 Guanqun Huang1,2†Shuting Chen1†Xiaoxia Chen1†
Guanqun Huang1,2†Shuting Chen1†Xiaoxia Chen1† Jiajun Zheng1Zhuoran Xu3
Jiajun Zheng1Zhuoran Xu3 Abolfazl Doostparast Torshizi3,4Siyi Gong1Qingpei Chen1Xiaokuang Ma2Jiandong Yu1
Abolfazl Doostparast Torshizi3,4Siyi Gong1Qingpei Chen1Xiaokuang Ma2Jiandong Yu1 Libing Zhou1Shenfeng Qiu2
Libing Zhou1Shenfeng Qiu2 Kai Wang3,4
Kai Wang3,4 Lingling Shi1,5*
Lingling Shi1,5*- 1Guangdong-Hongkong-Macau Institute of CNS Regeneration, Ministry of Education CNS Regeneration Collaborative Joint Laboratory, Jinan University, Guangzhou, China
- 2Department of Basic Medical Sciences, College of Medicine – Phoenix, The University of Arizona, Phoenix, AZ, United States
- 3Department of Biomedical Informatics, Columbia University, New York, NY, United States
- 4Children’s Hospital of Philadelphia, Philadelphia, PA, United States
- 5Co-innovation Center of Neuroregeneration, Nantong University, Nantong, China
SHANK3 mutations, including de novo deletions, have been associated with autism spectrum disorders (ASD). However, the effects of SHANK3 loss of function on neurodevelopment remain poorly understood. Here we generated human induced pluripotent stem cells (iPSC) in vitro, followed by neuro-differentiation and lentivirus-mediated shRNA expression to evaluate how SHANK3 knockdown affects the in vitro neurodevelopmental process at multiple time points (up to 4 weeks). We found that SHANK3 knockdown impaired both early stage of neuronal development and mature neuronal function, as demonstrated by a reduction in neuronal soma size, growth cone area, neurite length and branch numbers. Notably, electrophysiology analyses showed defects in excitatory and inhibitory synaptic transmission. Furthermore, transcriptome analyses revealed that multiple biological pathways related to neuron projection, motility and regulation of neurogenesis were disrupted in cells with SHANK3 knockdown. In conclusion, utilizing a human iPSC-based neural induction model, this study presented combined morphological, electrophysiological and transcription evidence that support that SHANK3 as an intrinsic, cell autonomous factor that controls cellular function development in human neurons.
Introduction
Autism spectrum disorders (ASD) are neurodevelopmental disorders characterized by impairments in social communication and interaction, and repetitive behaviors and restricted interests (Freitag et al., 2010; State and Levitt, 2011; Lyall et al., 2017; Rubeis et al., 2018). Multiple studies have indicated a strong connection between ASD and genetic variations of synapse-related genes and proteins, including neuroligins (NLGNs), postsynaptic density protein 95 (PSD-95), and SH3 and multiple ankyrin repeat domains proteins (SHANKs) (Xing et al., 2016; Nakanishi et al., 2017; Kathuria et al., 2018). In particular, genetic association studies have identified a significant role for SHANK3, which encodes a major scaffolding protein at postsynaptic densities (PSD). The SHANK3 protein contains multiple structural domains including ankyrin repeat, Src homologous, PDZ, proline-rich, Homer binding site, sterile alpha motif (Du et al., 1998; Grabrucker et al., 2011; Monteiro and Feng, 2017; Ponna et al., 2018), through which other PSD proteins extensively interact to form the post-synaptic protein signaling complex. Genomic sequencing and exon sequencing from ASD patients have indicated a strong connection between rare mutations in SHANK3 and ASD (Durand et al., 2007; Nemirovsky et al., 2015).
Currently, it is unclear how SHANK3 mutations confer ASD risks by affecting the developmental trajectory of the brain, particularly the excitatory glutamatergic synapses. Several studies have utilized mouse models (Peca et al., 2011; Wang et al., 2016, 2017; Chen et al., 2017; Harony-Nicolas et al., 2017; Luo et al., 2017; Zhao et al., 2017; Amal et al., 2018; Kerrisk Campbell and Sheng, 2018; Qin et al., 2018) and neuronal cell models (Bidinosti et al., 2016; Lu et al., 2016; Bey et al., 2018; Taylor et al., 2018) to explore the role of SHANK3 gene in synaptic function and animal behavior. The variation of SHANK3 at different loci resulted in distinct behavioral phenotypes when modeled in mice. Most of these SHANK3 mutant mice showed deficits in social interactions, with or without cognitive impairment, repetitive behavior, anxiety and motor deficit. Abnormal cortico-striatal circuits, disrupted excitability and inhibitory (E/I) balance, and synaptic dysfunction have been identified to be associated with the mechanism of ASD-like behavior (Bozdagi et al., 2010; Peca et al., 2011; Wang et al., 2011; Mei et al., 2016; Vicidomini et al., 2017; Bey et al., 2018). These analysis of synaptic physiology and mice behavior revealed a strong causal connection between SHANK3 mutations and ASD-like endophenotypes.
Despite the above cited studies, analyses on animal models cannot completely simulate human genetic background on neurodevelopment. In some cases, human brain tissue sample has been collected for neural development disease study (Konopka et al., 2012; Parikshak et al., 2016), however, the sample resource is very limited. More than 10 years ago, Takahashi and Yamanaka made a remarkable breakthrough in stem cell research when they generated ES-like cells from adult somatic cells using a cocktail of transcription factors (Takahashi et al., 2007). More recently, new methods have been developed to reprogram adult somatic cells (such as fibroblasts) into iPSC. Following the discovery of iPSC, several studies have fueled enthusiasm for their use in neurological disorders, and iPSC have been validated to develop into many kinds of neural subtype cells (Chambers et al., 2009; Shi et al., 2012; Kang et al., 2017; Lin et al., 2018). Creating neuronal cultures from iPSC has received wide attention for the potential to create translatable disease-in-a-dish models. This development has made it possible to mimic human neuron development defect following disease candidate gene dysfunction. One advantage of iPSC is that the cells possess genetic background that is distinctly human; the other is that stem cells could mimic certain aspects of human neural development courses in vitro. So far, iPSC have been increasingly recognized as a significant in vitro cellular model to study the function of susceptible genes in neurological diseases and neurodevelopment diseases. iPSC have successfully used to model the cellular physiology and guide therapeutic endeavors in Rett syndrome, Alzheimer’s disease and schizophrenia by revealing the functional effects of genetic mutations with single neuron resolution (Marchetto et al., 2010; Mitne-Neto et al., 2011; Israel et al., 2012). We have also previously used iPSC models to study the functional effects of NRXN1 deletion and NLGN4X deletions (Shi et al., 2013), in which we found that such synaptic genes as NLGN4X/NRXN1 deletion directly impacts neurodevelopmental process, synaptic adhesion and neuron differentiation during the formation of neurons and their connections. NLGNs, which bind presynaptic NRXNs, are anchored in scaffold protein SHANK3 indirectly. SHANK3 interacts with multiple key synaptic components including glutamate receptors and their anchoring proteins, ion channels (Du et al., 1998), thus serves as a master organizer of the PSD (Naisbitt et al., 2000; Hayashi et al., 2009). NLGNs/NRXNs/SHANK3 gene complex play an import role in synapse generation and neuron function formation.
To gain insights on the critical cellular and molecular effects of SHANK3 in human neuron development, we generated functional neurons derived from iPSC and transduced by shRNA-based lentivirus against SHANK3, as well as a control shRNA. Utilizing this iPSC-based in vitro model, we investigated the transcriptome alteration, coupled with morphology and electrophysiological analyses to determine the impacts of SHANK3 knockdown in the developing human neurons.
Materials and Methods
The Generation of iPSC-Derived Neural Development Model
Human urine epithelium-derived cells were reprogrammed into iPSC [generated by Dr. Pei’s lab (Zhou et al., 2011)]. The iPSC were cultured in Matrigel (BD MatrigelTM, hESC-qualified Matrix)-coated six-well plates. MTeSRTM medium (Stemcell Technologies) was added to each well (2 ml per well) and replaced once a day. After reaching 95% confluence, the cells were passaged with EDTA (1:3) in Matrigel-coated 12-well plates. Once the cells density reached almost 100%, the medium was switched to neural induction medium (N2B27 + 2 inhibitors (5 μmol/L Dorsomorphin and 5 μmol/L SB431542, Selleck). Dorsomorphin and SB431542 effectively inhibit SMAD signaling pathway by blocking phosphorylation of ALK4, ALK5, ALK7 receptors and successfully improve the efficiency of neural induction. At day 8, the cells were mechanically scraped to Matrigel-coated six-well plates with neural proliferation system I (N2B27, Thermo-Fisher Scientific) medium. At day 16, the cells in 6-well cell plates were mechanically scraped to T25 flasks and were cultured with neural proliferation system II (N2B27 + 20 ng/mL bFGF + 20 ng/mL EGF, Thermo-Fisher Scientific). NPCs were expanded for three passages with Accutase (StemPro Accutase cell dissociation reagent), single cells of NPCs were directly plated at a density of 105 cells per well on glass coverslips coated with glial cell feeder layer (prepared from rat astrocyte, P0–P3), and Matrigel in 24-well plates for morphology analysis. At the same time, the single cells of NPCs were directly plated at a density of 2.5 × 10ˆ6/well in Matrigel-coated 6-well plates for q-PCR and sequencing. The medium was switched to neuron differentiation medium (N2, B27, Thermo Fisher Scientific; 1 μM dibutyryl-cAMP, Sigma-Aldrich; 20 ng bdnf, PeproTech). The plates were kept at 37°C thereafter in a humidified incubator with 95% air and 5% CO2.
Virus Transduction and Quantitative Real-Time-PCR
Single cells of NPCs were directly cultured on Matrigel-coated plates. The next day the cells were infected by shControl and shSHANK3 lentivirus in proper titers. (TRIPZ Human SHANK3 shRNA, Dharmacon, Inc., ID: V2THS_264172, 1 × 108 TU/mL). Six hours after infection, the viruses were removed and replaced with NPCs proliferation system containing doxycycline (1 μg/mL). Three days after infection, medium was switch to NPCs proliferation system containing doxycycline (1 μg/mL) and puromycin (1 μg/mL). The cells were lysed with Trizol, total RNA was extracted using an RNeasy mini kit. RNA concentration (OD260/OD280) was measured using Nanodrop 2000C Spectrophotometer. Total RNA was reverse transcribed into cDNA using the PrimeScriptTM RT reagent Kit with gDNA Eraser (Perfect Real Time). Real-time quantitative PCR was performed with SYBR Premix Ex TaqTM II detection System and Inumin detection instrument. The primer sequences are as follows:
Human SHANK3-F, CAGGACGCGCTCAACTATG;
Human SHANK3-R, GCATAAACTCGCCGCTTGTA;
Human GAPDH-F, CATGTTCGTCATGGGTGTGAA;
Human GAPDH-R, AGTGATGGCATGGACTGTGGT.
All data was normalized to the GAPDH mRNA level and shSHANK3 knockdown efficiency were calculated.
Morphology Analysis
Neurons were sparsely infected with lentivirus (1 × 108 TU/mL) packed with a tetracycline-controlled red fluorescent protein (RFP) expression sequence to obtain fluorescent images. Neurons plated on glass coverslips were used for morphological construction at 3–28 days after plating. Standard immunohistochemistry protocols were used. The neurons were fixed using 4% paraformaldehyde for 30 min, washed with PBS, permeabilized with 0.3% Triton X (Thermo Scientific) in PBS and blocked with 1% BSA (VETECTM) in PBS. The neurons were stained by double immunofluorescence with DAPI (Thermo Scientific, D1306, 1:1000) and the following antibodies: Mouse-Oct4 (BD Pharmingen, 6765-100, 1:200), Mouse-SSEA4 (Invitrogen, 41-4000, 1:100), Mouse-SOX2 (R&D Systems, MAB2018, 1:500), Rabbit-Nestin (Millipore, ABD9, 1:1000), Mouse-TBR1(Abcam, ab31940, 1:800), Mouse-TUJ1 (Covance, Princeton, T8660, 1:1000), Tau1 (Millipore, MAB3420, 1:500), Ribbat-Map2 (2a + 2b) (Millipore, AB5622, 1:1000), Mouse-PSD95 (Thermo Fisher, MA1-045, 1:150), Rabbit-synapsin I (Millipore, AB1543, 1:1000), secondary antibodies used include: donkey anti-mouse Alexa-488 (Invitrogen, R37114, 1:1000), donkey anti-mouse Alexa-546 (Invitrogen, A10036, 1:1000). Images of neurons were visualized with 20, 40, and 60× objective (Imager Z2, Zeiss) and digitized using a Zeiss camera (Axiocam 506 mono, Zeiss). The soma area, growth cone area, neurite length and branches were analyzed and quantified with ImageJ. The complexity of neurite arborization was analyzed with the ImageJ/FIJI. Statistic results were analyzed by two-way ANOVA with GraphPad Prism 5.01. Growth cone was categorized into three types (blunt ended, filopodial and lamellipodial) as described by Khazaei et al. (2014).
Electrophysiological Recording
The RFP-expressing iPSC-derived neurons were perfused with artificial spinal cerebral fluid (pH 7.4, in mM: 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4 and 10 D-glucose) bubbled with 95% O2 and 5% CO2 at RT (25 ± 1°C). The neurons were visualized with a 40× water objective on an IR-DIC microscope (Nikon Eclipse FN-1 microscope) and recorded using an amplifier (MultiClamp 700B, Molecular Devices). Borosilicate microelectrodes with a resistance of 4–8 MΩ were pulled using a pipette puller (Narishige PC10) and the glass pipette was filled with filtered intracellular recording solution (pH 7.3, 290–310 mOsm, in mM: 126 K-gluconate, 4 KCl, 0.3 Na2-ATP, 4 Mg-GTP, 10 phosphocreatine, 10 HEPES). Recordings were filtered at 3 kHz (low pass) and digitized at 20 kHz (DigiData 1550A, Molecular Devices), and statistical analysis of electrophysiology data was collected and analyzed with pClamp10 (Molecular Devices). The input resistance of the cells (Rin) was recorded as the slope of linear fits of current-voltage plots responded to 1 s current injection steps (−10, 2, or 3 pA steps) in current-clamp mode. Cells were held at resting membrane potential (RMP), not corrected for a liquid junction potential. Spontaneous excitatory (EPSCs) and inhibitory (IPSCs) postsynaptic currents were recorded at a −70 and 0 mV, respectively, for 3 min in voltage-clamp mode. sEPSCs and sIPSCs were verified by complete blockade of kynurenic acid (KyA, 3 mM, Sigma-Aldrich) or SR95531 (10 μM, Tocris). The sodium currents (INa) were recorded at −70 mV in voltage-clamp mode and elicited by 200 ms voltage steps from −20 to 50 mV at 5 mV increments.
Illumina Transcriptome Library Preparation and Sequencing
The total RNA was subject to RNA-Seq analysis. RNA concentration was measured using Nanodrop 2000C Spectrophotometer and Qubit 3.0 (Invitrogen). RNA integrity number and the library length were detected by BioAnalyzer 2100 (Agilent). Library construction was performed with Truseq RNA Access library Pre Kit (Illumina, RS-301-2001). The library was sequenced on a HiSeq X platform (Illumina).
Western Blot Analysis
Cultured cells were washed twice with PBS (1 mL/well), then 200 μl cell lysis buffer (RIPA buffer (Thermo scientific) supplemented with protease inhibitor (Calbiochem, 50:1) were added to each well. Cells were collected with a cell scraper (NEST Biotechnology). Cell suspension was collected and allowed to stand on ice for 30 min, and then centrifuged at 4°C, 14,000 rpm for 15 min. After centrifugation, supernatant was transferred to a new tube and protein concentration was quantified using PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were mixed with 10× loading buffer (Takara) and boiled for 10 min 20 μg proteins were loaded each lane and separated by SDS-PAGE gel. The proteins were then transferred to PVDF membranes (Millipore), incubated with an antibody against SHANK3 protein (Santa Cruz Biotechnology), diluted in 5% BSA overnight at 4°C. The PVDF membranes were then washed three times, and further processed with HRP-conjugated secondary antibodies for 2 h at room temperature. Protein signals were developed using ChemiDoc Touch Imaging System. The optical density of immunoreactive bands was quantified by ImageJ.
Bioinformatics Analysis of RNA-Seq Data
All the sequencing data were obtained as FASTQ files for 16 samples with four different measurement times at days 0, 7, 9, and 28. To improve the quality of alignment, we used FastQC (Andrews, 2010) to investigate the quality of reads in the dataset and Cutadapt (Martin, 2011) for removing adapter sequences. The reads were then mapped to the reference human genome (GRCh38) and gene count data were produced using STAR (Dobin et al., 2013). Then, we proceeded with downstream analysis after assuring that the rates of uniquely mapped reads for all samples were higher than 80%.
Generalized linear model was used to identify differentially expressed genes between SHANK3 knockdown group and the control group over time using DESeq2 (Love et al., 2014). MA-plot (Supplementary Figure S6) and dispersion estimate plot (Supplementary Figure S7) were generated for quality control before further analysis. After performing regularized-logarithm transformation on the raw counts data using rlog function in DESeq2, we plotted a sample hierarchical clustering heatmap using Euclidian distance to assess the similarity among samples. The limma R package (Ritchie et al., 2015) was used for correcting batch effects among the samples produced from two different sequencing experiments. Benjamini and Hochberg correction methods (Benjamini and Hochberg, 1995) for multiple-testing adjustment were adopted in moderated t-test to identify differentially expressed genes. The genes with adjusted p-value less than 0.05 were selected as differentially expressed genes, which represent the ones at one or more time points with a gene-specific difference accounting for the difference at time 0. Then Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Gene Ontology (GO) enrichment analysis were performed for function annotation on WebGestalt (Wang et al., 2013), and FDR < 0.01 was used as the threshold to examine significant results.
Results
Generation of Neural Development Model in vitro
To study the effect of SHANK3 loss of function on neurodevelopment, we generated iPSC-derived neural development model in vitro with two-inhibitor culture system (Dorsomorphin and SB4315242) and collected neural cells for knockdown efficiency or morphology analysis at multiple time points (days 3, 7, 9, 14, 21, and 28). The iPSC were induced into neural progenitor cells (NPC), which were cultured in floating, and then differentiated into neurons on coverslips. The 1st day of induction on coverslip was defined as day 0 (D0).
The human iPSC used in this experiment were stained positive with specific iPSC marker proteins (OCT4 and SSEA4) (Figure 1A) and specific neural stem cells marker proteins (SOX2, nestin) (Figure 1B), indicating that the cells have the potential of differentiating into neurons. NPC were directly plated on glass coverslips coated with rat glial cell feeder layer and Matrigel in 24-well plates for sample collection in different time points. The neuron-specific marker protein TUJ-1, Tau-1, Map2, deep layer cortical neuron marker protein TBR1, and subtype neuron marker protein VGLUT1, GABA, TH were detected by immunohistochemistry (Figure 1C and Supplementary Figure S1). The results showed that neurons derived from iPSC could be differentiated into multiple CNS neuron types, reflecting a highly induction efficiency (estimated > 90%) using our method. We have further quantified that in this in vitro induced neural model, the proportion of glutamatergic neurons is about 10.7%, dopaminergic neurons is about 17.3%, GABAergic neurons is approximately 54.9%.
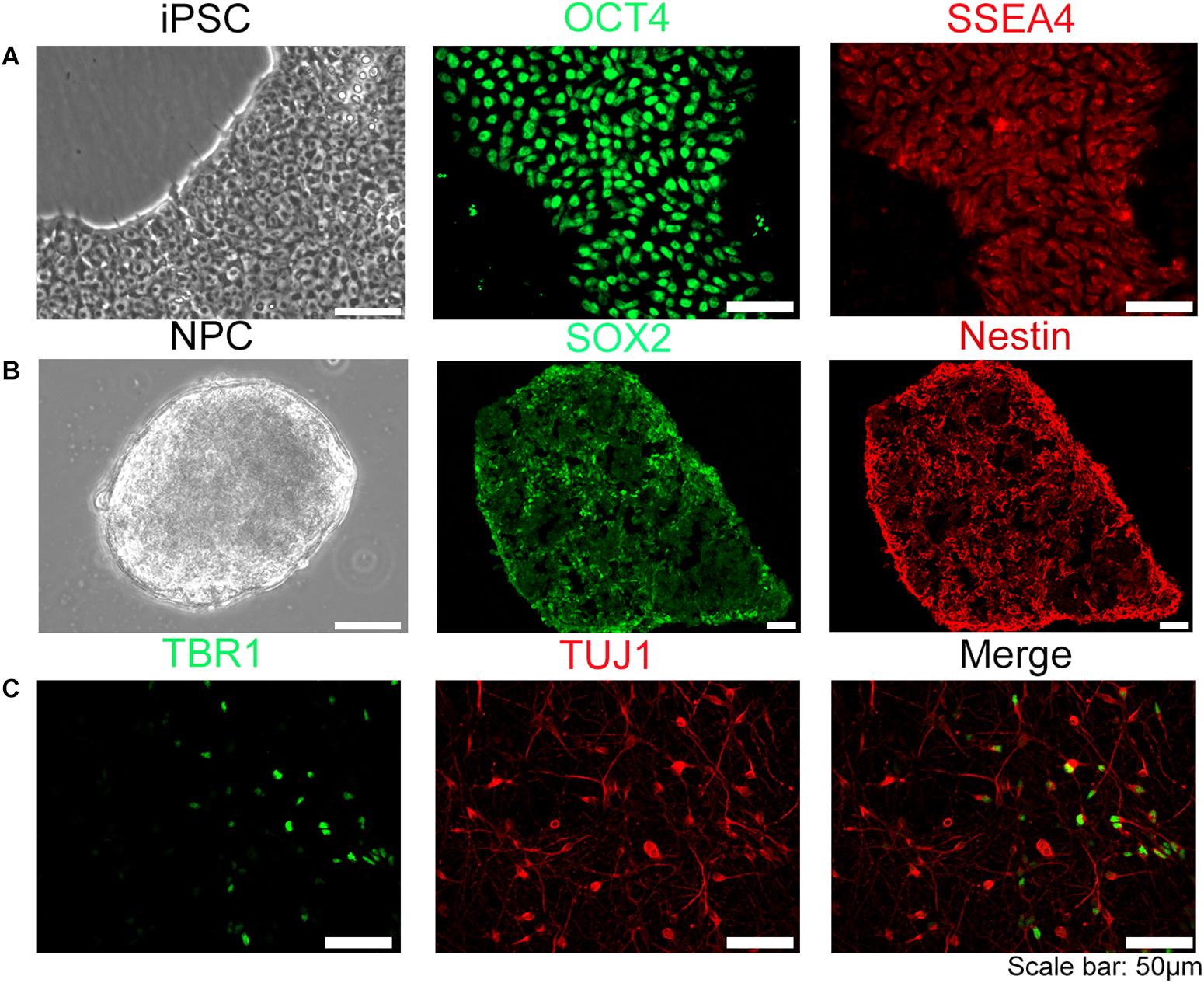
Figure 1. Establishment of human iPSC model. The iPSC were induced into neural progenitor cells (NPC) and further differentiated into neurons. (A) iPSC used in our experiment were OCT4, SSEA4 positive stain. (B) NPC induced form iPSC were SOX2, Nestin positive. (C) Neurons differentiated from NPC were TUJ-1 or TBR1 positive, suggesting that NPC can be differentiated into multiple categories of neural cells. Scale bar: 50 μm.
SHANK3 expression pattern data from Allan Brain Institute pointed to abundant expression in the developing brain at very early development stage1 (Supplementary Figure S2A), that is consistent with our Western blot result of SHANK3 (Supplementary Figure S2B). We also found that expression of SHANK3 emerged in early neuronal development stages (P0-7), and continue to increase into young adulthood (P42). This increased levels of SHANK3 protein may reflect the time course of cortical synapse maturation (Supplementary Figures S2A,B).
Morphological Effects of SHANK3 Knockdown on Neurodevelopment
We have generated two sets of neural induced model from two separate iPSC lines in this whole project, and we used one clone of each line to collect data for morphology analysis (Supplementary Figure S3). To knockdown SHANK3 expression in NPC, we infected NPC with shRNA-based lentivirus against SHANK3 or with a control lentivirus, here after referred as shSHANK3 and shControl, respectively. RFP signals were observed in 1 week post infection. Knockdown efficiency was verified at multiple levels, including qRT-PCR (Figure 2A), Western blot analysis (Figure 2B) and immunocytochemistry staining of cultured neurons (Figure 2C). These data suggest lentiviral-mediated SHANK3 knockdown was highly effective.
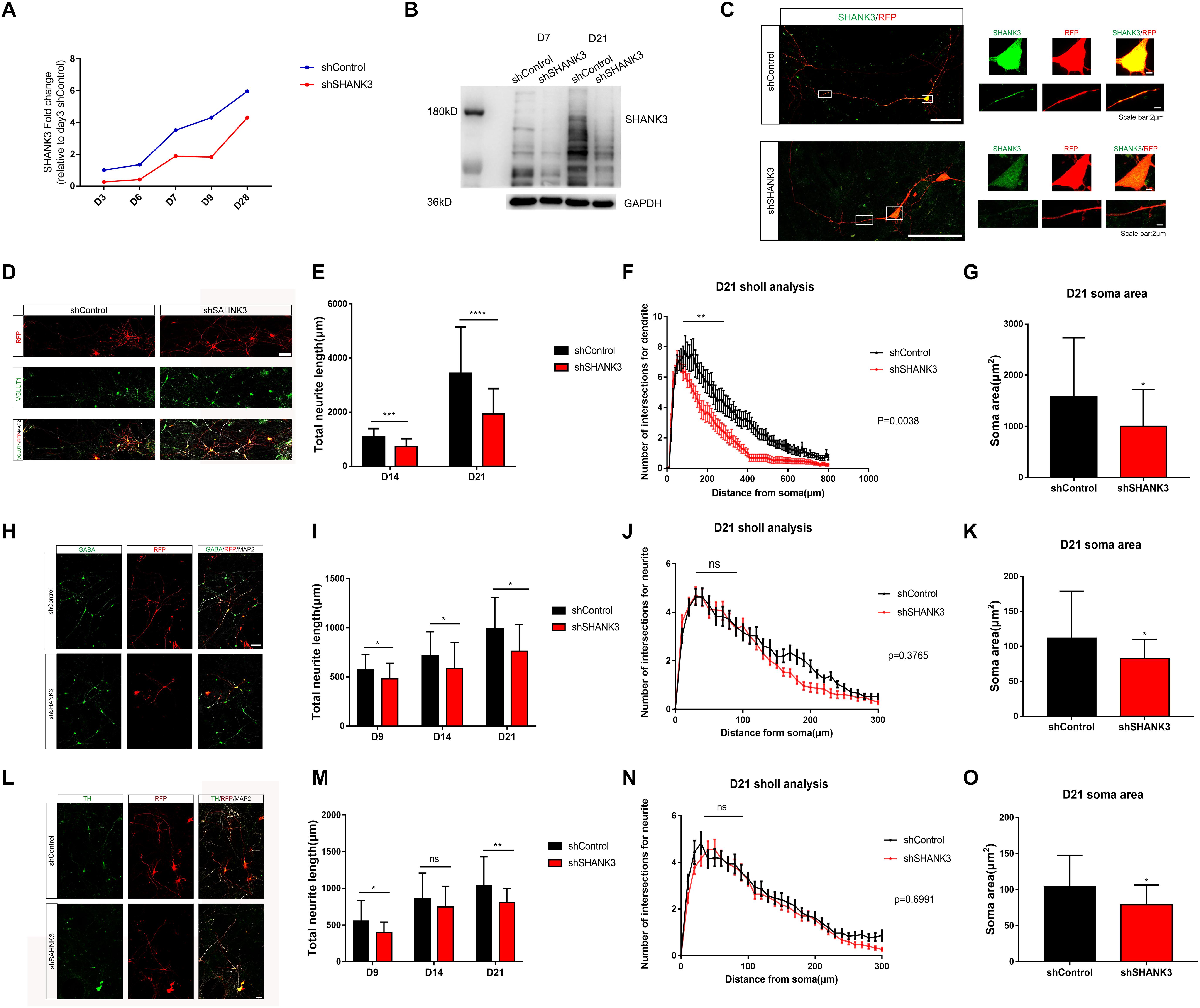
Figure 2. SHANK3 knockdown profoundly affects neurite length, complexity of neurite arborization and soma area. (A–C) The efficiency of SHANK3 knockdown in iPSC-derived neurons was verified by qRT-PCR (D3, D7, and D28), Western blot (D7 and D21), and immunofluorescence staining (D28). (D,H,L) Immunofluorescence staining of VGLUT1, GABA, TH, and MAP2 confirms generation of multiple types of differentiated neurons in our culture system. Scare bar: 50 μm. (E,I,M) Comparison of neurite length in neurons infected with shShank3 and shControl viruses. Data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. (D9: GABA: n = 33 control and 35 SHANK3 knockdown cells, p = 0.0181; TH: n = 33 control and 33 SHANK3 knockdown cells, p = 0.0159); D14: VGLUT1: n = 22 control and 22 SHANK3 knockdown cells, (p = 0.0003), GABA: n = 33 control and 34 SHANK3 knockdown cells (p = 0.0219), TH: n = 34 control and 37 SHANK3 knockdown cells (p = 0.2482); D21: VGLUT1: n = 22 control and 24 SHANK3 knockdown cells (p < 0.0001), GABA: n = 30 control and 29 SHANK3 knockdown cells (p = 0.0037), TH: n = 30 control and 30 SHANK3 knockdown cells, (p = 0.0035). (F,J,N) Analysis of the effect of SHANK3 gene knockdown on the complexity of neurite arborization. Statistical significance was evaluated by two-way ANOVA, p = 0.0038 (D21), p = 0.3765 (D21), p = 0.6991 (D21), data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (G,K,O) Statistic analysis of the effect of SHANK3 knockdown on neuronal soma area. Statistical significance was evaluated by two-way ANOVA, data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (D21: VGLUT1: n = 22 control and 24 SHANK3 knockdown cells, GABA: n = 30 control and 29 SHANK3 knockdown cells, TH: n = 30 control and 30 SHANK3 knockdown cells).
We next investigated the impact of SHANK3 knockdown on neurite length. Neuron morphology was traced and reconstructed by ImageJ. Sholl analysis was used to quantify neurite complexity. We further classified neurons into three groups in our in vitro model, excitatory neurons (VGLUT1+), inhibitory neurons (GABA+), and putative dopamine neurons (TH+) (Supplementary Figure S1). To distinguish axons and dendrites, we used MAP2 and Tau1 antibody to co-stain dendrites (MAP2+) and axons (Tau1+) (Supplementary Figure S1). Neuronal morphology was constructed base on RFP expression. We examined the effects of SHANK3 knockdown on neurite development (e.g., length and the number of branches) at different time points. We observed that, the total dendrites length of VGLUT1 positive neuron decreased significantly with SHANK3 knockdown on D14 (p = 0.0003), D21 (p < 0.0001) (Figures 2D,E); GABA positive neuron dendritic length decreased significantly on D9 (p = 0.0181), D14 (p = 0.0219), D21 (p = 0.0037) (Figures 2H,I); while TH positive neuron dendritic length decreased significantly on D9 (p = 0.0159), D14 (p = 0.2482), D21 (p = 0.0035) (Figures 2L,M). These results indicate that SHANK3 gene is required for the development of neurite length in iPSC derived human neurons.
To further assess of neurite complexity, Sholl analysis was used to quantify neurite branches number. We observed that SHANK3 knockdown had significant effect on neurite complexity in excitatory neurons at D21 (Figure 2F), especially on the neurite branches number close to the cell body area within 100 microns, while SHANK3 deficit had few effect on neurite complexity of inhibitory neurons and dopamine neurons (Figures 2J,N). These results indicate that SHANK3 gene is required for the development of excitatory neuronal branches in iPSC-derived human neurons.
We next investigated the morphological differences following SHANK3 knockdown in iPSC-derived human neurons, we analyzed soma and growth cone development in shControl and shShank3 groups. We observed that soma area of VGLUT1 positive neuron significantly decreased with SHANK3 knockdown at D21 (p = 0.0097), the soma area of GABA positive neurons decreased at D9 (p = 0.0254), D21 (p = 0.0108), the soma area of TH positive neurons decreased at D9 (p = 0.0294), D14 (p = 0.0002) (Figures 2G,K,O). We have identified TH and GABA positive neuronal soma size in D9, as VGLUT1 antibody specifically stained excitatory neuron at least in D14 in our study. The results indicate that SHANK3 gene knockdown affects the development of neuronal soma in early neuronal development of D9, at least in GABA and TH positive neurons. In addition, SHANK3 knockdown has a significant effect on VGLUT1 positive neuron in D21.
Next, we examined whether SHANK3 is required for growth cone formation. We found that the area of the growth cone significantly decreased with SHANK3 knockdown at all-time points (days 9, 12, 15, and 21) (Figure 3). These results indicate that SHANK3 knockdown affects the development of neuronal growth cone area.
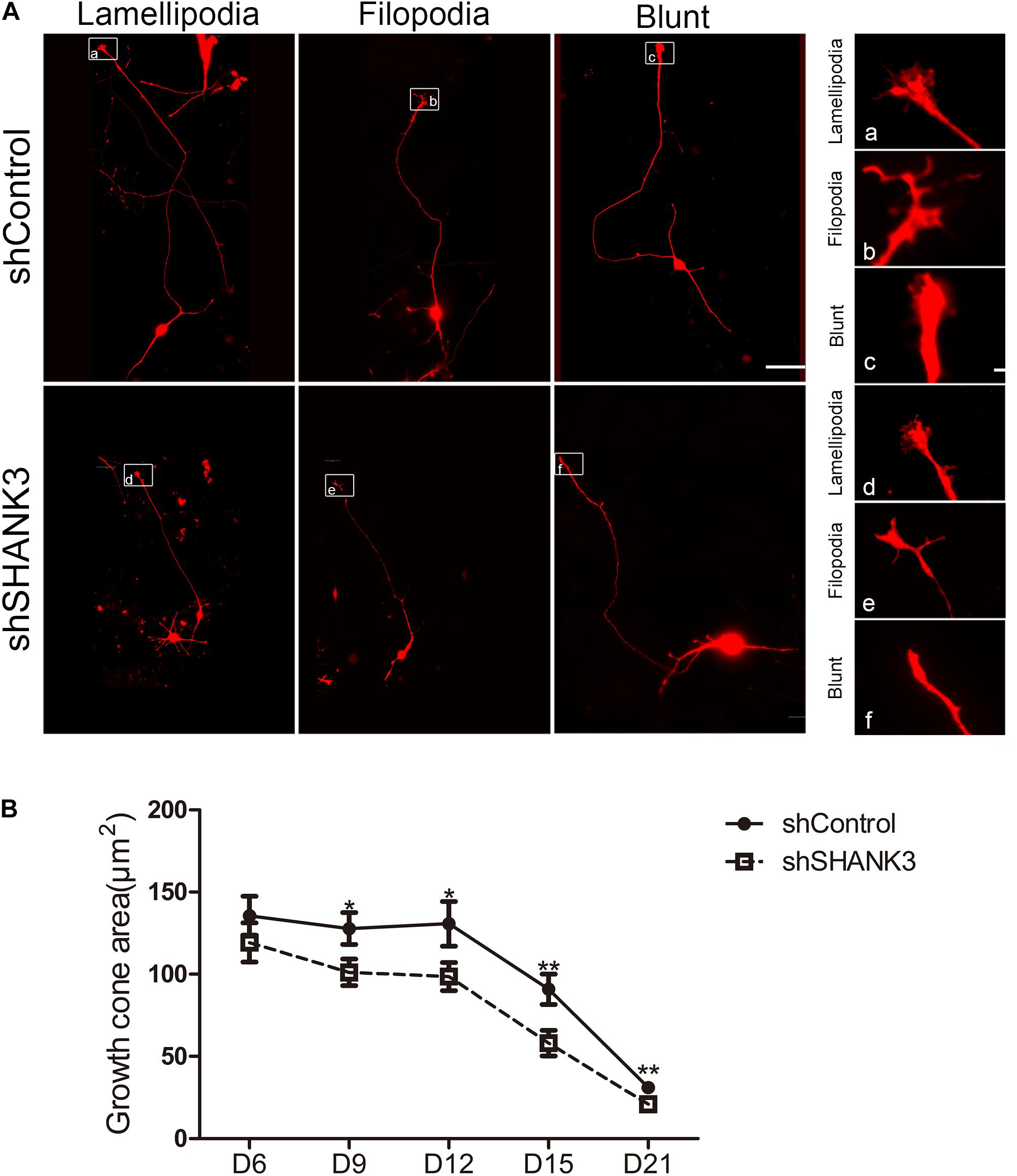
Figure 3. The effects of SHANK3 knockdown on neurons growth cone area. (A) Representative images of the growth cones of shControl and shSHANK3 transduced neurons. Scare bar: schematic diagram 50 μm; enlarged area 5 μm. (B) Analysis of the effect of SHANK3 knockdown on the growth cone area of neurons. Statistical significance was evaluated by two-way ANOVA, date are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01. (D6: n = 57; D9: n = 67 control and 62 SHANK3 knockdown cells; D12: n = 64 control and 63 SHANK3 knockdown cells; D15: n = 65 control and 61 SHANK3 knockdown cells; D21: n = 67 control and 64 SHANK3 knockdown cells; D28: n = 65).
SHANK3 Knockdown Affects the Electrophysiological Properties of Developing Human Neurons
To investigate functional outcomes of SHANK3 knockdown, we conducted whole cell patch clamp recording on RFP-expressing neurons at 3.5 and 5.5 weeks (Figure 4). To determine if excitatory synaptic transmission was altered in neurons with SHANK3 knockdown, were recorded spontaneous excitatory post-synaptic currents (sEPSCs). We found that the frequency of sEPSC from SHANK3 knockdown neurons was reduced relative to control neurons at 3.5 and 5.5 weeks (Figure 4C). In addition, spontaneous inhibitory post-synaptic currents (IPSCs) were recorded to determine the effect of SHANK3 down-regulation in inhibitory synaptic transmission. We found that the frequency of sIPSC recorded from SHANK3 knockdown neurons was reduced relative to control neurons at 3.5 and 5.5 weeks (Figure 4D). Taken together, these results suggest that both excitatory synaptic transmissions and inhibitory synaptic transmission are impaired in neurons with SHANK3 knockdown.
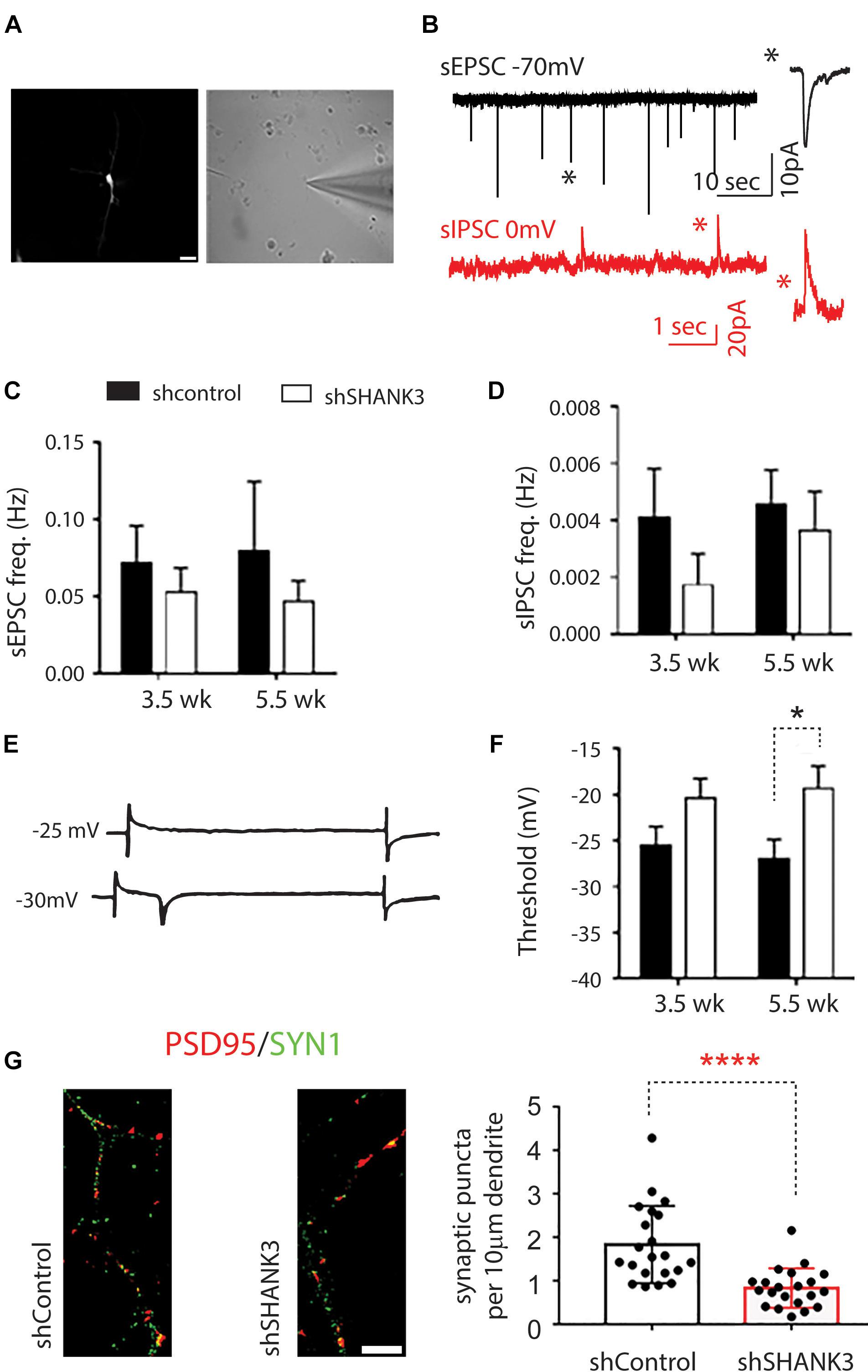
Figure 4. The effects of SHANK3 gene knockdown on neurons function. (A) RFP-expressing cell visualized under differential interference contrast microscopy. (B) Sample recordings of sEPSC (top panel) and sIPSC (bottom panel). (C) Averaged frequency of spontaneous EPSCs at 3.5 and 5.5 weeks in culture (3.5 weeks: n = 17 control and 12 SHANK3 knockdown cells; 5.5 weeks: n = 11 control and 15 SHANK3 knockdown cells). (D) Averaged frequency of spontaneous IPSCs (3.5 weeks: n = 10 control and 7 SHANK3 knockdown cells; 5.5 weeks: n = 15 control and 13 SHANK3 knockdown cells). (E) Illustration of sodium currents elicited by voltage step commands. (F) The threshold of sodium currents (3.5 weeks: n = 19 control and 12 SHANK3 knockdown cells; 5.5 weeks: n = 15 control and 15 SHANK3 knockdown cells). ∗p < 0.05. (G) Synapse number changes following SHANK3 knockdown: PSD95 (GFP) and Synapsin I (RFP) co-staining showed the synaptic puncta was reduced after SHANK3 knockdown (n = 21, ∗∗∗∗p < 0.0001).
Next, we investigated sodium currents threshold (INa). The results showed that neurons with SHANK3 knockdown exhibited an increase in the threshold of sodium currents at 3.5 weeks and increased significantly (p = 0.2245) relative to control neurons at 5.5 weeks (Figure 4F), suggesting that down-regulation of SHANK3 may extensively alter receptors for neurotransmission and voltage gated ion channels.
Effects of SHANK3 Knockdown on Transcriptome Regulation
We first examined the overall similarity between samples by using hierarchical clustering. It can be seen from the heatmap (Figure 5A and Supplementary Figure S8) that samples from the same measurement time points were clustered together regardless of the SHANK3 knockdown status. These results indicate that time exerted more important influence in determining gene expression variation comparing with SHANK3 knockdown.
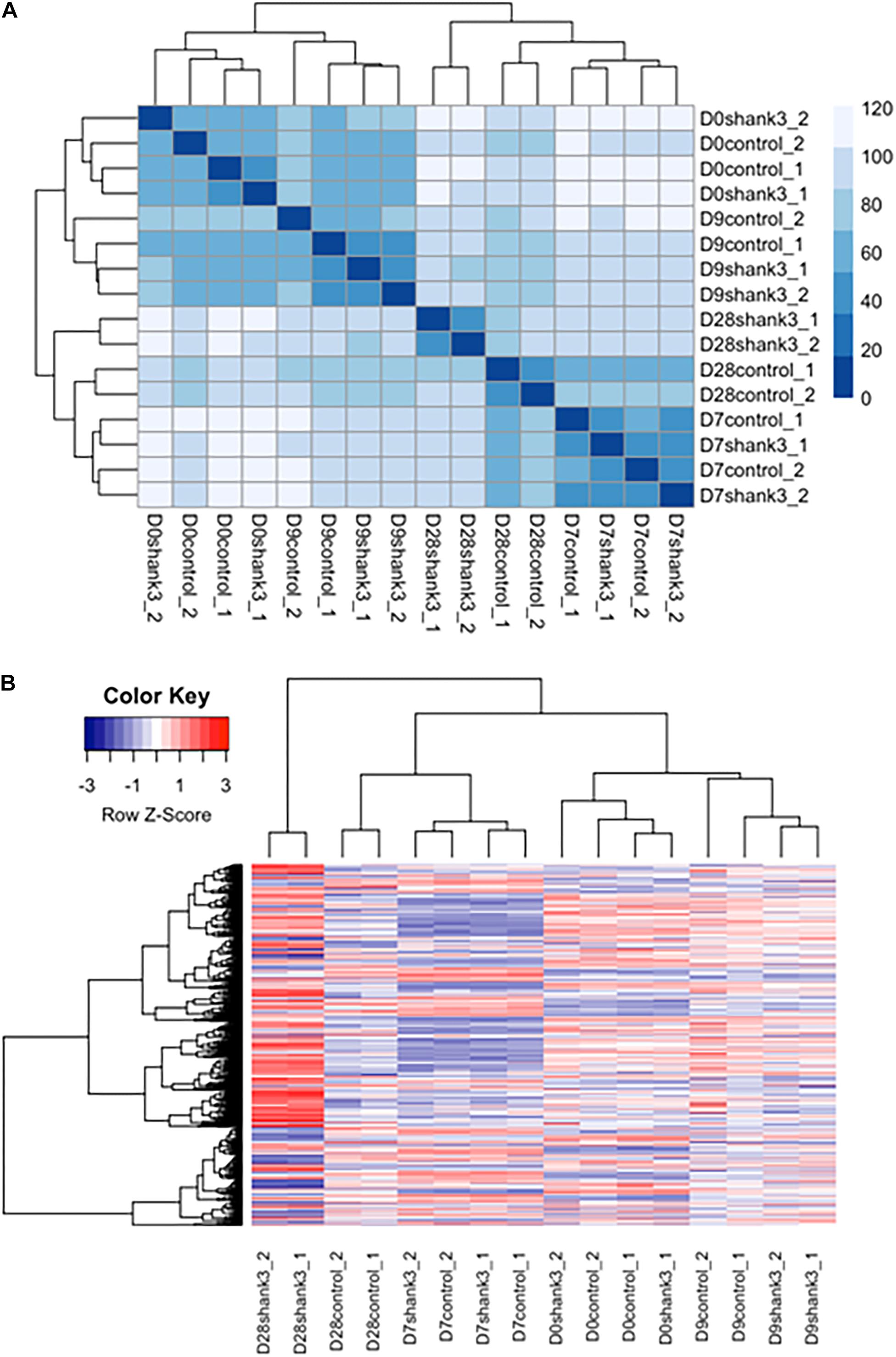
Figure 5. Heatmap of differentially expressed genes associated with SHANK3 loss of function. (A) Euclidian distances were used for constructing sample distance matrix. After eliminating batch effect that would separate the samples from two different experiments apart, samples measured at the same time points were clustered together over influence of gene knockdown. (B) Heatmap for 1576 differentially expressed genes (p-adj<0.05) over time. It can be seen obviously that the expression difference mainly occurred at day 28.
Next, differentially expressed genes were estimated between SHANK3 knockdown and control groups across four measurement time points (Supplementary Table S1). The main question is whether SHANK3 knockdown can alter gene expression over time and which genes would show this gene-specific differences. Considering the correlation of the response variable among different measurement time points and batch effects from two different experiments, we designed a generalized linear model with independent variables of batch, time, SHANK3 knockdown and an interaction term with SHANK3 and time. By testing the interaction term with likelihood ratio test, we identified 1576 differentially expressed genes (Supplementary Figure S5) over time with adjusted p-value less than 0.05 (p-adj < 0.05). Among those overall differentially expressed genes, 1078 genes showed significant difference at day 28, which can be seen obviously in the heatmap of differentially expressed genes (Figure 5B). The down regulated expression level of SHANK3 gene and 9 genes with smallest p-adj values are shown in Supplementary Figure S5.
Next, we examined whether these top differentially expressed genes shared common pathways or functional categories. We performed over representation enrichment analysis for pathways and gene ontology (Supplementary Figure S4). The results showed that SHANK3 down-regulation led to hippo signaling pathway (Hsa04390, FDR = 1.33e-05) and focal adhesion (Hsa04510, FDR = 1.84e-08) abnormalities. In addition, terms in GO analysis including neuron projection development (GO: 0031175, FDR = 0e+00, BP), regulation of neurogenesis (GO: 0050767, FDR = 0e+00, BP), central nervous system development (GO: 0007417, FDR = 0e+00, BP), focal adhesion (GO: 0005925, FDR = 1.2e-09, CC) and calcium ion binding (GO: 0005509, FDR = 4.7e-11, MF) were significantly enriched (Table 1). These transcriptome data, in combination with the morphological and functional alternations, suggest SHANK3 has a previously uncircumscribed role in developing neurons in addition to its function as a postsynaptic scaffold protein in mature synapse.
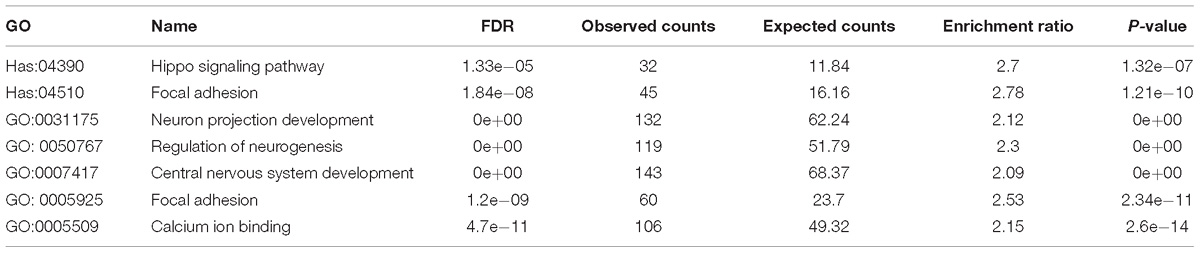
Table 1. Affected signaling pathways and gene ontology categories.
Discussion
In this study, we investigated the impact of SHANK3 loss of function in neurodevelopment by combining morphology, electrophysiology and transcriptomics analyses in neurons derived from iPSC. SHANK3 is a PSD components of glutamatergic synapses that plays a key role in excitatory synaptic transmission in adult brain (Rubeis et al., 2018). There is strong indication that SHANK3 gene mutations or variations are associated with increased autism risks, SHANK3 point mutations, truncations, and disruption by chromosome translocation have been all reported in ASD cases (Durand et al., 2007; Gauthier et al., 2009; Sykes et al., 2009; Peca et al., 2011; Boccuto et al., 2013). Consistently, mice mutant for SHANK3 display multiple neurological deficits including compulsive and repetitive behaviors, that are associated with deficits in corticostriatal circuits (Peca et al., 2011; Xiaoming et al., 2011; Bariselli et al., 2016; Sebastiano and Camilla, 2016; Zhou et al., 2016; Bariselli and Bellone, 2017; Jin et al., 2018). However, the molecular targets of SHANK3 that are causally linked to the ASD-like behavioral deficits, and how disruption of SHANK3 derails the normal neurodevelopmental trajectory in human brain remain largely unclear. In this study, we took advantage of an iPSC neuron induction model, and used shRNA to induce SHANK3 loss of function to investigate the neurodevelopmental role of SHANK3 protein in human neurons. Consistent with previous reports (Durand et al., 2012), we found that synapse numbers were reduced, and synaptic transmissions are impaired in SHANK3 knockdown neurons.
It’s worth noting that SHANK3 protein expressed in early developing stage from NPC to mature neuron based on our qRT-PCR and Western blot results. This is consistent with the curated data on the Allen Brain Atlas, which show that SHANK3 mRNA is abundant (Supplementary Figure S2). This early SHANK3 expression suggests a functional role in developing neuronal morphology and emergence of function, in addition to being a postsynaptic scaffold protein at the mature glutamatergic synapses at later developmental stages. We also found that SHANK3 deficiency affects the morphology at early development stages, before the synaptic transmission are established and mature. Our results show that SHANK3 knockdown reduces dendritic arborization in three major types of differentiated neurons (glutamatergic/GABAergic/dopamine). We also found that the soma size and growth cone area of glutamatergic/GABAergic/dopamine neuron were reduced as a result. We further confirmed SHANK3 protein did localize in dendrite and soma by immunofluorescence staining experiments. A few previous studies have reported that SHANK3 localization in neuron soma, dendrite, growth cone (Du et al., 1998; Boeckers et al., 2002; Durand et al., 2012; Halbedl et al., 2016) and the dendritic deficits (Durand et al., 2012) following SHANK3 deficiency in human neurons. SHANK3 protein is found to be located at the tip of actin filaments and promotes growth cone motility in developing neuron by enhancing actin polymerization (Durand et al., 2012; Halbedl et al., 2016). A de novo SHANK3 mutation in the ankyrin domain (Q312R) is associated with growth cone formation and motility in animal models (Durand et al., 2012; Kathuria et al., 2018). Kathuria et al. (2018) uncovered the SHANK3’s critical role in neuronal morphogenesis and the early defects which are associated with ASD-associated mutations. Our data are consistent with previous studies. Morphology analyses indicates strong correlation between SHANK3 knockdown and dendritic branching abnormalities; neuron soma area, neurite number and length, complexity of neurite arborization and growth cone area are all significantly affected by SHANK3 knockdown.
Another important finding from this study is that SHANK3 knockdown changes the transcriptome landscape across the time course of neural development. We identified genes with temporal changes of expression patterns across four time points between control shRNA and the shSHANK3 groups, and performed pathway enrichment analysis. We showed that even in early stage, the development-associated pathways can be affected by SHANK3 knockdown, which is consistent with the observed abnormality of neuronal morphology following SHANK3 knockdown in early stage of neurodevelopment. The PI3K associated pathway has been enriched in transcriptome analysis in our study, which also support the hypothesis that SHANKs mutation serves to cross-link further disease related signaling cascades (mTOR/PI3K). Altogether, these results indicate that SHANK3 has a previously unappreciated role in early neuronal development, in addition to its well established functional role as a postsynaptic scaffold protein in the mature synapse.
Our electrophysiology results are generally consistent with the morphological findings. We found that both excitatory synaptic transmissions and inhibitory synaptic transmission are impaired in SHANK3 knockdown neurons. However, the underlying mechanism of the synaptic transmission impairment in SHANK3 knockdown neurons should be investigated in future studies. Taken together, our study show SHANK3 loss of function profoundly derails the developmental trajectory of human neurons. Many functional domains of neural development are impaired, including dysplasia of neuronal soma, stunted neurite and growth cone, and altered of transcriptome. These changes suggest SHANK3 loss of function as an intrinsic, cell autonomous factor that impairs cellular development in human neurons both in early and in mature stages, which may account for the brain pathological changes in neurodevelopmental diseases such as ASD.
Author Contributions
LS designed the study and contributed to transcriptome data analysis, wrote and approved the final version of the manuscript. GH, SC, XC, QC, ZX, SG, and XM conducted cell culture experiments, neural morphology, and other data analysis. GH and SC co-wrote the final version of the manuscript. JZ and JY generated the electrophysiological data. KW and AT contributed to performing transcriptome data analysis. LZ and SQ contributed to the experimental design, data analyses, and preparation of the manuscript.
Funding
This project was supported by the National Natural Science Foundation of China (81771222) (LS), National Key Research and Development Program of China, Stem Cell and Translational Research (2017YFA0105102) (LS), Guangzhou Science and Technology Innovation Development special fund project (201804010212) (LS), Fundamental Research Funds for the Central Universities of China (11617435) (LS), and Program of Introducing Talents of Discipline to Universities (B14036) (LS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Duanqing Pei (Chinese Academy of Sciences) for providing the iPSC models used in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnana.2019.00023/full#supplementary-material
FIGURE S1 | Expression of MAP2 (GFP), Tau1 (RFP), VGLUT1 (GFP), GABA (GFP), and TH (GFP) in D28 neurons in our in vitro model, further supporting successful differentiation into various mature neuron types.
FIGURE S2 | Expression of SHANK3 mRNA and protein at very early developmental stages in mouse brain. (A) SHANK3 mRNA is abundantly expressed in the developing brain at early development stage (source: Allen Brain Atlas). (B) SHANK3 protein can be detected by Western blot in developing mouse cortex tissues as early as P0, but increase as the brain matures.
FIGURE S3 | Two sets of neural induced model from two separate iPSC lines have been generated in this whole project, and we used one clone of each line to collect data for morphology analysis. Both of these two clone showed significant decrease on dendritic length and soma size after SHANK3 knockdown. Comparison of neurite length in another iPSC line neurons infected with shShank3 and shControl viruses. Morphology reconstructed with ImageJ, statistical significance was evaluated by T-test, data are shown as mean ± SEM. ∗p < 0.05. (A) D9: GABA: n = 35 control and 33 SHANK3 knockdown cells, (p = 0.0037); (B) D9: TH: n = 30 control and 31 SHANK3 knockdown cells, (p = 0.0340); (C) Detailed Vector Map of the pTRIPZ lentiviral vector.
FIGURE S4 | Bar charts of gene ontology function categories.
FIGURE S5 | Effects of SHANK3 knockdown on gene expression levels. Expression level were transformed into log10 scale. Green lines represent SHANK3 knockdown group and red lines represent control group. (A) Expression level of SHANK3 gene over time between knockdown and control group. (B) Expression level of top 9 genes with smallest p-adj values.
FIGURE S6 | RNA sequence data MA plot. Each dot represents a gene. X-axis is the average expression over all samples. Y-axis is the log2 fold change between SHANK3 knockdown and control group. Genes with FDR<0.01 are shown in red. This plot demonstrates that only genes with a large average normalized count contain more information to yield a significant call.
FIGURE S7 | Dispersion estimation plot for raw counts data. The dispersion fit is an exponentially decaying curve where dispersion decreased as the counts increased of all genes. Each black point represents the dispersion estimates for each gene across all the eight samples. The red line is fitted trend line, which shows the dispersions’ dependence on the mean. Blue points are the final estimates from black points shrunk to the red fitted line. The blue circles are genes which are labeled as dispersion outliers and are not shrunk toward the fitted trend line. X-axis is the average expression over all eight samples. Y-axis is the dispersion value. The dispersion fit for all genes is an exponentially decaying curve where dispersion decreased with the mean of normalized counts.
FIGURE S8 | The list of top 150 differentially expressed (DE) at each time point between the knockdown and control samples.
TABLE S1 | Differential expression between SHANK3 knockdown and control groups.
Footnotes
References
Amal, H., Barak, B., Bhat, V., Gong, G., Joughin, B. A., Wang, X., et al. (2018). Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatry. doi: 10.1038/s41380-018-0113-6
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bariselli, S., and Bellone, C. (2017). VTA DA neuron excitatory synapses in SHANK3 Deltaex(4-9) mouse line. Synapse 71, doi: 10.1002/syn.21955
Bariselli, S., Tzanoulinou, S., Glangetas, C., Prévost-Solié, C., Pucci, L., Viguié, J., et al. (2016). SHANK3 controls maturation of social reward circuits in the VTA. Nat. Neurosci. 19, 926–934. doi: 10.1038/nn.4319
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bey, A. L., Wang, X., Yan, H., Kim, N., Passman, R. L., Yang, Y., et al. (2018). Brain region-specific disruption of SHANK3 in mice reveals a dissociation for cortical and striatal circuits in autism-related behaviors. Transl. Psychiatry 8:94. doi: 10.1038/s41398-018-0142-6
Bidinosti, M., Botta, P., Krüttner, S., Proenca, C. C., Stoehr, N., Bernhard, M., et al. (2016). CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 351, 1199–1203. doi: 10.1126/science.aad5487
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A., et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21, 310–316. doi: 10.1038/ejhg.2012.175
Boeckers, T. M., Bockmann, J., Kreutz, M. R., and Gundelfinger, E. D. (2002). ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 81, 903–910. doi: 10.1046/j.1471-4159.2002.00931.x
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated SHANK3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1:15. doi: 10.1186/2040-2392-1-15
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Mark, T., Michel, S., and Lorenz, S. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Chen, C. H., Chen, H. I., Liao, H. M., Chen, Y. J., Fang, J. S., Lee, K. F., et al. (2017). Clinical and molecular characterization of three genomic rearrangements at chromosome 22q13.3 associated with autism spectrum disorder. Psychiatr. Genet. 27, 23–33. doi: 10.1097/ypg.0000000000000151
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Du, Y., Weed, S. A., Xiong, W.-C., Marshall, T. D., and Parsons, J. T. (1998). Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol. Cell. Biol. 18, 5838–5851. doi: 10.1128/MCB.18.10.5838
Durand, C. M., Catalina, B., Boeckers, T. M., Juergen, B., Pauline, C., Fabien, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Durand, C. M., Perroy, J., Loll, F., Perrais, D., Fagni, L., Bourgeron, T., et al. (2012). SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 17, 7184. doi: 10.1038/mp.2011.57
Freitag, C. M., Staal, W., Klauck, S. M., Duketis, E., and Waltes, R. (2010). Genetics of autistic disorders: review and clinical implications. Eur. Child Adolesc. Psychiatry 19, 169–178. doi: 10.1007/s00787-009-0076-x
Gauthier, J., Spiegelman, D., Piton, A., Lafreniere, R. G., Laurent, S., St-Onge, J., et al. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 150B, 421–424. doi: 10.1002/ajmg.b.30822
Grabrucker, A. M., Schmeisser, M. J., Schoen, M., and Boeckers, T. M. (2011). Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21, 594–603. doi: 10.1016/j.tcb.2011.07.003
Halbedl, S., Schoen, M., Feiler, M. S., Boeckers, T. M., and Schmeisser, M. J. (2016). SHANK3 is localized in axons and presynaptic specializations of developing hippocampal neurons and involved in the modulation of NMDA receptor levels at axon terminals. J. Neurochem. 137, 26–32. doi: 10.1111/jnc.13523
Harony-Nicolas, H., Kay, M., Hoffmann, J. D., Klein, M. E., Bozdagi-Gunal, O., Riad, M., et al. (2017). Oxytocin improves behavioral and electrophysiological deficits in a novel Shank3-deficient rat. eLife 6:e18904. doi: 10.7554/eLife.18904
Hayashi, M. K., Tang, C., Verpelli, C., Narayanan, R., Stearns, M. H., Xu, R. M., et al. (2009). The postsynaptic density proteins homer and shank form a polymeric network structure. Cell 137, 159–171. doi: 10.1016/j.cell.2009.01.050
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Jin, C., Kang, H., Ryu, J. R., Kim, S., Zhang, Y., Lee, Y., et al. (2018). Integrative brain transcriptome analysis reveals region-specific and broad molecular changes in shank3-overexpressing mice. Front. Mol. Neurosci. 11:250. doi: 10.3389/fnmol.2018.00250
Kang, S., Chen, X., Gong, S., Yu, P., Yau, S., Su, Z., et al. (2017). Characteristic analyses of a neural differentiation model from iPSC-derived neuron according to morphology, physiology, and global gene expression pattern. Sci. Rep. 7:12233. doi: 10.1038/s41598-017-12452-x
Kathuria, A., Nowosiad, P., Jagasia, R., and Aigner, S. (2018). Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 23, 735–746. doi: 10.1038/mp.2017.185
Kerrisk Campbell, M., and Sheng, M. (2018). USP8 deubiquitinates SHANK3 to control synapse density and SHANK3 activity-dependent protein levels. J. Neurosci. 38, 5289–5301. doi: 10.1523/jneurosci.3305-17.2018
Khazaei, M. R., Girouard, M. P., Alchini, R., Ong Tone, S., Shimada, T., Bechstedt, S., et al. (2014). Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J. Biol. Chem. 289, 30133–30143. doi: 10.1074/jbc.M114.570440
Konopka, G., Wexler, E., Rosen, E., Mukamel, Z., Osborn, G. E., Chen, L., et al. (2012). Modeling the functional genomics of autism using human neurons. Mol. Psychiatry 17, 202–214. doi: 10.1038/mp.2011.60
Lin, H., Li, Q., Du, Q., Wang, O., Wang, Z., Akert, L., et al. (2018). Integrated generation of induced pluripotent stem cells in a low-cost device. Biomaterials 189, 23–36. doi: 10.1016/j.biomaterials.2018.10.027
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, C., Chen, Q., Zhou, T., Bozic, D., Fu, Z., Pan, J. Q., et al. (2016). Micro-electrode array recordings reveal reductions in both excitation and inhibition in cultured cortical neuron networks lacking Shank3. Mol. Psychiatry 21, 159–168. doi: 10.1038/mp.2015.173
Luo, J., Feng, Q., Wei, L., and Luo, M. (2017). Optogenetic activation of dorsal raphe neurons rescues the autistic-like social deficits in SHANK3 knockout mice. Cell Res. 27, 950–953. doi: 10.1038/cr.2017.52
Lyall, K., Schweitzer, J. B., Schmidt, R. J., Hertz-Picciotto, I., and Solomon, M. (2017). Inattention and hyperactivity in association with autism spectrum disorders in the CHARGE study. Res. Autism Spectr. Disord. 35, 1–12. doi: 10.1016/j.rasd.2016.11.011
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mei, Y., Monteiro, P., Zhou, Y., Kim, J. A., Gao, X., Fu, Z., et al. (2016). Adult restoration of SHANK3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Mitne-Neto, M., Machado-Costa, M., Marchetto, M. C., Bengtson, M. H., Joazeiro, C. A., Tsuda, H., et al. (2011). Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 20, 3642–3652. doi: 10.1093/hmg/ddr284
Monteiro, P., and Feng, G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157. doi: 10.1038/nrn.2016.183
Naisbitt, S., Valtschanoff, J., Allison, D. W., Sala, C., Kim, E., Craig, A. M., et al. (2000). Interaction of the postsynaptic density-95/guanylate kinase domain-associated protein complex with a light chain of myosin-V and dynein. J. Neurosci. 20, 4524–4534. doi: 10.1523/JNEUROSCI.20-12-04524.2000
Nakanishi, M., Nomura, J., Ji, X., Tamada, K., Arai, T., Takahashi, E., et al. (2017). Correction: functional significance of rare neuroligin 1 variants found in autism. PLoS Genet. 13:e1007035. doi: 10.1371/journal.pgen.1007035
Nemirovsky, S. I., Marta, C., Zaiat, J. J., Completa, S. P., Vega, P. A., Dolores, G. M., et al. (2015). Whole genome sequencing reveals a de novo SHANK3 mutation in familial autism spectrum disorder. PLoS One 10:e0116358. doi: 10.1371/journal.pone.0116358
Parikshak, N. N., Swarup, V., Belgard, T. G., Irimia, M., Ramaswami, G., Gandal, M. J., et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540:423. doi: 10.1038/nature20612
Peca, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). SHANK3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Ponna, S. K., Ruskamo, S., Myllykoski, M., Keller, C., Boeckers, T. M., and Kursula, P. (2018). Structural basis for PDZ domain interactions in the post-synaptic density scaffolding protein Shank3. J. Neurochem. 145, 449–463. doi: 10.1111/jnc.14322
Qin, L., Ma, K., Wang, Z. J., Hu, Z., and Matas, E. (2018). Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 21, 564–575. doi: 10.1038/s41593-018-0110-8
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Rubeis, S. D., Siper, P. M., Durkin, A., Weissman, J., Muratet, F., Halpern, D., et al. (2018). Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol. Autism 9:31. doi: 10.1186/s13229-018-0205-9
Sebastiano, B., and Camilla, B. (2016). VTA DA neuron excitatory synapses in SHANK3 Δex(4-9) mouse line. Synapse 71, doi: 10.1002/syn.21955
Shi, L., Chang, X., Zhang, P., Coba, M. P., Lu, W., and Wang, K. (2013). The functional genetic link of NLGN4X knockdown and neurodevelopment in neural stem cells. Hum. Mol. Genet. 22, 3749–3760. doi: 10.1093/hmg/ddt226
Shi, Y., Kirwan, P., and Livesey, F. J. (2012). Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846. doi: 10.1038/nprot.2012.116
State, M. W., and Levitt, P. (2011). The conundrums of understanding genetic risks for autism spectrum disorders. Nat. Neurosci. 14, 1499–1506. doi: 10.1038/nn.2924
Sykes, N. H., Toma, C., Wilson, N., Volpi, E. V., Sousa, I., Pagnamenta, A. T., et al. (2009). Copy number variation and association analysis of SHANK3 as a candidate gene for autism in the IMGSAC collection. Eur. J. Hum. Genet. 17, 1347–1353. doi: 10.1038/ejhg.2009.47
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Taylor, S. E., Taylor, R. D., Price, J., and Andreae, L. C. (2018). Single-molecule fluorescence in-situ hybridization reveals that human SHANK3 mRNA expression varies during development and in autism-associated SHANK3 heterozygosity. Stem Cell Res. Ther. 9:206. doi: 10.1186/s13287-018-0957-3
Vicidomini, C., Ponzoni, L., Lim, D., Schmeisser, M. J., Reim, D., Morello, N., et al. (2017). Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 22, 689–702. doi: 10.1038/mp.2016.30
Wang, J., Duncan, D., Shi, Z., and Zhang, B. (2013). WEB-based gene set analysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 41, W77–W83. doi: 10.1093/nar/gkt439
Wang, W., Li, C., Chen, Q., van der Goes, M.-S., Hawrot, J., Yao, A. Y., et al. (2017). Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Invest. 127, 1978–1990. doi: 10.1172/JCI87997
Wang, X., Bey, A. L., Katz, B. M., Badea, A., Kim, N., David, L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a SHANK3 complete knockout model of autism. Nat. Commun. 7:11459. doi: 10.1038/ncomms11459
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Xiaoming, W., Mccoy, P. A., Rodriguiz, R. M., Yanzhen, P., Shawn, J. H., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Xing, J., Kimura, H., Wang, C., Ishizuka, K., Kushima, I., Arioka, Y., et al. (2016). Resequencing and association analysis of Six PSD-95-related genes as possible susceptibility genes for schizophrenia and autism spectrum disorders. Sci. Rep. 6:27491. doi: 10.1038/srep27491
Zhao, H., Tu, Z., Xu, H., Yan, S., Yan, H., Zheng, Y., et al. (2017). Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res. 27:1293. doi: 10.1038/cr.2017.95
Zhou, T., Benda, C., Duzinger, S., Huang, Y., Li, X., Li, Y., et al. (2011). Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 22, 1221–1228. doi: 10.1681/ASN.2011010106
Keywords: induced pluripotent stem cells, neural stem cells, SHANK3, electrophysiology, RNA-Seq, autism, transcriptome
Citation: Huang G, Chen S, Chen X, Zheng J, Xu Z, Doostparast Torshizi A, Gong S, Chen Q, Ma X, Yu J, Zhou L, Qiu S, Wang K and Shi L (2019) Uncovering the Functional Link Between SHANK3 Deletions and Deficiency in Neurodevelopment Using iPSC-Derived Human Neurons. Front. Neuroanat. 13:23. doi: 10.3389/fnana.2019.00023
Received: 01 June 2018; Accepted: 07 February 2019;
Published: 13 March 2019.
Edited by:
Basilis Zikopoulos, Boston University, United StatesReviewed by:
Thomas Bourgeron, Institut Pasteur, FranceChiara Verpelli, Institute of Neuroscience (IN), Italy
Copyright © 2019 Huang, Chen, Chen, Zheng, Xu, Doostparast Torshizi, Gong, Chen, Ma, Yu, Zhou, Qiu, Wang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingling Shi, dGxpbmdzaGlAam51LmVkdS5jbg==
†These authors have contributed equally to this work