi: 10.1128/mcb.06159-11
An, H., and Harper, J. W. (2018). Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 20, 135–143. doi: 10.1038/s41556-017-0007-x
Behrends, C., Sowa, M. E., Gygi, S. P., and Harper, J. W. (2010). Network organization of the human autophagy system. Nature 466, 68–76. doi: 10.1038/nature09204
Bin Ibrahim, M. Z., Benoy, A., and Sajikumar, S. (2022). Long-term plasticity in the hippocampus: Maintaining within and ‘tagging’ between synapses. Febs J. 289, 2176–2201. doi: 10.1111/febs.16065
Bush, D. E., Caparosa, E. M., Gekker, A., and Ledoux, J. (2010). Beta-adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front. Behav. Neurosci. 4:154. doi: 10.3389/fnbeh.2010.00154
Cai, C. Z., Yang, C., Zhuang, X. X., Yuan, N. N., Wu, M. Y., Tan, J. Q., et al. (2021). NRBF2 is a RAB7 effector required for autophagosome maturation and mediates the association of APP-CTFs with active form of RAB7 for degradation. Autophagy 17, 1112–1130. doi: 10.1080/15548627.2020.1760623
Cao, Y., Wang, Y., Abi Saab, W. F., Yang, F., Pessin, J. E., and Backer, J. M. (2014). NRBF2 regulates macroautophagy as a component of Vps34 complex I. Biochem. J. 461, 315–322. doi: 10.1042/bj20140515
Compans, B., Camus, C., Kallergi, E., Sposini, S., Martineau, M., Butler, C., et al. (2021). NMDAR-dependent long-term depression is associated with increased short term plasticity through autophagy mediated loss of PSD-95. Nat. Commun. 12:2849. doi: 10.1038/s41467-021-23133-9
Flores, A. M., Li, L., and Aneskievich, B. J. (2004). Isolation and functional analysis of a keratinocyte-derived, ligand-regulated nuclear receptor comodulator. J. Invest. Dermatol. 123, 1092–1101. doi: 10.1111/j.0022-202X.2004.23424.x
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi: 10.1146/annurev-genet-102808-114910
Herzig, S., and Shaw, R. J. (2018). AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 19, 121–135. doi: 10.1038/nrm.2017.95
Hylin, M. J., Zhao, J., Tangavelou, K., Rozas, N. S., Hood, K. N., Macgowan, J. S., et al. (2018). A role for autophagy in long-term spatial memory formation in male rodents. J. Neurosci. Res. 96, 416–426. doi: 10.1002/jnr.24121
Johansen, J. P., Cain, C. K., Ostroff, L. E., and Ledoux, J. E. (2011). Molecular mechanisms of fear learning and memory. Cell 147, 509–524. doi: 10.1016/j.cell.2011.10.009
Kallergi, E., Daskalaki, A. D., Kolaxi, A., Camus, C., Ioannou, E., Mercaldo, V., et al. (2022). Dendritic autophagy degrades postsynaptic proteins and is required for long-term synaptic depression in mice. Nat. Commun. 13:680. doi: 10.1038/s41467-022-28301-z
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 13, 132–141. doi: 10.1038/ncb2152
Kuijpers, M., and Haucke, V. (2021). Neuronal autophagy controls the axonal endoplasmic reticulum to regulate neurotransmission in healthy neurons. Autophagy 17, 1049–1051. doi: 10.1080/15548627.2021.1893569
Lachance, V., Wang, Q., Sweet, E., Choi, I., Cai, C. Z., Zhuang, X. X., et al. (2019). Autophagy protein NRBF2 has reduced expression in Alzheimer’s brains and modulates memory and amyloid-beta homeostasis in mice. Mol. Neurodegener. 14:43. doi: 10.1186/s13024-019-0342-4
Li, K., Chen, H. S., Li, D., Li, H. H., Wang, J., Jia, L., et al. (2019). SAR405, a highly specific VPS34 inhibitor, disrupts auditory fear memory consolidation of mice via facilitation of inhibitory neurotransmission in basolateral amygdala. Biol. Psychiatry 85, 214–225. doi: 10.1016/j.biopsych.2018.07.026
Li, X., He, S., and Ma, B. (2020). Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19:12. doi: 10.1186/s12943-020-1138-4
Lu, J., He, L., Behrends, C., Araki, M., Araki, K., Jun Wang, Q., et al. (2014). NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat. Commun. 5:3920. doi: 10.1038/ncomms4920
Makkar, S. R., Zhang, S. Q., and Cranney, J. (2010). Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology 35, 1625–1652. doi: 10.1038/npp.2010.53
McGaugh, J. L. (2000). Memory–A century of consolidation. Science 287, 248–251. doi: 10.1126/science.287.5451.248
Nabavi, S., Fox, R., Proulx, C. D., Lin, J. Y., Tsien, R. Y., and Malinow, R. (2014). Engineering a memory with LTD and LTP. Nature 511, 348–352. doi: 10.1038/nature13294
Nader, K., and Hardt, O. (2009). A single standard for memory: The case for reconsolidation. Nat. Rev. Neurosci. 10, 224–234. doi: 10.1038/nrn2590
Nikoletopoulou, V., Sidiropoulou, K., Kallergi, E., Dalezios, Y., and Tavernarakis, N. (2017). Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 26, 230–242.e5. doi: 10.1016/j.cmet.2017.06.005
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Ohashi, Y. (2021). Class III phosphatidylinositol 3-kinase complex I subunit NRBF2/Atg38 - From cell and structural biology to health and disease. Autophagy 17, 3897–3907. doi: 10.1080/15548627.2021.1872240
Ouyang, X., Ahmad, I., Johnson, M. S., Redmann, M., Craver, J., Wani, W. Y., et al. (2020). Nuclear receptor binding factor 2 (NRBF2) is required for learning and memory. Lab. Invest. 100, 1238–1251. doi: 10.1038/s41374-020-0433-4
Painsipp, E., Herzog, H., and Holzer, P. (2008). Implication of neuropeptide-Y Y2 receptors in the effects of immune stress on emotional, locomotor and social behavior of mice. Neuropharmacology 55, 117–126. doi: 10.1016/j.neuropharm.2008.05.004
Pan, Y., He, X., Li, C., Li, Y., Li, W., Zhang, H., et al. (2021). Neuronal activity recruits the CRTC1/CREB axis to drive transcription-dependent autophagy for maintaining late-phase LTD. Cell Rep. 36:109398. doi: 10.1016/j.celrep.2021.109398
Pandey, K., Yu, X. W., Steinmetz, A., and Alberini, C. M. (2021). Autophagy coupled to translation is required for long-term memory. Autophagy 17, 1614–1635. doi: 10.1080/15548627.2020.1775393
Percie du Sert, N., Ahluwalia, A., Alam, S., Avey, M. T., Baker, M., et al. (2020). Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 18:e3000411. doi: 10.1371/journal.pbio.3000411
Ronan, B., Flamand, O., Vescovi, L., Dureuil, C., Durand, L., Fassy, F., et al. (2014). A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 10, 1013–1019. doi: 10.1038/nchembio.1681
Schlütermann, D., Skowron, M. A., Berleth, N., Böhler, P., Deitersen, J., Stuhldreier, F., et al. (2018). Targeting urothelial carcinoma cells by combining cisplatin with a specific inhibitor of the autophagy-inducing class III PtdIns3K complex. Urol. Oncol. 36, 160.e1–e13. doi: 10.1016/j.urolonc.2017.11.021
Shehata, M., Matsumura, H., Okubo-Suzuki, R., Ohkawa, N., and Inokuchi, K. (2012). Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci. 32, 10413–10422. doi: 10.1523/jneurosci.4533-11.2012
Shen, H., Zhu, H., Panja, D., Gu, Q., and Li, Z. (2020). Autophagy controls the induction and developmental decline of NMDAR-LTD through endocytic recycling. Nat. Commun. 11:2979. doi: 10.1038/s41467-020-16794-5
Shi, H., Zhang, X., Weng, Y. L., Lu, Z., Liu, Y., Lu, Z., et al. (2018). m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. doi: 10.1038/s41586-018-0666-1
Shoji, H., and Miyakawa, T. (2021). Effects of test experience, closed-arm wall color, and illumination level on behavior and plasma corticosterone response in an elevated plus maze in male C57BL/6J mice: A challenge against conventional interpretation of the test. Mol. Brain 14:34. doi: 10.1186/s13041-020-00721-2
Sun, L., Ma, S., Yu, Y., Li, X., Wei, Q., Min, L., et al. (2024). Transcutaneous auricular vagus nerve stimulation ameliorates adolescent depressive- and anxiety-like behaviors via hippocampus glycolysis and inflammation response. CNS Neurosci. Ther. 30:e14614. doi: 10.1111/cns.14614
Takeuchi, T., Duszkiewicz, A. J., and Morris, R. G. (2014). The synaptic plasticity and memory hypothesis: Encoding, storage and persistence. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369:20130288. doi: 10.1098/rstb.2013.0288
Tsokas, P., Ma, T., Iyengar, R., Landau, E. M., and Blitzer, R. D. (2007). Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J. Neurosci. 27, 5885–5894. doi: 10.1523/jneurosci.4548-06.2007
Wang, W., Liu, S. L., Li, K., Chen, Y., Jiang, B., Li, Y. K., et al. (2015). Leptin: A potential anxiolytic by facilitation of fear extinction. CNS Neurosci. Ther. 21, 425–434. doi: 10.1111/cns.12375
Wilensky, A. E., Schafe, G. E., Kristensen, M. P., and Ledoux, J. E. (2006). Rethinking the fear circuit: The central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J. Neurosci. 26, 12387–12396. doi: 10.1523/jneurosci.4316-06.2006
Wu, P. F., Guan, X. L., Wang, F., and Chen, J. G. (2022). N-acetylcysteine facilitates extinction of cued fear memory in rats via reestablishing basolateral amygdala glutathione homeostasis. Acta Pharmacol. Sin. 43, 260–272. doi: 10.1038/s41401-021-00661-0
Yang, C., Cai, C. Z., Song, J. X., Tan, J. Q., Durairajan, S. S. K., Iyaswamy, A., et al. (2017). NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 13, 2028–2040. doi: 10.1080/15548627.2017.1379633
Yasumo, H., Masuda, N., Furusawa, T., Tsukamoto, T., Sadano, H., and Osumi, T. (2000). Nuclear receptor binding factor-2 (NRBF-2), a possible gene activator protein interacting with nuclear hormone receptors. Biochim. Biophys. Acta 1490, 189–197. doi: 10.1016/s0167-4781(99)00244-4
Young, L. N., Cho, K., Lawrence, R., Zoncu, R., and Hurley, J. H. (2016). Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy. Proc. Natl. Acad. Sci. U S A. 113, 8224–8229. doi: 10.1073/pnas.1603650113
Keywords: Nrbf2, VPS34, autophagy, memory acquisition, LTP
Citation: Wu S, Zhuang H, Zhou X and Li K (2025) NRBF2 plays a crucial role in the acquisition process of learning and memory, independent of the Vps34 complex. Front. Behav. Neurosci. 19:1529522. doi: 10.3389/fnbeh.2025.1529522
Received: 28 November 2024; Accepted: 29 January 2025;
Published: 12 February 2025.
Edited by:
Xiangyou Hu, UConn Health, United StatesReviewed by:
Magdalena Natalia Wojtas, Biophysical Institute (IBF), SpainCopyright © 2025 Wu, Zhuang, Zhou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kuan Li, bGlfa3VhbjE5ODlAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.