 Brandon Emanuel León
Brandon Emanuel León Shinwoo Kang
Shinwoo Kang Gabriela Franca-Solomon
Gabriela Franca-Solomon Pei Shang
Pei Shang Doo-Sup Choi
Doo-Sup Choi- 1Regenerative Sciences Program, Center for Regenerative Medicine, Mayo Clinic, Rochester, MN, United States
- 2Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN, United States
- 3Neuroscience Program, Mayo Clinic College of Medicine and Science, Rochester, MN, United States
- 4Department of Psychiatry and Psychology, Mayo Clinic College of Medicine and Science, Rochester, MN, United States
Mitochondria are essential organelles central to various cellular functions such as energy production, metabolic pathways, signaling transduction, lipid biogenesis, and apoptosis. In the central nervous system, neurons depend on mitochondria for energy homeostasis to maintain optimal synaptic transmission and integrity. Deficiencies in mitochondrial function, including perturbations in energy homeostasis and mitochondrial dynamics, contribute to aging, and Alzheimer’s disease. Chronic and heavy alcohol use is associated with accelerated brain aging, and increased risk for dementia, especially Alzheimer’s disease. Furthermore, through neuroimmune responses, including pro-inflammatory cytokines, excessive alcohol use induces mitochondrial dysfunction. The direct and indirect alcohol-induced neuroimmune responses, including pro-inflammatory cytokines, are critical for the relationship between alcohol-induced mitochondrial dysfunction. In the brain, alcohol activates microglia and increases inflammatory mediators that can impair mitochondrial energy production, dynamics, and initiate cell death pathways. Also, alcohol-induced cytokines in the peripheral organs indirectly, but synergistically exacerbate alcohol’s effects on brain function. This review will provide recent and advanced findings focusing on how alcohol alters the aging process and aggravates Alzheimer’s disease with a focus on mitochondrial function. Finally, we will contextualize these findings to inform clinical and therapeutic approaches towards Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) constitutes 60%–80% of dementia cases worldwide. AD results in serious deteriorations in memory and cognitive abilities that lead to loss of independence and quality of life. As of 2021, 6.2 million Americans over 65 are living with AD and 72% are 75 years or older (Alzheimer’s Association, 2021). Studies over the past 25 years have suggested that the prevalence and incidence of AD in the United States and other high-income Western countries have declined. However, as the number of individuals aged 65 and older increases, the number of people diagnosed with AD is expected to quadruple by 2050 with 43% of those individuals needing high levels of care (Brookmeyer et al., 2007). Furthermore, global projections indicate that the majority of the prevalence and burden will take place in low- and middle-income countries (Alzheimer’s Association, 2021). These projections highlight the need to further our understanding of AD pathology and develop effective therapies to offset the financial and emotional burden. AD pathology is classically characterized by intracellular tau tangles and extracellular amyloid-β (Aβ) plaques ostensibly causing cognitive deficits and neurodegeneration (Kamal et al., 2020). However, numerous clinical drug trials targeting Aβ have failed to yield much success opening the door for novel intervention targets (Huang et al., 2020) including neuroinflammation and mitochondrial dysfunction (Calsolaro and Edison, 2016; Tyumentsev et al., 2018).
The greatest risk factor for AD is aging, but binge and chronic alcohol abuse has been identified as a risk factor for AD. Interestingly, mild-moderate alcohol use has been consistently associated with decreased risk for AD and dementia (Luchsinger and Mayeux, 2004), thus, we will focus on the more hazardous binge and chronic alcohol abuse. Heavy and chronic alcohol abuse is associated with changes in brain structure, cognitive deficits, and decreased brain volume (Rehm et al., 2019). Alcohol use disorder (AUD) is defined as the impaired ability to control or stop alcohol consumption despite negative social, occupational, or health consequences (American Psychiatric Association, 2013). AUD is among the most prevalent substance use disorders and disproportionately afflicts men compared to women (Rehm and Shield, 2019). However, since 2001, the instances of AUD have increased among women, racial and ethnic minorities, the socioeconomically disadvantaged, and, relevant to our review, older adults (Grant et al., 2017). AUD and hazardous alcohol abuse may contribute to the risk for AD development through neuroinflammatory response and mitochondrial dysfunction. Neuroinflammation is a hallmark feature of AD (Gomez-Nicola and Boche, 2015; Liu et al., 2015) and alcohol exposure induces a neuroinflammatory response (Crews et al., 2017). In addition, mitochondrial function, vital to neuronal function, is consistently impaired in AD (Wang et al., 2020) and with alcohol exposure (Tapia-Rojas et al., 2019).
Mitochondria are central to several cellular processes including energy production, metabolic pathways, signal transduction, lipid biogenesis, and apoptosis (Araiso et al., 2019). Neurons are energetically demanding; thus, proper mitochondrial energy homeostasis is vital to sustaining appropriate neural activity. Through oxidative phosphorylation (OXPHOS) and other metabolic pathways, mitochondria provide the substrates necessary to sustain neural activity. In addition, mitochondria are highly dynamic organelles that adapt to cellular demands. Through cycles of fission and fusion, mitophagy selectively disposes of dysfunctional mitochondria while biogenesis generates new mitochondria from pre-existing mitochondria. Mitochondria are also trafficked to dendrites and axons allowing direct energy supply in these subcellular components (Li et al., 2004; Chen and Chan, 2009). Therefore, deficiencies in mitochondrial bioenergetics and dynamics have dire consequences for the survival and health of its host cell and contribute to the pathogenesis of neurodegenerative diseases, especially AD (Wang et al., 2020).
In this review, we will summarize recent findings on hazardous alcohol-induced neuroinflammatory response and age-related mitochondrial dysfunction. We will then contextualize these findings to evaluate the mechanisms by which alcohol-induced neuroinflammatory response and mitochondrial dysfunction may contribute to the onset and progression of AD. Finally, we will summarize developments in novel therapeutics targeting neuroinflammation and mitochondria for AUD and AD.
Alcohol and Cell-Specific Neuroinflammatory Response
Alcohol is a known modulator of systemic immune signaling and perturbs the expression of several immune-related genes (Neupane, 2016). Alcohol increases gut permeability and bacterial translocation into the periphery (Adachi et al., 1995), induces an inflammatory response in the liver, and promotes the systemic release of pro-inflammatory cytokines (Crews et al., 2017). Additionally, circulating endotoxins may activate peripheral immune cells including monocytes, macrophages, T lymphocytes, and dendritic cells to further release pro-inflammatory cytokines and chemokines (Pascual et al., 2015). While the blood-brain barrier (BBB) maintains a relatively isolated environment from the body, systemic inflammation may cross over into the brain and induce neuroinflammation through three known routes: Vagal nerve afferents, pro-inflammatory factors crossing the BBB, and activated monocytes translocating into the CNS (Crews et al., 2017).
Microglia
Microglia, resident macrophages in the CNS, are the primary mediators of the neuroimmune response and are implicated in the pathology of AUD and AD. Microglia express cytokine, chemokine, and pattern recognition receptors (PPRs) that induce the expression of pro- or anti-inflammatory mediators when activated (Erickson et al., 2019). Classically, ramified, or resting microglia surveil their environment through cellular projections in search of infection or injury whereupon microglia adopt distinct activation profiles, M1 and M2, which have pro and anti-inflammatory effects respectively. M1 microglia adopt an amoeboid-like morphology and secrete pro-inflammatory molecules including interleukin (IL)-1, IL-6, and tumor necrosis factor-alpha (TNF-α). In contrast, M2 microglia secrete anti-inflammatory molecules including transforming growth factor-beta (TGF-β) and IL-10 (Tang and Le, 2016). However, recent transcriptional studies point to more nuanced and complex microglial activation profiles (Xue et al., 2014) that are dependent on the type of injury, insult, or brain region (Wes et al., 2016; De Biase et al., 2017). For example, in an AD mouse model, a distinct subpopulation of microglia demonstrated upregulated lipid metabolism, upregulated phagocytic-related genes, and spatially clustered around Aβ deposits (Keren-Shaul et al., 2017).
Alcohol exposure sensitizes the activation of microglia through toll-like receptors (TLRs), purinergic P2X receptors (PRXR), and cytokine signaling (Crews et al., 2017). TLRs are activated by both pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). In vitro, a high dose of alcohol exposure induced increases in TLR4, TLR2, IL-1β, TNF- α, and inducible nitric oxide synthetase (iNOS) expression. This is accompanied by an enhanced generation of reactive oxygen species (ROS; Fernandez-Lizarbe et al., 2013). It is well established that alcohol binds to gamma-aminobutyric acid (GABA) A receptor (GABAAR; Mihic et al., 1997). Thus, it is possible that alcohol increases TLR4 and TLR2 expression through activation of GABAAR although the exact mechanism remains unknown. AUD patient brains with BBB disruptions displayed enhanced microglial activation indicated by amplified ionized calcium-binding adaptor molecule 1 (IBA1) immunoreactivity. These findings were paralleled in mice exposed to binge-like alcohol drinking while the increase in IBA1 immunoreactivity was attenuated by TLR4 knockout (Rubio-Araiz et al., 2017). Transcriptomic studies of 2 bottle choice mice further demonstrated network perturbations in TLR and TGF-β signaling in microglia isolated from the prefrontal cortex (McCarthy et al., 2018).
P2XRs are activated by extracellular adenosine triphosphate (ATP) and are heavily expressed in microglia. P2X7R activation results in the release of pro-inflammatory cytokines IL-1β and IL-18 (He et al., 2017) which is mediated by the activation of nucleotide oligomerization domain containing protein 1 (NOD)-, leucine-rich repeat (LRR)-, and pyrin domain containing protein 3 (NLRP3) inflammasome complex (Iwata et al., 2016). Interestingly, NLRP3 inflammasome activation is a known pathogenic inflammatory response that occurs in AD (Halle et al., 2008; Heneka et al., 2013) Alcohol exposure of 100 mM, comparable to toxic levels in humans, in murine BV2 microglia had differential effects on P2X4R and P2X7R activity. Alcohol inhibited P2X4R activation while sparing P2X7R channel activity. Alcohol reduced P2X4R-mediated microglial migration but potentiated P2X7R pore formation. Alcohol exposure also enhanced P2X7R mediated IL-1β secretion and increased protein expression of both P2XR subtypes (Asatryan et al., 2018). Alcohol is implicated to act as a negative allosteric modulator for P2X4Rs having high and low-affinity sites corresponding to low and high concentrations of alcohol, respectively. In support of this, ivermectin (IVM), a positive allosteric modulator of P2X4R, reduces ethanol intake and operant self-administration in rodents (Franklin et al., 2014).
Besides playing a reactive role, microglial recruitment and activation may also contribute to the development and progression of AUD and AD. Depletion of microglia using PLX5622, a colony-stimulating factor-1 receptor antagonist, prevented increases in alcohol consumption and decreased anxiety-like behavior during abstinence during an alcohol vapor two-bottle choice drinking paradigm. Microglial depletion also rescued expression changes in genes related to inflammation, glutamatergic neurotransmission, and GABAergic neurotransmission in the prefrontal cortex and central nucleus of the amygdala (Warden et al., 2020). In a model of acute binge drinking, PLX5622 mediated depletion of microglia also resulted in increased expression of anti-inflammatory genes (Walter and Crews, 2017). Minocycline, an antibiotic that reduces microglia activation, reduced alcohol intake and preference in the two-bottle choice drinking paradigm in mice (Agrawal et al., 2011), and decreased withdrawal-induced anxiety and relapse in rats (Gajbhiye et al., 2018). These studies indicate that pharmacologically targeting microglia could be an effective strategy to prevent the development of AUD and withdrawal symptoms following abstinence. For AD, recent work demonstrated A2AR/N-methyl d aspartate ionotropic glutamate receptors (NMDAR) complexes are upregulated in activated microglia in the hippocampus of an AD mouse model. In neurons, while there was no difference in the number of complexes, activation of adenosine A2AR resulted in higher NMDAR functionality indicated by intracellular cyclic adenosine monophosphate levels and extracellular signal-regulated kinases 1/2 phosphorylation. Through A2AR antagonism, it may be possible to reduce glutamatergic excitotoxicity (Franco et al., 2020) and microglial release of pro-inflammatory mediators (Saura et al., 2005). Overall, these studies suggest that pharmacological targeting of microglia could be a novel mechanism to address AUD and AD.
Astrocytes
Astrocytes are involved in a myriad of physiological processes including maintaining appropriate ionic concentrations, supporting neurons bioenergetically, neurotransmitter clearance, bridging the BBB, and maintaining the integrity of synapses (Khakh and Sofroniew, 2015). Astrocytes are also a component of the immune system in the CNS. While astrocytes express chemokine and cytokine receptors, astrocytes partly depend on microglia for their activation. In response to LPS administration, microglial IL-1α, TNF, and complement component 1 subcomponent q (C1q) mediate A1 astrocytic activation. A1 astrocytes reduced neuronal survival, outgrowth, synaptogenesis, and were observed in post-mortem tissue from patients with neurodegenerative diseases including AD (Liddelow et al., 2017).
Astrocyte activities are essential for addictive behaviors (Scofield and Kalivas, 2014; Corkrum et al., 2020; Kang et al., 2020; Kang and Choi, 2021), especially for AUD (Ayers-Ringler et al., 2016; Erickson et al., 2018, 2021; Lindberg et al., 2018). Astrocytes regulate neuronal activities of glutamatergic neurons in the cortico-striatal and dopaminergic neurons in the ventral tegmental area (VTA)-nucleus accumbens (NAc) circuits (Scofield and Kalivas, 2014; Corkrum et al., 2020). Importantly, astrocyte-neuron interactions are reciprocal in regulating addictive behaviors (Durkee and Araque, 2019). Interestingly, astrocytes are also central mediators of adenosine signaling which is implicated in the pathology of AUD and neurodegenerative disorders (Kim et al., 2013). In the CNS, adenosine regulates neuronal activity by modulating the signaling of other neurotransmitters. For example, the adenosine receptors, A1 and A2A, often work in tandem dimerizing with each other and with dopamine and glutamate receptors to fine-tune dopaminergic and glutamatergic signaling. Astrocytes release ATP into the extracellular space, where ATP is converted to adenosine by ecto-nucleotidases. Astrocytes also release adenosine through bidirectional transporters especially equilibrative nucleoside transporter 1 (ENT1). ENT1 knockout (KO) mice display reduced ataxic and hypnotic responses to alcohol exposure and increased alcohol intake compared to wild-type littermates. This is associated with decreased adenosine tone on A1 receptors indirectly measured through decreased A1 receptor-mediated inhibition of glutamate excitatory postsynaptic currents in the nucleus accumbens (Choi et al., 2004). Furthermore, adenosine-mediated glutamatergic signaling reduces sensitivity to alcohol and may predispose individuals to AUD (Chen et al., 2010). In addition, pharmacological inhibition, and genetic deletion of ENT1 reduced the expression of glutamate transport 1 (GLT1) and astrocyte-specific aquaporin 4 (AQP4). Ceftriaxone administration, an antibiotic which is known to decrease alcohol consumption in rodents, elevated GLT1 and AQP4 expression in the striatum, and reduced alcohol consumption in mice indicating that adenosine signaling mediates GLT1 and AQP4 expression (Lee et al., 2013). ENT1 KO mice also displayed reduced glial fibrillary acidic protein (GFAP) expression in the dorsal striatum suggesting that ENT1 and adenosine regulate GFAP expression and astrocytic function (Hinton et al., 2014). The adenosine analog, N6-(4-hydroxybenzyl)-adenosine (NHBA), which activates A2AR and inhibits ENT1, reduced alcohol intake and alcohol reward seeking behavior in mice (Hong et al., 2019). Chronic alcohol exposure in mice also alters glucose and lactate concentrations in the brain and decreases the expression of monocarboxylate transporter indicating that alcohol disrupts bioenergetic homeostasis between neurons and glia (Lindberg et al., 2019). This suggests impaired alcohol-induced astrocytic-neuronal bioenergetic crosstalk may contribute to impaired energy metabolism seen in AD (Wang et al., 2020), and the important roles astrocytes and astrocytic signaling play in the development of AUD.
Mitochondria and Neuroinflammation
Mitochondrial are uniquely positioned as junctions of innate immunity by transducing immune signals, metabolic reprogramming, and generation of ROS and DAMPs (Figure 1). The outer mitochondrial membrane harbors mitochondrial antiviral signaling protein (MAVS) which signals the presence of cytosolic viral double-stranded RNA, leading to the activation of nuclear factor kappa B (NF-κB) and interferon regulator factor 3 (IRF3) to induce the expression of pro-inflammatory cytokines and type I interferon (IFN; Seth et al., 2005; Sun et al., 2006). Similarly, cytosolic DNA, detected by cyclic GMP-AMP synthase (cGAS), leads to the production of cyclic guanosine monophosphate-adenosine monophosphate (cGMP). cGMP binds to the stimulator of interferon genes (STING) located on the endoplasmic reticulum. STING then engages both IRF3 (Sun et al., 2013) and NF-κB to induce the expression of pro-inflammatory cytokines (Ishikawa and Barber, 2008).
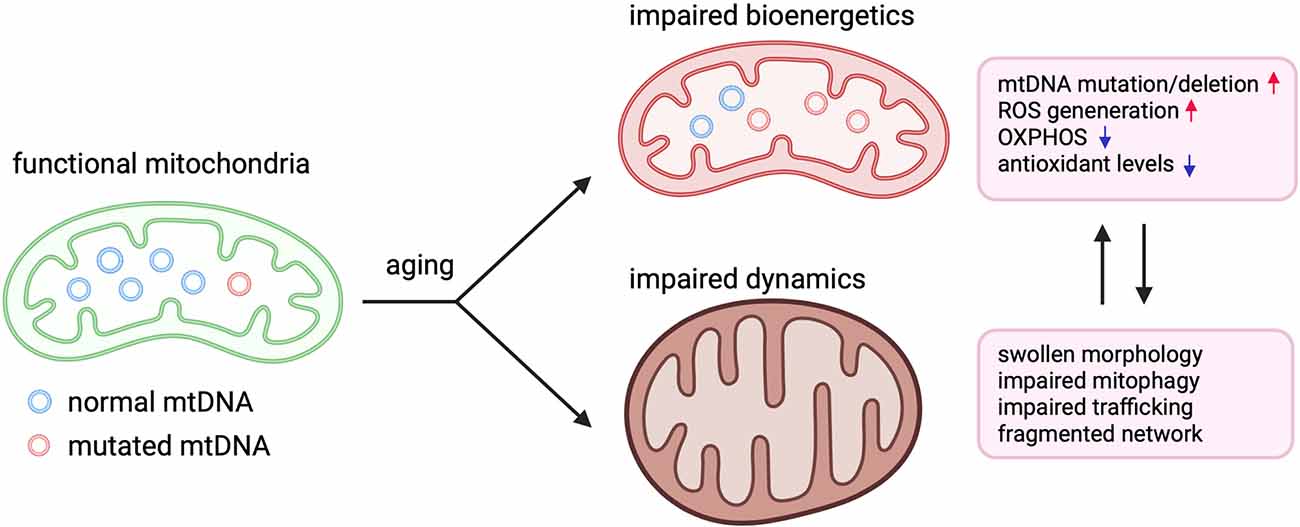
Figure 1. Perturbation of mitochondrial bioenergetics and dynamics in aging. Illustration demonstrating how aging negatively affects mitochondrial bioenergetics and dynamics in aging. In aged humans and animals, higher levels of mutations and deletions in mitochondrial DNA (mtDNA) lead to reduced mitochondrial protein expression and impairments in oxidative phosphorylation (OXPHOS), which exacerbates the generation of reactive oxygen species (ROS). Mitochondria also appear fragmented and swollen while also displaying impaired mitophagy and trafficking to neuronal compartments such dendrites and axons.
Mitochondrial metabolic reprogramming is central to microglial activation. Activation triggers a metabolic shift from OXPHOS to glycolysis followed by the adoption of the M1 phenotype and pro-inflammatory response (Voloboueva et al., 2013; Gimeno-Bayón et al., 2014; Orihuela et al., 2016). The mitochondrial toxins 3-nitropropionic acid and rotenone prevented IL-4 induced phenotype transition to M2 associated with healing and anti-inflammatory response (Ferger et al., 2010). Furthermore, inhibition of complex I promoted activation of microglia and production of pro-inflammatory cytokines via induction of NF-κB and mitogen-activated protein kinases pathways (MAPKs; Ye et al., 2016). These studies implicate mitochondrial metabolic programming in microglial activation and suggest that perturbations in mitochondrial energy homeostasis may enhance pro-inflammatory microglial phenotypes associated with AUD and AD (Rose et al., 2017).
Mitochondria also generate ROS primarily through oxidative phosphorylation (OXPHOS) where the majority of ATP is produced (Figure 1). Normal physiological levels of ROS participate in cellular signaling (Shadel and Horvath, 2015) including the modulation of immune receptors (TLRs, NLRs, MAVS) and transcription factors (NF-κB and IRFs; Weinberg et al., 2015). However, overaccumulation of ROS and reactive nitrogen species (RNS), such as nitric oxide (NO), can damage both cellular and mitochondrial nucleic acid, proteins, and lipids. The balance between ROS production and neutralization is maintained by antioxidant systems. When these systems are compromised, ROS accumulation can result in the assembly of the NLRP3 inflammasome, damage to mitochondrial constituents, and neuroinflammation (Vezzani et al., 2020). ROS also activate microglia which further secrete ROS, RNS, and cytokines (McElroy et al., 2017). In addition, ROS-induced mitochondrial damage leads to deficiencies in OXPHOS. In the brain, where ATP is critically needed to maintain highly energetic processes such as neuronal potential, deficiencies in OXPHOS are a central facet to neuroinflammation and neurodegeneration (Vezzani et al., 2020; Figure 2). Furthermore, alcohol is known to increase ROS concentrations and decrease endogenous antioxidants. Mitochondria in the CNS are devoid of alcohol dehydrogenase and must metabolize alcohol via cytochrome P450 (CYP450) which metabolizes alcohol into acetaldehyde while producing superoxide anion (O-2) and hydrogen peroxide (H2O2; García-Suástegui et al., 2017). Preclinical studies have demonstrated that chronic alcohol exposure decreases glutathione and superoxide dismutase, antioxidants crucial to maintaining oxidative homeostasis. Alcohol administration increased NO, iNOS, and neuronal nitric oxide synthase levels (nNOS). Furthermore, the activities of mitochondrial complexes I, III, IV, Na+/K+ ATPase were also decreased, and brain mitochondria were sensitized to apoptotic stimuli (Almansa et al., 2009; Reddy et al., 2013). Acute administration of alcohol also increases linearized forms of mtDNA which were attenuated by antioxidant treatment (Mansouri et al., 2001).
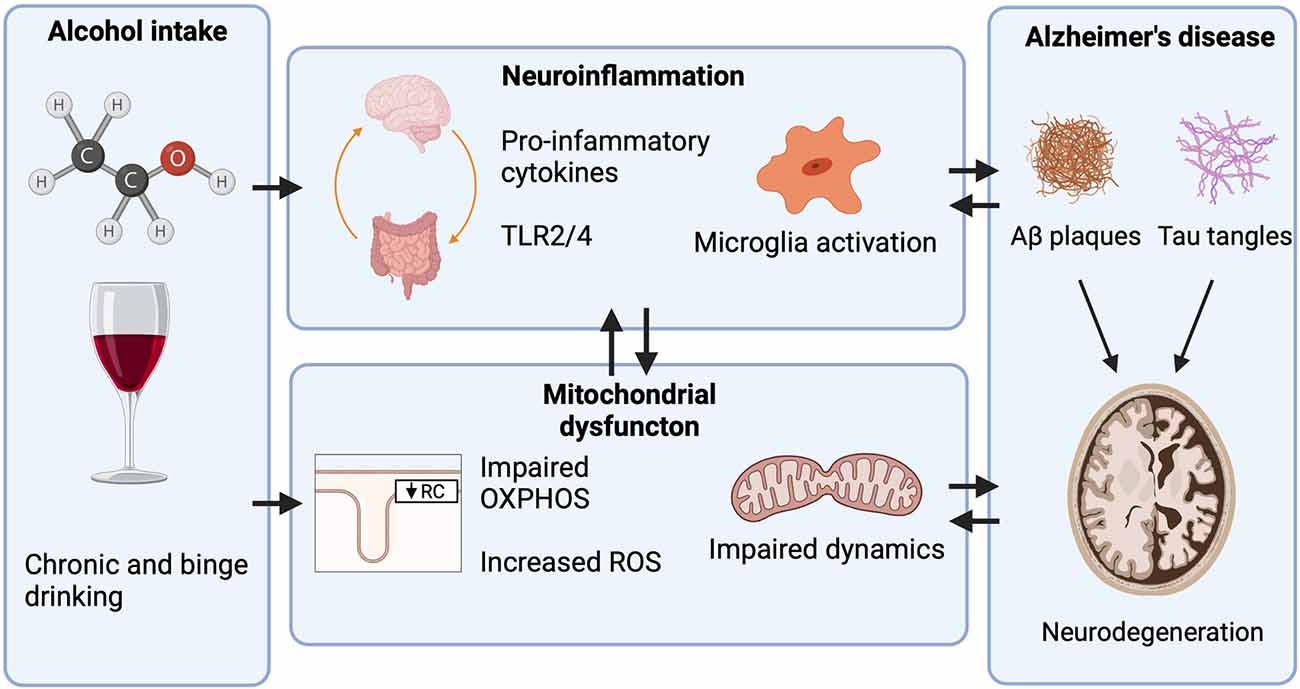
Figure 2. Mechanisms through which heavy alcohol abuse increases the risk for Alzheimer’s disease (AD). Only heavy alcohol abuse increases the risk for AD while light and moderate Alcohol use do not. Proposed mechanisms through which heavy alcohol abuse may increase the risk for AD. Alcohol increases peripheral inflammation by increasing the gut translocation of lipopolysaccharide (LPS) into circulation, where it activates peripheral immune cells and increases pro-inflammatory cytokine release. Pro-inflammatory cytokines then cross the blood-brain barrier (BBB) activating microglia and astrocytes. In the CNS, alcohol increases the expression of toll-like receptor 4 (TLR4) and High mobility group box 1 (HMGB1), a ligand for TLR4. TLR4 activation leads to NF-κB induction of pro-inflammatory cytokine release which further activates other microglia and astrocytes. Heavy alcohol abuse also impairs mitochondrial function by impairing OXPHOS, increasing the generation of ROS, and impairing mitochondrial dynamics. As neurons are energetically demanding cells, mitochondrial dysfunction is a contributing factor to neurodegeneration.
Because of their prokaryotic origins, mitochondria are potent sources of DAMPs, and damage to their outer membrane, mitochondrial permeability transition, or cell death can result in the cytoplasmic and/or extracellular release of mitochondrial DAMPs (Grazioli and Pugin, 2018). Mitochondrial DAMPs include mitochondrial DNA (mtDNA), ATP, mitochondrial transcription factor A (TFAM), N-foryml peptides (NFPs), succinate, cardiolipin, and cytochrome C (Grazioli and Pugin, 2018). Like bacterial DNA, mtDNA is characterized by low levels of methylation but is protected by packing DNA into complexes known as nucleoids. Nucleoid packaging is mediated by TFAM which is a member of the high mobility group box family. Intratracheal injection mtDNA in mice promoted infiltration of CD68+ macrophages, production of pro-inflammatory cytokines (IL-1β, Il-6, and TNF-α), and activated p38 MAPK through TLR9 signaling (Gu et al., 2015). Interestingly, melatonin deficiency in mice lacking the N-acetyltransferase gene in cerebro-cortical neurons was associated with increased mtDNA release, decreased mitochondrial membrane potential, and increased oxidative stress. Cytosolic mtDNA induced activation of cGAS-STING-IRF3 pathway and resulted in inflammatory cytokine induction (Jauhari et al., 2020). Interestingly, melatonin is a known free radical scavenger (Zhang and Zhang, 2014) and is produced in the mitochondrial matrix (Suofu et al., 2017). mtDNA can also initiate the assembly of inflammasomes especially the NLRP3 and is absent in melanoma-2 (AIM2) inflammasomes. Assembly and activation of inflammasomes induce the expression of IL-1β and triggers caspase-1-dependent mitochondrial damage. Caspase-1 activity inhibits mitophagy, leads to enhanced ROS production, perturbs mitochondrial membrane potential, and results in mitochondrial permeabilization and network fragmentation (Shimada et al., 2012; Yu et al., 2014).
The principle pathologic feature of AD, Aβ, has been observed to accumulate within the mitochondria of human AD patients and mouse models of AD by interacting with Aβ peptide-binding alcohol dehydrogenase (ABAD; Lustbader et al., 2004). ABAD- Aβ interacts with several mitochondrial constituents to induce dysfunction. Blockage of ABAD-Aβ interaction increased enzyme activity of respiratory chain constituents, decreased oxidative stress, and improved spatial memory in transgenic mutant amyloid precursor protein (mAPP) mice (Yao et al., 2011). ABAD- Aβ interactions also potentiated cyclophilin D (CypD) mediated mitochondrial permeability transition and induced neuronal death, while knockout of CypD attenuated these effects and improved behavioral and synaptic function in mAPP mice (Du et al., 2008). In Tau22 mice, loss of NLRP3 inflammasome decreased tau hyperphosphorylation and intracerebral injection of Aβ containing brain homogenates induced tau pathology in NLRP3 dependent manner (Ising et al., 2019).
Mitochondria, Energy Homeostasis, and Aging
Mitochondria are unique organelles due to their double membrane and own genome known as mitochondrial DNA (mtDNA). Besides rRNAs and tRNAs, mtDNA encodes for complexes I, III, IV, and V which are the essential constituents of the electron transport chain (ETC). Mitochondrial dysfunction is heavily implicated in the aging process and studies have demonstrated mitochondrial dysfunction occurs in the heart (Hoppel et al., 2017), muscles (Waltz et al., 2018), and livers (Ogrodnik et al., 2017) of older individuals. Interestingly, brains of older individuals displayed significant variation in the decline of OXPHOS suggesting that those with exacerbated declines in OXPHOS may be more prone to neuronal dysfunction (Ojaimi et al., 1999). In rat livers and spleens, the respiratory capacity of aged animals was significantly reduced compared to juveniles while there was no difference in the brain (Stocco and Hutson, 1978). Respiratory capacity also declines in the human liver, heart, and skeletal muscle tissue (Short et al., 2005). Mitochondrial respiratory chain complexes catalyze the oxidation of reducing equivalents, mainly nicotinamide adenine dinucleotide (NADH), using the terminal electron acceptor, oxygen, in the inner mitochondrial membrane (Genova and Lenaz, 2015). The activities of respiratory chain complexes I and IV decrease with age; however, the activities of complexes II, III, and IV remain unchanged (Navarro and Boveris, 2007). In a study in mice, protein levels of complexes I, II, IV, and V were increased in 12- and 18-month-old animals compared to 2-month-old animals. However, their mRNA expression in 24-month-old mice was decreased, indicating that age-related deficits in energy production are initially compensated by increased expression of ETC complexes. This compensation cannot be sustained for long and may be associated with the differences in the activity of ETC complexes with age (Manczak et al., 2005). Several decades of research have attempted to answer how age-related mitochondrial dysfunction occurs. The Free Radical Theory of Aging (MFRTA) and mtDNA insults (Chocron et al., 2019) have been proposed as mechanisms of age-related mitochondrial dysfunction. Importantly, the bioenergetics and dynamics of mitochondria are subject to extrinsic factors such as diet (Nisoli et al., 2005) and hormones (Cioffi et al., 2010) that may provide protection against oxidative damage and decelerate aging.
In contrast to nuclear DNA, mitochondria contain thousands of copies of mtDNA. Mutations and/or deletions of mtDNA can have deleterious effects on mitochondrial energy homeostasis. With age, wild-type and mutated mtDNA accumulate inside mitochondria and create a mosaic of functional and non-functional mtDNA. For mtDNA mutations to induce mitochondrial dysfunction, the ratio of mutated to wild type mtDNA must reach a threshold of 70%–90% (Rossignol et al., 2003). A study in healthy individuals demonstrated that low levels of mtDNA heteroplasmy are normal and are likely due to inheritance or single base substitutions (Payne et al., 2012). In women aged 17–85, those aged 70 years or older displayed increased mtDNA heteroplasmy (Zhang et al., 2017). In older men and women, high levels of mtDNA mutations in blood leukocytes were associated with reduced strength, cognitive abilities, and metabolic and cardiovascular function. Furthermore, participants with the highest levels of mutation load were at greater risk for developing dementia and stroke mortality (Tranah et al., 2018). Additionally, mutations in mtDNA are associated with an increased risk of cognitive decline and dementia (Kauppila et al., 2017). These studies suggest that reaching the 70%–90% threshold typically requires a lifetime to accumulate in enough cells but can cause bioenergetic deficiencies and are associated with aging phenotypes. The question remains whether mutation load correlates with age or drives aging phenotypes. Experimentally, the proofreading-deficient mtDNA polymerase (POLγ) mouse model provides interesting evidence demonstrating that abnormal accumulation of mtDNA mutations leads to reduced lifespan and aging-related phenotypes such as alopecia and osteoporosis. Briefly, the proofreading deficient POLγ leads to excessive accumulations of mtDNA mutations (Trifunovic et al., 2004). mtDNA mutations in these mice were not associated with signs of oxidative stress but displayed induction of apoptotic markers (Kujoth et al., 2005). Interestingly, offspring of mutator POLγ and wild type POLγ mice displayed mild signs of aging while offspring of mutator POLγ mice displayed severe brain malformations. These studies indicate that both inherited and somatic mtDNA mutations promote aging phenotypes (Ross et al., 2013). However, the levels of mtDNA mutations are much higher in these mice compared to aged controls and may not faithfully reflect what occurs in normal mice or humans. In contrast to mutations, mtDNA deletions are seldom inherited (Chinnery et al., 2004). Accumulation of mtDNA deletions has been reported in the brain (Corral-Debrinski et al., 1992; Taylor et al., 2014), muscle (Fayet et al., 2002), liver (Yen et al., 1991), and heart tissue of aged humans (Cortopassi and Arnheim, 1990). Additionally, Parkinson’s patients and age-matched controls displayed higher levels of mtDNA deletions in the substania nigra, especially in cells with respiratory chain deficiency (Bender et al., 2006, 2008; Kraytsberg et al., 2006). mtDNA deletions tend to be low in tissue homogenates; however, in individual cells, mtDNA deletions can be high where randomly distributed cells display respiratory chain deficiency (Baris et al., 2015). For example, muscle fibers of rats with electron transport chain deficiencies and atrophy displayed greater levels of deletions (Cao et al., 2001; Wanagat et al., 2001). Analogous to the mutator mouse model of mtDNA mutations, a mouse model expressing an inducible mitochondrially targeted restriction endonuclease demonstrated reduced levels of mtDNA encoded subunits of the electron transport chain and reduced oxidative phosphorylation activity (Fukui and Moraes, 2009). While the mechanisms by which mutations and deletions accumulate in mitochondria are still unclear, the wealth of studies has demonstrated that both mutations and deletions of mtDNA contribute to the aging phenotype.
Mitochondria are the primary sources of ROS (Figure 2). Respiratory chain complex activity results in the production of ROS as byproducts. Normal levels of ROS take part in physiological signal transduction such as regulating neural stem cell proliferation, self-renew, and neurogenesis (Le Belle et al., 2011). Hydrogen peroxide also modulates NF-κB by either stimulating or inhibiting NF-κB signaling depending on the context (Oliveira-Marques et al., 2009). To protect the cell and mitochondrial constituents, mitochondria deploy several ROS antioxidants that are adept at neutralizing ROS before oxidative damage can occur (Alexeyev, 2009). Several studies across different species have genetically overexpressed or knocked out antioxidant genes although no clear correlation was found between oxidant damage and lifespan (Bratic and Larsson, 2013). In Drosophila, for example, targeting catalase to the mitochondria nor overexpression of superoxide dismutase, catalase, and thioredoxin reductase extended lifespan (Mockett et al., 2003, 2010; Orr et al., 2003). Mice lacking superoxide dismutase and glutathione peroxidase-1 displayed increased DNA and protein oxidation without reduction of lifespan (Zhang et al., 2009). In C. elegans, the genetic deletion of superoxide dismutase yielded no obvious effects or extended lifespan (Doonan et al., 2008; Van Raamsdonk and Hekimi, 2009). These studies indicate that oxidative stress may not be the primary cause of aging. Nevertheless, under pathological conditions such as neurodegeneration or chronic heavy alcohol abuse, excessive oxidative stress may damage the molecular constituents of the brain. For example, the brain is highly concentrated with phospholipids and is vulnerable to lipid peroxidation (Kim et al., 2015). Excessive oxidative damage may also damage proteins and DNA which can induce mitochondrial dysfunction and cell death (García-Suástegui et al., 2017). Brains of older individuals and those diagnosed with neurodegenerative diseases displayed increased protein nitration and oxidation; decreased activity of antioxidant enzymes superoxide dismutase (SOD), catalase, and glutathione (GSH) reductase; and decreased complex I activity (Venkateshappa et al., 2012a, b). The peroxidation of lipids by ROS leads to signaling cascades that induce apoptosis and autophagy (Su et al., 2019).
Interestingly, increased ROS concentrations reduce the calcium concentration needed to open the mitochondrial permeability transition pore (Panel et al., 2018). The mitochondrial permeability transition has recently been proposed as a mechanism of aging and may contribute to the onset of neuronal injury in neurodegenerative disorders like AD (Figure 2). Briefly, the mitochondrial permeability transition is a sudden increase in the permeability of the inner mitochondrial membrane through a complexed nonspecific pore called the mitochondrial permeability transition pore (mPTP). The opening of the mPTP results in organelle swelling, drop in membrane potential, loss of mitochondrial substrates, and uncoupling of OXPHOS. The mPTP forms in response to increased matrix calcium levels and is sensitized by ROS, adenine nucleotide depletion, drop in membrane potential, and increased phosphate concentrations (Panel et al., 2018). In aged whole rat brains, mitochondrial susceptibility to mPTP opening was associated with impaired mitochondrial respiration and mitochondrial transmembrane electric potential (Marques-Aleixo et al., 2012; Krestinina et al., 2015); however, age-related mitochondrial impairments are not consistent in every brain region. For example, in aged Fischer 344 rats, striatal mitochondria were more sensitive to Ca2+ induced mPTP opening compared to cortical mitochondrial up to 24 months (LaFrance et al., 2005). Susceptibility to permeability transition may depend on CypD levels and neuron cell type. CypD has been implicated in the assembly of the mPTP. Interestingly, higher levels of cyclophilin D have been reported in neurons expressing glutamate decarboxylase or calbindin D-28k, GABAergic interneurons (Brustovetsky et al., 2003). Considering that these neurons are fast-spiking and energy demanding, brain regions with greater proportions of interneurons could explain the heterogenic susceptibility to Ca2+-induced mPTP formation (McCasland and Hibbard, 1997; Attwell and Laughlin, 2001; Brustovetsky et al., 2003).
Alcohol, Mitochondria, and Alzheimer’S Disease
Mitochondrial dynamics sustain cellular integrity and control the disposal of aberrant constituents in the mitochondrial network (Kim et al., 2007; Youle and Narendra, 2011). Alcohol is known to substantially alter mitochondrial morphology in a concentration-dependent manner. For example, mitochondria demonstrated morphological changes and metabolic dysfunction after alcohol exposure (Suen et al., 2008; Flores-Bellver et al., 2014). In addition, mitochondrial fission protein Drp1 levels are significantly elevated after alcohol treatment in a concentration-dependent manner (Bonet-Ponce et al., 2015). Our own studies have demonstrated both morphological and functional changes in mitochondria in the prefrontal cortex in response to alcohol exposure (Shang et al., 2020). Distinct types of fission have been observed that determine the fate of mitochondria. Division at the midzone leads to proliferation of mitochondria, while division at the periphery leads to mitophagy (Kleele et al., 2021) During fission, fragmented mitochondria release cytochrome c and other intermembrane proteins that affect neighboring mitochondria and induce apoptosis (Detmer and Chan, 2007). Proteomics studies have demonstrated that 40 mitochondrial proteins, including key enzymes involved in β-oxidation, the tricarboxylic acid cycle, and amino acid metabolism, were altered after alcohol consumption (Venkatraman et al., 2004).
Alcohol-induced mitochondrial energy homeostasis dysfunction may alter the plasticity and intrinsic excitability of neurons depending on the age, dose, duration of exposure, and brain region. In the cerebral cortex, for example, in PD6 to PD9 rat pups, alcohol induced a higher incidence of potentiation, while PD13 and PD15 rat pups displayed greater attenuation. These changes in the intrinsic excitability may be attributed to altered Ca2+ dynamics in mitochondria (Harrison et al., 2017; Cannady et al., 2018). For instance, selective inhibitors of Ca2+ uptake and release blocked post tetanic potentiation while endoplasmic reticulum Ca2+ pump inhibitors and activators showed no effects (Tang and Zucker, 1997). Synaptic plasticity may also depend on mitochondrial respiratory capacity. The complex I inhibitor, rotenone, significantly impaired long-term potential in hippocampal neurons of rats (Kimura et al., 2012). Furthermore, selective inhibition of mitochondrial dihydroorotate dehydrogenase reduced the respiratory capacity of mitochondria and attenuated the firing rates of hippocampal neurons (Styr et al., 2019). Compared with neurons in the prefrontal cortex and hippocampus, motor neurons in the brainstem and hypoglossal nucleus are more sensitive to changes in calcium load and hypoxia, especially during complex IV inhibition (Bergmann and Keller, 2004). During alcohol withdrawal, increased neuronal excitability was found in the infralimbic region of the medial prefrontal cortex in mice chronically exposed to alcohol vapor (Pleil et al., 2015). Therefore, depending on the factors mentioned above, alcohol differentially affects the intrinsic excitability and plasticity of neurons (Tu et al., 2007; Weitlauf and Woodward, 2008).
Harwood et al. (2010) demonstrated the correlation between alcohol consumption and elevated risk of AD. Reducing hazardous alcohol intake increases the quality of life, cognitive ability, and longevity (Venkataraman et al., 2017). Alcohol can directly cross the BBB (Szabo and Lippai, 2014) and thus, chronic hazardous alcohol use can damage the nervous system especially the cortex and hippocampus (Harper et al., 1985; de la Monte and Kril, 2014). In differentiated neuroblastoma cells, chronic alcohol exposure in the range of 125–500 mg/dl dose-dependently increased tau protein levels and decreased cell viability. Chronic alcohol also increased beta secretase-1 (BACE1) via the production of ROS, suggesting that it causes neuronal damage and death induced by Aβ (Gendron et al., 2008). Cyclin-dependent kinase 5 (Cdk-5) induces oxidative stress by inactivating peroxiredoxin-I (Prx-I) and peroxiredoxin-II (Prx-II), the cytoplasmic peroxidase enzymes that process ROS. These results show that chronic alcohol might trigger increased ROS due to Cdk-5 (Liu et al., 2016). In addition, excessive alcohol intake increases p25, increased p25 proceeds to the neurotoxic pathway through Cdk5 (Kusakawa et al., 2000). Increases in Cdk5 and glycogen synthase kinase-3 β (GSK3-β) results in tau hyperphosphorylation-induced cognitive decline (Camp et al., 2006). A recent study demonstrated binge alcohol exposure leads to GSK3β activation which results in neurodegeneration, indicated by reduced NeuN positive neurons and ultrastructural analysis, and impaired spatial learning and memory in the Morris water maze (Ji et al., 2018). GSK3β activity is inhibited by phosphorylation in serine 9 (Ser9) while alcohol exposure leads to dephosphorylation at Ser9. In vitro, overexpression of GSK3β resulted in cellular sensitivity to 400 mg/dl alcohol exposure via induction of caspase 3 ultimately resulting in apoptosis (Liu et al., 2009). These studies indicate that GSK3β may act as a mediator bridging hazardous alcohol abuse, neurodegeneration, and cognitive deficits.
Discussion
The increasing number of individuals diagnosed with AUD and AD highlights the need for novel therapeutics as many AD clinical trials fail to yield optimistic results (Huang et al., 2020). As discussed above, chronic neuroinflammatory response and mitochondrial dysfunction mechanistically link AUD and AD pathology and may be an avenue through which these disorders are addressed. Several anti-inflammatory and mitochondria targeting drugs are being developed and tested which are discussed in detail (Table 1). CP2 is a mitochondrial complex I inhibitor and AMP-activated protein kinase (AMPK) activator that improves mitochondrial bioenergetics and axonal trafficking, reduces inflammation and Aβ pathology, and improves synaptic activity (Zhang et al., 2015, 2019; Ray et al., 2018; Hara et al., 2019; Stojakovic et al., 2021). However, CP2 has not undergone clinical trials for AD. Additionally, CP2 has not been evaluated in the context of AUD. Minocycline is a tetracycline antibiotic that, like other antibiotics, reduces drinking in rodent models of AUD (Agrawal et al., 2011). Minocycline is thought to reduce drinking by inhibiting microglia activation (Regen et al., 2015; Clemens et al., 2018) and reducing levels of pro-inflammatory cytokines (Garcez et al., 2017). Interestingly, minocycline administrations improved spatial memory and provide protection against neuronal cell death in animal models of AD (Choi et al., 2007; Réus et al., 2015; Garcez et al., 2017). However, a clinical trial in which individuals with mild AD were treated with minocycline for over 2 years found no delay in the progress of cognitive or functional impairment (Howard et al., 2020). Phosphodiesterase 4 (PDE4) inhibitors are another drug class being investigated for AD and AUD. PDE4 inhibitor administrations reduce alcohol intake and preference in mice and rats (Blednov et al., 2014; Bell et al., 2015; Howard et al., 2020). Ibudilast also prevented Aβ1–42 induced memory impairments and reduced neuroinflammation and apoptotic responses (Wang et al., 2014). Ibudilast has been investigated in clinical trials for multiple sclerosis without any significant effects (Wang et al., 2014; Fox et al., 2021; Goodman et al., 2021). A promising novel Aβ antibody drug, aducanumab, received FDA approval under the accelerated approval pathway. Chronic administrations in aducanumab in Tg2576 mice rescued calcium homeostasis (Kastanenka et al., 2016). Aducanumab also demonstrated efficacy to reduce soluble and insoluble Aβ in transgenic mouse models of AD and patients with prodromal or mild AD. In patients with prodromal or mild AD, aducanumab also slowed cognitive decline in a phase 1b clinical trial (Sevigny et al., 2016). While promising, larger phase three trials, EMERGE and ENGAGE, initially failed to provide significant benefits from aducanumab. However, upon post hoc analysis, it was determined that there was sufficient clinical efficacy in a subset of participants. This has led to contention as many believe there is not enough evidence to conclude aducanumab is sufficiently efficacious (Howard and Liu, 2020; Schneider, 2020; Knopman et al., 2021). Finally, non-steroidal anti-inflammatory drugs (NSAIDs) are being tested as preventative therapies to slow the onset of AD. Specifically, cyclooxygenase-2 inhibitors (Zarghi and Arfaei, 2011) have demonstrated therapeutic effects in pre-clinical models of AD (Lim et al., 2000, 2001; Kotilinek et al., 2008). Chronic use of NSAIDs has demonstrated some benefit in prodromal stages of AD; however, their side effects pose concerns for widespread use (Imbimbo et al., 2010). More studies and investigations are needed to completely understand the underlying mechanisms through which these drugs exert improve mitochondrial function and anti-inflammatory properties. Realistically, a combination of therapies that target Aβ and Tau pathologies, chronic neuroinflammation, and mitochondrial dysfunction may be more effective than any of these alone.
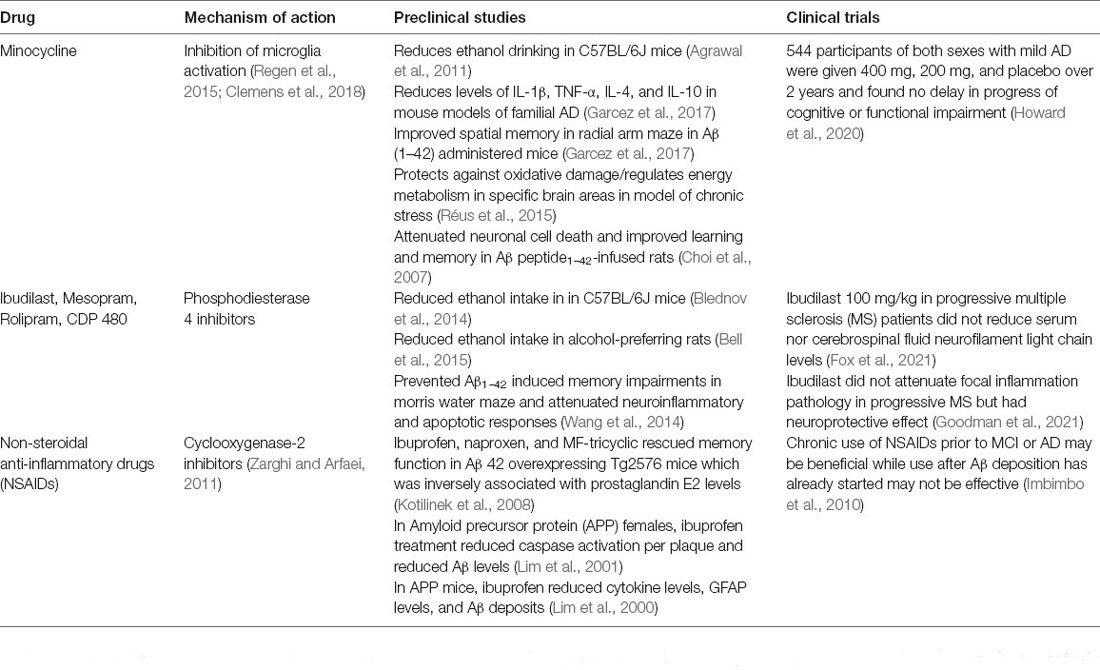
Table 1. Potential anti-inflammatory medications for alcohol use disorder and Alzheimer’s disease.
Finally, it is critical to better elucidate the molecular mechanisms underlying the alcohol-induced neuroinflammatory responses, which may impair mitochondrial function in AD. A clear understanding of these processes may yield novel therapeutic targets to mitigate the severity of AD symptoms associated with hazardous alcohol drinking.
Author Contributions
BEL, SK, GF-S, PS, and D-SC wrote the manuscript. BEL and D-SC prepared figures. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Samuel C. Johnson for Genomics of Addiction Program at Mayo Clinic, the Ulm Foundation, and the National Institute of Health (AA029258, AA028968, AG072898; National Institute on Alcohol Abuse and Alcoholism).
Conflict of Interest
D-SC is a scientific advisory board member to Peptron Inc. Peptron Inc had no role in the preparation, review, or approval of the manuscript; nor the decision to submit the manuscript for publication.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Choi lab members for their helpful comments and discussion.
References
Adachi, Y., Moore, L. E., Bradford, B. U., Gao, W., and Thurman, R. G. (1995). Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108, 218–224. doi: 10.1016/0016-5085(95)90027-6
Agrawal, R. G., Hewetson, A., George, C. M., Syapin, P. J., and Bergeson, S. E. (2011). Minocycline reduces ethanol drinking. Brain Behav. Immun. 25, S165–S169. doi: 10.1016/j.bbi.2011.03.002
Alexeyev, M. F. (2009). Is there more to aging than mitochondrial DNA and reactive oxygen species. FEBS J. 276, 5768–5787. doi: 10.1111/j.1742-4658.2009.07269.x
Almansa, I., Fernández, A., García-Ruiz, C., Muriach, M., Barcia, J. M., Miranda, M., et al. (2009). Brain mitochondrial alterations after chronic alcohol consumption. J. Physiol. Biochem. 65, 305–312. doi: 10.1007/BF03180583>
Alzheimer’s Association (2021). 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 17, 327–406. doi: 10.1002/alz.12328
Araiso, Y., Tsutsumi, A., Qiu, J., Imai, K., Shiota, T., Song, J., et al. (2019). Structure of the mitochondrial import gate reveals distinct preprotein paths. Nature 575, 395–401. doi: 10.1038/s41586-019-1680-7
Asatryan, L., Ostrovskaya, O., Lieu, D., and Davies, D. L. (2018). Ethanol differentially modulates P2X4 and P2X7 receptor activity and function in BV2 microglial cells. Neuropharmacology 128, 11–21. doi: 10.1016/j.neuropharm.2017.09.030
American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders-v. Arlington, VA: American Psychiatric Association, doi: 10.1176/appi.books.9780890425596
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow. Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Ayers-Ringler, J. R., Jia, Y. F., Qiu, Y. Y., and Choi, D. S. (2016). Role of astrocytic glutamate transporter in alcohol use disorder. World J. Psychiatry 6, 31–42. doi: 10.5498/wjp.v6.i1.31
Baris, O. R., Ederer, S., Neuhaus, J. F., von Kleist-Retzow, J. C., Wunderlich, C. M., Pal, M., et al. (2015). Mosaic deficiency in mitochondrial oxidative metabolism promotes cardiac arrhythmia during aging. Cell Metab. 21, 667–677. doi: 10.1016/j.cmet.2015.04.005
Bell, R. L., Lopez, M. F., Cui, C., Egli, M., Johnson, K. W., Franklin, K. M., et al. (2015). Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict. Biol. 20, 38–42. doi: 10.1111/adb.12106
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Bender, A., Schwarzkopf, R. M., McMillan, A., Krishnan, K. J., Rieder, G., Neumann, M., et al. (2008). Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J. Neurol. 255, 1231–1235. doi: 10.1007/s00415-008-0892-9
Bergmann, F., and Keller, B. U. (2004). Impact of mitochondrial inhibition on excitability and cytosolic Ca2+ levels in brainstem motoneurones from mouse. J. Physiol. 555, 45–59. doi: 10.1113/jphysiol.2003.053900
Blednov, Y. A., Benavidez, J. M., Black, M., and Harris, R. A. (2014). Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Front. Neurosci. 8:129. doi: 10.3389/fnins.2014.00129
Bonet-Ponce, L., Saez-Atienzar, S., da Casa, C., Flores-Bellver, M., Barcia, J. M., Sancho-Pelluz, J., et al. (2015). On the mechanism underlying ethanol-induced mitochondrial dynamic disruption and autophagy response. Biochim. Biophys. Acta 1852, 1400–1409. doi: 10.1016/j.bbadis.2015.03.006
Bratic, A., and Larsson, N. G. (2013). The role of mitochondria in aging. J. Clin. Invest. 123, 951–957. doi: 10.1172/JCI64125
Brookmeyer, R., Johnson, E., Ziegler-Graham, K., and Arrighi, H. M. (2007). Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191. doi: 10.1016/j.jalz.2007.04.381
Brustovetsky, N., Brustovetsky, T., Purl, K. J., Capano, M., Crompton, M., and Dubinsky, J. M. (2003). Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J. Neurosci. 23, 4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Camp, M. C., Mayfield, R. D., McCracken, M., McCracken, L., and Alcantara, A. A. (2006). Neuroadaptations of Cdk5 in cholinergic interneurons of the nucleus accumbens and prefrontal cortex of inbred alcohol-preferring rats following voluntary alcohol drinking. Alcohol. Clin. Exp. Res. 30, 1322–1335. doi: 10.1111/j.1530-0277.2006.00160.x
Cannady, R., Rinker, J. A., Nimitvilai, S., Woodward, J. J., and Mulholland, P. J. (2018). Chronic alcohol, intrinsic excitability and potassium channels: neuroadaptations and drinking behavior. Handb. Exp. Pharmacol. 248, 311–343. doi: 10.1007/164_2017_90
Cao, Z., Wanagat, J., McKiernan, S. H., and Aiken, J. M. (2001). Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: analysis by laser-capture microdissection. Nucleic. Acids Res. 29, 4502–4508. doi: 10.1093/nar/29.21.4502
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics—fusion, fission, movement and mitophagy—in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Chen, J., Nam, H. W., Lee, M. R., Hinton, D. J., Choi, S., Kim, T., et al. (2010). Altered glutamatergic neurotransmission in the striatum regulates ethanol sensitivity and intake in mice lacking ENT1. Behav. Brain Res. 208, 636–642. doi: 10.1016/j.bbr.2010.01.011
Chinnery, P. F., DiMauro, S., Shanske, S., Schon, E. A., Zeviani, M., Mariotti, C., et al. (2004). Risk of developing a mitochondrial DNA deletion disorder. Lancet 364, 592–596. doi: 10.1016/S0140-6736(04)16851-7
Chocron, E. S., Munkácsy, E., and Pickering, A. M. (2019). Cause or casualty: the role of mitochondrial DNA in aging and age-associated disease. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 285–297. doi: 10.1016/j.bbadis.2018.09.035
Choi, D. S., Cascini, M. G., Mailliard, W., Young, H., Paredes, P., McMahon, T., et al. (2004). The type1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nat. Neurosci. 7, 855–861. doi: 10.1038/nn1288
Choi, Y., Kim, H.-S., Shin, K. Y., Kim, E.-M., Kim, M., Kim, H.-S., et al. (2007). Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology 32, 2393–2404. doi: 10.1038/sj.npp.1301377
Cioffi, F., Lanni, A., and Goglia, F. (2010). Thyroid hormones, mitochondrial bioenergetics and lipid handling. Curr. Opin. Endocrinol. Diabetes Obes. 17, 402–407. doi: 10.1097/MED.0b013e32833cf354
Clemens, V., Regen, F., Le Bret, N., Heuser, I., and Hellmann-Regen, J. (2018). Anti-inflammatory effects of minocycline are mediated by retinoid signaling. BMC Neurosci. 19, 58–58. doi: 10.1186/s12868-018-0460-x
Corkrum, M., Covelo, A., Lines, J., Bellocchio, L., Pisansky, M., Loke, K., et al. (2020). Dopamine-evoked synaptic regulation in the nucleus accumbens requires astrocyte activity. Neuron 105, 1036–1047.e5. doi: 10.1016/j.neuron.2019.12.026
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., Flint Beal, M., and Wallace, D. C. (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 2, 324–329. doi: 10.1038/ng1292-324
Cortopassi, G. A., and Arnheim, N. (1990). Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 18, 6927–6933. doi: 10.1093/nar/18.23.6927
Crews, F. T., Lawrimore, C. J., Walter, T. J., and Coleman, L. G., Jr. (2017). The role of neuroimmune signaling in alcoholism. Neuropharmacology 122, 56–73. doi: 10.1016/j.neuropharm.2017.01.031
De Biase, L. M., Schuebel, K. E., Fusfeld, Z. H., Jair, K., Hawes, I. A., Cimbro, R., et al. (2017). Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron 95, 341–356.e6. doi: 10.1016/j.neuron.2017.06.020
de la Monte, S. M., and Kril, J. J. (2014). Human alcohol-related neuropathology. Acta Neuropathol. 127, 71–90. doi: 10.1007/s00401-013-1233-3
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879. doi: 10.1038/nrm2275
Doonan, R., McElwee, J. J., Matthijssens, F., Walker, G. A., Houthoofd, K., Back, P., et al. (2008). Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in caenorhabditis elegans. Genes Dev. 22, 3236–3241. doi: 10.1101/gad.504808
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Durkee, C. A., and Araque, A. (2019). Diversity and specificity of astrocyte-neuron communication. Neuroscience 396, 73–78. doi: 10.1016/j.neuroscience.2018.11.010
Erickson, E. K., DaCosta, A. J., Mason, S. C., Blednov, Y. A., Mayfield, R. D., and Harris, R. A. (2021). Cortical astrocytes regulate ethanol consumption and intoxication in mice. Neuropsychopharmacology 46, 500–508. doi: 10.1038/s41386-020-0721-0
Erickson, E. K., Farris, S. P., Blednov, Y. A., Mayfield, R. D., and Harris, R. A. (2018). Astrocyte-specific transcriptome responses to chronic ethanol consumption. Pharmacogenomics J. 18, 578–589. doi: 10.1038/s41397-017-0012-2
Erickson, E. K., Grantham, E. K., Warden, A. S., and Harris, R. A. (2019). Neuroimmune signaling in alcohol use disorder. Pharmacol. Biochem. Behav. 177, 34–60. doi: 10.1016/j.pbb.2018.12.007
Fayet, G., Jansson, M., Sternberg, D., Moslemi, A.-R., Blondy, P., Lombès, A., et al. (2002). Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul. Disord. 12, 484–493. doi: 10.1016/s0960-8966(01)00332-7
Ferger, A. I., Campanelli, L., Reimer, V., Muth, K. N., Merdian, I., Ludolph, A. C., et al. (2010). Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflammation 7:45. doi: 10.1186/1742-2094-7-45
Fernandez-Lizarbe, S., Montesinos, J., and Guerri, C. (2013). Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J. Neurochem. 126, 261–273. doi: 10.1111/jnc.12276
Flores-Bellver, M., Bonet-Ponce, L., Barcia, J. M., Garcia-Verdugo, J. M., Martinez-Gil, N., Saez-Atienzar, S., et al. (2014). Autophagy and mitochondrial alterations in human retinal pigment epithelial cells induced by ethanol: implications of 4-hydroxy-nonenal. Cell Death Dis. 5:e1328. doi: 10.1038/cddis.2014.288
Fox, R. J., Raska, P., Barro, C., Karafa, M., Konig, V., Bermel, R. A., et al. (2021). Neurofilament light chain in a phase 2 clinical trial of ibudilast in progressive multiple sclerosis. Mult. Scler. 27, 2014–2022. doi: 10.1177/1352458520986956
Franco, R., Rivas-Santisteban, R., Casanovas, M., Lillo, A., Saura, C. A., and Navarro, G. (2020). Adenosine A(2A) Receptor antagonists affects NMDA glutamate receptor function. potential to address neurodegeneration in Alzheimer’s disease. Cells 9:1075. doi: 10.3390/cells9051075
Franklin, K. M., Asatryan, L., Jakowec, M. W., Trudell, J. R., Bell, R. L., and Davies, D. L. (2014). P2X4 receptors (P2X4Rs) represent a novel target for the development of drugs to prevent and/or treat alcohol use disorders. Front. Neurosci. 8:176. doi: 10.3389/fnins.2014.00176
Fukui, H., and Moraes, C. T. (2009). Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 18, 1028–1036. doi: 10.1093/hmg/ddn437
Gajbhiye, S. V., Tripathi, R. K., Petare, A., Potey, A. V., and Shankar, A. (2018). Minocycline in alcohol withdrawal induced anxiety and alcohol relapse in rats. Curr. Clin. Pharmacol. 13, 65–72. doi: 10.2174/1574884713666180228110310
Garcez, M. L., Mina, F., Bellettini-Santos, T., Carneiro, F. G., Luz, A. P., Schiavo, G. L., et al. (2017). Minocycline reduces inflammatory parameters in the brain structures and serum and reverses memory impairment caused by the administration of amyloid β (1–42) in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 77, 23–31. doi: 10.1016/j.pnpbp.2017.03.010
García-Suástegui, W. A., Ramos-Chávez, L. A., Rubio-Osornio, M., Calvillo-Velasco, M., Atzin-Méndez, J. A., Guevara, J., et al. (2017). The role of CYP2E1 in the drug metabolism or bioactivation in the brain. Oxid. Med. Cell Longev. 2017:4680732. doi: 10.1155/2017/4680732
Gendron, T. F., McCartney, S., Causevic, E., Ko, L. W., and Yen, S. H. (2008). Ethanol enhances tau accumulation in neuroblastoma cells that inducibly express tau. Neurosci. Lett. 443, 67–71. doi: 10.1016/j.neulet.2008.07.052
Genova, M. L., and Lenaz, G. (2015). The interplay between respiratory supercomplexes and ROS in aging. Antioxid. Redox Signal. 23, 208–238. doi: 10.1089/ars.2014.6214
Gimeno-Bayón, J., López-López, A., Rodríguez, M. J., and Mahy, N. (2014). Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 92, 723–731. doi: 10.1002/jnr.23356
Gomez-Nicola, D., and Boche, D. (2015). Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimers Res. Ther. 7:42. doi: 10.1186/s13195-015-0126-1
Goodman, A. D., Fedler, J. K., Yankey, J., Klingner, E. A., Ecklund, D. J., Goebel, C. V., et al. (2021). Response to ibudilast treatment according to progressive multiple sclerosis disease phenotype. Ann. Clin. Transl. Neurol. 8, 111–118. doi: 10.1002/acn3.51251
Grant, B. F., Chou, S. P., Saha, T. D., Pickering, R. P., Kerridge, B. T., Ruan, W. J., et al. (2017). Prevalence of 12-month alcohol use, high-risk drinking and DSM-IV alcohol use disorder in the united states, 2001–2002 to 2012–2013: results from the national epidemiologic survey on alcohol and related conditions. JAMA Psychiatry 74, 911–923. doi: 10.1001/jamapsychiatry.2017.2161
Grazioli, S., and Pugin, J. (2018). Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front. Immunol. 9:832. doi: 10.3389/fimmu.2018.00832
Gu, X., Wu, G., Yao, Y., Zeng, J., Shi, D., Lv, T., et al. (2015). Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the TLR9–p38 MAPK pathway. Free Radic. Biol. Med. 83, 149–158. doi: 10.1016/j.freeradbiomed.2015.02.034
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hara, Y., McKeehan, N., and Fillit, H. M. (2019). Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 92:84. doi: 10.1212/WNL.0000000000006745
Harper, C. G., Kril, J. J., and Holloway, R. L. (1985). Brain shrinkage in chronic alcoholics: a pathological study. Br. Med. J. (Clin. Res. Ed.) 290, 501–504. doi: 10.1136/bmj.290.6467.501
Harrison, N. L., Skelly, M. J., Grosserode, E. K., Lowes, D. C., Zeric, T., Phister, S., et al. (2017). Effects of acute alcohol on excitability in the CNS. Neuropharmacology 122, 36–45. doi: 10.1016/j.neuropharm.2017.04.007
Harwood, D. G., Kalechstein, A., Barker, W. W., Strauman, S., St George-Hyslop, P., Iglesias, C., et al. (2010). The effect of alcohol and tobacco consumption and apolipoprotein E genotype, on the age of onset in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 25, 511–518. doi: 10.1002/gps.2372
He, Y., Taylor, N., Fourgeaud, L., and Bhattacharya, A. (2017). The role of microglial P2X7: modulation of cell death and cytokine release. J. Neuroinflammation 14:135. doi: 10.1186/s12974-017-0904-8
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hinton, D. J., Lee, M. R., Jang, J. S., and Choi, D.-S. (2014). Type 1 equilibrative nucleoside transporter regulates astrocyte-specific glial fibrillary acidic protein expression in the striatum. Brain Behav. 4, 903–914. doi: 10.1002/brb3.283
Hong, S. I., Peyton, L., Chern, Y., and Choi, D. S. (2019). Novel adenosine analog, N6–(4-Hydroxybenzyl)-adenosine, dampens alcohol drinking and seeking behaviors. J. Pharmacol. Exp. Ther. 371, 260–267. doi: 10.1042/bj2700605
Hoppel, C. L., Lesnefsky, E. J., Chen, Q., and Tandler, B. (2017). “Mitochondrial dysfunction in cardiovascular aging,” in Mitochondrial Dynamics in Cardiovascular Medicine, ed G. Santulli (Cham: Springer International Publishing), 451–464. doi: 10.1080/15548627.2020.1797280
Howard, R., and Liu, K. Y. (2020). Questions EMERGE as biogen claims aducanumab turnaround. Nat. Rev. Neurol. 16, 63–64. doi: 10.1038/s41582-019-0295-9
Howard, R., Zubko, O., Bradley, R., Harper, E., Pank, L., O’Brien, J., et al. (2020). Minocycline at 2 different dosages vs. placebo for patients with mild Alzheimer disease: a randomized clinical trial. JAMA Neurol. 77, 164–174. doi: 10.1001/jamaneurol.2019.3762
Huang, L.-K., Chao, S.-P., and Hu, C.-J. (2020). Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 27:18. doi: 10.1186/s12929-019-0609-7
Imbimbo, B. P., Solfrizzi, V., and Panza, F. (2010). Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment. Front. Aging Neurosci. 2:19. doi: 10.3389/fnagi.2010.00019
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. doi: 10.1038/nature07317
Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S. V., Vieira-Saecker, A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi: 10.1038/s41586-019-1769-z
Iwata, M., Ota, K. T., Li, X.-Y., Sakaue, F., Li, N., Dutheil, S., et al. (2016). Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 Receptor. Biol. Psychiatry 80, 12–22. doi: 10.1016/j.biopsych.2015.11.026
Jauhari, A., Baranov, S. V., Suofu, Y., Kim, J., Singh, T., Yablonska, S., et al. (2020). Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Invest. 130, 3124–3136. doi: 10.1172/JCI135026
Ji, Z., Yuan, L., Lu, X., Ding, H., Luo, J., and Ke, Z.-J. (2018). Binge alcohol exposure causes neurobehavioral deficits and GSK3β activation in the hippocampus of adolescent rats. Sci. Rep. 8:3088. doi: 10.1038/s41598-018-21341-w
Kamal, H., Tan, G. C., Ibrahim, S. F., Shaikh, M. F., Mohamed, I. N., Mohamed, R. M. P., et al. (2020). Alcohol use disorder, neurodegeneration, Alzheimer’s and Parkinson’s disease: interplay between oxidative stress, neuroimmune response and excitotoxicity. Front. Cell Neurosci. 14:282. doi: 10.3389/fncel.2020.00282
Kang, S., and Choi, D. S. (2021). Astrocyte adenosine signaling and neural mechanisms of goal-directed and habitual reward-seeking behaviors. Neuropsychopharmacology 46, 227–228. doi: 10.1038/s41386-020-0787-8
Kang, S., Hong, S.-I., Lee, J., Peyton, L., Baker, M., Choi, S., et al. (2020). Activation of astrocytes in the dorsomedial striatum facilitates transition from habitual to goal-directed reward-seeking behavior. Biol. Psychiatry 88, 797–808. doi: 10.1016/j.biopsych.2020.04.023
Kastanenka, K. V., Bussiere, T., Shakerdge, N., Qian, F., Weinreb, P. H., Rhodes, K., et al. (2016). Immunotherapy with Aducanumab Restores Calcium Homeostasis in Tg2576 Mice. J. Neurosci. 36, 12549–12558. doi: 10.1523/JNEUROSCI.2080-16.2016
Kauppila, T. E. S., Kauppila, J. H. K., and Larsson, N.-G. (2017). Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71. doi: 10.1016/j.cmet.2016.09.017
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
Khakh, B. S., and Sofroniew, M. V. (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952. doi: 10.1038/nn.4043
Kim, G. H., Kim, J. E., Rhie, S. J., and Yoon, S. (2015). The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340. doi: 10.5607/en.2015.24.4.325
Kim, I., Rodriguez-Enriquez, S., and Lemasters, J. J. (2007). Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253. doi: 10.1016/j.abb.2007.03.034
Kim, T., Vanderboom, P., Conard, P., and Choi, D.-S. (2013). Adenoine receptors in psychiatirc and neurological disorders. Nova Sci. 1808, 1380–1399. doi: 10.1016/j.bbamem.2010.12.001
Kimura, R., Ma, L. Y., Wu, C., Turner, D., Shen, J. X., Ellsworth, K., et al. (2012). Acute exposure to the mitochondrial complex I toxin rotenone impairs synaptic long-term potentiation in rat hippocampal slices. CNS Neurosci. Ther. 18, 641–646. doi: 10.1111/j.1755-5949.2012.00337.x
Kleele, T., Rey, T., Winter, J., Zaganelli, S., Mahecic, D., Perreten Lambert, H., et al. (2021). Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439. doi: 10.1016/j.micpath.2021.105347
Knopman, D. S., Jones, D. T., and Greicius, M. D. (2021). Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by biogen, december 2019. Alzheimers Dement. 17, 696–701. doi: 10.1002/alz.12213
Kotilinek, L. A., Westerman, M. A., Wang, Q., Panizzon, K., Lim, G. P., Simonyi, A., et al. (2008). Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain 131, 651–664. doi: 10.1093/brain/awn008
Kraytsberg, Y., Kudryavtseva, E., McKee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520. doi: 10.1038/ng1778
Krestinina, O., Azarashvili, T., Baburina, Y., Galvita, A., Grachev, D., Stricker, R., et al. (2015). In aging, the vulnerability of rat brain mitochondria is enhanced due to reduced level of 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNP) and subsequently increased permeability transition in brain mitochondria in old animals. Neurochem. Int. 80, 41–50. doi: 10.1016/j.neuint.2014.09.008
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress and apoptosis in mammalian aging. Science 309, 481–484. doi: 10.1126/science.1112125
Kusakawa, G., Saito, T., Onuki, R., Ishiguro, K., Kishimoto, T., and Hisanaga, S. (2000). Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J. Biol. Chem. 275, 17166–17172. doi: 10.1074/jbc.M907757199
LaFrance, R., Brustovetsky, N., Sherburne, C., Delong, D., and Dubinsky, J. M. (2005). Age-related changes in regional brain mitochondria from fischer 344 rats. Aging Cell 4, 139–145. doi: 10.1111/j.1474-9726.2005.00156.x
Le Belle, J. E., Orozco, N. M., Paucar, A. A., Saxe, J. P., Mottahedeh, J., Pyle, A. D., et al. (2011). Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8, 59–71. doi: 10.1016/j.stem.2010.11.028
Lee, M. R., Ruby, C. L., Hinton, D. J., Choi, S., Adams, C. A., Young Kang, N., et al. (2013). Striatal adenosine signaling regulates EAAT2 and astrocytic AQP4 expression and alcohol drinking in mice. Neuropsychopharmacology 38, 437–445. doi: 10.1038/npp.2012.198
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lim, G. P., Chu, T., Yang, F., Beech, W., Frautschy, S. A., and Cole, G. M. (2001). The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 21, 8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001
Lim, G. P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., et al. (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 20, 5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000
Lindberg, D., Andres-Beck, L., Jia, Y. F., Kang, S., and Choi, D. S. (2018). Purinergic signaling in neuron-astrocyte interactions, circadian rhythms and alcohol use disorder. Front. Physiol. 9:9. doi: 10.3389/fphys.2018.00009
Lindberg, D., Ho, A. M. C., Peyton, L., and Choi, D. S. (2019). Chronic ethanol exposure disrupts lactate and glucose homeostasis and induces dysfunction of the astrocyte-neuron lactate shuttle in the brain. Alcohol. Clin. Exp. Res. 43, 1838–1847. doi: 10.1111/acer.14137
Liu, Y., Chen, G., Ma, C., Bower, K. A., Xu, M., Fan, Z., et al. (2009). Overexpression of glycogen synthase kinase 3beta sensitizes neuronal cells to ethanol toxicity. J. Neurosci. Res. 87, 2793–2802. doi: 10.1002/jnr.22098
Liu, B., Le, K. X., Park, M. A., Wang, S., Belanger, A. P., Dubey, S., et al. (2015). in vivo Detection of age- and disease-related increases in neuroinflammation by 18F-GE180 TSPO MicroPET imaging in wild-type and Alzheimer’s transgenic mice. J. Neurosci. 35, 15716–15730. doi: 10.1523/JNEUROSCI.0996-15.2015
Liu, S. L., Wang, C., Jiang, T., Tan, L., Xing, A., and Yu, J. T. (2016). The role of Cdk5 in Alzheimer’s disease. Mol. Neurobiol. 53, 4328–4342. doi: 10.1007/s12035-015-9369-x
Luchsinger, J. A., and Mayeux, R. (2004). Dietary factors and Alzheimer’s disease. Lancet Neurol. 3, 579–587. doi: 10.1016/S1474-4422(04)00878-6
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Aß to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Manczak, M., Jung, Y., Park, B. S., Partovi, D., and Reddy, P. H. (2005). Time-course of mitochondrial gene expressions in mice brains: implications for mitochondrial dysfunction, oxidative damage and cytochrome c in aging. J. Neurochem. 92, 494–504. doi: 10.1111/j.1471-4159.2004.02884.x
Mansouri, A., Demeilliers, C., Amsellem, S., Pessayre, D., and Fromenty, B. (2001). Acute ethanol administration oxidatively damages and depletes mitochondrial dna in mouse liver, brain, heart and skeletal muscles: protective effects of antioxidants. J. Pharmacol. Exp. Ther. 298, 737–743.
Marques-Aleixo, I., Rocha-Rodrigues, S., Santos-Alves, E., Coxito, P. M., Passos, E., Oliveira, P. J., et al. (2012). in vitro salicylate does not further impair aging-induced brain mitochondrial dysfunction. Toxicology 302, 51–59. doi: 10.1016/j.tox.2012.07.018
McCarthy, G. M., Farris, S. P., Blednov, Y. A., Harris, R. A., and Mayfield, R. D. (2018). Microglial-specific transcriptome changes following chronic alcohol consumption. Neuropharmacology 128, 416–424. doi: 10.1016/j.neuropharm.2017.10.035
McCasland, J. S., and Hibbard, L. S. (1997). GABAergic neurons in barrel cortex show strong, whisker-dependent metabolic activation during normal behavior. J. Neurosci. 17:5509. doi: 10.1523/JNEUROSCI.17-14-05509.1997
McElroy, P. B., Liang, L.-P., Day, B. J., and Patel, M. (2017). Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 298, 13–22. doi: 10.1016/j.expneurol.2017.08.009
Mihic, S. J., Ye, Q., Wick, M. J., Koltchine, V. V., Krasowski, M. D., Finn, S. E., et al. (1997). Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature 389, 385–389. doi: 10.1038/38738
Mockett, R. J., Bayne, A.-C. V., Kwong, L. K., Orr, W. C., and Sohal, R. S. (2003). Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic. Biol. Med. 34, 207–217. doi: 10.1016/s0891-5849(02)01190-5
Mockett, R. J., Sohal, B. H., and Sohal, R. S. (2010). Expression of multiple copies of mitochondrially targeted catalase or genomic Mn superoxide dismutase transgenes does not extend the life span of Drosophila melanogaster. Free Radic. Biol. Med. 49, 2028–2031. doi: 10.1016/j.freeradbiomed.2010.09.029
Navarro, A., and Boveris, A. (2007). The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 292, C670–C686. doi: 10.1152/ajpcell.00213.2006
Neupane, S. P. (2016). Neuroimmune interface in the comorbidity between alcohol use disorder and major depression. Front. Immunol. 7, 655–655. doi: 10.3389/fimmu.2016.00655
Nisoli, E., Tonello, C., Cardile, A., Cozzi, V., Bracale, R., Tedesco, L., et al. (2005). Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310, 314–317. doi: 10.1126/science.1117728
Ogrodnik, M., Miwa, S., Tchkonia, T., Tiniakos, D., Wilson, C. L., Lahat, A., et al. (2017). Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 8:15691. doi: 10.1038/ncomms15691
Ojaimi, J., Masters, C. L., Opeskin, K., McKelvie, P., and Byrne, E. (1999). Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 111, 39–47. doi: 10.1016/s0047-6374(99)00071-8
Oliveira-Marques, V., Marinho, H. S., Cyrne, L., and Antunes, F. (2009). Role of hydrogen peroxide in NF-κB activation: from inducer to modulator. Antioxid. Redox Signal. 11, 2223–2243. doi: 10.1089/ars.2009.2601
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Orr, W. C., Mockett, R. J., Benes, J. J., and Sohal, R. S. (2003). Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase and thioredoxin reductase egnes on longevity in Drosophila melanogaster. J. Biol. Chem. 278, 26418–26422. doi: 10.1074/jbc.M303095200
Panel, M., Ghaleh, B., and Morin, D. (2018). Mitochondria and aging: a role for the mitochondrial transition pore. Aging Cell 17:e12793. doi: 10.1111/acel.12793
Pascual, M., Baliño, P., Aragón, C. M., and Guerri, C. (2015). Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: role of TLR4 and TLR2. Neuropharmacology 89, 352–359. doi: 10.1016/j.neuropharm.2014.10.014
Payne, B. A. I., Wilson, I. J., Yu-Wai-Man, P., Coxhead, J., Deehan, D., Horvath, R., et al. (2012). Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 22, 384–390. doi: 10.1093/hmg/dds435
Pleil, K. E., Lowery-Gionta, E. G., Crowley, N. A., Li, C., Marcinkiewcz, C. A., Rose, J. H., et al. (2015). Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology 99, 735–749. doi: 10.1016/j.neuropharm.2015.06.017
Ray, L. A., Bujarski, S., Roche, D. J. O., and Magill, M. (2018). Overcoming the “valley of death” in medications development for alcohol use disorder. Alcohol. Clin. Exp. Res. 42, 1612–1622. doi: 10.1111/acer.13829
Reddy, V. D., Padmavathi, P., Kavitha, G., Saradamma, B., and Varadacharyulu, N. (2013). Alcohol-induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Mol. Cell Biochem. 375, 39–47. doi: 10.1007/s11010-012-1526-1
Regen, F., Hildebrand, M., Le Bret, N., Herzog, I., Heuser, I., and Hellmann-Regen, J. (2015). Inhibition of retinoic acid catabolism by minocycline: evidence for a novel mode of action. Exp. Dermatol. 24, 473–476. doi: 10.1111/exd.12692
Rehm, J., Hasan, O. S. M., Black, S. E., Shield, K. D., and Schwarzinger, M. (2019). Alcohol use and dementia: a systematic scoping review. Alzheimers Res. Ther. 11:1. doi: 10.1186/s13195-018-0453-0
Rehm, J., and Shield, K. D. (2019). Global burden of disease and the impact of mental and addictive disorders. Curr. Psychiatry Rep. 21:10. doi: 10.1007/s11920-019-0997-0
Réus, G. Z., Abelaira, H. M., Maciel, A. L., Dos Santos, M. A., Carlessi, A. S., Steckert, A. V., et al. (2015). Minocycline protects against oxidative damage and alters energy metabolism parameters in the brain of rats subjected to chronic mild stress. Metab. Brain Dis. 30, 545–553. doi: 10.1007/s11011-014-9602-8
Rose, J., Brian, C., Woods, J., Pappa, A., Panayiotidis, M. I., Powers, R., et al. (2017). Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 391, 109–115. doi: 10.1016/j.tox.2017.06.011
Ross, J. M., Stewart, J. B., Hagström, E., Brené, S., Mourier, A., Coppotelli, G., et al. (2013). Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501, 412–415. doi: 10.1038/nature12474
Rossignol, R., Faustin, B., Rocher, C., Malgat, M., Mazat, J.-P., and Letellier, T. (2003). Mitochondrial threshold effects. Biochem. J. 370, 751–762. doi: 10.1042/BJ20021594
Rubio-Araiz, A., Porcu, F., Pérez-Hernández, M., García-Gutiérrez, M. S., Aracil-Fernández, M. A., Gutierrez-López, M. D., et al. (2017). Disruption of blood-brain barrier integrity in postmortem alcoholic brain: preclinical evidence of TLR4 involvement from a binge-like drinking model. Addict. Biol. 22, 1103–1116. doi: 10.1111/adb.12376
Saura, J., Angulo, E., Ejarque, A., Casadó, V., Tusell, J. M., Moratalla, R., et al. (2005). Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J. Neurochem. 95, 919–929. doi: 10.1111/j.1471-4159.2005.03395.x
Schneider, L. (2020). A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 19, 111–112. doi: 10.1016/S1474-4422(19)30480-6
Scofield, M. D., and Kalivas, P. W. (2014). Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist 20, 610–622. doi: 10.1177/1073858413520347
Seth, R. B., Sun, L., Ea, C.-K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56. doi: 10.1038/nature19323
Shadel, G. S., and Horvath, T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569. doi: 10.1016/j.cell.2015.10.001
Shang, P., Lindberg, D., Starski, P., Peyton, L., Hong, S.-I., Choi, S., et al. (2020). Chronic alcohol exposure induces aberrant mitochondrial morphology and inhibits respiratory capacity in the medial prefrontal cortex of mice. Front. Neurosci. 14, 561173–561173. doi: 10.3389/fnins.2020.561173
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Short, K. R., Bigelow, M. L., Kahl, J., Singh, R., Coenen-Schimke, J., Raghavakaimal, S., et al. (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. U S A 102, 5618–5623. doi: 10.1073/pnas.0501559102
Stocco, D. M., and Hutson, J. C. (1978). Quantitation of mitochondrial DNA and protein in the liver of Fischer 344 rats during aging. J. Gerontol. 33, 802–809. doi: 10.1093/geronj/33.6.802
Stojakovic, A., Trushin, S., Sheu, A., Khalili, L., Chang, S.-Y., Li, X., et al. (2021). Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Commun. Biol. 4:61. doi: 10.1038/s42003-020-01584-y
Styr, B., Gonen, N., Zarhin, D., Ruggiero, A., Atsmon, R., Gazit, N., et al. (2019). Mitochondrial regulation of the hippocampal firing rate set point and seizure susceptibility. Neuron 102, 1009–1024.e8. doi: 10.1016/j.neuron.2019.03.045
Su, L.-J., Zhang, J.-H., Gomez, H., Murugan, R., Hong, X., Xu, D., et al. (2019). Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy and ferroptosis. Oxid. Med. Cell Longev. 2019:5080843. doi: 10.1155/2019/5080843
Suen, D. F., Norris, K. L., and Youle, R. J. (2008). Mitochondrial dynamics and apoptosis. Genes. Dev. 22, 1577–1590. doi: 10.1101/gad.1658508
Sun, Q., Sun, L., Liu, H.-H., Chen, X., Seth, R. B., Forman, J., et al. (2006). The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24, 633–642. doi: 10.1016/j.immuni.2006.04.004
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. doi: 10.1126/science.1232458
Suofu, Y., Li, W., Jean-Alphonse, F. G., Jia, J., Khattar, N. K., Li, J., et al. (2017). Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. U S A 114, E7997–E8006. doi: 10.1073/pnas.1705768114
Szabo, G., and Lippai, D. (2014). Converging actions of alcohol on liver and brain immune signaling. Int. Rev. Neurobiol. 118, 359–380. doi: 10.1016/B978-0-12-801284-0.00011-7
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Tang, Y., and Zucker, R. S. (1997). Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18, 483–491. doi: 10.1016/s0896-6273(00)81248-9
Tapia-Rojas, C., Torres, A. K., and Quintanilla, R. A. (2019). Adolescence binge alcohol consumption induces hippocampal mitochondrial impairment that persists during the adulthood. Neuroscience 406, 356–368. doi: 10.1016/j.neuroscience.2019.03.018
Taylor, S. D., Ericson, N. G., Burton, J. N., Prolla, T. A., Silber, J. R., Shendure, J., et al. (2014). Targeted enrichment and high-resolution digital profiling of mitochondrial DNA deletions in human brain. Aging Cell 13, 29–38. doi: 10.1111/acel.12146
Tranah, G. J., Katzman, S. M., Lauterjung, K., Yaffe, K., Manini, T. M., Kritchevsky, S., et al. (2018). Mitochondrial DNA m.3243A > G heteroplasmy affects multiple aging phenotypes and risk of mortality. Sci. Rep. 8:11887. doi: 10.1038/s41598-018-30255-6
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Tu, Y., Kroener, S., Abernathy, K., Lapish, C., Seamans, J., Chandler, L. J., et al. (2007). Ethanol inhibits persistent activity in prefrontal cortical neurons. J. Neurosci. 27, 4765–4775. doi: 10.1523/JNEUROSCI.5378-06.2007
Tyumentsev, M. A., Stefanova, N. A., Muraleva, N. A., Rumyantseva, Y. V., Kiseleva, E., Vavilin, V. A., et al. (2018). Mitochondrial dysfunction as a predictor and driver of Alzheimer’s disease-like pathology in OXYS rats. J. Alzheimer’s Dis. 63, 1075–1088. doi: 10.3233/JAD-180065
Van Raamsdonk, J. M., and Hekimi, S. (2009). Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in caenorhabditis elegans. PLoS Genet. 5:e1000361. doi: 10.1371/journal.pgen.1000361
Venkateshappa, C., Harish, G., Mahadevan, A., Srinivas Bharath, M. M., and Shankar, S. K. (2012a). Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer’s disease. Neurochem. Res. 37, 1601–1614. doi: 10.1007/s11064-012-0755-8
Venkateshappa, C., Harish, G., Mythri, R. B., Mahadevan, A., Bharath, M. M., and Shankar, S. K. (2012b). Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: implications for Parkinson’s disease. Neurochem. Res. 37, 358–369. doi: 10.1007/s11064-011-0619-7
Venkataraman, A., Kalk, N., Sewell, G., Ritchie, C. W., and Lingford-Hughes, A. (2017). Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s disease. Alcohol Alcohol. 52, 151–158. doi: 10.1093/alcalc/agw092
Venkatraman, A., Landar, A., Davis, A. J., Chamlee, L., Sanderson, T., Kim, H., et al. (2004). Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J. Biol. Chem. 279, 22092–22101. doi: 10.1074/jbc.M402245200
Vezzani, B., Carinci, M., Patergnani, S., Pasquin, M. P., Guarino, A., Aziz, N., et al. (2020). The dichotomous role of inflammation in the CNS: a mitochondrial point of view. Biomolecules 10:1437. doi: 10.3390/biom10101437
Voloboueva, L. A., Emery, J. F., Sun, X., and Giffard, R. G. (2013). Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 587, 756–762. doi: 10.1016/j.febslet.2013.01.067
Walter, T. J., and Crews, F. T. (2017). Microglial depletion alters the brain neuroimmune response to acute binge ethanol withdrawal. J. Neuroinflammation 14:86. doi: 10.1186/s12974-017-0856-z
Waltz, T. B., Fivenson, E. M., Morevati, M., Li, C., Becker, K. G., Bohr, V. A., et al. (2018). Sarcopenia, aging and prospective interventional strategies. Curr. Med. Chem. 25, 5588–5596. doi: 10.2174/0929867324666170801095850
Wanagat, J., Cao, Z., Pathare, P., and Aiken, J. M. (2001). Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting and oxidative damage in sarcopenia. FASEB J. 15, 322–332. doi: 10.1096/fj.00-0320com
Wang, H., Mei, Z. L., Zhong, K. L., Hu, M., Long, Y., Miao, M. X., et al. (2014). Pretreatment with antiasthmatic drug ibudilast ameliorates Aβ1–42-induced memory impairment and neurotoxicity in mice. Pharmacol. Biochem. Behav. 124, 373–379. doi: 10.1016/j.pbb.2014.07.006
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15:30. doi: 10.1186/s13024-020-00376-6
Warden, A. S., Wolfe, S. A., Khom, S., Varodayan, F. P., Patel, R. R., Steinman, M. Q., et al. (2020). Microglia control escalation of drinking in alcohol-dependent mice: genomic and synaptic drivers. Biol. Psychiatry 88, 910–921. doi: 10.1016/j.biopsych.2020.05.011
Weinberg, S. E., Sena, L. A., and Chandel, N. S. (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. doi: 10.1016/j.immuni.2015.02.002
Weitlauf, C., and Woodward, J. J. (2008). Ethanol selectively attenuates NMDAR-mediated synaptic transmission in the prefrontal cortex. Alcohol. Clin. Exp. Res. 32, 690–698. doi: 10.1111/j.1530-0277.2008.00625.x
Wes, P. D., Holtman, I. R., Boddeke, E. W., Möller, T., and Eggen, B. J. (2016). Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 64, 197–213. doi: 10.1002/glia.22866
Xue, J., Schmidt, S. V., Sander, J., Draffehn, A., Krebs, W., Quester, I., et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. doi: 10.1016/j.immuni.2014.01.006
Yao, J., Du, H., Yan, S., Fang, F., Wang, C., Lue, L. F., et al. (2011). Inhibition of amyloid-β (Aβ) peptide-binding alcohol dehydrogenase-Aβ interaction reduces Aβ accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 31, 2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011
Ye, J., Jiang, Z., Chen, X., Liu, M., Li, J., and Liu, N. (2016). Electron transport chain inhibitors induce microglia activation through enhancing mitochondrial reactive oxygen species production. Exp. Cell Res. 340, 315–326. doi: 10.1016/j.yexcr.2015.10.026
Yen, T.-C., Su, J.-H., King, K.-L., and Wei, Y.-H. (1991). Ageing-associated 5 kb deletion in human liver mitochondrial DNA. Biochem. Biophys. Res. Commun. 178, 124–131. doi: 10.1016/0006-291x(91)91788-e
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Yu, J., Nagasu, H., Murakami, T., Hoang, H., Broderick, L., Hoffman, H. M., et al. (2014). Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. U S A 111, 15514–15519. doi: 10.1073/pnas.1414859111
Zarghi, A., and Arfaei, S. (2011). Selective COX-2 inhibitors: a review of their structure-activity relationships. Iran. J. Pharm. Res. 10, 655–683.
Zhang, Y., Ikeno, Y., Qi, W., Chaudhuri, A., Li, Y., Bokov, A., et al. (2009). Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J. Gerontol. A. Biol. Sci. Med. Sci. 64, 1212–1220. doi: 10.1093/gerona/glp132
Zhang, R., Wang, Y., Ye, K., Picard, M., and Gu, Z. (2017). Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genomics 18:890. doi: 10.1186/s12864-017-4287-0
Zhang, H.-M., and Zhang, Y. (2014). Melatonin: a well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 57, 131–146. doi: 10.1111/jpi.12162
Zhang, L., Zhang, S., Maezawa, I., Trushin, S., Minhas, P., Pinto, M., et al. (2015). Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease. EBioMedicine 2, 294–305. doi: 10.1016/j.ebiom.2015.03.009
Zhang, L., Zhang, S., Maezawa, I., Trushin, S., Minhas, P., Pinto, M., et al. (2019). Corrigendum to “Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease” [EBioMedicine 2 (2015) 294–305]. EBioMedicine 42:532. doi: 10.1016/j.ebiom.2019.03.062
Keywords: alcohol use disorder, mitochondria, morphology, aging, dementia, Alzheimer’s disease
Citation: León BE, Kang S, Franca-Solomon G, Shang P and Choi D-S (2022) Alcohol-Induced Neuroinflammatory Response and Mitochondrial Dysfunction on Aging and Alzheimer’s Disease. Front. Behav. Neurosci. 15:778456. doi: 10.3389/fnbeh.2021.778456
Received: 16 September 2021; Accepted: 07 December 2021;
Published: 10 February 2022.
Edited by:
Tiffany A. Wills, Louisiana State University, United StatesReviewed by:
Terrence Deak, Binghamton University, United StatesEmmanuel Moyse, Université de Tours, France
Copyright © 2022 León, Kang, Franca-Solomon, Shang and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Doo-Sup Choi, Y2hvaWRzQG1heW8uZWR1