
- Department of Psychiatry and Behavioral Sciences, Johns Hopkins University, Baltimore, MD, USA
DISC1 was discovered in a Scottish pedigree in which a chromosomal translocation that breaks this gene segregates with psychiatric disorders, mainly depression and schizophrenia. Linkage and association studies in diverse populations support DISC1 as a susceptibility gene to a variety of neuropsychiatric disorders. Many Disc1 mouse models have been generated to study its neuronal functions. These mouse models display variable phenotypes, some of them relevant to schizophrenia, others to depression. The Disc1 mouse models are popular genetic models for studying gene-environment interactions in schizophrenia. Five different Disc1 models have been combined with environmental factors. The environmental stressors employed can be classified as either early immune activation or later social paradigms. These studies cover major time points along the neurodevelopmental trajectory: prenatal, early postnatal, adolescence, and adulthood. Various combinations of molecular, anatomical and behavioral methods have been used to assess the outcomes. Additionally, three of the studies sought to rescue the resulting abnormalities. Here we provide background on the environmental paradigms used, summarize the results of these studies combining Disc1 mouse models with environmental stressors and discuss what we can learn and how to proceed. A major question is how the genetic and environmental factors determine which psychiatric disorder will be clinically manifested. To address this we can take advantage of the many Disc1 models available and expose them to the same environmental stressor. The complementary experiment would be to expose the same model to different environmental stressors. DISC1 is an ideal gene for this approach, since in the Scottish pedigree the same chromosomal translocation results in different psychiatric conditions.
Introduction
Both schizophrenia and depression have a strong genetic component but are also heavily influenced by the environment. Based on epidemiological studies, environmental schizophrenia risk factors include infections (especially during fetal development), obstetric complications (Mittal et al., 2008), advanced paternal age (Malaspina, 2001) adolescent cannabis use (Henquet et al., 2008), and several forms of stress (reviewed in Brown, 2011). In contrast to the wide variety of factors implicated in schizophrenia, stress in particular stands out as the major environmental risk factor for major depression. Early life stress, involving childhood maltreatment or traumatic events, can be particularly devastating (reviewed in Mann and Currier, 2006). Highly stressful life events, such as death of a family member or divorce are causally associated with onset of episodes of major depression (Kendler et al., 1999).
Here we review a group of studies that utilize mouse models of DISC1, a susceptibility gene for psychiatric disorders, to examine the interaction between genetic and environmental risk factors. We start by summarizing human data on the environmental factors most relevant to these studies, infections, and social stress, and their respective experimental paradigms in mice.
Environment
Environment: Infections (Reviewed in Brown, 2011)
Prenatal and early postnatal infections have been implicated in a number of major neurodevelopmental disorders. Direct infection of the fetus can cause serious congenital brain anomalies and mental retardation. Although schizophrenia typically arises in adolescence or young adulthood, it is increasingly regarded as neurodevelopmental in origin (Weinberger, 1996; Cannon et al., 2003; Allardyce et al., 2005; Jaaro-Peled et al., 2009; Shoji et al., 2012). This leads to the hypothesis that subtle perturbations in the developing brain, such as infection of the pregnant mother, may increase the risk of schizophrenia later on in the life of the offspring. Birth cohort studies have established an association between specific prenatal infections and elevated risk for schizophrenia. The largest effects have been attributed to influenza (Brown et al., 2004a), elevated maternal immunoglobulin G (IgG) to the parasite Toxoplasma gondii (Remington, 2006), and periconceptional genital or reproductive infection (Babulas et al., 2006).
A parsimonious explanation of how different viruses/parasites/bacteria increase the risk of the same disorder is suggested by the convergence of their pathogenic mechanism: stimulation of the maternal innate immune response and of cytokines in particular (Gilmore and Jarskog, 1997). Birth cohort studies support a role for excess maternal cytokines in the development of schizophrenia among offspring (Brown et al., 2004b). As with many proteins originally identified in the immune system, cytokines are also expressed in the nervous system and modulate many aspects of neural development and physiology (Szelenyi, 2001). Transplacental transfer of maternally produced cytokines and production of cytokines at the maternal-fetal interface both lead to an elevation of these molecules in the fetal brain (Meyer et al., 2009). Earlier inflammation is expected to have a more severe effect on brain development due to disruption of fundamental neurodevelopmental processes such as cell proliferation and differentiation, and may predispose the developing nervous system to fail in subsequent cell migration, target selection, and synapse maturation (Figure 6 in Meyer et al., 2007).
Although cytokines are the main suspects in mediating the detrimental effect of the pathogenic interaction between the maternal immune system and fetal brain, other mechanisms do exist. For example, while influenza is not believed to cross the placental barrier, it elicits IgG antibodies which do and subsequently cross-react with fetal brain antigens via molecular mimicry (Wright et al., 1999). Pathogens may also dysregulate brain development and function through a direct interaction between pathogen nucleic acids/proteins and (fetal) brain nucleic acids/proteins or modification of the fetal epigenome (Waterland and Michels, 2007). Some schizophrenia susceptibility genes may affect pathogen virulence, while the pathogens themselves may affect host genes and neural processes (Carter, 2009). For example, DISC1 regulates the microtubule network (Kamiya et al., 2005) that is exploited by viruses for intracellular trafficking.
Although both human and animal studies have mainly focused on maternal/prenatal immune activation, there is also evidence for postnatal infections increasing the risk of schizophrenia. Population-based studies have concluded that childhood viral infections of the central nervous system increase the risk of adult psychotic illness (Dalman et al., 2008; Khandaker et al., 2012).
Environment: Social Stress
Starting as early as prenatal development, environmental stress dysregulates the homeostatic physiological stress response by modulating the hypothalamic-pituitary-adrenal (HPA) axis. This system normally responds to stress by producing glucocorticoids, which can bind receptors throughout the brain and act as transcription factors (Lupien et al., 2009). Neurons in the hypothalamus release corticotrophin releasing hormone (CRH) in response to a stressful environment. CRH triggers secretion of adrenocorticotropic hormone (ACTH) from the pituitary gland, in turn leading to production of glucocorticoids (cortisol in humans, corticosterones in mice) by the adrenal gland. Once the stressful situation has passed, negative feedback loops shut down this response and return the HPA axis to a set homeostatic point (O'connor et al., 2000).
A highly stressful social environment can cause maladaptive brain development and function from the prenatal period all the way through old age, and as a result chronic stress is a critical factor in the epidemiology of major mental illness. van Os et al. suggested that “psychotic syndromes can be understood as disorders of adaptation to social context” (Van Os et al., 2010). For example, schizophrenia risk is increased for people raised in an urban environment (Pedersen and Mortensen, 2006). Sub-optimal social conditions have been identified as important mediating factors, in particular low social capital and high social fragmentation (Allardyce et al., 2005; March et al., 2008). Immigrants also suffer from higher risk of psychosis (reviewed in Cantor-Graae and Selten, 2005); two potential explanations of this phenomenon are immigrants' experiences of discrimination and exposure to social defeat, both chronically stressful conditions. Childhood trauma may also increase the risk for psychosis, but more research is needed to establish stronger correlation (Morgan and Fisher, 2007).
The negative effects of chronic environmental stress manifest first in prenatal development, since maternal glucocorticoids circulate freely through the fetal brain. Significantly elevated maternal glucocorticoid levels can result in increased basal HPA axis activity in the offspring up to at least 10 years of age (O'connor et al., 2005). This heightened homeostatic set point results in increased risk for neurological and behavioral disturbances in the offspring (Stott, 1973). Since the human brain undergoes extended postnatal development, the effects of prenatal stress are moderated by the quality of the offspring's environment in early life.
Early life stress, often as a result of childhood maltreatment or traumatic events, decreases mental health in a dose-response fashion (Edwards et al., 2003). It causes dysregulation of the aforementioned stress response systems, leading to increased sensitivity to stress throughout life (reviewed in Heim and Binder, 2012), and has been associated with increased risk for mood and anxiety disorders, posttraumatic stress disorder, unipolar depression, and schizophrenia (Heim et al., 2010). Physiologically, early-life disruption of the HPA axis manifests as an array of abnormalities such as glucocorticoid resistance (Charmandari et al., 2004), increased CRH activity (Cratty et al., 1995), and increased levels of inflammation (McEwen, 1998). Human imaging studies have revealed further effects of early life stress on brain anatomy and function (Papagni et al., 2011). Animal models of early life stress such as variable schedules of maternal separation have provided direct evidence for the causal link between early adverse experience and later brain and behavior abnormalities. Maternal separation has been shown to increase glucocorticoid levels in separated mouse pups and to permanently change glucocorticoid receptor gene expression resulting in excessive glucocorticoid release under stress in adulthood (Anisman et al., 1998).
Detrimental effects on brain and behavior induced by adult chronic stress are reversed after a few weeks of no stress, in contrast to the permanent effects of early life stress (Avital and Richter-Levin, 2005). Exposure to an environmental event during development results in a stable phenotypic alteration in adulthood. This developmental “programming” can alter gene transcription via transcription factors or epigenetic mechanisms (Weaver et al., 2004). In mice, more attentive maternal care of the offspring modulates the selective methylation/demethylation of specific CpG dinucleotides of the glucocorticoid receptor gene (Oberlander et al., 2008). The resulting permanent decrease in glucocorticoid receptor density, selective to the hippocampus and prefrontal cortex, can increase HPA axis sensitivity to glucocorticoid-mediated negative feedback and lead to a reduction in plasma glucocorticoid levels and stress reactivity throughout the offspring's life (Weaver et al., 2004).
Stress in Adolescence (Reviewed in Andersen and Teicher, 2008; Lupien et al., 2009)
Both animal and human studies show that adolescence is associated with higher basal and stress-induced activity of the HPA axis compared to both childhood (a stress hypo-responsive period) and adulthood (Vazquez and Akil, 1993; Gunnar et al., 2009). Adolescence is characterized by extensive changes in brain physiology (Andersen, 2003; Jaaro-Peled et al., 2009; Giedd and Rapoport, 2010; O'donnell, 2010), especially among regions involved in social function (Blakemore, 2008). Social stressors during this critical period can therefore potentially have significant long-term consequences on the “social brain” (Van Os et al., 2010). Notably, the brain areas undergoing development and hence most sensitive to stress during adolescence differ between rodents and humans. Rodent hippocampus continues to develop into adulthood, but in humans it is fully developed by 2 years of age (Giedd et al., 1996). While the frontal cortex and amygdala continue to develop in both, the process is more extensive in primates than in other species (Andersen, 2003). Adolescence is the peak age of onset for many psychiatric disorders, including anxiety, mood disorders, eating disorders, psychosis, and substance abuse (Paus et al., 2008).
Gene × Environment (G × E, Reviewed in Duncan and Keller, 2011)
Gene × environment (G × E) implies synergistic interplay between genes and environment in causing a particular phenotype, where the effect of one is conditional on the other. Genes may modulate sensitivity to the environment, exemplified by a landmark study which demonstrated that a specific serotonin transporter polymorphism moderates sensitivity to life stress contributing to the pathogenesis of depression (Caspi et al., 2003). The environment can impact DNA sequences themselves as well as epigenetic control of risk genes.
Human G × E studies of early life stress and depression have focused mostly on a number of logical candidate genes involved in the serotonin system, neurotrophins, and most of interest to this review, the HPA axis (reviewed in Nugent et al., 2011). Polymorphisms in the glucocorticoid receptor and its regulator FK506 binding protein 5 (FKBP5) as well as in the CRH type I receptor interact with early life stress in predicting depression (Bradley et al., 2008) or in the case of FKBP5, post-traumatic stress disorder (Binder et al., 2008).
First-generation epidemiological G × E studies of psychotic psychiatric disorders have used indirect measures of genetic risk, such as blood relation to a person with schizophrenia, thus representing the complete genetic load including gene × gene interactions. Environmental variables examined in G × E studies include migration, urbanicity, obstetric complications, cannabis use, and stress (Van Os et al., 2008). It is difficult to design human studies that include enough participants to be able to test genetic and environmental risk factors as independent variables alongside any putative gene-environment interaction; any such synergistic interaction might be masked by other genetic and environmental factors. Thus, animal models are utilized in order to rigorously examine the mechanisms and outcomes of G × E interactions.
Animal Models
Epidemiological studies detect correlations which point to potential mechanisms and public health interventions, but causality can only be determined in animal models. These enable the controlled combination of a specific genetic risk factor (on an otherwise uniform genetic background) with a specific environmental risk factor, at a specific developmental time, and detailed observation of the emerging phenotypes over time. Of course, these models have clear limitations of their own; for example, when modeling brain diseases in animals, it is critical to take into account interspecies differences in the timing of brain development. A further complication is that the time course of development differs between different brain regions (cortex, limbic, etc.) and constituents (neurons, glia, blood-brain-barrier, etc.). Most relevant to this review, a large part of gestation in the mouse corresponds to the first half of human pregnancy, and many of the brain maturation events which happen during the second half of human gestation occur postnatally in mice (Clancy et al., 2007).
Modeling Infections in Rodents
The effects of specific pathogens have been modeled in animals by prenatal infection with the influenza virus (Shi et al., 2003, 2005) and by adult infection with the Toxoplasma parasite (Vyas et al., 2007). More general effects of immune system activation have been modeled by injecting the bacterial endotoxin lipopolysaccharide (Miyazaki et al., 2004; Nogai et al., 2005) or proinflammatory cytokines (Nawa and Yamada, 2012). A strong innate immune response similar to that caused by viral infection can be induced by injection of a synthetic double-stranded RNA, polyriboinosinic-polyribocytidylic acid (polyI:C) (reviewed in Meyer and Feldon, 2012). Binding of polyI:C to Toll-like receptor 3 stimulates production of many pro-inflammatory cytokines and type I interferons (Okahira et al., 2005; Kumar et al., 2006). Maternal exposure to polyI:C alters cytokine levels in the three compartments of the maternal-fetal interface: placenta, amniotic fluid, and the fetus itself. PolyI:C has been widely used, as it has several advantages over live viruses; it is comparatively safe, and the intensity and timing of the immune response can be tightly controlled (Meyer and Feldon, 2012). However, polyI:C does not induce the full spectrum of immune responses induced by a virus. Prenatal polyI:C has been shown to induce distinct neuropathological and behavioral phenotypes depending on the gestational duration of exposure. For example, Meyer and Feldon hypothesize that the abnormalities induced by immune activation in early/middle pregnancy (embryonic day 9 in mice) are associated mainly with positive symptoms of schizophrenia, whereas the abnormalities caused by immune activation in late pregnancy (embryonic day 17 in mice) are more relevant to negative symptoms of schizophrenia (Bitanihirwe et al., 2010).
Modeling Social Stress in Rodents
As described above, social stress is particularly implicated in the pathogenesis of several major mental diseases. Maternal separation can be a devastating stressor, leading to an array of adult social and cognitive dysfunction (Schmidt et al., 2011). Mild chronic stress, entailed by serial exposure to mild but unpredictable stressors, is also a widely used model for depression (Hill et al., 2012). Social stress as applied to rodents can be subdivided to prenatal, early postnatal (until weaning), adolescent or adult.
Social isolation
Agonistic confrontations are not the only mediator of social stress in mammals. Positive social interactions are critical to healthy mental development. In humans, separation and social withdrawal are risk factors for psychiatric disorders (Allardyce et al., 2005). Both maternal separation and long-term social isolation in mice produce a phenotype characterized by cognitive and social-affective dysfunction, such as impaired prepulse inhibition (PPI) and novel object recognition as well as increased anxiety and aggressive behavior (Fone and Porkess, 2008; Niwa et al., 2011).
Social defeat
Social defeat paradigms involve agonistic interaction between two animals. Two mice (usually male) are put into a cage with a removable separator. Upon removal of the separator the two mice fight one another, and a clear “victor” is established in most cases. The defeated animal experiences social stress, and repeated exposures to this stressor have been linked to a robust social-affective dysfunction phenotype similar to depression, characterized by anhedonia, helplessness, hyperactivity, and social-avoidance (Kudryavtseva et al., 1991; Venzala et al., 2012). Exposure to social defeat can be one-time, intermittent, or chronic.
Gene × Environment × Timing
The timing of an environmental factor (whether immune activation or stress) is critical and must be taken into account in experimental design and data interpretation. Dynamic, rapid neurodevelopmental processes occur during the pre- and early post-natal periods and to some extent also in adolescence. Each brain region is maximally sensitive to a given environmental factor during critical periods of high plasticity, which differ between species. Analogous to the typical adolescent or adult onset of psychiatric symptoms, behavioral effects of early-life interventions in experimental animals may become evident only in adulthood.
Gene × Environment Studies Using DISC1 Mouse Models
G × E studies in psychiatry are a relatively new field of research, as discovery of risk genes has only started in this millennium and the mouse models based on the genetic findings are not yet fully established. One of the most extensively characterized risk genes for psychiatric disorders is DISC1. DISC1 was discovered in a Scottish pedigree in which a chromosomal translocation that breaks this gene segregates with psychiatric disorders (Millar et al., 2000). Although the acronym DISC1 means Disrupted in schizophrenia-1, members of the pedigree were diagnosed as suffering from a variety of psychiatric disorders: recurrent major depression, schizophrenia, bipolar disorder and adolescent conduct disorder with a combined LOD score of 7.1 (Blackwood et al., 2001). Subsequent association and linkage studies in a variety of populations have confirmed DISC1 as a susceptibility gene not only to those adult-onset disorders but also to autism (Chubb et al., 2008). DISC1 is a hub protein which regulates many aspects of development and function of the nervous system and as such mutations in it contribute to various brain disorders (Chubb et al., 2008; Brandon and Sawa, 2011; Porteous et al., 2011). The type of DISC1 mutation, mutations in other genes, and/or environmental factors may determine the specific symptoms.
DISC1 mouse models
One of the tools to study the biology of DISC1 and its relevance to psychiatric disorders is mouse models in which this gene has been manipulated. Numerous DISC1 mouse models have been generated and characterized for histological, anatomical, neurochemical, and behavioral phenotypes (for review, see Jaaro-Peled, 2009; Brandon and Sawa, 2011; Johnstone et al., 2011). Each model can be considered significantly unique from the others considering the genetic manipulation, the assays used to characterize it, and the particular lab environment; this diversity significantly complicates comparison between models. Theoretically similar assays may yield different results for different DISC1 models or even for the same model under different lab conditions. Still, each of these genetic models without any environmental intervention show some abnormalities relevant for schizophrenia and/or mood disorders such as: parvalbumin and synaptic deficits, enlarged lateral ventricles, disturbed dopaminergic system, cognitive and emotional behavioral deficits. When looking for a suitable genetic model for G × E studies, it may be useful select G and E conditions which do not individually produce impaired phenotypes in the hope of G × E interaction producing a novel, emergent phenotype. There is a substantial pool of Disc1 mouse models that could be potentially useful for such studies.
So far the DISC1 G × E studies have been done with either Disc1 point mutants (Clapcote et al., 2007) or with transgenic mice expressing C'-truncated (as in the original Scottish pedigree) dominant negative (DN) human DISC1 (on top of the endogenous wild-type mouse Disc1) under either the alpha calmodulin kinase II (CaMK) promoter or the prion protein promoter (PrP). The CaMK promoter induces expression of the DN-DISC1 in the pyramidal neurons of the forebrain (Tsien et al., 1996), whereas the PrP promoter induces its expression in most brain regions and cell types (Asante et al., 2002). Presumably due to DISC1 being one of the most established risk genes for psychiatric disorders, its clear involvement in brain development, and the availability of many mouse models, more G × E studies have been performed using DISC1 mouse models than with any other risk gene. Here we will review five studies, starting with the studies using DN-DISC1 mouse models (Table 1) and then moving to the studies using Disc1 point mutants (Table 2).
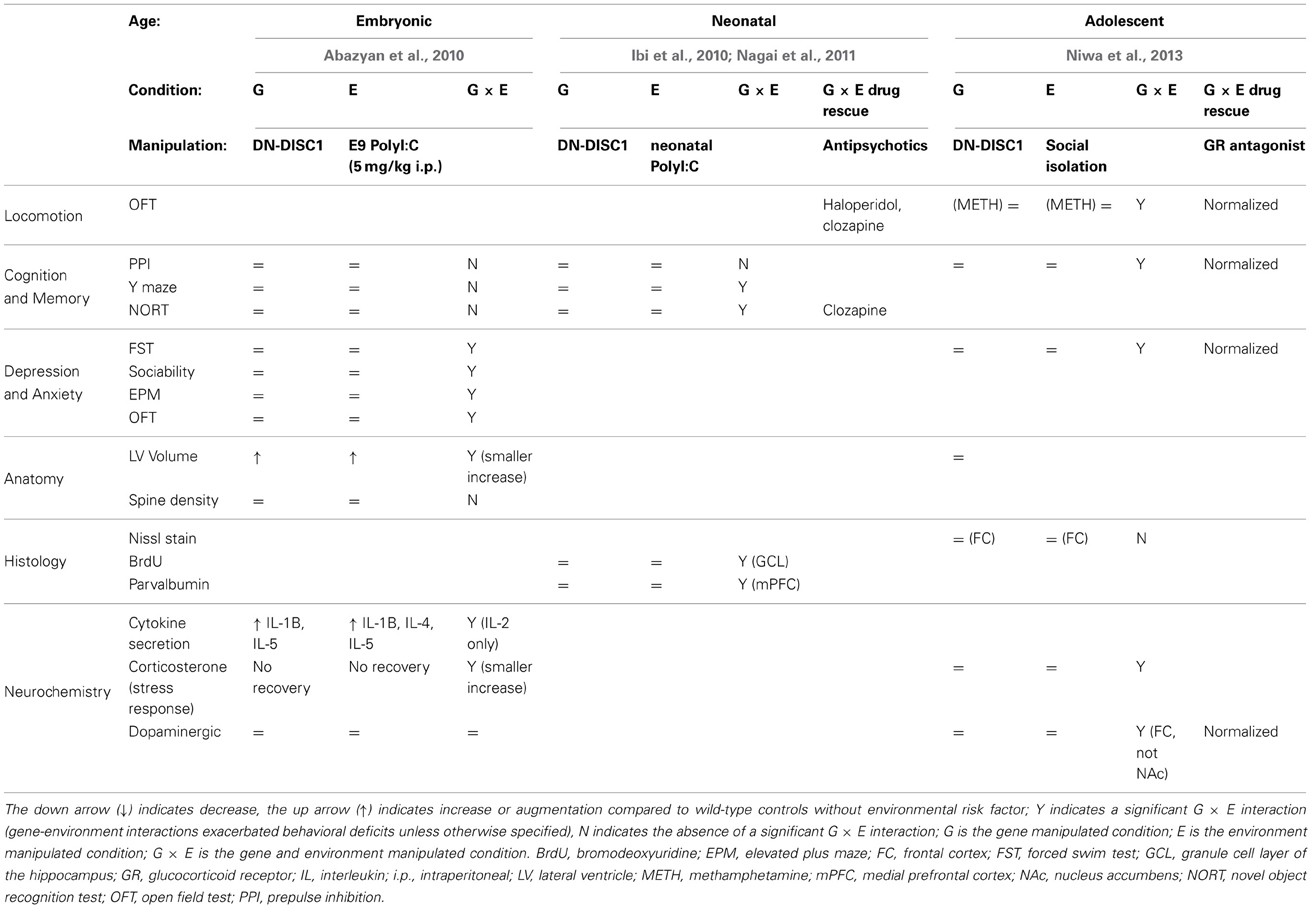
Table 1. Summary of G × E in DN-DISC1 mouse models.
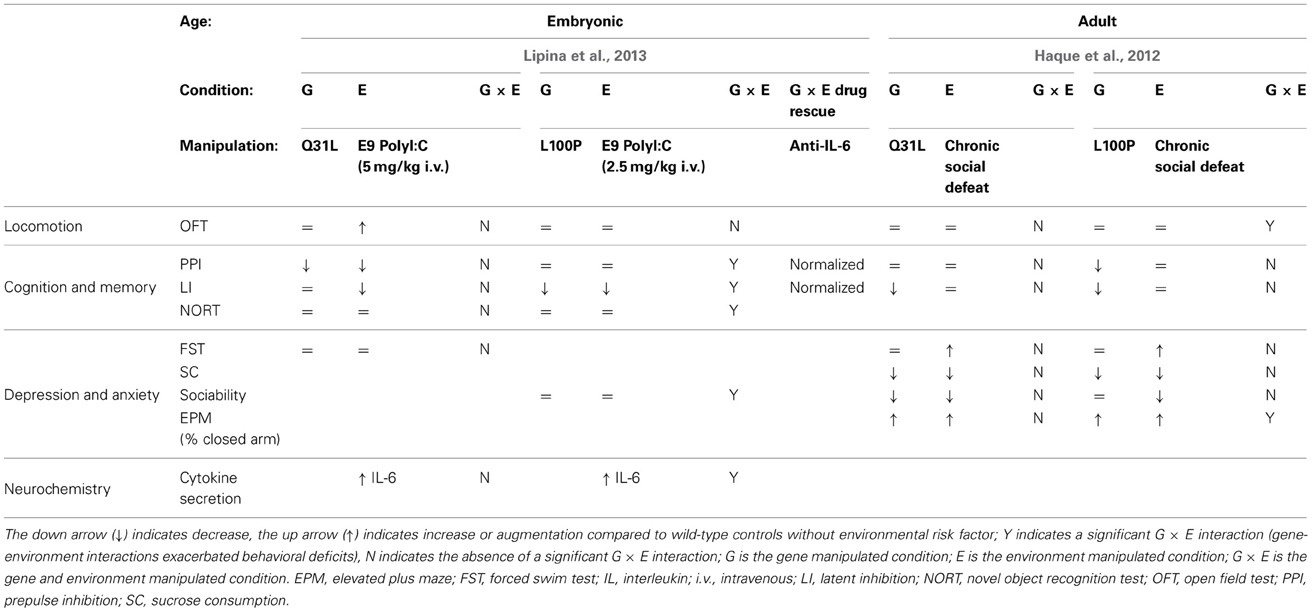
Table 2. Summary of G × E in point mutant Disc1 mouse models.
Inducible CaMK-DN-DISC1 × prenatal polyI:C (Abazyan et al., 2010)
The CaMK-DN-DISC1 genetic model displays brain and behavioral abnormalities including enlarged lateral ventricles, decreased protein levels of endogenous mouse DISC1, decreased levels of cortical dopamine and fewer parvalbumin-positive neurons in the cortex (Pletnikov et al., 2008; Ayhan et al., 2011). Behavioral abnormalities differ between males and females: males show both spontaneous and psychostimulant-induced hyperactivity in the open field as well as altered social interaction, whereas females exhibit impaired spatial memory.
PolyI:C (5 mg/kg) was injected to pregnant dams at embryonic day 9 (E9), modeling the complex scenario of infection affecting the maternal-fetal interface. This timing of polyI:C injection, corresponding to middle/end of first trimester of human pregnancy, has been widely used (Meyer et al., 2006). Abazyan et al. performed intraperitoneal injections—in contrast to the intravenous (i.v.) injections used by previous studies—in order to achieve a lower effective polyI:C dose. Indeed, no effect of the polyI:C alone could be detected in the behavioral tests used although the same i.v. dose has been shown to elicit extensive behavioral deficits (Meyer et al., 2005). DN-DISC1 altered basal levels of interleukin (IL)-1 beta and IL-5 and polyI:C induced secretion of IL-4 and IL-5 in fetal brains 6 h after injection of polyI:C. The other experiments were done on adult males only. The effect of the different manipulations on adult response to acute stress was measured by testing for corticosterone levels at baseline, after a 1 h restraint stress and following 1 h recovery. Although both polyI:C and DN-DISC1 on their own caused a trend of reduced stress-induced corticosterones and no recovery, only the G × E group had significantly lower corticosterones after stress compared to the control group. DN-DISC1 mice have an increased lateral ventricle volume (Pletnikov et al., 2008). Although polyI:C caused increased lateral ventricle volume in wild-type mice, it unexpectedly normalized lateral ventricle volume in the DN-DISC1 mice.
Combination of CaMK-DN-DISC1 × prenatal polyI:C produced new phenotypes not seen with either variable alone: anxiety, as measured by increased peripheral activity in the open field and by open arm time in the elevated plus maze; depression-like behavior, as measured by increased immobility in the forced swim test; and impaired sociability in the three-chamber social test. G × E had no effect on cognitive tests: spontaneous alternation in the Y-maze, object recognition, Morris water maze, PPI.
Finally the authors exploited the Tet-off inducibility of this CaMK-DN-DISC1 transgene to evaluate the effect of expression timing. In one group, expression of the transgene was induced until weaning (postnatal day 21) and in a second group, expression was induced from weaning onward. The G × E mice behaved normally, implying that continuous expression of the DN transgene is necessary for the G × E effect.
CaMK-DN-DISC1 × neonatal polyI:C (Ibi et al., 2010; Nagai et al., 2011)
The CaMK-DN-DISC1 mouse model is very similar to the one employed in the prenatal polyI:C study. The difference is that it expresses the transgene in a constitutive manner. The CaMK-DN-DISC1 genetic model on its own has enlarged lateral ventricles, reduced parvalbumin immunoreactivity, dopaminergic abnormalities and relatively mild behavioral deficits (Hikida et al., 2007; Jaaro-Peled et al., 2013). CaMK-DN-DISC1 mice were injected with polyI:C daily from postnatal day 2–6, which corresponds to the second trimester of human brain development (http://translatingtime.net). This paradigm of neonatal polyI:C injection on its own elicits behavioral abnormalities including impairments in PPI, novel object recognition and social behavior in mice tested at 10–12 weeks of age (Ibi et al., 2009). The DISC1 mutant mice (both males and females) were analyzed starting at 8 weeks of age, or young adulthood. In contrast to the previous study (Ibi et al., 2009), polyI:C on its own did not impair performance in the PPI or novel object recognition paradigms, perhaps due to the age difference. Only G × E resulted in significant reduction in parvalbumin-positive interneurons of the medial prefrontal cortex, reduction in spontaneous alternation in the Y-maze as well as in preference for a novel object in the novel object recognition test (Ibi et al., 2010). The other behavioral tests, sensitivity to MK801, fear conditioning and social interaction were not performed on the G or E groups alone, making it difficult to evaluate any potential G × E interaction. In a follow up paper, the same group tested whether antipsychotics could normalize the behavioral deficits in the CaMK-DN-DISC1 × neonatal polyI:C model. The atypical antipsychotic clozapine normalized the above-mentioned preference in the novel object recognition test, while the typical antipsychotic haloperidol did not (Nagai et al., 2011).
PrP-DN-DISC1 × adolescent social isolation (Niwa et al., 2013)
Niwa et al. (2013) is the first publication to introduce this DISC1 mouse model in which the C'-truncated DN-DISC1 is expressed under the control of the widely expressed prion protein promoter. Mice are typically weaned at 3 weeks of age and then housed 5/cage with same-sex littermates. Pre-weaning isolation is extremely stressful and complicated by the physical survival of the pups still dependent on nursing. PrP-DN-DISC1 mice were subjected to a new, milder social stress paradigm of 3 weeks of isolation from 5–8 weeks of age after which they were tested. This timing corresponds to middle and late adolescence, a sensitive period and may therefore model separation from family or general social isolation in adolescent humans. The combination of the DN-DISC1 gene (G) and mild social isolation (E) led to multiple behavioral, cellular and functional deficits whereas neither the environmental nor genetic factors produced deficits on their own. Both males and females were tested and showed similar phenotypes.
PrP-DN-DISC1 mice with 3-week isolation (G × E) displayed multiple behavioral deficits, including decreased prepulse inhibition, increased immobility in a forced swim test, and an augmented locomotion response to methamphetamine challenge when compared to the control, G, and E groups. These abnormal phenotypes resulted from synergistic gene-environment interaction at the cellular level, rather than any gross anatomical or histological mechanism. Body weight and lateral ventricle volume were similar to controls in the G, E, and G × E groups; Nissl staining and Glial fibrillary acidic protein immunostaining also failed to reveal any significant differences. However, the G × E mice displayed significant dopaminergic alterations at the cellular level. Tyrosine hydroxylase expression and total tissue dopamine levels were significantly decreased compared to control, G, and E groups, as was basal extracellular dopamine in the frontal cortex. Consistent with their abnormal locomotor behavior, methamphetamine challenge led to significantly higher dopamine release in the nucleus accumbens of the G × E mice compared to the control, G, and E groups.
Notably, treatment with the glucocorticoid receptor antagonist RU38486 normalized all behavioral and dopaminergic cellular abnormalities in the G × E group. The authors therefore hypothesized that an abnormally high stress-induced corticosterone response in the G × E mice might underlie their dopaminergic pathology. Glucocorticoids mediated projection-specific epigenetic changes in the dopaminergic midbrain of the G × E mice. Specifically, mesocortical dopaminergic projection neurons in the ventral tegmental area exhibited robust DNA methylation of the tyrosine hydroxylase gene promoter, while the mesolimbic projection was unaffected. Furthermore, these DNA methylation levels were maintained in the G × E mice until 20 weeks of age after the transient adolescent isolation. RU38486 normalized the increase in DNA methylation in G × E mice.
Only G × E produced neuronal and behavioral abnormalities in the PrP-DN-DISC1 model; neither the DISC1 mutation nor isolation stress produced any observed pathology on their own. As such, the PrP-DN-DISC1 mice are a promising genetic model for exploring synergistic gene-environment interactions underlying the mechanisms of psychiatric pathology.
L100P and Q31L × prenatal polyI:C (Lipina, 2013)
In addition to dominant-negative models, DISC1 point mutants have also been combined with environmental risk factors. These models, identified in a screen of ENU-mutagenized mice, express missense mutations in Disc1. The Q31L mutant mice showed depressive-like behavior (deficits in the forced swim test and other measures that were reversed by the antidepressant) whereas L100P mutants exhibited schizophrenia-like behavior (PPI and latent inhibition deficits which were reversed by antipsychotic treatment) (Clapcote et al., 2007). A subsequent study from another group did not reproduce most of these baseline behavioral abnormalities (Shoji et al., 2012) suggesting that the effect of the Disc1 point mutations is not very robust and may depend on the experimental conditions i.e., subtle environmental factors. The two G × E studies described here used heterozygous Disc1 point mutants. Heterozygotes exhibit relatively mild phenotypic impairments compared to homozygotes (Clapcote et al., 2007), and as such provide more room for exploring the synergistic interplay between genetic and environmental risk factors. PolyI:C 5 mg/kg i.v. at E9 has been routinely used in environment-only studies and shown to elicit robust phenotypes (Meyer et al., 2005). In this study the authors started with a dose of 5 mg/kg, but pregnant L100P heterozygotes administered polyI:C at 5mg/kg bore no offspring, dead or alive; as a result, they had to decrease the dose to 2.5 mg/kg in order to obtain offspring for behavioral analysis. Litter/offspring size in polyI:C-treated wild type and Q31L mice were unaffected. Thus, they studied Q31L mice with 5 mg/kg polyI:C and L100P mice with 2.5 mg/kg polyI:C. In the assays used, 2.5 mg/kg on its own produced a PPI deficit and 5 mg/kg on its own affected also locomotion in the open field and latent inhibition (LI). Even lower polyI:C dose of 1 mg/kg i.v. may be recommended for G × E studies (Giovanoli et al., 2013).
Since prenatal infection is one of the major epidemiological risk factors for schizophrenia, but not for depression, Lipina et al. (2013) hypothesized that G × E interactions between maternal immune activation produced by prenatal polyI:C would be significantly greater in the L100P mice than the Q31L mice. Heterozygous females were given a single injection of polyI:C on gestational day 9. The Q31L/polyI:C mice exhibited G × E interaction only in risk assessment behavior, meaning that the G × E spent more time scanning the EPM for potential threat than the G or E conditions alone. However, the L100P × polyI:C offspring exhibited a significant exacerbation of schizophrenia-related endophenotypes compared to L100P controls. Deficits in PPI, LI, object recognition, and sociability all demonstrated significant prenatal polyI:C × genotype interactions. Specifically, the L100P × polyI:C group showed a deficit in social motivation but not social recognition—correspondingly, their novel object recognition was intact while spatial object recognition was impaired.
IL-6, a cytokine critical for mediating the effects of prenatal polyI:C, was elevated in the fetal brains of all mice treated with polyI:C; the L100P group, however, exhibited a steeper dose-dependent increase compared to the WT and Q31L groups. It appears that at least PPI and LI behavioral deficits in the L100P mice were causally dependent on prenatal polyI:C, as coadministration of an antibody against IL-6 resulted in normal PPI and LI behavior.
L100P and Q31L × social defeat (Haque et al., 2012)
Haque et al. also used L100P or Q31L heterozygous mice for low baseline impairments to enable detection of G × E effect. The mice were subjected to chronic social defeat (CSD) as an environmental stressor. CSD on its own has been shown to induce a depressive/anxiety-like phenotype including hypoactivity in the open field, social avoidance, reduced sucrose consumption, and increased immobility time in the forced swim test (Kudryavtseva et al., 1991; Venzala et al., 2012). This makes it difficult to detect G × E effects in these behavioral tests.
CSD resulted in similar depressive phenotypes for all three genetic groups. L100P and Q31L mice did not display significant abnormality in either forced-swim test immobility or sucrose consumption similar to wild-type controls in both the naïve and defeated conditions. Anxiety-related endophenotypes, on the other hand, revealed significant differences between the two mutant lines and controls, as well as between the mutant lines themselves. L100P and Q31L mice spent less time exploring the open arm of the elevated plus maze than did controls. However, Q31L mice did not display any difference in exploration time in response to CSD, whereas CSD significantly decreased exploration time for wild-type and L100P groups. The L100P defeated mice displayed decreased vertical activity in the open field test compared to all G, E, and control groups. A CSD-related decrease in L100P horizontal activity was also observed during the first 5 min of the open field test, but the difference normalized in subsequent time bins. CSD diminished sociability and preference for social novelty in wild-type and L100P mice. Q31L were a social even without CSD, so no further effect could be detected by addition of CSD stress. CSD did not interact with cognitive deficits in the mutant lines. L100P mice displayed a deficit in PPI, and both L100P and Q31L mice displayed disruptions in LI, but CSD did not exacerbate either endophenotype for any group.
Discussion
A genetic model with too robust a phenotype may result in a “ceiling effect,” whereby the effects of any potential G × E interactions are occluded. It is therefore more practical to use genetic models with relatively weak phenotypes, such as heterozygotes instead of homozygotes (Ibi et al., 2010; Haque et al., 2012; Lipina et al., 2013; Niwa et al., 2013), or to at least focus on specific assays which are not affected by the G condition on its own (Abazyan et al., 2010). The same is true for the choice of the environmental stressor—to detect G × E effects it is useful to choose a regime which is sub-optimal on its own, such as changing the administration route (Abazyan et al., 2010) or reducing the dose (Lipina et al., 2013) of polyI:C, or developing a new, milder form of social stress paradigm (Niwa et al., 2013).
Interestingly, CaMK-DN-DISC1 × prenatal polyI:C synergistically induced anxiety and depression-like phenotypes, but cognitive function was not affected (Abazyan et al., 2010). In contrast, CaMK-DN-DISC1 × neonatal polyI:C significantly impacted on cognitive tasks (Ibi et al., 2010). Since these two DISC1 models are very similar, such results imply that specific timing of the polyI:C intervention determines which type of behavioral abnormalities emerge. To test this hypothesis, it would be useful to test a single DISC1 model against several polyI:C injections at different time points, with subsequent behavioral and histological analyses under identical experimental conditions.
Which biological changes underlie the behavioral abnormalities manifested exclusively as a result of G × E? Ibi et al. found decreased parvalbumin immunoreactivity in the prefrontal cortex but not the hippocampus, hinting at a specific fast-spiking interneuron deficiency (Ibi et al., 2010). Abazyan et al. observed decreased spine density in the dentate gyrus. Their most surprising result was that combining polyI:C with DISC1 suppressed the ventricular enlargement phenotype displayed by either the G or E factor alone (Abazyan et al., 2010). Abazyan et al. and Niwa et al. both looked at monoamines and plasma corticosterone response to environmental stress. In the CaMK-DN-DISC1 × neonatal polyI:C model, no changes were detected in dopamine content or turnover in the frontal cortex or hippocampus, while hippocampal serotonin content was increased (Abazyan et al., 2010). Stress-induced corticosterone levels were attenuated in the G × E condition more than in G or E alone. In the PrP-DN-DISC1 × isolation stress model, frontal cortex and caudate-putamen serotonin were not altered while several dopamine-related measures were lower in the frontal cortex but not in the nucleus accumbens (Niwa et al., 2013). These dopaminergic and behavioral phenotypes are at least in part affected by epigenetic silencing of the tyrosine hydroxylase promoter via the elevated glucocorticoids which were detected only in G × E. The isolation stress on its own was mild enough not to affect plasma corticosterones. Abazyan et al. and Lipina et al. detected effect of G × E on cytokines, but each tested for different cytokines (Abazyan et al., 2010; Lipina et al., 2013). Lipina et al. used anti-IL-6 to rescue polyI:C induced PPI and LI abnormalities in L100P mice, confirming the importance of IL-6 as a mediator of the detrimental effects of maternal immune activation.
Can G × E-induced abnormalities be reversed by pharmacological interventions regularly used in psychiatry? Nagai et al. focusing on schizophrenia, tested antipsychotics against the CaMK-DN-DISC1 × neonatal polyI:C model. Increased sensitivity to MK801, an NMDA receptor antagonist, was rescued by both haloperidol and clozapine. Novel object recognition deficit was normalized only by clozapine treatment, but the sociability deficit could not be rescued by either (Nagai et al., 2011). This model may be useful in exploratory screening for new drugs which are especially effective against social deficits. Based on the increased plasma corticosterone levels in the PrP-DN-DISC1 × social stress model, Niwa et al. treated the mice with the glucocorticoid receptor antagonist RU38486 which normalized all behavioral and dopaminergic cellular abnormalities in the G × E group, most likely via normalization of the tyrosine hydroxylase methylation. RU38486 is uniquely beneficial in psychotic depression, major depression with psychotic features (Flores et al., 2006), suggesting that the PrP-DN-DISC1 × social isolation model may be useful in finding better treatments for this disorder. The above drug treatment trials add predictive validity to their respective animal models and suggest that even abnormalities of neurodevelopmental origin may be reversed with appropriate treatment.
As discussed above, timing is critical. Abazyan et al. induced expression of the CaMK-DN-DISC1 transgene either until weaning or starting at weaning. Interestingly, neither group exhibited the depressive-like behavioral deficits detected with constitutive CaMK-DN-DISC1 expression. Elimination of the behavioral phenotypes after turning off expression during the late postnatal period is encouraging; it implies that these phenotypes are reversible despite being neurodevelopmental in origin (Abazyan et al., 2010). Niwa et al. asked whether adolescent stress-induced epigenetic changes in the tyrosine hydroxylase promoter persist into adulthood. They isolated mice from 5 to 8 weeks but then returned them to group housing until 20 weeks of age. The increase in tyrosine hydroxylase promoter methylation was maintained after 12 weeks of group housing supporting the idea that stress in adolescence can impact the brain for life (Niwa et al., 2013). Demonstrating the importance of the timing in which the mice are tested for potential effects of G × E, the detrimental effect of polyI:C on PPI was not detected at the end of adolescence (8 weeks) but only later in adulthood (16 weeks) (Lipina et al., 2013).
Sex differences are an important and under-researched facet of psychiatric disorders. Schizophrenia starts earlier and is generally more severe in men. The risk for depression is doubled in women compared to men. In depression there is human and animal data on differential effect of early life stress on females (Goel and Bale, 2009). In the CaMK-DN-DISC1 × prenatal polyI:C study (Abazyan et al., 2010), only males were tested, although the same group has shown sex-specific phenotypes in the genetic model alone (Ayhan et al., 2011). Ibi et al. tested both sexes together for CaMK-DN-DISC1 × postnatal polyI:C and did not mention anything about potential sex differences (Ibi et al., 2010). PrP-DN-DISC1 × social isolation was performed on both sexes separately and the results were notably similar for both, suggesting that this paradigm may not be useful to look at sex differences (Niwa et al., 2013). Chronic social defeat is designed to utilize aggressive interactions between males; therefore, only males were tested (Haque et al., 2012).
The recent study combining Disc1 point mutants with PolyI:C at E9 (Lipina et al., 2013) provides an opportunity to compare the effects of a similar environmental factor on different Disc1 models based on data obtained by the same lab. They found that maternal immune activation interacts with the L100P mutation that causes abnormalities related to schizophrenia while there was no interaction with the Q31L mutation that causes abnormalities related to depression. These results correspond nicely with the epidemiological studies pointing to prenatal infection as a risk factor for schizophrenia but not for depression. The two studies using Disc1 point mutants also enable us to start to compare the effects of different environmental risk factors on the same model based on data obtained by the same lab. Interestingly, prenatal polyI:C on its own had no effect on sociability, but in combination with L100P it eliminated preference for the stranger (Lipina et al., 2013). On the other hand, CSD reduced sociability both in WT and in L100P mice (Haque et al., 2012). So social impairment can either result from CSD, which is highly relevant for social behavior, or from Disc1 L100P × prenatal polyI:C interaction—two factors which have no individual effect on sociability.
Future Directions
Exploring a matrix of all risk genes for schizophrenia and their interactions with all environmental risk factors would be exceptionally difficult. Scientists can instead focus on major known susceptibility genes and combine them with environmental factors thought to increase risk, based on epidemiological studies. In order to rigorously dissect the complexity of G × E interactions, both factors must be manipulated independently and the results of G × E must be compared to G and E alone under identical experimental conditions. Ideally, the same genetic model can be exposed to different environmental factors at different times and tested for histological, neurochemical, and behavioral phenotypes without a priori assignment of a specific disease model and with equal value assigned to negative results. Such open-ended analysis, although laborious, could suggest how one risk gene can catalyze the emergence of a variety of psychiatric disorders depending on which environmental factor it interacts with (Harvey and Boksa, 2012). DISC1 is an especially attractive candidate for such an approach since it is a risk factor for many different mental disorders. Looking at genetic interactions with an array of environments will still be a simplified model, since additional G × G or E × E interactions probably determine exact outcomes in humans. To further refine our understanding of the DISC1 × E interaction paradigm, there is a need for greater emphasis on sex differences in mouse models, as well as examination of environmental risk factors such as Toxoplasma gondii exposure, adolescent cannabis abuse and advanced paternal age. In the future, the development of comprehensive “environment interaction profiles” for major risk genes like DISC1 would represent a significant advancement toward the scientific understanding of mental disease pathogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Minae Niwa and Lindsay Hayes for critical reading of the manuscript and the NARSAD Young Investigator Award for funding (Hanna Jaaro-Peled).
References
Abazyan, B., Nomura, J., Kannan, G., Ishizuka, K., Tamashiro, K. L., Nucifora, F., et al. (2010). Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol. Psychiatry 68, 1172–1181. doi: 10.1016/j.biopsych.2010.09.022
Allardyce, J., Gilmour, H., Atkinson, J., Rapson, T., Bishop, J., and McCreadie, R. G. (2005). Social fragmentation, deprivation and urbanicity: relation to first-admission rates for psychoses. Br. J. Psychiatry 187, 401–406. doi: 10.1192/bjp.187.5.401
Andersen, S. L. (2003). Trajectories of brain development: point of vulnerability or window of opportunity. Neurosci. Biobehav. Rev. 27, 3–18. doi: 10.1016/S0149-7634(03)00005-8
Andersen, S. L., and Teicher, M. H. (2008). Stress, sensitive periods and maturational events in adolescent depression. Trends Neurosci. 31, 183–191. doi: 10.1016/j.tins.2008.01.004
Anisman, H., Zaharia, M. D., Meaney, M. J., and Merali, Z. (1998). Do early-life events permanently alter behavioral and hormonal responses to stressors. Int. J. Dev. Neurosci. 16, 149–164. doi: 10.1016/S0736-5748(98)00025-2
Asante, E. A., Gowland, I., Linehan, J. M., Mahal, S. P., and Collinge, J. (2002). Expression pattern of a mini human PrP gene promoter in transgenic mice. Neurobiol. Dis. 10, 1–7. doi: 10.1006/nbdi.2002.0486
Avital, A., and Richter-Levin, G. (2005). Exposure to juvenile stress exacerbates the behavioural consequences of exposure to stress in the adult rat. Int. J. Neuropsychopharmacol. 8, 163–173. doi: 10.1017/S1461145704004808
Ayhan, Y., Abazyan, B., Nomura, J., Kim, R., Ladenheim, B., Krasnova, I. N., et al. (2011). Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Mol. Psychiatry 16, 293–306. doi: 10.1038/mp.2009.144
Babulas, V., Factor-Litvak, P., Goetz, R., Schaefer, C. A., and Brown, A. S. (2006). Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am. J. Psychiatry 163, 927–929. doi: 10.1176/appi.ajp.163.5.927
Binder, E. B., Bradley, R. G., Liu, W., Epstein, M. P., Deveau, T. C., Mercer, K. B., et al. (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299, 1291–1305. doi: 10.1001/jama.299.11.1291
Bitanihirwe, B. K., Peleg-Raibstein, D., Mouttet, F., Feldon, J., and Meyer, U. (2010). Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology 35, 2462–2478. doi: 10.1038/npp.2010.129
Blackwood, D. H., Fordyce, A., Walker, M. T., St Clair, D. M., Porteous, D. J., and Muir, W. J. (2001). Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am. J. Hum. Genet. 69, 428–433. doi: 10.1086/321969
Blakemore, S. J. (2008). The social brain in adolescence. Nat. Rev. Neurosci. 9, 267–277. doi: 10.1038/nrn2353
Bradley, R. G., Binder, E. B., Epstein, M. P., Tang, Y., Nair, H. P., Liu, W., et al. (2008). Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch. Gen. Psychiatry 65, 190–200. doi: 10.1001/archgenpsychiatry.2007.26
Brandon, N. J., and Sawa, A. (2011). Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 12, 707–722. doi: 10.1038/nrn3120
Brown, A. S. (2011). The environment and susceptibility to schizophrenia. Prog. Neurobiol. 93, 23–58. doi: 10.1016/j.pneurobio.2010.09.003
Brown, A. S., Begg, M. D., Gravenstein, S., Schaefer, C. A., Wyatt, R. J., Bresnahan, M., et al. (2004a). Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch. Gen. Psychiatry 61, 774–780. doi: 10.1001/archpsyc.61.8.774
Brown, A. S., Hooton, J., Schaefer, C. A., Zhang, H., Petkova, E., Babulas, V., et al. (2004b). Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am. J. Psychiatry 161, 889–895. doi: 10.1176/appi.ajp.161.5.889
Cannon, T. D., Van Erp, T. G., Bearden, C. E., Loewy, R., Thompson, P., Toga, A. W., et al. (2003). Early and late neurodevelopmental influences in the prodrome to schizophrenia: contributions of genes, environment, and their interactions. Schizophr. Bull. 29, 653–669. doi: 10.1093/oxfordjournals.schbul.a007037
Cantor-Graae, E., and Selten, J. P. (2005). Schizophrenia and migration: a meta-analysis and review. Am. J. Psychiatry 162, 12–24. doi: 10.1176/appi.ajp.162.1.12
Carter, C. J. (2009). Schizophrenia susceptibility genes directly implicated in the life cycles of pathogens: cytomegalovirus, influenza, herpes simplex, rubella, and Toxoplasma gondii. Schizophr. Bull. 35, 1163–1182. doi: 10.1093/schbul/sbn054
Caspi, A., Sugden, K., Moffitt, T. E., Taylor, A., Craig, I. W., Harrington, H., et al. (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301, 386–389. doi: 10.1126/science.1083968
Charmandari, E., Kino, T., and Chrousos, G. P. (2004). Familial/sporadic glucocorticoid resistance: clinical phenotype and molecular mechanisms. Ann. N.Y. Acad. Sci. 1024, 168–181. doi: 10.1196/annals.1321.014
Chubb, J. E., Bradshaw, N. J., Soares, D. C., Porteous, D. J., and Millar, J. K. (2008). The DISC locus in psychiatric illness. Mol. Psychiatry 13, 36–64. doi: 10.1038/sj.mp.4002106
Clancy, B., Finlay, B. L., Darlington, R. B., and Anand, K. J. (2007). Extrapolating brain development from experimental species to humans. Neurotoxicology 28, 931–937. doi: 10.1016/j.neuro.2007.01.014
Clapcote, S. J., Lipina, T. V., Millar, J. K., Mackie, S., Christie, S., Ogawa, F., et al. (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402. doi: 10.1016/j.neuron.2007.04.015
Cratty, M. S., Ward, H. E., Johnson, E. A., Azzaro, A. J., and Birkle, D. L. (1995). Prenatal stress increases corticotropin-releasing factor (CRF) content and release in rat amygdala minces. Brain Res. 675, 297–302. doi: 10.1016/0006-8993(95)00087-7
Dalman, C., Allebeck, P., Gunnell, D., Harrison, G., Kristensson, K., Lewis, G., et al. (2008). Infections in the CNS during childhood and the risk of subsequent psychotic illness: a cohort study of more than one million Swedish subjects. Am. J. Psychiatry 165, 59–65. doi: 10.1176/appi.ajp.2007.07050740
Duncan, L. E., and Keller, M. C. (2011). A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am. J. Psychiatry 168, 1041–1049. doi: 10.1176/appi.ajp.2011.11020191
Edwards, V. J., Holden, G. W., Felitti, V. J., and Anda, R. F. (2003). Relationship between multiple forms of childhood maltreatment and adult mental health in community respondents: results from the adverse childhood experiences study. Am. J. Psychiatry 160, 1453–1460. doi: 10.1176/appi.ajp.160.8.1453
Flores, B. H., Kenna, H., Keller, J., Solvason, H. B., and Schatzberg, A. F. (2006). Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology 31, 628–636. doi: 10.1038/sj.npp.1300884
Fone, K. C., and Porkess, M. V. (2008). Behavioural and neurochemical effects of post-weaning social isolation in rodents-relevance to developmental neuropsychiatric disorders. Neurosci. Biobehav. Rev. 32, 1087–1102. doi: 10.1016/j.neubiorev.2008.03.003
Giedd, J. N., and Rapoport, J. L. (2010). Structural MRI of pediatric brain development: what have we learned and where are we going. Neuron 67, 728–734. doi: 10.1016/j.neuron.2010.08.040
Giedd, J. N., Snell, J. W., Lange, N., Rajapakse, J. C., Casey, B. J., Kozuch, P. L., et al. (1996). Quantitative magnetic resonance imaging of human brain development: ages 4–18. Cereb. Cortex 6, 551–560. doi: 10.1093/cercor/6.4.551
Gilmore, J. H., and Jarskog, L. F. (1997). Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr. Res. 24, 365–367. doi: 10.1016/S0920-9964(96)00123-5
Giovanoli, S., Engler, H., Engler, A., Richetto, J., Voget, M., Willi, R., et al. (2013). Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 339, 1095–1099. doi: 10.1126/science.1228261
Goel, N., and Bale, T. L. (2009). Examining the intersection of sex and stress in modelling neuropsychiatric disorders. J. Neuroendocrinol. 21, 415–420. doi: 10.1111/j.1365-2826.2009.01843.x
Gunnar, M. R., Wewerka, S., Frenn, K., Long, J. D., and Griggs, C. (2009). Developmental changes in hypothalamus-pituitary-adrenal activity over the transition to adolescence: normative changes and associations with puberty. Dev. Psychopathol. 21, 69–85. doi: 10.1017/S0954579409000054
Haque, F. N., Lipina, T. V., Roder, J. C., and Wong, A. H. (2012). Social defeat interacts with Disc1 mutations in the mouse to affect behavior. Behav. Brain Res. 233, 337–344. doi: 10.1016/j.bbr.2012.05.037
Harvey, L., and Boksa, P. (2012). Prenatal and postnatal animal models of immune activation: relevance to a range of neurodevelopmental disorders. Dev. Neurobiol. 72, 1335–1348. doi: 10.1002/dneu.22043
Heim, C., and Binder, E. B. (2012). Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 233, 102–111. doi: 10.1016/j.expneurol.2011.10.032
Heim, C., Shugart, M., Craighead, W. E., and Nemeroff, C. B. (2010). Neurobiological and psychiatric consequences of child abuse and neglect. Dev. Psychobiol. 52, 671–690. doi: 10.1002/dev.20494
Henquet, C., Di Forti, M., Morrison, P., Kuepper, R., and Murray, R. M. (2008). Gene-environment interplay between cannabis and psychosis. Schizophr. Bull. 34, 1111–1121. doi: 10.1093/schbul/sbn108
Hikida, T., Jaaro-Peled, H., Seshadri, S., Oishi, K., Hookway, C., Kong, S., et al. (2007). Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. U.S.A. 104, 14501–14506. doi: 10.1073/pnas.0704774104
Hill, M. N., Hellemans, K. G., Verma, P., Gorzalka, B. B., and Weinberg, J. (2012). Neurobiology of chronic mild stress: parallels to major depression. Neurosci. Biobehav. Rev. 36, 2085–2117. doi: 10.1016/j.neubiorev.2012.07.001
Ibi, D., Nagai, T., Kitahara, Y., Mizoguchi, H., Koike, H., Shiraki, A., et al. (2009). Neonatal polyI:C treatment in mice results in schizophrenia-like behavioral and neurochemical abnormalities in adulthood. Neurosci. Res. 64, 297–305. doi: 10.1016/j.neures.2009.03.015
Ibi, D., Nagai, T., Koike, H., Kitahara, Y., Mizoguchi, H., Niwa, M., et al. (2010). Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav. Brain Res. 206, 32–37. doi: 10.1016/j.bbr.2009.08.027
Jaaro-Peled, H. (2009). Gene models of schizophrenia: DISC1 mouse models. Prog. Brain Res. 179, 75–86. doi: 10.1016/S0079-6123(09)17909-8
Jaaro-Peled, H., Hayashi-Takagi, A., Seshadri, S., Kamiya, A., Brandon, N. J., and Sawa, A. (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495. doi: 10.1016/j.tins.2009.05.007
Jaaro-Peled, H., Niwa, M., Foss, C. A., Murai, R., De Los Reyes, S., Kamiya, A., et al. (2013). Subcortical dopaminergic deficits in a DISC1 mutant model: a study in direct reference to human molecular brain imaging. Hum. Mol. Genet. 22, 1574–1580. doi: 10.1093/hmg/ddt007
Johnstone, M., Thomson, P. A., Hall, J., McIntosh, A. M., Lawrie, S. M., and Porteous, D. J. (2011). DISC1 in schizophrenia: genetic mouse models and human genomic imaging. Schizophr. Bull. 37, 14–20. doi: 10.1093/schbul/sbq135
Kamiya, A., Kubo, K., Tomoda, T., Takaki, M., Youn, R., Ozeki, Y., et al. (2005). A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 7, 1167–1178. doi: 10.1038/ncb1328
Kendler, K. S., Karkowski, L. M., and Prescott, C. A. (1999). Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry 156, 837–841.
Khandaker, G. M., Zimbron, J., Dalman, C., Lewis, G., and Jones, P. B. (2012). Childhood infection and adult schizophrenia: a meta-analysis of population-based studies. Schizophr. Res. 139, 161–168. doi: 10.1016/j.schres.2012.05.023
Kudryavtseva, N. N., Bakshtanovskaya, I. V., and Koryakina, L. A. (1991). Social model of depression in mice of C57BL/6J strain. Pharmacol. Biochem. Behav. 38, 315–320. doi: 10.1016/0091-3057(91)90284-9
Kumar, A., Zhang, J., and Yu, F. S. (2006). Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology 117, 11–21. doi: 10.1111/j.1365-2567.2005.02258.x
Lipina, T. V., Zai, C., Hlousek, D., Roder, J. C., and Wong, A. H. (2013). Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J. Neurosci. 33, 7654–7666. doi: 10.1523/JNEUROSCI.0091-13.2013
Lupien, S. J., McEwen, B. S., Gunnar, M. R., and Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10, 434–445. doi: 10.1038/nrn2639
Malaspina, D. (2001). Paternal factors and schizophrenia risk: de novo mutations and imprinting. Schizophr. Bull. 27, 379–393. doi: 10.1093/oxfordjournals.schbul.a006882
Mann, J. J., and Currier, D. (2006). Effects of genes and stress on the neurobiology of depression. Int. Rev. Neurobiol. 73, 153–189. doi: 10.1016/S0074-7742(06)73005-7
March, D., Hatch, S. L., Morgan, C., Kirkbride, J. B., Bresnahan, M., Fearon, P., et al. (2008). Psychosis and place. Epidemiol. Rev. 30, 84–100. doi: 10.1093/epirev/mxn006
McEwen, B. S. (1998). Protective and damaging effects of stress mediators. N. Engl. J. Med. 338, 171–179. doi: 10.1056/NEJM199801153380307
Meyer, U., and Feldon, J. (2012). To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology 62, 1308–1321. doi: 10.1016/j.neuropharm.2011.01.009
Meyer, U., Feldon, J., Schedlowski, M., and Yee, B. K. (2005). Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci. Biobehav. Rev. 29, 913–947. doi: 10.1016/j.neubiorev.2004.10.012
Meyer, U., Feldon, J., and Yee, B. K. (2009). A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr. Bull. 35, 959–972. doi: 10.1093/schbul/sbn022
Meyer, U., Nyffeler, M., Engler, A., Urwyler, A., Schedlowski, M., Knuesel, I., et al. (2006). The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J. Neurosci. 26, 4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006
Meyer, U., Yee, B. K., and Feldon, J. (2007). The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse. Neuroscientist 13, 241–256. doi: 10.1177/1073858406296401
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423. doi: 10.1093/hmg/9.9.1415
Mittal, V. A., Ellman, L. M., and Cannon, T. D. (2008). Gene-environment interaction and covariation in schizophrenia: the role of obstetric complications. Schizophr. Bull. 34, 1083–1094. doi: 10.1093/schbul/sbn080
Miyazaki, S., Ishikawa, F., Fujikawa, T., Nagata, S., and Yamaguchi, K. (2004). Intraperitoneal injection of lipopolysaccharide induces dynamic migration of Gr-1high polymorphonuclear neutrophils in the murine abdominal cavity. Clin. Diagn. Lab. Immunol. 11, 452–457.
Morgan, C., and Fisher, H. (2007). Environment and schizophrenia: environmental factors in schizophrenia: childhood trauma—a critical review. Schizophr. Bull. 33, 3–10. doi: 10.1093/schbul/sbl053
Nagai, T., Kitahara, Y., Ibi, D., Nabeshima, T., Sawa, A., and Yamada, K. (2011). Effects of antipsychotics on the behavioral deficits in human dominant-negative DISC1 transgenic mice with neonatal polyI:C treatment. Behav. Brain Res. 225, 305–310. doi: 10.1016/j.bbr.2011.07.049
Nawa, H., and Yamada, K. (2012). Experimental schizophrenia models in rodents established with inflammatory agents and cytokines. Methods Mol. Biol. 829, 445–451. doi: 10.1007/978-1-61779-458-2_28
Niwa, M., Jaaro-Peled, H., Tankou, S., Seshadri, S., Hikida, T., Matsumoto, Y., et al. (2013). Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science 339, 335–339. doi: 10.1126/science.1226931
Niwa, M., Matsumoto, Y., Mouri, A., Ozaki, N., and Nabeshima, T. (2011). Vulnerability in early life to changes in the rearing environment plays a crucial role in the aetiopathology of psychiatric disorders. Int. J. Neuropsychopharmacol. 14, 459–477. doi: 10.1017/S1461145710001239
Nogai, A., Siffrin, V., Bonhagen, K., Pfueller, C. F., Hohnstein, T., Volkmer-Engert, R., et al. (2005). Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J. Immunol. 175, 959–966.
Nugent, N. R., Tyrka, A. R., Carpenter, L. L., and Price, L. H. (2011). Gene-environment interactions: early life stress and risk for depressive and anxiety disorders. Psychopharmacology (Berl) 214, 175–196. doi: 10.1007/s00213-010-2151-x
Oberlander, T. F., Weinberg, J., Papsdorf, M., Grunau, R., Misri, S., and Devlin, A. M. (2008). Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3, 97–106. doi: 10.4161/epi.3.2.6034
O'connor, T. G., Ben-Shlomo, Y., Heron, J., Golding, J., Adams, D., and Glover, V. (2005). Prenatal anxiety predicts individual differences in cortisol in pre-adolescent children. Biol. Psychiatry 58, 211–217. doi: 10.1016/j.biopsych.2005.03.032
O'connor, T. M., O'halloran, D. J., and Shanahan, F. (2000). The stress response and the hypothalamic-pituitary-adrenal axis: from molecule to melancholia. QJM 93, 323–333. doi: 10.1093/qjmed/93.6.323
O'donnell, P. (2010). Adolescent maturation of cortical dopamine. Neurotox. Res. 18, 306–312. doi: 10.1007/s12640-010-9157-3
Okahira, S., Nishikawa, F., Nishikawa, S., Akazawa, T., Seya, T., and Matsumoto, M. (2005). Interferon-beta induction through toll-like receptor 3 depends on double-stranded RNA structure. DNA Cell Biol. 24, 614–623. doi: 10.1089/dna.2005.24.614
Papagni, S. A., Benetti, S., Arulanantham, S., McCrory, E., McGuire, P., and MeChelli, A. (2011). Effects of stressful life events on human brain structure: a longitudinal voxel-based morphometry study. Stress 14, 227–232.
Paus, T., Keshavan, M., and Giedd, J. N. (2008). Why do many psychiatric disorders emerge during adolescence. Nat. Rev. Neurosci. 9, 947–957.
Pedersen, C. B., and Mortensen, P. B. (2006). Are the cause(s) responsible for urban-rural differences in schizophrenia risk rooted in families or in individuals. Am. J. Epidemiol. 163, 971–978. doi: 10.1093/aje/kwj169
Pletnikov, M. V., Ayhan, Y., Nikolskaia, O., Xu, Y., Ovanesov, M. V., Huang, H., et al. (2008). Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 13, 173–186, 115. doi: 10.1038/sj.mp.4002079
Porteous, D. J., Millar, J. K., Brandon, N. J., and Sawa, A. (2011). DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol. Med. 17, 699–706. doi: 10.1016/j.molmed.2011.09.002
Remington, J. S. (2006). Infectious Diseases of the Fetus and Newborn Infant. Philadelphia, PA: Elsevier Saunders.
Schmidt, M. V., Wang, X. D., and Meijer, O. C. (2011). Early life stress paradigms in rodents: potential animal models of depression. Psychopharmacology (Berl.) 214, 131–140. doi: 10.1007/s00213-010-2096-0
Shi, L., Fatemi, S. H., Sidwell, R. W., and Patterson, P. H. (2003). Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 23, 297–302.
Shi, L., Tu, N., and Patterson, P. H. (2005). Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int. J. Dev. Neurosci. 23, 299–305. doi: 10.1016/j.ijdevneu.2004.05.005
Shoji, H., Toyama, K., Takamiya, Y., Wakana, S., Gondo, Y., and Miyakawa, T. (2012). Comprehensive behavioral analysis of ENU-induced Disc1-Q31L and -L100P mutant mice. BMC Res. Notes 5:108. doi: 10.1186/1756-0500-5-108
Stott, D. H. (1973). Follow-up study from birth of the effects of prenatal stresses. Dev. Med. Child Neurol. 15, 770–787. doi: 10.1111/j.1469-8749.1973.tb04912.x
Szelenyi, J. (2001). Cytokines and the central nervous system. Brain Res. Bull. 54, 329–338. doi: 10.1016/S0361-9230(01)00428-2
Tsien, J. Z., Chen, D. F., Gerber, D., Tom, C., Mercer, E. H., Anderson, D. J., et al. (1996). Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87, 1317–1326. doi: 10.1016/S0092-8674(00)81826-7
Van Os, J., Kenis, G., and Rutten, B. P. (2010). The environment and schizophrenia. Nature 468, 203–212. doi: 10.1038/nature09563
Van Os, J., Rutten, B. P., and Poulton, R. (2008). Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr. Bull. 34, 1066–1082. doi: 10.1093/schbul/sbn117
Vazquez, D. M., and Akil, H. (1993). Pituitary-adrenal response to ether vapor in the weanling animal: characterization of the inhibitory effect of glucocorticoids on adrenocorticotropin secretion. Pediatr. Res. 34, 646–653. doi: 10.1203/00006450-199311000-00017
Venzala, E., Garcia-Garcia, A. L., Elizalde, N., Delagrange, P., and Tordera, R. M. (2012). Chronic social defeat stress model: behavioral features, antidepressant action, and interaction with biological risk factors. Psychopharmacology (Berl.) 224, 313–325. doi: 10.1007/s00213-012-2754-5
Vyas, A., Kim, S. K., Giacomini, N., Boothroyd, J. C., and Sapolsky, R. M. (2007). Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc. Natl. Acad. Sci. U.S.A. 104, 6442–6447. doi: 10.1073/pnas.0608310104
Waterland, R. A., and Michels, K. B. (2007). Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 27, 363–388. doi: 10.1146/annurev.nutr.27.061406.093705
Weaver, I. C., Cervoni, N., Champagne, F. A., D'alessio, A. C., Sharma, S., Seckl, J. R., et al. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854. doi: 10.1038/nn1276
Weinberger, D. R. (1996). On the plausibility of “the neurodevelopmental hypothesis” of schizophrenia. Neuropsychopharmacology 14, 1S–11S. doi: 10.1016/0924-977X(96)88087-4
Keywords: DISC1, mouse models, gene-environment, schizophrenia, depression, immune activation, social stress
Citation: Cash-Padgett T and Jaaro-Peled H (2013) DISC1 mouse models as a tool to decipher gene-environment interactions in psychiatric disorders. Front. Behav. Neurosci. 7:113. doi: 10.3389/fnbeh.2013.00113
Received: 01 May 2013; Paper pending published: 08 June 2013;
Accepted: 07 August 2013; Published online: 03 September 2013.
Edited by:
Tim Karl, Neuroscience Research Australia, AustraliaReviewed by:
Francesca Cirulli, Istituto Superiore di Sanità, ItalyTsuyoshi Miyakawa, Fujita Health University, Japan
Copyright © 2013 Cash-Padgett and Jaaro-Peled. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanna Jaaro-Peled, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, 600 North Wolfe Street, Meyer 3-149, Baltimore, MD 21287, USA e-mail: hjaarop1@jhmi.edu