 C. C. Chen
C. C. Chen C. L. Williams*
C. L. Williams*- Department of Psychology, The University of Virginia, Charlottesville, VA, USA
It is well-established that exposure to emotionally laden events initiates secretion of the arousal-related hormone epinephrine in the periphery. These neuroendocrine changes and the subsequent increase in peripheral physiological output play an integral role in modulating brain systems involved in memory formation. The impermeability of the blood brain barrier to epinephrine represents an important obstacle in understanding how peripheral hormones initiate neurochemical changes in the brain that lead to effective memory formation. This obstacle necessitated the identity of a putative pathway capable of conveying physiological changes produced by epinephrine to limbic structures that incorporate arousal and affect related information into memory. A major theme of the proposed studies is that ascending fibers of the vagus nerve may represent such a mechanism. This hypothesis was tested by evaluating the contribution of ascending vagal fibers in modulating memory for responses learned under behavioral conditions that produce emotional arousal by manipulating appetitive stimuli. A combination of electrophysiological recording of vagal afferent fibers and in vivo microdialysis was employed in a second study to simultaneously assess how elevations in peripheral levels of epinephrine affect vagal nerve discharge and the subsequent potentiation of norepinephrine release in the basolateral amygdala. The final study used double immunohistochemistry labeling of c-fos and dopamine beta hydroxylase (DBH), the enzyme for norepinephrine synthesis to determine if epinephrine administration alone or stimulation of the vagus nerve at an intensity identical to that which improved memory in Experiment 1 produces similar patterns of neuronal activity in brain areas involved in processing memory for emotional events. Findings emerging from this collection of studies establish the importance of ascending fibers of the vagus nerve as an essential pathway for conveying the peripheral consequences of physiological arousal on brain systems that encode new information into memory storage.
Introduction
An extensive number of findings reveal that the emotional nature of learning experiences contributes to the strength that novel events are encoded and stored into memory. The influence of highly arousing events on memory is attributed to the impact salient stimuli have in initiating and maintaining heightened levels of neural activity in the amygdala (Cahill and McGaugh, 1995; Gerra et al., 1996; Canli et al., 2002; Chang et al., 2005; Pelletier et al., 2005). Emotional experiences influence the amydala and regulate how effective new events are converted into memory by their capacity to evoke epinephrine secretion from the adrenal glands (McCarty and Gold, 1981) which in turn initiates a long lasting and sustained release of norepinephrine in the amygdala (Williams et al., 1998; O'Carroll et al., 1999; Hurlemann et al., 2005). The contribution of norepinephrine activation of the amygdala to memory processing is revealed by studies showing that infusion of this transmitter or beta-noradrenergic agonists into the basolateral amygdala selectively improve memory for responses acquired in inhibitory avoidance (Ferry et al., 1999), contextual fear conditioning (LaLumiere et al., 2003; Huff et al., 2006), or spatial learning tasks (Hatfield and McGaugh, 1999). Although a great deal of attention has been devoted to understanding how activation of the amygdala modulates neuronal functioning in other limbic structures during memory formation, less emphasis has been placed on identifying how changes in peripheral autonomic activity produced by adrenal hormones, feedback to the brain to influence noradrenergic activation of the amygdala during this important process.
Elucidation of the mechanisms underlying these interactions will provide a more comprehensive understanding of memory formation since they have circumscribed effects on how effective new events are represented in memory. For example, the uniform enhancement in memory observed following heightened states of arousal produced by epinephrine release is abolished by experimental manipulations that disrupt amygdala functioning. Epinephrine's actions on memory are also ineffective when the amygdala is lesioned (Cahill and McGaugh, 1991) or if the major input and output pathways from the amygdala are severed (Liang et al., 1990; Roozendaal and McGaugh, 1996). Manipulations that produce more selective effects on neurotransmitter functioning such as blocking receptors that bind norepinephrine (Liang et al., 1986; Quirarte et al., 1997) or depleting amygdala norepinephrine concentrations with the selective neurotoxin DSP-4 (Liang et al., 1995) also block the memory enhancing actions of epinephrine. These two distinct neurochemical changes that follow emotional arousal have gained a great deal of attention for their role in modulating memory, however, the pathway that epinephrine secretion initiates and/or regulates a sustained increase in central noradrenergic output is not well understood. Such transmission is not likely to be a direct process because epinephrine does not gain access to the CNS due to the restrictive properties of the blood-brain barrier (BBB) (Weil-Malherbe et al., 1959; Bradbury, 1993).
In light of the restricted access of epinephrine to the brain, reports from anatomical, electrophysiological, and pharmacological experiments suggest that epinephrine's actions on memory and in potentiating norepinephrine output in the amygdala may be mediated by the initial activation of peripheral vagal fibers that project to the brain. The vagus nerve is considered a potential candidate to mediate the actions of epinephrine and by implication, emotional arousal on the brain because dorsal and ventral branches of the nerve innervate the adrenals (Coupland et al., 1989), electrical stimulation of the adrenal nerve, which results in epinephrine release evokes action potentials in the vagus (Niijima, 1992) and ascending fibers of the vagus are densely embedded with beta-adrenergic receptors that bind epinephrine (Schreurs et al., 1986; Lawrence et al., 1995). Blocking peripheral beta-adrenergic receptors abolishes arousal-induced changes in memory, peripheral autonomic functioning or epinephrine induced increases in vagal nerve firing (van den Buuse et al., 2001; van den Buuse, 2002; Carrive, 2006; Miyashita and Williams, 2006; King and Williams, 2009). The vagus nerve is also an ideal candidate to subserve this function because peripheral endings of the nerve innervate organs such as the heart and lungs that display heightened activity in humans that show enhanced retention in response to viewing emotionally arousing slides or animals displaying similar cognitive improvements following epinephrine administration (Shapiro and Miselis, 1985; Coupland et al., 1989; Paton, 1998a,b).
Information regarding changes in the functioning of the adrenals and other organs after exposure to arousing stimuli are transmitted via afferent fibers of the vagus nerve (Coupland et al., 1989; Paton, 1998a,b) to a region of the brainstem known as the nucleus tractus solitarius (NTS: Kalia and Mesulam, 1980; Sumal et al., 1983). The projection of vagal afferents to the NTS is important in understanding how vagal activation may affect distinct neuronal circuits to regulate norepinephrine release in the amygdala. For instance, ascending fibers of the vagus synapse on the A2 norepineprhine-containing neurons in the NTS (Sumal et al., 1983) that course through the brainstem to innervate and release norepinephrine in the amygdala (Fallon et al., 1978; Bjorklund and Lindvall, 1986; Riche et al., 1990; Petrov et al., 1993). A2 neurons in the NTS also project directly to locus coeruleus neurons (LC; Van Bockstaele et al., 1999) that provide the major source of noradrenergic innervation to the basolateral amygdala (Fallon et al., 1978). Interestingly, electrical stimulation of ascending vagal fibers or epinephrine injection alone, produces significant and long lasting increases in amygdala norepinephrine levels that are attenuated by inactivating the site of termination of vagal axons in the brainstem, the NTS (Williams et al., 1998, 2000; Hassert et al., 2004). Thus, vagal activation in response to epinephrine secretion may represent one mechanism by which emotionally arousing events facilitate memory processing by initiating norepinephrine release in the amygdala.
However, no findings to date have demonstrated simultaneously, that vagal nerve firing consequent to elevations in peripheral epinephrine levels, influence noradrenergic functioning in the amygdala to facilitate memory processing. The following studies were conducted to address this shortcoming with three separate approaches. These experiments examined this hypothesis by first determining if stimulating vagal afferent fibers at an intensity that augments norepinephrine release in the amygdala (Hassert et al., 2004), enhances memory for responses learned under behavioral conditions that produce emotional arousal by manipulating appetitive stimuli as opposed to administering noxious stimuli such as footshock. A second study utilized electrophysiological recording of vagal afferent fibers in conjunction with in vivo microdialysis to assess simultaneously, how elevations in peripheral levels of epinephrine affect vagal nerve discharge and the subsequent potentiation of norepinephrine release in the basolateral amygdala. The final study used double immunohistochemistry labeling of c-fos and dopamine beta hydroxylase (DBH), the enzyme for norepinephrine synthesis to determine if epinephrine administration alone or stimulation of the vagus nerve in separate groups of subjects produce similar patterns of neuronal activity in brain areas that process memory for emotional events. Findings emerging from this collection of studies are expected to reveal the contribution of ascending fibers of the vagus nerve in serving as an essential pathway for conveying the peripheral consequences of emotional arousal induced by epinephrine secretion on brain systems that encode new experiences into memory storage.
Experiment 1
The objectives of the first experiment were two-fold. This study determined if reducing the quantity of expected rewards produces emotional changes associated with frustration (i.e., the “Crespi Effect”) that are manifested by decreases in motivation and behavioral responding. These changes are indexed by increased latencies to run the length of the runway to receive the reduced quantity of food rewards. The second goal was to determine whether retention of the emotional experience involving reward reduction is improved by increasing noradrenergic drive within the amygdala by post-training stimulation of vagal afferent fibers with an intensity that has been shown to produce significant and long lasting elevations in norepinephrine release in this structure.
Methods
Subjects
Twenty-three male Sprague-Dawley rats (275–300 g) obtained from Charles River Laboratories (Wilmington, MA) were used in Experiment 1. The rats were individually housed in polypropylene cages with corncob bedding and maintained on a standard 12:12 h light–dark cycle with lights on at 7:00 a.m. Food and water were available ad libitum during the seven day adaptation period to the vivarium.
Surgery to Implant Vagal Stimulation Electrodes
Atropine sulfate (0.4 mg/kg) was given ip 10 min before anesthesia with sodium pentobarbital (50 mg/kg). Each rat was implanted with a stimulating electrode on the left vagus nerve at the cervical level that was constructed from two 30-gauge, 7 cm long insulated silver wires, stripped 1 mm on one end (the electrode end), and 1 mm on the other end (the current source end). As shown in Figure 1, polyvinyl chloride (PVC) tubes were placed over the 7 mm exposed section of each wire and contact was achieved by forming the electrode into a helix that, when implanted, would encircle the nerve. The two wires were inserted into a 3.5 cm piece of PVC and the electrode was completed by soldering metal female pins to the 1 mm stripped leads. The electrode leads from the vagus nerve were then drawn under the skin through the neck to the dorsal surface just posterior to the skull and affixed to a head post constructed from an Amphenol miniature strip connector and cemented to the skull with dental acrylic. The male pins were later attached to the female pins via 30 gauge silver wires to deliver electrical stimulation supplied by a current generator (Model 82400, Lafeyette Instruments, Lafeyette, IN). Electrode placement along the vagus was verified visually and by monitoring resistance between the two stimulating electrodes with a digital voltmeter. The resistance was constantly monitored to ensure the effectiveness of the vagus nerve stimulation (VNS).

Figure 1. Time line for the training and testing regimen in the straight alley learning task and dimensions of the training apparatus. Illustration of the electrode implanted along the vagus nerve to produce vagal nerve stimulation.
Straight Alley Food Reward Task
Apparatus
As shown in Figure 1, the training apparatus consisted of a 72-inch long straight alley that was 15 inches in height, 10 inches wide and constructed of wood. The apparatus included a start compartment ( 6 inches long) that was blocked by a sliding door connected to a digital timer. The alley also included three hurdles (5 cm high) that were placed 2, 12, and 24 inches from the start box to ensure that a standard level of effort and motivation develops to obtain the food rewards. The goal cup containing the food rewards was 1 inch wide and placed at the end of the alley.
Training
Rats were placed on a weight maintenance schedule seven days before training to reduce weights to 15% below ad-lib feeding weights. Each animal was pre-exposed for two days to sucrose pellets (45 mg; Research Diets, Inc, New Brunswick, NJ) used for rewards throughout the study. Animals were randomly assigned to groups that would be given either sham or vagal stimulation following a shift or reduction in food reward from the normally expected 10 pellets to a small reward of only one sucrose pellet on Day 24. Training began by placing each rat in the start box facing away from the closed sliding door. The door was opened and the rat was given 120 s to jump over the three hurdles to reach and consume the sucrose pellet rewards placed in the food cup at the end of the six-foot long alley. As soon as the reward was consumed, the trial ended and the rat was placed in a holding cage for an inter-trial interval (ITI) of 30 s. The latency to consume the food reward was measured on each of the four daily trials using a digital timer. The animals were trained on Days 1–11 prior to surgery (Figure 2A) to implant the vagal stimulating electrode. They were then given seven days of postoperative recovery (Days 12–18) before training resumed on Day 19 and continued for 5 days (See Figure 1 for timeline).
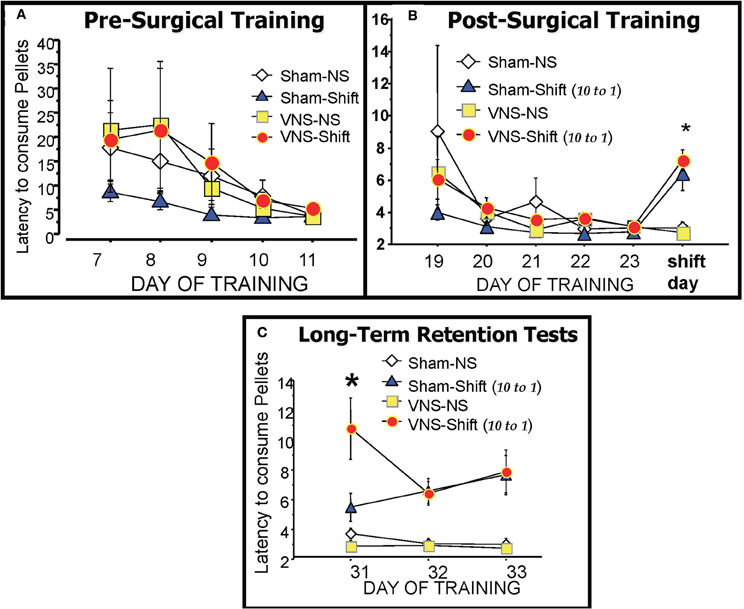
Figure 2. The runway latencies of each training group during pre-surgical training (A), post-surgical training (B) and during the three days of retention testing (C). There were no significant differences between groups during the training periods. The reduction in reward magnitude from 10 pellets to only 1 pellet on the day of the Shift resulted in significantly longer latencies in the two shifted groups (Sham-Shift and VNS-Shift) relative to the Non-Shifted controls (Sham-NS and VNS-NS). The VNS-Shift group received vagal stimulation after the six training trials with the reduced reward whereas the Sham-Shift group was connected to the stimulator but no current was applied. Retention for the frustrating experience of reward reduction produced by the Shift was assessed one-week later on three daily retention tests. Animals in the vagal stimulation Shift group displayed enhanced retention on the 7 day delayed retention test as evidenced by their continued long latencies to traverse the maze to consume the reduced reward of 1 pellet. Details included in the text.
Induction of emotional frustration by shifting reward magnitude
The experimental or “Shift” day in which emotional frustration was produced by the reduction in reward magnitude from 10 to 1 food pellets occurred on Day 24. On this day, animals assigned to the “No-Shift” group continued to receive 10 pellets while those in the “Shift Group” received only one, rather than 10 pellets on 6, as opposed to the normal four daily training trials. Each rat was removed from the alley after the sixth trial and those in the vagus nerve stimulation group received three, 10 s stimulations (0.4 mA current separated by 10 s intervals) along the left cervical vagus. The stimulation parameters were selected from those previously shown to produce long lasting elevations in amygdala concentrations of norepinephrine (Hassert et al., 2004) after VNS. Animals in the sham groups received no stimulation but were connected to the stimulating device. The four groups in Experiment 1 consisted of a Sham-No Shift (Sham-NS), VNS-No Shift (VNS-NS), Sham-Shift, and VNS-Shift. The subjects remained on the weight maintenance schedule and were not disturbed during the seven day interval between shift in reward magnitude and the first retention test (Days 24–31). This extended delay interval was imposed to examine whether memory of the frustrating experience of reward reduction is encoded more effectively by activating the vagus nerve following training since retention for this type of change in reward contingency decays in control animals after a one-week delay but remains intact in groups administered memory modulating treatments (Salinas et al., 1996).
Retention test
During the three days of retention testing (Days 31–33), animals in the shifted groups continued to receive a 1 pellet food reward on each of the six retention trials while those in the non-shift groups received the standard 10 pellets upon reaching the goal box. The latency to run the length of the alley and cross the three hurdles to consume the 10 pellets (non-shift groups) or the reduced quantity of one pellet (shifted groups) was recorded and served as an index of memory for the reduction in food reward.
Statistical analysis
Behavioral measures from the straight alley task are expressed as mean ± SE. A One-Way analysis of variance (ANOVA) followed by post hoc tests were used to detect differences in the latency to consume the food rewards between Shifted Sham and VNS groups as well as the No-Shift controls. Repeated measure ANOVA's were used to analyze differences in the latency to consume the reward between treatment groups during the training days, the shift day, and the first trial of the retention test.
Results
Effects of Reducing the Magnitude of Expected Rewards
As depicted in Figure 2B, performance was statistically similar in all groups during the 5 days of post-operative training that preceded the shift in reward magnitude on Day 24. On this day however, decreasing the expected value in reward from 10 to only 1 pellet caused a significant overall change in the runway performance of the shifted groups as revealed by a One-Way ANOVA F(3, 19) = 9.137, p < 0.01. F(3, 21) = 8.44, p < 0.01. As shown in (Figure 2B), Post-hoc analysis revealed that the two shifted groups took significantly longer to cross the three hurdles and run the length of the six foot long alley to consume the new quantity of one reward pellet compared to the two non-shifted groups (Sham-Shift vs. Sham—NS, p < 0.01; Sham-Shift vs. VNS-NS, p < 0.01; VNS-Shift vs. Sham-NS, p < 0.01; VNS-Shift vs. VNS-NS, p < 0.01). These findings demonstrate the procedures used to induce emotional frustration through reward reduction produce reliable changes in behavior that are reflected in increased latencies and lower motivational levels to approach and consume the food rewards.
Effects of VNS on Long Term Retention
A comparison of the mean runway latencies recorded during the six daily trials on the three days of retention testing is shown in Figure 2C. Post-training VNS produced a significant enhancement in memory for reward reduction on the first retention test given 7 days after the Shift. A One-Way ANOVA revealed a significant group difference [F(3, 19) = 9.575; p < 0.01]. Post-hoc comparisons with Fisher's PLSD tests showed that the runway latencies of the VNS-shifted group were significantly longer than both non-shifted groups (VNS-Shift vs Sham-NS, p < 0.01; VNS-Shift vs. VNS-NS, p < 0.01) as well as the Sham-Shift group that also experienced a reward reduction (p < 0.01). The lasting consequences of reward reduction were evident on each of the three retention tests given on Days 31–33. A One-Way ANOVA on retention day 2 [F(3, 19) = 9.613; p < 0.01] and 3 [F(3, 19) = 7.172, p < 0.01] revealed that both the shifted groups exhibited longer latencies to consume the reward than the unshifted groups (Sham-Shift vs. Sham- NS, p < 0.01; Sham-Shift vs. VNS-NS, p < 0.01; VNS-Shift vs. Sham-NS, p < 0.01; VNS-Shift vs. VNS-NS, p < 0.01). The results demonstrate that increasing neural transmission from the vagus to the brain following an emotional event enhances memory and these effects are evident even when assessments of retention are delayed for as long as 7 days after the experience.
Experiment 2
Our previous findings demonstrated that neural activity recorded along the cervical vagus increased significantly following epinephrine injection and these excitatory actions were not observed in groups given an identical dose of the hormone after peripheral β-adrenergic receptor blockade with sotalol (Miyashita and Williams, 2006). These findings demonstrate that neural discharge in vagal afferent fibers is increased by elevations in peripheral concentrations of epinephrine and the effects are mediated in part by epinephrine binding to peripheral β-adrenergic receptors along the vagus nerve. This study also revealed the time course the nerve maintains high levels of firing following epinephrine coincides with the time course norepinephrine levels in the amygdala remain elevated after an identical dose epinephrine (Williams et al., 1998, 2000) or following stimulation of the vagus nerve at an intensity that improves memory (Hassert et al., 2004). When considered together, these findings raise a fundamental question as to whether neural impulses propagated along the vagus following emotional arousal and/or epinephrine secretion serve an important function in maintaining high-levels of noradrenergic transmission in the brain during the period of memory consolidation. However, no study to date has examined simultaneously, whether elevations in peripheral levels of epinephrine that lead to increased firing along ascending fibers of the vagus nerve, parallel the sustained changes in norepinephrine output that are observed in the amygdala. The present study combined electrophysiological and in vivo microdialysis approaches that were employed separately in the studies discussed above, to specifically test whether heightened activity in ascending vagal fibers following epinephrine administration regulates the release of norepinephrine in the amygdala.
Methods
Subjects
Forty-four male Sprague-Dawley rats purchased from Harlan Laboratories weighing approximately 250–300 g were used in this study. They were singly housed and maintained on a 12-h light-dark cycle (lights on at 6:00 a.m.) with water and food available ad libitum. The rats were handled daily for one week prior to the initiation of all experiments.
Surgery
Two separate surgical procedures were performed in one-stage. The first involved severing the efferent rootlets of the left vagus nerve at the level of the cervix. This procedure was crucial to insure that electrophysiological recordings in response to epinephrine administration derived solely from the intact ascending branches of the nerve and did not reflect impulses propagated along the cut descending fibers. The second procedure involved stereotaxic implantation of a microdialysis guide cannula above the left basolateral amygdala to measure extracellular concentrations of norepinephrine in response to epinephrine-induced changes in vagal nerve firing. The methods used to severe the efferent branch of the vagus nerve was adopted and modified from those of Norgren and Smith (1994). Each rat was anesthetized with an intramuscular injection of ketamine (100 mg/kg). A 3–4 cm incision was made from the chin to the thorax and the salivary gland, and lymph nodes were retracted laterally to expose the sternohyoid and omohyoid muscles. These two muscles were then separated from the underlying muscles to expose the superior laryngeal nerve and thyroid artery. A #15 blade in a scalpel handle was used to create an opening in the occipital bone to expose the efferent rootlets of the vagus nerve. The efferent rootlets were severed using ultra-fine forceps and the cavity was packed with bone wax. The retracted muscles, salivary gland, lymph nodes, and skin were repositioned and the skin was closed with suture.
Following deafferentation, each rat was placed in a stereotaxic apparatus and a midline incision on the skull was made to expose bregma. A single microdialysis guide cannula was lowered 2 mm above the basolateral amygdala (AP −3.0, L +5.0, DV −6.7) according to the atlas of Paxinos and Watson (1986). Following implantation, the guide cannula was cemented to the skull by two anchoring screws using dental acrylic. Animals were then allowed seven days to recover from both surgeries prior to electrophysiological recordings combined with in vivo microdialysis collection of norepinephrine.
Simultaneous vagal recording and microdialysis sample collection
On the experiment day, the rat was anesthetized with urethane (1.2 g/kg, i.p.) and a 4 cm incision was made 5 mm lateral to of the midline of the cervix to identify the internal carotid artery and cervical branch of the vagus nerve. Approximately 1 cm of the nerve was separated from connective tissue and placed onto two 30 gauge silver wire hook electrodes connected to a recording device. After the surgical preparation and recording settings were established, a 1:1 petroleum jelly and mineral oil mixture was used to cover the vagus nerve and electrodes, preventing dryness and the interference of tissue fluids during recording. The animal was left undisturbed for a 45 min period of nerve stabilization before any recordings were measured.
During recording, multifiber neural activity in the form of mV was collected from the electrodes, amplified 10–20,000 times using AC amplifiers (Grass, P511; Astromed: West Warwick, RI) with low (100 Hz) and high (1 kHz) frequency cutoffs. The signals were relayed to a spike processor to separate signals generated from action potentials from those with variable amplitudes derived from background noise. The window discriminator was set above the level of noise on a computer screen and the output of the spike processor representing action potentials from only the nerve fiber were averaged using the Chart data acquisition software (ADI instrument: Mountain view, CA) and monitored with an audio amplifier. For data analysis, the amplified signals were passed through an integrator with a time constant of 1 s and the summated electrical activity from action potentials recorded from the whole-nerve was stored on a PC computer and analyzed with the Chart program.
Concomitant microdialysis collection and electrophysiological recording from the vagus nerve
During the first hour pre-collection period, a microdialysis probe (CMA/12; Carnegie /Medicin, Acton, MA) was inserted into the left basolateral amygdala. After the vagal surgical procedures were carried out, the rat was left undisturbed until the pre-collection period was over. During this period, no brain samples or nerve activity was recorded. At the start of the second hour, dialysate samples of norepinephrine were collected every 20 min over a period of 60 min. The concentration of norepinephrine in these three samples was averaged to represent baseline levels of this transmitter in the amygdala. Electrical recordings of vagal basal activity were initiated during the final 30 min of this period and consisted of 15 minutes of baseline collection that was followed by i.p. injection of saline or sotalol to block peripheral adrenergic receptors in some groups and a second period of 15 min of collection to ensure the drug injections alone, did not affect vagal nerve firing.
The rats were randomly assigned to one of seven treatment groups. Four of these groups received an ip injection 15 min prior to the start of the experiment of either saline, 4.0 mg/kg of the peripherally acting beta-adrenergic antagonist Sotalol or 2.0 mg/kg of this antagonist (two groups). These groups would also be given an ip injection at the very beginning of the experiment at time period (0) of saline or epinephrine. This combination of treatments resulted in the following four groups (SAL, n = 7); sotalol 2.0 mg/kg + saline (SOT2 + SAL, n = 6); sotalol 4.0 mg/kg + saline (SOT4 + SAL, n = 4); sotalol 2.0 mg/kg + epinephrine 0.3 mg/kg (SOT2 + EPI, n = 4). Three additional groups received initial injections of 0.3 mg/kg of epinephrine at time period (0) and a second injection of 2.0 or 4.0 mg/kg of sotalol that was delayed by 60 min. These groups were included to determine whether blocking peripheral adrenergic receptors after vagal firing had reached asymptote attenuates neural impulses recorded from the vagus nerve. The groups consisted of epinephrine 0.3 mg/kg alone (EPI, n = 6); epinephrine 0.3 mg/kg + sotalol 2.0 mg/kg (EPI + SOT2, n = 6); and epinephrine 0.3 mg/kg + sotalol 4.0 mg/kg (EPI + SOT4, n = 6). The recording period lasted for 3 h after the first injection.
Norepinephrine assay with HPLC
Dialysate samples (35 ul) of norepinephrine were assayed by an HPLC system with a Waters 510 pump, Waters 717 autosampler, Atlantis T3 column (3 micron ODS, 4.6 × 100 mm) and a Waters 2465 electrochemical detector. The mobile phase consisted of 50 mg disodium EDTA, 13.8 mg monobasic sodium phosphate, and 58 mg octane sulfonate adjusted to pH 3.2 by adding 85% phosphoric acid. The flow rate was adjusted to 1.0 ml per minute.
Histology
The rat was deeply anesthetized with Euthosol (Virbac) and perfused with 0.9% saline and 10% buffered Formalin intracardially. The perfused brains were stored in 10% buffered Formalin, sectioned at the thickness of 50 um using a Vibratome, mounted onto gelatin-coated slides, stained with cresyl violet, coverslipped, and then dried. The cannula tracts were verified using a microscope Olympus CX41. Three animals were excluded from the study due to incorrect cannula placement and electrophysiological data from two animals were discarded due to technical problems. This yielded 6, 6, 6, 7, 4, 4, and 6 animals in the EPI, EPI + SOT2, EPI + SOT4, SAL, SAL + SOT4, SOT2 + EPI, and SOT2 + SAL groups respectively for electrophysiological recording.
Statistical analysis
Electrophysiology. Two separate sets of analyses were performed to characterize the data collected from the vagal recording. Repeated measure and factorial ANOVAs were used respectively, to assess fluctuations in vagal neural activity across and within treatment groups during the 15 min drug free baseline period. Data points in the analysis were taken by averaging electrical activity of the vagus nerve in millivolts in 5 min bins. Any change in electrical activity measured in each subsequent 5 min bin during the 15 min drug free baseline period is expressed as the percentage change relative to this value. A repeated measure ANOVA was used to detect changes in whole nerve neural activity across the 180 min collection period. Factorial ANOVA's with post-hoc Fisher's tests were used to detect differences between individual treatment groups (i.e., SAL, SOT2 + SAL, SOT2 + EPI, EPI, SAL + SOT4, EPI + SOT2 EPI + SOT4). a level of p < 0.05 was deemed significant in all analysis.
In vivo microdialysis. The levels of norepinephrine (in picograms per microliter) collected from the basolateral amygdala during the initial 60 min baseline period was averaged to yield a standard baseline value of 100%. The concentration of norepinephrine collected from dialysate samples throughout the experiment is expressed as the percent change above or below the baseline value. A Two-Way ANOVA was used to evaluate the overall effect of treatment for each group relative to baseline levels. This resulted in a mixed factorial analysis with one between-subjects factor (treatments), one within-subjects factor (time), and one interaction (Treatments × Time).
Results
Vagal recording
Baseline comparisons
There were no between group differences in vagal neural activity during the 15 min baseline period or the 15 min period that followed saline or sotalol injection. This tendency was consistent within and across each of the experimental groups. A Two-Way repeated measure ANOVA for changes in vagal neural acitivity during the 15 min drug free baseline period revealed no significant overall effects for treatment [F(6, 32) = 0.37, p = 0.89] or time [F(2, 64) = 1.42, p = 0.24] or interaction between time and treatment [F(12, 64) = 0.36, p = 0.97]. Additional factorial ANOVAs used to detect overall differences in neural activity between the seven treatment groups during each of the three 5 min bins following saline or sotalol administration also yielded no significant effects.
Post-injection within group comparisons
Figure 3 shows that over the 3 h recording period following drug treatment, groups given epinephrine injection at time period (0) and sotalol 60 min later (i.e., EPI, EPI + SOT2, and EPI + SOT4) exhibited increases in vagal firing discharge that remained statistically significant above baseline during the 10–60 min post-epinephrine injection. For the remaining time of recording, vagal activity in the EPI and EPI + SOT2 groups gradually decreased to approximately 20% above the values measured at baseline. In contrast, vagal activity in the EPI + SOT4 group dropped steeply at minute 90 (sotalol was given at 60 min after epinephrine injection), and was reduced by 50% relative to baseline at the end of recording. In addition, neural activity recorded from the vagus nerve in the SAL, SAL + SOT4, SOT2 + EPI, and SOT2 + SAL groups showed a tendency to decrease over time with a reduction rate of 50% relative to baseline at the end. A Two-Way repeated measure ANOVA indicated that drug treatment caused a main group difference [F(6, 32) = 10.21, p < 0.0001]. Additionally, time was another factor that contributed to the overall change in vagal neural activity [F(19, 608) = 42.28, p < 0.0001]. An interaction between treatment drug and time was also observed [F(114, 608) = 2.47, p < 0.0001].
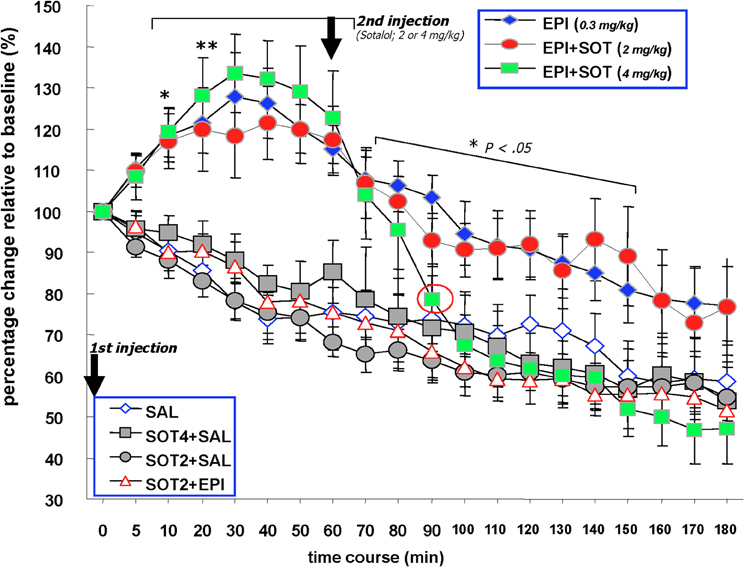
Figure 3. The effects of systemically administered saline, epinephrine (0.3 mg/kg), sotalol (2.0 or 4.0 mg/kg) or the combination of epinephrine and sotalol on vagal nerve firing in Millivolts. Animals received sotalol 15 min prior to minute (0) and epinephrine at this time point. The first injection at 0 min. Two of the epinephrine injected groups received a second injection at 60 min that consisted of sotalol at 2 mg/kg or 4 mg/kg. One of the saline injected groups received a second injection at 60 min of sotalol at 4 mg/kg. The EPI, EPI + SOT2, and EPI + SOT4 groups exhibited significant increases in vagal firing discharge above baseline for 10–60 min post-epinephrine injection. For the remaining time of recording, vagal activity in the EPI and EPI + SOT2 groups gradually decreased to approximately 20% above basal values. In contrast, vagal activity in the EPI + SOT4 group dropped steeply at 40 min post-sotalol injectionand was reduced by approximately 50% relative to baseline at the end of recording. Neural activity recorded in the form of Millivolts from the vagus nerve in the SAL, SAL + SOT4, SOT2 + EPI, and SOT2 + SAL groups decreased over time with a reduction of approximately 50% relative to baseline at the end.
Individual repeated measure ANOVAs comparing baseline levels of vagal activity with that occurring during the post-injection period also revealed significant differences across time in all seven treatment groups [EPI: F(5, 19) = 7.9, p < 0.0001, EPI + SOT2: F(5, 19) = 3.6, p < 0.0001, EPI + SOT4: F(5, 19) = 18.2, p < 0.0001, SAL: F(6, 19) = 5.9, p < 0.0001, SOT2 + SAL: F(5, 19) = 8.3, p < 0.0001, SAL + SOT4: F(3, 19) = 5.1, p < 0.0001, and SOT2 + EPI: F(3, 19) = 13.0, p < 0.0001]. With the exception of the EPI, EPI + SOT2, and EPI + SOT4 groups, the significant changes identified in this analysis were attributed to reductions in neural activity recorded from this preparation during the 180 min post-injection period relative to baseline levels. In contrast, post-hoc analysis revealed that neural activity recorded from the vagus of animals given EPI, EPI + SOT2, EPI + SOT4 increased significantly above baseline values 10 min post-injection (p < 0.05) and remained significantly elevated for each of the next five periods of data collection (i.e., from 10 to 60 min).
Between group comparisons
From 10 to 60 min post epinephrine injection, neural activity along vagal ascending fibers were significantly higher in the EPI, EPI + SOT2, and EPI + SOT4 groups than any of the SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups. A factorial ANOVA with post-hoc Fisher's tests run at each post-injection data collection point revealed significantly higher levels of vagal activity in the EPI, EPI + SOT2, EPI + SOT4 relative to groups treated with SAL, SOT2 + SAL, SOT2 + EPI, or SAL + SOT4 at each of the data collection points 10–60 minutes post-injection [10 min: F(6, 32) = 8.01, p < 0.0001; 20 min: F(6, 32) = 8.11, p < 0.0001; 30 min: F(6, 32) = 9.43, p < 0.0001; 40 min: F(6, 32) = 12.70, p < 0.0001; 50 min: F(6, 32) = 11.32, p < 0.0001; 60 min: F(6, 32) = 9.58, p < 0.0001; for post-hoc comparisons between any of EPI, EPI + SOT2, and EPI + SOT4 groups vs. any of SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups, p-values < 0.05].
Factorial ANOVAs at 80–150 min post-injection continued to reveal significant overall differences between treatment groups. Post-hoc tests indicated that during this period, the EPI group continued to show a significantly higher-level of vagal activity than any of the SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups (p < 0.05). During 110–150 min post-injection, vagal discharge of the EPI group remained higher than the SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups (p < 0.1). For the remaining recording period (i.e., 160–180 min), vagal activity in the EPI group was not different from any of the control groups.
During 70–80 min post epinephrine injection, vagal activity of the EPI + SOT4 group was not different from the EPI and EPI + SOT2 groups. During 90–150 min post-epinephrine injection, the EPI + SOT4 group showed significantly lower vagal activity compared to the EPI group (EPI vs. EPI + SOT4, p < 0.05). During 90–180 min post-epinephrine injection, the EPI + SOT4 exhibited a level of vagal activity that was not different from that of any of the control groups, i.e., SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 (p > 0.1).
Norepinephrine release in the basolateral amygdala
Baseline comparisons. As shown in Figure 4, the baseline levels of norepinephrine were similar across treatment groups during the 60 min drug free baseline period. A two-way repeated measure anova for changes in norepinephrine release for the 60 min baseline period showed no main effect by group [F(6, 31) = 0.1, p = 0.99], time [F(2, 62) = 0.4, p = 0.66], or their interaction [F(12, 62) = 0.3, p = 0.99]. individual factorial anovas at each baseline collection point did not detect any significant group effect during the drug free period.
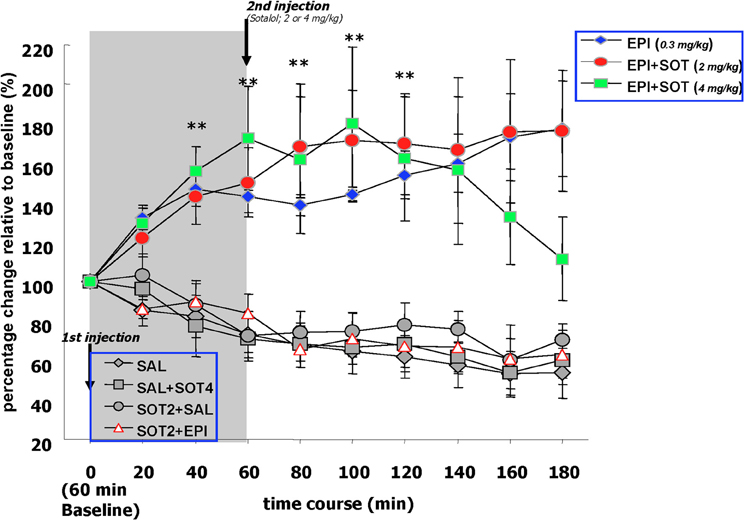
Figure 4. The effects of systemically administered saline, epinephrine (0.3 mg/kg), sotalol (2.0 or 4.0 mg/kg), or the combination of epinephrine and sotalol on norepinephrine release in the amygdala. Basal levels of norepinephrine were similar across treatment groups during the 60 min drug-free baseline period. Norepinephrine concentrations sampled from the amygdala of the EPI and EPI + SOT2 groups showed consistent and sustained increases throughout the collection period whereas the norepinephrine levels of EPI + SOT4 became comparable to control groups at 180 min post-epinephrine injection. Blocking peripheral beta-adrenergic receptors with sotalol blocked epinephrine induced augmentation of norepinephrine output in the amygdala.
Post-injection comparaisons. During the 3 h post-epinephrine collection period, norepinephrine levels in the basolateral amygdala fluctuated differently depending upon a given drug treatment. Norepinephrine concentrations sampled from the amygdala of the EPI, EPI + SOT2 and EPI + SOT4 groups showed significantly consistent and sustained increases throughout the collection period whereas the levels sampled from the control groups, (i.e., SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4) showed reductions over time that were approximately 40% lower than the baseline values. A Two-Way repeated measure ANOVA comparing norepinephrine levels over the 180 min duration indicated significant differences between the seven treatment groups [F(6, 31) = 9.65, p < 0.0001] as well as an interaction between treatment and time [F(54, 279) = 3.1, p < 0.0001].
Individual repeated measure ANOVAs comparing norepinephrine levels at each post-injection collection time point with baseline values detected that there were increases of norepinephrine release over time in the EPI, EPI + SOT2, and EPI + SOT4 groups but decreases over time in the SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups [EPI: F(9, 45) = 3.9, p < 0.001; EPI + SOT2: F(9, 36) = 3.0, p < 0.01; SAL: F(9, 54) = 8.6, p < 0.0001; SOT2 + EPI: F(9, 45) = 3.90, p < 0.01; SOT2 + SAL: F(9, 27) = 2.4, p < 0.05; SAL + SOT4: F(9, 27) = 3.8, p < 0.01]. Post-hoc analysis revealed that norepinephrine release in the EPI, and EPI + SOT2 groups increased significantly above initial baseline values at 20 min post-injection (p < 0.05) and remained significantly elevated for each of the following eight periods of data collection (i.e., from 40 to 180 min) (p < 0.05). In addition, significant changes identified in the SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups were attributed to reductions in norepinephrine release during the 180 min post-injection period relative to baseline levels.
Between group comparison. Factorial ANOVAs detected group difference at each collection time point after epinephrine injection. post-hoc tests comparing the EPI groups with the controls groups at each post-epinephrine collection point, revealed that levels of norepinephrine sampled from the amygdala were significantly higher than those of the SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups (all p-values < 0.05). the EPI + SOT2 groups showed significantly higher levels of amygdala norepinephrine than any control groups over the 180 min post-epinephrine injection (i.e., p < 0.05) vs. SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4. The EPI + SOT4 group exhibited significantly higher norepinephrine levels up to 140 min post-epinephrine injection and then declined for the remaining two periods of collection. At 180 min post-injection, the norepinephrine levels of the EPI + SOT4 group were significantly lower than those of the epi and EPI + SOT2 groups. Post-hoc tests comparing the EPI + SOT4 with other groups at each post-epinephrine collection point, revealed that the levels of norepinephrine release in the amygdala was significantly higher than the SAL, SOT2 + SAL, SOT2 + EPI, and SAL + SOT4 groups during 40–140 min post-epinephrine injection (all p-values < 0.05). After 160 minutes, norepinephrine levels in the EPI + SOT4 group declined and was not different from any of the control groups (p = 0.11). Post-hoc tests revealed no significant difference in norepinephrine levels between any control group at each of the 180 min time periods. Additionally, norepinephrine release in these groups was reduced by approximately 40% at the end of collection.
Experiment 3
The capacity for emotionally arousing events to affect central structures that process memory may require the integration of peripheral epinephrine secretion, vagal neural transmission, activation of noradrenergic neurons in the brainstem, and noradrenergic release in important limbic structures. The current study will identify which components of this system are influenced by the two mechanisms that are known to be the peripheral substrates (i.e., increased epinephrine levels and vagal firing discharge) of an arousal related system that is involved in the modulation of memory. The patterns of activation induced by either epinephrine administration or VNS will be assessed using Fos immunocytochemistry. Additionally, activation of noradrenergic cells will be examined using Fos coupled with DBH immunocytochemistry. The immediate early gene c-fos is known to play a role in transcriptional regulation that leads to molecular changes for hours to days (Chiasson et al., 1997). At cellular levels, the expression of Fos protein is rapidly induced by the activity of neurotransmitters or neuromodulators, and electrical excitation at the cell surface (Morgan and Curran, 1986). Therefore, the detection of Fos protein is often used as a neuronal maker to label cells with activity changes in response to exogenous stimuli.
Findings from the current study will reveal whether or not increased levels of epinephrine and VNS produce similar patterns of neuronal activation within memory associated areas of the brain. Furthermore, localization of activated noradrenergic neurons with those that express Fos protein will reveal the contribution of noradrenergic nuclei in driving limbic areas that respond to peripheral physiological arousal. The current study would be the first to provide a comprehensive assessment on activation patterns in memory associated structures induced by different peripheral arousal mechanisms. If the patterns of activation following the two treatments are convergent, the findings will suggest that up-regulated peripheral adrenergic activity induced by emotional arousal increases neural transmission along ascending vagal fibers that in turn, generates cellular activity within central noradrenergic systems that modulate memory formation.
Methods
Subjects
Thirty-one male Sprague-Dawley rats purchased from Charles River Laboratories weighing approximately 250–300 g were used in this study. They were singly housed, and maintained on a 12-h light–dark cycle (lights on at 6:00 a.m.) with water and food available ad libitum.
Experimental Treatments
Surgery
The procedures described in Experiment 1 to implant stimulation electrodes along the left cervical vagus were also followed for this study. After a one-week period of postoperative recovery, animals implanted with a stimulation electrode or unoperated controls were handled for 5 min on six consecutive days. On the 7th day of handling, animals in the VNS groups were stimulated in the handling room with an intensity of either 0.0 mA (sham) or 0.4 mA current (10 s on/off, six times) by connecting the vagal electrode with wires to a current source. The 0.4 mA current was selected because this level of intensity has been shown to improve retention performance in behavioral learning tasks (Clark et al., 1998, 1999). Animals in the epinephrine injection groups were handled in an identical manner but were given saline, 0.1 mg/kg, or 0.5 mg/kg epinephrine intraperitoneally. The doses were selected from those that have been shown to affect plasma epinephrine levels (McCarty and Gold, 1981) and memory performance (Gold and van Buskirk, 1975; Williams and McGaugh, 1993; Clayton and Williams, 2000).
Immunohistochemistry
Ninety minutes after VNS or epinephrine injection, each rat was deeply anesthetized with 1.3 ml/kg pentobarbital (Nembutal) and then perfused with 100 ml 0.01 M phosphate buffer saline (PBS) and then 2000 ml fresh 4% paraformaldehyde. The brain was then removed, post-fixed overnight at 4°C, stored in 30% sucrose sliced at the thickness of 50 um and stored in 0.1 M PBS with 0.1% sodium azide till the processing of immunostaining. Free floating immunostaining was started with 30 min of blocking endogenous hydrogen peroxidase by 0.3% H2O2 and 0.1% NaN3 in 0.01 M PBS. After rinses, sections were then incubated in solution with 1:1000 goat anti-rat, 1% normal goat serum (NGS), 0.5% Triton X100, 0.1% NaN3 in 0.01 M PBS for two hours at room temperature. After rinses, sections were then incubated in the primary antibody, 1:50,000 polyclonal rabbit anti-fos (ab5, Oncogene), with 1% NGS 0.5% Triton X100, 0.1% NaN3 in 0.01 M PBS for 48 h at room temperature. After rinses, the sections were incubated in solution with 1:1000 goat anti-rabbit, 1% NGS, 0.5% Triton X100, 0.1% NaN3 in 0.01 M PBS for overnight at room temperature. After rinses, sections were incubated with 1:500 avidin-biotin-peroxidase complex (ABC kit, Vector Laboratories) in 0.5% Triton X100 in 0.01 M PBS for 3–4 h at room temperature. After rinses in 0.05 M Tris buffer, all sections were simultaneously stained in 0.02% nickel-DAB (diaminobenzidine), 0.04% ammonium chloride sulfate, 0.15% nickel ammonium sulfate, 0.25% 3% H2O2 in Tris buffer for 5–10 min till the desired color, dark purple, developed. The reaction was stopped by rinsing the sections in 0.01 M PBS.
A second set of sections were used for double immunostaining of Fos and DBH. For the DBH immunostaining, the sections were processed as described above except for the primary antibody, mouse anti-DBH (Chemicon), for 36–48 h (1:30,000 mouse anti-DBH, 1% NGS, 0.5% Triton X100, 0.1% NaN3 in 0.01 M PBS), and the DAB staining [0.02% DAB (diaminobenzidine), 0.04% ammonium chloride sulfate, 0.25% 3% H2O2 in Tris buffer] for 5–10 min until the desired brown color developed. After the completion of staining, sections were mounted on gelatin coated slides, dried overnight, cleaned in 75%, 95%, 100%, 100% ethanol, and Histoclear for 5 min in each solution, and then coverslipped with DPX.
Fos quantification
The number of Fos labeled cells were quantified in six brain regions, including the shell region of the NAC, CEA, BLA, HIP (dorsal portion), LC, VLM, and NTS. Additionally, the hypoglossal nucleus and the core region of NAC were selected as control areas to ensure that Fos was not expressed nonspecifically. Images were captured using an Olympus CX41 microscope at the magnification of 40X and a Nikon Coolpix 4500 digital camera. The quantification was conducted using the Scion Image program. To obtain representative estimates of Fos counts, each image underwent standard procedures using the Scion Image features described as follows: (1) defining the boundary of each brain region with reference to the Brain Atlas (Paxinos and Watson, 1997), (2) background subtraction to equalize the brightness and contrast, (3) computing Fos positive cells in the defined area using Scion Image tools. Fos counts in each brain region were obtained from a fixed number of coronal levels, Figure 5 illustrates the specific levels counted in each of the examined brain regions.
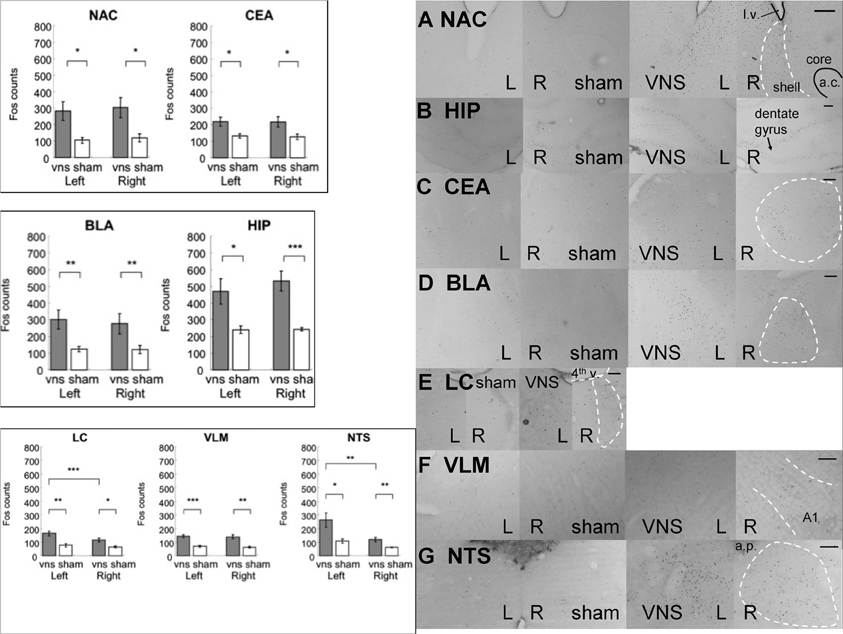
Figure 5. Electrical stimulation of the left vagus nerve significantly increased Fos-like immunoreactivity in each of the examined brain regions bilaterally: NAC, CEA, BLA, HIP, LC, VLM and NTS. In addition, the stimulation also induced an ipsilateral predominance of Fos expression in the LC and NTS. (*, **, and *** denotes significant difference between the compared groups with a p-value less than 0.05, 0.01, and 0.001, respectively). Photomicrographs of Fos labeling in the NAC (A), HIP (B), CEA (C), BLA (D), LC (E), VLM (F), and NTS (G) following sham or vagus nerve stimulation. Scale bar = 200 um. (l.v.: lateral ventricle; a.c.: anterior commisure; shell: the shell region of the nucleus accumbens; core: the core region of the nucleus accumbens; 4th v.: the fourth ventricle; a.p.: area postrema.
Double labeled (Fos+ and DBH +) cells quantification
The number of double labeled cells was quantified in three brain regions including LC, VLM, and NTS. An Olympus CX41 microscope was used to visualize and quantify double labeled cells. Two criteria were developed to designate a given neuron as a double labeled cell: (1) a solid, purple/black nucleus and (2) brown cytoplasm around the nucleus. During quantification, brightness, and contrast of each image was adjusted to an optimal level. The observer was blind to the treatment condition.
Statistical analysis
Individual ANOVAs followed by Fisher's post hoc tests were used to compare Fos expression between the treatment and control groups within each brain region following VNS or epinephrine treatment. Because stimulation of the vagus nerve was unilateral, Two-Way repeated measure ANOVAs using treatment (sham or 0.4 mA) as the between subjects factor and hemispheres (ipsilateral or contralateral to vagal stimulation) as the within subjects factor were used to analyze data from the vagus nerve treatment. For epinephrine groups, One-Way ANOVAs were used to determine differences between the two epinephrine doses and saline. Statistical significance was achieved when a level of p < 0.05 was obtained.
Results
Fos Induction in Response to Vagus Nerve Stimulation
In the control areas (the hypoglossal nucleus and the NAC core region), there were no significant differences in the expression of Fos positive cells between the VNS and sham groups [hypoglossal nucleus: F(1, 11) = 1.98, p > 0.1; NAC core: F(1, 11) = 3.42, p = 0.09]. Figure 5 shows that electrical stimulation of the left vagus nerve significantly increased Fos-like immunoreactivity in each of the following brain regions bilaterally: NAC, CEA, BLA, HIP, LC, VLM, and NTS. Individual Two-Way ANOVAs using treatment (sham, 0.4mA) as the between subjects factor and hemisphere (left vs. right) as the within subjects factor indicated significant differences in Fos expression between the sham and VNS groups (NAC: F(1, 11) = 7.29, p < 0.05; CEA: F(1, 11) = 9.36, p < 0.01; BLA: F(1, 11) = 13.77, p < 0.01; HIP: F(1, 11) = 21.88, p < 0.001; LC: F(1, 11) = 12.2, p < 0.01; VLM: F(1, 11) = 25.93, p < 0.001; NTS: F(1, 11) = 10.79, p < 0.01). Post-hoc tests comparing vagal stimulation and sham groups revealed that stimulation resulted in significantly more Fos expression within bilateral NAC (Left: p < 0.05; Right: p < 0.05), CEA (Left: p < 0.05; Right: p < 0.05), BLA (Left: p < 0.01; Right: p < 0.01), HIP (Left: p < 0.05; Right: p < 0.001), LC (Left: p < 0.01; Right: p < 0.05), VLM (Left: p < 0.001; Right: p < 0.01), and NTS (Left: p < 0.05; Right: p < 0.01) compared to sham stimulated animals.
Stimulation Side Predominance of Fos Expression
Figure 5 also illustrates that electrical stimulation of the left vagus nerve not only caused bilateral activation in the above mentioned brain regions, but also induced a ipsilateral predominance of Fos expression in the LC and NTS. Within the LC, the Two-Way ANOVA revealed a hemisphere difference [F(1, 11) = 26.48, p < 0.001] and interaction between treatment and hemisphere [F(1, 11) = 9.73, p < 0.01]. Post-hoc tests comparing the two hemispheres detected a significant predominance of Fos expression in the left over right hemisphere of the vagus nerve stimulated animals (p < 0.001) but not of the sham controls. Within the NTS, the Two-Way ANOVA indicated a hemisphere difference [F(1, 11) = 8.47, p < 0.05]. Post-hoc tests comparing the two hemispheres showed that both simulation and sham groups exhibited ipsilateral predominance of Fos expression (0.4 mA: p < 0.05; sham: p < 0.05). No other areas sampled exhibited side preferences in the expression of Fos.
Double Labeled (Fos+ /DBH+) Cells in LC, VLM, and NTS
Figure 6 shows that left VNS activated noradrenergic activity in bilateral LC, bilateral VLM, and ipsilateral NTS. The photomicrographs are shown in Figure 6. Two-Way ANOVAs indicated that significant more double labeled (Fos+/DBH+) cells were induced by VNS in the LC [F(1, 11) = 10.09, p < 0.01], VLM [F(1, 11) = 18.44, p < 0.01], and NTS [F(1, 11) = 13.43, p < 0.01]. Post-hoc tests revealed that VNS induced significant increase of the number of double labeled cells in the bilateral LC (Left: p < 0.01; Right: p < 0.05), bilateral VLM (Left: p < 0.01; Right: p < 0.001), and only left NTS (Left: p < 0.01) following left VNS.
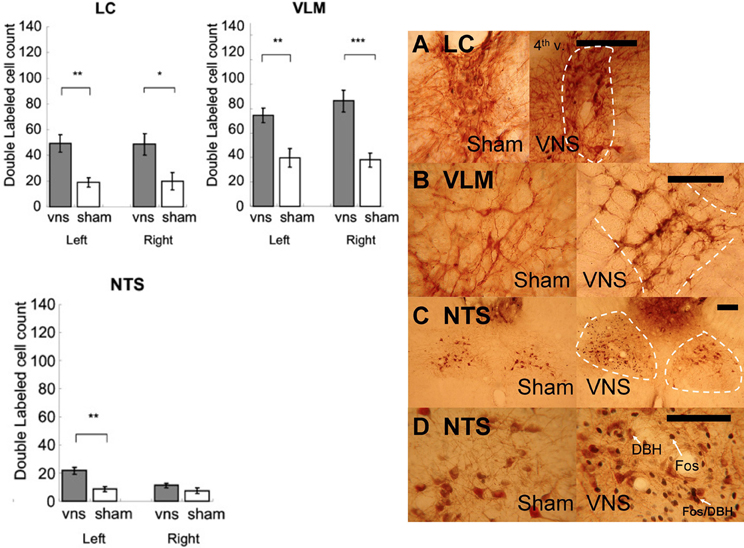
Figure 6. Left vagus nerve stimulation activated noradrenergic cells in bilateral LC, bilateral VLM, and ipsilateral NTS. (*, **, and *** denotes significant difference between the compared groups with a p-value less than 0.05, 0.01, and 0.001, respectively). Photomicrographs of Fos/DBH labeling in the LC (A), VLM (B), NTS (C,D) following sham or vagus nerve stimulation. Scale bar = 200 um. (4th v.: the fourth ventricle).
Fos Induction in the NTS in Response to Epinephrine Administration
Injection of 0.1 or 0.5 mg/kg epinephrine induced strong Fos expression along five coronal levels of the NTS. A Two-Way ANOVA, with treatment (saline, 0.1 mg/kg, 0.5 mg/kg) as the between subjects factor and five NTS coronal levels as the within subjects factor showed that systemic epinephrine injection caused significant increases in the expression of Fos-like immunoreactivity within the NTS [F(2, 15) = 9.34, p < 0.01]. Post-hoc tests found that both doses of epinephrine increased Fos expression significantly across all five levels of the NTS (all p-values < 0.05). This confirmed the effects of systemic injection of epinephrine in this study. In the control areas (the hypoglossal nucleus and the NAC core region), there were no differences in the expression of Fos positive cells between the epinephrine injected and saline groups [hypoglossal nucleus: F(2, 15) = 1.26, p > 0.1; NAC core: F(2, 15) = 1.48, p > 0.1].
Fos Induction in Memory Associated Areas Following Epinephrine Administration
Figure 7 shows that systemic administration of epinephrine induced significantly more Fos-like immunoreactivity in the following brain regions: NAC, CEA, BLA, HIP, LC, VLM, AP, and NTS. Because injection was given systemically and no hemisphere difference was found, Fos counts from both hemispheres were collapsed. Individual ANOVAs indicated significantly higher Fos expression in response to epinephrine injection (NAC [F(2, 15) = 7.12, p < 0.01], CEA [F(2, 15) = 10.73, p < 0.01], BLA [F(2, 15) = 16.69, p < 0.001], HIP [F(2, 15) = 5.19, p < 0.05], LC [F(2, 15) = 7.25, p < 0.01], VLM [F(2, 15) = 10.92, p < 0.01], and NTS [F(2, 15) = 9.34, p < 0.01]. Post-hoc tests further indicated that (1) within the NAC, both doses of epinephrine caused more Fos-like immunoreactivity than in the saline group with no significant differences between the two doses (0.1 vs. saline, p < 0.05; 0.5 vs. saline, p < 0.01), (2) within CEA and BLA, both doses of epinephrine induced more Fos expression than saline. Additionally, 0.5 mg/kg caused more Fos expression than 0.1 mg/kg (CEA: 0.5 mg/kg vs. 0.1 mg/kg, p < 0.05; 0.1 mg/kg vs. saline, p < 0.05; 0.5 mg/kg vs. saline, p < 0.001; BLA: 0.5 mg/kg vs. 0.1 mg/kg, p < 0.01; 0.1 mg/kg vs. saline, p < 0.05; 0.5 mg/kg vs. saline, p < 0.001), (3) within HIP, only 0.5 mg/kg induced more Fos-like immunoreactivity compared to saline (0.5 mg/kg vs. saline, p < 0.01), (4) within the LC, VLM, and NTS, both doses induced more Fos-like immunoreactivity compared with the saline group, with no significant difference between the two doses (LC: 0.5 mg/kg vs. saline, p < 0.01; 0.1 mg/kg vs. saline, p < 0.05; VLM: 0.5 mg/kg vs. saline, p < 0.01; 0.1 mg/kg vs. saline, p < 0.01; NTS: 0.5 mg/kg vs. saline, p < 0.01; 0.1 mg/kg vs. saline, p < 0.01).
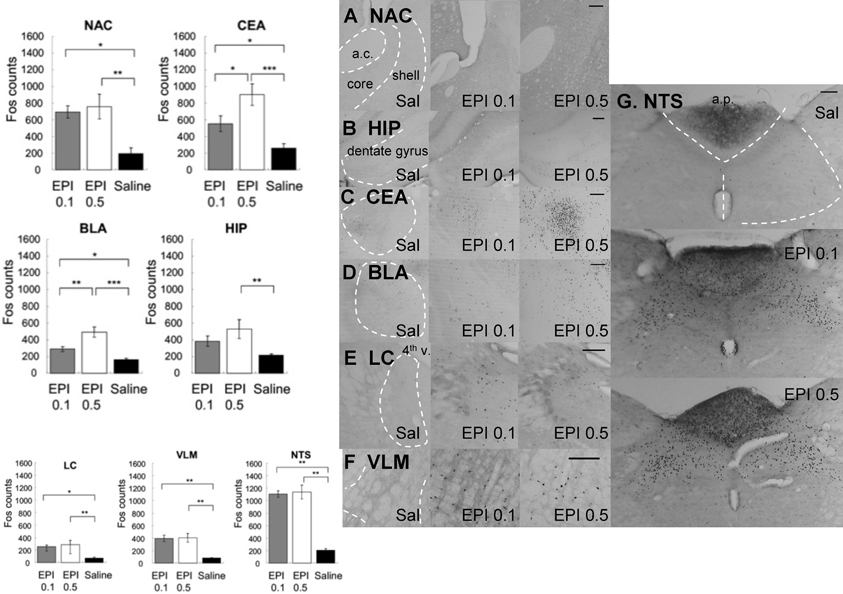
Figure 7. Systemic injection of epinephrine induced significantly more Fos-like immunoreactivity in the NAC, CEA, BLA, HIP, LC, VLM, AP, and NTS. Because injection was given systemically and no hemisphere difference was found, Fos counts from both hemisphere were collapsed. (*, **, and *** denotes significant difference between the compared groups with a p-value less than 0.05, 0.01, and 0.001, respectively). Photomicrographs of Fos labeling in the NAC (A), HIP (B), CEA (C), BLA (D), LC (E), VLM (F), and NTS (G) following saline, 0.1 mg/kg or 0.5 mg/kg epinephrine injection. Scale bar = 200 um (a.c.: anterior commisure; shell: the shell region of the nucleus accumbens; core: the core region of the nucleus accumbens; 4th v.: the fourth ventricle; a.p.: area postrema).
Double Labeled (Fos+ /DBH+) Cells in LC, VLM, and NTS
Figure 8 shows that systemic administration of epinephrine activated noradrenergic cells in the LC, VLM, and NTS. One-Way ANOVAs indicated that epinephrine injection significantly increased the number of double labeled cells in the LC [F(2, 15) = 5.28, p < 0.05], VLM [F(2, 15) = 12.29, p < 0.001], and NTS [F(2, 15) = 33.59, p < 0.0001]. Post-hoc tests showed that both doses of epinephrine induced more double labeled cells in the LC (0.1 mg/kg vs. saline, p < 0.05; 0.5 mg/kg vs. saline, p < 0.05) and VLM (0.1 mg/kg vs. saline, p < 0.001; 0.5 mg/kg vs. saline, p < 0.01). However, in the NTS, although both doses of epinephrine induced significantly more double labeled cells than saline (0.1 mg/kg vs. saline, p < 0.0001; 0.5 mg/kg vs. saline, p < 0.0001), the 0.1 mg/kg dose induced significantly more double labeled cells than 0.5 mg/kg (0.5 mg/kg vs. 0.1 mg/kg, p < 0.05).
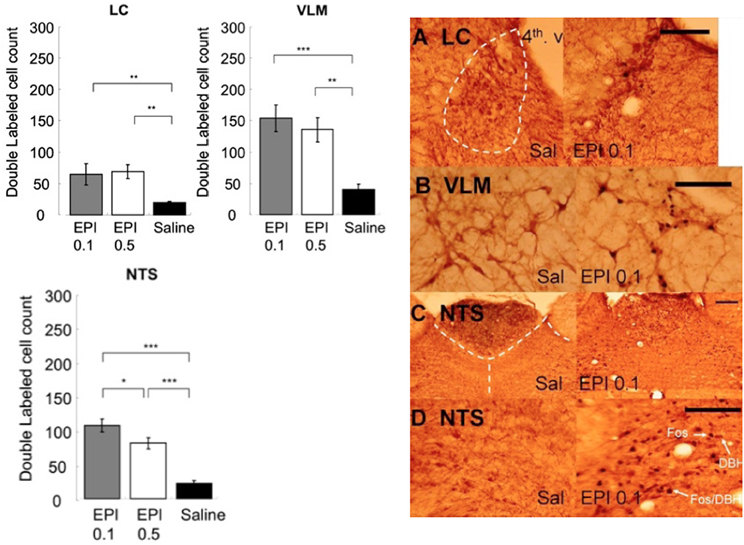
Figure 8. Systemic injection of epinephrine activated noradrenergic cells in the LC, VLM, and NTS. (*, **, and *** denotes significant difference between the compared groups with a p-value less than 0.05, 0.01, and 0.001, respectively). Photomicrographs of Fos/DBH labeling in the LC (A), VLM (B), NTS (C,D) following saline or epinephrine injection (0.1 mg/kg). Scale bar = 200 um. (4th v.: the fourth ventricle).
Double Labeled Cell Percentage of Total Fos+ Labeled Cells in LC, VLM, and NTS
Figure 9A illustrates that VNS induces noradrenergic activation that accounts for 44% of the neuronal activity in the LC, 56% in the VLM, and 8.2% in the NTS. In addition, 0.1 mg/kg epinephrine injection induces noradrenergic activation that accounts for 25% of the neuronal activity in the LC, 39% in the VLM, and 9.6% in the NTS. Moreover, 24% of the Fos activation in the LC, 33% in the VLM, and 7.1% in the NTS are attributed to activated noradrenergic cells induced by 0.5 mg/kg epinephrine.
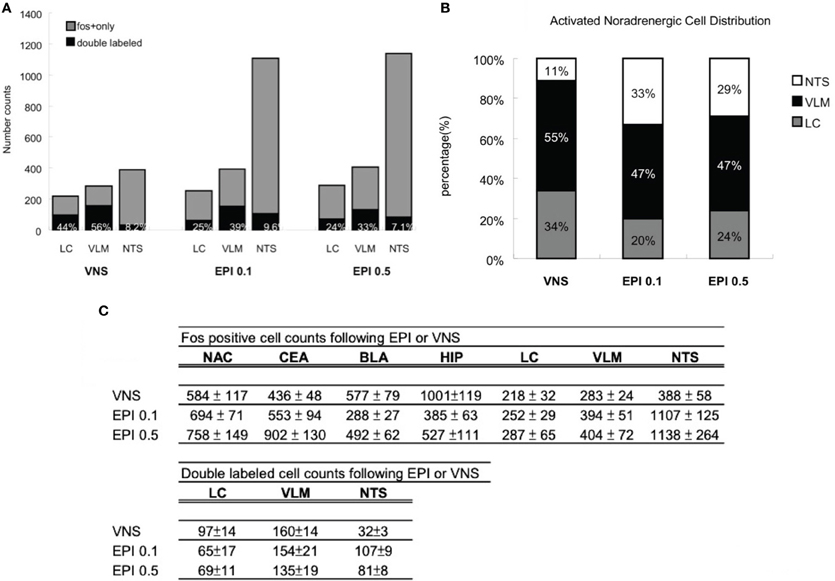
Figure 9. (A) Percentage of noradrenergic of cell activation. Vagus nerve stimulation induces noradrenergic activation that accounts for 44% of the neuronal activity in the LC, 56% in the VLM, and 8.2% in the NTS. In addition, 0.1 mg/kg epinephrine injection induces noradrenergic activation that accounts for 25% of the neuronal activity in the LC, 39% in the VLM, and 9.6% in the NTS. Moreover, 24% of the Fos activation in the LC, 33% in the VLM, and 7.1% in the NTS are attributed to activated noradrenergic cells induced by 0.5 mg/kg epinephrine. (B) Activated noradrenergic cell distribution. In the vagus nerve stimulation group, out of the total double labeled cells of the three noradrenergic nuclei, 34% was contributed by the LC, 55% by the VLM, and 11% by the NTS. In the epinephrine 0.1 mg/kg group, 20% was contributed by the LC, 47% by the VLM, and 33% by the NTS. In the epinephrine 0.5 mg/kg group, 24% was contributed by the LC, 47% by the VLM, and 29% by the NTS. (C) A numerical summary of the different activation patterns induced by vagus nerve stimulation or systemic epinephrine injection. The findings indicate that heightened levels of plasma epinephrine and increased vagal activity are capable of affecting in a similar fashion, the cellular activity of noradrenergic neurons and its output areas. Epinephrine injection activates high levels of NTS noradrenergic neurons whereas the vagus nerve stimulation recruits more LC noradrenergic activity. As a consequence, epinephrine injected animals exhibited more cell activation in NTS noradrenergic output areas (CEA, NAC) and vagus nerve stimulation animals showed more cell activation in LC noradrenergic output areas (HIP, BLA).
Percentage Distribution of Double Labeled Cells in the LC, VLM, and NTS
Figure 9B illustrates that, in the VNS group, out of the total double labeled cells of the three noradrenergic nuclei, 34% was contributed by the LC, 55% by the VLM, and 11% by the NTS. In the epinephrine 0.1 mg/kg group, 20% was contributed by the LC, 47% by the VLM, and 33% by the NTS. In the epinephrine 0.5 mg/kg group, 24% was contributed by the LC, 47% by the VLM, and 29% by the NTS. The numerical summary of the immunostaining results from the two treatment groups is given as in Figure 9C.
Discussion
Findings from this series of studies reveal an integral relationship between the arousal related hormone epinephrine and ascending fibers of the vagus nerve in influencing memory processing through interactions with central noradrenergic systems. The results from Experiment 1 demonstrated that increasing neural transmission along ascending vagal fibers following learning produces a significant and long lasting improvement in memory for emotional experiences. In the present experiment, altering the contingencies between expected rewards and experienced observations induced a level of emotional arousal in the Sham or VNS shifted groups that was reflected in increased latencies to traverse the length of the maze to receive only 1 as opposed to the expected 10 food pellets on the day of the Shift. A reduction in the expected level of reinforcement produces heightened levels of emotional frustration and changes in neuroendocrine functioning that influence the encoding of memory for these events (Crespi, 1942; Levine et al., 1972; Goldman et al., 1973). The vagus nerve plays an important role in this process by conveying to the brain, increased peripheral endocrine and autonomic activity that transpired as a result of the emotionally frustrating experience. Consequently, vagal nerve stimulation may produce beneficial effects on retention performance because increased vagal output to the brain may represent a natural consequence of elevated peripheral activity resulting from exposure to environmental events that are salient enough to induce adrenal secretion of epinephrine.
This view is supported by the finding that retention performance, as reflected by latencies to approach the food cup and consume the single food pellet reward 7 days following the Shift in reward magnitude, remained significantly longer than the No-Shift groups. In contrast, the latencies of the Sham-stimulated groups that also experienced the Shift and reduced food rewards were no different than those of No-Shift controls on this delayed retention test. Moreover, the latencies of the VNS group were also significantly longer on the 7 day delayed test than the Shifted Sham-stimulated group. These as well as other findings demonstrate that similar to epinephrine injections, VNS produces bimodal effects on retention performance. For example, whereas only moderate doses of epinephrine improve memory, higher doses of this hormone such as those that result from traumatic experiences that are not well retained in humans produce amnesia (Gold et al., 1977; Izquierdo and McGaugh, 1987). Similarly, the intensity of current applied to the vagus nerve in the current study (0.4 mA) has been shown to improve memory in one-trial avoidance tasks in rodents and declarative memory in humans (i.e., 0.5 mA) whereas higher levels of stimulation are known to produce memory impairments (Clark et al., 1998). Thus, the consequences on mnemonic processing in response to graded levels of vagal input to the brain mirror the dose-dependent inverted-U actions of peripheral hormones such as epinephrine on memory formation. Consistent with this view, our findings indicate that up-regulation of vagal neural activity improves memory formation as evidenced by the enhanced retention performance observed in the vagal stimulated groups up to seven days after learning.
Increasing impulse flow from the vagus nerve to the brain is an effective method of influencing brain processes that not only encode memory for experiences that are emotionally arousing but this treatment has also been shown to improve both recognition and recall performance in humans trained on serial word lists tasks (Clark et al., 1999; Ghacibeh et al., 2006), increase non-associative learning in animals (Wang et al., 2005) and to facilitate attention and alertness in children implanted with vagal electrodes (Kossoff and Pyzik, 2004). The findings from the current study corroborate previous reports and extend these results by revealing that the capacity for vagal input to influence mnemonic processing is not limited to learning conditions involving noxious stimuli but activity along this nerve is an essential component of the processes involved in modulating a broad range of new emotional learning experiences into long term memory.
The results from Experiment 2 demonstrated that epinephrine increases firing discharge along afferent fibers of the vagus nerve and this change persisted for up to one hour post-injection. The epinephrine-induced increase in vagal firing was accompanied by significant elevations in extracellular concentrations of norepinephrine in the basolateral amygdala that remained high for up to 120 min post-injection. Additionally, the results suggest that the excitatory actions of epinephrine on vagal nerve discharge are mediated through influences on β-adrenergic receptors. The β-adrenergic blocker sotalol (2 mg/kg) given with epinephrine was effective in preventing epinephrine related changes in vagal activity and also blocked the capacity for this hormone to produce in appreciable changes in amygdala norepinephrine release. However, peripheral adrenergic blockade with sotalol was delayed to 60 min post-epinephrine injection, only the higher dose (4 mg/kg in the EPI + SOT4 group) was effective in reversing the effects of epinephrine on vagal nerve discharge. But the counteracting effects were not present immediately. That is, epinephrine-induced vagal activity and amygdala norepinephrine levels were not reduced to control levels until 40 and 120 min, respectively post-sotalol injection. Furthermore, in groups where no increases in vagal discharge were observed, no increases in norepinephrine release were detected in the basolateral amygdala. For convenience, these groups (SAL, SAL + SOT4, SOT2 + SAL, SOT2 + EPI) were referred to as the control groups. Collectively, our results indicate that changes in vagal discharge and amygdala norepinephrine release are mediated in part by β-adrenergic receptors in response to the arousal related hormone epinephrine. Once adrenergic activity is enhanced by epinephrine, it requires a higher dose of an adrenergic antagonist to counteract epinephrine effects.
Epinephrine vs. Saline
Findings from the current study show that a single injection of the hormone at a dose of 0.3 mg/kg significantly increased neural impulses propagated along ascending vagal fibers for approximately 50 min. The magnitude of the increase peaked at 30% above baseline 30 min post-epinephrine injection. The concomitant microdialysis data show that peripheral epinephrine elevates noradrenergic output in the basolateral amygdala in a pattern that is similar to the changes this hormone induces initially on the firing rates of vagal fibers. Unlike vagal activity however, amygdala norepinephrine release induced by epinephrine remained elevated for the entire collection period (i.e., 180 min). Norepinephrine levels were 44% higher than baseline at 40 min post injection, 77% higher at 180 min post injection indicating an increasing elevation of central noradrenergic activity in response to peripheral epinephrine. Consequently, we hypothesize that the role of ascending vagal fibers may play an integral role in initiating, as opposed to maintaining brain noradrenergic activation in response to increased peripheral adrenergic activity. Therefore, central noradrenergic activation, induced by systemic epinephrine injection may be initiated by increased neural impulses carried by ascending vagal fibers and then maintained by other mechanisms.
Neural activity propagated along vagal afferent fibers may recruit additional mechanisms that affect long-term changes in CNS functioning. In fact, cAMP has been reported to accumulate in the vagus nerve following incubation in a medium containing epinephrine (Roch and Salamin, 1977). The change in vagal concentrations of cAMP has been reported to increase as much as 10–50-fold following incubation in solutions containing the β adrenergic agonist isopreteronol (Schreurs et al., 1986). Given the importance of cAMP as an important second messenger essential for ligand gated neurotransmission, the accumulated cAMP molecules might be trafficked to areas that undergo cAMP-PKA-dependent transmission, such as the central nervous system. It is known that vagal afferents influence the level of cAMP in central sites of termination such as the NTS, which then determine the state of activation in post-synaptic NTS neurons (Browning and Travagli, 2006). Noradrenergic cells are one major type of post-synaptic NTS neurons. Given the fact that noradrenergic neurotransmission is mostly cAMP PKA-dependent, accumulated cAMP in the vagus nerve following epinephrine treatment might be utilized to promote noradrenergic activity in the NTS. In fact, in severely stressful conditions that are known to enhance peripheral adrenergic activity, NTS neurons are up-regulated with PKA, Fos, and tyrosine hydroxylase immunoreactivity (Benavides et al., 2005). This is consistent with the finding that noradrenergic neurons are activated in response to increased vagal activity or peripheral adrenergic activity. With the activation of NTS noradrenergic neurons, norepinephrine release in the basolateral amygdala can then be initiated and maintained.
Findings emerging from the final study reveal that systemic epinephrine injection and VNS induce similar patterns of neuronal activation within brain regions associated with memory. Our findings indicate that treatments that raise circulating levels of epinephrine in the periphery or increase discharge along ascending fibers of the vagus nerve produce similar changes in the expression of Fos proteins in noradrenergic neurons in the NTS, LC, VLM, and their areas of termination. The results show that central noradrenergic nuclei represent a primary target of peripheral systems that are engaged by exposure to emotionally arousing events. In turn, activation of noradrenergic release in each of the limbic circuits identified in this study constitutes areas wherein the beneficial effects of heightened levels of arousal in the periphery influence the effective storage of events into memory.
When comparing the number of Fos expressing cells observed in response to either vagal stimulation or epinephrine injections, the two doses of epinephrine recruited nearly three times as many Fos neurons as that produced by vagal stimulation (EPI 0.1 mg/kg: 1107 ± 125; EPI 0.5 mg/kg: 1128 ± 264; VNS: 388.58 ± 58). The difference in treatments most likely accounts for this observation. For example, in contrast to the unilateral stimulation of the vagus nerve, both vagal nerves are most likely activated in response to the systemic epinephrine injections and the observed effects may reflect the central consequences of increasing discharge along both vagi.
Even though NTS neurons were activated to a different degree by the separate treatments, the patterns of increased activation within other more rostral structures were uniformly bilateral. Systemic injection of epinephrine produced equivalent activation in both hemispheres within all of the examined areas, i.e., NTS, VLM, LC, BLA, CEA, HIP, and NAC. Similar findings were observed after vagal stimulation, with the exception of a clear ipsilateral predominance of Fos expression in the NTS and LC. Consistent with previous studies (Gieroba and Blessing, 1994; Osharina et al., 2006), this observation suggests that the propagation of ascending information from the vagus bifurcates at the level of the NTS and LC. Vagal afferents first decussate at the spinal trigeminal tract before entering the NTS (Kalia and Mesulam, 1980; Ranson et al., 1993; Norgren and Smith, 1994). NTS neurons then project to a wide range of medullary and pontine areas bilaterally (Otake et al., 1992). Based upon these observations, VLM projecting neurons in the NTS send out nearly equivalent innervations to both hemispheres whereas LC projecting neurons show an ipsilateral predominance. LC neurons subsequently project to several forebrain areas bilaterally with an ipsilateral predominance (Jones and Moore, 1977; Espana and Berridge, 2006).
Animal studies that investigated the effects of VNS on forebrain areas are relatively sparse. According to currently available evidence, noradrenergic output is up-regulated in the amygdala (Hassert et al., 2004), hippocampus, and cortex (Roosevelt et al., 2006). Additionally, the Roosevelt et al. study (2006) reported a bilateral activation pattern following VNS at an intensity of 0.5 mA in the hippocampus, or of 1.0 mA in both the hippocampus and cortex. These findings are consistent with the Fos activation patterns obtained in our study. That is, we observed equivalent increases in both hemispheres following VNS at an intensity of 0.4 mA in noradrenergic output areas, including NAC, CEA, BLA, and HIP.
The findings from neuroimaging studies that investigated the clinical therapeutic effects of VNS in treating epilepsy and depression are generally congruent with our findings. Significant increases in cerebral blood flow after acute left vagal stimulation was noted bilaterally in the hippocampus, amygdala, orbitofrontal gyri, cingulate, thalami, hypothalami, and the anterior insula (Henry et al., 1998). However, changes observed after vagal stimulation in a fMRI study of patients with intractable depression involved increased blood oxygenation level in bilateral orbitofrontal and parieto-occipital cortex, left temporal cortex, amygdala, and the hypothalamus (Bohning et al., 2001). In patients with refractory epilepsy, VNS-induced activation was detected in bilateral thalamus, insular cortices, postcentral gyri and inferomedial occipital gyri, left basal ganglia, and right posterior superior temporal gyrus (Narayanan et al., 2002). Although side predominance was observed in some of the neuroimaging studies, neuroimaging evidence is still in agreement with our findings that VNS induces equivalently bilateral activation in brain areas that are secondary to the stimulation site, i.e., limbic areas in our study.
Noradrenergic Activation Patterns
Although the activation patterns (indicated by Fos data) within the lower brain regions appear similar following the two treatments, double labeling data found that each of the noradrenergic nuclei may have different roles in enhancing central noradrenergic activity. That is, VNS induces a higher percentage of noradrenergic activation in the LC (44%) and VLM (56%) than epinephrine injection whereas epinephrine injection recruits a higher percentage (9.6% by 0.1 mg/kg) of noradrenergic activation in the NTS. The contribution of each nucleus is best shown by the distribution of activated noradrenergic (double labeled) neurons within the three noradrenergic regions. Results indicate that the LC noradrenergic neurons are more involved in the VNS activation whereas the NTS is more important in the epinephrine injection activation. VLM A1 cells, however, account for nearly half of the noradrenergic activation induced by either treatment, with a higher predominance following VNS.
In conclusion, the collective findings demonstrate an integral role of epinephrine and the vagus nerve in influencing central noradrenergic systems ascribed a role in processing memory for emotionally arousing experiences. The findings also illustrate how interactions between these two peripheral endocrine and autonomic systems serve an important function in elevating and maintaining heightened levels of central activity in response to emotionally salient events. This comprehensive analysis of vagal nerve functioning in memory, in regulating norepinephrine output in the amygdala and in influence activity in norepinephrine containing cell bodies in the CNS reveals the important contribution of this nerve in facilitating “peripheral to central” communication during critical states of emotional arousal.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Benavides, M., Laorden, M. L., and Milanes, M. V. (2005). Involvement of 3′, 5′-cyclic adenosine monophosphate-dependent protein kinase in regulation of Fos expression and tyrosine hydroxylase levels during morphine withdrawal in the hypothalamic paraventricular nucleus and medulla oblongata catecholaminergic cell groups. J. Neurochem. 92, 246–254.
Bjorklund, A., and Lindvall, O. (1986). “Catecholaminergic brain stem regulatory systems,” in Handbook of Physiology, A Critical, Comprehensive Presentation of Physiological Knowledge and Concepts, Section, I, The Nervous System, ed V. B. Mountcastle (Bethesda: American Physiological Society), 155–235.
Bohning, D. E., Lomarev, M. P., Denslow, S., Nahas, Z., Shastri, A., and George, M. S. (2001). Feasibility of vagus nerve stimulation-synchronized blood oxygenation level-dependent functional MRI. Invest. Radiol. 36, 470–479.
Browning, K. N., and Travagli, R. A. (2006). Short-term receptor trafficking in the dorsal vagal complex: an overview. Auton. Neurosci. 126–127, 2–8.
Cahill, L., and McGaugh, J. L. (1991). Amygdaloid complex lesions differentially affect retention of tasks using appetitive and aversive reinforcement. Behav. Neurosci. 104, 532–543.
Cahill, L., and McGaugh, J. L. (1995). A novel demonstration of enhanced memory associated with emotional arousal. Conscious. Cogn. 4, 410–421.
Canli, T., Desmond, J. E., Zhao, Z., and Gabrieli, J. D. (2002). Sex differences in the neural basis of emotional memories. Proc. Natl. Acad. Sci. U.S.A. 99, 10789–10794.
Carrive, P. (2006). Dual activation of cardiac sympathetic and parasympathetic components during conditioned fear to context in the rat. Clin. Exp. Pharmacol. Physiol. 33, 1251–1254.
Chang, C. H., Liang, K. C., and Yen, C. T. (2005). Inhibitory avoidance learning altered ensemble activity of amygdaloid neurons in rats. Eur. J. Neurosci. 21, 210–218.
Chiasson, B. J., Hong, M. G., and Robertson, H. A. (1997). Putative roles for the inducible transcription factor c-fos in the central nervous system: studies with antisense oligonucleotides. Neurochem. Int. 31, 459–475.
Clark, K. B., Naritoku, D. K., Smith, D. C., Browning, R. A., and Jensen, R. A. (1999). Enhanced recognition memory following vagus nerve stimulation in human subjects. Nat. Neurosci. 2, 94–98.
Clark, K. B., Smith, D. C., Hassert, D. L., Browning, R. A., Naritoku, D. K., and Jensen, R. A. (1998). Posttraining electrical stimulation of vagal afferents with concomitant vagal efferent inactivation enhances memory storage processes in the rat. Neurobiol. Learn. Mem. 70, 364–373.
Clayton, E. C., and Williams, C. L. (2000). Noradrenergic receptor blockade of the NTS attenuates the mnemonic effects of epinephrine in an appetitive light-dark discrimination learning task. Neurobiol. Learn. Mem. 74, 135–145.
Coupland, R. E., Parker, T. L., Kesse, W. K., and Mohamed, A. A. (1989). The innervation of the adrenal gland. III. Vagal innervation. J. Anat. 163, 173–181.
Crespi, L. P. (1942). Quantitative variation in incentive and performance in the white rat. Am. J. Psychol. 40, 467–517.
de Boer, S. F., de Beun, R., Slangen, J. L., and van der Gugten, J. (1990). Dynamics of plasma catecholamine and corticosterone concentrations during reinforced and extinguished operant behavior in rats. Physiol. Behav. 47, 691–698.
Espana, R. A., and Berridge, C. W. (2006). Organization of noradrenergic efferents to arousal-related basal forebrain structures. J. Comp. Neurol. 496, 668–683.
Fallon, J. H., Koziell, D. A., and Moore, R. Y. (1978). Catecholamine innervation of the basal forebrain. II., Amygdala, suprarhinal cortex and entorhinal cortex. J. Comp. Neurol. 180, 509–532.
Feldman, S., Conforti, N., and Weidenfeld, J. (1995). Limbic pathways and hypothalamic neurotransmitters mediating adrenocortical responses to neural stimuli. Neurosci. Biobehav. Rev. 19, 235–240.
Ferry, B., Roozendaal, B., and McGaugh, J. L. (1999). Role of norepinephrine in mediating stress hormone regulation of long-term memory storage: a critical involvement of the amygdala. Biol. Psychiatry 46, 1140–1152.
Gabriel, M., and Seller, H. (1970). Interaction of baroreceptor afferents from carotid sinus and aorta at the nucleus tractus solitarii. Pflügers Arch. 318, 7–20.
Gerra, G., Fertomani, G., Zaimovic, A., Caccavari, R., Reali, N., Maestri, D., Avanzini, P., Monica, C., Delsignore, R., and Brambilla, F. (1996). Neuroendocrine responses to emotional arousal in normal women. Neuropsychobiology 33, 173–181.
Ghacibeh, G. A., Shenker, J. I., Shenal, B., Uthman, B. M., and Heilman, K. M. (2006). The influence of vagus nerve stimulation on memory. Cogn. Behav. Neurol. 19, 119–122.
Gieroba, Z. J., and Blessing, W. W. (1994). Fos-containing neurons in medulla and pons after unilateral stimulation of the afferent abdominal vagus in conscious rabbits. Neuroscience 59, 851–858.
Gold, P. E., and van Buskirk, R. B. (1975). Facilitation of time-dependent memory processes with posttrial epinephrine injections. Behav. Biol. 13, 145–153.
Gold, P. E., and van Buskirk, R. B. (1978). Effects of alpha and beta adrenergic receptor antagonists on post-trial epinephrine modulation of memory: relationship to posttraining brain norepinephrine concentrations. Behav. Biol. 24, 168–184.
Gold, P. E., van Buskirk, R., and Haycock, J. W. (1977). Effects of posttraining epinephrine injections on retention of avoidance training in mice. Behav. Biol. 20, 197–204.
Goldman, L., Coover, G. O., and Levine, S. (1973). Bidirectional effects of reinforcement shifts on pituitary adrenal activity. Physiol. Behav. 10, 209–214.
Groenewegen, H. J., and Uylings, H. B. (2000). The prefrontal cortex and the integration of sensory, limbic and autonomic information. Prog. Brain Res. 126, 3–28.
Hassert, D., Miyashita, T., and Williams, C. L. (2004). The effects of peripheral vagal nerve stimulation at a memory modulating intensity on norepinephrine output in the basolateral amygdala. Behav. Neurosci. 118, 79–88.
Hatfield, T., and McGaugh, J. L. (1999). Norepinephrine infused into the basolateral amygdala posttraining enhances retention in a spatial water maze task. Neurobiol. Learn. Mem. 71, 232–239.
Henry, T. R., Bakay, R. A., Votaw, J. R., Pennell, P. B., Epstein, C. M., Faber, T. L., Grafton, S. T., and Hoffman, J. M. (1998). Brain blood flow alterations induced by therapeutic vagus nerve stimulation in partial epilepsy: I. Acute effects at high and low levels of stimulation. Epilepsia 39, 983–990.
Huff, N. C., Wright-Hardesty, K. J., Higgins, E. A., Matus-Amat, P., and Rudy, J. W. (2006). Amygdala regulation of immediate-early gene expression in the hippocampus induced by contextual fear conditioning. J. Neurosci. 26, 1616–1623.
Hurlemann, R., Hawellek, B., Matusch, A., Kolshc, H., Wollersen, H., Madea, B., Vogeley, K., Maier, W., and Dolan, R. J. (2005). Noradrenergic modulation of emotion-induced forgetting and remembering. J. Neurosci. 25, 6343–6349.
Irwin, J., Ahluwalia, P., Zacharko, R. M., and Anisman, H. (1986). Central norepinephrine and plasma corticosterone following acute and chronic stressors: influence of social isolation and handling. Pharmacol. Biochem. Behav. 24, 1151–1154.
Izquierdo, I., and McGaugh, J. L. (1987). Retention impairment by posttraining epinephrine: role of state dependency and of endogenous opioid mechanisms. Behav. Neurosci. 101, 778–781.
Jones, B. E., and Moore, R. Y. (1977). Ascending projections of the LC in the rat. II. Autoradiographic study. Brain Res. 127, 25–53.
Kalia, M., and Mesulam, M. M. (1980). Brain stem projections of sensory and motor components of the vagus complex in the cat: I. The cervical vagus and nodose ganglion. J. Comp. Neurol. 193, 435–465.
King, S. O., and Williams, C. L. (2009). Novelty-induced arousal enhances memory for cued classical fear conditioning: interactions between peripheral adrenergic and brainstem glutamatergic systems. Learn. Mem. 16, 625–634.
Kossoff, E. H., and Pyzik, P. L. (2004). Improvement in alertness and behavior in children treated with combination topiramate and vagus nerve stimulation. Epilepsy behav. 5, 256–259.
LaLumiere, R. T., Buen, T. V., and McGaugh, J. L. (2003). Postraining intra-basolateral amygdala infusions of norepinephrine enhance consolidation of memory for contextual fear conditioning. J. Neurosci. 23, 6754–6758.
Lawrence, A. J., Watkins, D., and Jarrott, B. (1995). Visualization of beta-adrenoceptor binding sites on human inferior vagal ganglia and their axonal transport along the rat vagus nerve. J. Hypertens. 13, 631–635.
Levine, S., Goldman, L., and Coover, G. D. (1972). Expectancy and the pituitary-adrenal system. Ciba Found. Symp. 8, 281–291.
Liang, K. C., Chen, L. L., and Huang, T. E. (1995). The role of amygdala norepinephrine in memory formation: involvement in the memory enhancing effect of peripheral epinephrine. Chin. J. Physiol. 38, 81–91.
Liang, K. C., and McGaugh, J. L. (1983). Lesions of the stria terminalis attenuate the enhancing effect of post-training epinephrine on retention of an inhibitory avoidance response. Behav. Brain Res. 9, 49–58.
Liang, K. C., Juler, R. G., and McGaugh, J. L. (1986). Modulating effects of posttraining epinephrine on memory: involvement of the amygdala noradrenergic system. Brain Res. 368, 125–133.
Liang, K. C., McGaugh, J. L., and Yao, H. Y. (1990). Involvement of amygdala pathways in the influence of post-training intra-amygdala norepinephrine, and peripheral epinephrine on memory storage. Brain Res. 508, 225–233.
McAllen, R. M., May, C. N., and Shafton, A. D. (1995). Functional anatomy of sympathetic premotor cell groups in the medulla. Clin. Exp. Hypertens. 17, 209–221.
McCarty, R., and Gold, P. E. (1981). Plasma catecholamines: effects of footshock level and hormonal modulators of memory storage. Horm. Behav. 15, 168–182.
Miyashita, T., and Williams, C. L. (2006). Epinephrine administration increases neural impulses propagated along the vagus nerve: role of peripheral beta-adrenergic receptors. Neurobiol. Learn. Mem. 85, 116–124.
Morgan, J. I., and Curran, T. (1986). Role of ion flux in the control of c-fos expression. Nature 322, 552–555.
Narayanan, J. T., Watts, R., Haddad, N., Labar, D. R., Li, P. M., and Filippi, C. G. (2002). Cerebral activation during vagus nerve stimulation: a functional MR study. Epilepsia 43, 1509–1514.
Naritoku, D. K., Terry, W. J., and Helfert, R. H. (1995). Regional induction of fos immunoreactivity in the brain by anticonvulsant stimulation of the vagus nerve. Epilepsy Res. 22, 53–62.
Niijima, A. (1992). Electrophysiological study on the vagal innervation of the adrenal gland in the rat. J. Auton. Nerv. Syst. 41, 87–92.
Norgren, R., and Smith, G. P. (1994). A method for selective section of vagal afferent or efferent axons in the rat. Am. J. Physiol. 267, R1136–R1141.
O'Carroll, R. E., Drysdale, E., Cahill, L., Shajahan, P., and Ebmeier, K. P. (1999). Stimulation of the noradrenergic system enhances and blockade reduces memory for emotional material in man. Psychol. Med. 29, 1083–1088.
Osharina, V., Bagaev, V., Wallois, F., and Larnicol, N. (2006). Autonomic response and Fos expression in the NTS following intermittent vagal stimulation: importance of pulse frequency. Auton. Neurosci. 126–127, 72–80.
Otake, K., Ezure, K., Lipski, J., and Wong She, R. B. (1992). Projections from the commissural subnucleus of the nucleus of the solitary tract: an anterograde tracing study in the cat. J. Comp. Neurol. 324, 365–378.
Packard, M. G., and McGaugh, J. L. (1992). Double dissociation of fornix and caudate nucleus lesions on acquisition of two water maze tasks: further evidence for multiple memory systems. Behav. Neurosci. 106, 439–446.
Paton, J. F. (1998a). Convergence properties of solitary tract neurones driven synaptically by cardiac vagal afferents in the mouse. J. physiol. 508(Pt 1), 237–252.
Paton, J. F. (1998b). Pattern of cardiorespiratory afferent convergence to solitary tract neurons driven by pulmonary vagal C-fiber stimulation in the mouse. J. Neurophysiol. 79, 2365–2373.
Paxinos, G., and Watson, C. (1986). The Rat Brain in Stereotaxic Coordinates, 2nd Edn. New York, NY: Academic Press.
Paxinos, G., and Watson, C. (1997). The Rat Brain in Stereotaxic Coordinates, 2nd Edn. San Diego, CA: Academic Press.
Pelletier, J. G., Likhtik, E., Filali, M., and Pare, D. (2005). Lasting increases in basolateral amygdala activity after emotional arousal: implications for facilitated consolidation of emotional memories. Learn. Mem. 12, 96–102.
Peper, M., Herpers, M., Spreer, J., Hennig, J., and Zentner, J. (2006). Functional neuroimaging of emotional learning and autonomic reactions. J. Physiol. Paris 99, 342–354.
Petrov, T., Krukoff, T. L., and Jhamandas, J. H. (1993). Branching projections of catecholaminergic brainstem neurons to the paraventricular hypothalamic nucleus and the central nucleus of the amygdala in the rat. Brain Res. 609, 81–92.
Quirarte, G. L., Roozendaal, B., and McGaugh, J. L. (1997). Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 94, 14048–14053.
Ranson, R. N., Butler, P. J., and Taylor, E. W. (1993). The central localization of the vagus nerve in the ferret (Mustela putorius furo) and the mink (Mustela vison). J. Auton. Nerv. Syst. 43, 123–137.
Riche, D., de Pommery, J., and Menetrey, D. (1990). Neuropeptides and catecholamines in efferent projections of the nuclei of the solitary tract in the rat. J. Comp. Neurol. 293, 399–424.
Roch, P., and Salamin, A. (1977). Effect of beta-adrenergic drugs on adenosine 3′, 5′-monophosphate in rabbit vagus nerve. J. Neurochem. 28, 947–950.
Roosevelt, R. W., Smith, D. C., Clough, R. W., Jensen, R. A., and Browning, R. A. (2006). Increased extracellular concentrations of norepinephrine in cortex and HIP following vagus nerve stimulation in the rat. Brain Res. 1119, 124–132.
Roozendaal, B., and McGaugh, J. L. (1997). Glucocorticoid receptor agonist and antagonist administration into the basolateral but not central amygdala modulates memory storage. Neurobiol. Learn. Mem. 67, 176–179.
Roozendaal, B., Quirarte, G. L., and McGaugh, J. L. (2002). Glucocorticoids interact with the basolateral amygdala beta-adrenoceptor–cAMP/cAMP/PKA system in influencing memory consolidation. Eur. J. Neurosci. 15, 553–560.
Roozendaal, B., and McGaugh, J. L. (1996). Amygdaloid nuclei lesions differentially affect glucocorticoid-induced memory enhancement in an inhibitory avoidance task. Neurobiol. Learn. Mem. 65, 1–8.
Salinas, J. A., Williams, C. L., and McGaugh, J. L. (1996). Peripheral post-training administration of 4-OH amphetamine enhances retention of a reduction in reward magnitude. Neurobiol. Learn. Mem. 65, 192–195.
Sawchenko, P. E., Li, H. Y., and Ericsson, A. (2000). Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog. Brain Res. 122, 61–78.
Schneider, F., Grodd, W., Weiss, U., Klose, U., Mayer, K. R., Nägele, T., and Gur, R. C. (1997). Functional MRI reveals left amygdala activation during emotion. Psychiatry Res. 76, 75–82.
Schreurs, J., Seelig, T., and Schulman, H. (1986). Beta 2-adrenergic receptors on peripheral nerves. J. Neurochem. 46, 294–296.
Shapiro, R. E., and Miselis, R. R. (1985). The central organization of the vagus nerve innervating the stomach of the rat. J. Comp. Neurol. 238, 473–488.
Sumal, K. K., Blessing, W. W., Joh, T. H., Reis, D. J., and Pickel, V. M. (1983). Synaptic interaction of vagal afferents and catecholaminergic neurons in the rat nucleus tractus solitarius. Brain Res. 277, 31–40.
Torres, C., Candido, A., Escarabajal, M. D., de la Torre, L., Maldonado, A., Tobena, A., and Fernandez-Teruel, A. (2005). Successive negative contrast in one-way avoidance learning in female Roman rats. Physiol. Behav. 85, 377–382.
Van Bockstaele, E. J., Peoples, J., and Telegan, P. (1999). Efferent projections of the nucleus of the solitary tract to perio-locus coeruleus dendrites in the rat brain: evidence for a monosynaptic pathway. J. Comp. Neurol. 412, 410–428.
van den Buuse, M. (2002). Effect of atropine or atenolol on cardiovascular responses to novelty stress in freely-moving rats. Stress 5, 227–231.
van den Buuse, M., Van Acker, S. A., Fluttert, M., and De Kloet, E. R. (2001). Blood pressure, heart rate, and behavioral responses to psychological “novelty” stress in freely moving rats. Psychophysiology 38, 490–499.
Wang, G. M., Song, G., and Zhang, H. (2005). Phenomenon of non-associative learning in Hering-Breuer reflex simulated by electrical vagal stimulation in rabbits. Sheng Li Xue Bao 57, 511–516.
Weidenfeld, J., Itzik, A., and Feldman, S. (1997). Effect of glucocorticoids on the adrenocortical axis responses to electrical stimulation of the amygdala and the ventral noradrenergic bundle. Brain Res. 754, 187–194.
Weidenfeld, J., Newman, M. E., Itzik, A., and Feldman, S. (2005). Adrenocortical axis responses to adrenergic and glutamate stimulation are regulated by the amygdala. Neuroreport 16, 1245–1249.
Weil-Malherbe, H., Axelrod, J., and Tomchick, R. (1959). Blood-brain barrier for adrenaline. Science 129, 1226–1227.
Williams, C. L., and McGaugh, J. L. (1993). Reversible lesions of the nucleus of the solitary tract attenuate the memory-enhancing effects posttraining epinephrine. Behav. Neurosci. 107, 955–962.
Williams, C. L., Men, D., and Clayton, E. C. (2000). The effects of noradrenergic activation of the nucleus tractus solitarius on memory and in potentiating norepinephrine release in the amygdala. Behav. Neurosci. 114, 1131–1144.
Williams, C. L., Men, D., Clayton, E. C., and Gold, P. E. (1998). Norepinephrine release in the amygdala after systemic injection of epinephrine or escapable footshock: contribution of the nucleus of the solitary tract. Behav. Neurosci. 112, 1414–1422.
Woodson, J. C., Macintosh, D., Fleshner, M., and Diamond, D. M. (2003). Emotion-induced amnesia in rats: working memory-specific impairment, corticosterone-memory correlation, and fear versus arousal effects on memory. Learn. Mem. 10, 326–336.
Keywords: amygdala, emotional arousal, epinephrine, learning, memory, vagus nerve
Citation: Chen CC and Williams CL (2012) Interactions between epinephrine, ascending vagal fibers, and central noradrenergic systems in modulating memory for emotionally arousing events. Front. Behav. Neurosci. 6:35. doi: 10.3389/fnbeh.2012.00035
Received: 14 March 2012; Paper pending published: 23 April 2012;
Accepted: 12 June 2012; Published online: 28 June 2012.
Edited by:
Antonella Gasbarri, University of L'Aquila, ItalyReviewed by:
Claude Messier, University of Ottawa, CanadaEdwin J. Barea, University of Texas at San Antonio, USA
Copyright: © 2012 Chen and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: C. L. Williams, Department of Psychology, The University of Virginia, 102 Gilmer Hall, P.O. Box 400400 Charlottesville, VA 22904-4400, USA. e-mail:Y2x3M2JAdmlyZ2luaWEuZWR1