

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Behav. Neurosci. , 08 November 2011
Sec. Learning and Memory
volume 5 - 2011 | https://doi.org/10.3389/fnbeh.2011.00076
This article is part of the Research Topic Neuronal cell signaling and behavior View all 14 articles
In the last decade, a plethora of studies utilizing pharmacological, biochemical, and genetic approaches have shown that precise translational control is required for long-lasting synaptic plasticity and the formation of long-term memory. Moreover, more recent studies indicate that alterations in translational control are a common pathophysiological feature of human neurological disorders, including developmental disorders, neuropsychiatric disorders, and neurodegenerative diseases. Finally, translational control mechanisms are susceptible to modification by psychoactive drugs. Taken together, these findings point to a central role for translational control in the regulation of synaptic function and behavior.
The critical importance of de novo protein synthesis as a molecular mechanism involved in the process of consolidating storage of information in the brain has been shown in numerous experimental systems using a variety of pharmacological and genetic approaches (Neves et al., 2008). For instance, one of the first behavioral studies performed in rodents treated with the protein synthesis inhibitor puromycin demonstrated that loss of memory for avoidance discrimination learning was caused by blockade of protein synthesis (Flexner et al., 1963).
Information is encoded and stored in the brain at the level of individual synapses, which represent the “cellular unit of memories” (Hoeffer and Klann, 2010). Synapses are “plastic” in that their physiological efficacy (strength) changes and outlasts the events that trigger them. These persistent, activity-dependent changes in synaptic strength are triggered by de novo protein synthesis (Klann and Sweatt, 2008). Evidence indicating that protein synthesis can occur at specific synaptic sites comes from electron microscopy studies that demonstrated the presence of polyribosomes in dendrites of dentate granule neurons (Steward and Levy, 1982; Steward and Schuman, 2001). Accordingly, electrical stimulation of CA1 pyramidal cell dendrites led to the rapid incorporation of radiolabeled amino acids into newly synthesized proteins (Feig and Lipton, 1993). The functional role of dendritic protein synthesis then was demonstrated in hippocampal slices where newly synthesized proteins were required to enhance synaptic strength induced by brain-derived neurotrophic factor (BDNF; Kang and Schuman, 1996). In addition, dendritic protein synthesis is necessary for long-lasting decreases in synaptic strength induced by activation of group I metabotropic glutamate receptors (Huber et al., 2000). These findings indicate that protein synthesis is triggered at synaptic locations and is required for several forms of synaptic plasticity, which in turn is thought to be essential for higher brain functions, including memory formation.
Biochemical signaling mechanisms coordinating the process of neuronal protein synthesis are highly regulated and can be separated into three general phases: initiation, elongation and termination. The vast majority of the known translation regulation occurs at the level of the initiation phase and involves the coordinate activities of numerous molecular factors (Costa-Mattioli et al., 2009a; Richter and Klann, 2009; Sossin and Lacaille, 2010). Critical to the regulation of translation initiation is the activity of mammalian target of rapamycin (mTOR), a kinase ubiquitously expressed in eukaryotes (Hoeffer and Klann, 2010). In this review, we describe molecular mechanisms that control translation initiation and discuss examples of how perturbations of this regulation are a common pathophysiological feature of human neurological disorders.
Mammalian target of rapamycin function is regulated by the activity state of several neuronal surface receptors and channels (i.e., TrkB, NMDAR, AMPA, D1R, D2R, mGluRs) and by diverse signaling cascades activated by these receptors, including PDK1, PI3K, Akt, and TSC1/2 (Averous and Proud, 2006; Wang and Proud, 2006). mTOR is a serine–threonine kinase and exists as one component of protein complexes termed mTORCs (Figure 1) that include numerous factors regulating mTOR signaling and controlling the specificity for its diverse effectors targets, as well as directing its subcellular localization (Cutler et al., 1999; Martin and Blenis, 2002).
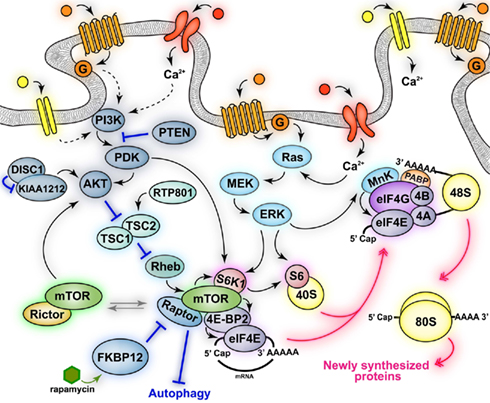
Figure 1. Schematic of mTOR signaling pathways. Activation of neuronal receptors and channels (mGluRs, NMDARs, TrkB, D1Rs, and D2Rs) leads to activation of mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 activation increases some neuronal processes (protein synthesis) while inhibiting others (autophagy). mTORC1 regulates the activity of downstream effectors involved in translation (S6K1, 4E-BP2), some of which are also directly phosphorylated via convergent activation of the MAPK signaling pathway (S6K1, MnK, S6). mTORC1-dependent phosphorylation of 4E-BP2 results in the association of eIF4E with eIF4G and the formation of the active eIF4F (eIF4E–eIF4A–eIF4G) complex. eIF4F recruits eIF4B and MnK and promotes the binding of mRNAs to the 43S pre-initiation complex to form the 48S initiation complex. The eIF4F complex and the poly(A) tail act synergistically together with MnK-dependent phosphorylation of eIF4E to stimulate cap-dependent translation initiation. The signaling pathways that activate mTORC2 and its downstream effectors, as well as its physiological functions are currently unknown. Black solid arrows indicate direct phosphorylation/activation, pink double arrows represent molecular association/dissociation and blue lines indicate inhibition.
mTORCs are defined primarily by the key scaffold proteins interacting with mTOR, Raptor, and Rictor. mTOR, when found in a complex with Raptor is known as mTORC1, whereas mTOR bound to Rictor is known as mTORC2 (Beretta et al., 1996; Kim et al., 2002; Sarbassov et al., 2004). These two complexes are thought to participate in the regulation of diverse signaling pathways that mediate different neuronal functions. Indeed, mTORC1 is primarily involved in the regulation of cap-dependent translation initiation, whereas mTORC2 is implicated in structural modifications such as cytoskeletal rearrangements (Jacinto and Hall, 2003; Hay and Sonenberg, 2004). Furthermore, these two complexes have different sensitivity to rapamycin, which does not inhibit mTOR kinase activity per se, but rather interferes with the binding of mTOR to Raptor, thereby disrupting mTORC1 and the phosphorylation of its substrates (Figure 1). Thus, mTORC1, but not mTORC2, is inhibited by rapamycin (Beretta et al., 1996; Kim et al., 2002), although prolonged treatment with rapamycin in cultured cells may also indirectly inhibit mTORC2 (Sarbassov et al., 2006).
Critical downstream targets of mTORC1 that are involved in translation include the p70 ribosomal S6 kinases 1 and 2 (S6K1 and S6K2) and the eIF4E-binding proteins (4E-BP; Klann and Dever, 2004). S6K1 is involved in the phosphorylation of the ribosomal protein S6 (Figure 1), an essential component of the 40S ribosomal subunit, and regulates its synthesis. The role of S6 in the regulation of translation is not well understood. S6 is located in close proximity to mRNA- and tRNA-binding sites, and genetic deletion of S6 impairs ribosome biogenesis and cell proliferation (Volarevic et al., 2000). S6K1 also mediates the phosphorylation of initiation factor 4B (eIF4B), which increases the catalytic activity of eIF4A (Rogers et al., 2001b; Raught et al., 2004), an RNA helicase critical for the translation of mRNA with complex 5′UTR secondary structures (Rogers et al., 1999, 2001a). Furthermore, S6K1 is also involved in translation elongation via phosphorylation of elongation factor 2 (eEF2) kinase. Phosphorylation of eEF2 kinase by S6K1 enhances the elongation rate and presumably protein synthesis by inhibiting its kinase activity, thereby decreasing the phosphorylation of eEF2 (Wang et al., 2001).
4E-BPs, which will be referred to from this point on as 4E-BP2 (Figure 1) because it is the predominant isoform in the mammalian brain (Banko et al., 2005), bind to the cap-binding protein eIF4E and repress its interaction with eIF4G and the other factors involved in formation of the initiation complex (Pause et al., 1994; Haghighat et al., 1995; Fletcher et al., 1998; Poulin et al., 1998). Thus, 4E-BP2 blocks eIF4F formation and inhibits protein synthesis by competing with eIF4G for the binding of eIF4E (Figure 1). The repressor activity of 4E-BP2 is regulated by sequential phosphorylation of its numerous phosphorylation sites (Bidinosti et al., 2010). The various 4E-BP2 residues are phosphorylated by diverse kinases in a sequential manner, with the last phosphorylation event occurring by mTORC1 (Gingras et al., 1999, 2001). Thus, mTORC1-dependent phosphorylation of 4E-BP2 releases eIF4E, which in turn associates with eIF4G. The binding of eIF4E to eIF4G and eIF4A forms the eIF4F initiation complex, which also recruits eIF4B (Figure 1). Formation of eIF4F complex is the crucial molecular event that triggers translation initiation. The formation of the eIF4F complex leads to the circularization of the mRNA through the interaction between cap-eIF4E, eIF4G, and poly A-binding proteins (PABP; Figure 1), which recruits ribosomes and promotes the unwinding of the secondary structure of 5′UTR mRNA, thereby facilitating the recognition of the start codon (Gingras et al., 1999; Klann and Dever, 2004). The eIF4F complex is further regulated through phosphorylation of some of its factors, such as eIF4G and eIF4E, which are phosphorylated by mTORC1 and Mnk1/2, respectively (Figure 1). Currently, the functional effect of the phosphorylation of eIF4G is not clear, whereas phospho-eIF4E is thought to facilitate translation initiation (Gingras et al., 1999; Pyronnet et al., 1999; Waskiewicz et al., 1999; Raught et al., 2000).
Initially, the role of mTORC1 signaling in the physiology of the nervous system was studied to understand the causal relationship between mTOR-dependent translation and either brain synaptic plasticity or memory formation (Costa-Mattioli et al., 2009a; Banko and Klann, 2008; Costa-Mattioli and Sonenberg, 2008). The availability of specific pharmacological inhibitors of upstream kinases, together with genetically modified mice that have deletions in either upstream activators or downstream effectors have greatly accelerated our understanding about the role of mTORC1 in nervous system function. In general, these studies have revealed that mTORC1 critically modulates protein synthesis-dependent synaptic plasticity and memory (Tang et al., 2002; Cammalleri et al., 2003; Tischmeyer et al., 2003; Hou and Klann, 2004; Tsokas et al., 2005; Dash et al., 2006; Gelinas et al., 2007; Antion et al., 2008a,b; Hoeffer et al., 2011). In addition, deletion of negative regulators of mTORC1 results in aberrant synaptic plasticity and altered memory function (Banko et al., 2005, 2006, 2007; Costa-Mattioli et al., 2009b; Banko and Klann, 2008; Swiech et al., 2008; Hoeffer and Klann, 2010). Thus, unbalanced mTORC1 signaling that perturbs the precise regulation of protein synthesis appears to be detrimental and have pathological consequences for nervous system function. These findings are important in light of the increasing amount of evidence linking dysregulated mTOR signaling as a common molecular mechanism involved in the etiology of several neurological disorders. In the next few sections we will discuss recent studies implicating mTORC1-dependent protein synthesis in disorders of the nervous system.
Neurodevelopmental disorders are defined as diseases caused by abnormal development of the brain during the first two decades of life. These disorders have a strong and complex genetic basis with diverse expression and thus, their clinical diagnosis is complicated by a broad array of symptoms (Grice and Buxbaum, 2006; Persico and Bourgeron, 2006; Levitt and Campbell, 2009). However, it is possible to define a subgroup of these disorders that are characterized by single-gene mutations and behavioral impairments similar to those reported in autistic patients. Autism is diagnosed based on the appearance of behavioral abnormalities that include deficits in social interaction skills, impaired ability to communicate, and ritualistic-like repetitive behaviors (Hill and Frith, 2003). A common molecular abnormality in several of these autism-like disorders is loss-of-function mutations and/or deletion of genes that encode proteins that normally repress mTORC1 signaling pathway. These observations led to the hypothesis that dysregulation of protein synthesis may be the cause of altered synaptic development and plasticity, resulting in autistic-like phenotypes in humans (Kelleher and Bear, 2008; Bourgeron, 2009; Hoeffer and Klann, 2010).
Fragile X syndrome (FXS) is an X-linked form of metal impairment caused by transcriptional silencing of the FMR1 gene. The fragile X mental retardation protein (FMRP), encoded by the FMR1 gene, is a RNA-binding protein that represses the translation of specific mRNAs, such as MAP1B Arc/Arg3.1, αCaMKII, PSD-95, SAPAP3 (Todd et al., 2003; Zalfa et al., 2003; Hou et al., 2006; Muddashetty et al., 2007; Narayanan et al., 2008). Consistent with this idea, ablation of FMRP has been shown to increase the levels of the proteins encoded by these specific mRNAs. Thus, the etiology of FXS, resulting from the absence of functional FMRP, is an exaggerated synthesis of specific proteins. However, it also has been shown that general protein synthesis is exaggerated in FXS model mice (Qin et al., 2005; Dölen et al., 2007; Osterweil et al., 2010). It has been proposed that the molecular mechanism mediating FMRP-dependent repression of translation initiation is inhibition of interactions of eIF4E and eIF4G (Figure 2). Indeed, FMRP interacts with a non-canonical 4E-BP, named cytoplasmic FMRP Interacting protein 1 (CYFIP1; Napoli et al., 2008). In addition, it has been demonstrated that FXS model mice have exaggerated mTORC1 signaling in the hippocampus resulting from increased expression of PIKE. Indeed, PIKE mRNAs are repressed by FMRP and encode for a GTPase that enhances PI3K activity. Thus, PIKE is abnormally synthesized in absence of FMRP resulting in enhanced PI3K–Akt–mTORC1 signaling (Figure 2). These observations are consistent with the idea that aberrant cap-dependent translation is involved in FXS (Gross et al., 2010; Sharma et al., 2010). The molecular abnormalities of FXS model mice are accompanied by several behavioral deficits, which include impairments in hippocampus-dependent learning and memory in the Morris water maze and terrestrial radial arm maze paradigms (Kooy et al., 1996; D’Hooge et al., 1997; Mineur et al., 2002). In addition, FXS model mice showed increased latency in finding the new platform position in the reversal phase of the Morris water maze test, where the position of the hidden platform is suddenly changed (D’Hooge et al., 1997). This suggests that FXS model mice not only have learning and memory deficits, but also behavioral inflexibility in switching to a new searching strategy. FXS model mice also exhibit impaired contextual fear conditioning (Van Dam et al., 2000), altered sensorimotor integration (Chen and Toth, 2001; Nielsen et al., 2002), and increased susceptibility to audiogenic epileptic seizures (Musumeci et al., 2000; Chen and Toth, 2001).
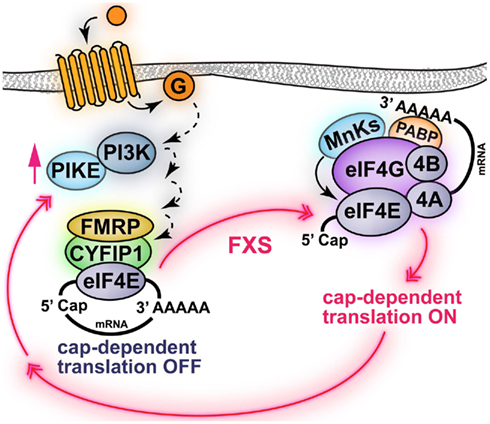
Figure 2. Schematic of FMRP regulation of cap-dependent translation initiation. FMRP represses the translation of specific mRNAs via interaction with a 4E-BP-like protein termed CYFIP1, which also binds eIF4E. One of the mRNAs regulated by FMRP is encoding for PIKE, which enhances PI3K signaling. In fragile X syndrome (FXS), the absence of functional FMRP results in the abnormal translation of PIKE and other mRNAs. Indeed, in FXS model mice PI3K/mTORC1 signaling is enhanced. Note that activation of surface receptors (i.e., group I mGluRs) promotes PI3K signaling via PIKE and activation of mTORC1-dependent protein synthesis. Activation of mTORC1 may also induce translation of FMRP-dependent mRNAs via an unknown mechanism. Black solid arrows indicate direct phosphorylation/activation, dashed arrows represent events mediated by unknown molecular effectors.
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder caused by mutations in either hamartin (TSC1) or tuberin (TSC2), which negatively regulate the mTORC1 signaling pathway, and is clinically characterized by a high prevalence of autism and cognitive impairments (Wiznitzer, 2004; Kwiatkowski and Manning, 2005; Wullschleger et al., 2006). TSC1 is a chaperone protein and TSC2 is a GTPase-activating protein (GAP) with catalytic activity directed toward the small G protein Rheb (Inoki et al., 2003). Loss-of-function mutations affecting TSC1/TSC2 result in the abolishment of the GAP activity of TSC2 (Garami et al., 2003). mTORC1 activation is typically regulated by TSC2 (Figure 1), which mediates the binding of GTP–Rheb (Ras homologs enriched in the brain) to the mTORC1 kinase domain (Zhang et al., 2003). Thus, TSC1/TSC2 mutations result in the exaggerated activation of mTORC1 signaling pathway (Manning and Cantley, 2003a,b).
Mouse models utilized to study TSC have been engineered with heterozygous mutations in the genes for either TSC1 or TSC2. TCS model mice have been observed to display several hippocampus-dependent learning and memory deficits, including impaired spatial learning and memory, and abnormalities in contextual fear conditioning (Goorden et al., 2007; Ehninger et al., 2008b) reminiscent of behavioral phenotypes displayed by FXS model mice. In addition, TSC1 heterozygous knockout mice and dominant/negative TSC2 mutant mice also displayed impaired social behaviors (Goorden et al., 2007; Chevere-Torres et al., 2012), whereas TSC2 heterozygous knockout mouse pups exhibited abnormalities in social communication as demonstrated by changes in ultrasonic vocalizations following maternal separation (Young et al., 2010). Together, these observations suggest that mutation of only one of the two alleles encoding for the TSC proteins is sufficient to induce cognitive deficits. Importantly, the mTORC1 inhibitor rapamycin has been shown to be effective in ameliorating learning and memory deficits in TSC2 heterozygous knockout mice (Ehninger et al., 2008b), suggesting that uncontrolled mTORC1 signaling is a core molecular mechanism involved in the behavioral abnormalities. Moreover, the TSC model mice display behavioral abnormalities in the absence of neuropathological features such as brain tumors (tubers) and abnormalities in neuronal spine density, which were assumed to be responsible for the cognitive deficits displayed by TSC patients (Goorden et al., 2007; Ehninger et al., 2008a; Chevere-Torres et al., 2012). These findings imply that the pathological brain abnormalities in TSC are not necessary for the occurrence of behavioral deficits, suggesting that their appearance results from the activity of other molecular factors.
Phosphatase and tensin homolog on chromosome 10 (PTEN) is a tumor suppressor gene that is mutated in many human cancers (Ali et al., 1999) as well as in brain disorders such as autism, mental impairment, and macrocephaly (Zori et al., 1998; Goffin et al., 2001; Waite and Eng, 2002; Butler et al., 2005). PTEN has lipid phosphatase activity and antagonizes the phosphatidylinositol 3-kinase (PI3K)-dependent signaling (Figure 1) by dephosphorylating the lipid targets of PI3K (Maehama and Dixon, 1999). Thus, loss of PTEN function in neurons results in hyperactivation of mTORC1, which is a downstream target of the PI3K/Akt signaling pathway (Kwon et al., 2006a). Mice with genetic deletion of PTEN restricted to a subset of differentiated hippocampal and cortical neurons were shown to exhibit behavioral impairments consistent with clinically relevant autistic behaviors. For example, the conditional PTEN mutant mice exhibited abnormalities in social behavior accompanied by deficits in pre-pulse inhibition, anxiety-like behaviors, and seizures (Kwon et al., 2006a,b). The behavioral abnormalities of PTEN mutant mice were observed in the absence of dramatic brain pathologies, which is similar to what was observed in TSC heterozygous knockout mice (Kwon et al., 2006a).
Phosphatase and tensin homolog on chromosome 10 mutant mice have been shown to develop macrocephaly as well as abnormalities in dendritic arborization and axonal growth (Kwon et al., 2006a). Importantly, pharmacological attenuation of mTORC1 activity with rapamycin reduced the impairments in social interactions and the duration and frequency of spontaneous seizures as well as neuronal hypertrophy responsible for the macrocephaly. This suggests that exaggerated mTORC1 signaling pathway is responsible for the behavioral abnormalities observed in PTEN mutant mice (Zhou et al., 2009). Thus, these findings are consistent with the notion that upregulated mTORC1 signaling results in behavioral abnormalities in both TSC model mice and PTEN mutant mice.
Genetic studies demonstrate that chromosomal deletions immediately flanking the gene encoding for FK506-binding protein 12 (FKBP12) are present in autistic patients (Sebat et al., 2007). FKBP12 is an immunophilin that binds rapamycin and regulates the phosphorylation of mTORC1 substrates by blocking the formation of the mTORC1 complex (Jacinto and Hall, 2003). Thus, genetic deletion of FKBP12 should result in exaggerated mTORC1 activity (Figure 1). The hypothesis that increased mTORC1 signaling is involved in the onset of autism-like syndromes has been tested with conditional knockout mice in which the FKBP12 gene was removed in the forebrain at approximately 3 weeks of age (Hoeffer et al., 2008). Deletion of FKBP12 was associated with increased activity of mTORC1 as shown by an augmented interaction between mTOR and Raptor and enhanced phopshorylation of the mTORC1 target S6K1 (Hoeffer et al., 2008). The changes in mTORC1 signaling in the FKBP12 conditional knockout mice were accompanied by cognitive abnormalities, including enhanced associative contextual fear conditioning, preference for the familiar object in the novel object recognition test, and behavioral inflexibility in several reversal learning paradigms (Hoeffer et al., 2008). These findings suggest that exaggerated mTORC1 activation leads to multiple behavioral abnormalities, including increased perseveration. Furthermore, these results imply that autistic behaviors in syndromes with upregulated mTORC1 signaling are not entirely predetermined during development because dysregulation of mTORC1 signaling in FKBP12 conditional knockout mice occurs postnatally.
Depression is a neuropsychiatric disorder characterized by anhedonia, irritability, depressed mood, difficulties in concentrating, and abnormalities in appetite and sleep (Krishnan and Nestler, 2008). Complications in the current pharmacotherapy for depression are related mainly to the long duration of time required by the treatment to produce a therapeutic response and the abundance of treatment-resistant patients (Clark et al., 2009). Clinical studies revealed that low doses of ketamine, a non-selective NMDA receptor (NMDAR) antagonist, produces a rapid and sustained antidepressant effects that are also observed in patients with treatment-resistant depression (Berman et al., 2000; Zarate et al., 2006; Price et al., 2009). Thus, NMDARs may play an important role not only in the pathophysiology of depression, but also as a novel molecular target for the mechanism of action of antidepressant drugs. However, the molecular connection between antagonism of NMDARs and antidepressant behavioral effects is unknown. Recently, two independent studies demonstrated that administration of a non-anesthetic dose of ketamine produced antidepressant-like behavior in rodents but the molecular mechanisms responsible for this effect appear to be different.
Nanxin et al. (2010) showed that ketamine transiently activates mTORC1 signaling in the prefrontal cortex (PFC) of rats. This treatment results in mTORC1-dependent synthesis of new pre- and postsynaptic proteins, such as Arc, synapsin, PSD-95, and GluR1, that are required for plastic changes at new spines and synapses. Accordingly, the enhanced mTORC1-dependent synthesis of synapse-associated proteins was accompanied by a rapid increase in spine density and increased excitatory postsynaptic currents (EPSCs) in layer V pyramidal neurons in the PFC (Nanxin et al., 2010). Thus, acute ketamine administration results in enhanced mTORC1-dependent protein synthesis, which leads to long-term changes in PFC neurons, such as an increase in synapses and spine formation, and enhanced synaptic strengthening. Importantly, intracerebroventricular (ICV) infusion of rapamycin blocked all the physiological effects induced by ketamine, which strongly suggests that the formation of new spines and increases in synaptic strength and antidepressant-like behaviors were functional consequences of activation of mTORC1. Furthermore, “depressed rats” exhibit reduced levels of GluR1, PSD-95, and synapsin I in the PFC, and a single dose of ketamine normalized the levels of these proteins in an mTORC1-dependent manner (Nanxin et al., 2010). These results indicate that depression may be the pathophysiological consequence of altered spine remodeling and synaptic plasticity in the PFC.
In contrast, Autry et al. (2011) demonstrated that the antidepressant effects of ketamine are mediated by the synthesis of BDNF in the hippocampus. Indeed, increased BDNF expression was blocked by anisomycin, which is a general inhibitor of protein synthesis. Reduction of BDNF levels in the hippocampus also was accompanied by blockade of antidepressant-like behavior. Furthermore, BDNF and neurotrophic tyrosine kinase receptor type 2 (TrkB receptor) null mice were resistant to the antidepressant effects of ketamine, strongly suggesting that newly translated BDNF mediates the positive, antidepressant effects of NMDA receptor blockade. The authors also demonstrated that acute blockade of NMDA receptor decreased phosphorylation of elongation factor 2 (eEF2), which normally promotes the ribosomal translocation of the nascent polypeptide chain. Phosphorylation of eEF2 inhibits its catalytic activity and blocks ribosomal translocation, thereby halting ribosomes and blocking protein synthesis. Mice pharmacologically treated with inhibitors of eEF2 phosphorylation showed antidepressant-like behavior accompanied by increased BDNF levels in the hippocampus (Autry et al., 2011). These findings suggest that reduction of eEF2 phosphorylation is sufficient to disinhibit protein synthesis, stimulate translation of BDNF, and promote antidepressant-like behaviors. Thus, drugs targeting eEF2 kinase activity are a potential novel pharmacological treatment for depression.
It appears that antidepressant drugs share the ability of increase protein synthesis. Indeed, this is a common molecular mechanism of drugs antagonizing NMDA receptors (ketamine; Nanxin et al., 2010; Autry et al., 2011) as well as inhibiting the reuptake of serotonin (fluoxetine; Dagestad et al., 2006). Thus, one could reasonably conclude that dysregulation of translational control is a molecular mechanism involved in the onset of depressive syndromes.
The involvement of mTORC1 signaling in schizophrenia has been suggested by genetic and biochemical studies conducted in schizophrenic patients. Indeed, mutations of the Akt1 gene on chromosome 14q22–32 are associated with genetic form of schizophrenia (Harrison, 1999). Diminished Akt expression and kinase activity accompanied by reduced phosphorylation at Ser473 was found in postmortem brains of patients with schizophrenia (Kalkman, 2006; Zhao et al., 2006). Moreover, decreased expression of the catalytic subunit of PI3K was detected in lymphocytes of schizophrenic individuals (Middleton et al., 2005). These studies suggest that schizophrenia may be a consequence of decreased activity of the PI3K–Akt signaling cascade. Accordingly, some antipsychotic drugs result in the correction of dysfunctional PI3K–Akt signaling pathway (Chalecka-Franaszek and Chuang, 1999; Lu and Dwyer, 2005; Kalkman, 2006). Both PI3K and Akt are upstream regulators of mTORC1 (Kalkman, 2006), but whether mTORC1 is downregulated due to decreased activity of PI3K and/or Akt has not been clearly demonstrated in schizophrenia. Recently, the consequences of reduced Akt activity were investigated in a mouse model in which phosphorylation of Ser473 was abrogated selectively in neurons by genetic deletion of Rictor, the key regulatory subunit of mTORC2 (Siuta et al., 2010). Mice that lack mTORC2-dependent phosphorylation of Akt on Ser473 exhibited decreased pre-pulse inhibition, consistent with impairments in sensorimotor integration. These behavioral impairments were accompanied by alterations in cortical catecholamine content, as evidenced by decreased levels of dopamine (DA) and increased levels of norepinephrine (NE) in the PFC (Siuta et al., 2010). This finding is consistent with experimental models of schizophrenia that suggest that dysregulation in cortical DA content contribute both to cognitive deficits and negative symptoms (Weinberger et al., 1988; Davis et al., 1991; Howes and Kapur, 2009). The molecular link between decreased mTORC2–Akt signaling and abnormalities in DA levels in the PFC of Rictor null mice was explained by an Akt-dependent increase in surface expression and function of the NE transporter (NET), resulting in enhanced uptake of DA in noradrenergic neurons and its conversion to NE (Siuta et al., 2010). Finally, administration of nisoxetine, a NET inhibitor, reversed the sensorimotor deficits and the DA content in the PFC of the Rictor null mice, suggesting that the behavioral deficits displayed by the mice are a consequence of an Akt-dependent imbalance in cortical levels of DA (Siuta et al., 2010).
Other evidence of the critical involvement of Akt signaling in schizophrenia comes from studies in which Akt activity is increased. For example, suppression of DISC1 activity results in increased phosphorylation of Akt and ribosomal protein S6, which is a downstream effector of mTORC1, during adult neurogenesis in the hippocampus (Kim et al., 2009). DISC1 is a gene that was identified at the breakpoint of a chromosomal translocation (Blackwood et al., 2001; Chubb et al., 2008). Disruption of DISC1, due to genetic rearrangement, co-segregates with several psychiatric disorders, including schizophrenia (Millar et al., 2000; Blackwood et al., 2001). DISC1 regulates Akt by blocking KIAA1212, which is a binding partner of Akt (Figure 1). Therefore, deletion of DISC1 results in uncontrolled binding of KIAA1212 to Akt, leading to enhanced Akt signaling (Kim et al., 2009). Genetic manipulations that increase Akt activity resemble the defects caused by DISC1 suppression; importantly, these defects were rescued by inhibition of mTORC1 activity with rapamycin (Kim et al., 2009). These findings identified Akt–mTORC1 as a critical downstream target of DISC1 in neuronal development. It will be important to determine how these molecules, which have been indicated as schizophrenia-related genes, can contribute to the etiology of schizophrenia.
Parkinson’s disease is a progressive neurodegenerative disorder that results from loss of the dopaminergic (DAergic) neurons located in the substantia nigra pars compacta that project to the striatum (nigrostriatal DAergic pathway). The loss of striatal DA content leads to motor symptoms such as bradykinesia, rigidity, and tremor (Parkinson, 2002). Recent studies addressing the mechanism of neurodegeneration in PD demonstrate the involvement of the mTORC1 signaling pathway in the survival mechanism of DAergic neurons.
In vivo and in vitro studies show that degeneration induced by treatment with PD toxins, such as 6-OHDA and MPTP, leads to upregulation of RTP801, a protein encoded by a RTP801 stress-responsive gene, which in turn reduces mTOR kinase activity. Importantly, enhanced RTP801 was also found in nigral DAergic neurons of postmortem human PD brains. Accordingly, neurons with inhibited expression of RTP801 or overexpression of mTOR were protected from the insults induced by the toxins and showed high survival rate (Malagelada et al., 2006). These studies suggest that neurodegeneration induced by PD toxins is dependent on upregulated RTP801 and the subsequent reduction of mTOR signaling.
It has been proposed that the molecular mechanism, linking high levels of RTP801 to mTORC1 inhibition and neurodegeneration involves TSC2 and Akt (Deyoung et al., 2008; Malagelada et al., 2008; Figure 1). Either genetic manipulations that interfere with TSC2 or increase the expression of a constitutively active form of Akt protected against the PD toxins and prevented the increase in RTP801 (Malagelada et al., 2008). These findings suggest that PD toxins induce neurodegeneration through inhibition of Akt, elevation of RTP801 levels, and TSC1/2-dependent suppression of mTORC1 activity. Importantly, DAergic neurons of PD patients also exhibited diminished levels of Akt phosphorylation together with increased expression of RTP801 (Malagelada et al., 2010). Thus, treatments that inhibit mTORC1 signaling should lead to an enhanced toxicity and even a greater DAergic loss. Surprisingly, rapamycin was reported as neuroprotective agent both in cell culture and in a MPTP mouse model. The beneficial effect of rapamycin, which inhibits mTORC1, has been explained through selective maintenance of mTORC1-dependent activities responsible for neuronal survival whereas RTP801, which acts upstream of mTORC1, would completely block all of its downstream physiological effects (Malagelada et al., 2010). For example, inhibition of mTORC1 may reduce the synthesis of new RTP801, thereby relieving RTP801-dependent inhibition of mTORC1 and stimulating the phosphorylation of the survival promoting kinase Akt (Malagelada et al., 2010). Alternatively, rapamycin may enhance Akt activity through inhibition of mTORC1-dependent activation of S6K1 and the subsequent reduction of phospho-IRS-1, which is a scaffold protein involved in the activation of PI3K and Akt (Shah et al., 2004).
The mTORC1 signaling cascade also was shown to be involved in the onset of levodopa-induced dyskinesia (LID), a debilitating side effect associated with the pharmacotherapy of PD (Santini et al., 2009b). LID consists of dystonic and choreic motor disturbances that are induced by repetitive and discontinuous administration of L-DOPA, the main therapy of choice for the treatment of PD (Bezard et al., 2001). A single administration of L-DOPA induces transient activation of mTORC1 signaling in a mouse model of PD (Santini et al., 2009b). Upregulated mTORC1 activity, measured by increased phosphorylation of downstream effectors, occurred in the dopamine D1 receptor (D1R) expressing subset of striatal neurons, specifically medium spiny neurons (MSNs), which were previously implicated as an important site for the onset and development of LID (Gerfen et al., 2002; Westin et al., 2007; Santini et al., 2008, 2009a; Darmopil et al., 2009). Accordingly, the ability of L-DOPA to activate mTORC1 signaling in D1R-bearing MSNs was maintained with repetitive administration of the drug and was correlated with the severity of the degree of dyskinesia. Treatment with rapamycin together with L-DOPA relieved the dyskinetic side effects without reducing the positive, anti-parkinsonian efficacy of drug (Santini et al., 2009b). These results suggest that inhibitors of mTORC1 may be used clinically as antidyskinetic agents. In summary, rapamycin and other inhibitors of mTORC1 have promising therapeutic potential for the treatment of PD, both as neuroprotective and antidyskinetic drugs.
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by an expansion mutation of the trinucleotide CAG in exon 1 of the huntingtin (HTT) gene. The expansion mutation is translated into an abnormally long polyglutamine tract at the N terminus of huntingtin (MacDonald et al., 1993). The pathogenic mechanism by which huntingtin with an extended polyglutamine tract induces neuronal dysfunction and neurodegeneration is not fully understood. However, mutated huntingtin localizes in intraneuronal aggregates together with other aggregate-prone proteins, and these inclusions appear to be deleterious for the neurons (Davies et al., 1997; Saudou et al., 1998; Rubinsztein, 2002). Turnover of huntingtin was found to be impaired after inhibition of the autophagy–lysosome pathway (Ravikumar et al., 2002) suggesting that this pathway is involved in the clearance of the intracellular inclusions. Activation of mTORC1 also inhibits autophagy (Figure 1) in cells from yeast to humans (Schmelzle and Hall, 2000). Consistent with this observation, induction of autophagy by inhibition of mTORC1 with rapamycin resulted in protection against mutated huntingtin-induced toxicity and enhanced clearance of aggregates. In the same cell line, the ability of rapamycin to inhibit mTORC1 and subsequently reduce inclusions was impaired after prolonged expression of mutated huntingtin (Ravikumar et al., 2002). The decreased effectiveness of rapamycin was explained with the localization of mTOR in the intracellular inclusions after prolonged expression of mutated huntingtin (Ravikumar et al., 2004). Co-localization of mTOR and mutated huntingtin to the inclusions was also described in the brain of transgenic mice expressing mutant huntingtin and, most importantly, in human patients affected by HD (Ravikumar et al., 2004). The interaction between mTOR and mutated huntingtin also was confirmed with co-immunoprecipitation in cultured cells and in brain lysates from mutant huntingtin transgenic mice (Ravikumar et al., 2004). Thus, mTOR and mutated huntingtin are sequestered together in intracellular inclusions, resulting in inactivation of mTORC1 as evidenced by reduced levels of phosphorylation of S6K1, 4E-BP2, and S6 both in cell culture and in the brains of the mutant huntingtin transgenic mice. In these conditions, mTORC1-dependent protein synthesis and autophagy were impaired and were not further reduced by treatment with rapamycin (Ravikumar et al., 2004). These findings suggest that inhibition of mTORC1 is effective in preventing toxicity induced by mutant huntingtin only in earlier stages of disease, possibly when mTOR is not yet sequestered and inactivated in the intracellular inclusions. Consistent with this idea, chronic administration of rapamycin to HD model mice before disease onset rescued the behavioral deficits associated with the progression of the disease (Ravikumar et al., 2004). HD model mice treated with rapamycin exhibited improvement in performance in four different behavioral paradigms used to test motor dysfunction: rotarod test, grip strength test, wire maneuver, and presence of tremors (Ravikumar et al., 2004). Moreover, the behavioral recovery in these mice was accompanied by a reduction in the number and size of striatal and cortical inclusions (Ravikumar et al., 2004). Taken together, these findings suggest that early pharmacological intervention to inhibit mTORC1 signaling effectively reduces neurological phenotypes in animal models of HD.
Alzheimer’s disease (AD) is characterized by the formation of amyloid plaques and neurofibrillary tangles (NFTs), which are correlated with neurodegeneration in brain areas implicated in acquisition and storage of declarative memory, such as the hippocampus, entorhinal cortex, amygdala, and associative regions of the cortex (Nestor et al., 2006; Götz and Ittner, 2008). The major component of plaques is the polypeptide Aβ that is derived from the amyloid precursor protein (APP), whereas NFTs are composed of hyperphosphorylated forms of the microtubule-associated protein tau (Glenner and Wong, 1984; Kang et al., 1987; Tanzi et al., 1987; Binder et al., 2005).
Studies on the regulation of the mTORC1 signaling pathway in AD has revealed a complicated picture, mainly due to the lack of agreement about the direction of mTORC1 dysfunction (increase vs. decrease) and the pathogenic mechanism leading to neurodegeneration (Swiech et al., 2008). Aβ and tau exert toxicity through distinct mechanisms but there is now evidence from human, rodent, and in vitro studies demonstrating a link between Aβ and tau in neurotoxicity, although the molecular nature of this link remains elusive (Ittner and Götz, 2011). Studies of Aβ-induced neurotoxicity report increased phosphorylation of double-stranded RNA-dependent protein kinase (PKR; Chang et al., 2002a,b; Peel and Bredesen, 2003; Onuki et al., 2004), eukaryotic initiation factor 2α (eIF2α; Chang et al., 2002a,b) and S6K1 (An et al., 2003) in mouse models of AD and AD patients. For example, the Aβ peptide is shown to activate PKR and Aβ-induced neurotoxicity was reduced in neuronal cultures from PKR knockout mice. (Chang et al., 2002b) Furthermore, increased levels of phosphorylated PKR were found in APP/PS1 mutant mice, a rodent model of AD expressing human APP with the Swedish and London mutation, as well as presenilin 1 (PS1) carrying two different mutations known to cause a familial, early onset form of the disease (Page et al., 2006). These studies suggest a correlation between Aβ-induced neurotoxicity and a decrease in translation, as PKR attenuates protein synthesis through eIF2α phosphorylation. In support of the idea that AD is accompanied by reduction of protein translation, the mTORC1 signaling cascade was found to be impaired in a mouse model of AD and was correlated with impaired synaptic plasticity (Ma et al., 2010). It was possible to mimic these impairments by treating hippocampal slices of wild-type mice with soluble Aβ. Importantly, pharmacological and genetic manipulation leading to increased mTORC1 signaling restored the synaptic plasticity deficits of the AD model mice and prevented Aβ-induced impairments in synaptic plasticity in wild-type mice (Ma et al., 2010). Altogether, these data indicate that there is an association between Aβ-induced toxicity, enhanced eIF2α phosphorylation, and deficits in mTORC1 signaling pathway.
On the other hand, several groups have reported increases in the phosphorylation of S6K1, mTOR, eIF4E, and 4E-BP2 in AD brains (Li et al., 2004, 2005). This led to the hypothesis that enhanced mTORC1 signaling could be responsible for the high levels of toxic proteins such as tau (Pei et al., 2008). In addition, hyperactivated mTORC1 signaling was also positively correlated with the levels of total and phosphorylated tau. Thus, the continuous synthesis of tau in degenerating neurons could be the result of upregulated mTORC1 signaling (Pei and Hugon, 2008). Furthermore, increased levels of phosphorylated mTOR and S6K1 were also found in some of the brain areas affected in AD, such as cortex, in the same double APP/PS1 transgenic mice (Lafay-Chebassier et al., 2005). Importantly, chronic treatment with rapamycin rescued learning and memory deficits in two different mouse models of AD (Caccamo et al., 2010; Spilman et al., 2010) suggesting that enhanced mTORC1 activity is involved in the cognitive impairments.
The use of the term psychoactive when describing drugs refers to a diverse group of chemicals that act primarily at the level of the central nervous system and affect brain function, resulting in changes of mood, cognition, perception, and consciousness. These substances are often used recreationally even if originally synthesized for the therapeutic use, namely to treat both physical and neuropsychiatric disorders (Di Chiara and North, 1992; Nestler, 2004). Recently, studies have demonstrated that discrete behavioral effects induced by some of these drugs are caused by activation of the mTORC1 signaling pathway in the brain.
Cannabis sativa is included in marijuana, hashish, bhang, and other well-known illicit psychoactive drugs that are largely used recreationally (Pierce and Kumaresan, 2006). The primary psychoactive ingredient of cannabis is Δ9-tetrahydrocannabinol (THC; Gaoni and Mechoulam, 1964), which is responsible for multiple behavioral effects including euphoria, enhanced sensory perception, increased appetite, analgesia, memory impairment, anxiety, paranoia and, at higher doses, sedation (Ameri, 1999; Piomelli, 2003; Pierce and Kumaresan, 2006). Experimental mouse models are utilized to study behaviors induced by THC (Hampson and Deadwyler, 1999; Maldonado and Rodriguez De Fonseca, 2002). For example, acute administration of THC to mice immediately after training in the novel object recognition task produced impairment in long-term memory, as indicated by the inability of THC-treated mice to discriminate between familiar and novel objects (Puighermanal et al., 2009). Moreover, the same dose of THC activated the mTORC1 signaling cascade in the hippocampus, as indicated by increased phosphorylation of downstream targets, such as S6K1, S6, eIF4E, eIF4G, and 4E-BP2 (Puighermanal et al., 2009). The behavioral and molecular changes induced by THC were abolished in CB1 receptor knockout mice and in mice with a conditional deletion of the CB1 receptor in GABAergic neurons. Moreover, pretreatment of mice with either rapamycin or anisomycin abolished the amnesic-like effects induced by THC, suggesting that the memory impairment is mediated by mTORC1-dependent translation (Puighermanal et al., 2009). Surprisingly, the THC-induced activation of the mTORC1 cascade was localized in glutamatergic pyramidal neurons and was dependent on NMDA receptors (Puighermanal et al., 2009). This indicates that the amnesiac-like effect induced by THC is most likely mediated via a heterosynaptic mechanism because CB1 and NMDA receptors are localized in different hippocampal neurons (GABAergic basket-cells and glutamatergic pyramidal neurons, respectively). It is also interesting to note that the same molecular process of protein translation occurring in hippocampal pyramidal neurons, is involved in opposing behavioral processes; memory consolidation vs. THC-induced memory loss. A possible explanation for the contradictory functional effects of translation may be due to the different synaptic locations where it occurs (Mackie and Katona, 2009). Indeed, in pyramidal neurons THC-induced activation of mTORC1, and potentially translation, occurs in the stratum pyramidale, a cellular region with a high density of GABAergic synapses (Papp et al., 2001). Thus, it is possible that enhanced translation in the soma of pyramidal neurons results in memory impairments, whereas enhanced translation in dendritic spines at excitatory synapses is required for memory formation (Costa-Mattioli et al., 2009b; Richter and Klann, 2009).
Cocaine recently was shown to activate the mTORC1 signaling cascade in brain regions that undergo long-lasting neuroadaptation in response to drug exposure, including the mesolimbic dopamine pathway (VTA) and its forebrain targets, especially the nucleus accumbens (NAc; Nestler, 2005; Mameli et al., 2007; Wang et al., 2010; Wu et al., 2011). Specifically, systemic administration of cocaine in mice increased the phosphorylation of the mTORC1 downstream effector S6 in the VTA and NAc (Wu et al., 2011). Sensitization to cocaine is often measured in behavioral experiments as an increase in locomotor activity. Behavioral sensitization consists of an initial induction phase, in which repeated injections of cocaine leads to an increase in locomotor activity. Induction is followed by drug withdrawal and then re-exposure to the drug for the expression phase of sensitization, which results in an even greater enhancement in locomotor activation (Berridge and Robinson, 1998). Rapamycin effectively reduced both induction and expression of cocaine-induced locomotor sensitization (Wu et al., 2011), suggesting that mTORC1 signaling may underlie the different adaptive changes elicited by cocaine exposure during induction and expression phases of locomotor sensitization. Notably, it also has been shown that cocaine induces altered synaptic plasticity in DA neurons of the VTA in an mTORC1- and protein synthesis-dependent manner (Mameli et al., 2007). These findings provide additional evidence for cocaine-induced activation of mTORC1.
Another aspect of cocaine addiction linked to activation of mTORC1 signaling is relapse, which is experienced by addicts after a prolonged period of abstinence and induced by craving-provoking environmental cues paired with the rewarding properties of the drug (Gawin and Kleber, 1986a,b; O’Brien, 1997; Childress et al., 1999; Wang et al., 2010). Cue-induced relapse is modeled in rodents with a reinstatement paradigm, in which rats are trained to self-administer a drug reward paired with a discrete cue. Lever pressing is then extinguished in the absence of the discrete cue. During a reinstatement test, exposure to the discrete cue reinstates drug-seeking behavior as measured by increased lever pressing (Meil and See, 1997; Kruzich and See, 2001). Reinstatement of drug-seeking behavior in rats exposed to a cue previously associated with the drug was accompanied by an increase of mTORC1 activation in the NAc, as measured by increased phosphorylation of S6K1 and S6 (Wang et al., 2010). Infusion of rapamycin in the NAc core before the reinstatement test significantly decreased the cue-induced seeking behavior (Wang et al., 2010). These behavioral effects are NMDA-dependent as NMDA agonist infusions into the NAc core enhanced the reinstatement of cue-induced cocaine seeking by increasing the activation of the mTORC1 cascade, and co-infusions of rapamycin blocked both the molecular and behavioral effects (Wang et al., 2010). These findings suggest that mTORC1-dependent protein synthesis in the NAc core is involved in reinstatement of cue-induced seeking behavior. Because rapamycin administered acutely before the reinstatement test reduces the cue-induced cocaine seeking, it could used therapeutically to treat relapse in cocaine addicts (Wang et al., 2010). Importantly, it has been shown that rapamycin reduced the craving induced by a drug-related cue in abstinent heroin addicts (Shi et al., 2009).
It is clear that mTORC1 is a critical regulator of multiple functions of the central nervous system. In this review, we have described a series of disorders in which there is a clear association between neuropathology and regulation of mTORC1-dependent translation (see Table 1 for a summary).
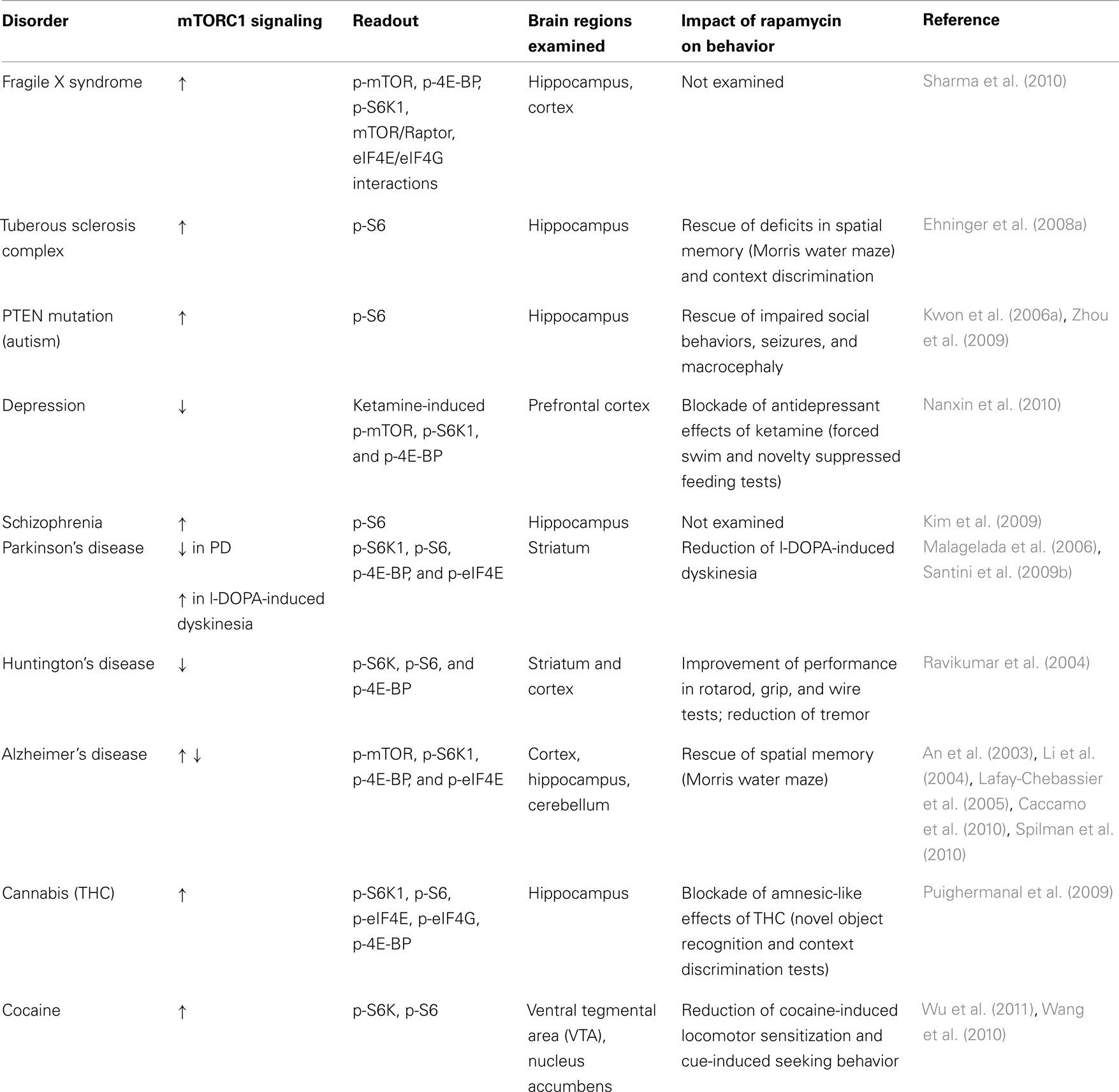
Table 1. Summary of altered mTORC1 signaling in brain disorders and in response to psychoactive drugs.
However, mTORC1 has several other physiological functions that either are not fully understood or have not yet been explored. For instance, the role of dysregulated mTORC1 signaling in autophagy in neurons, and its association with diseases has the potential to open a completely new line of research. Furthermore, there are aspects of cap-dependent protein synthesis that are regulated via mTORC1 that deserve more attention and/or need to be addressed in more detail. For example, the molecular machinery that is involved in the sorting and localization of particular mRNAs at specific neuronal subregions is poorly understood, as is the identity of the mRNAs whose translation is regulated by mTORC1.
An open question that needs to be addressed in greater detail is the role that mTORC1 plays in the pathophysiology of the CNS. The ubiquitous distribution and the fine control of mTORC1 signaling render it difficult to ascertain whether mTORC1 is the cause and/or one of the consequences of various brain disorders discussed in this review. At minimum, we would argue in favor of a causative role of mTORC1 in the etiology of brain disorders where behavioral abnormalities are corrected either by pharmacological or genetic manipulations of mTORC1.
Most research investigating mTOR signaling in the brain is focused on mTORC1, with very little known about mTORC2. The contribution of mTORC2 to the functions of the central nervous system needs to be clarified. Moreover, since mTOR is a component of both mTORC1 and mTORC2, there may be a dynamic equilibrium regarding the relative amount of mTOR engaged in the two mTORCs. Furthermore, it will be important to understand whether there is interplay between mTORC1 and mTORC2 during normal physiological processes in the brain, in neurological disorders and/or in response to psychoactive drugs.
Finally, the mTORC1 signaling pathway and its impact on protein synthesis is well characterized in some brain regions and cell types such as glutamatergic pyramidal neurons of the hippocampus, but it has not been rigorously explored in other regions. For example, mechanisms of translational control in striatal GABArgic neurons and/or glial cells have not been studied intensely. Thus, in the future, more thorough studies of the mechanisms that regulate translation will be necessary in diverse brain regions and cell types in order to acquire a better understanding of how proteins are synthesized throughout the brain in response to diverse stimuli.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ali, I. U., Schriml, L. M., and Dean, M. (1999). Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J. Natl. Cancer Inst. 91, 1922–1932.
An, W.-L., Cowburn, R. F., Li, L., Braak, H., Alafuzoff, I., Iqbal, K., Iqbal, I.-G., Winblad, B., and Pei, J.-J. (2003). Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 163, 591–607.
Antion, M. D., Hou, L., Wong, H., Hoeffer, C. A., and Klann, E. (2008a). mGluR-dependent long-term depression is associated with increased phosphorylation of S6 and synthesis of elongation factor 1A but remains expressed in S6K-deficient mice. Mol. Cell. Biol. 28, 2996–3007.
Antion, M. D., Merhav, M., Hoeffer, C. A., Reis, G., Kozma, S. C., Thomas, G., Schuman, E. M., Rosenblum, K., and Klann, E. (2008b). Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn. Mem. 15, 29–38.
Autry, A. E., Adachi, M., Nosyreva, E., Na, E. S., Los, M. F., Cheng, P. F., Kavalali, E. T., and Monteggia, L. M. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95.
Averous, J., and Proud, C. G. (2006). When translation meets transformation: the mTOR story. Oncogene 25, 6423–6435.
Banko, J. L., Hou, L., Poulin, F., Sonenberg, N., and Klann, E. (2006). Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 26, 2167–2173.
Banko, J. L., and Klann, E. (2008). Cap-dependent translation initiation and memory. Prog. Brain Res. 169, 59–80.
Banko, J. L., Merhav, M., Stern, E., Sonenberg, N., Rosenblum, K., and Klann, E. (2007). Behavioral alterations in mice lacking the translation repressor 4E-BP2. Neurobiol. Learn. Mem. 87, 248–256.
Banko, J. L., Poulin, F., Hou, L., Demaria, C. T., Sonenberg, N., and Klann, E. (2005). The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J. Neurosci. 25, 9581–9590.
Beretta, L., Gingras, A. C., Svitkin, Y. V., Hall, M. N., and Sonenberg, N. (1996). Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 15, 658–664.
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., Charney, D. S., and Krystal, J. H. (2000). Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354.
Berridge, K. C., and Robinson, T. E. (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 28, 309–369.
Bezard, E., Brotchie, J. M., and Gross, C. E. (2001). Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat. Rev. Neurosci. 2, 577–588.
Bidinosti, M., Ran, I., Sanchez-Carbente, M. R., Martineau, Y., Gingras, A.-C., Gkogkas, C., Raught, B., Bramham, C. R., Sossin, W. S., Costa-Mattioli, M., Desgroseillers, L., Lacaille, J.-C., and Sonenberg, N. (2010). Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission. Mol. Cell 37, 797–808.
Binder, L. I., Guillozet-Bongaarts, A. L., Garcia-Sierra, F., and Berry, R. W. (2005). Tau, tangles, and Alzheimer’s disease. Biochim. Biophys. Acta 1739, 216–223.
Blackwood, D. H., Fordyce, A., Walker, M. T., St Clair, D. M., Porteous, D. J., and Muir, W. J. (2001). Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am. J. Hum. Genet. 69, 428–433.
Butler, M. G., Dasouki, M. J., Zhou, X.-P., Talebizadeh, Z., Brown, M., Takahashi, T. N., Miles, J. H., Wang, C. H., Stratton, R., Pilarski, R., and Eng, C. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321.
Caccamo, A., Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120.
Cammalleri, M., Lutjens, R., Berton, F., King, A. R., Simpson, C., Francesconi, W., and Sanna, P. P. (2003). Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc. Natl. Acad. Sci. U.S.A. 100, 14368–14373.
Chalecka-Franaszek, E., and Chuang, D. M. (1999). Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. U.S.A. 96, 8745–8750.
Chang, R. C.-C., Suen, K.-C., Ma, C.-H., Elyaman, W., Ng, H.-K., and Hugon, J. (2002a). Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J. Neurochem. 83, 1215–1225.
Chang, R. C. C., Wong, A. K. Y., Ng, H.-K., and Hugon, J. (2002b). Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13, 2429–2432.
Chen, L., and Toth, M. (2001). Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103, 1043–1050.
Chevere-Torres, I., Maki, J. M., Santini, E., and Klann, E. (2012). Impaired social interactions and motor learning in tubero sclerosis complex model mice. Neurobiol. Dis. (in press).
Childress, A. R., Mozley, P. D., Mcelgin, W., Fitzgerald, J., Reivich, M., and O’Brien, C. P. (1999). Limbic activation during cue-induced cocaine craving. Am. J. Psychiatry 156, 11–18.
Chubb, J. E., Bradshaw, N. J., Soares, D. C., Porteous, D. J., and Millar, J. K. (2008). The DISC locus in psychiatric illness. Mol. Psychiatry 13, 36–64.
Clark, L., Chamberlain, S. R., and Sahakian, B. J. (2009). Neurocognitive mechanisms in depression: implications for treatment. Annu. Rev. Neurosci. 32, 57–74.
Costa-Mattioli, M., and Sonenberg, N. (2008). Translational control of gene expression: a molecular switch for memory storage. Prog. Brain Res. 169, 81–95.
Costa-Mattioli, M., Sonenberg, N., and Richter, J. D. (2009a). Chapter 8 – translational regulatory mechanisms in synaptic plasticity and memory storage. Prog. Mol. Biol. Transl. Sci. 90, 293–311.
Costa-Mattioli, M., Sossin, W. S., Klann, E., and Sonenberg, N. (2009b). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26.
Cutler, N. S., Heitman, J., and Cardenas, M. E. (1999). TOR kinase homologs function in a signal transduction pathway that is conserved from yeast to mammals. Mol. Cell. Endocrinol. 155, 135–142.
Dagestad, G., Kuipers, S. D., Messaoudi, E., and Bramham, C. R. (2006). Chronic fluoxetine induces region-specific changes in translation factor eIF4E and eEF2 activity in the rat brain. Eur. J. Neurosci. 23, 2814–2818.
Darmopil, S., Martín, A., Diego, I., Ares, S., and Moratalla, R. (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613.
Dash, P. K., Orsi, S. A., and Moore, A. N. (2006). Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J. Neurosci. 26, 8048–8056.
Davies, S. W., Turmaine, M., Cozens, B. A., Difiglia, M., Sharp, A. H., Ross, C. A., Scherzinger, E., Wanker, E. E., Mangiarini, L., and Bates, G. P. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548.
Davis, K. L., Kahn, R. S., Ko, G., and Davidson, M. (1991). Dopamine in schizophrenia: a review and reconceptualization. Am. J. Psychiatry 148, 1474–1486.
Deyoung, M. P., Horak, P., Sofer, A., Sgroi, D., and Ellisen, L. W. (2008). Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 22, 239–251.
D’Hooge, R., Nagels, G., Franck, F., Bakker, C. E., Reyniers, E., Storm, K., Kooy, R. F., Oostra, B. A., Willems, P. J., and De Deyn, P. P. (1997). Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76, 367–376.
Di Chiara, G., and North, R. (1992). Neurobiology of opiate abuse. Trends Pharmacol. Sci. 13, 185–193.
Dölen, G., Osterweil, E., Rao, B. S. S., Smith, G. B., Auerbach, B. D., Chattarji, S., and Bear, M. F. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962.
Ehninger, D., Han, S., Shilyansky, C., Zhou, Y., Li, W., Kwiatkowski, D. J., Ramesh, V., and Silva, A. J. (2008a). Reversal of learning deficits in a Tsc2± mouse model of tuberous sclerosis. Nat. Med. 14, 843–848.
Ehninger, D., Li, W., Fox, K., Stryker, M. P., and Silva, A. J. (2008b). Reversing neurodevelopmental disorders in adults. Neuron 60, 950–960.
Feig, S., and Lipton, P. (1993). Pairing the cholinergic agonist carbachol with patterned Schaffer collateral stimulation initiates protein synthesis in hippocampal CA1 pyramidal cell dendrites via a muscarinic, NMDA-dependent mechanism. J. Neurosci. 13, 1010–1021.
Fletcher, C. M., Mcguire, A. M., Gingras, A. C., Li, H., Matsuo, H., Sonenberg, N., and Wagner, G. (1998). 4E binding proteins inhibit the translation factor eIF4E without folded structure. Biochemistry 37, 9–15.
Flexner, J. B., Flexner, L. B., and Stellar, E. (1963). Memory in mice as affected by intracerebral puromycin. Science 141, 57–59.
Gaoni, Y., and Mechoulam, R. (1964). Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 86, 1646–1647.
Garami, A., Zwartkruis, F. J. T., Nobukuni, T., Joaquin, M., Roccio, M., Stocker, H., Kozma, S. C., Hafen, E., Bos, J. L., and Thomas, G. (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466.
Gawin, F., and Kleber, H. (1986a). Pharmacologic treatments of cocaine abuse. Psychiatr. Clin. North Am. 9, 573–583.
Gawin, F. H., and Kleber, H. D. (1986b). Abstinence symptomatology and psychiatric diagnosis in cocaine abusers. Clinical observations. Arch. Gen. Psychiatry 43, 107–113.
Gelinas, J. N., Banko, J. L., Hou, L., Sonenberg, N., Weeber, E. J., Klann, E., and Nguyen, P. V. (2007). ERK and mTOR signaling couple beta-adrenergic receptors to translation initiation machinery to gate induction of protein synthesis-dependent long-term potentiation. J. Biol. Chem. 282, 27527–27535.
Gerfen, C. R., Miyachi, S., Paletzki, R., and Brown, P. (2002). D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J. Neurosci. 22, 5042–5054.
Gingras, A. C., Raught, B., Gygi, S. P., Niedzwiecka, A., Miron, M., Burley, S. K., Polakiewicz, R. D., Wyslouch-Cieszynska, A., Aebersold, R., and Sonenberg, N. (2001). Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 15, 2852–2864.
Gingras, A. C., Raught, B., and Sonenberg, N. (1999). eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68, 913–963.
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890.
Goffin, A., Hoefsloot, L. H., Bosgoed, E., Swillen, A., and Fryns, J. P. (2001). PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 105, 521–524.
Goorden, S. M. I., Van Woerden, G. M., Van Der Weerd, L., Cheadle, J. P., and Elgersma, Y. (2007). Cognitive deficits in Tsc1± mice in the absence of cerebral lesions and seizures. Ann. Neurol. 62, 648–655.
Götz, J., and Ittner, L. M. (2008). Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 9, 532–544.
Grice, D. E., and Buxbaum, J. D. (2006). The genetics of autism spectrum disorders. Neuromolecular Med. 8, 451–460.
Gross, C., Nakamoto, M., Yao, X., Chan, C. B., Yim, S. Y., Ye, K., Warren, S. T., and Bassell, G. J. (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638.
Haghighat, A., Mader, S., Pause, A., and Sonenberg, N. (1995). Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 14, 5701–5709.
Hampson, R. E., and Deadwyler, S. A. (1999). Cannabinoids, hippocampal function and memory. Life Sci. 65, 715–723.
Harrison, P. J. (1999). The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain 122(Pt 4), 593–624.
Hill, E. L., and Frith, U. (2003). Understanding autism: insights from mind and brain. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 281–289.
Hoeffer, C. A., Cowansage, K. K., Arnold, E. C., Banko, J. L., Moerke, N. J., Rodriguez, R., Schmidt, E. K., Klosi, E., Chorev, M., Lloyd, R. E., Pierre, P., Wagner, G., Ledoux, J. E., and Klann, E. (2011). Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proc. Natl. Acad. Sci. U.S.A. 108, 3383–3388.
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75.
Hoeffer, C. A., Tang, W., Wong, H., Santillan, A., Patterson, R. J., Martinez, L. A., Tejada-Simon, M. V., Paylor, R., Hamilton, S. L., and Klann, E. (2008). Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron 60, 832–845.
Hou, L., Antion, M. D., Hu, D., Spencer, C. M., Paylor, R., and Klann, E. (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454.
Hou, L., and Klann, E. (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361.
Howes, O. D., and Kapur, S. (2009). The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr. Bull. 35, 549–562.
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257.
Inoki, K., Li, Y., Xu, T., and Guan, K.-L. (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834.
Ittner, L. M., and Götz, J. (2011). Amyloid-(ß and tau–a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 65–72.
Jacinto, E., and Hall, M. N. (2003). Tor signalling in bugs, brain and brawn. Nat. Rev. Mol. Cell Biol. 4, 117–126.
Kalkman, H. O. (2006). The role of the phosphatidylinositide 3-kinase-protein kinase B pathway in schizophrenia. Pharmacol. Ther. 110, 117–134.
Kang, H., and Schuman, E. M. (1996). A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402–1406.
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., Multhaup, G., Beyreuther, K., and Müller-Hill, B. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736.
Kelleher, R. J., and Bear, M. F. (2008). The autistic neuron: troubled translation? Cell 135, 401–406.
Kim, D.-H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., Tempst, P., and Sabatini, D. M. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175.
Kim, J. Y., Duan, X., Liu, C. Y., Jang, M.-H., Guo, J. U., Pow-Anpongkul, N., Kang, E., Song, H., and Ming, G.-L. (2009). DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63, 761–773.
Klann, E., and Dever, T. E. (2004). Biochemical mechanisms for translational regulation in synaptic plasticity. Nat. Rev. Neurosci. 5, 931–942.
Klann, E., and Sweatt, J. D. (2008). Altered protein synthesis is a trigger for long-term memory formation. Neurobiol. Learn. Mem. 89, 247–259.
Kooy, R. F., D’Hooge, R., Reyniers, E., Bakker, C. E., Nagels, G., De Boulle, K., Storm, K., Clincke, G., De Deyn, P. P., Oostra, B. A., and Willems, P. J. (1996). Transgenic mouse model for the fragile X syndrome. Am. J. Med. Genet. 64, 241–245.
Krishnan, V., and Nestler, E. J. (2008). The molecular neurobiology of depression. Nature 455, 894–902.
Kruzich, P. J., and See, R. E. (2001). Differential contributions of the basolateral and central amygdala in the acquisition and expression of conditioned relapse to cocaine-seeking behavior. J. Neurosci. 21, RC155.
Kwiatkowski, D. J., and Manning, B. D. (2005). Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 14, R251–R258.
Kwon, C.-H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., Li, Y., Baker, S. J., and Parada, L. F. (2006a). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388.
Kwon, C.-H., Zhou, J., Li, Y., Kim, K. W., Hensley, L. L., Baker, S. J., and Parada, L. F. (2006b). Neuron-specific enolase-cre mouse line with cre activity in specific neuronal populations. Genesis 44, 130–135.
Lafay-Chebassier, C., Paccalin, M., Page, G., Barc-Pain, S., Perault-Pochat, M. C., Gil, R., Pradier, L., and Hugon, J. (2005). mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 94, 215–225.
Levitt, P., and Campbell, D. B. (2009). The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J. Clin. Invest. 119, 747–754.
Li, X., Alafuzoff, I., Soininen, H., Winblad, B., and Pei, J.-J. (2005). Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 272, 4211–4220.
Li, X., An, W.-L., Alafuzoff, I., Soininen, H., Winblad, B., and Pei, J.-J. (2004). Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport 15, 2237–2240.
Lu, X.-H., and Dwyer, D. S. (2005). Second-generation antipsychotic drugs, olanzapine, quetiapine, and clozapine enhance neurite outgrowth in PC12 cells via PI3K/AKT, ERK, and pertussis toxin-sensitive pathways. J. Mol. Neurosci. 27, 43–64.
Ma, T., Hoeffer, C. A., Capetillo-Zarate, E., Yu, F., Wong, H., Lin, M. T., Tampellini, D., Klann, E., Blitzer, R. D., and Gouras, G. K. (2010). Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS ONE 5, e12845.
MacDonald, M. E., Barnes, G., Srinidhi, J., Duyao, M. P., Ambrose, C. M., Myers, R. H., Gray, J., Conneally, P. M., Young, A., and Penney, J. (1993). Gametic but not somatic instability of CAG repeat length in Huntington’s disease. J. Med. Genet. 30, 982–986.
Maehama, T., and Dixon, J. E. (1999). PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 9, 125–128.
Malagelada, C., Jin, Z. H., and Greene, L. A. (2008). RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 28, 14363–14371.
Malagelada, C., Jin, Z. H., Jackson-Lewis, V., Przedborski, S., and Greene, L. A. (2010). Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 30, 1166–1175.
Malagelada, C., Ryu, E. J., Biswas, S. C., Jackson-Lewis, V., and Greene, L. A. (2006). RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci. 26, 9996–10005.
Maldonado, R., and Rodriguez De Fonseca, F. (2002). Cannabinoid addiction: behavioral models and neural correlates. J. Neurosci. 22, 3326–3331.
Mameli, M., Balland, B., Luján, R., and Lüscher, C. (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317, 530–533.
Manning, B. D., and Cantley, L. C. (2003a). Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 28, 573–576.
Manning, B. D., and Cantley, L. C. (2003b). United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem. Soc. Trans. 31, 573–578.
Martin, K. A., and Blenis, J. (2002). Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv. Cancer Res. 86, 1–39.
Meil, W. M., and See, R. E. (1997). Lesions of the basolateral amygdala abolish the ability of drug associated cues to reinstate responding during withdrawal from self-administered cocaine. Behav. Brain Res. 87, 139–148.
Middleton, F. A., Pato, C. N., Gentile, K. L., Mcgann, L., Brown, A. M., Trauzzi, M., Diab, H., Morley, C. P., Medeiros, H., Macedo, A., Azevedo, M. H., and Pato, M. T. (2005). Gene expression analysis of peripheral blood leukocytes from discordant sib-pairs with schizophrenia and bipolar disorder reveals points of convergence between genetic and functional genomic approaches. Am. J. Med. Genet. B Neuropsychiatr. Genet. 136B, 12–25.
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., Devon, R. S., St Clair, D. M., Muir, W. J., Blackwood, D. H., and Porteous, D. J. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423.
Mineur, Y. S., Sluyter, F., De Wit, S., Oostra, B. A., and Crusio, W. E. (2002). Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus 12, 39–46.
Muddashetty, R. S., Kelic, S., Gross, C., Xu, M., and Bassell, G. J. (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 27, 5338–5348.
Musumeci, S. A., Bosco, P., Calabrese, G., Bakker, C., De Sarro, G. B., Elia, M., Ferri, R., and Oostra, B. A. (2000). Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41, 19–23.
Nanxin, L., Lee, B., Liu, R.-J., Banasr, M., Dwyer, J. M., Iwata, M., Li, X.-Y., Aghajanian, G., and Duman, R. S. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964.
Napoli, I., Mercaldo, V., Boyl, P. P., Eleuteri, B., Zalfa, F., De Rubeis, S., Di Marino, D., Mohr, E., Massimi, M., Falconi, M., Witke, W., Costa-Mattioli, M., Sonenberg, N., Achsel, T., and Bagni, C. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054.
Narayanan, U., Nalavadi, V., Nakamoto, M., Thomas, G., Ceman, S., Bassell, G. J., and Warren, S. T. (2008). S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J. Biol. Chem. 283, 18478–18482.
Nestler, E. (2004). Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol. Sci. 25, 210–218.
Nestor, P. J., Fryer, T. D., and Hodges, J. R. (2006). Declarative memory impairments in Alzheimer’s disease and semantic dementia. Neuroimage 30, 1010–1020.
Neves, G., Cooke, S. F., and Bliss, T. V. P. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75.
Nielsen, D. M., Derber, W. J., Mcclellan, D. A., and Crnic, L. S. (2002). Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 927, 8–17.
O’Brien, C. P. (1997). A range of research-based pharmacotherapies for addiction. Science 278, 66–70.
Onuki, R., Bando, Y., Suyama, E., Katayama, T., Kawasaki, H., Baba, T., Tohyama, M., and Taira, K. (2004). An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. EMBO J. 23, 959–968.
Osterweil, E. K., Krueger, D. D., Reinhold, K., and Bear, M. F. (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 30, 15616–15627.
Page, G., Rioux Bilan, A., Ingrand, S., Lafay-Chebassier, C., Pain, S., Perault Pochat, M. C., Bouras, C., Bayer, T., and Hugon, J. (2006). Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 139, 1343–1354.
Papp, E., Leinekugel, X., Henze, D. A., Lee, J., and Buzsáki, G. (2001). The apical shaft of CA1 pyramidal cells is under GABAergic interneuronal control. Neuroscience 102, 715–721.
Parkinson, J. (2002). An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 14, 223–236; discussion 222.
Pause, A., Belsham, G. J., Gingras, A. C., Donzé, O., Lin, T. A., Lawrence, J. C., and Sonenberg, N. (1994). Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5’-cap function. Nature 371, 762–767.
Peel, A. L., and Bredesen, D. E. (2003). Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice. Neurobiol. Dis. 14, 52–62.
Pei, J.-J., Björkdahl, C., Zhang, H., Zhou, X., and Winblad, B. (2008). p70 S6 kinase and tau in Alzheimer’s disease. J. Alzheimers Dis. 14, 385–392.
Pei, J.-J., and Hugon, J. (2008). mTOR-dependent signalling in Alzheimer’s disease. J. Cell. Mol. Med. 12, 2525–2532.
Persico, A. M., and Bourgeron, T. (2006). Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 29, 349–358.
Pierce, R., and Kumaresan, V. (2006). The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci. Biobehav. Rev. 30, 215–238.
Piomelli, D. (2003). The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4, 873–884.
Poulin, F., Gingras, A. C., Olsen, H., Chevalier, S., and Sonenberg, N. (1998). 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J. Biol. Chem. 273, 14002–14007.
Price, R. B., Nock, M. K., Charney, D. S., and Mathew, S. J. (2009). Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol. Psychiatry 66, 522–526.
Puighermanal, E., Marsicano, G., Busquets-Garcia, A., Lutz, B., Maldonado, R., and Ozaita, A. (2009). Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat. Neurosci. 12, 1152–1158.
Pyronnet, S., Imataka, H., Gingras, A. C., Fukunaga, R., Hunter, T., and Sonenberg, N. (1999). Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 18, 270–279.
Qin, M., Kang, J., Burlin, T. V., Jiang, C., and Smith, C. B. (2005). Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J. Neurosci. 25, 5087–5095.
Raught, B., Gingras, A. C., Gygi, S. P., Imataka, H., Morino, S., Gradi, A., Aebersold, R., and Sonenberg, N. (2000). Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 19, 434–444.
Raught, B., Peiretti, F., Gingras, A.-C., Livingstone, M., Shahbazian, D., Mayeur, G. L., Polakiewicz, R. D., Sonenberg, N., and Hershey, J. W. B. (2004). Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23, 1761–1769.
Ravikumar, B., Duden, R., and Rubinsztein, D. C. (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117.
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., Scaravilli, F., Easton, D. F., Duden, R., O’Kane, C. J., and Rubinsztein, D. C. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595.
Richter, J. D., and Klann, E. (2009). Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 23, 1–11.
Rogers, G. W., Lima, W. F., and Merrick, W. C. (2001a). Further characterization of the helicase activity of eIF4A. Substrate specificity. J. Biol. Chem. 276, 12598–12608.
Rogers, G. W., Richter, N. J., Lima, W. F., and Merrick, W. C. (2001b). Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J. Biol. Chem. 276, 30914–30922.
Rogers, G. W., Richter, N. J., and Merrick, W. C. (1999). Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem. 274, 12236–12244.
Rubinsztein, D. C. (2002). Lessons from animal models of Huntington’s disease. Trends Genet. 18, 202–209.
Santini, E., Alcacer, C., Cacciatore, S., Heiman, M., Hervé, D., Greengard, P., Girault, J.-A., Valjent, E., and Fisone, G. (2009a). l-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 108, 621–633.
Santini, E., Heiman, M., Greengard, P., Valjent, E., and Fisone, G. (2009b). Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2, ra36.
Santini, E., Valjent, E., and Fisone, G. (2008). Parkinson’s disease: levodopa-induced dyskinesia and signal transduction. FEBS J. 275, 1392–1399.
Sarbassov, D. D., Ali, S. M., Kim, D.-H., Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., Tempst, P., and Sabatini, D. M. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302.
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J.-H., Hsu, P. P., Bagley, A. F., Markhard, A. L., and Sabatini, D. M. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168.
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66.
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., Yamrom, B., Yoon, S., Krasnitz, A., Kendall, J., Leotta, A., Pai, D., Zhang, R., Lee, Y.-H., Hicks, J., Spence, S. J., Lee, A. T., Puura, K., Lehtimäki, T., Ledbetter, D., Gregersen, P. K., Bregman, J., Sutcliffe, J. S., Jobanputra, V., Chung, W., Warburton, D., King, M.-C., Skuse, D., Geschwind, D. H., Gilliam, T. C., Ye, K., and Wigler, M. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449.
Shah, O. J., Wang, Z., and Hunter, T. (2004). Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14, 1650–1656.
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., Mcbride, S. M., Klann, E., and Zukin, R. S. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702.
Shi, J., Jun, W., Zhao, L.-Y., Xue, Y.-X., Zhang, X.-Y., Kosten, T. R., and Lu, L. (2009). Effect of rapamycin on cue-induced drug craving in abstinent heroin addicts. Eur. J. Pharmacol. 615, 108–112.
Siuta, M. A., Robertson, S. D., Kocalis, H., Saunders, C., Gresch, P. J., Khatri, V., Shiota, C., Kennedy, J. P., Lindsley, C. W., Daws, L. C., Polley, D. B., Veenstra-Vanderweele, J., Stanwood, G. D., Magnuson, M. A., Niswender, K. D., and Galli, A. (2010). Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice. PLoS Biol. 8, e1000393.
Sossin, W. S., and Lacaille, J. C. (2010). Mechanisms of translational regulation in synaptic plasticity. Curr. Opin. Neurobiol. 20, 450–456.
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., Richardson, A., Strong, R., and Galvan, V. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 5, e9979.
Steward, O., and Levy, W. B. (1982). Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J. Neurosci. 2, 284–291.
Steward, O., and Schuman, E. M. (2001). Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci. 24, 299–325.
Swiech, L., Perycz, M., Malik, A., and Jaworski, J. (2008). Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 1784, 116–132.
Tang, S. J., Reis, G., Kang, H., Gingras, A. C., Sonenberg, N., and Schuman, E. M. (2002). A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 99, 467–472.
Tanzi, R. E., St George-Hyslop, P. H., Haines, J. L., Polinsky, R. J., Nee, L., Foncin, J. F., Neve, R. L., Mcclatchey, A. I., Conneally, P. M., and Gusella, J. F. (1987). The genetic defect in familial Alzheimer’s disease is not tightly linked to the amyloid beta-protein gene. Nature 329, 156–157.
Tischmeyer, W., Schicknick, H., Kraus, M., Seidenbecher, C. I., Staak, S., Scheich, H., and Gundelfinger, E. D. (2003). Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur. J. Neurosci. 18, 942–950.
Todd, P. K., Mack, K. J., and Malter, J. S. (2003). The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl. Acad. Sci. U.S.A. 100, 14374–14378.
Tsokas, P., Grace, E. A., Chan, P., Ma, T., Sealfon, S. C., Iyengar, R., Landau, E. M., and Blitzer, R. D. (2005). Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J. Neurosci. 25, 5833–5843.
Van Dam, D., D’Hooge, R., Hauben, E., Reyniers, E., Gantois, I., Bakker, C. E., Oostra, B. A., Kooy, R. F., and De Deyn, P. P. (2000). Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav. Brain Res. 117, 127–136.
Volarevic, S., Stewart, M. J., Ledermann, B., Zilberman, F., Terracciano, L., Montini, E., Grompe, M., Kozma, S. C., and Thomas, G. (2000). Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288, 2045–2047.
Wang, X., Li, W., Williams, M., Terada, N., Alessi, D. R., and Proud, C. G. (2001). Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20, 4370–4379.
Wang, X., Luo, Y.-X., He, Y.-Y., Li, F.-Q., Shi, H.-S., Xue, L.-F., Xue, Y.-X., and Lu, L. (2010). Nucleus accumbens core mammalian target of rapamycin signaling pathway is critical for cue-induced reinstatement of cocaine seeking in rats. J. Neurosci. 30, 12632–12641.
Wang, X., and Proud, C. G. (2006). The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 21, 362–369.
Waskiewicz, A. J., Johnson, J. C., Penn, B., Mahalingam, M., Kimball, S. R., and Cooper, J. A. (1999). Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol. 19, 1871–1880.
Weinberger, D. R., Berman, K. F., and Illowsky, B. P. (1988). Physiological dysfunction of dorsolateral prefrontal cortex in schizophrenia. III. A new cohort and evidence for a monoaminergic mechanism. Arch. Gen. Psychiatry 45, 609–615.
Westin, J. E., Vercammen, L., Strome, E. M., Konradi, C., and Cenci, M. A. (2007). Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol. Psychiatry 62, 800–810.
Wu, J., Mccallum, S. E., Glick, S. D., and Huang, Y. (2011). Inhibition of the mammalian target of rapamycin pathway by rapamycin blocks cocaine-induced locomotor sensitization. Neuroscience 172, 104–109.
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484.
Young, D. M., Schenk, A. K., Yang, S.-B., Jan, Y. N., and Jan, L. Y. (2010). Altered ultrasonic vocalizations in a tuberous sclerosis mouse model of autism. Proc. Natl. Acad. Sci. U.S.A. 107, 11074–11079.
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., Oostra, B., and Bagni, C. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327.
Zarate, C. A., Singh, J. B., Carlson, P. J., Brutsche, N. E., Ameli, R., Luckenbaugh, D. A., Charney, D. S., and Manji, H. K. (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864.
Zhang, Y., Gao, X., Saucedo, L. J., Ru, B., Edgar, B. A., and Pan, D. (2003). Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 5, 578–581.
Zhao, Z., Ksiezak-Reding, H., Riggio, S., Haroutunian, V., and Pasinetti, G. M. (2006). Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr. Res. 84, 1–14.
Zhou, J., Blundell, J., Ogawa, S., Kwon, C.-H., Zhang, W., Sinton, C., Powell, C. M., and Parada, L. F. (2009). Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 29, 1773–1783.
Keywords: protein synthesis, translation initiation, mTORC1 signaling, S6K1, eIF4E, developmental disorders, neurodegenerative diseases, psychoactive drugs
Citation: Santini E and Klann E (2011) Dysregulated mTORC1-dependent translational control: from brain disorders to psychoactive drugs. Front. Behav. Neurosci. 5:76. doi: 10.3389/fnbeh.2011.00076
Received: 27 July 2011; Accepted: 21 October 2011;
Published online: 08 November 2011.
Edited by:
Gilberto Fisone, Karolinska Institutet, SwedenReviewed by:
Clive R. Bramham, University of Bergen, NorwayCopyright: © 2011 Santini and Klann. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Eric Klann, Center for Neural Science, New York University, New York, NY 10003, USA. e-mail:ZWtsYW5uQGNucy5ueXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.