
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Nanotechnol. , 26 October 2022
Sec. Nanomaterials
Volume 4 - 2022 | https://doi.org/10.3389/fnano.2022.1034795
 Qian Mao1
Qian Mao1 Yuwei Zhang2Malgorzata Kowalik1
Yuwei Zhang2Malgorzata Kowalik1 Nadire Nayir1,3,4
Nadire Nayir1,3,4 Michael Chandross5Adri C. T. van Duin1,2,4*
Michael Chandross5Adri C. T. van Duin1,2,4*An atomistic modeling tool is essential to an in-depth understanding upon surface reactions of transition metal dichalcogenides (TMDs), such as molybdenum disulfide (MoS2), with the presence of compositing agents, including Ti and Au, under different environmental exposures. We report a new ReaxFF reactive force field parameter set for Mo, Ti, Au, O, S, and H interactions. We apply the force field in a series of molecular dynamics (MD) simulations to unravel the impact of the Ti dopant on the oxidation/hydrogenation behaviors of MoS2 surface. The simulation results reveal that, in the absence of Ti clusters, the MoS2 surface is ruptured and oxidized at elevated temperatures through a process of adsorption followed by dissociation of the O2 molecules on the MoS2 surface during the temperature ramp. When the MoS2 surface is exposed to H2O molecules, surface hydrogenation is most favored, followed by oxidation, then hydroxylation. The introduction of Ti clusters to the systems mitigates the oxidation/hydrogenation of MoS2 at a low or intermediate temperature by capturing the O2/H2O molecules and locking the O/H-related radicals inside the clusters. However, OH− and H3O+ are emitted from the Ti clusters in the H2O environment as temperature rises, and the accelerating hydrogenation of MoS2 is consequently observed at an ultra-high temperature. These findings indicate an important but complex role of Ti dopants in mitigating the oxidation and hydrogenation of MoS2 under different environmental exposures. The possible mechanisms of oxidation and hydrogenation revealed by MD simulations can give an insight to the design of oxidation resistant TMDs and can be useful to the optical, electronic, magnetic, catalytic, and energy harvesting industries.
Lamellar solids exhibit low friction due to the low shear strength resulting from weak van der Waals interactions between lamellae (Spalvins, 1987; Scharf and Prasad, 2013; Ouyang et al., 2022). Lamellar solids, such as transition metal dichalcogenides (TMDs), are not only candidates for solid lubricants utilized in extreme environments, but also considered promising semiconductor materials (Street, 2009; Liu et al., 2011; Radisavljevic et al., 2011; Marian et al., 2022). The stability of the two-dimensional (2D) structure of lamellar solids is important in terms of maintaining their desired properties and long-term functionalities, e.g., tribological response (Hilton and Fleischauer, 1992; Curry et al., 2016; Vazirisereshk et al., 2019). However, the mechanism of lamellar degradation is still not well understood. This work aims to reveal the mechanisms of the thermal degradation of molybdenum disulfide (MoS2), a typical solid lubricant material, in the presence of oxygen (O2) and water molecules (H2O).
Several thermal etching mechanisms have been proposed to explain the oxidation of MoS2 crystals (Szoszkiewicz, 2021). Oxidation products, such as MoO3, were detected at room temperature at the edges of layered MoS2 flakes (Walter et al., 2017). In the basal planes of MoS2, the O atoms could directly bond with Mo atoms at temperatures higher than 300°C (Yamamoto et al., 2013; Wu et al., 2013; Zhou et al., 2013; Ukegbu and Szoszkiewicz, 2019). In the presence of H2O, however, the volatile species, such as MoO2(OH)2, were expected to emerge at temperatures higher than 300°C, leading to more rapid MoS2 oxidation compared to that in dry air conditions (Walter et al., 2017). In addition to volatile oxidation products, the presence of H2SO4 as well as sulfur oxides at temperatures above 400°C was proposed (Ross and Sussman, 1955). The passivation layer of MoO3 forms at temperatures lower than 500°C, while amorphous Mo oxides and MoOx clusters appeared above 500°C (Szoszkiewicz et al., 2020; Rogala et al., 2021; Park et al., 2021a). These alterations to the MoS2 surface upon oxidation in humid environments were detrimental to desired tribological behaviors of MoS2 thin films. Curry et al. (2017) investigated the oxidation behavior of various microstructures of MoS2 thin films and the effects on the shear strength between MoS2 basal planes. It was shown that films with ordered microstructures were resistant to both O atoms and O2 molecules at even elevated temperatures, while amorphous microstructures provided more edge sites for O atoms and O2 to penetrate and react throughout the depth of the film. Such findings imply that controlling the number of edges during the epitaxial growth of TMD thin films leads to robust stability of as-grown 2D materials. Apart from growth control, the introduction of compositing agents, such as Ti and Au, has been demonstrated to mitigate the detrimental effects of oxidation to TMD thin films in humid environments (Colas et al., 2015; Li et al., 2017). The underlying mechanism responsible for the improved resistance to the oxidation and humidity has been reported to be a sacrificial effect of dopant metals, protecting the MoS2 from interacting with O2 and H2O molecules.
Since the microstructure of MoS2 as well as the presence of Ti in the system, plays an important role in resistance to oxidation and humidity, we developed a new ReaxFF parameter set describing Mo, Ti, O, S, and H interactions. A series of molecular dynamics (MD) simulations were performed at different temperatures with the presence of O2 and H2O molecules. The simulation results reveal several possible kinetic pathways of the oxidation of the MoS2 surface when exposed to O2 molecules, and the hydrogenation or hydroxylation when exposed to H2O molecules. The same series of simulations were also performed with the introduction of Ti clusters to the systems to identify the mechanisms responsible for the prevention of oxidation/hydrogenation by the Ti clusters.
Reactive force field (ReaxFF) uses the bond order formalism to treat reactive chemical systems, where bond order is described as a continuous function of interatomic distance (van Duin et al., 2001). Unlike classical force fields that adopt a static bond assumption, ReaxFF updates bond orders at every iteration and allows energetic contributions to disappear upon bond dissociation. Thus, ReaxFF can describe reactive events involving bond formation and breakage with no energy discontinuities over the course of simulations. The potential energy of the system is described as a sum of several partial terms (Senftle et al., 2016),
The simplest form of
Recent ReaxFF force fields have been validated and extensively applied to modeling carbon materials (Mao et al., 2022; Mao et al., 2020; Kowalik et al., 2019; Rajabpour et al., 2021a; Zhang et al., 2020; Vashisth et al., 2020), 2D materials (Curry et al., 2021; Rajabpour et al., 2021b; Nayir et al., 2020; Nayir et al., 2021a; Nayir et al., 2021b; Ostadhossein et al., 2017; Lele et al., 2022; Mojtabavi et al., 2021; Ganeshan et al., 2020), oxidation kinetics of MXenes (Zhao et al., 2020a; Zhao et al., 2019; Lotfi et al., 2018; Zheng et al., 2021) and multiple other materials.
We developed a new ReaxFF reactive force field for C/H/O/S/Mo/Ti/Au interactions to study the thermal degradation, oxidation, and hydrogenation of MoS2 in the presence of compositing agents, i.e., Ti and Au. Prior to the force field training, three ReaxFF force fields were combined for an initial attempt at parameterization. Particularly, the Ti/O/H interactions from the force field ReaxFF Ti/O/H (Raju et al., 2013) and the Au/O/S/C/H interactions from the force field ReaxFF C/H/O/S/Au (Monti et al., 2016) were incorporated into a refitted force field based on ReaxFF Mo/S/O/H (Islam et al., 2014) on the aqueous branch. Then, we trained the C/H/O/S/Mo/Ti/Au parameter set against non-periodic DFT data using Jaguar (Bochevarov et al., 2013) at the B3LYP/LACV3P++** level of theory due to the presence of heavy atoms, i.e., Mo, Au, specifically on bond dissociations of Ti-S, Au-S, and S-H as well as angular distortions of S-Ti-S, Ti-S-H, Ti-S-Ti, Ti-S-Mo, Au-S-Mo, Au-S-H, and H-S-H, drawn from the single molecules of Ti(SH)4, Ti(SH)3-S-Ti(SH)3, Ti(SH)3-S-Mo(SH)3, AuSH, Au-S-Mo(SH)3, and H2S. In addition, the dissociation energy of Ti(SH)3 and SH radicals from Ti(SH)4 and the reaction energies of Ti(SH)3-S-Ti(SH)3, Ti(SH)3-S-Mo(SH)3, and Au-S-Mo(SH)3 with H2S were computed at the DFT level by using Eqs 2–5
During the parameterization, a single-parameter search technique was utilized to minimize the error of the sum of squares (Chenoweth et al., 2008). The comparisons of relative bond and angle energies between the parameterized ReaxFF data and DFT data are depicted in Figure 1, showing good agreement around their equilibrium positions. The ReaxFF and DFT predicted dissociation and reaction relative energies are shown in Supplementary Figure S1, where the energy difference between DFT and ReaxFF falls within 5 kcal/mol. It should be noted that the basis set superposition error (BSSE) may be a problem when small basis sets, such as LACV3P++** for our systems, are used. However, as reported by Schneebeli et al. (Schneebeli et al., 2011), the average BSSE on a gigantic data set is ∼4 kcal/mol for B3LYP/LACVP. For this reason, we typically assume that DFT methods can overestimate/underestimate the energies within the range of 5 kcal/mol due to intrinsically present errors. Since we reach an energy difference between ReaxFF and DFT calculated data within 5 kcal/mol, we assume the energies meet sufficient accuracy.
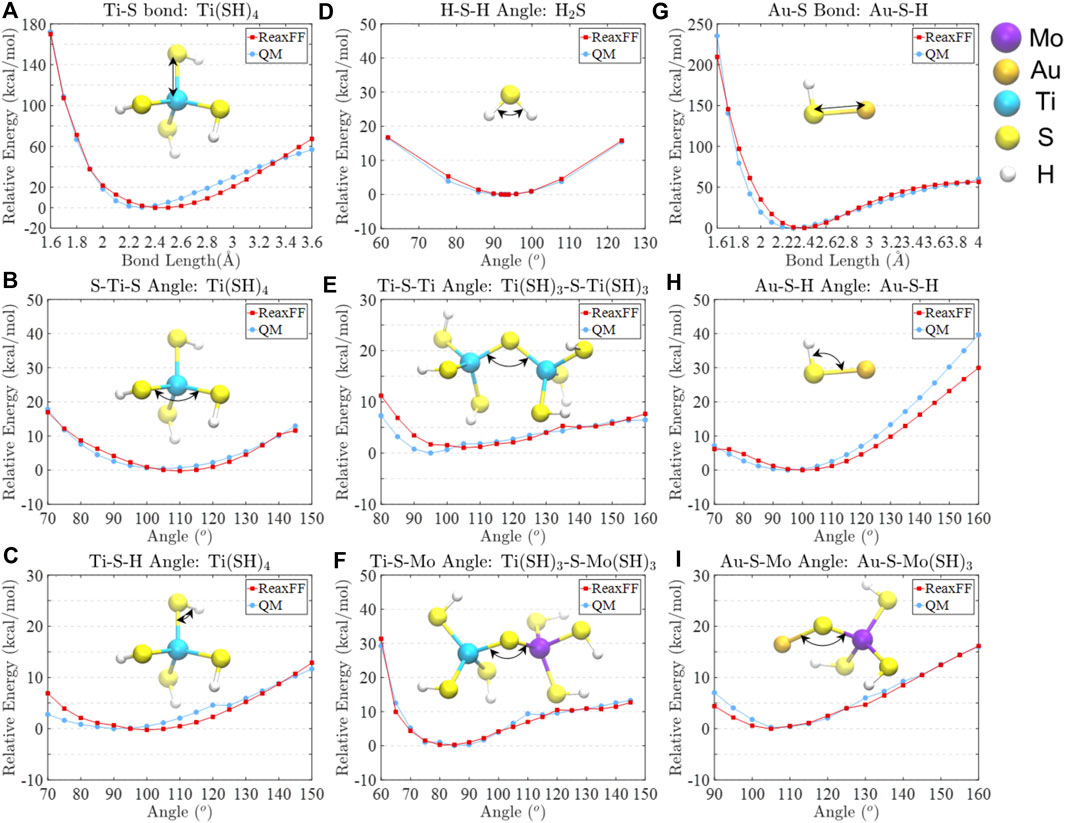
FIGURE 1. Comparisons of relative bond and angle energies between ReaxFF and QM computed data for (A) Ti-S bond in Ti(SH)4, (B) S-Ti-S angle in Ti(SH)4, (C) Ti-S-H angle in Ti(SH)4, (D) H-S-H angle in H2S, (E) Ti-S-Ti angle in Ti(SH)3-S-Ti(SH)3, (F) Ti-S-Mo angle in Ti(SH)3-S-Mo(SH)3, (G) Au-S bond in Au-S-H, (H) Au-S-H angle in Au-S-H, and (I) Au-S-Mo angle in Au-S-Mo(SH)3.
The accuracy and transferability of this newly developed force field were validated by benchmarking small MD simulations on surface reactions of MoS2 with compositing Ti and Au clusters at ambient conditions in order to compare with experimental measurements and DFT calculations in the literature (Wu et al., 2019; Freedy et al., 2019; Szoszkiewicz, 2021; Merida et al., 2018; Liu et al., 2020). In addition, the developed force field exhibits better performance of describing M/O crystalline systems than that using the previously reported parameters (Ostadhossein et al., 2017; Hong et al., 2017; Chen et al., 2020).
In this work, we investigated the surface reactions of monolayer MoS2 with Ti clusters exposed to gas-phase molecules at elevated temperatures. We used two MoS2 substrates, pristine MoS2 (P-MoS2) and defective MoS2 with 5% S-vacancies (D-MoS2). The defects we created are neutral S-vacancies, which are the most stable MoS2 defects experimentally measured in low-pressure conditions. Note that although ReaxFF allows for charge transfer between defected sites and gas phase molecules or compositing agents with Electronegativity Equalization Method, it is typically used to describe neutral defects in MD simulations.
We designed the following eight systems to study the impact of defects, gas-phase molecules, i.e., O2 and H2O, and Ti clusters on the oxidation and hydrogenation behaviors of MoS2. Details of the eight systems and the nomenclatures are given in Table 1.
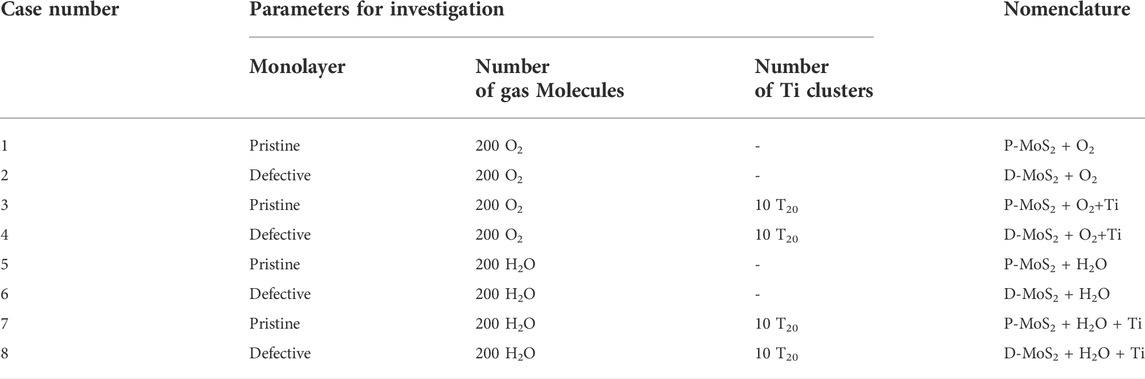
TABLE 1. The eight systems designed to study the oxidation and hydrogenation mechanisms of the MoS2 surface with respect to the effects of defects, gas-phase environments, and compositing agents (Ti clusters).
The monolayer 2H-phase MoS2 contained 225 Mo atoms and 450 S atoms in a periodic hexagonal supercell (α = β = 90°, γ = 120°) with the lattice parameters of a = b = 49.8 Å, c = 80.0 Å for the systems without Ti clusters and c = 97.0 Å for the systems with Ti clusters. The length of c for the systems with Ti clusters was elongated in order to ensure consistency of gas densities among the systems. We deleted 22 S atoms from the P-MoS2 to create a D-MoS2, with 11 S atoms randomly removed from the top surface and another 11 S atoms randomly removed from the bottom surface, amounting to 5% vacancies. The 10 Ti20 clusters were placed near the MoS2 surface, with 5 clusters randomly distributed within a 5–10 Å spacing from the top surface and another 5 clusters randomly distributed within a 5–10 Å spacing from the bottom surface. The initial configuration of a Ti20 cluster was obtained via an annealing treatment. For this, we randomly placed 20 elemental Ti atoms in an orthogonal box (a = b = c = 80.0 Å, α = β = γ = 90°), heated at 1,800 K for 200 ps in an NVT ensemble, allowing the Ti atoms to cluster and reorganize, and finally we ran an energy minimization after cooling to 300 K, to release internal stresses. We introduced 200 O2 or H2O gas molecules to the systems, evenly distributed on the top and bottom surfaces of the monolayer MoS2. For the systems without Ti clusters, the gas density was kept at ∼0.062 g/cm3 for O2-containing systems and at ∼0.035 g/cm3 for H2O-containing systems. These densities of O2 and H2O are one order of magnitude higher than the atmospheric air density (at 101.325 kPa and 15°C). However, since we aim to understand the MoS2 surface reactions individually with O2 or H2O, it is crucial to place the MoS2 in each gas-phase environment with a relatively high density to enable more extensive surface-gas interactions in our nanosecond-scale simulations.
After energy minimization, we equilibrated the systems at 300 K in an NVT ensemble for 500 ps so that they could be fully relaxed. Subsequently, these eight systems were heated to 3,000 K with a rate of 10 K/ps in an NVT ensemble. The time step was set to be 0.1 fs in order to prevent unphysically wide oscillations of covalent bonds at high temperatures. It should be noted that the temperature-dependent measurements of oxidation of monolayer MoS2 usually take place at a temperature below 1,500 K in experiments (Park et al., 2021b; Grønborg et al., 2019; Walter et al., 2017). However, exposure to extreme space-terrestrial environments or operating conditions can lead to instantaneous ultra-high temperatures for MoS2 (Voevodin and Zabinski, 2005; Li et al., 2021). On the other hand, such a wide temperature range provides a better insight into temperature-dependent oxidation and hydrogenation mechanisms of MoS2, as MoS2 readily reacts with O2 but does not volatilize above 1,500 K from MD simulations (Rahman et al., 2021; Farigliano et al., 2020; Jo et al., 2020). For the statistical calculations, the six configurations, represented by different coordinates and velocities, were saved every 10 ps prior to the temperature ramp for each of the eight systems. In post-processing, we computed the mean values and the standard deviations from the forty-eight trajectories that were recorded during the temperature ramp.
We also performed size-dependent studies of the Ti clusters, with an aim of understanding the mechanisms mitigating oxidation and hydrogenation in O2- and H2O-rich environments. We designed six models by randomly inserting an increased number of O2 or H2O molecules around a Ti cluster with varying size. Details of the six models and the nomenclatures are given in Table 2. The six Ti clusters were placed in an orthogonal box, with a1,4 = b1,4 = c1,4 = 20.0 Å, a2,5 = b2,5 = c2,5 = 50.0 Å, a3,6 = b3,6 = c3,6 = 100.0 Å, and α = β = γ = 90°, so that all systems have a comparable density. The six systems were separately annealed at 300 K for 500 ps in an NVT ensemble and then heated up from 300 K to 1,000 K with a rate of 10 K/ps. Lastly, these systems were annealed at 1,000 K for another 500 ps in an NVT ensemble. All courses of ReaxFF MD simulations were carried out with the use of Amsterdam Modeling Suite (AMS) (ReaxFF 2022.1 and SCM, 2022).
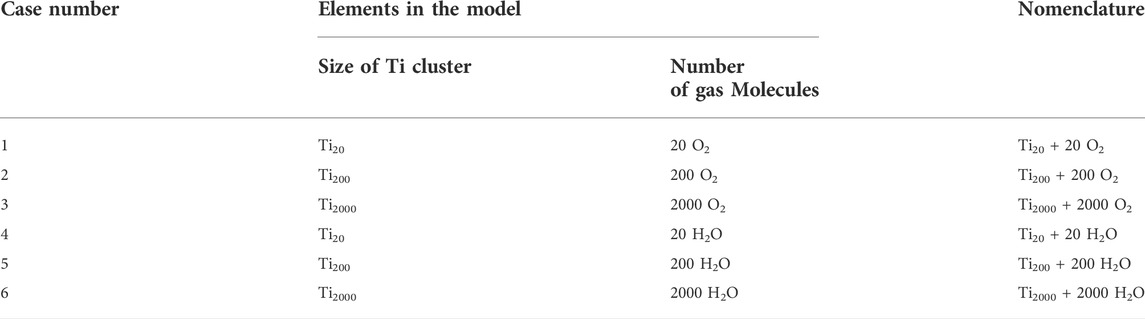
TABLE 2. The six models designed to study the anti-oxidation and anti-hydrogenation mechanisms of Ti clusters of varying size.
From the ReaxFF literature, a bond order cutoff of 0.3–0.5 is commonly used for the recognition of molecule fragmentation (Wang et al., 2020; Wang et al., 2022; Hahn et al., 2018; Liu et al., 2021). We determine the cutoff bond lengths through the bond scan technique with an increment of 0.1 Å, where a cutoff bond length is computed as the bond order between the two target atoms is reduced to 0.5. Therefore, the cutoff bond lengths for Mo-S, Mo-O, Mo-H, Ti-S, Ti-O, S-H, and O-H can be derived and applied to data post-processing. Namely, cutoffMo-S = 3.0 Å, cutoffMo-O = 2.1 Å, cutoffMo-H = 2.3 Å, cutoffTi-S = 3.5 Å, cutoffTi-O = 2.2 Å, cutoffS-H = 2.1 Å, and cutoffO-H = 1.5 Å. The Visual Molecular Dynamics (VMD) (Humphrey et al., 1996) and the Open Visualization Tool (OVITO) (Stukowski, 2009) were used to display molecular structures and perform trajectory analyses.
We investigate the roles of O2 and H2O individually in MoS2 surface reactions, both with and without Ti20 clusters. Mo, S, O, and H atoms are tracked over a temperature ramp course, from 300 K to 3,000 K. We present top-down views of the eight models listed in Table 1 to illustrate the MoS2 lattice degradation, as also evidenced by the changes in the coordination number of Mo atoms (Figures 2, 3, Supplementary Figures S2, S3), Mo-S bond dissociation and formation, and Mo-O and Mo-H bond formation (Supplementary Figures S4, S5).
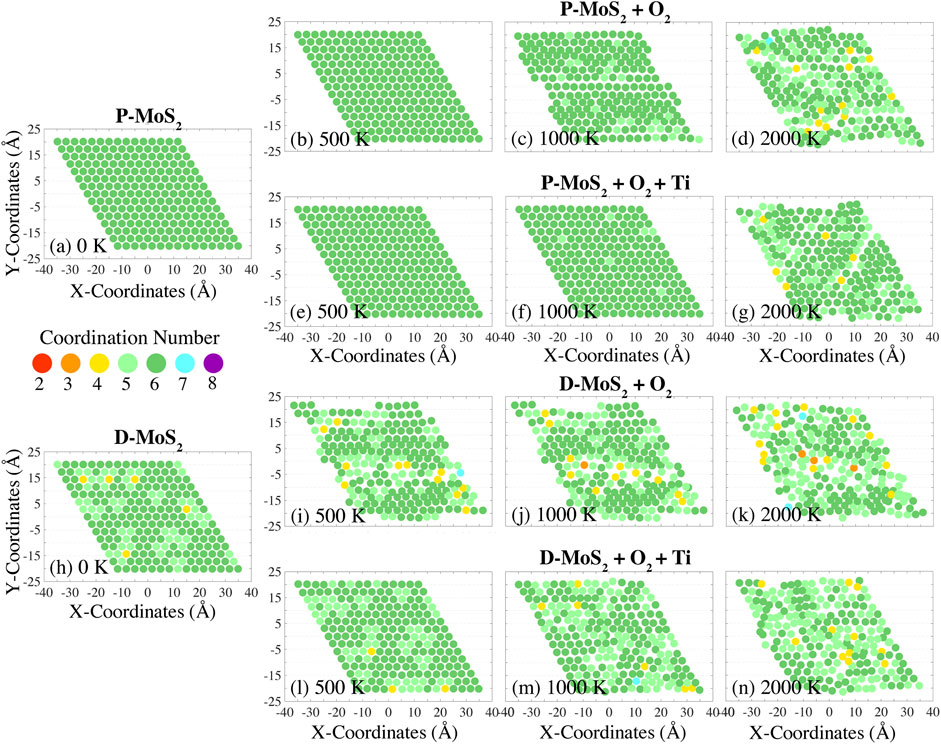
FIGURE 2. Variation of Mo atom coordination numbers during the temperature ramp with a rate of 10 K/ps, for pristine MoS2 with O2 (P-MoS2 + O2) at (B) 500 K, (C) 1,000 K, and (D) 2,000 K; pristine MoS2 with O2 and Ti clusters (P-MoS2 + O2 + Ti) at (E) 500 K, (F) 1,000 K, and (G) 2,000 K; defective MoS2 with O2 (D-MoS2 + O2) at (I) 500 K, (J) 1,000 K, and (K) 2,000 K; and defective MoS2 with O2 and Ti clusters (D-MoS2 + O2 + Ti) at (L) 500 K, (M) 1,000 K, and (N) 2,000 K. The coordination numbers for the initial configurations (at 0 K) of P-MoS2 and D-MoS2 are presented in (A) and (H) for reference.
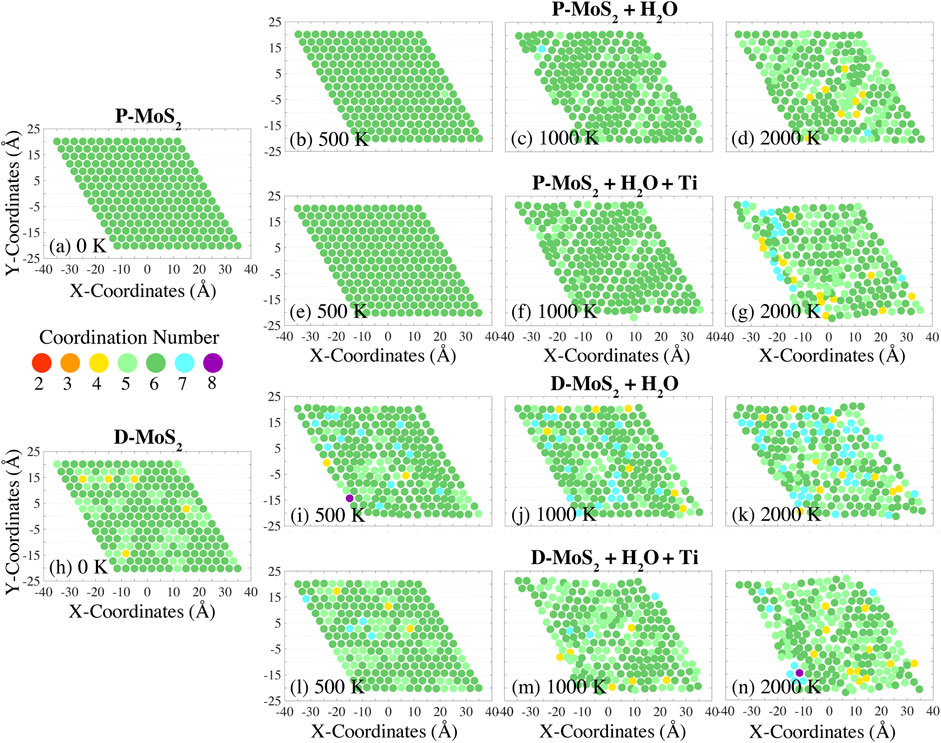
FIGURE 3. Variation of Mo atom coordination numbers during the temperature ramp with a rate of 10 K/ps, for pristine MoS2 with H2O (P-MoS2 + H2O) at (B) 500 K, (C) 1,000 K, and (D) 2,000 K; pristine MoS2 with H2O and Ti clusters (P-MoS2 + H2O+ Ti) at (E) 500 K, (F) 1,000 K, and (G) 2,000 K; defective MoS2 with H2O (D-MoS2 + H2O) at (I) 500 K, (J) 1,000 K, and (K) 2,000 K; and defective MoS2 with H2O and Ti clusters (D-MoS2 + H2O+ Ti) at (L) 500 K, (M) 1,000 K, and (N) 2,000 K. The coordination numbers for the initial configurations (at 0 K) of P-MoS2 and D-MoS2 are presented in (A) and (H) for reference.
In Figure 2, we show the lattice distortion and the coordination number variation of Mo atoms in a monolayer of MoS2 exposed to O2 at temperatures of 500, 1,000, and 2,000 K. The coordination numbers of the initial configurations P-MoS2 and D-MoS2 (i.e., prior to gas-phase exposure) are given in Figures 2A,H for comparison. The distribution of coordination numbers of Mo atoms at these temperatures can be viewed in Supplementary Figures S6A,B,D. The equilibrated monolayer MoS2 (2H-MoS2) lattice has hexagonal symmetry, where Mo atoms have a coordination number of six and form trigonal prismatic geometry with nearby S atoms. Thus, under-coordination (coordination number below 6) of Mo atoms indicates distortion of the MoS2 lattice as a consequence of the Mo-S bond dissociation. On the other hand, over-coordination (coordination number above 6) of Mo atoms is used to identify reactions such as chemisorption accompanying bond formation. The variation of Mo atom coordination numbers of P-MoS2 and D-MoS2 in vacuum and in the absence of compositing agents is also provided as a base line for comparison, as shown in Supplementary Figures S2(a1)–(a10) and Supplementary Figures S3(a1)–(a10) respectively. The P-MoS2 and D-MoS2 systems present lower degree of miscoordination and better structure integrity at low and intermediate temperatures compared to the systems with gas phase molecules and Ti clusters. However, P-MoS2 + O2 + Ti system exhibits more 6-coordinated Mo atoms and higher crystallinity than P-MoS2 at 900 K, where the MoS2 plane stays flat in the former system yet the rippling of MoS2 is found in the latter system, resulting from the dissipation of internal stress of MoS2 with the presence of TixOy clusters. The miscoordination of Mo atoms and the lattice distortion are similar in all cases when temperature reaches 2,400 K, regardless of the presence of gas phase molecules or Ti clusters.
From Figures 2B,I, we observe that P-MoS2 outperforms D-MoS2 in sustaining lattice integrity in the O2 environment at 500 K. Specifically, D-MoS2 in Figure 2I exhibits disturbed atomic arrangement with grouped under-coordinated Mo atoms at 500 K, indicating that initially isolated S-vacancies migrate into line defects or vacancy clustering, which is energetically favored (Zhao et al., 2020b; Garcia-Esparza et al., 2022). The presence of Ti20 clusters, as shown in Figure 2L, significantly reduces the oxidation-related lattice distortion, leading to the recovery of the coordination number of Mo atoms in the initial configuration of D-MoS2. This suggests that Ti20 clusters alleviate S-vacancy driven lattice degradation of MoS2 and help keep the original Mo coordination in the O2 environment. The higher binding energies of O and O2 adsorbed to a Ti20 cluster than to a mono S-vacancy from a monolayer MoS2 (Supplementary Table S1) also implies that O and O2 molecules are more prone to capture by Ti clusters than binding to S-vacancies on MoS2. From Figures 2B–D and Figures 2I–K, we note that the lattice structure is further disturbed as temperature increases for both P-MoS2 and D-MoS2. The presence of Ti20 clusters also mitigates thermal degradation of the lattice and the miscoordination of Mo atoms at low or intermediate temperatures (<2,000 K), as shown by the comparisons of Figures 2E–G to Figures 2B–D and Figures 2L–N to Figures 2I–K, or as can be seen more clearly in Supplementary Figures S2(c1)–(c6) compared with Supplementary Figures S2(b1)–(b6) and Supplementary Figures S3(c1)–(c6) compared with Supplementary Figures S3(b1)–(b6). However, when the temperature is raised to 2,700 K, the lattice distortion and the miscoordination of Mo atoms are similar in all cases with the presence of O2, as shown in Supplementary Figures S2, S3(b9), (b10), (c9), and (c10).
The conclusions are similar for the H2O environment, namely that 1) the migration and clustering behaviors of S-vacancies occur at elevated temperatures; 2) Ti20 clusters are capable of constraining the fluctuation of Mo coordination number; 3) Ti20 clusters maintain the lattice structure of P-MoS2 and attenuate the miscoordination of Mo atoms when temperature does not exceed 2,000 K, as shown both by the comparison of Figures 3E–G to Figures 3B–D and more clearly from Supplementary Figures S2(d1)–(d6) and Supplementary Figures S2(e1)–(e6). Additionally, the distribution of coordination numbers of Mo atoms below 2,000 K in the H2O environment can be viewed in Supplementary Figures S6E–H. Other crucial findings drawn from the simulations are that 1) D-MoS2 has more over-coordinated Mo atoms in the H2O environment than that in the O2 environment below 2,000 K, as shown by the comparison of Figures 3I–K to Figures 2I–K and Figures 3L–N to Figures 2L–N. 2) the over-coordination of Mo atoms in the H2O environment due to hydrogenation barely disturbs the ordered structures, while the under-coordination in the O2 environment degrades the ordered structures below 2,000 K. We find that the under-coordination of Mo atoms at low or intermediate temperatures primarily results from crystallographic defects. O2 molecules stay inert and do not decompose to O-radicals on the surface during the temperature ramp process even in the proximity of S-vacancies. Nevertheless, one should expect the oxidation of Mo atoms to happen extensively in just a few hundred picoseconds of annealing or at much higher applied temperatures. 3) The presence of Ti20 clusters distorts the ordered structures of MoS2 in the H2O environment below 2,000 K, shown by the comparison of Supplementary Figures S2(e1)–(e6) to Supplementary Figures S2(d1)–(d6) or by the comparison of Supplementary Figures S3(e1)–(e6) to Supplementary Figures S3(d1)–(d6); this is ascribed to attack by H2 emitted from TixHyOz clusters with increased temperatures. The addition of Ti20 clusters, however, helps sustain the ordered structures of MoS2 in the O2 environment, shown by the comparison of Figures 2E–G to Figures 2B–D and Figures 2L–N to Figures 2I–K, or by the comparison of Supplementary Figures S2(c1)–(c6) to Supplementary Figures S2(b1)–(b6) and Supplementary Figures S3(c1)–(c6) to Supplementary Figures S3(b1)–(b6). These results suggest that 1) H atoms form extra bonds with Mo atoms in the H2O environment that are initially 6-coordinated at low temperatures; 2) the interaction between H2O and S-vacancies is less detrimental to the lattice structure than that with O2; 3) H atoms barely affect the S-Mo-S substructure and can even stabilize the original atomic arrangement.
From the previous analyses, there is a clear increase in the number of under-coordinated Mo atoms for the systems in the O2 environment below 2,000 K, as a consequence of the thermally driven S-vacancy migration in the basal plane of MoS2. By contrast, over-coordination of Mo atoms for systems in the H2O environment below 2,000 K originates from the hydrogenation of Mo atoms, while the hydroxylation of Mo atoms is barely observed regardless of the presence of S-vacancy. In this section, we discuss the temperature-dependent effects of crystallographic defects as well as the associated reactions at the MoS2-gas interface at elevated temperatures. As such, we present the bonding of S, O, and H with Mo atoms in Supplementary Figures S4, S5. The O and H atoms that bond with the MoS2 surface are magnified, so as to highlight the locally oxidized and hydrogenated regions of MoS2 in the O2 and H2O environments from 300 K to 3,000 K with a temperature increment of 300 K. The representative O- and H-containing products drawn from the onset of oxidation for each system are displayed by the side, as shown in Supplementary Figures S4(z1)–(z4) and Supplementary Figures S5(z1)–(z4).
In Supplementary Figures S4(a3)–(a6), there are buckled areas in triangular- and chain-shape in the free-standing monolayer MoS2, which can be ascribed to the out-of-plane movements of atoms to release the thermal and defect-induced lattice strain. It is also noteworthy that we do not observe a 2H-1T phase transition in these buckled areas, for their lattice still displays distorted hexagonal pattern and comprises trigonal prismatic coordinated Mo atoms. Oxidation occurs particularly in the buckled areas, where the initiation as well as propagation of cracks increase the chemical reactivity of the surface exposed to gas-phase molecules. For instance, the oxidation of Mo atoms in Supplementary Figure S4(a8) occurs on the wrinkle ridges of chain-shape buckled regions, and the crack formed on the top left of Supplementary Figure S4(a8) leads to the further oxidation at higher temperatures, as can be seen in Supplementary Figures S4(a9)–(a10). In comparison with Supplementary Figures S4(a7)–(a10), Supplementary Figures S4(b7)–(b10) demonstrate that the oxidation of D-MoS2 triggers collaborative atomic displacement and further disorganizes the atomic arrangement. The presence of Ti20 clusters constrains O2 from disintegrating ordered structures for both P-MoS2 and D-MoS2 at all selected temperatures, as can be seen from the comparisons of Supplementary Figures S4(c1)–(c10) to Supplementary Figures S4(a1)–(a10) and Supplementary Figures S4(d1)–(d10) to Supplementary Figures S4(b1)–(b10). In other words, Ti20 clusters can effectively seize the O2 molecules that are detrimental to the original S-Mo-S substructure, so that the collaborative displacement zones and buckled areas in both P-MoS2 and D-MoS2 maintain their shape. In addition to the formation of Mo-O bonds, shown by Supplementary Figures S4(z1) and (z2), sulfur oxides such as SO, SO2, SO3 are detected near the MoS2 surface, as shown in Supplementary Figures S4(z3) and (z4). These products are qualitatively comparable with those reported in the experiment over elevated temperatures (Walter et al., 2017; Szoszkiewicz, 2021).
For systems in the H2O environment, oxidation and hydrogenation reactions of Mo atoms take place due to the decomposition of H2O molecules, whereas hydroxylation is rarely found in our simulations. This is because the binding strength of the OH radical to the MoS2 surface is weaker than that of H or O radical. The binding energy of an OH radical to a surface Mo atom of P-MoS2 is 65.30 and 59.07 kcal/mol lower than those of H and O radicals, drawn from the ReaxFF minimization results. The hydroxylation of an Mo atom can be seen in Supplementary Figure S5(z1). Comparing Supplementary Figures S5(b1)–(b10) with Supplementary Figures S4(b1)–(b10), we find that D-MoS2 is better able to maintain its lattice structure in the H2O environment than that in the O2 environment at the same temperature. Meanwhile, we observe that hydrogenation occurs in D-MoS2 over the entire temperature range, while oxidation occurs to a much smaller extent only as temperature reaches the intermediate level, which signifies that oxidation degrades the lattice structure more than hydrogenation. Hydrogenation even appears to stabilize ordered structures of D-MoS2 from 600 K to 1,500 K, as can be seen from the comparison of Supplementary Figures S5(b2)–(b5) and Supplementary Figures S5(d2)–(d5). When Ti20 clusters interact with H2O and reduce the hydrogenation of Mo atoms, the D-MoS2 in Supplementary Figures S5(d2)–(d6) exhibits less ordered atomic arrangement than without the presence of Ti20, as shown in Supplementary Figures S5(b2)–(b6). Similarly, we observe S-containing products such as S2 and SO, as shown in Supplementary Figure S5(z3) at 2,100 K. From Supplementary Figures S4(z3) and (z4) and Supplementary Figure S5(z3), we conclude that oxidation of S usually occurs in the high temperature range (>2,000 K) regardless of environment, when the systems undergo thermal shock that severely ruptures the MoS2 lattice structure, causing S to dissociate from the Mo and bond with O or the neighboring S atoms.
In the previous discussion, we can understand the role of Ti clusters in anti-oxidation and anti-hydrogenation for the monolayer MoS2 systems. For systems in the O2 environment, Ti clusters effectively capture O2 and thus prevent the MoS2 lattice from O2 attack and oxidation. For systems in the H2O environment, on the other hand, Ti clusters lock the dissociated OH and H radicals inside the clusters, so that the MoS2 appears less ordered than without Ti clusters. The absence of Ti clusters allows faster hydrogenation of Mo atoms, inducing a conversion to a trigonal planar geometry (1 H atom with 3 Mo atoms) and helping to stabilize the original atomic arrangement of MoS2. Here, we study the effects of the size of Ti clusters on the prevention of oxidation and hydrogenation. To this end, we investigate the adsorption of O2 and H2O onto a Ti cluster for the six models described in Table 2. The snapshots of these models at the end of 300 K annealing are presented in Figures 4A–F. The numbers of Ti atoms that bond with O or OH are color projected to the x-y plane, as can be seen in Figures 4G–L, for a better visualization of the oxidation states from core to shell.
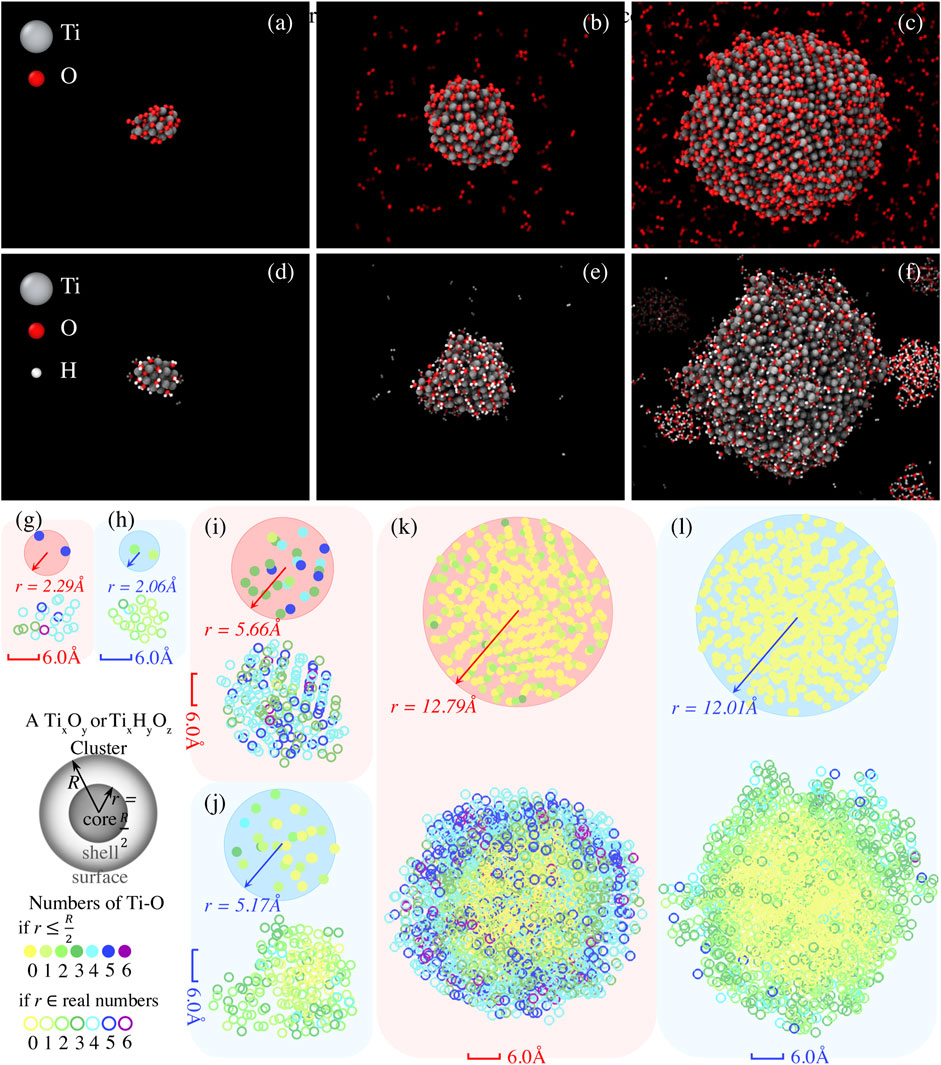
FIGURE 4. Adsorption of O2 and H2O molecules on Ti cluster with varying size. The molecular snapshots showing Ti20 cluster with (A) 20 O2 molecules (Ti20 + 20 O2) and (D) 20 H2O molecules (Ti20 + 20 H2O), Ti200 cluster with (B) 200 O2 molecules (Ti200 + 200 O2) and (E) 200 H2O molecules (Ti200 + 200 H2O), as well as Ti2000 cluster with (C) 2000 O2 molecules (Ti 2000 + 2000 O2) and (F) 2000 H2O molecules (Ti 2000 + 2000 H2O), are extracted at t = 500 ps during NVT simulations at 300 K. Accordingly, Ti atoms constituting Ti-O or Ti-OH that are projected onto the x-y plane are given to identify the size-dependent oxidation states of core and shell of Ti clusters, as can be seen in (G) for Ti20 + 20 O2, (H) for Ti20 + 20 H2O, (I) for Ti200 + 200 O2, (J) Ti200 + 200 H2O, (K) Ti 2000 + 2000 O2, and (L) Ti 2000 + 2000 H2O. The solid circles represent the numbers of Ti atoms constituting Ti-O or Ti-OH within the core (i.e., r
As depicted in Figures 4A,G, the smaller Ti20 clusters effectively react with all gas-phase molecules in the environment by consuming 20 O2 or 20 H2O within 500 ps, leading to the compositional mixing of Ti20Oy and Ti20OyHz, respectively. In addition, there is one Ti atom coordinated with 6 O atoms for the Ti20 + 20 O2 system, and two Ti atoms coordinated with 5 O atoms fall within the core, defined as a sphere with a radius r
In this section, we statistically evaluate the bond formation and the gas production for the eight systems (Table 1) exposed to the O2 and H2O environments to quantify the observations presented in Section 3.1. To this end, Figure 5 and Supplementary Figure S8 show the numbers of oxidized Mo, S, and Ti for the eight systems in the O2 and H2O environments and the numbers of hydrogenated Mo and hydroxylated Ti only for the systems in the H2O environment as a function of temperature. Similarly, the numbers of Mo-O, S-O, and Ti-O bonds as well as Mo-H bonds are profiled in Supplementary Figure S9 for the respective systems. In addition, we plot the numbers of volatile gas molecules, O2, H2, and H2O in Figure 6, in order to shed light on the interaction mechanisms between O2 or H2O gas phase molecules and the monolayer MoS2.
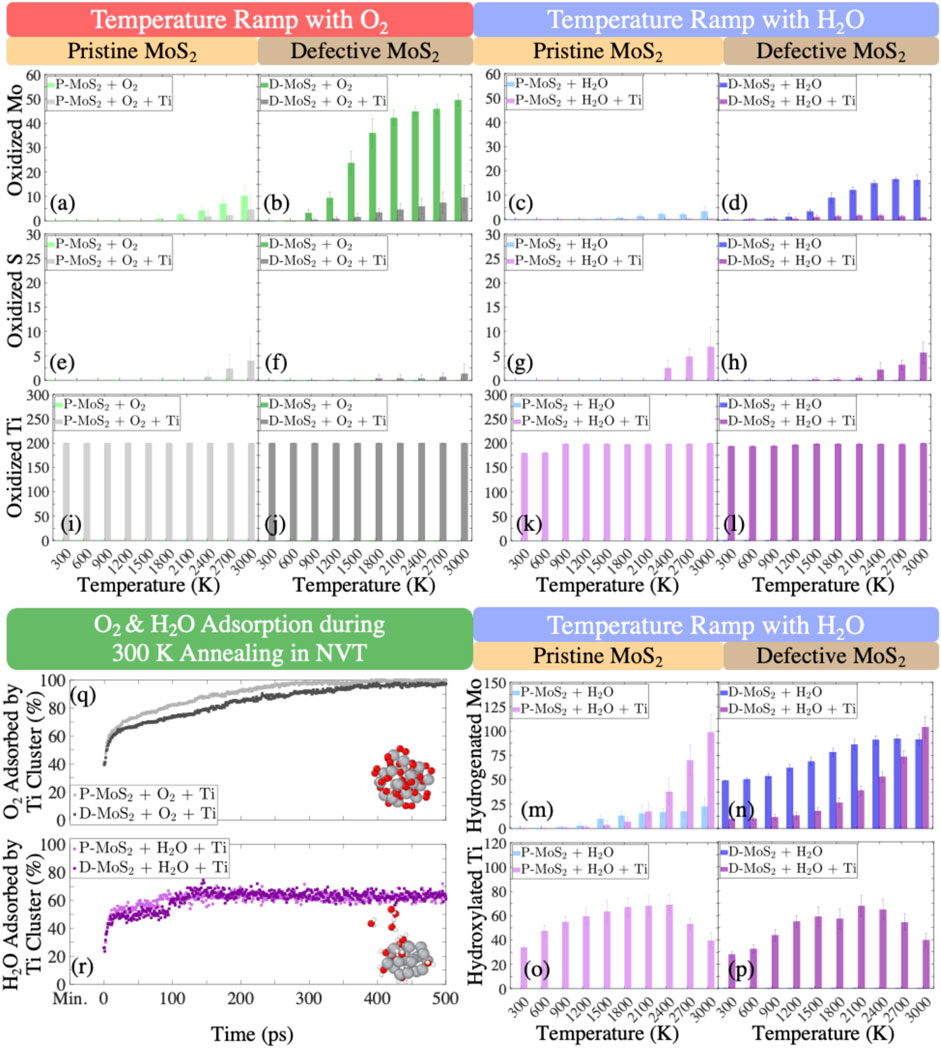
FIGURE 5. Evolution of oxidized Mo, S, and Ti atoms in the systems of (A), (E), and (I) pristine MoS2 with O2 (P-MoS2 + O2); (B), (F), and (J) defective MoS2 with O2 (D-MoS2 + O2); (C), (G), and (K) pristine MoS2 with H2O (P-MoS2 + H2O); (D), (H), and (L) defective MoS2 with H2O (D-MoS2 + H2O) quantifying the reactions during the temperature ramp. Evolution of hydrogenated Mo and hydroxylated Ti atoms in the systems of (M,O) P-MoS2 + H2O as well as (N,P) D-MoS2 + H2O quantifying hydrogenation and hydroxylation reactions during the temperature ramp. The percentages of O2 and H2O molecules adsorbed by Ti clusters during the 300 K NVT annealing are shown in (Q,R). The heights of bars and the error bars represent the mean values and the standard deviations derived from six configurations for each system, respectively.
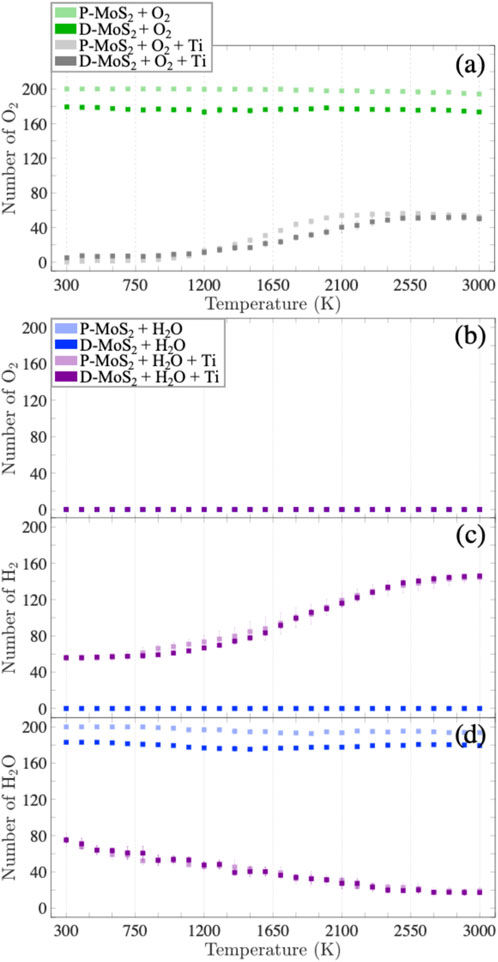
FIGURE 6. Evolution of the number of (A) O2 gas molecules for the systems with O2 at elevated temperatures; evolution of the numbers of (B) O2 gas molecules, (C) H2 gas molecules, and (D) H2O gas molecules for the systems with H2O at elevated temperatures. The light green, dark green, light gray, dark gray, light blue, dark blue, light purple, and dark purple squares represent the mean values of gas molecule numbers for the cases of P-MoS2 + O2, D-MoS2 + O2, P-MoS2 + O2 + Ti, D-MoS2 + O2 + Ti, P-MoS2 + H2O, D-MoS2 + H2O, P-MoS2 + H2O+ Ti, D-MoS2 + H2O+ Ti, respectively. The error bars indicate the standard deviations. The mean values and the standard deviations are derived from six configurations for each system, respectively.
In Figure 5, the numbers of oxidized Mo, S, and Ti at the ramped temperatures from 300 K to 3,000 K are presented. Figures 5A,B indicate that in the O2 environment, detectable oxidation of Mo starts to happen to P-MoS2, P-MoS2 + Ti, D-MoS2, and D-MoS2 + Ti systems around 2,100, 2,400, 900, and 1,500 K, respectively. The number of oxidized Mo in D-MoS2 is much larger than the other systems at the same temperature, whereas the number of oxidized Mo in D-MoS2 is reduced to a level similar to that of P-MoS2, as Ti20 clusters are introduced. This is consistent with the conclusion from Section 3.1, that Ti20 clusters mitigate lattice degradation by attenuating the oxidation of D-MoS2.
Figures 5I,J indicate a stable oxidation state of Ti over the whole temperature range for both P-MoS2 and D-MoS2 systems, where all Ti atoms are bonded with O atoms. Further, from Supplementary Figure S8C and Supplementary Figure S9C, we can conclude that most Ti atoms are coordinated with 4 O atoms on average, aligning well with the oxidation state studies of small and medium-sized Ti clusters presented in Figures 4G,I and in Supplementary Figures S7A,C. The snapshots shown in Supplementary Figures S10A–D indicate a partial transformation of TixOy to a rutile crystal (Ti in 6-fold coordination and O in 3-fold coordination), where the tetragonal crystal formation can be seen in Supplementary Figures S10B,D. Unlike the metal oxides that are major oxidation products over a wide temperature range, we only observe sulfur oxides when the temperature is above 2,000 K. Particularly, Figure 5E shows that the oxidation of S occurs from 2,400 K to 3,000 K. This is consistent with the analysis in Section 3.1 that the distortion of S layer is not caused by S-O bonding but caused by the interference of O2 in the S-Mo-S substructure.
In the H2O environment, comparing Figures 5C,D with Figures 5A,B shows that the oxidation of Mo is largely suppressed over the whole temperature range. However, Ti atoms still form bonds with 1.5–2.5 O atoms on average throughout the temperature ramp, as shown in Supplementary Figure S8F and Supplementary Figure S9F. The snapshots in Supplementary Figures S10F,H further confirm the occurrence of tetragonal Ti2O (O atoms in 6-fold coordination and Ti atoms in 3-fold coordination), in good agreement with the oxidation state studies of small and medium-sized Ti clusters presented in Figures 4H,J and in Supplementary Figures S7B,D. Moreover, Mo atoms prefer to bond with H atoms. Even though the D-MoS2 is extensively hydrogenated throughout the process, the hydrogenation of Mo is greatly reduced by the presence of Ti20 below 1,500 K, as shown in Figure 5N. One interesting observation from Figures 5M,N is that the presence of Ti20 clusters favors the hydrogenation of Mo above 1500 K, for both P-MoS2 and D-MoS2. For instance, P-MoS2 + Ti experiences an increased number of hydrogenated Mo with a slope steeper than that of P-MoS2 above 1,500 K. At 3,000 K, both P-MoS2+Ti and D-MoS2+Ti have more hydrogenated Mo atoms than those without Ti clusters.
Figure 6A indicates that for the systems exposed to O2, the number of O2 gas molecules stays stable in the absence of Ti-clusters, where D-MoS2 has fewer O2 gas molecules than P-MoS2. Recall that in Supplementary Figure S9A, there are very few Mo-O bonds forming below 900 K in D-MoS2, yet the number of Mo-O bonds increases significantly from 900 to 2,100 K and then reaches a plateau. The discrepancy between the trend of Mo-O bond formation and the trend of changing amount of O2 molecules suggests that at low temperatures, O2 molecules are physiosorbed to the D-MoS2 surface; whereas, as temperature increases, the adsorbed O2 molecules dissociate to O radicals and start to bond with Mo atoms until the initially adsorbed O2 molecules are fully consumed. For P-MoS2 + O2 + Ti and D-MoS2 + O2 + Ti systems, the number of O2 grows slowly from 0 at 300 K yet increases rapidly at 1,200 K and then reaches a plateau at 2,100 K. This suggests that as temperature goes above 1,200 K, O atoms are released from TixOy clusters and trigger the detectable oxidation of MoS2.
On the other hand, D-MoS2 in the H2O environment in the absence of Ti clusters shows different chemical behavior than that in the O2 environment, i.e., MoS2 is functionalized by H2O with no O2 gas produced, as shown in Figure 5N and Figures 6B–D. The near-constant difference of H2O molecules between D-MoS2 and P-MoS2 in Figure 6D suggests that in the proximity of the surface, H atoms dissociate from H2O and saturate Mo atoms by leaving OmHn radicals at low and intermediate temperatures. Nevertheless, the OmHn radicals interact further with the surface to hydrogenate and oxidize Mo atoms above 1,500 K.
When Ti clusters are introduced, the number of H2O molecules decreases constantly, which indicates that the presence of Ti-clusters catalyzes the H2 gas formation from H2O, while the remaining OmHn radicals are absorbed by Ti clusters, forming TixHyOz clusters, as shown in Figures 6B–D. Moreover, it is found from Figures 5M–P and Figures 6C,D that, in the early stage of H2O reduction, the hydroxylation of Ti increases, yet the hydrogenation of Mo is suppressed to a stable lower level until 1,500 K. At a temperature below 2,100 K, Ti clusters extensively react with H2O molecules, resulting in the emission of H2 gas molecules, OmHn radicals, as well as TixHyOz clusters. Meanwhile, the TixHyOz clusters are more reactive towards the radicals of low molecular weight than D-MoS2, thus inhibiting the hydrogenation and oxidation of Mo atoms. As temperature increases to 2,100 K and above, the number of Ti-OH bonds starts to decrease because of the H and OpHq radicals released from TixHyOz clusters, leading to a rapid increase in the hydrogenation/oxidation for Mo atoms. From these observations, we can suggest the following reaction mechanisms in MoS2 + Ti system in the H2O environment: (i) H2O interacts with Ti-clusters to produce H2 and OmHn radicals, i.e., (z + m) H2O+ x Ti → TixHyOz + [z + m-(y + n)/2] H2 + OmHn. (ii) The TixHyOz clusters decompose to TixHbOc, OpHq, and H2, i.e., TixHyOz → TixHbOc + [(y-b-(z-c) q/p)/2] H2 + (z-c)/p OpHq. As such, OmHn and OpHq radicals can functionalize Mo atoms gradually as temperature rises.
In order to substantiate the reaction mechanisms we suggested in the previous subsections, we inspect the MD trajectories for the D-MoS2 + Ti systems in the O2 and H2O environments. The three representative reactions of (i) oxidation of Mo atoms in the O2 environment, (ii) hydrogenation of Mo atoms in H2O environment, and (iii) hydrogen gas formation near the MoS2 surface in H2O environment are visualized and detailed in Figure 7.
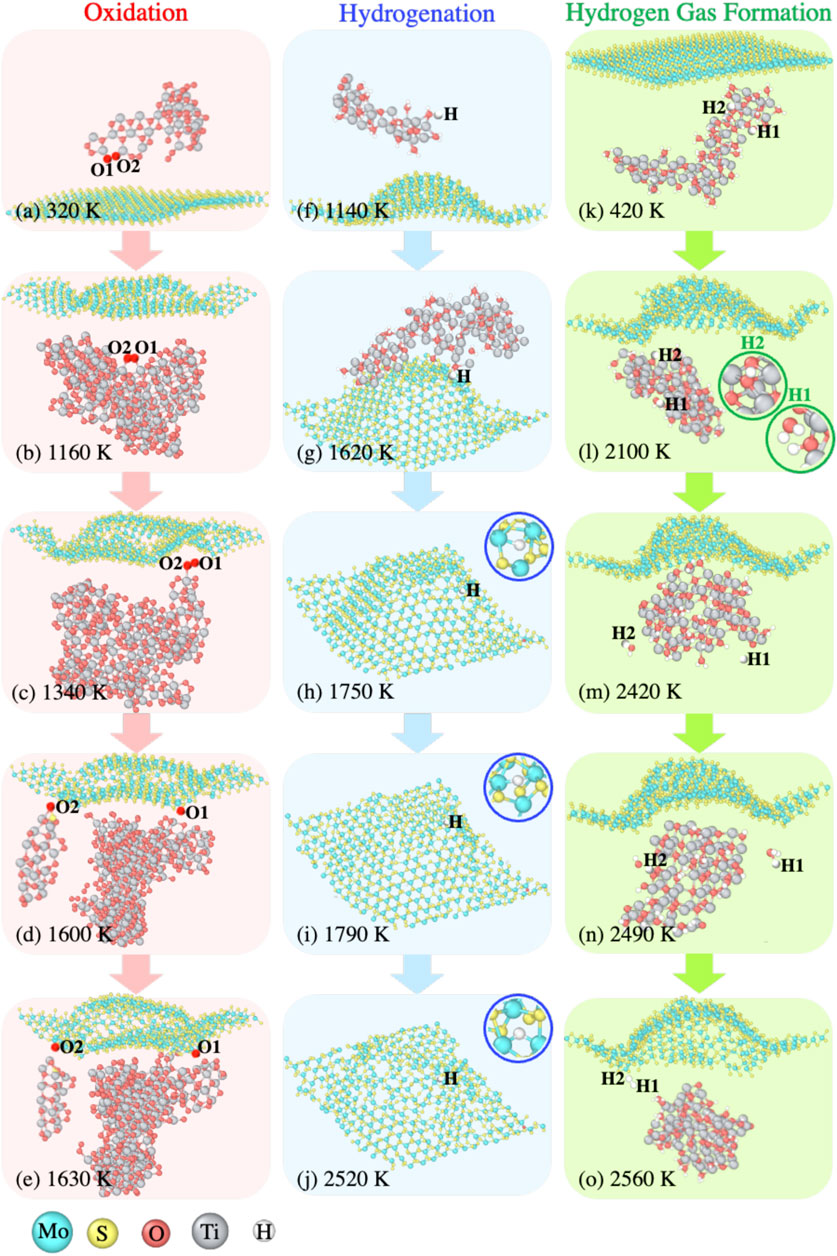
FIGURE 7. Representative reaction pathways during the temperature ramp, such as (A–E) oxidation of Mo atoms with TixOy clusters delivering O radicals drawn from the D-MoS2 + O2 + Ti system, (F–J) hydrogenation of Mo atoms with TixHyOz clusters delivering H radicals drawn from the D-MoS2 + H2O+ Ti system, and (K–O) hydrogen gas formation with the presence of TixHyOz clusters drawn from the D-MoS2 + H2O+ Ti system.
The oxidation of Mo atoms for the systems without Ti clusters typically occurs at a region with S-vacancies or a region with locally distorted S-Mo-S lattice structures due to thermal degradation. The presence of Ti clusters effectively inhibits oxidation of the MoS2 substrate at low temperatures, yet at sufficiently high temperatures, we can still detect the oxidation of Mo for the systems with Ti clusters. Figures 7A–E show that the MoS2 surface oxidation results from O-transfer from TixOy clusters to the S-defects in the vicinity of Mo atoms. During the 300 K annealing, prior to the temperature ramp, the formation of TixOy clusters takes place as the majority of volatile O2 gas molecules are captured by Ti clusters. Thus, when temperature is increased to 320 K, as shown in Figure 7A, the dangling O1 and O2 atoms from a large TixOy cluster start to bond with each other. As temperature is increased to 1,160 K, the buckling of MoS2 plane is observed, yielding chemically reactive local regions facing towards O1 and O2, as shown in Figure 7B. At 1,340 K, shown by Figure 7C, O1 and O2 approach the MoS2 surface and interact with dangling Mo atoms on the surface. At 1,600 K, shown by Figure 7D, O1 is the first to bind to a 4-coordinated Mo atom, followed by O2 binding to a 3-coordinated Mo atom at 1,630 K, shown by Figure 7E.
The presence of dangling Mo bonds drives the surface hydrogenation for the D-MoS2 systems even at a temperature as low as 300 K. Nonetheless, the Mo-H bonds are barely observed for the P-MoS2 systems in the H2O environment at low temperatures. Figures 7F–J depict the H-transfer from a TixHyOz cluster to the MoS2 surface, with the H atom travelling along the path paved by the adjacent S-defects.
At 1,140 K, an H atom dissociating from an H2O molecule bonds with a small TixHyOz cluster, as shown in Figure 7F. In Figure 7G at 1,620 K, the small TixHyOz cluster and the other clusters coalesce into a larger TixHyOz cluster, with the H atom moving towards the S-defects. At 1,750 K shown by Figure 7H, the H radical passivates an S-defect site and bond with two Mo atoms. In Figure 7I, at the temperature of 1,790 K, the two Mo-H bonds break, and the H atom shifts slightly upwards and connects with two under-coordinated Mo atoms. The Mo-H bonds are not as stable as Mo-O bonds, presented in Figures 7D,E. Lastly, the H atom moves downwards and dwells at the new S-defect connecting with two Mo atoms, as shown in Figure 7J.
In Figure 6C, we detect an increase in the number of H2 molecules during the temperature ramp for systems with Ti clusters in the H2O environment. In an effort to reveal the mechanism of H2 formation in the presence of Ti clusters, we examine the reaction pathway of two H atoms, H1 and H2 from the D-MoS2 + H2O+ Ti system, as shown in Figures 7K–O. At the temperature of 420 K, as shown in Figure 7K, H1 and H2 are bonded with a TixHyOz cluster. When temperature ramps up to 2,100 K, shown by Figure 7L, H1 dissociates from the TixHyOz cluster and evolves to an H2O molecule, while H2 remains bound to the TixHyOz cluster. As can be seen in Figure 7M, at 2,420 K, H1 dissociates from the H2O molecule into an H radical, and H2 is eventually released from the TixHyOz cluster, forming another H2O molecule due to the high temperature. Further, at 2,490 K, shown by Figure 7N, H1 transforms into an H3O radical, whereas H2 forms an OH radical as a consequence of the decomposition of the H2O molecule. Lastly, at 2,560 K, H1 and H2 bind together to form an H2 molecule. In the meantime, the residual OH and H radicals are adsorbed by the TixHyOz cluster, as shown in Figure 7O.
In this work, we have developed a new ReaxFF reactive force field for Mo, Ti, Au, O, S, and H (ReaxFF Mo/Ti/Au/O/S/H-2022) to investigate the role of Ti clusters in preventing the oxidation and hydrogenation of a monolayer MoS2 surface in O2- and H2O-rich environments, respectively. Our ReaxFF MD simulations reveal the oxidation and the hydrogenation mechanisms for the MoS2 surfaces exposed to O2 and H2O environments with and without the presence of Ti clusters.
The coordination number analysis for Mo atoms in the monolayer MoS2 demonstrates that D-MoS2 under H2O exposure possesses more over-coordinated Mo atoms than that under O2 exposure, and that the H2O-induced over-coordination does not disturb the S-Mo-S lattice but rather stabilizes the original atomic arrangement at low or intermediate temperatures. The presence of Ti20 clusters can significantly mitigate the defect-induced lattice degradation of MoS2 in the O2 environment and sustain the original Mo coordination in both the O2 and H2O environments. In addition, the study of the effects of size of the Ti clusters indicates that Ti clusters regardless of size are promising antioxidants or anti-hydrogenation agents and can effectively capture O2 or H2O molecules within a wide temperature range. Finally, the statistical calculations on the bond formation of Mo-O, Mo-H, S-O, Ti-O, and Ti-H as well as the gas formation of O2, H2, and H2O reveal the stoichiometry of oxidation and hydrogenation in response to elevated temperatures. We suggest three reaction pathways: (i) oxidation of Mo atoms with O-transfer by TixOy clusters; (ii) hydrogenation of Mo atoms with H-transfer by TixHyOz clusters; (iii) hydrogen gas formation near the MoS2 surface due to the catalytic role of TixHyOz clusters. In other words, in the O2 environment, the O radicals or O2 gas molecules are released from TixOy clusters, leading to thermal degradation and oxidation of MoS2 at elevated temperatures. On the other hand, in the H2O environment, the H and HmOn radicals that dissociate from TixHyOz clusters initiate more extensive hydrogenation and oxidation of Mo atoms as temperature rises. In particular, the pathway of H-transfer along the S-vacancies in the MoS2 due to the reversible adsorption of H and HmOn radicals from TixHyOz clusters is identified during the hydrogenation reaction.
To conclude, this work offers a new ReaxFF force field to describe the Mo/Ti/Au/O/S/H interactions for the materials modeling society with an interest in 2D materials and nanotechnologies. Our MD simulations uncover the impact of the Ti dopant on the oxidation/hydrogenation behaviors of MoS2 surface. These results provide a theoretical basis for engineering the oxidation and anti-oxidation properties of MoS2 thin films and other TMDs, which can benefit 2D material designs in automotive, aerospace, semiconductor, and nanotechnology industries.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
QM: modeling, investigation, data curation, visualization, writing, and editing; YZ: modeling, writing, and editing; MK: literature review, Introduction drafting, and reviewing; NN: reviewing and editing; MC: reviewing, editing, and supervision; AD: reviewing, editing, project supervision, funding acquisition, project administration.
The authors acknowledge the funding from the National Science Foundation 2D Crystal Consortium Materials Innovation Platform (NSF 2DCC-MIP) under cooperative agreement DMR-1539916. Computations for this research were performed on the PSU’s Institute for Cyber Science Advanced Cyber Infrastructure (ICS-ACI). This work was funded by the Laboratory Directed Research and Development program at Sandia National Laboratories, a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the US. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the US. Department of Energy or the United States Government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnano.2022.1034795/full#supplementary-material
Bochevarov, A. D., Harder, E., Hughes, T. F., Greenwood, J. R., Braden, D. A., Philipp, D. M., et al. (2013). Jaguar: A high‐performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 113, 2110–2142. doi:10.1002/qua.24481
Chen, R., Konicek, A. R., Jusufi, A., Kliewer, C. E., Jaishankar, A., Schilowitz, A., et al. (2020). Limiting domain size of MoS2: Effects of stoichiometry and oxygen. J. Phys. Chem. C 124, 27571–27579. doi:10.1021/acs.jpcc.0c08981
Chenoweth, K., van Duin, A. C. T., and Goddard, W. A. (2008). ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 112, 1040–1053. doi:10.1021/jp709896w
Colas, G., Saulot, A., Regis, E., and Berthier, Y. (2015). Investigation of crystalline and amorphous MoS2 based coatings: Towards developing new coatings for space applications. Wear 330, 448–460. doi:10.1016/j.wear.2015.01.011
Curry, J. F., Argibay, N., Babuska, T., Nation, B., Martini, A., Strandwitz, N. C., et al. (2016). Highly oriented MoS2 coatings: Tribology and environmental stability. Tribol. Lett. 64, 11–19. doi:10.1007/s11249-016-0745-0
Curry, J. F., Ohta, T., DelRio, F. W., Mantos, P., R Jones, M., F Babuska, T., et al. (2021). Structurally driven environmental degradation of friction in MoS2 films. Tribol. Lett. 69, 96–10. doi:10.1007/s11249-021-01453-7
Curry, J. F., Wilson, M. A., Luftman, H. S., Strandwitz, N. C., Argibay, N., Chandross, M., et al. (2017). Impact of microstructure on MoS2 oxidation and friction. ACS Appl. Mat. Interfaces 9, 28019–28026. doi:10.1021/acsami.7b06917
Farigliano, L. M., Paredes-Olivera, P. A., and Patrito, E. M. (2020). Initial steps of oxidative etching of MoS2 basal plane induced by O2. J. Phys. Chem. C 124, 13177–13186. doi:10.1021/acs.jpcc.0c02141
Freedy, K. M., Zhang, H., Litwin, P. M., Bendersky, L. A., Davydov, A. V., and McDonnell, S. (2019). Thermal stability of titanium contacts to MoS2. ACS Appl. Mat. Interfaces 11, 35389–35393. doi:10.1021/acsami.9b08829
Ganeshan, K., Shin, Y. K., Osti, N. C., Sun, Y., Prenger, K., Naguib, M., et al. (2020). Structure and dynamics of aqueous electrolytes confined in 2D-TiO2/Ti3C2T2 MXene heterostructures. ACS Appl. Mat. Interfaces 12, 58378–58389. doi:10.1021/acsami.0c17536
Garcia-Esparza, A. T., Park, S., Abroshan, H., Paredes Mellone, O. A., Vinson, J., Abraham, B., et al. (2022). Local structure of sulfur vacancies on the basal plane of monolayer MoS2. ACS Nano 16, 6725–6733. doi:10.1021/acsnano.2c01388
Grønborg, S. S., Thorarinsdottir, K., Kyhl, L., Rodriguez-Fernández, J., Sanders, C. E., Bianchi, M., et al. (2019). Basal plane oxygen exchange of epitaxial MoS2 without edge oxidation. 2D Mat. 6, 045013. doi:10.1088/2053-1583/ab2d00
Hahn, S. H., Rimsza, J., Criscenti, L., Sun, W., Deng, L., Du, J., et al. (2018). Development of a ReaxFF reactive force field for NaSiOx/water systems and its application to sodium and proton self-diffusion. J. Phys. Chem. C 122, 19613–19624. doi:10.1021/acs.jpcc.8b05852
Hilton, M. R., and Fleischauer, P. D. (1992). Applications of solid lubricant films in spacecraft. Surf. Coat. Technol. 54, 435–441. doi:10.1016/S0257-8972(07)80062-4
Hong, S., Krishnamoorthy, A., Rajak, P., Tiwari, S., Misawa, M., Shimojo, F., et al. (2017). Computational synthesis of MoS2 layers by reactive molecular dynamics simulations: Initial sulfidation of MoO3 surfaces. Nano Lett. 17, 4866–4872. doi:10.1021/acs.nanolett.7b01727
Humphrey, W., Dalke, A., and Schulten, K. (1996). Vmd: Visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi:10.1016/0263-7855(96)00018-5
Islam, M. M., Bryantsev, V. S., and van Duin, A. C. T. (2014). ReaxFF reactive force field simulations on the influence of teflon on electrolyte decomposition during Li/SWCNT anode discharge in lithium-sulfur batteries. J. Electrochem. Soc. 161, E3009–E3014. doi:10.1149/2.005408jes
Jo, S. S., Singh, A., Yang, L., Tiwari, S. C., Hong, S., Krishnamoorthy, A., et al. (2020). Growth kinetics and atomistic mechanisms of native oxidation of ZrSx Se2–x and MoS2 crystals. Nano Lett. 20, 8592–8599. doi:10.1021/acs.nanolett.0c03263
Kowalik, M., Ashraf, C., Damirchi, B., Akbarian, D., Rajabpour, S., and van Duin, A. C. T. (2019). Atomistic scale analysis of the carbonization process for C/H/O/N-based polymers with the ReaxFF reactive force field. J. Phys. Chem. B 123, 5357–5367. doi:10.1021/acs.jpcb.9b04298
Lele, A., Krstic, P., and van Duin, A. C. T. (2022). ReaxFF force field development for gas-phase hBN nanostructure synthesis. J. Phys. Chem. A 126, 568–582. doi:10.1021/acs.jpca.1c09648
Li, B., Xv, W., Liu, P., Huang, D., Zhou, X., Zhao, R., et al. (2021). Novel green lubricated materials: Aqueous PAI-MoS2-graphite bonded solid lubricating coating. Prog. Org. Coat. 155, 106225. doi:10.1016/j.porgcoat.2021.106225
Li, H., Li, X., Zhang, G., Wang, L., and Wu, G. (2017). Exploring the tribophysics and tribochemistry of MoS2 by sliding MoS2/Ti composite coating under different humidity. Tribol. Lett. 65, 38–10. doi:10.1007/s11249-017-0824-x
Liu, H., Grasseschi, D., Dodda, A., Fujisawa, K., Olson, D., Kahn, E., et al. (2020). Spontaneous chemical functionalization via coordination of Au single atoms on monolayer MoS2. Sci. Adv. 6, eabc9308. doi:10.1126/sciadv.abc9308
Liu, L., Kumar, S. B., Ouyang, Y., and Guo, J. (2011). Performance limits of monolayer transition metal dichalcogenide transistors. IEEE Trans. Electron Devices 58, 3042–3047. doi:10.1109/TED.2011.2159221
Liu, Y., Wei, X., Sun, W., and Zhao, L. (2021). ReaxFF MD investigation of the high temperature combustion of six octane isomers. Energy fuels. 35, 16778–16790. doi:10.1021/acs.energyfuels.1c02462
Lotfi, R., Naguib, M., Yilmaz, D. E., Nanda, J., and Van Duin, A. C. (2018). A comparative study on the oxidation of two-dimensional Ti3C2 MXene structures in different environments. J. Mat. Chem. A 6, 12733–12743. doi:10.1039/C8TA01468J
Mao, Q., Rajabpour, S., Kowalik, M., and van Duin, A. C. T. (2020). Predicting cost-effective carbon fiber precursors: Unraveling the functionalities of oxygen and nitrogen-containing groups during carbonization from ReaxFF simulations. Carbon 159, 25–36. doi:10.1016/j.carbon.2019.12.008
Mao, Q., Rajabpour, S., Talkhoncheh, M. K., Zhu, J., Kowalik, M., and van Duin, A. C. T. (2022). Cost-effective carbon fiber precursor selections of polyacrylonitrile-derived blend polymers: Carbonization chemistry and structural characterizations. Nanoscale 14, 6357–6372. doi:10.1039/D2NR00203E
Marian, M., Berman, D., Rota, A., Jackson, R. L., and Rosenkranz, A. (2022). Layered 2D nanomaterials to tailor friction and wear in machine elements—a review. Adv. Mat. Interfaces 9, 2101622. doi:10.1002/admi.202101622
Merida, C. S., Le, D., Echeverria, E. M., Nguyen, A. E., Rawal, T. B., Naghibi Alvillar, S., et al. (2018). Gold dispersion and activation on the basal plane of single-layer MoS2. J. Phys. Chem. C 122, 267–273. doi:10.1021/acs.jpcc.7b07632
Mojtabavi, M., VahidMohammadi, A., Ganeshan, K., Hejazi, D., Shahbazmohamadi, S., Kar, S., et al. (2021). Wafer-scale lateral self-assembly of mosaic Ti3C2Tx MXene monolayer films. ACS Nano 15, 625–636. doi:10.1021/acsnano.0c06393
Monti, S., Carravetta, V., and Ågren, H. (2016). Simulation of gold functionalization with cysteine by reactive molecular dynamics. J. Phys. Chem. Lett. 7, 272–276. doi:10.1021/acs.jpclett.5b02769
Nayir, N., Shin, Y. K., Wang, Y., Sengul, M. Y., Hickey, D. R., Chubarov, M., et al. (2021). A ReaxFF force field for 2D-WS2 and its interaction with sapphire. J. Phys. Chem. C 125, 17950–17961. doi:10.1021/acs.jpcc.1c03605
Nayir, N., Wang, Y., Ji, Y., Choudhury, T. H., Redwing, J. M., Chen, L. Q., et al. (2021). Theoretical modeling of edge-controlled growth kinetics and structural engineering of 2D-MoSe2. Mater. Sci. Eng. B 271, 115263. doi:10.1016/j.mseb.2021.115263
Nayir, N., Wang, Y., Shabnam, S., Hickey, D. R., Miao, L., Zhang, X., et al. (2020). Modeling for structural engineering and synthesis of two-dimensional WSe2 using a newly developed Reaxff reactive force field. J. Phys. Chem. C 124, 28285–28297. doi:10.1021/acs.jpcc.0c09155
Ostadhossein, A., Rahnamoun, A., Wang, Y., Zhao, P., Zhang, S., Crespi, V. H., et al. (2017). ReaxFF reactive force-field study of molybdenum disulfide (MoS2). J. Phys. Chem. Lett. 8, 631–640. doi:10.1021/acs.jpclett.6b02902
Ouyang, J. H., Li, Y. F., Zhang, Y. Z., Wang, Y. M., and Wang, Y. J. (2022). High-temperature solid lubricants and self-lubricating composites: A critical review. Lubricants 10, 177. doi:10.3390/lubricants10080177
Park, S., Garcia‐Esparza, A. T., Abroshan, H., Abraham, B., Vinson, J., Gallo, A., et al. (2021). Operando study of thermal oxidation of monolayer MoS2. Adv. Sci. 8, 2002768. doi:10.1002/advs.202002768
Park, S., Garcia‐Esparza, A. T., Abroshan, H., Abraham, B., Vinson, J., Gallo, A., et al. (2021). Operando study of thermal oxidation of monolayer MoS2. Adv. Sci. 8, 2002768. doi:10.1002/advs.202002768
Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V., and Kis, A. (2011). Single-layer MoS2 transistors. Nat. Nanotechnol. 6, 147–150. doi:10.1038/nnano.2010.279
Rahman, M. H., Chowdhury, E. H., and Hong, S. (2021). High temperature oxidation of monolayer MoS2 and its effect on mechanical properties: A ReaxFF molecular dynamics study. Surf. Interfaces 26, 101371. doi:10.1016/j.surfin.2021.101371
Rajabpour, S., Mao, Q., Gao, Z., Talkhoncheh, M. K., Zhu, J., Schwab, , et al. (2021). Low-temperature carbonization of polyacrylonitrile/graphene carbon fibers: A combined ReaxFF molecular dynamics and experimental study. Carbon 174, 345–356. doi:10.1016/j.carbon.2020.12.038
Rajabpour, S., Mao, Q., Nayir, N., Robinson, J. A., and van Duin, A. C. T. (2021). Development and applications of ReaxFF reactive force fields for group-III gas-phase precursors and surface reactions with graphene in metal–organic chemical vapor deposition synthesis. J. Phys. Chem. C 125, 10747–10758. doi:10.1021/acs.jpcc.1c01965
Raju, M., Kim, S. Y., van Duin, A. C. T., and Fichthorn, K. A. (2013). ReaxFF reactive force field study of the dissociation of water on titania surfaces. J. Phys. Chem. C 117, 10558–10572. doi:10.1021/jp402139h
Rogala, M., Sokołowski, S., Ukegbu, U., Mierzwa, A., and Szoszkiewicz, R. (2021). Direct identification of surface bound MoO3 on single MoS2 flakes heated in dry and humid air. Adv. Mat. Interfaces 8, 2100328. doi:10.1002/admi.202100328
Ross, S., and Sussman, A. (1955). Surface oxidation of molybdenum disulfide. J. Phys. Chem. 59, 889–892. doi:10.1021/j150531a020
Scharf, T. W., and Prasad, S. V. (2013). Solid lubricants: A review. J. Mat. Sci. 42, 511–531. doi:10.1007/s10853-012-7038-2
Schneebeli, S. T., Bochevarov, A. D., and Friesner, R. A. (2011). Parameterization of a B3LYP specific correction for noncovalent interactions and basis set superposition error on a gigantic data set of CCSD (T) quality noncovalent interaction energies. J. Chem. Theory Comput. 7, 658–668. doi:10.1021/ct100651f
Senftle, T. P., Hong, S., Islam, M. M., Kylasa, S. B., Zheng, Y., Shin, Y. K., et al. (2016). The ReaxFF reactive force-field: Development, applications and future directions. Npj Comput. Mat. 2, 15011–15014. doi:10.1038/npjcompumats.2015.11
Spalvins, T. (1987). A review of recent advances in solid film lubrication. J. Vac. Sci. Technol. A Vac. Surfaces Films 5, 212–219. doi:10.1116/1.574106
Stukowski, A. (2009). Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mat. Sci. Eng. 18, 015012. doi:10.1088/0965-0393/18/1/015012
Szoszkiewicz, R. (2021). Local interactions of atmospheric oxygen with MoS2 crystals. Materials 14, 5979. doi:10.3390/ma14205979
Szoszkiewicz, R., Rogala, M., and Dąbrowski, P. (2020). Surface-bound and volatile Mo oxides produced during oxidation of single MoS2 crystals in air and high relative humidity. Materials 13, 3067. doi:10.3390/ma13143067
Ukegbu, U., and Szoszkiewicz, R. (2019). Microscopic kinetics of heat-induced oxidative etching of thick MoS2 crystals. J. Phys. Chem. C 123, 22123–22129. doi:10.1021/acs.jpcc.9b02739
van Duin, A. C. T., Dasgupta, S., Lorant, F., and Goddard, W. A. (2001). ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 105, 9396–9409. doi:10.1021/jp004368u
Vashisth, A., Kowalik, M., Gerringer, J. C., Ashraf, C., van Duin, A. C. T., and Green, M. J. (2020). ReaxFF simulations of laser-induced graphene (LIG) formation for multifunctional polymer nanocomposites. ACS Appl. Nano Mat. 3, 1881–1890. doi:10.1021/acsanm.9b02524
Vazirisereshk, M. R., Martini, A., Strubbe, D. A., and Baykara, M. Z. (2019). Solid lubrication with MoS2: A review. Lubricants 7, 57. doi:10.3390/lubricants7070057
Voevodin, A. A., and Zabinski, J. S. (2005). Nanocomposite and nanostructured tribological materials for space applications. Compos. Sci. Technol. 65, 741–748. doi:10.1016/j.compscitech.2004.10.008
Walter, T. N., Kwok, F., Simchi, H., Aldosari, H. M., and Mohney, S. E. (2017). Oxidation and oxidative vapor-phase etching of few-layer MoS2. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 35, 021203. doi:10.1116/1.4975144
Wang, Y., Qin, J., Xu, J., Sun, J., Chen, L., Qian, L., et al. (2022). Definition of atomic-scale contact: What dominates the atomic-scale friction behaviors. Langmuir 38, 11699–11706. doi:10.1021/acs.langmuir.2c01786
Wang, Y., Su, Y., Zhang, J., Chen, Q., Xu, J., Bai, S., et al. (2020). Reactive molecular dynamics simulations of wear and tribochemical reactions of diamond like carbon interfaces with nanoscale asperities under H2 gas: Implications for solid lubricant coatings. ACS Appl. Nano Mat. 3, 7297–7304. doi:10.1021/acsanm.0c01775
Wu, J., Li, H., Yin, Z., Li, H., Liu, J., Cao, X., et al. (2013). Layer thinning and etching of mechanically exfoliated MoS2 nanosheets by thermal annealing in air. Small 9, 3314–3319. doi:10.1002/smll.201301542
Wu, R. J., Udyavara, S., Ma, R., Wang, Y., Chhowalla, M., Birol, T., et al. (2019). Visualizing the metal-MoS2 contacts in two-dimensional field-effect transistors with atomic resolution. Phys. Rev. Mat. 3, 111001. doi:10.1103/PhysRevMaterials.3.111001
Yamamoto, M., Einstein, T. L., Fuhrer, M. S., and Cullen, W. G. (2013). Anisotropic etching of atomically thin MoS2. J. Phys. Chem. C 117, 25643–25649. doi:10.1021/jp410893e
Zhang, L., Kowalik, M., Gao, Z., Ashraf, C. M., Rajabpour, S., Bumgardner, C., et al. (2020). Converting PBO fibers into carbon fibers by ultrafast carbonization. Carbon 159, 432–442. doi:10.1016/j.carbon.2019.12.067
Zhao, X., Vashisth, A., Blivin, J. W., Tan, Z., Holta, D. E., Kotasthane, V., et al. (2020). pH, nanosheet concentration, and antioxidant affect the oxidation of Ti3C2Tx and Ti2CTx MXene dispersions. Adv. Mat. Interfaces 7, 2000845. doi:10.1002/admi.202000845
Zhao, X., Vashisth, A., Prehn, E., Sun, W., Shah, S. A., Habib, T., et al. (2019). Antioxidants unlock shelf-stable Ti3C2T (MXene) nanosheet dispersions. Matter 1, 513–526. doi:10.1016/j.matt.2019.05.020
Zhao, Y., Tang, M. T., Wu, S., Geng, J., Han, Z., Chan, K., et al. (2020). Rational design of stable sulfur vacancies in molybdenum disulfide for hydrogen evolution. J. Catal. 382, 320–328. doi:10.1016/j.jcat.2019.12.028
Zheng, P., Zhang, X., Yan, M., Ma, Y., Jiang, Y., and Li, H. (2021). The eruption of carbon chains in the oxidation of 2D Tin+1Cn (n=1, 2, 3) MXenes. Appl. Surf. Sci. 550, 149310. doi:10.1016/j.apsusc.2021.149310
Keywords: transition metal dichalcogenides, MoS2 oxidation, MoS2 hydrogenation, Ti cluster oxidation, catalytic role of Ti clusters, molecular dynamics simulations, ReaxFF force field development, environmental exposures
Citation: Mao Q, Zhang Y, Kowalik M, Nayir N, Chandross M and van Duin ACT (2022) Oxidation and hydrogenation of monolayer MoS2 with compositing agent under environmental exposure: The ReaxFF Mo/Ti/Au/O/S/H force field development and applications. Front. Nanotechnol. 4:1034795. doi: 10.3389/fnano.2022.1034795
Received: 02 September 2022; Accepted: 14 October 2022;
Published: 26 October 2022.
Edited by:
Taron Makaryan, Murata Manufacturing Co., Ltd., JapanReviewed by:
Ilia Ponomarev, Czech Technical University in Prague, CzechiaCopyright © 2022 Mao, Zhang, Kowalik, Nayir, Chandross and van Duin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adri C. T. van Duin, YWN2MTNAcHN1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.