
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Behav. Neurosci. , 03 June 2016
Sec. Pathological Conditions
Volume 10 - 2016 | https://doi.org/10.3389/fnbeh.2016.00104
This article is part of the Research Topic Intellectual disabilities in Down syndrome from birth and throughout life: assessment and treatment View all 14 articles
 Arnaud Duchon1,2,3,4
Arnaud Duchon1,2,3,4 Yann Herault1,2,3,4,5*
Yann Herault1,2,3,4,5*Down syndrome (DS) is one of the leading causes of intellectual disability, and patients with DS face various health issues, including learning and memory deficits, congenital heart disease, Alzheimer’s disease (AD), leukemia, and cancer, leading to huge medical and social costs. Remarkable advances on DS research have been made in improving cognitive function in mouse models for future therapeutic approaches in patients. Among the different approaches, DYRK1A inhibitors have emerged as promising therapeutics to reduce DS cognitive deficits. DYRK1A is a dual-specificity kinase that is overexpressed in DS and plays a key role in neurogenesis, outgrowth of axons and dendrites, neuronal trafficking and aging. Its pivotal role in the DS phenotype makes it a prime target for the development of therapeutics. Recently, disruption of DYRK1A has been found in Autosomal Dominant Mental Retardation 7 (MRD7), resulting in severe mental deficiency. Recent advances in the development of kinase inhibitors are expected, in the near future, to remove DS from the list of incurable diseases, providing certain conditions such as drug dosage and correct timing for the optimum long-term treatment. In addition the exact molecular and cellular mechanisms that are targeted by the inhibition of DYRK1A are still to be discovered.
Since Down (1866) described patients with mental retardation and characteristic faces, Down Syndrome (DS) has been recognized as one of the most common genetic disorders leading to intellectual disability. DS results from the presence of an extra copy of all or part of chromosome 21 (Lejeune et al., 1959). The clinical presentation of DS is complex and variable. A few features occur to some degree in every individual with trisomy 21, including 100% of patients with intellectual disability, hypotonia, and cranio-facial dysmorphology, 75% with brachycephaly or 60% with epicanthic fold, 40% with congenital heart disease, and an increased incidence of leukemia in DS that is 10- to 20-fold higher than that in the general population (Antonarakis et al., 2004). The most disabling phenotype for patients is the impaired intellectual and adaptive functioning, strongly contributed by defects in hippocampal- and prefrontal cortex-dependent functions (Pennington et al., 2003; Dierssen et al., 2009; Contestabile et al., 2010; Lott and Dierssen, 2010). These deficits are associated with both learning and short-term and long-term memory, resulting in a delay in cognitive development (Nelson et al., 2005) along with various aspects of language acquisition and comprehension (Chapman and Hesketh, 2000; Abbeduto et al., 2007). In the last decades, with better care and medical follow-up, the life expectancy of people with DS is increasing and is quickly approaching 60 years (Puri et al., 1995; Covelli et al., 2016). However, because DS patients show accelerated aging, including early onset dementia similar to Alzheimer’s disease (AD) (Zigman, 2013), and the increase in life expectancy does not follow the one observed in the general population or other groups with intellectual disability (Coppus, 2013).
Down syndrome features have largely been attributed to the overexpression of specific trisomic genes (Antonarakis et al., 2004), even if non-coding element (Elton et al., 2010) or DNA methylation (Lu and Sheen, 2013) could have significant effect on DS phenotypes. Transcriptome analyses showed that between 29 and 62% of trisomic genes are overexpressed by a factor of 1.5 with some variability and depending on the cell type (Saran et al., 2003; Aït Yahya-Graison et al., 2007; Prandini et al., 2007; Sultan et al., 2007; Laffaire et al., 2009; Moldrich et al., 2009) and that trisomy has an impact on large number of genes located in domains all over the genome (Letourneau et al., 2014). A few candidate genes have been selected for therapeutic approaches because of their brain related functions, their specific pattern of expression/localization and/or their contribution to signaling pathways involved in cognitive functions. In parallel, investigation of the relationship between phenotype and genotype using a panel of rare DS patients with only partial duplication of the chromosome 21 segment led to the hypothesis of the DS chromosomal region (DCR), in which a small set of genes (between D21S55-MX1) plays a major role in the determination of DS phenotypes including the development of cognitive disabilities (McCormick et al., 1989; Rahmani et al., 1989; Korenberg, 1990; Delabar et al., 1993; Sinet et al., 1994; Korbel et al., 2009; Lyle et al., 2009). Among the 33 genes in the DCR, dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A), which has been always found overexpressed in DS patients and mouse models (Guimera et al., 1999; Dowjat et al., 2007), has received considerable attention because of its involvement in brain functions and processes that are altered in DS, and in the early onset of neurofibrillary degeneration, β-amyloidosis, neuronal loss and AD-like phenotypes in DS (Liu et al., 2008; Wegiel et al., 2011).
DYRK1A is the homologue of the Drosophila minibrain (mnb) which was named from the description of the brain phenotype observed in hypomorphic mutant flies (Tejedor et al., 1995). The DYRK family includes four additional mammalian subtypes including DYRK1B, DYRK2, DYRK3, and DYRK4. DYRK proteins show little sequence homology with other kinases outside of their catalytic domains but are highly conserved across species (Becker et al., 1998). DYRK1A can catalyze its own activation through auto-phosphorylation of a single tyrosine residue in its activation loop (Lochhead et al., 2005; Soundararajan et al., 2013; Soppa and Becker, 2015).
DYRK1A is expressed during embryonic neurogenesis. First, DYRK1A is transiently detected in preneurogenic region during early mouse embryogenesis from 8 to 10.5 days postcoitum (dpc). Then Dyrk1a expression was observed in cycling neuronal progenitor cells of the ventricular and subventricular zones at 14.5 dpc (Hammerle et al., 2008). The authors proposed that DYRK1A controls the mouse neuronal precursor exit from differentiation, leaving the cells in a quiescent state ready to differentiate while its expression is reduced (Hammerle et al., 2011). Finally, DYRK1A is expressed and translocated from the cytoplasm to the nucleus while the dendritic tree differentiated independently in several neuronal populations (Hammerle et al., 2003, 2008). In the adult mouse, the expression of Dyrk1a is found in several brain regions both in the cytoplasm and the nucleus (Marti et al., 2003).
The majority of the DYRK1A protein (almost 80%) is found associated with the cytoskeletal fraction in human and mouse brain, and the remaining protein is located in the cytosolic and nuclear fractions (Marti et al., 2003; Kaczmarski et al., 2014). The phosphorylated forms of DYRK1A are specific to subcellular localization in human and mouse brain. With only one residue phosphorylated (the conserved autophosphorylation site Y321) in the cytosolic DYRK1A and multiple heterogeneous phosphorylated sites found in the cytoskeletal and nuclear DYRK1A (Kaczmarski et al., 2014). Thus the function of DYRK1A could be regulated by the action of specific kinase(s) that will influence its stability or its ability to localize to nuclear, cytosolic or cytoskeletal compartments and thus to interact with specific substrates. Indeed the nuclear accumulation of DYRK2 is controlled by the “ataxia telangiectasia mutated” (ATM) dependent phosphorylation. When phosphorylated by ATM, DYRK2 dissociates from MDM2 (“transformed mouse 3T3 cell double minute 2”) and is no more degraded in the nucleus through a MDM2-dependent ubiquitination and thus could accumulate (Taira et al., 2010). This finding affecting DYRK localisation raises several questions such as whether a similar mechanism exists for DYRK1A and how it will be perturbed if there is an overdosage of the protein.
DYRK1A phosphorylates different targets depending upon its cellular localization (Marti et al., 2003; Park et al., 2009; Kaczmarski et al., 2014). It acts on a multitude of exogenous protein substrates, including transcription factors [CREB, NFAT (nuclear factor of activated T-cells), STAT3, FKHR, GLI1, RNApol2], splicing factors (cyclin L2, SF2, SF3), a translation factor (eIF2Be), miscellaneous proteins (glycogen synthase, caspase-9, Notch) or cytoskeletal target (TAU and MAP1B) and synaptic proteins (dynamin I, amphiphysin I, synaptojanin I; Table 1). Several targets listed here, might contribute to the role of DYRK1A in neuronal synaptic plasticity (Wegiel et al., 2004; Aranda et al., 2008; Murakami et al., 2009, 2012). Recent studies in Cos7 cells suggest that DYRK1A is involved in the regulation of dendritic spine formation through Neural Wiskott–Aldrich syndrome protein phosphorylation (Park et al., 2012). At the synaptic level, DYRK1A could regulate synaptic vesicle endocytosis via phosphorylation of AP180, dynamin I, amphiphysin I, and synaptojanin I, as demonstrated in isolated rat brain clathrin coated vesicle (Murakami et al., 2009, 2012). Clathrin-mediated endocytosis is essential for the recycling of membrane after neurotransmitter release (Saheki and De Camilli, 2012). DYRK1A is found in the pre-synaptic compartment of the neuromuscular synapse (Arque et al., 2013). Conversely, in the Drosophila neuromuscular junction, MNB acts as a synaptic kinase that promotes efficient synaptic vesicle recycling (Chen C.K. et al., 2014). Moreover, DYRK1A phosphorylation of GRIN2A modifies the biophysical properties of GRIN1/GRIN2A, two subunits of N-methyl-D-aspartate receptors (NMDAR) and controls NMDAR activity in neurons, which are involved in neural development, survival, synaptic plasticity and memory processes (Grau et al., 2014).
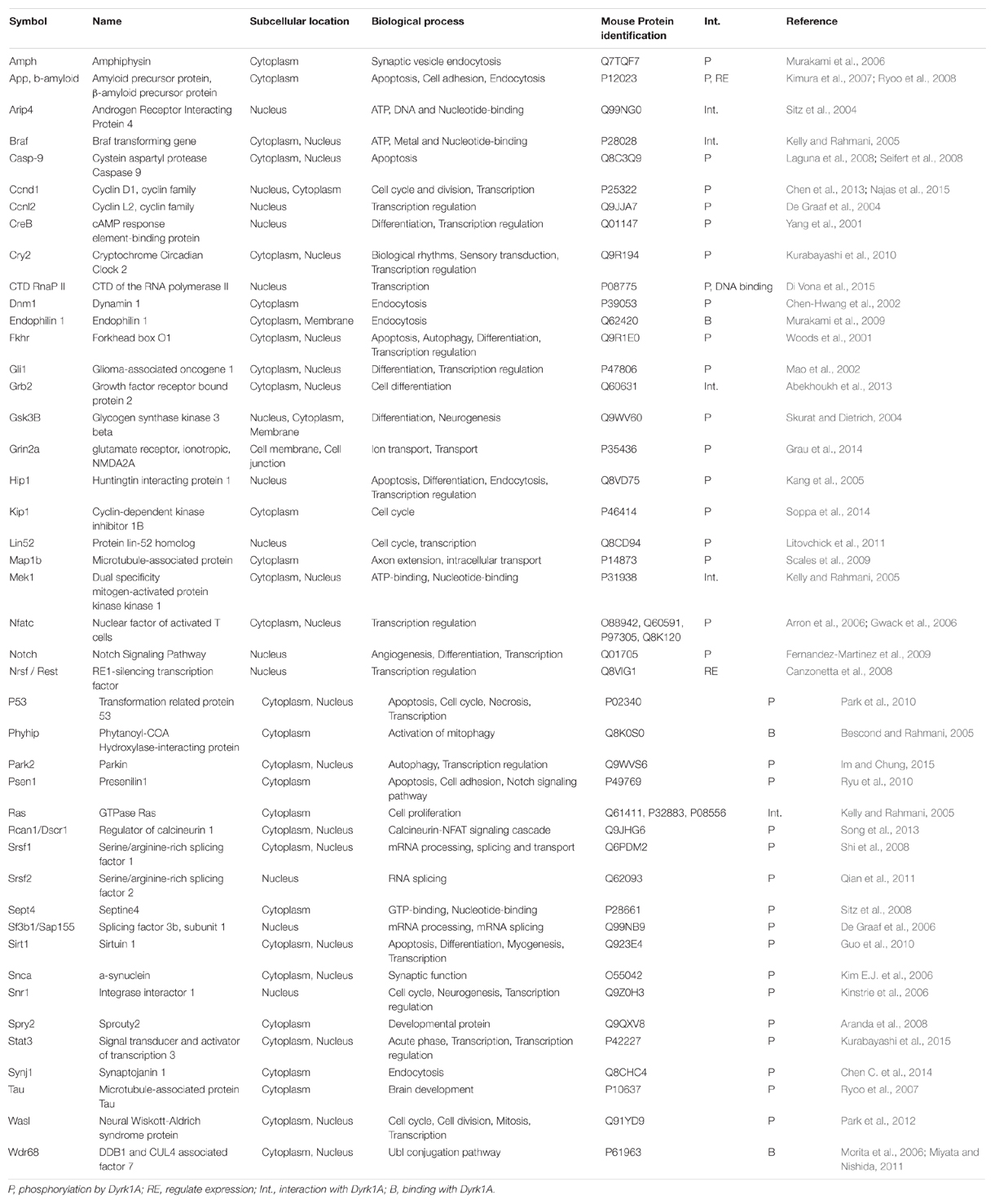
TABLE 1. Proteins that interact with MNB/DYRK1A.
DYRK1A is a paradigm of a dosage sensitive gene with its underexpression, caused by heterozygous disruption or loss-of-function mutations, leading to MRD7 and its overexpression contributing to DS cognitive dysfunction. Such a variation in gene dosage could have major impact on multiprotein complex at the level of enzymatic activities or transcriptional regulation (Figure 1) (Veitia et al., 2008). The first MRD7 cases were translocation disrupting DYRK1A in two patients with microcephaly, severe mental retardation without speech, anxious autistic behavior, or dysmorphic features (Møller et al., 2008). Then, a second study identified patients isolated from large screen of 3,009 intellectually disabled individuals who presented a de novo heterozygous deletion of the last three exons of DYRK1A (van Bon et al., 2011). Several reports showed heterozygous mutations in DYRK1A in patients with multiple phenotypes, including developmental delays or intellectual disability with autism spectrum disorder, microcephaly, epileptic seizures, facial dysmorphisms and cardiac defects (Courcet et al., 2012; O’Roak et al., 2012; Bronicki et al., 2015; Ruaud et al., 2015; Van Bon et al., 2015) defining a new syndromic condition (Courcet et al., 2012; Bronicki et al., 2015; Van Bon et al., 2015). Similar neuro-developmental phenotypes (delay in eyelid and ear opening, in the appearance of the righting reflex and of the Preyer’s reflex), microcephaly, locomotor activity and coordination (sensorimotor tets, balance, gait analysis, rotarod deficiency) cognition defects (delay in the startle response, spatial memory deficit in the Morris water maze and in the radial-arm water maze, memory recognition in the object recognition paradigm) and reduced dendritic tree of the layer III pyramidal cells were observed in mouse heterozygous knockout models (Fotaki et al., 2002; Benavides-Piccione et al., 2005; Dierssen and Martinez de Lagran, 2006; Arqué et al., 2008, 2009). Homozygous Dyrk1a knock-out (KO) mice die in utero with growth retardation, reduced body size and morphological developmental delay of the primitive organs. In an inbred genetic background, newborn heterozygous KO mice have reduced neonatal viability and decreased body size (Fotaki et al., 2002). Nevertheless, approximately 30% of the heterozygous KO mutants can survive in a mixed background. Their adult brains present increased neuronal densities in some brain regions and a specific decrease in the number of neurons in the superior colliculus, which exhibits a significant size reduction (Fotaki et al., 2002).
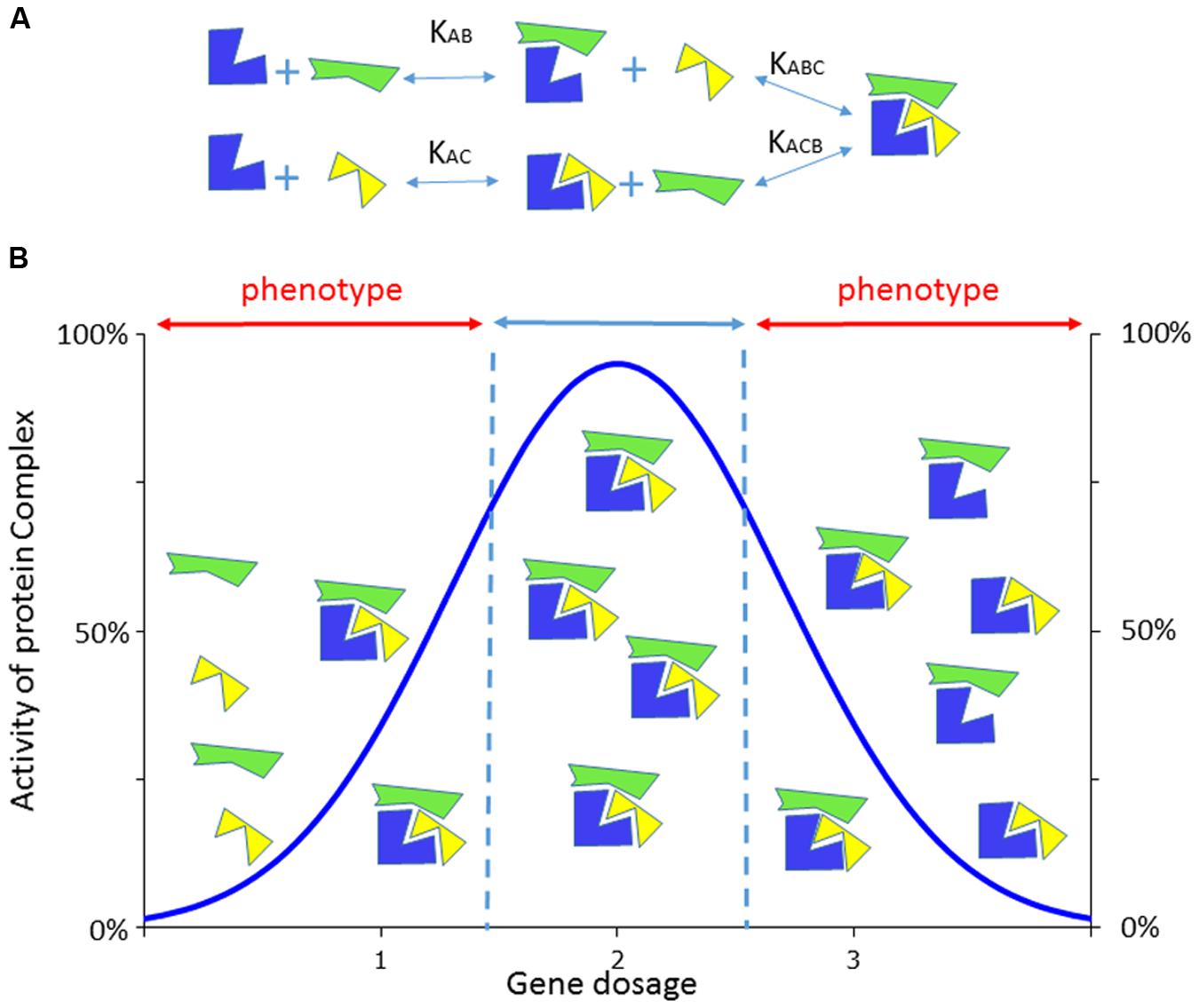
FIGURE 1. Consequence of the dosage effect on the activity of a multiprotein complex. (A) Example of a dosage sensitive gene whose encoded protein (in blue) is able to form a tripartite complex with two partners (in yellow or green), using different constant of association/dissociation. (B) The formation of the complex will be altered by the level of expression of the blue protein compared to the yellow or green ones which are not dosage dependent here. With two copies of the dosage sensitive genes, complexes will be formed with the expected 100% level of activity while in only one (or three copies) are expressed the corresponding complexes will be disrupted delivering only partial activity that would lead when below (or above) a certain threshold to phenotypes. Adapted from (Veitia et al., 2008).
The role of DYRK1A in DS is deduced from several studies in mouse models. Mainly, mouse models overexpressing Dyrk1a or trisomic for genomic segments, homologous to Hsa21 and encompassing Dyrk1a, showed that Dyrk1a overdosage was sufficient to impair cognition with defects similar to DS people (For review see Ahn et al., 2006; Dierssen and Martinez de Lagran, 2006; Dierssen et al., 2009; Guedj et al., 2012; Herault et al., 2012). Several strategies to reduce the expression of Dyrk1a in DS mouse models have shown that Dyrk1a overdosage was necessary for DS related phenotypes. First, the normalization of Dyrk1a expression was achieved via adeno-associated viral delivery of a shRNA sequence specific for Dyrk1a. If injected in the hippocampal region, hippocampal-dependent defects in long term potentiation (LTP), a persistent increase in synaptic strength following high-frequency stimulation of a chemical synapse, and related memory performance (Morris water maze and fear conditioning) were restored in Ts65Dn mice (Altafaj et al., 2013). In addition, motor alterations were reduced after striatal injection of AAV-shRNA in transgenic mice overexpressing Dyrk1a (Ortiz-Abalia et al., 2008). These results demonstrated that normalization of Dyrk1a expression in the adult brain when neuro-developmental alterations have already occurred is sufficient to ameliorate synaptic plasticity changes via its effect on the Erk/CREB signaling pathway, that participates in the LTP induction (Thomas and Huganir, 2004), considered to be one of the major cellular mechanism sustaining learning and memory (Bliss and Collingridge, 1993; Cooke and Bliss, 2006). Second, the normalization of Dyrk1a copy number in trisomic mice was shown to improve hippocampal-dependent learning, presumably due to the recovery of synaptic plasticity, the enhancement of adult hippocampal cell proliferation and differentiation and/or the improvement of the balance between inhibitory and excitatory synaptic markers (Garcia-Cerro et al., 2014; Jiang et al., 2015). In addition, three copies of Dyrk1a was also shown to cause retinal structural and functional alterations in trisomic mice, and normalization of Dyrk1a copy number completely rescued both the morphological and functional visual phenotypes (Laguna et al., 2013).
Overall the phenotypes observed in MRD7 and DS affect similar areas and functions. Brain function is altered in both conditions but with different impact on intellectual abilities. As mentioned previously DS defects includes both short- and long-term memory with delay in learning and in language acquisition and comprehension (Chapman and Hesketh, 2000; Pennington et al., 2003; Nelson et al., 2005; Abbeduto et al., 2007; Dierssen et al., 2009; Contestabile et al., 2010; Lott and Dierssen, 2010) whereas in MRD7 autistic traits, repetitive behavior, feeding difficulties are found associated with more severe intellectual disabilities, speech delay or absence (Courcet et al., 2012; O’Roak et al., 2012; Bronicki et al., 2015; Ruaud et al., 2015). The vision is affected in both conditions but a more precise phenotypic characterization is needed in MRD7. Microcephaly is observed in MRD7 and in Dyrk1a heterozygous mice. Macrocephaly with more precise regional morphological changes are detected in transgenic mice overexpressing Dyrk1a alone (Guedj et al., 2012) but the macrocephaly is not observed in DS mouse models such as the Ts65Dn, suggesting that other genes are contributing to the DS phenotypes (Jiang et al., 2015). Accordingly DS people have a more reduced brain size, with a particular impact on the cerebellum (Pinter et al., 2001; Guihard-Costa et al., 2006), than the normal population. The microcephaly observed in MRD7 should be a direct consequence of abnormal neuronal progenitor proliferation and differentiation due to the loss-of-function of DYRK1A, and induce delay in brain development. On the opposite developmental changes do not seem to be causing the defects rescued by normalizing DYRK1A in DS models. Indeed treatment targeting DYRK1A in adult preclinical model demonstrate that the increase in DYRK1A gene dosage seems perturbed more the physiological function of DYRK1A even though the situation should be more complex in both diseases.
Cognitive dysfunctions observed in MRD7 and in DS result from DYRK1A misdosage during in utero development or in the adult brain. As in MRD7, the DS brain volume is smaller (Guihard-Costa et al., 2006), and this difference persists at post-natal stages (Pinter et al., 2001). Development of the superior temporal neocortex is abnormal with defect in axonal and dendritic arborization (Becker et al., 1986; Golden and Hyman, 1994). Several studies conducted in humans and DS mouse models have suggested the presence of a defect in neurogenesis and increased cell death in the hippocampus. This defect is probably due to alterations of the cell cycle in neuronal progenitors (Contestabile et al., 2007; Esposito et al., 2008; Guidi et al., 2008).
A number of studies have been conducted and demonstrate that DYRK1A dosage is key role for neurogenesis and neuronal maturation (for review see Park et al., 2009; Tejedor and Haemmerle, 2011). DYRK1A regulates proliferation and neuronal differentiation through the phosphorylation of p27Kip1 and Cyclin D1 (CCND1) (Hammerle et al., 2011; Soppa et al., 2014). Dyrk1a gain of function experiments were shown to stop cell proliferation with an increased number of cells expressing p27Kip1, a cyclin dependent kinase inhibitor acting as a main negative regulator of the cell cycle of neurons. Conversely its loss-of-function triggered proliferation and cell death through a p27Kip1 downregulation (Hammerle et al., 2011). Dyrk1a transient expression control the neuronal precursor exit from differentiation through CCND1 and P27Kip1, leaving the cells in a quiescent state ready to differentiate while its expression reduced (Hammerle et al., 2011). DYRK1A can also regulate the G1-phase of the cell cycle of fibroblast cells through the direct phosphorylation of CCND1 (at T286), its nuclear export, degradation, and relative level with p21, all together determining the cycle entry versus exit decision. As such the G1 phase is extended in trisomic fibroblasts due to a lower CCND1 level that could be counteracted by inhibiting DYRK1A (Chen et al., 2013). The same mechanism has been found while DYRK1A controls the cell exit of ventricular neuronal progenitor cells in mouse embryos. In this compartment, Dyrk1a overexpression, through in utero electroporation of mouse embryos, inhibits cell cycle progression and induces premature neuronal differentiation without affecting the capacity to migrate or to differentiate in the post-natal cortex (Yabut et al., 2010). Accordingly in transgenic Dyrk1a mouse embryos, the G1 phase is increased in progenitor cortical stem cells due to CCND1 phosphorylation by DYRK1A, producing a deficit in cortical projection neurons that persists in postnatal stages (Najas et al., 2015). This phenotype is also present in the Ts65Dn DS mouse model which are trisomic for a large region homologous to the human chromosome 21.
Nevertheless additional mechanisms are also perturbed by change in DYRK1A dosage. DYRK1A interferes with the NFATc pathway that is critical for the regulation of vertebrate development, organogenesis and neuronal development (Graef et al., 2001; Nguyen and Di Giovanni, 2008). Overexpression of Dyrk1a acts synergistically with GSK3, as a priming kinase, to inhibit NFAT-dependent transcription in cortical neurons stimulated by FGF8 and heart valve elongation during development of the Ts1Cje DS models, through increase in NFAT nuclear export (Arron et al., 2006). Conversely DYRK1A and DYRK2 were found to phosphorylate NFAT regulatory domain in Drosophila (Gwack et al., 2006). As expected overexpression of both Dyrk1a and Rcan1 results in delayed differentiation of neuronal progenitor cells, with a marked cell-cycle re-entry and alteration of laminar positioning. This phenomenon leads to a reduction of nuclear NFAT localization and can be rescued by expressing a constitutively active form of NFAT (Kurabayashi and Sanada, 2013). As expected interfering with Rcan1 or Dyrk1a overdosage in the Ts1Cje models, or expressing the same constitutive active form of NFAT, rescue the differentiation process, found affected in the trisomic mice. As such the deregulation of the DYRK1A/RCAN1/NFAT pathway leads to developmental alterations which should impact brain size and neuronal density, two traits altered in DS.
In addition, the aberrant enhancement of astrocytic differentiation of cortical progenitor cells has been described in the Ts1Cje mouse model. It is promoted by DYRK1A acting on STAT3, a transcription factor critical for astrogliogenesis (Kurabayashi et al., 2015). This defect could be related to phenotypes observed in DS astroglia. Indeed, differentiation of human induced Pluripotent Stem cells (hiPSc)-from DS patients revealed defect in the neuronal maturation, synapse formation and neurogenesis due to specific overexpression of S100B in medium derived from hIPSc-derived astrocytes (Chen C. et al., 2014). Unfortunately, no S100b-dependent and specific phenotypes have been found so far in DS trisomic models (Yu et al., 2010a,b; Duchon et al., 2011; Belichenko et al., 2015; Jiang et al., 2015) (Duchon and Herault, personal communication) but further analysis in older individuals or to address the function of the tripartite synapse (Pereira and Furlan, 2010) might unravel additional changes.
Overall the variation in DYRK1A activity may impact the proliferation of progenitors cells, the function of the REST complex, leading to the microcephaly observed in MRD7 while in DS premature differentiation will lead to a reduced pool of mature neurons. Presumably both mechanism will contribute to the decreased brain size observed in DS, and cell density alterations found in animal models.
DYRK1A controls neuronal morphogenesis by regulating cytoskeletal dynamics, and overexpression of Dyrk1a in mice is sufficient to recapitulate the dendritic alterations observed in DS patients (Martinez de Lagran et al., 2012). In the adult, connectivity and plasticity are also impacted at different levels. In trisomic mice, dendritic arbor size of neocortical pyramidal cells is smaller, and the peak branching in the arbor was less complex (Dierssen et al., 2003). Total synapse density and synapse-to-neuron ratios are significantly lower in trisomic context (Kurt et al., 2004). Pre-synaptic and post-synaptic elements are significantly enlarged in the hippocampus, the motor and somatosensory cortex, the entorhinal cortex, and the medial septum (Belichenko et al., 2004). Moreover, there is a significant alteration of inhibitory synapses in the fascia dentata (Belichenko et al., 2009). These defects are probably responsible of changes in LTP and long-term depression (LTD), which have been observed in DS mouse models (Siarey et al., 1997, 1999, 2005; Kleschevnikov et al., 2004; Belichenko et al., 2007; Fernandez et al., 2007). Among other possible mechanisms, reduced N-methyl-D-aspartate (NMDA) receptor activation may contribute with an excessive GABAergic inhibition (Kleschevnikov et al., 2004; Costa and Grybko, 2005; Belichenko et al., 2007). In addition increase in GABAergic neurons and changes in the excitatory/inhibitory balance are observed in DS models (Altafaj et al., 2008; Grau et al., 2014; Souchet et al., 2014). Both observations are important but nothing is known at present linking DYRK1A to the glutamatergic synapse or to the GABAergic neurons.
Finally, in the later stages of life, DS patients present a neuropathology similar to AD that will evolve in dementia for 80% of patients after the age of 65 (Strydom et al., 2010; McCarron et al., 2014). This degenerative modification appears in conjunction with the presence of Aβ and tau lesions in several brain regions (Wisniewski et al., 1985). Additionally, a deficit in cholinergic neurons similar to the deficit that occurs in AD has been observed in DS during aging (Contestabile and Ciani, 2008). Even if there is a high prevalence of dementia, not every DS patient develops the accompanying clinical symptoms (Nieuwenhuis-Mark, 2009). Interestingly, overexpression of DYRK1A and some direct targets, such as APP and MAPT, contributes to the early onset of neurofibrillary degeneration, β-amyloidosis, neuronal loss and dementia in DS (Rovelet-Lecrux et al., 2006, 2010; Salehi et al., 2006; Liu et al., 2008; Wegiel et al., 2011; Rovelet-Lecrux and Campion, 2012; Park and Chung, 2013).
DYRK1A is expressed in the nucleus and can also control the expression and interact transcription factor such as the RE1-silencing transcription factor/neuron-restrictive silencer factor REST/NRSF–SWI/SNF chromatin remodeling complex (Canzonetta et al., 2008). Through these mechanisms, increase of Dyrk1a gene dosage induces a SWI/SNF-linked deregulation of gene clusters involved in the neuronal phenotypic traits of DS (Lepagnol-Bestel et al., 2009). Dyrk1a overexpression also leads to alteration in the transcriptome, and these changes affect the NMDA type glutamate receptor with an alteration of NMDA-induced calcium dynamics (Grau et al., 2014). More recently DYRK1A has been showed to act as a transcription factor, phosphorylating the carboxy terminal domain of the RNA polymerase 2 in HeLa cells (Di Vona et al., 2015). Nevertheless no evidence have been found yet for an impact of DYRK1A dosage on the direct interaction of DYRK1A with the RNApol 2 in MRD7 or DS models.
Inhibition of DYRK1A activity represents a new field of study, and a growing number of active molecules to target this protein have been isolated over the last 5–10 years (for review see Becker and Sippl, 2011; Ionescu et al., 2012; Smith et al., 2012; Tell and Hilgeroth, 2013; Becker et al., 2014; Abbassi et al., 2015).
Historically, several compounds were isolated that exhibit dual inhibition of both DYRK family members and cell division cycle-like kinase (CLK family kinases). Effectively, the CLK and DYRK kinase families are part of the CMGC group of the eukaryotic kinome; this group, named after the initials of some members, exhibits auto-phosphorylation activity in addition to phosphorylating serine and tyrosine residues of specific substrates (Aranda et al., 2011). The CLK family has been targeted for the regulation of alternative splicing. CLK family kinases are involved in controlling HIV-1 gene expression (Wong et al., 2011), hepatic gluconeogenesis (Rodgers et al., 2010), cancer (Liu et al., 2013) and neurodegenerative disease such as AD, and their inhibition can be used as a therapeutic strategy (Jain et al., 2014). Thus, many inhibitors have been developed in recent years and are still under investigation for the treatment of DS.
Several compounds with inhibitory activity were originally isolated from natural sources. For example, harmine, a β-carboline alkaloid with a pyrido[3,4-b]indole ring structure was first isolated from the South American vine Banisteriopsis caapi. This molecule has been shown to be an ATP-competitive inhibitor of DYRK1A in vitro, with less potency against other DYRK family members (Laguna et al., 2008; Gockler et al., 2009; Adayev et al., 2011). Tg(Dyrk1a) mice, which overexpress Dyrk1a like people with DS, treated with harmine showed a significant decrease of homocysteine and liver ERK1/2 phosphorylation (Noll et al., 2012). Additionally, harmine prevents premature maturation of neuronal progenitors isolated from Ts65Dn (Mazur-Kolecka et al., 2012). Finally, harmine or its derivatives have been shown to reduce the levels of multiple phosphorylated forms of tau protein in tau overexpressing H4 neuroglioma cells, which are important in the pathological progression of AD (Frost et al., 2011). However, β-carboline analogs possess additional properties such as the inhibition of monoamine oxidase A, a target for depression (Kim et al., 1997), but with significant drawbacks, such as their hallucinogenic properties and a plethora of psychoactive effects, which limit their use in vivo (Fuentes and Longo, 1971). The flavonoid epigallocatechin-3-gallate (EGCG) was the second compound used but the first to be shown to improve cognition in DS models and in humans. Flavanoids, are characterized by having a benzopyrane skeleton, with a pyrane ring bearing at least one aromatic ring. EGCG is also a natural polyphenol and is a major catechin component of green tea leaves (Camellia sinensis) and was identified as a non-competitive ATP inhibitor of DYRK1A (Bain et al., 2003; Adayev et al., 2006). Both EGCG and harmine can fully restore the endocytic defects of hippocampal neurons in mouse models that overexpress Dyrk1a (Kim et al., 2010). In human DS- iPSCs, DYRK1A inhibition by EGCG treatment during neural induction and neuronal differentiation induces an improvement in the number of neurons and promotes dendritic development (Hibaoui et al., 2014). In vivo and in vitro studies of DS mouse models treated with a green tea extract or EGCG extract demonstrated an improvement in brain structure, adult neurogenesis, synaptic plasticity and learning and memory (Xie et al., 2008; Guedj et al., 2009; Pons-Espinal et al., 2013; De la Torre et al., 2014). EGCG also regulates the expression of REST, which is a modulator of genes that encode fundamental neuronal functions, via inhibition of DYRK1A in embryonic stem cells and in the mouse cortex (Canzonetta et al., 2008). Moreover, a pilot study of EGCG in young adults with DS demonstrated effects on memory recognition, working memory and quality of life (De la Torre et al., 2014). EGCG has been demonstrated to be a safe substance after repeated administration in humans and can be easily found as a dietary supplement because of its anti-oxidant properties (Kanwar et al., 2012). A second trial (phase II) has already been conducted by Dierssen and coworkers, and the results are highly anticipated. However, some problems with this compound remain, such as its complex pharmacokinetic properties, poor bioavailability, multiple and heterogeneous effects on signaling pathways and the degree of purity of the commercially available compound (Lambert et al., 2007; Kanwar et al., 2012; Lorenz, 2013). In addition, EGCG was found to have a low inhibitory effect on cannabinoid receptor 1 (CNR1) activity (Korte et al., 2010), which could have negative consequences for long-term treatment as the well-known rimonabant, an inverse agonist directing CNR1. Rimonabant was used as an anorectic and antiobesity drug and was removed from the market after reports of severe depression and suicide in treated people (Thomas et al., 2014). Thus, it is essential to concentrate future efforts on finding a more specific DYRK1A inhibitor, with no interference with CNR1.
Other inhibitors were isolated from natural sources. This is the case for several marine alkaloids, such as variolins, a family containing a central pyrido[3′,2′:4,5]pyrrolo[1,2-c] pyrimidine core substituted with a 2-aminopyrimidine ring and meridianins, and a family of 3-(2-amino-pyrimidine)indoles (Gompel et al., 2004; Giraud et al., 2011; Tahtouh et al., 2012) as well as meriolins, which are a chemical hybrid between the natural products meridianins and variolins (Echalier et al., 2008). Olomoucine is one of the first CDK inhibitors to be developed, and two of its derivatives, family members of 2,6,9-trisubstituted purines roscovitine and purvalanol, also act on DYRK1A (Vesely et al., 1994; Gray et al., 1999; Bain et al., 2003). Staurosporine belongs to the class of indolocarbazoles, which bear a single sugar residue bound to both indole nitrogens, and was originally isolated from the bacterium Streptomyces stauroporus. This compound has exhibited potential as an inhibitor of DYRK1A, but its inhibitory activity was not selective enough among a large array of kinases (Sanchez et al., 2009).
Many inhibitors of DYRK1A kinase activity that have been developed so far are type I inhibitors; i.e., they target the ATP binding site of the kinase in its active conformation when the activation loop is phosphorylated. They have a chemical structure close to the adenine nucleus like many nitrogen heterocyclic compounds such as quinoline, quinazolines, pyrimidines, pyrrolopyrimidines, pyrrolopyridines, and pyrazolopyrimidines. These heteroaromatic cores are often capable of highly efficient binding to proteins because of their shape and hydrophobic nature. Their advantages are their lack of flexibility combined with hydrogen bonding potential from their heteroatoms that can provide a level of target selectivity. Additionally, a rapid exploration of the effect of adding different substituents is facilitated by the applicability of parallelizable reactions, and finally, two or more substitution positions can be explored without the complication of introducing a stereocenter. Their major disadvantages are their hydrophobic nature and flat shape, resulting in low aqueous solubility (Pitt et al., 2009). These inhibitors have higher potency on substrate phosphorylation than on autophosphorylation. Serine and threonine phosphorylation of substrates is inhibited with lower impact on autophosphorylation and the remaining tyrosine kinase activity. Such a difference may reflect the accessibility of the inhibitor target site. DYRK1A kinase activity is closely regulated, with two successive conformational states proposed: the first immature state allows the enzyme to act on the tyrosine residue, while the second more mature state, being irreversible, reacts with the serine/threonine residue (R(X1-2)S/TP) (Lochhead et al., 2005). The two-states model was enriched with the two conformations having slight differences in reacting with tyrosine or serine/threonine, with the immature state having lower catalytic activity and different equilibrium for the amino acid than the mature state (Walte et al., 2013). Additionally, several inhibitors have been synthesized, such as Tg003 (Muraki et al., 2004) and INDY (Ogawa et al., 2010) two benzothiazole derivatives. For example, DANDY, a new 3,5-diaryl-7-azaindole that demonstrates potent inhibition against DYRK1A kinase activity (Gourdain et al., 2013). HCD160 and its derivatives (Kim N.D. et al., 2006; Koo et al., 2009), a series of substituted 6-arylquinazolin-4-amines (Mott et al., 2009; Rosenthal et al., 2011), a new 3-(6-hydroxyindol-2-yl)-5-(phenyl)pyridine (Kassis et al., 2011), a series of aryl-substituted aminopyrimidines (Coombs et al., 2013), a new 7-substituted pyrido[2′,3′:4,5]furo[3,2-d]pyrimidin-4-amines (Deau et al., 2013), an 8-arylpyrido[30′20′:4,5]thieno[3,2-d]pyrimidin-4-amines (Loidreau et al., 2015), and a series of hydroxybenzothiophene ketones (Smith et al., 2012; Schmitt et al., 2014). To date, no DYRK-specific or CLK-specific inhibitors have been reported, certainly because there is a high degree of conservation of the ATP binding site inside the CLK and DYRK kinase families. Nevertheless novel interesting compounds are in development (Rüben et al., 2015). Even if some cellular side effects have been reported for several of the listed compounds, none of them have yet been tested in DS models. Moreover, the activity of these compounds is usually stronger on CLK kinases, excluding varioline B, leucettine 41 and harmine (Tahtouh et al., 2012).
Recent progress on more specific and selective inhibitor has already been made. For example, we note the development of a new bioluminescent reporter assay for evaluation of DYRK1A inhibitors. This system led to the identification of (Z)-5-[(2,3-dihydrobenzofuran-5-yl)methylene]-2-iminothiazolidin-4-one (referred to as CaNDY: CDC37 association inhibitor for DYRK1A) as a strong inhibitor of DYRK family kinases. In addition to inhibition potential, CaNDY decreases DYRK1A molecules in cells, thus efficiently suppressing DYRK1A activity compared to simple inhibition of kinase activity (Sonamoto et al., 2015). Substituted quinazolines are a common pharmacophore for ATP-competitive kinase inhibitors, and the 5 novel thiazolo[5,4-f]quinazoline derivatives that have been synthesized (EHT 5372, 6840, 1610, 9851 and 3356) are among the most potent DYRK1A/1B inhibitors disclosed to date. In particular, they are more potent than NCGC-00189310 (Rosenthal et al., 2011) and leucettine L41 (Tahtouh et al., 2012), the two most active reference inhibitors tested during the screening campaign (Foucourt et al., 2014; Coutadeur et al., 2015). In addition, an indirubin derivative was inactive toward cyclin-dependent kinase 5 (CDK5), GSK3β, and casein kinase 1 (CK1) while exhibiting good selectivity and affinity for DYRK kinases (Myrianthopoulos et al., 2013). Consequently, the development of type II or III inhibitors, non-ATP-mimetics that are anchored to more diverse regions of the ATP binding site, may result in more selective inhibitors while also targeting the premature DYRK1A kinase. For example KHCB19 is a dichloroindolyl enaminonitrile derived from bauerine C, a β-carboline alkaloid originally isolated from the blue-green alga Dichothrix baueriana. This compound exhibits a unique ‘non-ATP mimetic like’ binding mode to CLK1 and also acts on DYRK1A (Fedorov et al., 2011). The non-ATP competitive inhibitors, called type II and type III inhibitors act by inducing a conformational shift in the target enzyme such that the kinase is no longer able to function. This unusual binding mode highlights the opportunity to develop very potent and specific inhibitors with new chemical profiles because they offer the possibility to overcome the major problem of the type I inhibitor (Garuti et al., 2010). Nevertheless even if a more potent and specific inhibitor would be better, too much inhibition may be deleterious as in the case of loss-of-function mutation of DYRK1A in MRD7. In addition DYRK1A specific inhibition on substrates phosphorylation versus autophosphorylation would be interesting to determine. A specific substrate kinase inhibition may have a more beneficial effect rather than targeting the autophosphorylation. The situation is even more complex considering that the target site of the inhibitor might have different accessibility depending on the two conformations of DYRK1A.
Modulation of DYRK1A in MRD7 by increasing its activity would be a good strategy to alleviate the severe cognitive deficits present in the disease, in particular for loss-of-function mutations. Nevertheless only a few proteins are known to increase DYRK1A activity (see Table 2) and no DYRK1A compound activator has been described to date. Thus further work is needed to investigate such a strategy for MRD7.
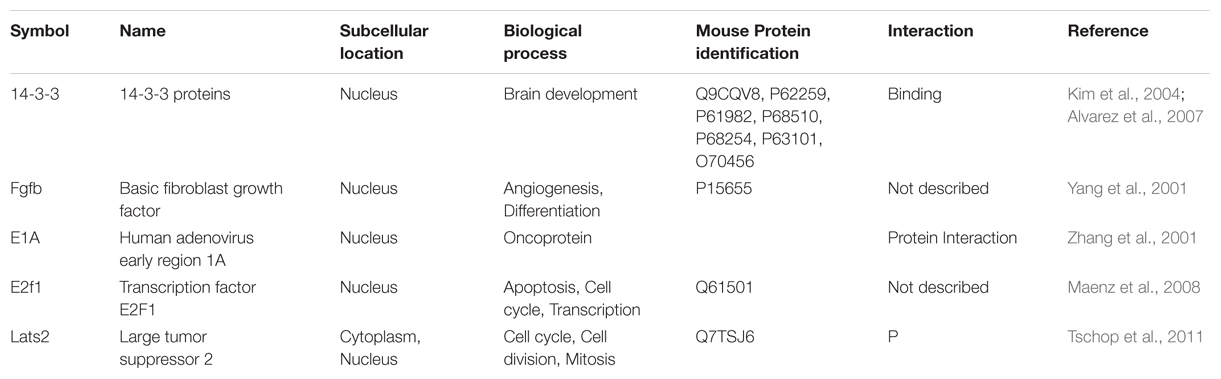
TABLE 2. Proteins that regulate Dyrk1A activity.
Major steps have been achieved to establish the role of DYRK1A in intellectual disabilities such as DS and MRD7 syndromic condition involving heterozygous loss-of-function mutations affecting DYRK1A. These two neurodevelopmental disorders share common features resulting from the alteration of DYRK1A-controlled mechanisms such as neuronal proliferation and differentiation. However, many questions on DYRK1A inhibitors remain unanswered. Are type 1 DYRK1A inhibitors blocking the ATP binding site better suited than type 2 or 3 inhibitors selective for a particular conformation of DYRK1A? What are the cellular targets of DYRK1A: GABAergic or glutamatergic neurons, glial cells?
Undeniably DYRK1A inhibitors represent a promising class of molecules to improve cognitive deficits in people with DS. The pilot clinical trials in adults treated with EGCG is promising, showing a modest gain of cognition (De la Torre et al., 2014). Perinatal treatment during earlier phases of brain development is attractive for improving synaptic plasticity, keeping in mind that DYK1A plays an important role in brain growth by controlling neuronal proliferation and differentiation. Additional studies will be needed to evaluate the use of DYRK1A inhibitors during perinatal periods.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the members of the research group, to the IGBMC laboratory for their helpful discussion and for support from the National Centre for Scientific Research (CNRS), the French National Institute of Health and Medical Research (INSERM), the “Université de Strasbourg” (UDS), the “Centre Européen de Recherche en Biologie et en Médecine (CERBM),” and the ANR Therapeutics-21 project. We also received support from French state funds through the “Agence Nationale de la Recherche” under the frame programs “Investissements d’Avenir” labeled ANR-10-IDEX-0002-02, ANR-10-LABX-0030-INRT, and ANR-10-INBS-07 PHENOMIN, from the EU FP7 large-scale integrated project GENCODYS (FP7-COLLABORATION PROJECT-2009-2.1.1-1/241995) and the Fondation Jerome Lejeune and the Foundation Bettencourt Schueller. The funders had no role in the decision to publish, or preparation of the manuscript.
Abbassi, R., Johns, T. G., Kassiou, M., and Munoz, L. (2015). DYRK1A in neurodegeneration and cancer: molecular basis and clinical implications. Pharmacol. Therapeut. 151, 87–98. doi: 10.1016/j.pharmthera.2015.03.004
Abbeduto, L., Warren, S. F., and Conners, F. A. (2007). Language development in down syndrome: from the prelinguistic period to the acquisition of literacy. Ment. Retard. Dev. Disabil. Res. 13, 247–261. doi: 10.1002/mrdd.20158
Abekhoukh, S., Planque, C., Ripoll, C., Urbaniak, P., Paul, J. L., Delabar, J. M., et al. (2013). Dyrk1A, a serine/threonine kinase, is involved in ERK and Akt activation in the brain of hyperhomocysteinemic mice. Mol. Neurobiol. 47, 105–116. doi: 10.1007/s12035-012-8326-1
Adayev, T., Chen-Hwang, M. C., Murakami, N., Wegiel, J., and Hwang, Y. W. (2006). Kinetic properties of a MNB/DYRK1A mutant suitable for the elucidation of biochemical pathways. Biochemistry 45, 12011–12019. doi: 10.1021/bi060632j
Adayev, T., Wegiel, J., and Hwang, Y. W. (2011). Harmine is an ATP-competitive inhibitor for dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). Arch. Biochem. Biophys. 507, 212–218. doi: 10.1016/j.abb.2010.12.024
Ahn, K. J., Jeong, H. K., Choi, H. S., Ryoo, S. R., Kim, Y. J., Goo, J. S., et al. (2006). DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol. Dis. 22, 463–472. doi: 10.1016/j.nbd.2005.12.006
Aït Yahya-Graison, E., Aubert, J., Dauphinot, L., Rivals, I., Prieur, M., Golfier, G., et al. (2007). Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am. J. Hum. Genet. 81, 475–491. doi: 10.1086/520000
Altafaj, X., Martin, E. D., Ortiz-Abalia, J., Valderrama, A., Lao-Peregrin, C., Dierssen, M., et al. (2013). Normalization of Dyrk1A expression by AAV2/1-shDyrk1A attenuates hippocampal-dependent defects in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 52, 117–127. doi: 10.1016/j.nbd.2012.11.017
Altafaj, X., Ortiz-Abalia, J., Fernandez, M., Potier, M. C., Laffaire, J., Andreu, N., et al. (2008). Increased NR2A expression and prolonged decay of NMDA-induced calcium transient in cerebellum of TgDyrk1A mice, a mouse model of Down syndrome. Neurobiol. Dis. 32, 377–384. doi: 10.1016/j.nbd.2008.07.024
Alvarez, M., Altafaj, X., Aranda, S., and De La Luna, S. (2007). DYRK1A autophosphorylation on serine residue 520 modulates its kinase activity via 14-3-3 binding. Mol. Biol. Cell 18, 1167–1178. doi: 10.1091/mbc.E06-08-0668
Antonarakis, S. E., Lyle, R., Dermitzakis, E. T., Reymond, A., and Deutsch, S. (2004). Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat. Rev. Genet. 5, 725–738. doi: 10.1038/nrg1448
Aranda, S., Alvarez, M., Turro, S., Laguna, A., and De La Luna, S. (2008). Sprouty2-mediated inhibition of fibroblast growth factor signaling is modulated by the protein kinase DYRK1A. Mol. Cell. Biol. 28, 5899–5911. doi: 10.1128/MCB.00394-08
Aranda, S., Laguna, A., and De La Luna, S. (2011). DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. Faseb J. 25, 449–462. doi: 10.1096/fj.10-165837
Arque, G., Casanovas, A., and Dierssen, M. (2013). Dyrk1A Is Dynamically expressed on subsets of motor neurons and in the neuromuscular junction: possible role in down syndrome. PLoS ONE 8:e54285. doi: 10.1371/journal.pone.0054285
Arqué, G., De Lagrán, M. M., Arbonés, M. L., and Dierssen, M. (2009). Age-associated motor and visuo-spatial learning phenotype in Dyrk1A heterozygous mutant mice. Neurobiol. Dis. 36, 312–319. doi: 10.1016/j.nbd.2009.07.027
Arqué, G., Fotaki, V., Fernández, D., De Lagrán, M., Arbonés, M. L., and Dierssen, M. (2008). Impaired spatial learning strategies and novel object recognition in mice haploinsufficient for the dual specificity tyrosine-regulated kinase-1A (Dyrk1A). PLoS ONE 3:e2575. doi: 10.1371/journal.pone.0002575
Arron, J. R., Winslow, M. M., Polleri, A., Chang, C. P., Wu, H., Gao, X., et al. (2006). NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441, 595–600. doi: 10.1038/nature04678
Bain, J., Mclauchlan, H., Elliott, M., and Cohen, P. (2003). The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204. doi: 10.1042/bj20021535
Becker, L. E., Armstrong, D. L., and Chan, F. (1986). Dendritic atrophy in children with Down’s syndrome. Ann. Neurol. 20, 520–526. doi: 10.1002/ana.410200413
Becker, W., and Sippl, W. (2011). Activation, regulation, and inhibition of DYRK1A. FEBS J. 278, 246–256. doi: 10.1111/j.1742-4658.2010.07956.x
Becker, W., Soppa, U., and Tejedor, F. J. (2014). DYRK1A: a potential drug target for multiple down syndrome neuropathologies. CNS Neurol. Disord. Drug Targets 13, 26–33. doi: 10.2174/18715273113126660186
Becker, W., Weber, Y., Wetzel, K., Eirmbter, K., Tejedor, F. J., and Joost, H. G. (1998). Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J. Biol. Chem. 273, 25893–25902. doi: 10.1074/jbc.273.40.25893
Belichenko, P. V., Kleschevnikov, A. M., Becker, A., Wagner, G. E., Lysenko, L. V., Yu, Y. E., et al. (2015). Down syndrome cognitive phenotypes modeled in mice trisomic for All HSA 21 homologues. PLoS ONE 10:e0134861. doi: 10.1371/journal.pone.0134861
Belichenko, P. V., Kleschevnikov, A. M., Salehi, A., Epstein, C. J., and Mobley, W. C. (2007). Synaptic and cognitive abnormalities in mouse models of down syndrome: exploring genotype-phenotype relationships. J. Comp. Neurol. 504, 329–345. doi: 10.1002/cne.21433
Belichenko, P. V., Klescrevnikov, A. M., Masliah, E., Wu, C. B., Takimoto-Kimura, R., Salehi, A., et al. (2009). Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 512, 453–466. doi: 10.1002/cne.21895
Belichenko, P. V., Masliah, E., Kleschevnikov, A. M., Villar, A. J., Epstein, C. J., Salehi, A., et al. (2004). Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. J. Comp. Neurol. 480, 281–298. doi: 10.1002/cne.20337
Benavides-Piccione, R., Dierssen, M., Ballesteros-Yanez, I., De Lagran, M. M., Arbones, M. L., Fotaki, V., et al. (2005). Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/- mouse. Neurobiol. Dis. 20, 115–122. doi: 10.1016/j.nbd.2005.02.004
Bescond, M., and Rahmani, Z. (2005). Dual-specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A) interacts with the phytanoyl-CoA alpha-hydroxylase associated protein I (PAHX-AP1), a brain specific protein. Int. J. Biochem. Cell Biol. 37, 775–783. doi: 10.1016/j.biocel.2004.12.006
Bliss, T. V. P., and Collingridge, G. L. (1993). A synaptic model of memory - long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Bronicki, L. M., Redin, C., Drunat, S., Piton, A., Lyons, M., Passemard, S., et al. (2015). Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. Eur. J. Hum. Genet. 23, 1482–1487. doi: 10.1038/ejhg.2015.29
Canzonetta, C., Mulligan, C., Deutsch, S., Ruf, S., O’doherty, A., Lyle, R., et al. (2008). DYRK1A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic stem cell fate in Down syndrome. Am. J. Hum. Genet. 83, 388–400. doi: 10.1016/j.ajhg.2008.08.012
Chapman, R. S., and Hesketh, L. J. (2000). Behavioral phenotype of individuals with Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 6, 84–95. doi: 10.1002/1098-2779(2000)6
Chen, C., Jiang, P., Xue, H., Peterson, S. E., Tran, H. T., Mccann, A. E., et al. (2014). Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 5:4430. doi: 10.1038/ncomms5430
Chen, C.-K., Bregere, C., Paluch, J., Lu, J. F., Dickman, D. K., and Chang, K. T. (2014). Activity-dependent facilitation of Synaptojanin and synaptic vesicle recycling by the Minibrain kinase. Nat. Commun. 5:4246. doi: 10.1038/ncomms5246
Chen, J. Y., Lin, J. R., Tsai, F. C., and Meyer, T. (2013). Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol. Cell. 52, 87–100. doi: 10.1016/j.molcel.2013.09.009
Chen-Hwang, M. C., Chen, H. R., Elzinga, M., and Hwang, Y. W. (2002). Dynamin is a minibrain kinase/dual specificity Yak1-related kinase 1A substrate. J. Biol. Chem. 277, 17597–17604. doi: 10.1074/jbc.M111101200
Contestabile, A., Benfenati, F., and Gasparini, L. (2010). Communication breaks-Down: from neurodevelopment defects to cognitive disabilities in Down syndrome. Prog. Neurobiol. 91, 1–22. doi: 10.1016/j.pneurobio.2010.01.003
Contestabile, A., and Ciani, E. (2008). The place of choline acetyltransferase activity measurement in the “Cholinergic hypothesis” of neurodegenerative diseases. Neurochem. Res. 33, 318–327. doi: 10.1007/s11064-007-9497-4
Contestabile, A., Fila, T., Ceccarelli, C., Bonasoni, P., Bonapace, L., Santini, D., et al. (2007). Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with down syndrome and in Ts65Dn mice. Hippocampus 17, 665–678. doi: 10.1002/hipo.20308
Cooke, S. F., and Bliss, T. V. P. (2006). Plasticity in the human central nervous system. Brain 129, 1659–1673. doi: 10.1093/brain/awl082
Coombs, T. C., Tanega, C., Shen, M., Wang, J. L., Auld, D. S., Gerritz, S. W., et al. (2013). Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: development of chemical probe ML315. Bioorg. Med. Chem. Lett. 23, 3654–3661. doi: 10.1016/j.bmcl.2013.02.096
Coppus, A. M. W. (2013). People with intellectual disability: what do we know about adulthood and life expectancy? Dev. Disabil. Res. Rev. 18, 6–16. doi: 10.1002/ddrr.1123
Costa, A. C. S., and Grybko, M. J. (2005). Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci. Lett. 382, 317–322. doi: 10.1016/j.neulet.2005.03.031
Courcet, J.-B., Faivre, L., Malzac, P., Masurel-Paulet, A., Lopez, E., Callier, P., et al. (2012). The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet. 49, 731–736. doi: 10.1136/jmedgenet-2012-101251
Coutadeur, S., Benyamine, H., Delalonde, L., De Oliveira, C., Leblond, B., Foucourt, A., et al. (2015). A novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: effect on Tau and amyloid pathologies in vitro. J. Neurochem. 133, 440–451. doi: 10.1111/jnc.13018
Covelli, V., Raggi, A., Meucci, P., Paganelli, C., and Leonardi, M. (2016). Ageing of people with Down’s syndrome: a systematic literature review from 2000 to 2014. Int. J. Rehabil. Res. 39, 20–28. doi: 10.1097/MRR.0000000000000147
De Graaf, K., Czajkowska, H., Rottmann, S., Packman, L. C., Lilischkis, R., Lüscher, B., et al. (2006). The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434 ,a novel in vivo phosphorylation site. BMC Biochem. 7:7. doi: 10.1186/1471-2091-7-7
De Graaf, K., Hekerman, P., Spelten, O., Herrmann, A., Packman, L. C., Bussow, K., et al. (2004). Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain - Phosphorylation by DYRK1A and colocalization with splicing factors. J. Biol. Chem. 279, 4612–4624. doi: 10.1074/jbc.M310794200
De la Torre, R., De Sola, S., Pons, M., Duchon, A., De Lagran, M. M., Farre, M., et al. (2014). Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutrit. Food Res. 58, 278–288. doi: 10.1002/mnfr.201300325
Deau, E., Loidreau, Y., Marchand, P., Nourrisson, M. R., Loaec, N., Meijer, L., et al. (2013). Synthesis of novel 7-substituted pyrido 2′,3′:4,5 furo 3,2-d pyrimidin-4-amines and their N-aryl analogues and evaluation of their inhibitory activity against Ser/Thr kinases. Bioorg. Med. Chem. Lett. 23, 6784–6788. doi: 10.1016/j.bmcl.2013.10.019
Delabar, J. M., Theophile, D., Rahmani, Z., Chettouh, Z., Blouin, J. L., Prieur, M., et al. (1993). Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur. J. Hum. Genet. 1, 114–124.
Di Vona, C., Bezdan, D., Islam, A., Salichs, E., Lopez-Bigas, N., Ossowski, S., et al. (2015). Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA Polymerase II CTD Kinase. Mol. Cell. 57, 506–520. doi: 10.1016/j.molcel.2014.12.026
Dierssen, M., Benavides-Piccione, R., Martinez-Cue, C., Estivill, X., Florez, J., Elston, G. N., et al. (2003). Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cereb. Cortex 13, 758–764. doi: 10.1093/cercor/13.7.758
Dierssen, M., Herault, Y., and Estivill, X. (2009). Aneuploidy: from a physiological mechanism of variance to Down syndrome. Physiol. Rev. 89, 887–920. doi: 10.1152/physrev.00032.2007
Dierssen, M., and Martinez de Lagran, M. (2006). DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A): a gene with dosage effect during development and neurogenesis. Sci. World J. 6, 1911–1922. doi: 10.1100/tsw.2006.319
Dowjat, W. K., Adayev, T., Kuchna, I., Nowicki, K., Palminiello, S., Hwang, Y. W., et al. (2007). Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci. Lett. 413, 77–81. doi: 10.1016/j.neulet.2006.11.026
Down, J. L. H. (1866). Observations on an ethnic classification of idiots. Clin. Lecture Rep. Lond. Hospital 3, 259–262.
Duchon, A., Pothion, S., Brault, V., Sharp, A. J., Tybulewicz, V. L. J., Fisher, E. M. C., et al. (2011). The telomeric part of the human chromosome 21 from Cstb to Prmt2 is not necessary for the locomotor and short-term memory deficits observed in the Tc1 mouse model of Down syndrome. Behav. Brain Res. 217, 271–281. doi: 10.1016/j.bbr.2010.10.023
Echalier, A., Bettayeb, K., Ferandin, Y., Lozach, O., Clement, M., Valette, A., et al. (2008). Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J. Med. Chem. 51, 737–751. doi: 10.1021/jm700940h
Elton, T. S., Sansom, S. E., and Martin, M. M. (2010). Trisomy-21 gene dosage overexpression of miRNAs results in the haploinsufficiency of specific target proteins. RNA Biol. 7, 540–547. doi: 10.4161/rna.7.5.12685
Esposito, G., Imitola, J., Lu, J., De Filippis, D., Scuderi, C., Ganesh, V. S., et al. (2008). Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Hum. Mol. Genet. 17, 440–457. doi: 10.1093/hmg/ddm322
Fedorov, O., Huber, K., Eisenreich, A., Filippakopoulos, P., King, O., Bullock, A. N., et al. (2011). Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 18, 67–76. doi: 10.1016/j.chembiol.2010.11.009
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 10, 411–413.
Fernandez-Martinez, J., Vela, E. M., Tora-Ponsioen, M., Ocana, O. H., Nieto, M. A., and Galceran, J. (2009). Attenuation of Notch signalling by the Down-syndrome-associated kinase DYRK1A. J. Cell Sci. 122, 1574–1583. doi: 10.1242/jcs.044354
Fotaki, V., Dierssen, M., Alcantara, S., Martinez, S., Marti, E., Casas, C., et al. (2002). Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 22, 6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002
Foucourt, A., Hedou, D., Dubouilh-Benard, C., Girard, A., Taverne, T., Casagrande, A. S., et al. (2014). Design and synthesis of thiazolo 5,4-f quinazolines as DYRK1A inhibitors. Part II Mol. 19, 15411–15439. doi: 10.3390/molecules191015411
Frost, D., Meechoovet, B., Wang, T., Gately, S., Giorgetti, M., Shcherbakova, I., et al. (2011). Beta-Carboline compounds, including harmine, inhibit DYRK1A and tau phosphorylation at multiple Alzheimer’s disease-related sites. PLoS ONE 6:e19264. doi: 10.1371/journal.pone.0019264
Fuentes, J. A., and Longo, V. G. (1971). An investigation on central effects of harmine, harmaline and related beta-carboline. Neuropharmacology 10:15. doi: 10.1016/0028-3908(71)90004-9
Garcia-Cerro, S., Martinez, P., Vidal, V., Corrales, A., Florez, J., Vidal, R., et al. (2014). Overexpression of dyrk1a is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of down syndrome. PLoS ONE 9:e106572. doi: 10.1371/journal.pone.0106572
Garuti, L., Roberti, M., and Bottegoni, G. (2010). Non-ATP competitive protein kinase inhibitors. Curr. Med. Chem. 17, 2804–2821. doi: 10.2174/092986710791859333
Giraud, F., Alves, G., Debiton, E., Nauton, L., Thery, V., Durieu, E., et al. (2011). Synthesis, protein kinase inhibitory potencies, and in vitro antiproliferative activities of meridianin derivatives. J. Med. Chem. 54, 4474–4489. doi: 10.1021/jm200464w
Gockler, N., Jofre, G., Papadopoulos, C., Soppa, U., Tejedor, F. J., and Becker, W. (2009). Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. Febs J. 276, 6324–6337. doi: 10.1111/j.1742-4658.2009.07346.x
Golden, J. A., and Hyman, B. T. (1994). Development of the superior temporal neocortex is anomalous in trisomy 21. J. Neuropathol. Exp. Neurol. 53, 513–520. doi: 10.1097/00005072-199409000-00011
Gompel, M., Leost, M., Joffe, E. B. D., Puricelli, L., Franco, L. H., Palermo, J., et al. (2004). Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 14, 1703–1707. doi: 10.1016/j.bmcl.2004.01.050
Gourdain, S., Dairou, J., Denhez, C., Bui, L. C., Rodrigues-Lima, F., Janel, N., et al. (2013). Development of DANDYs, New 3,5-Diaryl-7-azaindoles demonstrating potent DYRK1A kinase inhibitory activity. J. Med. Chem. 56, 9569–9585. doi: 10.1021/jm401049v
Graef, I. A., Chen, F., and Crabtree, G. R. (2001). NFAT signaling in vertebrate development. Curr. Opin. Genet. Dev. 11, 505–512. doi: 10.1016/S0959-437X(00)00225-2
Grau, C., Arato, K., Fernandez-Fernandez, J. M., Valderrama, A., Sindreu, C., Fillat, C., et al. (2014). DYRK1A-mediated phosphorylation of GluN2A at Ser(1048) regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front. Cell. Neurosci. 8:331. doi: 10.3389/fncel.2014.00331
Gray, N., Detivaud, L., Doerig, C., and Meijer, L. (1999). ATP-site directed inhibitors of cyclin-dependent kinases. Curr. Med. Chem. 6, 859–875.
Guedj, F., Pereira, P. L., Najas, S., Barallobre, M. J., Chabert, C., Souchet, B., et al. (2012). DYRK1A: a master regulatory protein controlling brain growth. Neurobiol. Dis. 46, 190–203. doi: 10.1016/j.nbd.2012.01.007
Guedj, F., Sébrié, C., Rivals, I., Ledru, A., Paly, E., Bizot, J. C., et al. (2009). Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS ONE 4:e4606. doi: 10.1371/journal.pone.0004606
Guidi, S., Bonasoni, P., Ceccarelli, C., Santini, D., Gualtieri, F., Ciani, E., et al. (2008). Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 18, 180–197.
Guihard-Costa, A. M., Khung, S., Delbecque, K., Menez, F., and Delezoide, A. L. (2006). Biometry of face and brain in fetuses with trisomy 21. Pediatr. Res. 59, 33–38. doi: 10.1203/01.pdr.0000190580.88391.9a
Guimera, J., Casas, C., Estivill, X., and Pritchard, M. (1999). Characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome of the human minibrain homologue (MNBH/DYRK1). Cytogenet. Cell Genet. 86:15.
Guo, X., Williams, J. G., Schug, T. T., and Li, X. (2010). DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 285, 13223–13232. doi: 10.1074/jbc.M110.102574
Gwack, Y., Sharma, S., Nardone, J., Tanasa, B., Iuga, A., Srikanth, S., et al. (2006). A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 441, 646–650. doi: 10.1038/nature04631
Hammerle, B., Carnicero, A., Elizalde, C., Ceron, J., Martinez, S., and Tejedor, F. J. (2003). Expression patterns and subcellular localization of the Down syndrome candidate protein MNB/DYRK1A suggest a role in late neuronal differentiation. Eur. J. Neurosci. 17, 2277–2286. doi: 10.1046/j.1460-9568.2003.02665.x
Hammerle, B., Elizalde, C., and Tejedor, F. J. (2008). The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur. J. Neurosci. 27, 1061–1074. doi: 10.1111/j.1460-9568.2008.06092.x
Hammerle, B., Ulin, E., Guimera, J., Becker, W., Guillemot, F., and Tejedor, F. J. (2011). Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27(KIP1) expression and suppressing NOTCH signaling. Development 138, 2543–2554. doi: 10.1242/dev.066167
Herault, Y., Duchon, A., Velot, E., Maréchal, D., and Brault, V. (2012). The in vivo Down syndrome genomic library in mouse. Prog. Brain Res. 197, 169–197. doi: 10.1016/B978-0-444-54299-1.00009-1
Hibaoui, Y., Grad, I., Letourneau, A., Sailani, M. R., Dahoun, S., Santoni, F. A., et al. (2014). Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. Embo Mol. Med. 6, 259–277. doi: 10.1002/emmm.201302848
Im, E., and Chung, K. C. (2015). Dyrk1A phosphorylates parkin at Ser-131 and negatively regulates its ubiquitin E3 ligase activity. J. Neurochem. 134, 756–768. doi: 10.1111/jnc.13164
Ionescu, A., Dufrasne, F., Gelbcke, M., Jabin, I., Kiss, R., and Lamoral-Theys, D. (2012). DYRK1A kinase inhibitors with emphasis on cancer. Mini Rev. Med. Chem. 12, 1315–1329. doi: 10.2174/13895575112091315
Jain, P., Karthikeyan, C., Moorthy, N., Waiker, D. K., Jain, A. K., and Trivedi, P. (2014). Human CDC2-like kinase 1 (CLK1): a novel target for Alzheimer’s Disease. Curr. Drug Targets 15, 539–550. doi: 10.2174/1389450115666140226112321
Jiang, X., Liu, C., Yu, T., Zhang, L., Meng, K., Xing, Z., et al. (2015). Genetic dissection of the Down syndrome critical region. Hum. Mol. Genet. 24, 6540–6551. doi: 10.1093/hmg/ddv364
Kaczmarski, W., Barua, M., Mazur-Kolecka, B., Frackowiak, J., Dowjat, W., Mehta, P., et al. (2014). Intracellular distribution of differentially phosphorylated dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A). J. Neurosci. Res. 92, 162–173. doi: 10.1002/jnr.23279
Kang, J. E., Choi, S. A., Park, J. B., and Chung, K. C. (2005). Regulation of the proapoptotic activity of huntingtin interacting protein 1 by Dyrk1 and caspase-3 in hippocampal neuroprogenitor cells. J. Neurosci. Res. 81, 62–72. doi: 10.1002/jnr.20534
Kanwar, J., Taskeen, M., Mohammad, I., Huo, C., Chan, T. H., and Dou, Q. P. (2012). Recent advances on tea polyphenols. Front. Biosci. 4:131. doi: 10.2741/e363
Kassis, P., Brzeszcz, J., Beneteau, V., Lozach, O., Meijer, L., Le Guevel, R., et al. (2011). Synthesis and biological evaluation of new 3-(6-hydroxyindol-2-yl)-5-(Phenyl) pyridine or pyrazine V-Shaped molecules as kinase inhibitors and cytotoxic agents. Eur. J. Med. Chem. 46, 5416–5434. doi: 10.1016/j.ejmech.2011.08.048
Kelly, P. A., and Rahmani, Z. (2005). DYRK1A enhances the mitogen-activated protein kinase cascade in PC12 cells by forming a complex with Ras, B-Raf, and MEK1. Mol. Biol. Cell 16, 3562–3573. doi: 10.1091/mbc.E04-12-1085
Kim, D., Won, J., Shin, D. W., Kang, J., Kim, Y. J., Choi, S. Y., et al. (2004). Regulation of Dyrk1A kinase activity by 14-3-3. Biochem. Biophys. Res. Commun. 323, 499–504. doi: 10.1016/j.bbrc.2004.08.102
Kim, E. J., Sung, J. Y., Lee, H. J., Rhim, H., Hasegawa, M., Iwatsubo, T., et al. (2006). Dyrk1A phosphorylates alpha-synuclein and enhances intracellular inclusion formation. J. Biol. Chem. 281, 33250–33257. doi: 10.1074/jbc.M606147200
Kim, N. D., Yoon, J., Kim, J. H., Lee, J. T., Chon, Y. S., Hwang, M. K., et al. (2006). Putative therapeutic agents for the learning and memory deficits of people with Down syndrome. Bioorg. Med. Chem. Lett. 16, 3772–3776. doi: 10.1016/j.bmcl.2006.04.042
Kim, H., Sablin, S. O., and Ramsay, R. R. (1997). Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch. Biochem. Biophys. 337, 137–142. doi: 10.1006/abbi.1996.9771
Kim, Y., Park, J., Song, W. J., and Chang, S. (2010). Overexpression of Dyrk1A causes the defects in synaptic vesicle endocytosis. Neurosignals 18, 164–172. doi: 10.1159/000321994
Kimura, R., Kamino, K., Yamamoto, M., Nuripa, A., Kida, T., Kazui, H., et al. (2007). The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 16, 15–23. doi: 10.1093/hmg/ddl437
Kinstrie, R., Lochhead, P. A., Sibbet, G., Morrice, N., and Cleghon, V. (2006). dDYRK2 and Minibrain interact with the chromatin remodelling factors SNR1 and TRX. Biochem. J. 398, 45–54. doi: 10.1042/BJ20060159
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 24, 8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004
Koo, K. A., Kim, N. D., Chon, Y. S., Jung, M. S., Lee, B. J., Kim, J. H., et al. (2009). QSAR analysis of pyrazolidine-3,5-diones derivatives as Dyrk1A inhibitors. Bioorg. Med. Chem. Lett. 19, 2324–2328. doi: 10.1016/j.bmcl.2009.02.062
Korbel, J. O., Tirosh-Wagner, T., Urban, A. E., Chen, X. N., Kasowski, M., Dai, L., et al. (2009). The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U.S.A. 106, 12031–12036. doi: 10.1073/pnas.0813248106
Korenberg, J. R. (1990). Molecular mapping of the Down syndrome phenotype. Prog. Clin. Biol. Res. 360, 105–115.
Korte, G., Dreiseitel, A., Schreier, P., Oehme, A., Locher, S., Geiger, S., et al. (2010). Tea catechins’ affinity for human cannabinoid receptors. Phytomedicine 17, 19–22. doi: 10.1016/j.phymed.2009.10.001
Kurabayashi, N., Hirota, T., Sakai, M., Sanada, K., and Fukada, Y. (2010). DYRK1A and glycogen synthase kinase 3 beta, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol. Cell. Biol. 30, 1757–1768. doi: 10.1128/MCB.01047-09
Kurabayashi, N., Nguyen, M. D., and Sanada, K. (2015). DYRK1A overexpression enhances STAT activity and astrogliogenesis in a Down syndrome mouse model. EMBO Report 2015, e201540374. doi: 10.15252/embr.201540374
Kurabayashi, N., and Sanada, K. (2013). Increased dosage of DYRK1A and DSCR1 delays neuronal differentiation in neocortical progenitor cells. Genes Dev. 27, 2708–2721. doi: 10.1101/gad.226381.113
Kurt, M. A., Kafa, M. I., Dierssen, M., and Davies, D. C. (2004). Deficits of neuronal density in CA1 and synaptic density in the dentate gyrus, CA3 and CA1, in a mouse model of Down syndrome. Brain Res. 1022, 101–109. doi: 10.1016/j.brainres.2004.06.075
Laffaire, J., Rivals, I., Dauphinot, L., Pasteau, F., Wehrle, R., Larrat, B., et al. (2009). Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genomics 10:138. doi: 10.1186/1471-2164-10-138
Laguna, A., Aranda, S., Barallobre, M. J., Barhoum, R., Fernandez, E., Fotaki, V., et al. (2008). The Protein Kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev. Cell 15, 841–853. doi: 10.1016/j.devcel.2008.10.014
Laguna, A., Barallobre, M. J., Marchena, M. A., Mateus, C., Ramirez, E., Martinez-Cue, C., et al. (2013). Triplication of DYRK1A causes retinal structural and functional alterations in Down syndrome. Hum. Mol. Genet. 22, 2775–2784. doi: 10.1093/hmg/ddt125
Lambert, J. D., Sang, S. M., and Yang, C. S. (2007). Biotransformation of green tea polyphenols and the biological activities of those metabolites. Mol. Pharmaceut. 4, 819–825. doi: 10.1021/mp700075m
Lejeune, J., Gauthier, M., and Turpin, R. (1959). Etude des chromosomes somatiques de neuf enfants mongoliens. C. R. Acad. Sci. 248:1721.
Lepagnol-Bestel, A. M., Zvara, A., Maussion, G., Quignon, F., Ngimbous, B., Ramoz, N., et al. (2009). DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum. Mol. Genet. 18, 1405–1414. doi: 10.1093/hmg/ddp047
Letourneau, A., Santoni, F. A., Bonilla, X., Sailani, M. R., Gonzalez, D., Kind, J., et al. (2014). Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 508, 345–350. doi: 10.1038/nature13200
Litovchick, L., Florens, L. A., Swanson, S. K., Washburn, M. P., and Decaprio, J. A. (2011). DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 25, 801–813. doi: 10.1101/gad.2034211
Liu, F., Liang, Z. H., Wegiel, J., Hwang, Y. W., Iqbal, K., Grundke-Iqbal, I., et al. (2008). Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. Faseb J. 22, 3224–3233. doi: 10.1096/fj.07-104539
Liu, Y. Y., Conaway, L., Bethard, J. R., Al-Ayoubi, A. M., Bradley, A. T., Zheng, H., et al. (2013). Phosphorylation of the alternative mRNA splicing factor 45 (SPF45) by Clk1 regulates its splice site utilization, cell migration and invasion. Nucleic Acids Res. 41, 4949–4962. doi: 10.1093/nar/gkt170
Lochhead, P. A., Sibbet, G., Morrice, N., and Cleghon, V. (2005). Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 121, 925–936. doi: 10.1016/j.cell.2005.03.034
Loidreau, Y., Deau, E., Marchand, P., Nourrisson, M. R., Loge, C., Coadou, G., et al. (2015). Synthesis and molecular modelling studies of 8-arylpyrido 3′,2′:4,5 thieno 3,2-d pyrimidin-4-amines as multitarget Ser/Thr kinases inhibitors. Eur. J. Med. Chem. 92, 124–134. doi: 10.1016/j.ejmech.2014.12.038
Lorenz, M. (2013). Cellular targets for the beneficial actions of tea polyphenols. Am. J. Clin. Nutrit. 98, 1642S–1650S. doi: 10.3945/ajcn.113.058230
Lott, I. T., and Dierssen, M. (2010). Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 9, 623–633. doi: 10.1016/S1474-4422(10)70112-5
Lu, J., and Sheen, V. (2013). “Genetic and epigenetic mechanisms in down syndrome brain,” in Down Syndrome, ed. S. Kumar Dey (Rijeka: InTech). doi: 10.5772/52807
Lyle, R., Béna, F., Gagos, S., Gehrig, C., Lopez, G., Schinzel, A., et al. (2009). Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur. J. Hum. Genet. 17, 454–466. doi: 10.1038/ejhg.2008.214
Maenz, B., Hekerman, P., Vela, E. M., Galceran, J., and Becker, W. (2008). Characterization of the human DYRK1A promoter and its regulation by the transcription factor E2F1. BMC Mol. Biol. 9:16. doi: 10.1186/1471-2199-9-30
Mao, J. H., Maye, P., Kogerman, P., Tejedor, F. J., Toftgard, R., Xie, W., et al. (2002). Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 277, 35156–35161. doi: 10.1074/jbc.M206743200
Marti, E., Altafaj, X., Dierssen, M., De La Luna, S., Fotaki, V., Alvarez, M., et al. (2003). Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res. 964, 250–263. doi: 10.1016/S0006-8993(02)04069-6
Martinez de Lagran, M., Benavides-Piccione, R., Ballesteros-Yanez, I., Calvo, M., Morales, M., Fillat, C., et al. (2012). Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb. Cortex 22, 2867–2877. doi: 10.1093/cercor/bhr362
Mazur-Kolecka, B., Golabek, A., Kida, E., Rabe, A., Hwang, Y. W., Adayev, T., et al. (2012). Effect of DYRK1A activity inhibition on development of neuronal progenitors isolated from Ts65Dn mice. J. Neurosci. Res. 90, 999–1010. doi: 10.1002/jnr.23007
McCarron, M., Mccallion, P., Reilly, E., and Mulryan, N. (2014). A prospective 14-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 58, 61–70. doi: 10.1111/jir.12074
McCormick, M. K., Schinzel, A., Petersen, M. B., Stetten, G., Driscoll, D. J., Cantu, E. S., et al. (1989). Molecular genetic appoach to the characterization of the ‘Down syndrome region’ of chromosome 21. Genomics 5, 325–331. doi: 10.1016/0888-7543(89)90065-7
Miyata, Y., and Nishida, E. (2011). DYRK1A binds to an evolutionarily conserved WD40-repeat protein WDR68 and induces its nuclear translocation. Biochim. Biophys. Acta Mol. Cell Res. 1813, 1728–1739. doi: 10.1016/j.bbamcr.2011.06.023
Moldrich, R. X., Dauphinot, L., Laffaire, J., Vitalis, T., Herault, Y., Beart, P. M., et al. (2009). Proliferation deficits and gene expression dysregulation in down’s syndrome (Ts1Cje) neural progenitor cells cultured from neurospheres. J. Neurosci. Res. 87, 3143–3152. doi: 10.1002/jnr.22131
Møller, R. S., Kübart, S., Hoeltzenbein, M., Heye, B., Vogel, I., Hansen, C. P., et al. (2008). Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 82, 1165–1170. doi: 10.1016/j.ajhg.2008.03.001
Morita, K., Lo Celso, C., Spencer-Dene, B., Zouboulis, C. C., and Watt, F. M. (2006). HAN11 binds mDia1 and controls GLI1 transcriptional activity. J. Dermatol. Sci. 44, 11–20. doi: 10.1016/j.jdermsci.2006.06.001
Mott, B. T., Tanega, C., Shen, M., Maloney, D. J., Shinn, P., Leister, W., et al. (2009). Evaluation of substituted 6-arylquinazolin-4-amines as potent and selective inhibitors of cdc2-like kinases (Clk). Bioorg. Med. Chem. Lett. 19, 6700–6705. doi: 10.1016/j.bmcl.2009.09.121
Murakami, N., Bolton, D., and Hwang, Y.-W. (2009). Dyrk1A binds to multiple endocytic proteins required for formation of clathrin-coated vesicles. Biochemistry 48, 9297–9305. doi: 10.1021/bi9010557
Murakami, N., Bolton, D. C., Kida, E., Xie, W., and Hwang, Y. W. (2012). Phosphorylation by Dyrk1A of clathrin coated vesicle-associated proteins: identification of the substrate proteins and the effects of phosphorylation. PLoS ONE 7:e34845. doi: 10.1371/journal.pone.0034845
Murakami, N., Xie, W., Lu, R. C., Chen-Hwang, M. C., Wieraszko, A., and Hwang, Y. W. (2006). Phosphorylation of amphiphysin I by minibrain kinase/dual-specificity tyrosine phosphorylation-regulated kinase, a kinase implicated in Down syndrome. J. Biol. Chem. 281, 23712–23724. doi: 10.1074/jbc.M513497200
Muraki, M., Ohkawara, B., Hosoya, T., Onogi, H., Koizumi, J., Koizumi, T., et al. (2004). Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 279, 24246–24254. doi: 10.1074/jbc.M314298200
Myrianthopoulos, V., Kritsanida, M., Gaboriaud-Kolar, N., Magiatis, P., Ferandin, Y., Durieu, E., et al. (2013). Novel inverse binding mode of indirubin derivatives yields improved selectivity for DYRK kinases. Acs Med. Chem. Lett. 4, 22–26. doi: 10.1021/ml300207a
Najas, S., Arranz, J., Lochhead, P. A., Ashford, A. L., Oxley, D., Delabar, J. M., et al. (2015). DYRK1A-mediated Cyclin D1 degradation in neural stem cells contributes to the neurogenic cortical defects in down syndrome. EBioMed. 2, 120–134. doi: 10.1016/j.ebiom.2015.01.010
Nelson, L., Johnson, J. X., Freedman, M., Lott, I., Groot, J., Chang, M., et al. (2005). Learning and memory as a function of age in Down syndrome: a study using animal-based tasks. Progr. Neuro Psychopharmacol. Biol. Psychiatry 29, 443–453. doi: 10.1016/j.pnpbp.2004.12.009
Nguyen, T., and Di Giovanni, S. (2008). NFAT signaling in neural development and axon growth. Int. J. Dev. Neurosci. 26, 141–145. doi: 10.1016/j.ijdevneu.2007.10.004
Nieuwenhuis-Mark, R. E. (2009). Diagnosing Alzheimer’s dementia in Down syndrome: problems and possible solutions. Res. Dev. Disabil. 30, 827–838. doi: 10.1016/j.ridd.2009.01.010
Noll, C., Tlili, A., Ripoll, C., Mallet, L., Paul, J. L., Delabar, J. M., et al. (2012). Dyrk1a activates antioxidant NQO1 expression through an ERK1/2-Nrf2 dependent mechanism. Mol. Genet. Metab. 105, 484–488. doi: 10.1016/j.ymgme.2011.11.194
Ogawa, Y., Nonaka, Y., Goto, T., Ohnishi, E., Hiramatsu, T., Kii, I., et al. (2010). Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 1:9. doi: 10.1038/ncomms1090
O’Roak, B. J., Vives, L., Fu, W. Q., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
Ortiz-Abalia, J., Sahun, I., Altafaj, X., Andreu, N., Estivill, X., Dierssen, M., et al. (2008). Targeting Dyrk1A with AAVshRNA Attenuates Motor Alterations in TgDyrk1A, a Mouse Model of Down Syndrome. Am. J. Hum. Genet. 83, 479–488. doi: 10.1016/j.ajhg.2008.09.010
Park, J., and Chung, K. C. (2013). New perspectives of Dyrk1A Role in neurogenesis and neuropathologic features of down syndrome. Exp. Neurobiol. 22, 244–248. doi: 10.5607/en.2013.22.4.244
Park, J., Oh, Y., Yoo, L., Jung, M. S., Song, W. J., Lee, S. H., et al. (2010). Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J. Biol. Chem. 285, 31895–31919. doi: 10.1074/jbc.M110.147520
Park, J., Song, W. J., and Chung, K. C. (2009). Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell. Mol. Life Sci. 66, 3235–3240. doi: 10.1007/s00018-009-0123-2
Park, J., Sung, J. Y., Song, W. J., Chang, S., and Chung, K. C. (2012). Dyrk1A negatively regulates the actin cytoskeleton through threonine phosphorylation of N-WASP. J. Cell Sci. 125, 67–80. doi: 10.1242/jcs.086124
Pennington, B. F., Moon, J., Edgin, J., Stedron, J., and Nadel, L. (2003). The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Dev. 74, 75–93. doi: 10.1111/1467-8624.00522
Pereira, A., and Furlan, F. A. (2010). Astrocytes and human cognition: modeling information integration and modulation of neuronal activity. Prog. Neurobiol. 92, 405–420. doi: 10.1016/j.pneurobio.2010.07.001
Pinter, J. D., Eliez, S., Schmitt, J. E., Capone, G. T., and Reiss, A. L. (2001). Neuroanatomy of Down’s syndrome: a high-resolution MRI study. Am. J. Psychiatry 158, 1659–1665. doi: 10.1176/appi.ajp.158.10.1659
Pitt, W. R., Parry, D. M., Perry, B. G., and Groom, C. R. (2009). Heteroaromatic rings of the future. J. Med. Chem. 52, 2952–2963. doi: 10.1021/jm801513z
Pons-Espinal, M., De Lagran, M. M., and Dierssen, M. (2013). Environmental enrichment rescues DYRK1A activity and hippocampal adult neurogenesis in TgDyrk1A. Neurobiol. Dis. 60, 18–31. doi: 10.1016/j.nbd.2013.08.008
Prandini, P., Deutsch, S., Lyle, R., Gagnebin, M., Delucinge Vivier, C., Delorenzi, M., et al. (2007). Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am. J. Hum. Genet. 81, 252–263. doi: 10.1086/519248
Puri, B. K., Lekh, S. K., Langa, A., Zaman, R., and Singh, I. (1995). Mortality in a hospitalized mentally-handicapped population: a 10-year survey. J. Intellect. Disabil. Res. 39, 442–446. doi: 10.1111/j.1365-2788.1995.tb00549.x
Qian, W., Liang, H., Shi, J., Jin, N., Grundke-Iqbal, I., Iqbal, K., et al. (2011). Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 39, 6161–6171. doi: 10.1093/nar/gkr195
Rahmani, Z., Blouin, J. L., Creau-Goldberg, N., Watkins, P. C., Mattei, J. F., Poissonnier, M., et al. (1989). Critical role of the D21S55 region on chromosome 21 in the pathogenesis of Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 86, 5958–5962. doi: 10.1073/pnas.86.15.5958
Rodgers, J. T., Haas, W., Gygi, S. P., and Puigserver, P. (2010). Cdc2-like Kinase 2 is an insulin-regulated suppressor of hepatic gluconeogenesis. Cell Metabolism 11, 23–34. doi: 10.1016/j.cmet.2009.11.006
Rosenthal, A. S., Tanega, C., Shen, M., Mott, B. T., Bougie, J. M., Nguyen, D. T., et al. (2011). Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk). Bioorg. Med. Chem. Lett. 21, 3152–3158. doi: 10.1016/j.bmcl.2011.02.114
Rovelet-Lecrux, A., and Campion, D. (2012). Copy number variations involving the microtubule-associated protein tau in human diseases. Biochem. Soc. Trans. 40, 672–676. doi: 10.1042/BST20120045
Rovelet-Lecrux, A., Hannequin, D., Guillin, O., Legallic, S., Jurici, S., Wallon, D., et al. (2010). Frontotemporal dementia phenotype associated with MAPT gene duplication. J. Alzheimers. Dis. 21, 897–902. doi: 10.3233/JAD-2010-100441
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerriere, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi: 10.1038/ng1718
Ruaud, L., Mignot, C., Guet, A., Ohl, C., Nava, C., Heron, D., et al. (2015). DYRK1A mutations in two unrelated patients. Eur. J. Med. Genet. 58, 168–174. doi: 10.1016/j.ejmg.2014.12.014
Rüben, K., Wurzlbauer, A., Walte, A., Sippl, W., Bracher, F., and Becker, W. (2015). Selectivity profiling and biological activity of novel β-carbolines as potent and selective DYRK1 kinase inhibitors. PLoS ONE 10:e0132453. doi: 10.1371/journal.pone.0132453
Ryoo, S. R., Cho, H. J., Lee, H. W., Jeong, H. K., Radnaabazar, C., Kim, Y. S., et al. (2008). Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 104, 1333–1344. doi: 10.1111/j.1471-4159.2007.05075.x
Ryoo, S. R., Jeong, H. K., Radnaabazar, C., Yoo, J. J., Cho, H. J., Lee, H. W., et al. (2007). DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 282, 34850–34857.
Ryu, Y. S., Park, S. Y., Jung, M. S., Yoon, S. H., Kwen, M. Y., Lee, S. Y., et al. (2010). Dyrk1A-mediated phosphorylation of Presenilin 1: a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 115, 574–584. doi: 10.1111/j.1471-4159.2010.06769.x
Saheki, Y., and De Camilli, P. (2012). Synaptic vesicle endocytosis. Cold Spring Harb. Perspect. Biol. 2012:a005645. doi: 10.1101/cshperspect.a005645
Salehi, A., Delcroix, J. D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42. doi: 10.1016/j.neuron.2006.05.022
Sanchez, C., Salas, A. P., Brana, A. F., Palomino, M., Pineda-Lucena, A., Carbajo, R. J., et al. (2009). Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem. Commun. 27, 4118–4120. doi: 10.1039/b905068j
Saran, N. G., Pletcher, M. T., Natale, J. E., Cheng, Y., and Reeves, R. H. (2003). Global disruption of the cerebellar transcriptome in a Down syndrome mouse model. Hum. Mol. Genet. 12, 2013–2019. doi: 10.1093/hmg/ddg217
Scales, T. M. E., Lin, S., Kraus, M., Goold, R. G., and Gordon-Weeks, P. R. (2009). Nonprimed and DYRK1A-primed GSK3 beta-phosphorylation sites on MAP1B regulate microtubule dynamics in growing axons. J. Cell Sci. 122, 2424–2435. doi: 10.1242/jcs.040162
Schmitt, C., Miralinaghi, P., Mariano, M., Hartmann, R. W., and Engel, M. (2014). Hydroxybenzothiophene ketones are efficient Pre-mRNA splicing modulators due to dual inhibition of Dyrk1A and Clk1/4. ACS Med. Chem. Lett. 5, 963–967. doi: 10.1021/ml500059y
Seifert, A., Allan, L. A., and Clarke, P. R. (2008). DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. Febs J. 275, 6268–6280. doi: 10.1111/j.1742-4658.2008.06751.x
Shi, J. H., Zhang, T. Y., Zhou, C. L., Chohan, M. O., Gu, X. S., Wegiel, J., et al. (2008). Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 283, 28660–28669. doi: 10.1074/jbc.M802645200
Siarey, R. J., Carlson, E. J., Epstein, C. J., Balbo, A., Rapoport, S. I., and Galdzicki, Z. (1999). Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 38, 1917–1920. doi: 10.1016/S0028-3908(99)00083-0
Siarey, R. J., Stoll, J., Rapoport, S. I., and Galdzicki, Z. (1997). Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down Syndrome. Neuropharmacology 36, 1549–1554. doi: 10.1016/S0028-3908(97)00157-3
Siarey, R. J., Villar, A. J., Epstein, C. J., and Galdzicki, Z. (2005). Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology 49, 122–128. doi: 10.1016/j.neuropharm.2005.02.012
Sinet, P. M., Theophile, D., Rahmani, Z., Chettouh, Z., Blouin, J. L., Prieur, M., et al. (1994). Mapping of the Down syndrome phenotype on chromosome 21 at the molecular level. Biomed. Pharmacother. 48, 247–252. doi: 10.1016/0753-3322(94)90140-6
Sitz, J. H., Baumgartel, K., Hammerle, B., Papadopoulos, C., Hekerman, P., Tejedor, F. J., et al. (2008). The Down syndrome candidate dual-specificity tyrosine phosphorylation-regulated kinase 1A phosphorylates the neurodegeneration-related Septin 4. Neuroscience 157, 596–605. doi: 10.1016/j.neuroscience.2008.09.034
Sitz, J. H., Tigges, M., Baumgartel, K., Khaspekov, L. G., and Lutz, B. (2004). Dyrk1A potentiates steroid hormone-induced transcription via the chromatin remodeling factor Arip4. Mol. Cell. Biol. 24, 5821–5834. doi: 10.1128/MCB.24.13.5821-5834.2004
Skurat, A. V., and Dietrich, A. D. (2004). Phosphorylation of Ser(640) in muscle glycogen synthase by DYRK family protein kinases. J. Biol. Chem. 279, 2490–2498. doi: 10.1074/jbc.M301769200
Smith, B., Medda, F., Gokhale, V., Dunckley, T., and Hulme, C. (2012). Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: a new avenue for a disease modifying treatment of Alzheimer’s? ACS Chem. Neurosci. 3, 857–872. doi: 10.1021/cn300094k
Sonamoto, R., Kii, I., Koike, Y., Sumida, Y., Kato-Sumida, T., Okuno, Y., et al. (2015). Identification of a DYRK1A inhibitor that induces degradation of the target kinase using co-chaperone CDC37 fused with Luciferase nanoKAZ. Sci. Rep. 5:13. doi: 10.1038/srep12728
Song, W. J., Song, E. A. C., Choi, S. H., Baik, H. H., Jin, B. K., Kim, J. H., et al. (2013). Dyrk1A-mediated phosphorylation of RCAN1 promotes the formation of insoluble RCAN1 aggregates. Neurosci. Lett. 554, 135–140. doi: 10.1016/j.neulet.2013.08.066
Soppa, U., and Becker, W. (2015). DYRK protein kinases. Curr. Biol. 25, R488–R489. doi: 10.1016/j.cub.2015.02.067
Soppa, U., Schumacher, J., Ortiz, V. F., Pasqualon, T., Tejedor, F. J., and Becker, W. (2014). The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 13, 2084–2100. doi: 10.4161/cc.29104
Souchet, B., Guedj, F., Sahun, I., Duchon, A., Daubigney, F., Badel, A., et al. (2014). Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol. Dis. 69, 65–75. doi: 10.1016/j.nbd.2014.04.016
Soundararajan, M., Roos, A. K., Savitsky, P., Filippakopoulos, P., Kettenbach, A. N., Olsen, J. V., et al. (2013). Structures of down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure 21, 986–996. doi: 10.1016/j.str.2013.03.012
Strydom, A., Shooshtari, S., Lee, L., Raykar, V., Torr, J., Tsiouris, J., et al. (2010). Dementia in older adults with intellectual disabilities-epidemiology, presentation, and diagnosis. J. Policy Practice Intel. Disabil. 7, 96–110. doi: 10.1111/j.1741-1130.2010.00253.x
Sultan, M., Piccini, I., Balzereit, D., Herwig, R., Saran, N. G., Lehrach, H., et al. (2007). Gene expression variation in ‘Down syndrome’ mice allows to prioritize candidate genes. Genome Biol. 8, R91. doi: 10.1186/gb-2007-8-5-r91
Tahtouh, T., Elkins, J. M., Filippakopoulos, P., Soundararajan, M., Burgy, G., Durieu, E., et al. (2012). Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 55, 9312–9330. doi: 10.1021/jm301034u
Taira, N., Yamamoto, H., Yamaguchi, T., Miki, Y., and Yoshida, K. (2010). ATM augments nuclear stabilization of DYRK2 by Inhibiting MDM2 in the apoptotic response to DNA damage. J. Biol. Chem. 285, 4909–4919. doi: 10.1074/jbc.M109.042341
Tejedor, F., Zhu, X. R., Kaltenbach, E., Ackermann, A., Baumann, A., Canal, I., et al. (1995). minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 14, 287–301. doi: 10.1016/0896-6273(95)90286-4
Tejedor, F. J., and Haemmerle, B. (2011). MNB/DYRK1A as a multiple regulator of neuronal development. Febs J. 278, 223–235. doi: 10.1111/j.1742-4658.2010.07954.x
Tell, V., and Hilgeroth, A. (2013). Recent developments of protein kinase inhibitors as potential AD therapeutics. Front. Cell. Neurosci. 7:189. doi: 10.3389/fncel.2013.00189
Thomas, G. M., and Huganir, R. L. (2004). Mapk cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183. doi: 10.1038/nrn1346
Thomas, K. H., Martin, R. M., Potokar, J., Pirmohamed, M., and Gunnell, D. (2014). Reporting of drug induced depression and fatal and non-fatal suicidal behaviour in the UK from 1998 to 2011. BMC Pharmacol. Toxicol. 15:54. doi: 10.1186/2050-6511-15-54
Tschop, K., Conery, A. R., Litovchick, L., Decaprio, J. A., Settleman, J., Harlow, E., et al. (2011). A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev. 25, 814–830. doi: 10.1101/gad.2000211
Van Bon, B. W., Coe, B. P., Bernier, R., Green, C., Gerdts, J., Witherspoon, K., et al. (2015). Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21, 126–132. doi: 10.1038/mp.2015.5
van Bon, B. W. M., Hoischen, A., Hehir-Kwa, J., De Brouwer, A. P. M., Ruivenkamp, C., Gijsbers, A. C. J., et al. (2011). Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 79, 296–299. doi: 10.1111/j.1399-0004.2010.01544.x
Veitia, R. A., Bottani, S., and Birchler, J. A. (2008). Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet. 24, 390–397. doi: 10.1016/j.tig.2008.05.005
Vesely, J., Havlicek, L., Strnad, M., Blow, J. J., Donelladeana, A., Pinna, L., et al. (1994). Inhibition of cyclin dependent kinases by purine analogs. Eur. J. Biochem. 224, 771–786. doi: 10.1111/j.1432-1033.1994.00771.x
Walte, A., Rüben, K., Birner-Gruenberger, R., Preisinger, C., Bamberg-Lemper, S., Hilz, N., et al. (2013). Mechanism of dual specificity kinase activity of DYRK1A. FEBS J. 280, 4495–4511. doi: 10.1111/febs.12411
Wegiel, J., Gong, C.-X., and Hwang, Y.-W. (2011). The role of DYRK1A in neurodegenerative diseases. FEBS J. 278, 236–245. doi: 10.1111/j.1742-4658.2010.07955.x
Wegiel, J., Kuchna, I., Nowicki, K., Frackowiak, J., Dowjat, K., Silverman, W. P., et al. (2004). Cell type- and brain structure-specific patterns of distribution of minibrain kinase in human brain. Brain Res. 1010, 69–80. doi: 10.1016/j.brainres.2004.03.008
Wisniewski, K. E., Dalton, A. J., Mclachlan, D. R. C., Wen, G. Y., and Wisniewski, H. M. (1985). Alzheimers disease in Down syndrome - clinicopathologic studies. Neurology 35, 957–961. doi: 10.1212/WNL.35.7.957
Wong, R., Balachandran, A., Mao, A. Y. Q., Dobson, W., Gray-Owen, S., and Cochrane, A. (2011). Differential effect of CLK SR Kinases on HIV-1 gene expression: potential novel targets for therapy. Retrovirology 8:47. doi: 10.1186/1742-4690-8-47
Woods, Y. L., Rena, G., Morrice, N., Barthel, A., Becker, W., Guo, S. D., et al. (2001). The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser(329) in vitro, a novel in vivo phosphorylation site. Biochem. J. 355, 597–607. doi: 10.1042/bj3550597
Xie, W., Ramakrishna, N., Wieraszko, A., and Hwang, Y. W. (2008). Promotion of neuronal plasticity by (-)-epigallocatechin-3-gallate. Neurochem. Res. 33, 776–783. doi: 10.1007/s11064-007-9494-7
Yabut, O., Domogauer, J., and D’arcangelo, G. (2010). Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J. Neurosci. 30, 4004–4014. doi: 10.1523/JNEUROSCI.4711-09.2010
Yang, E. J., Ahn, Y. S., and Chung, K. C. (2001). Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J. Biol. Chem. 276, 39819–39824. doi: 10.1074/jbc.M104091200
Yu, T., Li, Z., Jia, Z., Clapcote, S., Liu, C., Li, S., et al. (2010a). A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum. Mol. Genet. 19, 2780–2791. doi: 10.1093/hmg/ddq179
Yu, T., Liu, C. H., Belichenko, P., Clapcote, S. J., Li, S. M., Pao, A. N., et al. (2010b). Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 1366, 162–171. doi: 10.1016/j.brainres.2010.09.107
Zhang, Z., Smith, M. M., and Mymryk, J. S. (2001). Interaction of the E1A oncoprotein with Yak1p, a novel regulator of yeast pseudohyphal differentiation, and related mammalian kinases. Mol. Biol. Cell 12, 699–710. doi: 10.1091/mbc.12.3.699
Keywords: trisomy 21, neurodevelopmental disorder, mouse model, cognition, learning and memory, clinical trial, DYRK1A and kinase inhibitors
Citation: Duchon A and Herault Y (2016) DYRK1A, a Dosage-Sensitive Gene Involved in Neurodevelopmental Disorders, Is a Target for Drug Development in Down Syndrome. Front. Behav. Neurosci. 10:104. doi: 10.3389/fnbeh.2016.00104
Received: 30 July 2015; Accepted: 17 May 2016;
Published: 03 June 2016.
Edited by:
Marie-Claude Potier, Centre National de la Recherche Scientifique, FranceReviewed by:
Mara Dierssen, Centre for Genomic Regulation, SpainCopyright © 2016 Duchon and Herault. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yann Herault, aGVyYXVsdEBpZ2JtYy5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.