 Ayako Kawatake-Kuno
Ayako Kawatake-Kuno Toshiya Murai
Toshiya Murai Shusaku Uchida
Shusaku Uchida
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Behav. Neurosci. , 30 September 2021
Sec. Emotion Regulation and Processing
Volume 15 - 2021 | https://doi.org/10.3389/fnbeh.2021.749180
This article is part of the Research Topic Neural Plasticity and Behavioral Responses to Adversity View all 7 articles
Major depressive disorder (MDD) is a debilitating disease characterized by depressed mood, loss of interest or pleasure, suicidal ideation, and reduced motivation or hopelessness. Despite considerable research, mechanisms underlying MDD remain poorly understood, and current advances in treatment are far from satisfactory. The antidepressant effect of ketamine is among the most important discoveries in psychiatric research over the last half-century. Neurobiological insights into the ketamine’s effects have shed light on the mechanisms underlying antidepressant efficacy. However, mechanisms underlying the rapid and sustained antidepressant effects of ketamine remain controversial. Elucidating such mechanisms is key to identifying new therapeutic targets and developing therapeutic strategies. Accumulating evidence demonstrates the contribution of the glutamatergic pathway, the major excitatory neurotransmitter system in the central nervous system, in MDD pathophysiology and antidepressant effects. The hypothesis of a connection among the calcium signaling cascade stimulated by the glutamatergic system, neural plasticity, and epigenetic regulation of gene transcription is further supported by its associations with ketamine’s antidepressant effects. This review briefly summarizes the potential mechanisms of ketamine’s effects with a specific focus on glutamatergic signaling from a multiscale perspective, including behavioral, cellular, molecular, and epigenetic aspects, to provide a valuable overview of ketamine’s antidepressant effects.
Major depressive disorder (MDD) is the leading cause of disability worldwide. Despite considerable research, biological mechanisms underlying MDD pathophysiology remain unclear, with significant unmet needs for treatment. Typical antidepressants, including selective serotonin reuptake inhibitors (SSRIs) and serotonin and noradrenaline reuptake inhibitors, increase monoamine concentration in the synaptic cleft, resulting in antidepressant effects (Berton and Nestler, 2006). However, although increased monoamine concentration in the synapse occurs relatively quickly as an acute pharmacological action, recovery from depression takes several weeks to months in clinical practice (Krishnan and Nestler, 2008). Electroconvulsive therapy (ECT) is also an effective treatment for drug-resistant depression, although achieving clinically meaningful or sustained remission with ECT required at least 1 month (Yamasaki et al., 2020). Such substantial time lags are a major concern since patients with depression are at high risk for suicide. Thus, there is an urgent need to develop antidepressants with rapid onset and sustained effectiveness.
Ketamine, a non-competitive glutamate N-methyl-D-aspartate receptor (NMDAR) antagonist, has gained considerable interest in the neuropsychiatric field. A single administration of ketamine elicits rapid and sustained antidepressant effects for 1–2 weeks in both humans and animals (Berman et al., 2000; Zarate et al., 2006; Li et al., 2010; Autry et al., 2011). This discovery offered new insight into the investigation of a whole new class of agents beyond the monoamine system to treat depression (Chaki, 2017). Esketamine, an enantiomer of (R,S)-ketamine, has been approved by the U.S. Food and Drug Administration (USFDA) for treating patients with treatment-resistant depression. Thus, research on pathophysiology and drug discovery for MDD has transitioned from the monoaminergic to the glutamatergic system. Recently, the importance of multiscale neuroscience to study cross-scale interactions at genetic, molecular, cellular, and macroscale levels of brain circuitry, connectivity, and behavior has been emphasized to establish a comprehensive understanding of neuropsychiatric disease (Van Den Heuvel et al., 2019). This mini-review aims to update the current knowledge regarding ketamine effect on the brain, focusing on the glutamatergic signaling pathway from a multiscale perspective at the behavioral, cellular, molecular, and epigenetic levels.
Glutamate is the major excitatory neurotransmitter in the brain, and increasing evidence indicates that dysfunction in glutamatergic signaling contributes to MDD pathophysiology (Popoli et al., 2011; Duman and Aghajanian, 2012; Thompson et al., 2015; Duman et al., 2019; Xia et al., 2021). The glutamatergic system is modulated by both ionotropic [NMDARs, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), and kainate receptors] and metabotropic glutamate receptors (mGluRs). NMDARs are found throughout the central nervous system and contribute to synaptic calcium (Ca2+) influx, which is required for activity-dependent synaptic plasticity (Koester and Sakmann, 1998; Reid et al., 2001; Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007; Carter et al., 2007). NMDAR function is tightly linked to AMPAR, which gates sodium and mediates fast excitatory transmission. Increased AMPAR density in the postsynaptic membrane causes NMDAR-dependent long-term potentiation (LTP) (Huganir and Nicoll, 2013). AMPARs can also have several direct effects on synaptic transmission (i.e., LTP) and intracellular signals without the proper functioning of NMDARs. This NMDAR-independent and AMPAR-dependent intracellular signaling pathway is also hypothesized to underlie ketamine’s antidepressant actions (Zanos et al., 2016; Duman et al., 2019; Wei et al., 2021).
Ca2+ influx into the postsynaptic neuron stimulates a signaling-cascade, such as calcium/calmodulin-dependent kinases [CAMKs; e.g., calcium/calmodulin-dependent kinase II (CaMKIIs), eukaryotic elongation factor 2 (eEF2) kinase]. Brain-derived neurotrophic factor (BDNF) and its receptor, neurotrophic receptor tyrosine kinase 2 (TrkB), also plays a key role in synaptic plasticity (Minichiello, 2009). TrkB activation stimulates phospholipase Cγ1 (PLCγ1), which results in CaMK activation (Minichiello, 2009). Calcium-signaling activation further sends its signal toward downstream epigenetic and transcription modulators, such as MEF2, MeCP2, and HDAC5. These pathways modulate gene expression that affects dendritic growth, synaptic development, and neuronal plasticity (Greer and Greenberg, 2008; Graff and Tsai, 2013; Takemoto-Kimura et al., 2017; Uchida and Shumyatsky, 2018a,b; Figure 1). Taken together, calcium-signaling stimulation through NMDARs and/or AMPARs activates multiple downstream nucleocytoplasmic pathways; it induces activity-dependent epigenetic genetic expression, contributing to depression and antidepressant action.
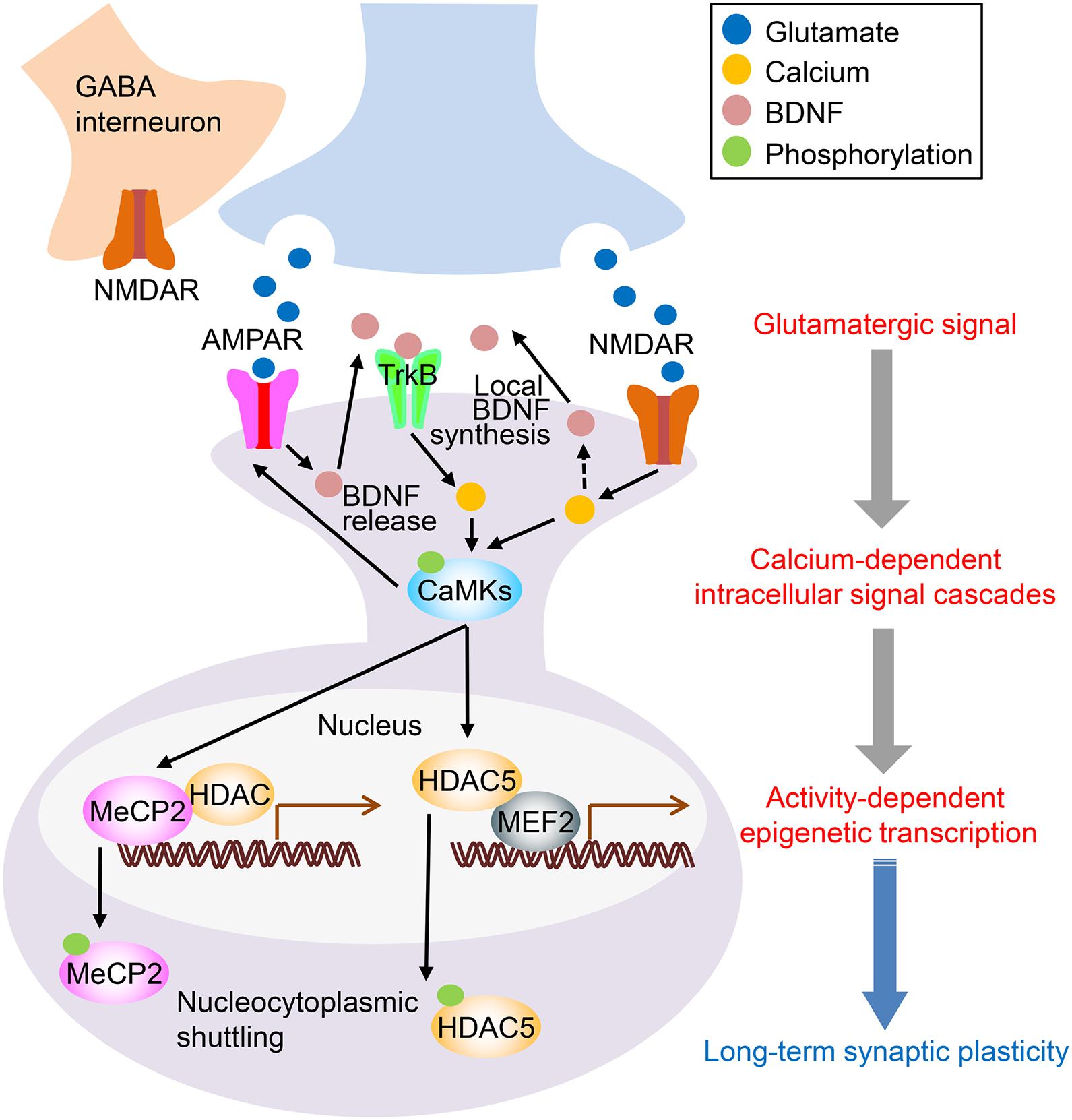
Figure 1. Proposed mechanisms of ketamine’s antidepressant action. The binding of ketamine to N-methyl-D-aspartate receptors (NMDARs) on GABAergic interneurons disinhibits glutamatergic neurons, which results in increased synaptic glutamate release. AMPAR activation by glutamate increases brain-derived neurotrophic factor (BDNF) levels. Although the exact source of BDNF is yet to be determined, local release of BDNF is thought to stimulate TrkB receptors. This activation activates intracellular signaling, such as the Ca2+ pathway. Another mechanism is the direct inhibition of NMDAR by ketamine. Inhibiting postsynaptic NMDARs reduces eEF2 via the inactivation of CaMK (eEF2 kinase), which leads to enhanced local protein synthesis of BDNF. Increased intracellular Ca2+ stimulates CaMKs and their downstream targets, including MeCP2, MEF2, and HDAC5. MeCP2, a transcriptional regulator, binds to methylated CpG sites on the genomic region and interacts with other transcription repressors, including HDACs. CaMKII phosphorylates MeCP2, promotes its nuclear export, and increases activity-dependent transcription. MEF2 recruits HDAC5 and removes activating acetyl groups from histones, which results in a silenced or repressed state of transcription. CaMKII phosphorylates HDAC5, which promotes nuclear export and increases activity-dependent transcription. Ketamine is known to increase the phosphorylation of CaMKII, MeCP2, and HDAC5 (see detail in the main text). Thus, ketamine-mediated enhancement of intracellular Ca2+ signaling is linked to epigenetic regulation of transcription, which leads to long-term synaptic plasticity and, consequently, prolonged antidepressant-like effects.
Chronic stress initiates and exacerbates several psychiatric illnesses. Indeed, adverse stressful environments are associated with the pathophysiology of major psychiatric disorders, including mood and anxiety disorders (Mcewen, 2007; Krishnan and Nestler, 2008; Duman and Aghajanian, 2012). There are several evidences demonstrating alterations in the expression and/or function of glutamatergic signaling and its downstream molecules (e.g., NMDARs, AMPARs, CaMKIIs, MEF2, MeCP2, and HDAC5), which is associated with plasticity and behaviors induced by chronic stress, traditional antidepressant drugs, and/or ketamine (Table 1). Moreover, molecular dysregulation associated with glutamatergic system is visible in postmortem brain tissues of patients with MDD (Table 1). Thus, such clinical and preclinical evidences suggest that calcium-signaling is a downstream target of the glutamatergic system in MDD pathophysiology and antidepressant effects.
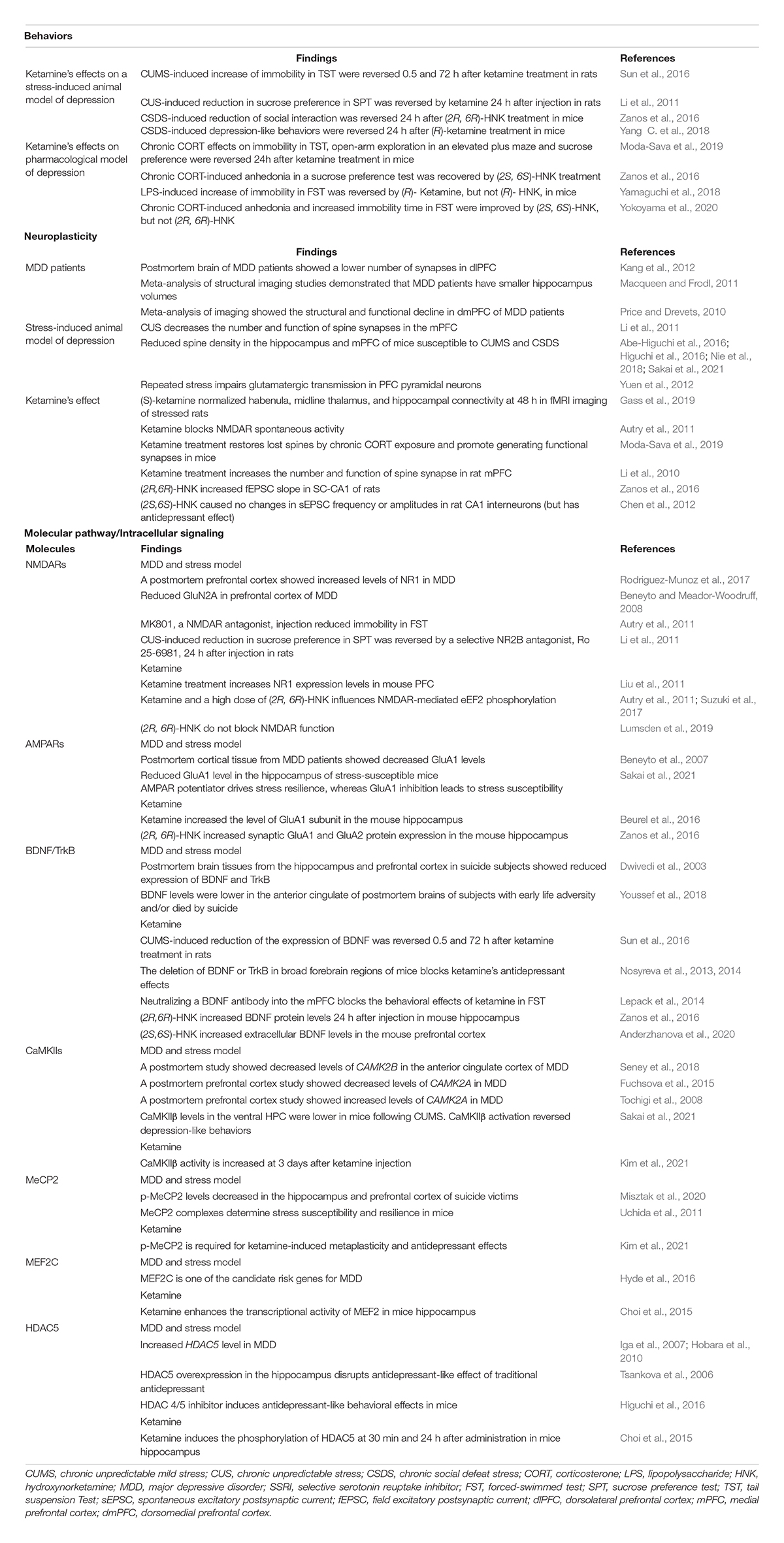
Table 1. Example evidence indicates alterations in behavior, glutamatergic signaling, and its downstream pathways regarding depression, chronic stress, and antidepressants: translational and multiscale views.
Less than one-third of patients with MDD achieve remission using traditional antidepressant pharmacotherapy (Trivedi et al., 2006). Treatment resistance occurs in up to 30% of patients with MDD (Fava, 2003). However, a single subanesthetic dose of ketamine produces a therapeutic response within a few hours that lasts for several days in patients with depression (Berman et al., 2000; Zarate et al., 2006). Intravenous infusion of ketamine results in clinical response and remission in 70 and 30% of treatment-resistant patients with MDD, respectively (Zarate et al., 2006). Additionally, Ketamine reduces suicidal ideation (Krystal et al., 2013). In 2020, esketamine was approved by the USFDA for treating depressive symptoms in adults with MDD having acute suicidal ideation or behavior.
Ketamine elicits robust unwanted side effects, including prepulse-inhibition deficits, cognitive deficits, and schizophrenia-like psychotic symptoms in humans (Lahti et al., 1995; Chan et al., 2013; Giorgetti et al., 2015). Recent preclinical data indicate that ketamine’s enantiomer (R)-ketamine (Hashimoto, 2019; Wei et al., 2021) and its metabolites (2R, 6R)-hydroxynorketamine (HNK) (Zanos et al., 2016) exert antidepressant effects with fewer adverse effects than do ketamine or (S)-ketamine. Since potential mechanisms underlying the rapid antidepressant actions of ketamine and its metabolites have been reviewed elsewhere (Fukumoto et al., 2017; Yang C. et al., 2018; Duman et al., 2019; Krystal et al., 2019; Sial et al., 2020; Highland et al., 2021; Shinohara et al., 2021; Wei et al., 2021; Xia et al., 2021), we review the recent progress in deciphering mechanisms underlying ketamine’s sustained antidepressant effects, with a particular focus on the role of calcium signaling from a multiscale perspective.
Several animal studies have demonstrated antidepressant-like responses to ketamine. A single intraperitoneal injection of ketamine or its metabolites produces rapid (30 min–1 h) and long-lasting (24 h–7 days) antidepressant effects (Autry et al., 2011; Koike et al., 2011; Zhou et al., 2014; Sun et al., 2016; Zanos et al., 2016; Yang C. et al., 2018; Kim et al., 2021). Moreover, such ketamine antidepressant effects have been observed in not only naïve, non-stressed animals but also in animals subjected to adverse stressful life events. Animals exposed to chronic stress show despair-like behavior, anhedonia, anxiety, and/or social avoidance, whereas a single injection of ketamine or its metabolites rapidly reverses these deleterious effects and exerts long-term effects (Li et al., 2011; Zanos et al., 2016; Duman et al., 2019; Wei et al., 2021).
Neuroimaging studies have shown structural and functional alterations in the hippocampus and dorsomedial prefrontal cortex (dmPFC) of patients with MDD (Price and Drevets, 2010; Macqueen and Frodl, 2011). Human functional magnetic resonance imaging (MRI) studies have demonstrated that a single dose of ketamine ameliorates reductions in functional connectivity in the prefrontal cortex (PFC), which is associated with the alleviation of depressive symptoms (Abdallah et al., 2017). Interestingly, a recent MRI study in animals demonstrated short- and long-term effects of ketamine on distinct brain circuitry. Gass et al. (2019) found in an animal model of depression that ketamine causes a rapid response in the amygdala, anterodorsal hippocampus, and ventral pallidum, which are related to cognitive, sensory, emotional, and reward functions. However, 48 h after administration, ketamine showed a long-term normalization of the habenula, midline thalamus, and hippocampal connectivity. They mediate cognitive flexibility for processing contextual information, distinguish contextual cues in safe versus threatening situations, and modulate fear and emotional responses in non-threatening environments (Gass et al., 2019).
There is increasing evidence suggesting altered neuronal and structural plasticity in animal models of depression as well as in patients with MDD (Duman and Aghajanian, 2012; Kang et al., 2012; Abe-Higuchi et al., 2016; Higuchi et al., 2016; Nie et al., 2018; Uchida et al., 2018; Sakai et al., 2021). Ketamine rapidly increases the number and function of spine synapses. Furthermore, Li et al. found that ketamine increases the number and function of spine synapses in the medial PFC (mPFC) and rapidly reverses synaptic abnormalities caused by chronic stress exposure (Li et al., 2010). Although this evidence suggests an association between ketamine-induced spinogenesis and antidepressant-like behavior, the causal relationship is unclear. However, a recent report by Moda-Sava et al. has addressed this issue. They used a photoactivable proof to selectively reverse ketamine effects on spine formation in the PFC. They found that newly formed spines are necessary for and play a specific role in the sustained antidepressant-like behavior induced by ketamine treatment (Moda-Sava et al., 2019).
Brain-derived neurotrophic factor and its receptor TrkB play key roles in synaptic plasticity, stress, and depression (Duman and Monteggia, 2006; Minichiello, 2009; Castren and Monteggia, 2021). A recent report discovered that several antidepressants, including fluoxetine, imipramine, and ketamine, directly bind to TrkB, facilitating BDNF action and plasticity (Casarotto et al., 2021). In addition, increased BDNF-TrkB signaling in rodent frontocortical/hippocampal circuits has been observed following acute treatment with ketamine (Li et al., 2010; Autry et al., 2011).
Clinical evidence suggests that repeated ketamine administration allows cumulative and sustained antidepressant effects and that it is more effective than a single injection in patients with MDD (Aan Het Rot et al., 2010; Murrough et al., 2013; Phillips et al., 2019). The threshold and sensitivity of the persistent increase and decrease of synaptic strength are subject to activity-dependent regulation. This type of plasticity, called “metaplasticity,” is important for stabilizing synaptic strength and preventing LTP saturation and long-term depression, leading to homeostatic alternations of synaptic activation (Bienenstock et al., 1982; Turrigiano et al., 1998; Kavalali and Monteggia, 2020). Notably, a preclinical study suggested that ketamine administration elicits metaplastic effects on LTP modulation and potentially other processes for long term. Kim et al. (2021) reported that, by using slice recordings of the Schaffer collateral-CA1 pathway in the hippocampus, ketamine induces AMPAR-mediated synaptic potentiation. Interestingly, this effect was more than two-fold higher in brain slices of mice that had received ketamine 7 days earlier, suggesting a priming effect of ketamine treatment such that subsequent ketamine augments synaptic potentiation. Further experiments to understand the mechanisms of this metaplasticity will provide critical insight into mechanisms underlying ketamine’s potent and prolonged antidepressant effects.
N-methyl-D-aspartate receptors activate eEF2 via CaMKs (eEF2 kinases) and depress BDNF levels (Scheetz et al., 2000). Ketamine-induced suppression of postsynaptic NMDARs deactivates eEF2 kinase, leading to reduced eEF2 phosphorylation and increased translation of BDNF in the hippocampus (Autry et al., 2011; Suzuki and Monteggia, 2020). This signaling pathway then potentiates synaptic AMPAR responses through the insertion of GluA1/2 subunits (Autry et al., 2011). In contrast, ketamine’s metabolite (2R, 6R)-HNK has NMDAR inhibition-independent antidepressant actions (Zanos et al., 2016; Lumsden et al., 2019), whereas other reports have shown that NMDAR inhibition at a high dose of (2R, 6R)-HNK triggers intracellular signaling via eEF2 (Suzuki et al., 2017).
A transient burst of glutamate via NMDAR blockade on GABAergic interneurons by ketamine activates postsynaptic AMPARs in excitatory neurons. This activation induces depolarization and activation of NMDARs that trigger Ca2+ influx, releasing BDNF (Krystal et al., 2019). Local release of BDNF is thought to activate TrkB on the postsynaptic membrane, stimulating the ERK and PI3K-Akt signaling pathways and mammalian target of rapamycin complex 1 (mTORC1) phosphorylation to promote synapse formation by stimulating synaptic proteins, such as GluA1 and PSD-95, which are required for synaptic plasticity (Cavalleri et al., 2018). Recently, mTORC1 effectors 4E-BP2 and 4-EB2 in excitatory or inhibitory neurons underlie behavioral and neurobiological responses to ketamine (Aguilar-Valles et al., 2021). Ketamine-induced activation of TrkB increases GSK-3β phosphorylation via the ERK signaling pathway, decreasing PSD-95 phosphorylation and internalizing the AMPA GluA1 subunit, which upregulates signaling through the GluA1 to promote synapse formation (Liu et al., 2013; Beurel et al., 2016). Ketamine-dependent changes in dendritic arborization and soma size are abolished by AMPAR antagonists or mTOR complex/signaling inhibitors (Cavalleri et al., 2018). Intracellular molecular signaling cascades stimulated by the glutamatergic pathway may be associated with ketamine-induced structural and synaptic plasticity and its antidepressant effects.
As mentioned earlier, CaMKIIs are major downstream target for the glutamatergic pathway and might be involved in stress and depression. TrkB activation stimulates phospholipase Cγ1 (PLCγ1) and also results in the activation of CaMKs (Minichiello, 2009). Activated CaMKIIs further stimulate MeCP2 phosphorylation (Zhou et al., 2006), allowing the transcription of downstream target genes. A recent study showed that MeCP2 phosphorylation at S421 (p-MeCP2) is essential for the expression of metaplasticity and the sustained, but not acute, antidepressant effects of ketamine (Kim et al., 2021). Hippocampal BDNF protein levels were shown to increase rapidly 30 min after ketamine administration but returned to baseline 3 days after injection. In contrast, hippocampal p-MeCP2 levels increased 3 and 7 days, but not 30 min, after ketamine injection. CaMKIIβ were elevated at 3 days after ketamine injection but returned to baseline at 7 days. These findings indicate that CaMKIIβ plays a role in the intermediary process between BDNF activation and MeCP2 phosphorylation required for the sustained antidepressant effects of ketamine. This hypothesis is also supported, at least in part, by a recent finding that hippocampal CaMKIIβ is downregulated in chronic stress-susceptible mice and that short-term (within 4 days) CaMKIIβ activation ameliorates depression-like behaviors (Sakai et al., 2021).
The interplay between genetic and environmental factors underlies depression pathophysiology, and epigenetic mechanisms might contribute to these interactions (Nestler et al., 2016; Uchida et al., 2018; Kawatake-Kuno et al., 2021). Although accumulating evidence demonstrated altered epigenetic functioning in animal models of depression and postpartum MDD-patient brains, few studies have used ketamine-induced transcriptome and epigenome analyses to characterize ketamine’s antidepressant effects. Genome-wide transcriptome and epigenome mapping offer a template for several strategies to identify novel drug targets in unbiased ways to develop more effective treatments for MDD (Bagot et al., 2017). Here we summarize how ketamine-induced activation of Ca2+ signal influences epigenetic regulation of gene transcription.
MeCP2, MEF2, and HDAC5 functions are regulated by Ca2+ signaling and are associated with stress and depression (Table 1). As mentioned above, p-MeCP2 is necessary for sustained antidepressant response to ketamine (Kim et al., 2021). MeCP2 is a methylated cytosine reader that impacts chromatin organization with any change in DNA methylation. A previous report showed that chronic stress differentially modulates MeCP2 activity in stress-resilient and -susceptible mice and subsequent epigenetic gene transcription (Uchida et al., 2011). Thus, ketamine-induced enhancement of p-MeCP2 may be associated with the formation of chromatin-remodeling complexes on target genes and, thus, transcription regulation. HDAC5 is a histone deacetylase, and its phosphorylation by CaMKs is associated with transcription repression (Mckinsey et al., 2000). Hippocampal HDAC5 is associated with behavioral response to chronic stress and traditional antidepressants (e.g., imipramine and SSRIs) (Tsankova et al., 2006; Higuchi et al., 2016). A recent study suggested that ketamine rapidly induces HDAC5 phosphorylation and nuclear export through CaMKII-dependent pathways, which leads to enhanced MEF2 transcription that regulates neuronal structural and functional plasticity (Choi et al., 2015). Correspondingly, HDAC5 knockdown occludes the actions of ketamine. Moreover, MeCP2 is considered as a master regulator of metaplasticity (Chen et al., 2012). Ca2+-signal-mediated modulation of MeCP2, HDAC5, and MEF2 functions may be involved in the sustained antidepressant response of ketamine through epigenetic transcription.
This mini-review highlights that the glutamatergic pathway is associated with behavioral, neuroplastic, neurobiological, molecular, and epigenetic effects of ketamine, focusing on Ca2+ signaling wherein its dysfunction is involved in depression pathophysiology according to both clinical and animal studies. Such (reverse) translational implications for bridging the research gap between human depression and animal models will provide a better understanding of how ketamine affects and modulates depression pathophysiology and ultimately contribute to the clinical application of ketamine or the development of related compounds for wide range of psychiatric disorders. Glutamatergic transmission and monoaminergic systems induce rapid biological changes that induce fast antidepressant effects. In contrast, ketamine’s sustained antidepressant actions are likely mediated by intracellular Ca2+ signaling cascades that affect neurobiological processes, including dendritic spine formation, epigenetic modifications, and long-term synaptic plasticity, and consequently, maintain physiological functioning.
In this mini-review, we particularly focused on the hippocampus and prefrontal cortex, key brain regions associated with MDD pathophysiology and ketamine’s antidepressant effect. However, other brain regions were suggested to also be involved in these processes, such as the lateral habenula. Emerging evidence from preclinical and clinical studies identified an important role of the lateral habenula in depression and ketamine’s antidepressant effect through a glutamatergic pathway (Li et al., 2013; Cui et al., 2018a,b, 2019; Yang et al., 2018; Hu, 2019, Hu et al., 2020). In addition, dynamic molecular changes were observed in the nucleus accumbens of animal models of depression and ketamine-treated animals (Bagot et al., 2017). Thus, future studies are warranted to clarify how ketamine impacts neuronal circuit activity and identify underlying molecular and epigenetic mechanisms.
In summary, ketamine has great potential in the development of groundbreaking neuropsychiatric therapies. Our current understanding of depression pathophysiology and ketamine’s action suggests that diverse drug actions converge around Ca2+-signaling-mediated neural plasticity. However, ketamine plays diverse roles in the glutamatergic pathway and other neurotransmitter systems, neurogenesis, inflammation, and even body–brain crosstalk. Furthermore, several studies have suggested the distinct roles of ketamine enantiomers ([S]-ketamine and [R]-ketamine) and their metabolites ([2R,6R]-HNK and [2S,6S]-HNK) in plasticity and behavior (Zanos et al., 2016; Yamaguchi et al., 2018; Hashimoto, 2019; Lumsden et al., 2019; Yokoyama et al., 2020; Highland et al., 2021; Wei et al., 2021). Thus, mechanisms underlying ketamine’s actions remain controversial. Moreover, ketamine effects at the mesoscale of neural architecture and macroscale of neural connectivity, cognition, and behavior are poorly understood. Further investigations at both the multiscale and multisystem levels are necessary to comprehensively understand mechanisms underlying ketamine’s antidepressant effects and develop novel drugs for treating MDD.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by JSPS KAKENHI Grant Numbers JP21K19707 and JP18H02750, by MEXT KAKENHI Grant Number JP21H00198, and by AMED under Grant Numbers JP21ak0101136 and JP21dm0307102h0003.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aan Het Rot, M., Collins, K. A., Murrough, J. W., Perez, A. M., Reich, D. L., Charney, D. S., et al. (2010). Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 67, 139–145. doi: 10.1016/j.biopsych.2009.08.038
Abdallah, C. G., Averill, L. A., Collins, K. A., Geha, P., Schwartz, J., Averill, C., et al. (2017). Ketamine Treatment and Global Brain Connectivity in Major Depression. Neuropsychopharmacology 42, 1210–1219. doi: 10.1038/npp.2016.186
Abe-Higuchi, N., Uchida, S., Yamagata, H., Higuchi, F., Hobara, T., Hara, K., et al. (2016). Hippocampal Sirtuin 1 Signaling Mediates Depression-like Behavior. Biol. Psychiatry 80, 815–826. doi: 10.1016/j.biopsych.2016.01.009
Aguilar-Valles, A., De Gregorio, D., Matta-Camacho, E., Eslamizade, M. J., Khlaifia, A., Skaleka, A., et al. (2021). Antidepressant actions of ketamine engage cell-specific translation via eIF4E. Nature 590, 315–319. doi: 10.1038/s41586-020-03047-0
Anderzhanova, E., Hafner, K., Genewsky, A. J., Soliman, A., Pohlmann, M. L., Schmidt, M. V., et al. (2020). The stress susceptibility factor FKBP51 controls S-ketamine-evoked release of mBDNF in the prefrontal cortex of mice. Neurobiol. Stress 13:100239. doi: 10.1016/j.ynstr.2020.100239
Autry, A. E., Adachi, M., Nosyreva, E., Na, E. S., Los, M. F., Cheng, P. F., et al. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. doi: 10.1038/nature10130
Bagot, R. C., Cates, H. M., Purushothaman, I., Vialou, V., Heller, E. A., Yieh, L., et al. (2017). Ketamine and Imipramine Reverse Transcriptional Signatures of Susceptibility and Induce Resilience-Specific Gene Expression Profiles. Biol. Psychiatry 81, 285–295. doi: 10.1016/j.biopsych.2016.06.012
Beneyto, M., Kristiansen, L. V., Oni-Orisan, A., Mccullumsmith, R. E., and Meador-Woodruff, J. H. (2007). Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 32, 1888–1902. doi: 10.1038/sj.npp.1301312
Beneyto, M., and Meador-Woodruff, J. H. (2008). Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 33, 2175–2186. doi: 10.1038/sj.npp.1301604
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., Charney, D. S., et al. (2000). Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354. doi: 10.1016/s0006-3223(99)00230-9
Berton, O., and Nestler, E. J. (2006). New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 7, 137–151. doi: 10.1038/nrn1846
Beurel, E., Grieco, S. F., Amadei, C., Downey, K., and Jope, R. S. (2016). Ketamine-induced inhibition of glycogen synthase kinase-3 contributes to the augmentation of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor signaling. Bipolar. Disord. 18, 473–480. doi: 10.1111/bdi.12436
Bienenstock, E. L., Cooper, L. N., and Munro, P. W. (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982
Bloodgood, B. L., and Sabatini, B. L. (2007). Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53, 249–260. doi: 10.1016/j.neuron.2006.12.017
Carter, A. G., Soler-Llavina, G. J., and Sabatini, B. L. (2007). Timing and location of synaptic inputs determine modes of subthreshold integration in striatal medium spiny neurons. J. Neurosci. 27, 8967–8977. doi: 10.1523/JNEUROSCI.2798-07.2007
Casarotto, P. C., Girych, M., Fred, S. M., Kovaleva, V., Moliner, R., Enkavi, G., et al. (2021). Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313e1219. doi: 10.1016/j.cell.2021.01.034
Castren, E., and Monteggia, L. M. (2021). Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 90, 128–136. doi: 10.1016/j.biopsych.2021.05.008
Cavalleri, L., Merlo Pich, E., Millan, M. J., Chiamulera, C., Kunath, T., Spano, P. F., et al. (2018). Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol. Psychiatry 23, 812–823. doi: 10.1038/mp.2017.241
Chaki, S. (2017). Beyond Ketamine: New Approaches to the Development of Safer Antidepressants. Curr. Neuropharmacol. 15, 963–976. doi: 10.2174/1570159X15666170221101054
Chan, K. W., Lee, T. M., Siu, A. M., Wong, D. P., Kam, C. M., Tsang, S. K., et al. (2013). Effects of chronic ketamine use on frontal and medial temporal cognition. Addict. Behav. 38, 2128–2132. doi: 10.1016/j.addbeh.2013.01.014
Chen, S. X., Cherry, A., Tari, P. K., Podgorski, K., Kwong, Y. K., and Haas, K. (2012). The transcription factor MEF2 directs developmental visually driven functional and structural metaplasticity. Cell 151, 41–55. doi: 10.1016/j.cell.2012.08.028
Choi, M., Lee, S. H., Wang, S. E., Ko, S. Y., Song, M., Choi, J. S., et al. (2015). Ketamine produces antidepressant-like effects through phosphorylation-dependent nuclear export of histone deacetylase 5 (HDAC5) in rats. Proc. Natl. Acad. Sci. U S A 112, 15755–15760. doi: 10.1073/pnas.1513913112
Cui, Y., Hu, S., and Hu, H. (2019). Lateral Habenular Burst Firing as a Target of the Rapid Antidepressant Effects of Ketamine. Trends Neurosci. 42, 179–191. doi: 10.1016/j.tins.2018.12.002
Cui, Y., Yang, Y., Dong, Y., and Hu, H. (2018a). Decoding Depression: Insights from Glial and Ketamine Regulation of Neuronal Burst Firing in Lateral Habenula. Cold Spr. Harb. Symp. Quant. Biol. 83, 141–150. doi: 10.1101/sqb.2018.83.036871
Cui, Y., Yang, Y., Ni, Z., Dong, Y., Cai, G., Foncelle, A., et al. (2018b). Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554, 323–327. doi: 10.1038/nature25752
Duman, R. S., and Aghajanian, G. K. (2012). Synaptic dysfunction in depression: potential therapeutic targets. Science 338, 68–72. doi: 10.1126/science.1222939
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127. doi: 10.1016/j.biopsych.2006.02.013
Duman, R. S., Shinohara, R., Fogaca, M. V., and Hare, B. (2019). Neurobiology of rapid-acting antidepressants: convergent effects on GluA1-synaptic function. Mol. Psychiatry 24, 1816–1832. doi: 10.1038/s41380-019-0400-x
Dwivedi, Y., Rizavi, H. S., Conley, R. R., Roberts, R. C., Tamminga, C. A., and Pandey, G. N. (2003). Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 60, 804–815. doi: 10.1001/archpsyc.60.8.804
Fava, M. (2003). Diagnosis and definition of treatment-resistant depression. Biol. Psychiatry 53, 649–659. doi: 10.1016/s0006-3223(03)00231-2
Fuchsova, B., Alvarez Julia, A., Rizavi, H. S., Frasch, A. C., and Pandey, G. N. (2015). Altered expression of neuroplasticity-related genes in the brain of depressed suicides. Neuroscience 299, 1–17. doi: 10.1016/j.neuroscience.2015.04.057
Fukumoto, K., Toki, H., Iijima, M., Hashihayata, T., Yamaguchi, J. I., Hashimoto, K., et al. (2017). Antidepressant Potential of (R)-Ketamine in Rodent Models: Comparison with (S)-Ketamine. J. Pharmacol. Exp. Ther. 361, 9–16. doi: 10.1124/jpet.116.239228
Gass, N., Becker, R., Reinwald, J., Cosa-Linan, A., Sack, M., Weber-Fahr, W., et al. (2019). Differences between ketamine’s short-term and long-term effects on brain circuitry in depression. Transl. Psychiatry 9:172. doi: 10.1038/s41398-019-0506-6
Giorgetti, R., Marcotulli, D., Tagliabracci, A., and Schifano, F. (2015). Effects of ketamine on psychomotor, sensory and cognitive functions relevant for driving ability. Forensic. Sci. Int. 252, 127–142. doi: 10.1016/j.forsciint.2015.04.024
Graff, J., and Tsai, L. H. (2013). Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111. doi: 10.1038/nrn3427
Greer, P. L., and Greenberg, M. E. (2008). From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. doi: 10.1016/j.neuron.2008.09.002
Hashimoto, K. (2019). Rapid-acting antidepressant ketamine, its metabolites and other candidates: A historical overview and future perspective. Psychiatry Clin. Neurosci. 73, 613–627. doi: 10.1111/pcn.12902
Highland, J. N., Zanos, P., Riggs, L. M., Georgiou, P., Clark, S. M., Morris, P. J., et al. (2021). Hydroxynorketamines: Pharmacology and Potential Therapeutic Applications. Pharmacol Rev 73, 763–791. doi: 10.1124/pharmrev.120.000149
Higuchi, F., Uchida, S., Yamagata, H., Abe-Higuchi, N., Hobara, T., Hara, K., et al. (2016). Hippocampal MicroRNA-124 Enhances Chronic Stress Resilience in Mice. J. Neurosci. 36, 7253–7267. doi: 10.1523/JNEUROSCI.0319-16.2016
Hobara, T., Uchida, S., Otsuki, K., Matsubara, T., Funato, H., Matsuo, K., et al. (2010). Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatr. Res. 44, 263–270. doi: 10.1016/j.jpsychires.2009.08.015
Hu, H. (2019). Advances in Molecular and Circuitry Mechanisms of Depressive Disorder-A Focus on Lateral Habenula. Adv. Exp. Med. Biol. 1180, 135–146. doi: 10.1007/978-981-32-9271-0_7
Hu, H., Cui, Y., and Yang, Y. (2020). Circuits and functions of the lateral habenula in health and in disease. Nat. Rev. Neurosci. 21, 277–295. doi: 10.1038/s41583-020-0292-4
Huganir, R. L., and Nicoll, R. A. (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. doi: 10.1016/j.neuron.2013.10.025
Hyde, C. L., Nagle, M. W., Tian, C., Chen, X., Paciga, S. A., Wendland, J. R., et al. (2016). Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 48, 1031–1036. doi: 10.1038/ng.3623
Iga, J., Ueno, S., Yamauchi, K., Numata, S., Kinouchi, S., Tayoshi-Shibuya, S., et al. (2007). Altered HDAC5 and CREB mRNA expressions in the peripheral leukocytes of major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 628–632. doi: 10.1016/j.pnpbp.2006.12.014
Kang, H. J., Voleti, B., Hajszan, T., Rajkowska, G., Stockmeier, C. A., Licznerski, P., et al. (2012). Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 18, 1413–1417. doi: 10.1038/nm.2886
Kavalali, E. T., and Monteggia, L. M. (2020). Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron 106, 715–726. doi: 10.1016/j.neuron.2020.05.015
Kawatake-Kuno, A., Murai, T., and Uchida, S. (2021). The Molecular Basis of Depression: Implications of Sex-Related Differences in Epigenetic Regulation. Front. Mol. Neurosci. 14:708004. doi: 10.3389/fnmol.2021.708004
Kim, J. W., Autry, A. E., Na, E. S., Adachi, M., Bjorkholm, C., Kavalali, E. T., et al. (2021). Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat. Neurosci. 2021:8. doi: 10.1038/s41593-021-00868-8
Koester, H. J., and Sakmann, B. (1998). Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc. Natl. Acad. Sci. U S A 95, 9596–9601. doi: 10.1073/pnas.95.16.9596
Koike, H., Iijima, M., and Chaki, S. (2011). Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav. Brain Res. 224, 107–111. doi: 10.1016/j.bbr.2011.05.035
Krishnan, V., and Nestler, E. J. (2008). The molecular neurobiology of depression. Nature 455, 894–902. doi: 10.1038/nature07455
Krystal, J. H., Abdallah, C. G., Sanacora, G., Charney, D. S., and Duman, R. S. (2019). Ketamine: A Paradigm Shift for Depression Research and Treatment. Neuron 101, 774–778. doi: 10.1016/j.neuron.2019.02.005
Krystal, J. H., Sanacora, G., and Duman, R. S. (2013). Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol. Psychiatry 73, 1133–1141. doi: 10.1016/j.biopsych.2013.03.026
Lahti, A. C., Koffel, B., Laporte, D., and Tamminga, C. A. (1995). Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13, 9–19. doi: 10.1016/0893-133X(94)00131-I
Lepack, A. E., Fuchikami, M., Dwyer, J. M., Banasr, M., and Duman, R. S. (2014). BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 18:33. doi: 10.1093/ijnp/pyu033
Li, K., Zhou, T., Liao, L., Yang, Z., Wong, C., Henn, F., et al. (2013). betaCaMKII in lateral habenula mediates core symptoms of depression. Science 341, 1016–1020. doi: 10.1126/science.1240729
Li, N., Lee, B., Liu, R. J., Banasr, M., Dwyer, J. M., Iwata, M., et al. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964. doi: 10.1126/science.1190287
Li, N., Liu, R. J., Dwyer, J. M., Banasr, M., Lee, B., Son, H., et al. (2011). Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 69, 754–761. doi: 10.1016/j.biopsych.2010.12.015
Liu, F., Paule, M. G., Ali, S., and Wang, C. (2011). Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr. Neuropharmacol. 9, 256–261. doi: 10.2174/157015911795017155
Liu, R. J., Fuchikami, M., Dwyer, J. M., Lepack, A. E., Duman, R. S., and Aghajanian, G. K. (2013). GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology 38, 2268–2277. doi: 10.1038/npp.2013.128
Lumsden, E. W., Troppoli, T. A., Myers, S. J., Zanos, P., Aracava, Y., Kehr, J., et al. (2019). Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc. Natl. Acad. Sci. U S A 116, 5160–5169. doi: 10.1073/pnas.1816071116
Macqueen, G., and Frodl, T. (2011). The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol. Psychiatry 16, 252–264. doi: 10.1038/mp.2010.80
Mcewen, B. S. (2007). Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 87, 873–904. doi: 10.1152/physrev.00041.2006
Mckinsey, T. A., Zhang, C. L., Lu, J., and Olson, E. N. (2000). Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111. doi: 10.1038/35040593
Minichiello, L. (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 10, 850–860. doi: 10.1038/nrn2738
Misztak, P., Panczyszyn-Trzewik, P., Nowak, G., and Sowa-Kucma, M. (2020). Epigenetic marks and their relationship with BDNF in the brain of suicide victims. PLoS One 15:e0239335. doi: 10.1371/journal.pone.0239335
Moda-Sava, R. N., Murdock, M. H., Parekh, P. K., Fetcho, R. N., Huang, B. S., Huynh, T. N., et al. (2019). Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364:8078. doi: 10.1126/science.aat8078
Murrough, J. W., Perez, A. M., Pillemer, S., Stern, J., Parides, M. K., Aan Het, et al. (2013). Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol. Psychiatry 74, 250–256. doi: 10.1016/j.biopsych.2012.06.022
Nestler, E. J., Pena, C. J., Kundakovic, M., Mitchell, A., and Akbarian, S. (2016). Epigenetic Basis of Mental Illness. Neuroscientist 22, 447–463. doi: 10.1177/1073858415608147
Ngo-Anh, T. J., Bloodgood, B. L., Lin, M., Sabatini, B. L., Maylie, J., and Adelman, J. P. (2005). SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 8, 642–649. doi: 10.1038/nn1449
Nie, X., Kitaoka, S., Tanaka, K., Segi-Nishida, E., Imoto, Y., Ogawa, A., et al. (2018). The Innate Immune Receptors TLR2/4 Mediate Repeated Social Defeat Stress-Induced Social Avoidance through Prefrontal Microglial Activation. Neuron 99, 464–479e467. doi: 10.1016/j.neuron.2018.06.035
Nosyreva, E., Autry, A. E., Kavalali, E. T., and Monteggia, L. M. (2014). Age dependence of the rapid antidepressant and synaptic effects of acute NMDA receptor blockade. Front. Mol. Neurosci. 7:94. doi: 10.3389/fnmol.2014.00094
Nosyreva, E., Szabla, K., Autry, A. E., Ryazanov, A. G., Monteggia, L. M., and Kavalali, E. T. (2013). Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J. Neurosci. 33, 6990–7002. doi: 10.1523/JNEUROSCI.4998-12.2013
Phillips, J. L., Norris, S., Talbot, J., Birmingham, M., Hatchard, T., Ortiz, A., et al. (2019). Single, Repeated, and Maintenance Ketamine Infusions for Treatment-Resistant Depression: A Randomized Controlled Trial. Am. J. Psychiatry 176, 401–409. doi: 10.1176/appi.ajp.2018.18070834
Popoli, M., Yan, Z., Mcewen, B. S., and Sanacora, G. (2011). The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 13, 22–37. doi: 10.1038/nrn3138
Price, J. L., and Drevets, W. C. (2010). Neurocircuitry of mood disorders. Neuropsychopharmacology 35, 192–216. doi: 10.1038/npp.2009.104
Reid, C. A., Fabian-Fine, R., and Fine, A. (2001). Postsynaptic calcium transients evoked by activation of individual hippocampal mossy fiber synapses. J. Neurosci. 21, 2206–2214. doi: 10.1523/JNEUROSCI.21-07-02206.2001
Rodriguez-Munoz, M., Sanchez-Blazquez, P., Callado, L. F., Meana, J. J., and Garzon-Nino, J. (2017). Schizophrenia and depression, two poles of endocannabinoid system deregulation. Transl. Psychiatry 7:1291. doi: 10.1038/s41398-017-0029-y
Sakai, Y., Li, H., Inaba, H., Funayama, Y., Ishimori, E., Kawatake-Kuno, A., et al. (2021). Gene-environment interactions mediate stress susceptibility and resilience through the CaMKIIbeta/TARPgamma-8/AMPAR pathway. iScience 24:102504. doi: 10.1016/j.isci.2021.102504
Scheetz, A. J., Nairn, A. C., and Constantine-Paton, M. (2000). NMDA receptor-mediated control of protein synthesis at developing synapses. Nat. Neurosci. 3, 211–216. doi: 10.1038/72915
Seney, M. L., Huo, Z., Cahill, K., French, L., Puralewski, R., Zhang, J., et al. (2018). Opposite Molecular Signatures of Depression in Men and Women. Biol. Psychiatry 84, 18–27. doi: 10.1016/j.biopsych.2018.01.017
Shinohara, R., Aghajanian, G. K., and Abdallah, C. G. (2021). Neurobiology of the Rapid-Acting Antidepressant Effects of Ketamine: Impact and Opportunities. Biol. Psychiatry 90, 85–95. doi: 10.1016/j.biopsych.2020.12.006
Sial, O. K., Parise, E. M., Parise, L. F., Gnecco, T., and Bolanos-Guzman, C. A. (2020). Ketamine: The final frontier or another depressing end? Behav. Brain Res. 383:112508. doi: 10.1016/j.bbr.2020.112508
Sun, H. L., Zhou, Z. Q., Zhang, G. F., Yang, C., Wang, X. M., Shen, J. C., et al. (2016). Role of hippocampal p11 in the sustained antidepressant effect of ketamine in the chronic unpredictable mild stress model. Transl. Psychiatry 6:e741. doi: 10.1038/tp.2016.21
Suzuki, K., and Monteggia, L. M. (2020). The role of eEF2 kinase in the rapid antidepressant actions of ketamine. Adv. Pharmacol. 89, 79–99. doi: 10.1016/bs.apha.2020.04.005
Suzuki, K., Nosyreva, E., Hunt, K. W., Kavalali, E. T., and Monteggia, L. M. (2017). Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546, E1–E3. doi: 10.1038/nature22084
Takemoto-Kimura, S., Suzuki, K., Horigane, S. I., Kamijo, S., Inoue, M., Sakamoto, M., et al. (2017). Calmodulin kinases: essential regulators in health and disease. J. Neurochem. 141, 808–818. doi: 10.1111/jnc.14020
Thompson, S. M., Kallarackal, A. J., Kvarta, M. D., Van Dyke, A. M., Legates, T. A., and Cai, X. (2015). An excitatory synapse hypothesis of depression. Trends Neurosci. 38, 279–294. doi: 10.1016/j.tins.2015.03.003
Tochigi, M., Iwamoto, K., Bundo, M., Sasaki, T., Kato, N., and Kato, T. (2008). Gene expression profiling of major depression and suicide in the prefrontal cortex of postmortem brains. Neurosci. Res. 60, 184–191. doi: 10.1016/j.neures.2007.10.010
Trivedi, M. H., Rush, A. J., Wisniewski, S. R., Nierenberg, A. A., Warden, D., Ritz, L., et al. (2006). Evaluation of outcomes with citalopram for depression using measurement-based care in STAR∗D: implications for clinical practice. Am. J. Psychiatry 163, 28–40. doi: 10.1176/appi.ajp.163.1.28
Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., and Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9, 519–525. doi: 10.1038/nn1659
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. doi: 10.1038/36103
Uchida, S., Hara, K., Kobayashi, A., Otsuki, K., Yamagata, H., Hobara, T., et al. (2011). Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69, 359–372. doi: 10.1016/j.neuron.2010.12.023
Uchida, S., and Shumyatsky, G. P. (2018a). Epigenetic regulation of Fgf1 transcription by CRTC1 and memory enhancement. Brain Res. Bull. 141, 3–12. doi: 10.1016/j.brainresbull.2018.02.016
Uchida, S., and Shumyatsky, G. P. (2018b). Synaptically Localized Transcriptional Regulators in Memory Formation. Neuroscience 370, 4–13. doi: 10.1016/j.neuroscience.2017.07.023
Uchida, S., Yamagata, H., Seki, T., and Watanabe, Y. (2018). Epigenetic mechanisms of major depression: Targeting neuronal plasticity. Psychiatry Clin. Neurosci. 72, 212–227. doi: 10.1111/pcn.12621
Van Den Heuvel, M. P., Scholtens, L. H., and Kahn, R. S. (2019). Multiscale Neuroscience of Psychiatric Disorders. Biol. Psychiatry 86, 512–522. doi: 10.1016/j.biopsych.2019.05.015
Wei, Y., Chang, L., and Hashimoto, K. (2021). Molecular mechanisms underlying the antidepressant actions of arketamine: beyond the NMDA receptor. Mol. Psychiatry 2021:1. doi: 10.1038/s41380-021-01121-1
Xia, C. Y., He, J., Du, L. D., Yan, Y., Lian, W. W., Xu, J. K., et al. (2021). Targeting the dysfunction of glutamate receptors for the development of novel antidepressants. Pharmacol. Ther. 226:107875. doi: 10.1016/j.pharmthera.2021.107875
Yamaguchi, J. I., Toki, H., Qu, Y., Yang, C., Koike, H., Hashimoto, K., et al. (2018). (2R,6R)-Hydroxynorketamine is not essential for the antidepressant actions of (R)-ketamine in mice. Neuropsychopharmacology 43, 1900–1907. doi: 10.1038/s41386-018-0084-y
Yamasaki, S., Aso, T., Miyata, J., Sugihara, G., Hazama, M., Nemoto, K., et al. (2020). Early and late effects of electroconvulsive therapy associated with different temporal lobe structures. Transl. Psychiatry 10:344. doi: 10.1038/s41398-020-01025-8
Yang, C., Ren, Q., Qu, Y., Zhang, J. C., Ma, M., Dong, C., et al. (2018). Mechanistic Target of Rapamycin-Independent Antidepressant Effects of (R)-Ketamine in a Social Defeat Stress Model. Biol. Psychiatry 83, 18–28. doi: 10.1016/j.biopsych.2017.05.016
Yang, Y., Cui, Y., Sang, K., Dong, Y., Ni, Z., Ma, S., et al. (2018). Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554, 317–322. doi: 10.1038/nature25509
Yokoyama, R., Higuchi, M., Tanabe, W., Tsukada, S., Naito, M., Yamaguchi, T., et al. (2020). (S)-norketamine and (2S,6S)-hydroxynorketamine exert potent antidepressant-like effects in a chronic corticosterone-induced mouse model of depression. Pharmacol. Biochem. Behav. 191:172876. doi: 10.1016/j.pbb.2020.172876
Youssef, M. M., Underwood, M. D., Huang, Y. Y., Hsiung, S. C., Liu, Y., Simpson, N. R., et al. (2018). Association of BDNF Val66Met Polymorphism and Brain BDNF Levels with Major Depression and Suicide. Int. J. Neuropsychopharmacol. 21, 528–538. doi: 10.1093/ijnp/pyy008
Yuen, E. Y., Wei, J., Liu, W., Zhong, P., Li, X., and Yan, Z. (2012). Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73, 962–977. doi: 10.1016/j.neuron.2011.12.033
Zanos, P., Moaddel, R., Morris, P. J., Georgiou, P., Fischell, J., Elmer, G. I., et al. (2016). NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486. doi: 10.1038/nature17998
Zarate, C. A. Jr., Singh, J. B., Carlson, P. J., Brutsche, N. E., Ameli, R., Luckenbaugh, D. A., et al. (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864. doi: 10.1001/archpsyc.63.8.856
Zhou, W., Wang, N., Yang, C., Li, X. M., Zhou, Z. Q., and Yang, J. J. (2014). Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry 29, 419–423. doi: 10.1016/j.eurpsy.2013.10.005
Keywords: ketamine, antidepressant action, neuroplasticity, epigenetics, gene expression, stress, glutamate receptor, calcium signaling
Citation: Kawatake-Kuno A, Murai T and Uchida S (2021) A Multiscale View of the Mechanisms Underlying Ketamine’s Antidepressant Effects: An Update on Neuronal Calcium Signaling. Front. Behav. Neurosci. 15:749180. doi: 10.3389/fnbeh.2021.749180
Received: 29 July 2021; Accepted: 14 September 2021;
Published: 30 September 2021.
Edited by:
Luisa Pinto, University of Minho, PortugalReviewed by:
He Liu, Zhejiang University School of Medicine and Huzhou Central Hospital, ChinaCopyright © 2021 Kawatake-Kuno, Murai and Uchida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shusaku Uchida, VWNoaWRhLnNodXNha3UuM25Aa3lvdG8tdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.