 Anett Futácsi
Anett Futácsi Kitti Rusznák
Kitti Rusznák Gergely Szarka
Gergely Szarka Béla Völgyi
Béla Völgyi Ove Wiborg
Ove Wiborg Boldizsár Czéh
Boldizsár Czéh- 1Szentágothai Research Centre, University of Pécs, Pécs, Hungary
- 2Department of Laboratory Medicine, Medical School, University of Pécs, Pécs, Hungary
- 3Imaging Core Facility, Szentágothai Research Centre, University of Pécs, Pécs, Hungary
- 4Department of Neurobiology, Faculty of Sciences, University of Pécs, Pécs, Hungary
- 5Department of Health Science and Technology, Aalborg University, Aalborg, Denmark
Background: Animal models of Alzheimer’s disease (AD) are essential tools for investigating disease pathophysiology and conducting preclinical drug testing. In this study, we examined neuronal and glial alterations in the hippocampus and medial prefrontal cortex (mPFC) of young TgF344-AD rats and correlated these changes with cognitive decline and amyloid-β plaque load.
Methods: We compared TgF344-AD and non-transgenic littermate rats aged 7–8 months of age. We systematically quantified β-amyloid plaques, astrocytes, microglia, four different subtypes of GABAergic interneurons (calretinin-, cholecystokinin-, parvalbumin-, and somatostatin-positive neurons), and newly generated neurons in the hippocampus. Spatial learning and memory were assessed using the Barnes maze test.
Results: Young TgF344-AD rats had a large number of amyloid plaques in both the hippocampus and mPFC, together with a pronounced increase in microglial cell numbers. Astrocytic activation was significant in the mPFC. Cholecystokinin-positive cell numbers were decreased in the hippocampus of transgenic rats, but calretinin-, parvalbumin-, and somatostatin-positive cell numbers were not altered. Adult neurogenesis was not affected by genotype. TgF344-AD rats had spatial learning and memory impairments, but this cognitive deficit did not correlate with amyloid plaque number or cellular changes in the brain. In the hippocampus, amyloid plaque numbers were negatively correlated with cholecystokinin-positive neuron and microglial cell numbers. In the mPFC, amyloid plaque number was negatively correlated with the number of astrocytes.
Conclusion: Pronounced neuropathological changes were found in the hippocampus and mPFC of young TgF344-AD rats, including the loss of hippocampal cholecystokinin-positive interneurons. Some of these neuropathological changes were negatively correlated with amyloid-β plaque load, but not with cognitive impairment.
1 Introduction
Alzheimer’s disease (AD) is the most common form of dementia, affecting more than 50 million people worldwide and causing severe social and economic burden (Deuschl et al., 2020; Scheltens et al., 2021). Despite extensive research on disease pathophysiology, the exact pathogenic processes are still not fully understood. One of the most influential theories on the pathophysiology of AD is the “amyloid cascade hypothesis,” which postulates that cerebral amyloid sets neurotoxic events into motion that precipitate Alzheimer dementia (Hardy and Allsop, 1991; Selkoe and Hardy, 2016). Other complementary theories emphasize the importance of chronic neuroinflammation (Kinney et al., 2018), or the progressive loss of limbic and neocortical cholinergic innervation (Hampel et al., 2018). Imbalance of other neurotransmitter systems has also been implicated to contribute to the pathophysiology, such as glutamate (Wang and Reddy, 2017) or GABA (Carello-Collar et al., 2023; Melgosa-Ecenarro et al., 2023; Tang et al., 2023). A growing body of evidence indicates that the GABAergic system is vulnerable to AD pathology as several components of the GABAergic system are reduced in patients with AD (Reid et al., 2021). A recent meta-analysis reported that patients with AD display lower GABA levels in their brain and cerebrospinal fluid, and GAD65/67, GABAA receptors, and GABA transporters were also lower in the AD brains (Carello-Collar et al., 2023). Therefore, the GABAergic system has been proposed as a potential target for developing pharmacological strategies and novel AD biomarkers (Calvo-Flores Guzmán et al., 2018). In harmony with this concept, several studies reported reduced numbers of GABAergic interneurons in the hippocampus of transgenic mouse models of AD and found profound changes in cells immunopositive for parvalbumin (PV+), somatostatin (SST+), calretinin (CR+), and neuropeptide Y (NPY+) (Ramos et al., 2006; Popović et al., 2008; Takahashi et al., 2010; Stanley et al., 2012; Albuquerque et al., 2015; Giesers and Wirths, 2020; Xu et al., 2020; Ali et al., 2023), while negative findings exist as well (e.g., Sos et al., 2020). In harmony with the neuroanatomical data, electrophysiological studies have demonstrated dysfunctional GABAergic interneurons in animal models for AD (Palop and Mucke, 2016; Schmid et al., 2016; Hijazi et al., 2023; Shu et al., 2023) and altered cortical and hippocampal oscillatory network dynamics have also been reported in the TgF344-AD transgenic rat model (Bazzigaluppi et al., 2018; Stoiljkovic et al., 2019; van den Berg et al., 2023). Finally, the clinical findings are in line with these observations, as loss of PV+ and SST+ neurons has been documented in the neocortex and hippocampus of AD patients (Brady and Mufson, 1997; Waller et al., 2020).
Numerous experimental models of AD exist, and these models are invaluable tools for gaining a better understanding of AD pathogenesis and testing novel therapeutic approaches (Drummond and Wisniewski, 2017). The most widely used animal models are transgenic mice, but transgenic rat models are also available (Götz and Ittner, 2008; Van Dam and De Deyn, 2011; Novati et al., 2022) and rat models have distinctive advantages over mice (Leon et al., 2010; Cohen et al., 2013; Do Carmo and Cuello, 2013; Agca et al., 2016; Pang et al., 2022). For example, the amyloid precursor protein (APP) gene knock-in rat model for Alzheimer’s disease exhibits pathologies and disease progression resembling more closely to the human condition compared to transgenic mice overexpressing the APP gene (Pang et al., 2022). Furthermore, rats are typically more suitable for behavioral studies, as most rodent behavioral tests have been originally developed for rats enabling a more robust assessment of behavioral phenotypes in rat models (Paul et al., 2009; Novati et al., 2022). Rats are behaviorally better characterized and display a more complex behavioral repertoire than mice and as they are terrestrial, aquatic and arboreal mammals therefore, they often perform better in maze tasks assessing spatial cognition (Do Carmo and Cuello, 2013). Furthermore, transgenic rats offer great potential to read subtle and early aspects of AD pathology (Do Carmo and Cuello, 2013). Consequently, rat models have significant advantage for in vivo electrophysiology, neuroimaging, epigenetic and optogenetic studies, therefore represent an important asset for research on neuropathology (Do Carmo and Cuello, 2013).
Currently, one of the best rodent models to mimic the complete spectrum of Alzheimer’s disease neuropathology without insertion of a human tau transgene is the TgF344-AD rat line (Cohen et al., 2013). The TgF344-AD rat expresses human APP with the Swedish mutation and human presenilin 1 with the ΔE9 mutation on the Fischer 344 rat background and displays all major hallmarks of AD pathology, i.e., progressive amyloid deposition, tauopathy, cognitive dysfunction, neurodegeneration, and neuroinflammation with gliosis (Cohen et al., 2013; Pentkowski et al., 2018; Fowler et al., 2022; Bac et al., 2023), and most likely, these hallmarks develop sequentially over time (Chaney et al., 2021; Fowler et al., 2022; van den Berg et al., 2023; Fang et al., 2023). Overall, this model is a very attractive tool for research on AD pathophysiology and preclinical drug testing.
In this study, we performed an extensive quantitative histopathological analysis of GABAergic neurons and glial cells in the hippocampus and medial prefrontal cortex of TgF344-AD rats. Based on unbiased stereological principles, we quantified the number of PV, CR, SST, and cholecystokinin (CCK) positive interneurons, as well as Iba-1-positive cells, most of which are microglia, and GFAP-positive astrocytes. In addition to that, we assessed the number of amyloid-β (Aβ) plaques. Furthermore, we investigated adult hippocampal neurogenesis by quantifying doublecortin (DCX) positive immature neurons in the dentate gyrus. To evaluate the spatial learning capacities of the rats, we performed behavioral assessments using the Barnes maze apparatus. Our hypothesis was to find: (1) reduced number of GABAergic interneurons; (2) an increased number of glial cells; (3) impaired adult hippocampal neurogenesis; (4) the cellular changes will correlate with the cognitive abilities of the rats and with the β-amyloid load.
2 Materials and methods
2.1 Animals
TgF344-AD rats were generated on a Fischer 344 background by co-injecting rat pronuclei with two human genes driven by the mouse prion promoter; ‘Swedish’ mutant human amyloid precursor protein (APP) and a deleted exon 9 mutant human presenilin (APPsw, PS1ΔE9, Cohen et al., 2013). Both constructs were previously described (Jankowsky et al., 2001). Homo-and hemizygous TgF344-AD, and WT littermate rats, were bred in the animal facility of the Faculty of Medicine, Aalborg University from hemizygous founders purchased from Rat Resource and Research Center Columbia, MO, USA. Animals were group housed under a standard 12-h light/dark cycle at 24 ± 2°C with relative humidity of 50–60%. Food and water were available ad libitum in the home cages. Genotyping was verified by digital PCR and both sexes were included in this study, which is a common practice in experiments with TgF344-AD rats (e.g., Cohen et al., 2013; Bazzigaluppi et al., 2018; Pentkowski et al., 2018; Morrone et al., 2020). No sex differences were observed during phenotyping, therefore behavioral data were combined for the two sexes. Homozygous transgenic (TG/TG) TgF344-AD (n = 6) and homozygous wild-type (WT/WT, n = 7) littermate rats, at an age of 7–8 month, were included in the study.
Experiments complied with the ARRIVE guidelines and were approved by the National Danish Animal Research Committee (2019-15-0201-00215), adhering to the EU Directive 2010/63/EU for animal experiments.
2.2 The Barnes maze
The Barnes maze was applied to address hippocampal-dependent spatial reference learning and memory. The maze (Panlab/Harvard Bioscience, Inc.) consisted of a circular platform (diameter 122 cm) with 18 holes (diameter 10 cm) in the perimeter. The platform was elevated 90 cm from the floor. A dark escape box was located beneath one of the holes while the remaining holes were blinded. The platform was brightly illuminated as an aversive stimulus. The proximal surroundings of the maze were to remain constant to provide as visual cues for the rats to navigate around the platform.
2.3 Assessment of spatial learning and memory
Three days before experimental initiation rats were habituated for 30 min/day to stay in an empty transit cage. At day 0 rats were adapted to the maze by being placed in a transparent cylinder in the center of the platform for 30 s. and then guided to the escape box by slowly sliding the cylinder toward the escape hole. The rat was confined to the escape box for 2 min. On consecutive acquisition trials, on days 1 and 2, rats were confined to an opaque cylinder in the center of the platform for 15 s. After removing the cylinder, rats were allowed 3 min to explore the platform to locate the escape box. Each day 4 trials with 1 h interval was executed for each rat.
Short-term and long-term spatial memory was assessed using two probe trials on days 3 and 10, respectively. The escape box was replaced by a disc similar to the blinded holes. Acquisition and probe trials were recorded and subsequently latency to enter the hole, or nose poke on the disc for the first time was scored manually.
2.4 Brain tissue fixation and processing for immunohistochemistry
After an overdose of sodium pentobarbital (200 mg/mL dissolved in 10% ethanol), animals were transcardially perfused with 0.9% physiological saline followed by 4% paraformaldehyde (pH = 7.4). Serial coronal sections were cut throughout the entire brain using a Vibratome (Leica VT1200S). Fifty micrometer thick sections were collected in series and stored in 0.1 M phosphate buffer (pH = 7.4) with 0.5% sodium azide at 4°C until staining. Nine different primary antibodies (Table 1) were used to identify four types of GABAergic cells, astrocytes, Iba-1-positive microglia and macrophages, immature neurons in the dentate gyrus, and to visualize amyloid plaques. Samples from the wild-type and transgenic groups were always processed in parallel to eliminate any nonspecific effect of the staining procedure.
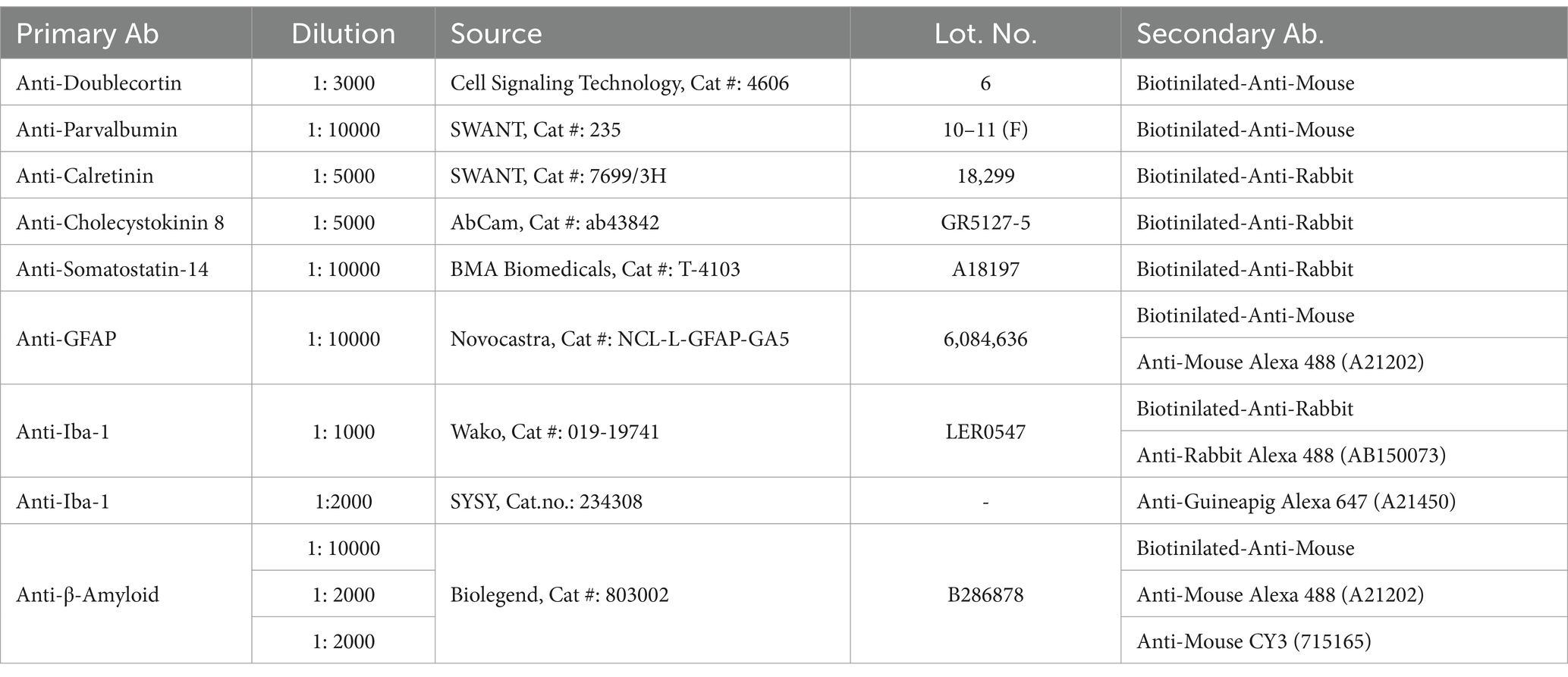
Table 1. Primary and secondary antibodies used in this study.
2.5 Immunohistochemistry procedures
Immunolabeling of GABAergic neurons, glial cells and immature dentate granule cells was performed as previously reported (Czéh et al., 2006, 2015, 2018; Rusznák et al., 2022). Briefly, a general immunohistochemistry protocol was performed as follows: Free-floating sections were thoroughly washed and treated with 1% H2O2 for 20 min. Nonspecific binding was prevented by incubating the sections for 1 h in 5% normal goat serum (Vector Laboratories, Burlingame, CA, USA). Subsequently, the sections were incubated overnight at 4°C with various primary antibodies. After thorough rinsing, the sections were incubated with a corresponding biotinylated secondary antibody for 2 h and labeling was visualized using an avidin-biotin-horseradish peroxidase kit (1:200; Vectastaine Elite ABC Kit, Vector Laboratories), and developed with diaminobenzidine (1:200; DAB Peroxidase Substrate Kit, Vector Laboratories).
Fluorescence labeling was performed using similar procedures except that we used fluorescent secondary antibodies (Table 1) furthermore the sections were labeled afterwards with DAPI. Fluorescent samples were scanned using a Zeiss LSM-710 confocal microscope with 20× (Z = 1 μm; Zeiss W Plan-Apochromat 20/1.0) and 63× objectives (Z = 0.5 μm; Zeiss Plan Apochromat 63/1.4) at high resolution and normalized laser intensity.
2.6 Cell and amyloid-β plaque quantification
Two experimenters (AF and KR), who were blinded to group identification, collected data. All cell counting was performed manually using either the Neurolucida or the StereoInvestigator reconstruction systems (Version 7, Microbrightfield, Colchester, VT, USA) attached to a Nikon Eclipse Ti-U bright field microscope, using 20× and 40× objectives. Quantitative analysis was performed based on a modified unbiased stereology protocol that has been reported to successfully quantify hippocampal neurons appearing in low densities (Czéh et al., 2015). The different subtypes of interneurons were counted in a systematic manner in a complete series of 50 μm thick sections starting at a random position along the entire septo-temporal axis of hippocampal formation (from −1.80 to −6.60 relative to Bregma, according to the atlas of Paxinos and Watson, 1998). With each primary antibody, we labeled every eight serial sections from the complete series, resulting in 10–12 hippocampal sections for each antibody. We also focused on the regional distribution of the neurons; thus, cells were counted in the three main hippocampal subareas (dentate gyrus, CA2-3, CA1) separately. To quantify the GABAergic interneurons in the hippocampus, first, the contours of the different hippocampal subareas were traced under low magnification, and then the GABAergic cells were examined and counted using an objective of 20× magnification, omitting labeled profiles in the outermost focal plane. The total number of labeled neurons in the hippocampus – including both hemispheres – was estimated by multiplying the number of cells counted in every eight sections by eight. The same procedure was used to quantify β-amyloid plaques and to quantify DCX+ immature granule cells in the dentate gyrus.
Glial cells were counted using the stereology method (Gundersen et al., 1988) as described in detail previously (Czéh et al., 2006). Stereology was performed using MBF StereoInvestigator software (Version 7) and a Nikon Eclipse Ti-U microscope utilizing the optical fractionator probe and systematic analysis of randomly placed counting frames (size, 75 × 50 μm) on a counting grid (size of 100 × 100 μm) and sampled (25 μm optical dissector with 3 μm upper and lower guard zones) to obtain unbiased counts of Iba-1-positive cells and astrocytes. All cell counts had a Gundersen coefficient (m = 1) of <0.10 to ensure accuracy and consistency (Gundersen et al., 1999).
In the mPFC, we quantified GABAergic neurons and Aβ plaques using a similar systemic quantification protocol as above, and as described in detail before (Czéh et al., 2018). Cell and plaque numbers in the mPFC are presented as densities (cell number/mm3).
2.7 Statistical analysis
Results are expressed as mean ± SEM. Data analysis was performed using GraphPad Prism, Version 7 (San Diego, California, USA). β-amyloid plaque numbers and cell quantification data were analyzed using two-way ANOVA (genotype × brain area) followed by Šídák’s multiple comparisons post hoc test. Behavioral data were analyzed using two-way repeated measures ANOVA (time × genotype), or unpaired Student’s t-test. Correlation analysis was performed using the Pearson’s correlation coefficient. The level of significance was set at p < 0.05.
3 Results
3.1 TgF344-AD rats had large number of amyloid-β plaques both in the hippocampus and medial prefrontal cortex
Transgenic rats had numerous Aβ plaques in all hippocampal subareas and layers (Figures 1A,B). Similarly, in the prefrontal cortex, Aβ plaques were scattered evenly and were found in all cortical layers, all over the anterior cingulate (aCg), pre-limbic (PrL), and infralimbic (IL) cortices (Figures 1C,D). Quantification of plaque numbers revealed a significant genotype effect in both the hippocampus and neocortex. Two-way ANOVA (genotype × brain area) had significant genotype effect [F(1, 40) = 193.0, p < 0.0001] in the hippocampus (Figure 1B), as well as in the mPFC [F(1, 40] = 360.5, p < 0.0001, Figure 1D).
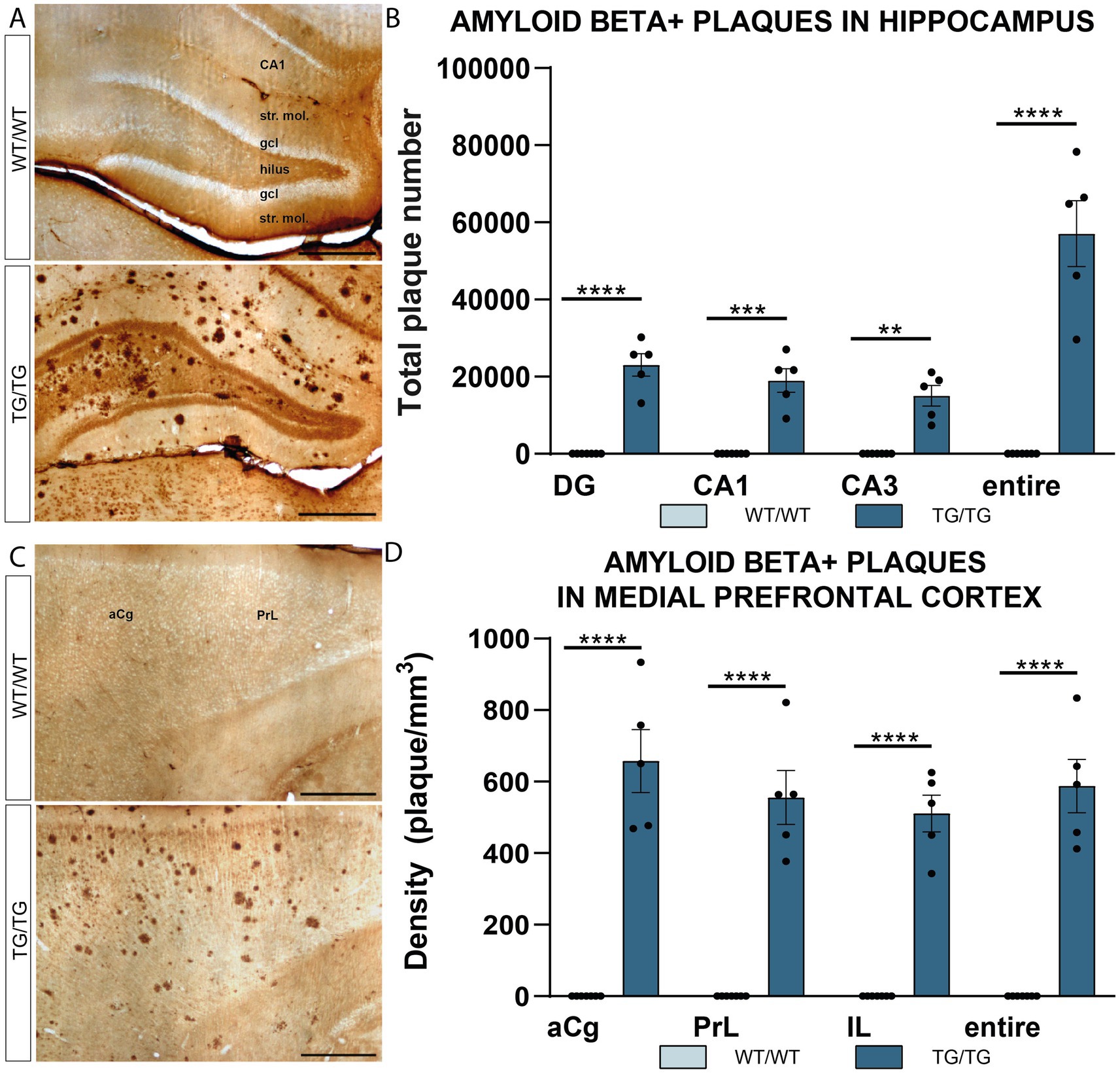
Figure 1. Amyloid β plaques in the hippocampus and prefrontal cortex of TgF344-AD rats. (A) Representative images showing immunolabeled β-amyloid plaques in the hippocampus of wild-type (WT/WT) and transgenic (TG/TG) rats. (B) Quantification of plaque numbers confirmed an even distribution of amyloid β plaques in all hippocampal subareas (DG, dentate gyrus; CA, Cornu Ammonis). (C) Aβ plaques in the medial prefrontal cortex. (D) Similar to the hippocampus, plaque densities showed an equal distribution in all subareas of the medial prefrontal cortex (anterior cingulate (aCg), pre-limbic (PrL), and infralimbic (IL) cortices). Statistical analysis: Two-way ANOVA (genotype × brain area) followed by Šídák’s multiple comparisons post-hoc test (****p < 0.0001). Scale bars represent 500 μm for all images.
We also performed fluorescence labeling and confocal microscope imaging to investigate the spatial relationship between glial cells and Aβ aggregates. As shown in Figure 2, Aβ deposits were typically surrounded both by Iba-1-positive cells and GFAP+ astrocytes. Both types of glial cells encircled the plaque deposits indicating an inflammatory response triggered by the amyloid-β plaques (Figures 2B,E).
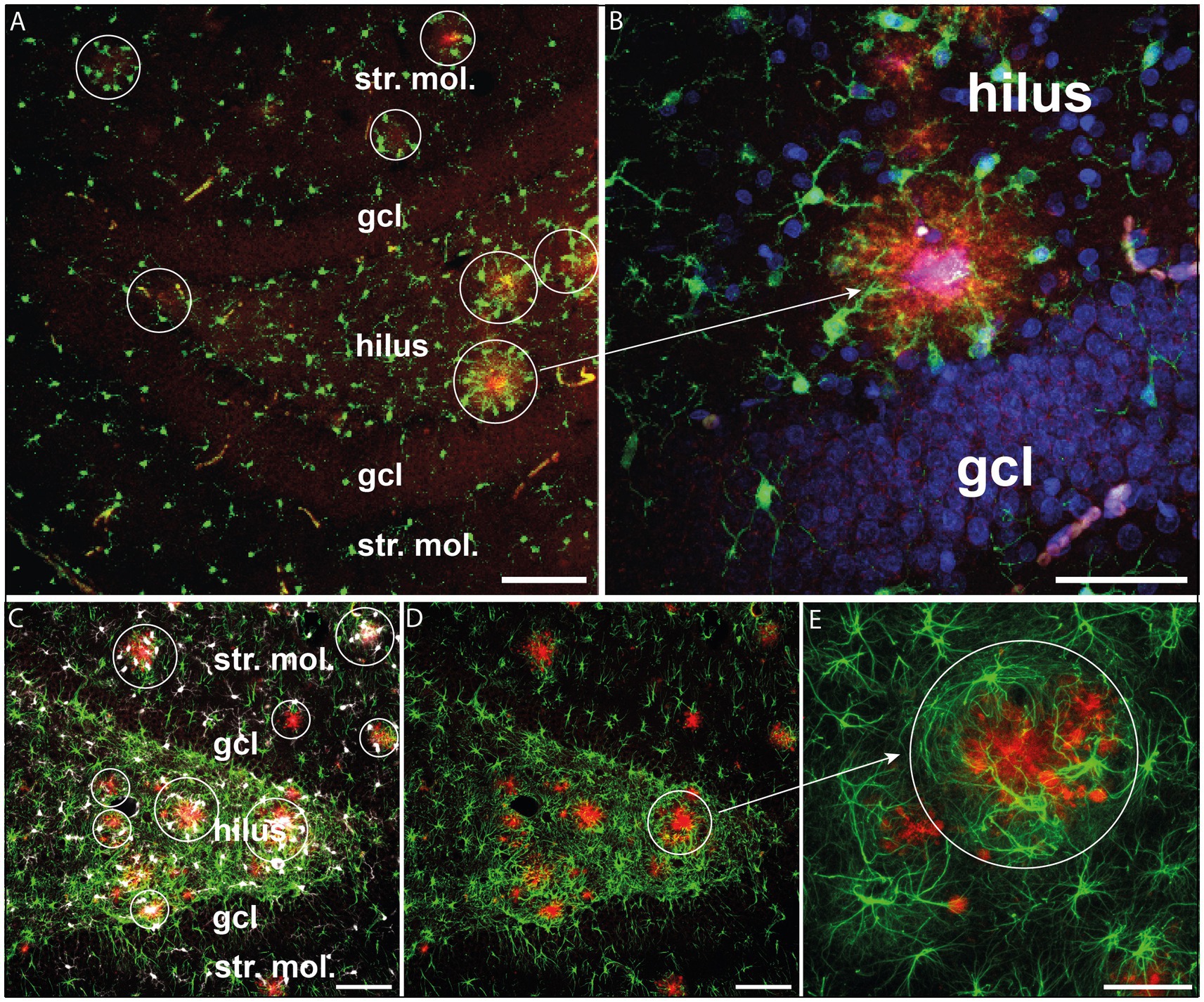
Figure 2. Glial activation surrounding β-amyloid plaques. (A) β-amyloid plaques (red) surrounded by activated Iba-1+ microglia (green) in the hippocampal dentate gyrus. (B) The same phenomenon is observed at a higher magnification. Iba-1+ microglial processes (green) are oriented towards β-amyloid plaques (red). Cell nuclei were labeled with DAPI (blue). (C) β-amyloid plaques (red) surrounded by Iba-1+ microglia (white) and GFAP+ astrocytes (green) in the hippocampal dentate gyrus. (D) The same hilar area where GFAP+ astrocytes surrounded the β-amyloid plaques (red). (E) Enlarged detail of D displaying GFAP+ astrocytes encircling the plaques (red). Scale bars: 100 μm on A, C, D, E and 50 μm on B. gcl, granule cell layer; str. mol., stratum moleculare.
3.2 TgF344-AD rats had pronounced gliosis both in the hippocampus and medial prefrontal cortex
Iba-1+ cells in the hippocampus of wild type and transgenic rats are displayed on Figure 3A. Most of these Iba-1+ cells are microglia, although Iba-1 is also expressed by peripheral myeloid cells, such as macrophages, that may infiltrate the brain upon injury. The number of hippocampal Iba-1 cells were significantly increased [t(12) = 5.90, p < 0.0001] in the TgF344-AD rats (Figure 3B). Activated Iba-1+ cells were also seen in the mPFC of the Tg/Tg rats (Figure 3C), where these cells were present in significantly higher numbers [t(12) = 4.30, p = 0.001] compared to WT/WT littermates (Figure 3D). On the high magnification images of Iba-1+ cells (Figures 3E,F), it is clearly visible that transgenic rats had numerous activated microglia in the neocortex.
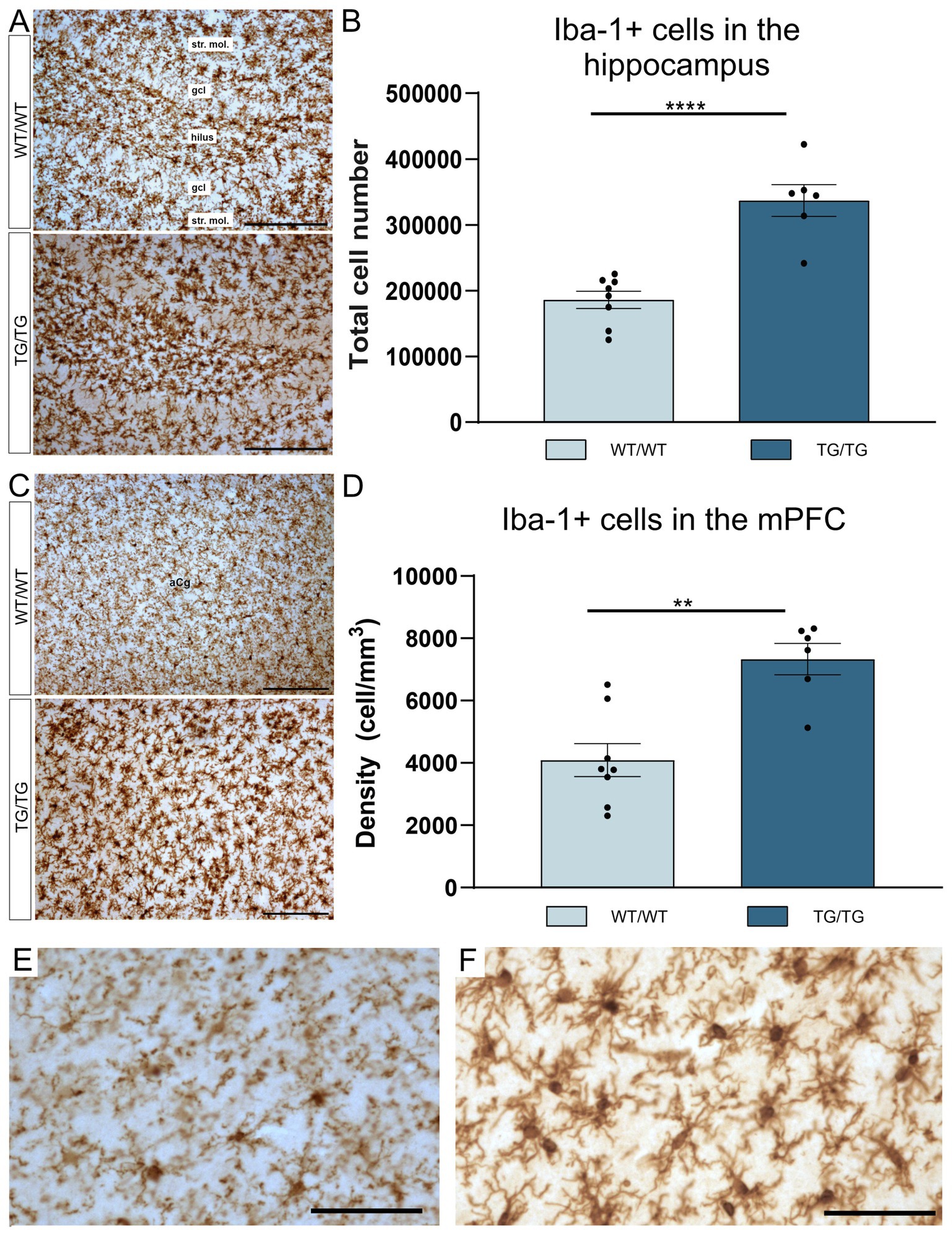
Figure 3. TgF344-AD rats had pronounced activation of Iba-1+ glia in both the hippocampus and mPFC. (A) Representative images of Iba-1+ cells in the hippocampi of wild-type and transgenic rats. (B) Quantitative data for Iba-1+ cells in the hippocampus. TgF344-AD rats had a significantly higher number of Iba-1+ cells in the hippocampus. Cell numbers indicate cell counts from both hemispheres. Statistical analysis: unpaired Student’s t-test; ****p < 0.0001. (C) Iba-1+ cells in the mPFC. (D) TgF344-AD rats have a significantly higher number of Iba-1+ cells in the mPFC. Statistical analysis: unpaired Student’s t-test; ** p = 0.001. (E) A high magnification image of Iba-1+ cells in the mPFC of a wild-type rat. (F) High magnification image of activated Iba-1+ cells in the mPFC of a TgF344-AD rat. gcl, granule cell layer; str. mol., stratum moleculare. Scale bars represent 200 μm in (A,C), and 50 μm in (E,F).
GFAP+ astrocytes are shown it the hippocampi of the WT/WT and Tg/Tg rats on Figure 4A. Results of the cell quantification indicated that hippocampal GFAP+ cell numbers were similar in both groups and were not affected by the genotype (Figure 4B). However, we found activated GFAP+ astrocytes in the mPFC of the TgF344-AD rats (Figure 4C). In the mPFC, the number of the GFAP+ cells were significantly increased [t(11) = 4.93, p = 0.0004], as it is seen on Figure 4D. High magnification images of GFAP+ astrocytes are shown in Figures 4E,F.
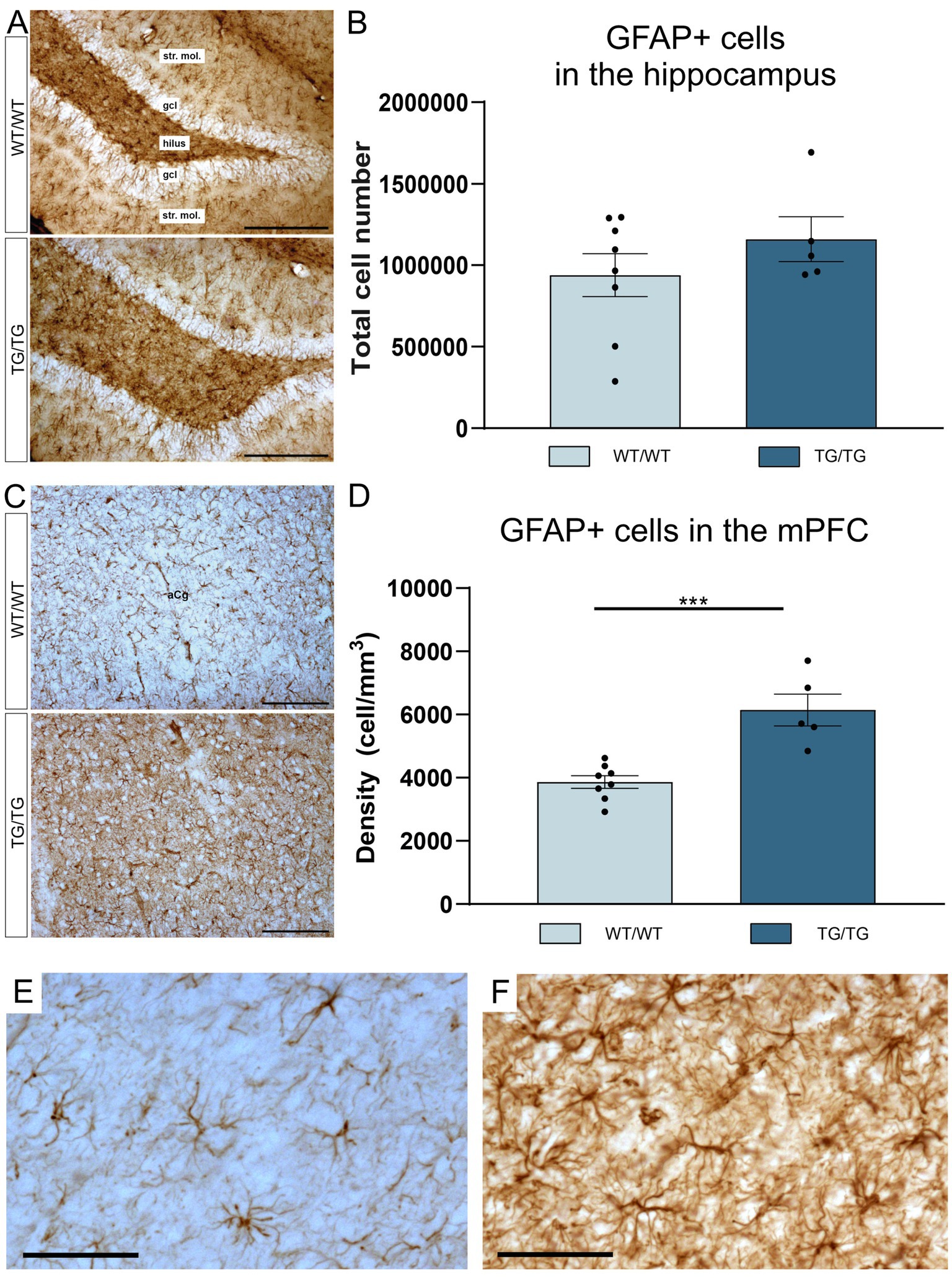
Figure 4. TgF344-AD rats had GFAP+ gliosis in the mPFC. (A) Representative images of GFAP+ astrocytes in the hippocampi of wild-type and transgenic rats. (B) Quantitative data for GFAP+ cells in the hippocampus. In the hippocampus, GFAP+ cell numbers were not affected by genotype. Cell numbers indicate cell counts from both hemispheres. (C) GFAP+ glia in the mPFC. (D) In the mPFC, TgF344-AD rats had significantly higher density of GFAP+ astrocytes. Statistics: unpaired Student’s t-test, *** p < 0.0004. (E) A high magnification image of GFAP+ cells in the mPFC of a wild-type rat and activated GFAP+ cells in the mPFC of a TgF344-AD rat (F). gcl, granule cell layer; str. mol., stratum moleculare. Scale bars represent 200 μm in (A,C), and 50 μm in (E,F).
3.3 TgF344-AD rats had reduced number of CCK+ neurons in the hippocampus
In this study, we have quantified the number of four types of GABAergic neurons: PV+, CR+, SST+, and CCK+ interneurons because previous experiments typically report on changes affecting these cell types. We could not observe any change in cell numbers of the PV+, CR+, SST+ neurons in the young TgF344-AD rats. On Figure 5, representative images are presented for parvalbumin immunoreactive interneurons in the hippocampus and mPFC, together with the results of the corresponding cell quantification data. On Figure 6, representative images are presented for calretinin immunoreactive neurons in the hippocampus and mPFC, together with the results of the corresponding cell quantification data. On Figure 7, representative images are presented for somatostatin immunoreactive neurons in the hippocampus and mPFC, together with the results of the corresponding cell quantification data.
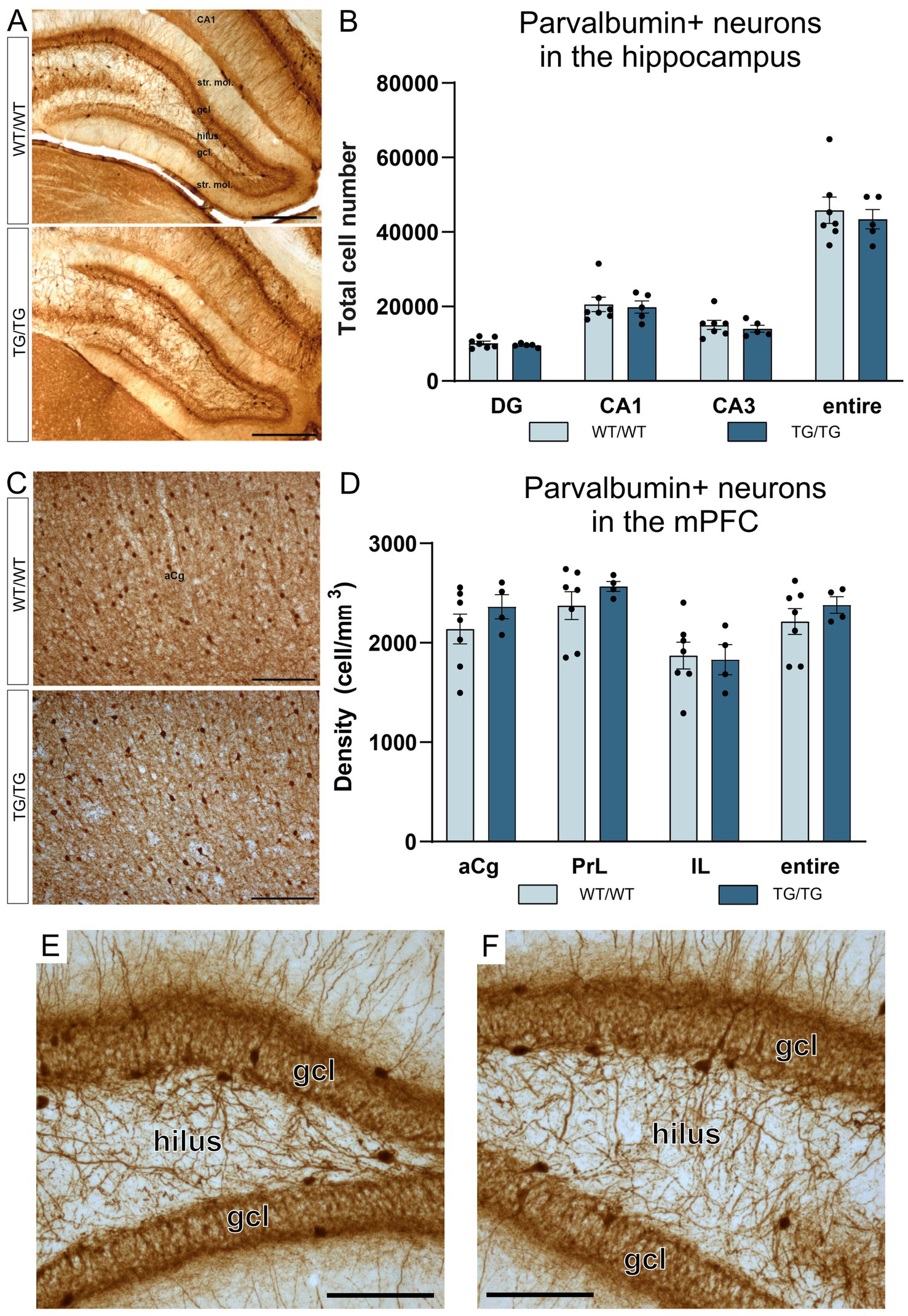
Figure 5. Parvalbumin-positive interneurons were not affected by the genotype. Representative images of hippocampal interneurons expressing parvalbumin (A), and the corresponding cell quantification data on cell numbers (B). Parvalbumin-positive neurons in the mPFC (C), and PV+ cell densities in the anterior cingulate (aCg), prelimbic (PrL) and infralimbic (IL) cortices (D). The number of PV+ neurons was not altered in the transgenic rats. (E) A high magnification image of PV+ interneurons in the dentate gyrus of a wild-type rat and PV+ cells in the dentate gyrus of a TgF344-AD rat (F). DG, dentate gyrus; gcl, granule cell layer; str. mol., stratum moleculare; aCg, anterior cingulate; IL, infralimbic; mPFC, medial prefrontal cortex; PrL, prelimbic. Scale bars represent 500 μm in A, 200 μm in (C), and 50 μm in (E,F).
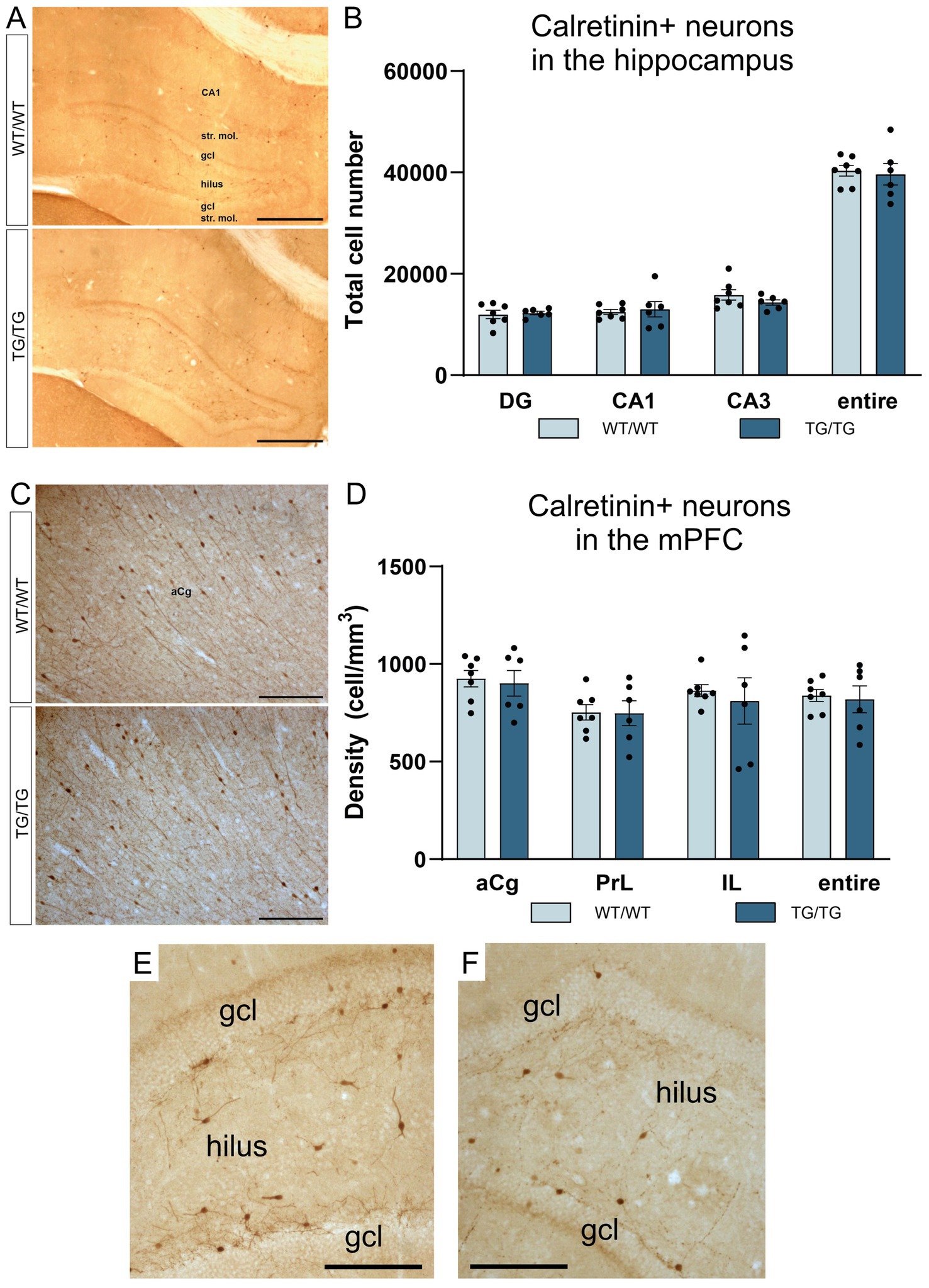
Figure 6. The number of calretinin-positive neurons were not affected by genotype. Calretinin immunopositive cells in the hippocampus (A), and CR+ cell numbers in the hippocampal subareas (B). Calretinin immunoreactive neurons in the mPFC (C), and CR+ cell densities in the mPFC (D). The number of CR+ neurons was not altered in the transgenic rats. (E) A high magnification image of CR+ interneurons in the dentate gyrus of a wild-type rat. (F) A high magnification image of CR+ neurons in the dentate gyrus of a TgF344-AD rat. DG, dentate gyrus; gcl, granule cell layer; str. mol., stratum moleculare; aCg, anterior cingulate; IL, infralimbic; mPFC, medial prefrontal cortex; PrL, prelimbic. Scale bars represent 500 μm in A, 200 μm in (C), and 50 μm in (E,F).
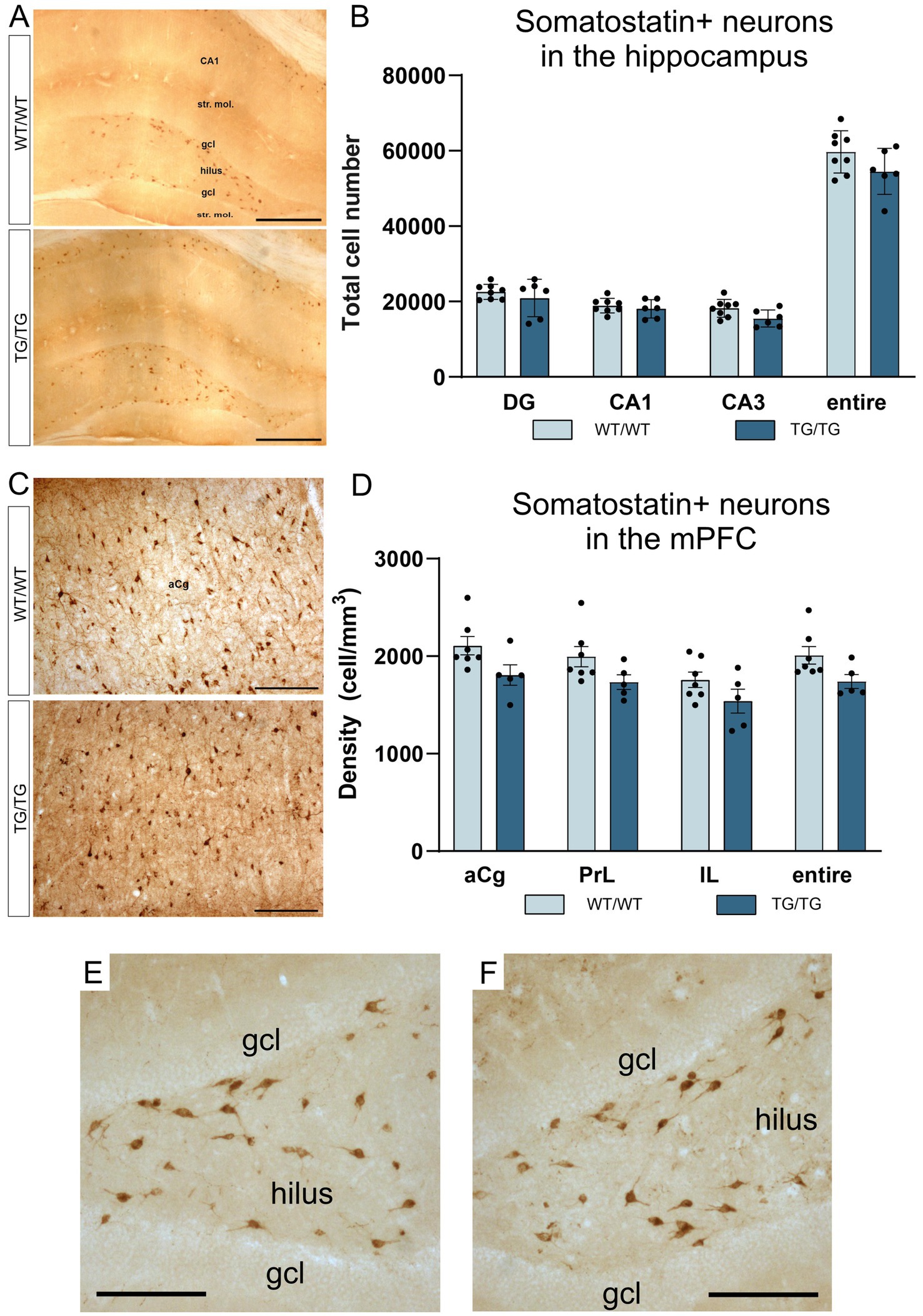
Figure 7. Somatostatin-positive neurons were not altered by genotype. Somatostatin-positive neurons in the hippocampus (A), and the corresponding cell quantification data on hippocampal SST+ cell numbers (B). Somatostatin-positive cells in the mPFC (C), and a graph depicting SST+ cell densities in the anterior cingulate, prelimbic and infralimbic cortices (D). The number of SST+ neurons was not altered in the transgenic rats. (E) A high magnification image of SST+ interneurons in the dentate gyrus of a wild-type rat. (F) A high magnification image of SST+ neurons in the dentate gyrus of a TgF344-AD rat. DG, dentate gyrus; gcl, granule cell layer; str. mol., stratum moleculare; aCg, anterior cingulate; IL, infralimbic; mPFC, medial prefrontal cortex; PrL, prelimbic. Scale bars represent 500 μm in (A), 200 μm in (C), and 50 μm in (E,F).
The only GABAergic cell type which was affected by the genotype was the cholecystokinin-positive interneurons. On Figure 8, we present representative images of CCK immunoreactive neurons in the hippocampus and neocortex. One should emphasize here that the visualization of the CCK immunoreaction is difficult, especially in the cortex. We have tested several primary anti-CCK antibodies before, and we decided to use the AbCam Anti-Cholecystokinin-8 primary antibody (Table 1) which gives a specific labeling and always worked reliably in our previous studies (Czéh et al., 2015, 2018; Varga et al., 2017). With this primary antibody, we could visualize CCK+ interneurons in the hippocampus of wild-type (Figure 8A) and transgenic rats (Figure 8B). This antibody typically labeled only the cell bodies and dendrites were only rarely visible, as shown in higher magnification images of Figures 8C,D. In the anterior cingulate cortex, CCK+ neurons were rather few (Figure 8E) and in the transgenic rats we could observe quite a few CCK+ plaques as well (Figure 8F). A plausible explanation for the presence of CCK+ plaques is that the CCK peptide is incorporated in the plaques, or the plaques were expressing certain epitopes which were recognized by the CCK antibody. We did not observe comparable immunoreactive plaques with any other immunostaining, only with CCK-immunohistochemistry.
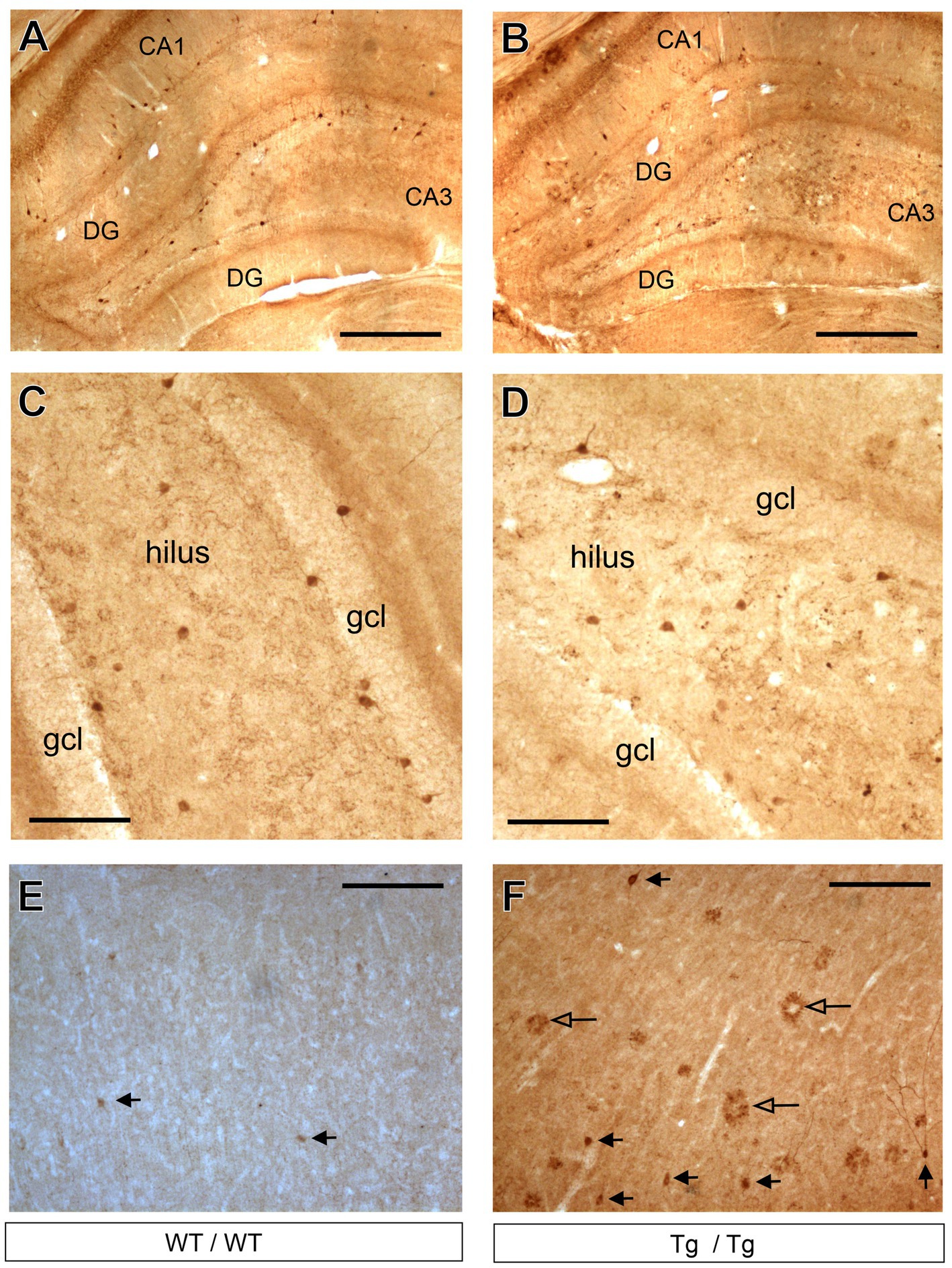
Figure 8. Cholecystokinin-positive neurons in the hippocampus and prefrontal cortex. (A) Representative images of CCK+ GABAergic neurons in the hippocampus of wild-type rats. (B) CCK+ interneurons in the hippocampus of transgenic rats. (C) Higher magnification images of CCK+ neurons in the dentate gyrus of wild-type rats. CCK + cells were clearly identifiable. (D) Higher magnification images of CCK+ neurons in the dentate gyrus of transgenic rats. (E) CCK+ cells in the frontal cortex of wild-type rats. (F) CCK+ neurons and CCK+ plaques in the frontal cortex of transgenic rats. Black arrowheads indicate CCK+ neurons, whereas open arrowheads point to CCK+ plaques. CCK+ plaques were present mainly in the neocortex of the transgenic rats and these objects were most likely staining artefacts. CA, Cornu Ammonis; DG, dentate gyrus; gcl, granule cell layer. Scale bars represent 500 μm in (A,B), and 100 μm in (C–F).
The systemic cell quantification revealed that CCK+ neuron numbers were significantly reduced in the hippocampus (Figure 9A). Two-way ANOVA (genotype × brain area) revealed significant genotype effect [F(1, 44) = 72.92, p < 0.0001] and CCK+ cell numbers were significantly reduced both in the CA1 and CA3 subareas as well as in the entire hippocampus, but not in the dentate gyrus (Figure 9A). Pairwise comparisons using Šídák’s multiple comparisons post hoc test indicated that the TgF344-AD rats had significantly fewer CCK+ interneurons in the CA3 (p = 0.0014) and CA1 (p = 0.002) hippocampal subareas as well as in their entire hippocampus (p < 0.0001) (Figure 9A). In the mPFC, we observed a tendency for increased CCK+ cell numbers, but statistically this was not significant (Figure 9B).
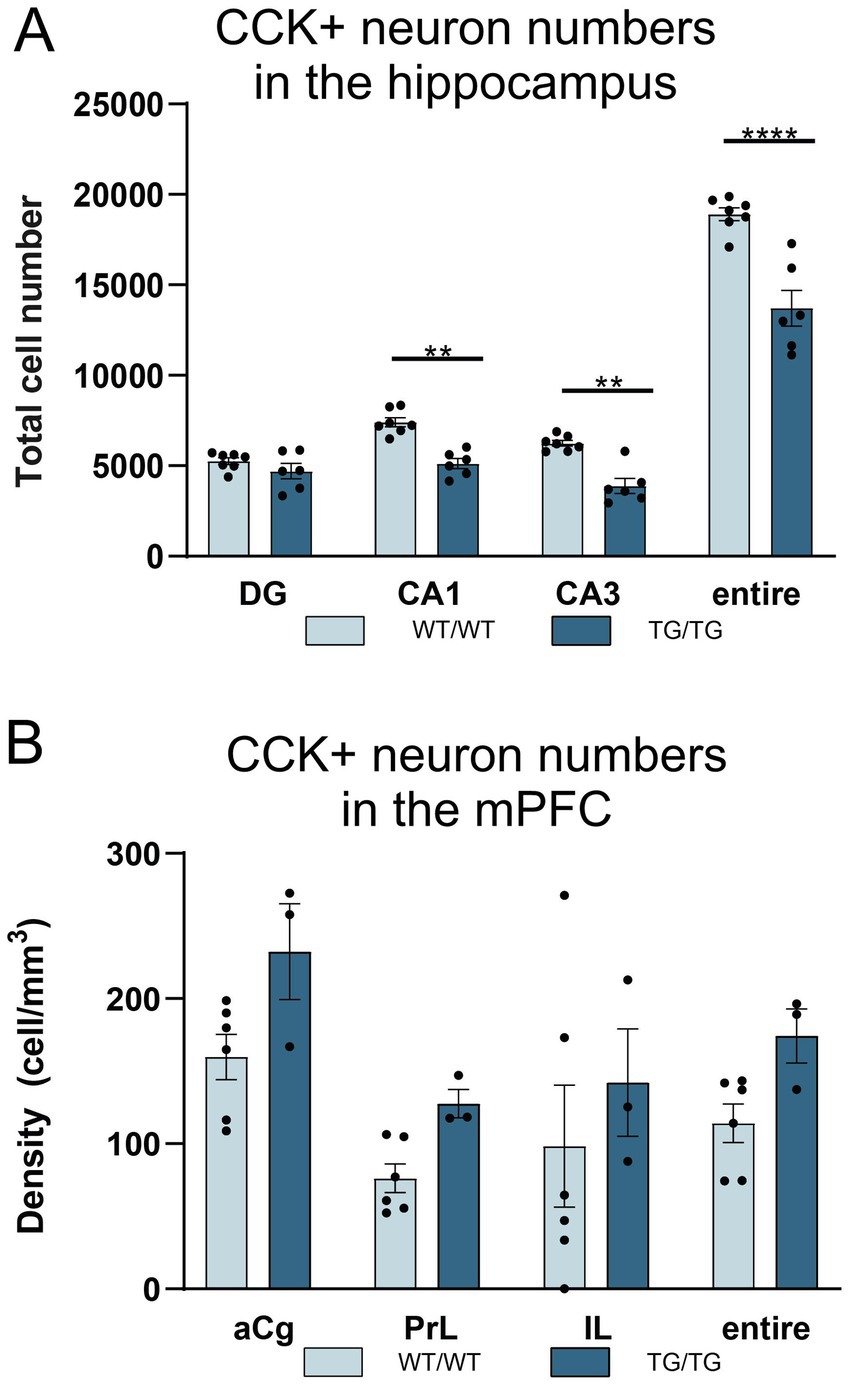
Figure 9. TgF344-AD rats had reduced numbers of cholecystokinin-positive (CCK+) interneurons in the hippocampus. (A) Quantification of CCK+ cells revealed that TgF344-AD rats had a reduced number of CCK+ cells in the CA1 and CA3 areas, as well as in the entire hippocampus. Cell numbers indicate cell counts from both hemispheres. Statistical analysis: Two-way ANOVA (genotype × brain area) followed by Šídák’s multiple comparisons post-hoc test CA1: ** p = 0.0014; CA3 ** p = 0.0020; entire hippocampus: ****p < 0.0001. (B) Cell quantification data revealed no genotype effect on CCK+ cell densities in the mPFC. CA, Cornu Ammonis; DG, dentate gyrus; aCg, anterior cingulate; PrL, prelimbic; IL, infralimbic.
3.4 The number of immature neurons in the dentate gyrus was not altered in the young TgF344-AD rats
To quantify newly generated neurons in the adult hippocampal dentate gyrus, we used doublecortin-immunolabeling because doublecortin (DCX+) is expressed by immature neurons and therefore it is a widely used marker to assess adult neurogenesis (Couillard-Despres et al., 2005; von Bohlen Und Halbach, 2007). TgF344-AD rats had less DCX+ cells in their dentate gyrus, but this difference was statistically not significant (Figure 10).
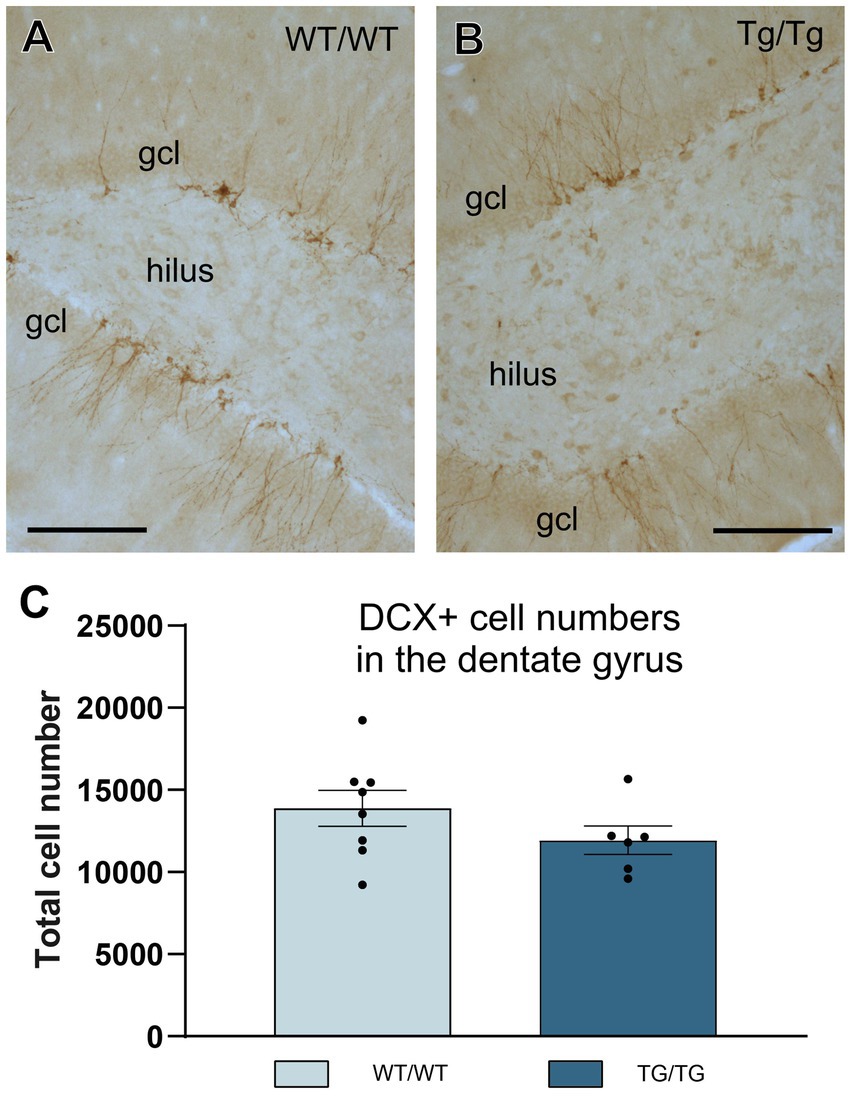
Figure 10. Adult hippocampal neurogenesis was not altered in the young TgF344-AD rats. (A,B) Representative images of doublecortin-positive (DCX+) immature neurons in the hippocampal dentate gyrus of wild-type and transgenic rats. (C) Systematic cell-count data. We found a small, but non-significant reduction of DCX+ cells numbers in the transgenic rats. Cell numbers indicate cell counts from both hemispheres. DG, dentate gyrus; gcl, granule cell layer. Scale bars represent 200 μm.
3.5 TgF344-AD rats had impaired spatial learning and memory performance in the Barnes maze
On Figure 11, we present behavioral data on the performance of the rats in the Barnes maze. On the first two days, when the rats were subjected to the spatial learning trials, TgF344-AD rats were impaired finding the escape box, especially on the first day (Figures 11A,B). Repeated measures two-way ANOVA (time × genotype) revealed a significant effect of time (F = 20.57, p < 0.0001), and genotype (F = 16.97, p = 0.001). Šídák’s multiple comparisons post-hoc test detected a significant difference between the wild-type and transgenic rats in the first trial of day 1 (p < 0.05).
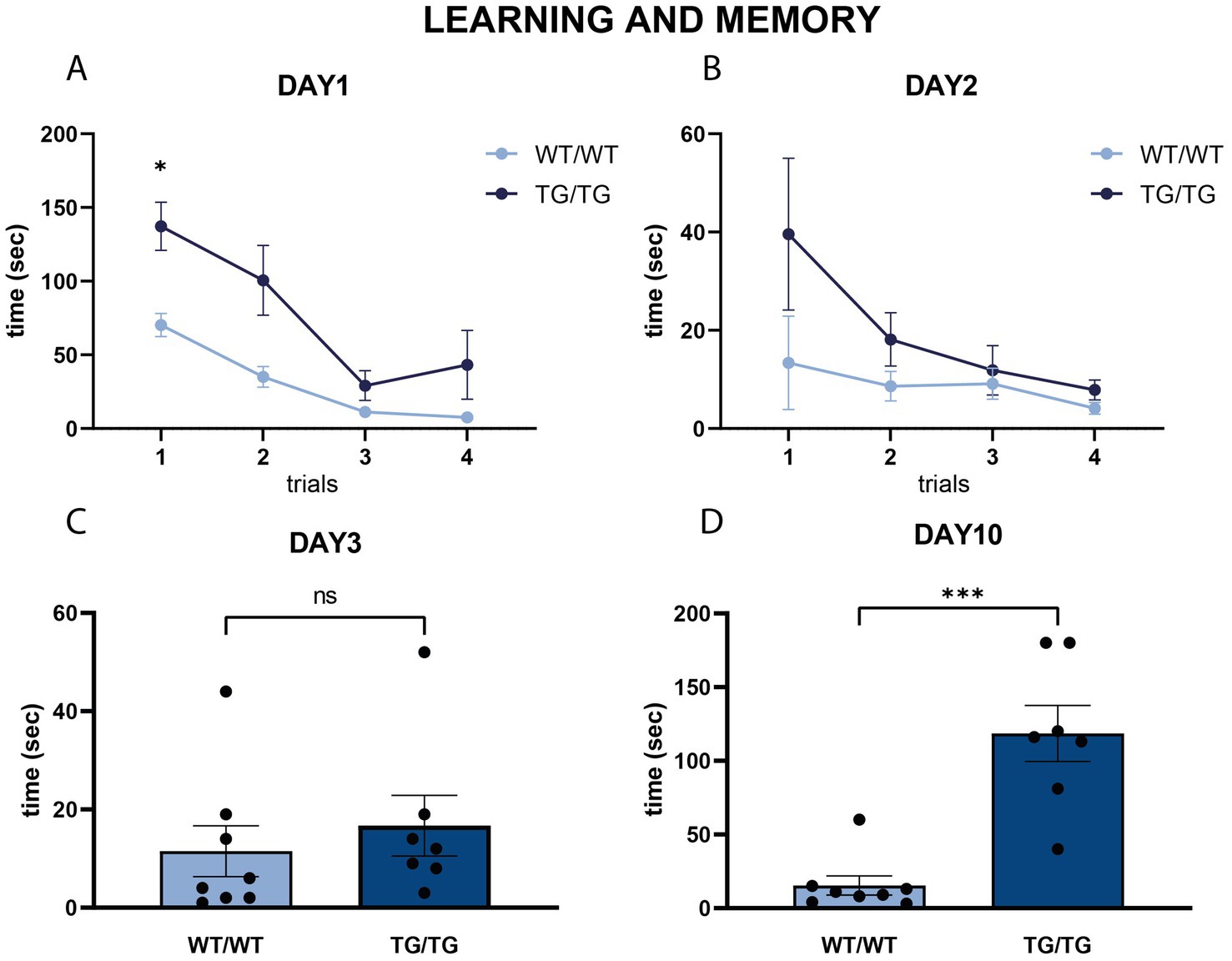
Figure 11. Learning and memory in the Barnes maze. (A,B) Line diagrams represent the learning curves in the Barnes maze task, where rats were tested in four subsequent trials on day 1 (A) and day 2 (B). Spatial learning of the wild-type rats was significantly faster in the trials of the first day. Repeated measures two-way ANOVA (time × genotype) detected significant difference between the learning curves of the wild-type and transgenic rats on the first training day (p < 0.01), and Šídák’s multiple comparisons post-hoc test detected a significant difference between the first trial of day 1 (* p < 0.05). (C,D) Results of the two probe trials on Day 3 (C) and day 10 (D) indicating spatial memory competences. On Day 3, no difference was present between the WT/WT and Tg/Tg animals, but on Day 10 transgenic rats spent significantly more time finding the escape box, indicating impaired spatial memory Statistics: unpaired Student’s t-test, ***p < 0.0001.
Short-term and long-term spatial memory was assessed using two probe trials of the Barnes maze on days 3 and 10, respectively. On day 3, TgF344-AD rats had similar short-term memory as the wild-type rats (Figure 11C). However, on day 10, TgF344-AD rats had a significantly impaired long-term spatial memory, as it took much longer for them to locate where the escape box was [t(13) = 5.42, p < 0.0001, Figure 11D].
3.6 Correlation analysis comparing behavioral performance, Aβ plaque numbers, and cellular changes in the brain
As a final step in our data analysis, we performed correlation analyses comparing the behavioral data with Aβ load, and cellular changes in the brain. Since the cognitive impairment of the transgenic rats were the most pronounced in the second probe trial of the Barnes maze, which was carried out on day-10 (Figure 11D), we took these values for the correlation analysis, but we could not detect any correlation between memory impairment and the neuropathological changes in the brain.
Correlation analysis comparing Aβ load with the cellular changes, however, yielded a few positive results which are presented on Figure 12. In the hippocampus, we found a negative correlation between Aβ plaque numbers and Iba-1+ cell numbers (Pearson r = −0.916, p < 0.05, Figure 12A), as well as a negative correlation between Aβ plaque numbers and CCK+ interneuron numbers (Pearson r = −0.720, p < 0.05, Figure 12B). In the mPFC, we found a negative correlation between Aβ plaque density and astrocyte density (Pearson r = −0.954, p < 0.05, Figure 12C).
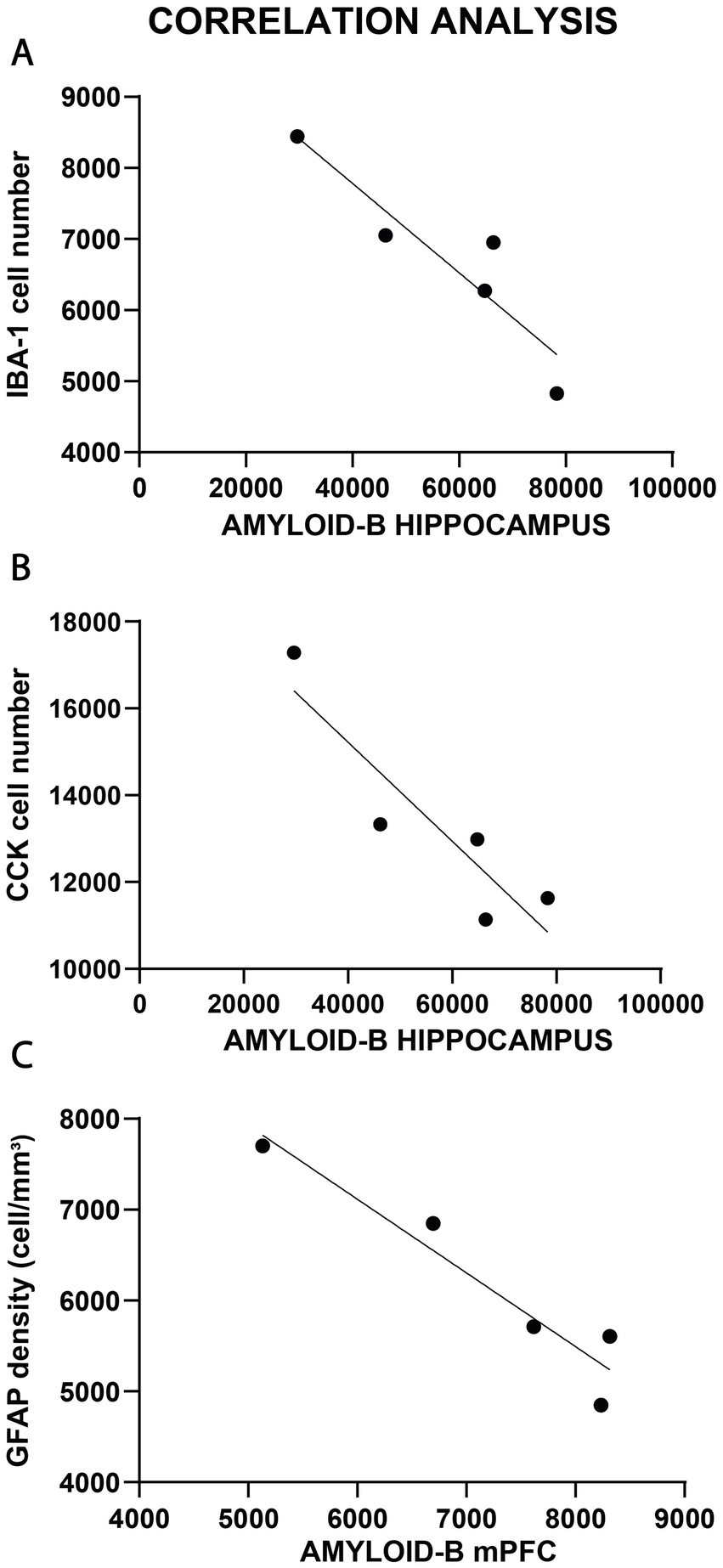
Figure 12. Correlations analysis between Aβ plaque numbers and cellular changes. (A) In the hippocampus, we found a negative correlation between Aβ plaque numbers and Iba-1+ cell numbers (p < 0.05). (B) Similarly, negative correlation was found between Aβ plaque numbers and CCK+ interneuron numbers (p < 0.05) of the hippocampus. (C) In the mPFC, there was a negative correlation between Aβ plaque density and astrocyte density (p < 0.05).
4 Discussion
In this study, we report that relative to age-matched controls, young adult TgF344-AD rats have pronounced activation of glial cells in both the hippocampus and frontal cortex. We found reduced number of cholecystokinin-positive cells in the hippocampus of transgenic rats, while other types of GABAergic neurons were not affected. Young TgF344-AD rats had spatial learning and memory impairments, but this cognitive deficit did not correlate with amyloid plaque number or cellular changes in the brain. In the hippocampus, amyloid plaque numbers were negatively correlated with cholecystokinin-positive neuron, and Iba-1-positive cell numbers. In the mPFC, amyloid plaque number was negatively correlated with the number of astrocytes.
4.1 Glial cell activation showed negative correlation with the number of β-amyloid plaques
We observed a pronounced increase in Iba-1-positive cell numbers in both the hippocampus and the mPFC of TgF344-AD rats. This inflammatory reaction was accompanied by the activation of astrocytes, especially in the mPFC, where the number of GFAP+ cells was significantly increased. Typically, the β-amyloid plaques were surrounded by both types of glia. Several studies have documented similar pronounced gliosis in the brains of TgF344-AD rats (e.g., Cohen et al., 2013; Rorabaugh et al., 2017; Anckaerts et al., 2019; Ceyzériat et al., 2021; Bac et al., 2023; Hernandez et al., 2024). We also found a negative correlation between the number of β-amyloid plaques and Iba-1+ cell numbers in the hippocampus, as well as a negative correlation between the number of β-amyloid plaques and GFAP+ astrocyte numbers in the mPFC.
The relationship between glial activation and protein aggregation is still debated since glial cells seem to participate both in the formation and the clearing up of Aβ plaques (Nagele et al., 2004). Numerous studies performed a correlation analysis between glial cells and Aβ plaques, but the available results are ambiguous. These studies typically report that both astrocytes and microglia are spatially associated with the plaques (Serrano-Pozo et al., 2013; Dani et al., 2018), but the microglial response and the activation of astrocytes are often different and may depend on, e.g., plaque size (Serrano-Pozo et al., 2013). While some studies report on no correlation between microglia activation and Aβ load in AD patients (Arends et al., 2000; Hayes et al., 2002), a more recent in vivo imaging study found positive correlation between microglial activation and amyloid deposition (Dani et al., 2018). Yet another study found that brain accumulation of soluble small oligomeric species of Aβ is an early event that predates by months the classic fibrillar amyloid plaque deposition, and the amount of such oligomeric Aβ deposits showed positive correlation with GFAP-positive astrocytes in the entorhinal cortex and hippocampal CA1 (DaRocha-Souto et al., 2011).
One possible explanation for these conflicting data might be that activated microglia and astrocytes develop into heterogenous phenotypes and some may act as neuroprotective, while others neurotoxic, furthermore, their phenotypic distribution may change, based on the progression of the disease (Kwon and Koh, 2020; Gerrits et al., 2021; Ferrari-Souza et al., 2022). Regulation of Aβ levels in the brain is one of the most important functions of glial cells in AD. Currently, both microglial and astrocyte activation are regarded as a “double-edged sword,” as they actively participate in the pathogenesis of the disease, but they also play a central role as moderators of Aβ clearance and degradation (Ries and Sastre, 2016; Chun and Lee, 2018; Wu and Eisel, 2023). Importantly, both astrocytes and microglia play essential roles in phagocytosing Aβ plaques (Xin et al., 2018; Giusti et al., 2024).
Originally, we expected to find a positive correlation between Aβ load and glial activation, but to our surprise, we found the opposite. One may speculate that what we observed here was the early, acute phase of neuroinflammation, when microglial recruitment can promote Aβ clearance and hinder the pathologic progression in AD (Cai et al., 2014). Overall, our present data suggests that in the young TgF344-AD rats the presence of larger number of microglia and astrocytes was protective, as more glial cells were associated with fewer Aβ plaques, either because the glial cells were clearing them up, or because they prevented the formation of Aβ deposits. In line with this explanation, there is experimental evidence that microglia can exert neuroprotective function and clear Aβ peptides from the brain (Mandrekar et al., 2009; Fu et al., 2012; Griciuc et al., 2013). A negative correlation between microglial cells and Aβ deposits, has been shown in an experiment, where repeated injection of the macrophage colony-stimulating factor to Swedish beta-amyloid precursor protein (APPSwe)/PS1 transgenic mice, increased the number of microglia and decreased the number of Aβ deposits (Boissonneault et al., 2009).
4.2 Reduced number of cholecystokinin-positive interneurons in the hippocampus
Numerous neurotransmitter systems, including the GABAergic system, have been implicated in the pathophysiology of AD. Numerous studies have shown GABAergic neural network abnormalities in the early stages of AD, which may exist decades earlier than clinical symptoms (Palop and Mucke, 2016; Tang et al., 2023; Li et al., 2024). A recent systematic review with meta-analysis found a global reduction in various GABAergic system components in the AD brain and concluded that the GABAergic system is vulnerable to AD pathology (Carello-Collar et al., 2023). Furthermore, it has been shown that the endogenous amyloid precursor protein (APP) is highly expressed in a heterogeneous subset of GABAergic interneurons in the entire hippocampus, and during the early stages of plaque deposition, interneurons contribute to approximately 30% of the total plaque load in the hippocampus, therefore these cells are likely to have a profound contribution to AD plaque pathology (Rice et al., 2020). Based on these findings recent theories emphasize the importance of GABAergic neurons in the pathogenesis of AD and propose a concept that excitatory and inhibitory imbalance together with the structural and functional remodeling of neural networks drives the pathogenesis of Alzheimer’s disease (Bi et al., 2020; Li et al., 2024).
Many studies focusing on animal models have reported a reduced number of GABAergic interneurons in the brains of various transgenic mouse models of AD (reviewed by Xu et al., 2020). These studies typically document a pronounced reduction in the number of PV+, SST+, and CR+ neurons in the hippocampus and neocortex of transgenic mice (Ramos et al., 2006; Andrews-Zwilling et al., 2010; Baglietto-Vargas et al., 2010; Takahashi et al., 2010; Huh et al., 2016; Zallo et al., 2018; Cheng et al., 2020; Shi et al., 2020). Not only cell loss, but changes in cellular morphology have also been documented. For example, in the human amyloid precursor protein (hAPP) transgenic mouse line, a compensatory sprouting of GABAergic fibers has been found in response to the spontaneous nonconvulsive seizure activity in the hippocampus (Palop et al., 2007). In other mouse models, such as the hAPP-J20 and Mutated Tau VLW line, the selective loss of GABAergic septo-hippocampal axons has been observed which results in dysfunctional hippocampal network activities (Rubio et al., 2012; Soler et al., 2017). Selective degeneration of entorhinal-CA1 synapses onto parvalbumin+ neurons together with the loss of CA1 PV-positive spines have also been reported (Shu et al., 2016; Yang et al., 2018).
Human studies have also revealed changes in GABAergic cell number. In postmortem samples of AD brains, a substantial decrease in the number of PV+ interneurons has been found in all hippocampal subareas except in the CA3 subfield (Brady and Mufson, 1997). A significant loss of CR+ neurons has been reported in the dentate gyrus of AD patients (Takahashi et al., 2010). Reduced somatostatin-like immunoreactivity, potentially indicating loss of SST+ neurons, has been reported in several neocortical areas of patients with AD, including the frontal lobe (Davies et al., 1980; Candy et al., 1985; Beal et al., 1986; Mazurek and Beal, 1991).
In our present study, the transgenic rats did not have any alterations in the number of PV+, SST+, or CR+ neurons; instead, the number of CCK+ interneurons was reduced in all hippocampal areas, except the dentate gyrus. Furthermore, we found a negative correlation between the number of β-amyloid plaques and CCK+ neuron numbers, indicating that a more pronounced amyloid-β load was associated with fewer CCK+ cells.
CCK is a neuromodulator neuropeptide which facilitates hippocampal glutamate release and gates GABAergic basket cell activity, and by that modulates learning and memory. More importantly, as recent studies suggest, it seem to exert neuroprotective effects in AD (Reich and Hölscher, 2024). An early human postmortem study reported significant reduction of CCK-like immunoreactivity in several cortical areas of patients with AD, indicating that CCK+ neurons are affected by the AD process (Mazurek and Beal, 1991). Another clinical study measuring cerebrospinal fluid levels of CCK found that higher CCK was associated with a decreased likelihood of having mild cognitive impairment or AD, suggesting that CCK may serve as a biomarker of neural integrity, and cognitive performance in AD (Plagman et al., 2019). Furthermore, a growing number of evidence suggest that a CCK analog (CCK-8 L) exert neuroprotective effect in mouse models of AD. For example, in a recent study, a CCK analog effectively improved spatial learning and memory, reduced amyloid plaque load in the brain, enhanced synaptic plasticity in the hippocampus, and normalized synapse number and morphology in APP/PS1 mice (Zhang et al., 2023). In a follow-up study, the same group reported that the CCK analogue ameliorated cognitive deficits and regulated mitochondrial dynamics by activating the CCKB receptor and the AMPK/Drp1 pathway in APP/PS1 mice (Hao et al., 2024).
Animal studies have reported diminished cerebral CCK expression and the number of binding sites in the aging rat hippocampus (Harro and Oreland, 1992) and reduced hippocampal CCK mRNA levels in APP/PS1 mice, suggesting that a lack of CCK might predispose to neurodegeneration in AD (Liu et al., 2021). In a knock-in mouse model of AD (App NL-F/NL-F), the CCK+ neurons showed aberrant hyperexcitability in the early stage of AD together with a gradual decline in the expression level of CCK, whereas in aged animals a significant decrease in the number of CCK+ cells was observed in the hippocampal CA1 area (Shi et al., 2020). Furthermore, this study proposed that early hyperactivity of CCK+ cells may facilitate increased Αβ cleavage and accumulation that subsequently leads to the destruction of the CCK+ cells (Shi et al., 2020). A similar cascade of cellular events may account for the decreased number of CCK+ cells found in our present study.
4.3 Adult hippocampal neurogenesis in AD
Neurogenesis persists in the adult mammalian hippocampus, even in humans (Boldrini et al., 2018; Tobin et al., 2019; Moreno-Jiménez et al., 2021), but it declines with advanced age (Culig et al., 2022). A growing body of evidence suggests that it is impaired in AD, and decreased neurogenesis contributes to cognitive decline in aging (Babcock et al., 2021). Reduced neurogenesis has been documented in various transgenic mouse models of AD (Babcock et al., 2021), even in young animals by the age of 1.5–3 months (e.g., Wang et al., 2004; Wen et al., 2004; Demars et al., 2010; Moon et al., 2014; Zeng et al., 2016; Scopa et al., 2020). However, other studies have reported an increased incidence of neurogenesis in AD mouse models (Babcock et al., 2021).
Human studies focusing on adult hippocampal neurogenesis in patients with AD have yielded contradictory results. The first report by Jin et al. documented increased neurogenesis in the brains of AD patients (Jin et al., 2004). More precisely, they found increased expression of immature neuronal marker proteins, such as doublecortin, polysialylated nerve cell adhesion molecule, neurogenic differentiation factor, and TUC-4, which signal the birth of new neurons (Jin et al., 2004). Another study that examined various markers expressed at different stages of neurogenesis found that markers for hippocampal stem cells decreased, while markers for cell proliferation increased, whereas markers for the differentiation/migration phase remained virtually unchanged (Perry et al., 2012). The authors concluded that neurogenic abnormalities in AD differ between various phases of neurogenesis (Perry et al., 2012). A more recent study reported marked impairment of adult neurogenesis in patients with AD (Moreno-Jiménez et al., 2019). Similarly, another study found that the number of DCX+ cells was reduced in individuals with mild cognitive impairment (Tobin et al., 2019).
Adult hippocampal neurogenesis in the TgF344-AD rat model has been investigated before. A study by Morrone et al. (2020) examined rats at 13 months of age and they found a significant reduction of adult hippocampal neurogenesis in the transgenic rats. In our case, we found a small, but statistically insignificant reduction in the number of DCX+ immature neurons in the dentate gyrus of young TgF344-AD transgenic rats. Most likely this was an early sign for an impairment which amplifies as the animals grow older.
4.4 TgF344-AD rats exhibited cognitive deficits during a spatial navigation task, but their behavioral performance did not correlate with the neuropathological changes
In the present study, transgenic rats at the age–7-8 months showed clear spatial learning and memory deficits in the Barnes maze. Cognitive impairment is one of the most consistent findings in TgF344-AD rats, which gradually emerge at 7–8 months and then becomes pronounced at 10–11 months of age (Berkowitz et al., 2018). This has been repeatedly demonstrated using various spatial navigation tasks, such as the Barnes maze (Cohen et al., 2013; Fowler et al., 2022), Morris water maze (Rorabaugh et al., 2017; Berkowitz et al., 2018; Bernaud et al., 2022; Kelberman et al., 2022; Bac et al., 2023), water radial-arm maze (Bernaud et al., 2022), and in an active allothetic place avoidance task (Proskauer Pena et al., 2021). Some studies have reported cognitive impairment in transgenic rats as early as four months of age (Proskauer Pena et al., 2021; Fowler et al., 2022), but negative findings on cognitive deficits are also available (for example, Pentkowski et al., 2018).
A few studies have investigated the correlation between biomarker changes in the brain and cognitive impairment of TgF344-AD rats. For example, Bac et al. analyzed proteomic differences in the dorsal hippocampal CA1 region and correlated them with learning impairments in the Morris water maze (Bac et al., 2023). They found no correlation between amyloid plaque Aβ peptide levels and learning impairment; however, soluble Aβ peptides, phosphorylated tau, and proinflammatory cytokine levels were correlated with performance in the water maze (Bac et al., 2023). Furthermore, GFAP levels, reactive astrocytes, and microglial numbers are also correlated with learning impairment (Bac et al., 2023). More recently, Hernandez et al. employed a battery of cognitive and emotional tests and correlated the performance of young adult (aged 5–7 months) TgF344-AD rats with protein markers of inflammation and AD-like pathology in the prelimbic cortex of the mPFC, basolateral amygdala, and nucleus accumbens (Hernandez et al., 2024). Transgenic rats display maladaptive decision-making, greater apathy, and impaired working memory, and numerous associations have been found between these cognitive and emotional deficits and AD-like pathology in the relevant brain structures (Hernandez et al., 2024). However, in the present study, we could not detect any correlation between impaired spatial memory in TgF344-AD rats and neuropathological changes in the hippocampus and mPFC.
4.5 Limitations
The present study has several limitations. We systematically quantified Aβ plaques in the hippocampus and mPFC, but this approach does not provide a precise estimate on amyloid load because the size of the plaques was rather diverse. Measuring amyloid burden at a protein level with Western blotting or ELISA and differentiating between Sarkosyl-soluble and-insoluble fractions may yield a more precise quantitative result. Another limiting factor was the relatively low number of animals involved in this study. Furthermore, tissue fixation was not sufficient in case of some rats, that explains why we had fewer number or rats in the correlation analyses compared to the rat numbers used for the behavioral experiments. Finally, we used both male and female rats in this study, since we did not detect any differences between the sexes during the behavioral, and later in the neuropathological phenotyping. This is not unusual as experiments with TgF344-AD rats often use rats with mixed sexes (e.g., Cohen et al., 2013; Bazzigaluppi et al., 2018; Pentkowski et al., 2018; Morrone et al., 2020). Even the original Cohen et al. (2013) study stated that: “We did not observe gender differences on any of the measures reported, and therefore males and females were combined for all analyses.” Since then, however, several studies reported sex differences in the behavior and neuropathology of TgF344-AD rats (e.g., Saré et al., 2020; Chaudry et al., 2022; Srivastava et al., 2023). Therefore, a study using only male or female rats would be more rigorous, and may yield different results.
4.6 Conclusion
We report here that in young animals, pronounced neuropathological changes are present in the hippocampus and mPFC of TgF344-AD rats, which include marked gliosis and loss of CCK+ GABAergic interneurons. These cellular changes were negatively correlated with amyloid-β plaque load, but we found no correlation between the neuropathological changes and the cognitive impairment of the animals. Dysfunctional hippocampal CCK-positive interneurons seem to contribute to AD pathophysiology and deserve further investigations.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by National Danish Animal Research Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AF: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft. KR: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Visualization. GS: Investigation, Methodology, Writing – original draft, Visualization. BV: Funding acquisition, Resources, Supervision, Writing – review & editing, Methodology. OW: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – review & editing, Data curation. BC: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing, Visualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was financed by the following programs and grant agencies: (1) the Hungarian Brain Research Program 3 (NAP-3) to BC; (2) the Thematic Excellence Program 2021 Health Sub-programme of the Ministry for Innovation and Technology in Hungary, within the framework of the EGA-16 project of the Pécs of University to BC and BV; (3) support from the NKFI and the European Union under the action of the ERA-NET COFUND (2019-2.1.7-ERANET-2021-00018); NEURON (NEURON-066 Rethealthsi) to BV; (4) the NKFI (OTKA NN128293) to BV and (5) the EKÖP-24-4-I-PTE-11 to GS from the New National Excellence Program of the Ministry of Human Capacities.
Acknowledgments
This research was performed in collaboration with the Imaging Core Facility at the Szentágothai Research Centre of the University of Pécs. We are grateful to Krisztina Vudi-Garai for her technical assistance during the histological procedures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Aβ, Amyloid-β; aCg, Anterior cingulate; AD, Alzheimer’s disease; CR, Calretinin; CCK, Cholecystokinin; DCX, Doublecortin; DG, Dentate gyrus; gcl, Granule cell layer; IL, Infralimbic; mPFC, Medial prefrontal cortex; PV, Parvalbumin; NPY, Neuropeptide Y; PrL, Prelimbic; SST, Somatostatin; str. mol., Stratum moleculare; TG/TG, Homozygous transgenic; WT/WT, Homozygous wild type.
References
Agca, C., Klakotskaia, D., Schachtman, T. R., Chan, A. W., Lah, J. J., and Agca, Y. (2016). Presenilin 1 transgene addition to amyloid precursor protein overexpressing transgenic rats increases amyloid beta 42 levels and results in loss of memory retention. BMC Neurosci. 17:46. doi: 10.1186/s12868-016-0281-8
Albuquerque, M. S., Mahar, I., Davoli, M. A., Chabot, J. G., Mechawar, N., Quirion, R., et al. (2015). Regional and sub-regional differences in hippocampal GABAergic neuronal vulnerability in the TgCRND8 mouse model of Alzheimer’s disease. Front. Aging Neurosci. 7:30. doi: 10.3389/fnagi.2015.00030
Ali, A. B., Islam, A., and Constanti, A. (2023). The fate of interneurons, GABAA receptor sub-types and perineuronal nets in Alzheimer’s disease. Brain Pathol. 33:e13129. doi: 10.1111/bpa.13129
Anckaerts, C., Blockx, I., Summer, P., Michael, J., Hamaide, J., Kreutzer, C., et al. (2019). Early functional connectivity deficits and progressive microstructural alterations in the TgF344-AD rat model of Alzheimer’s disease: a longitudinal MRI study. Neurobiol. Dis. 124, 93–107. doi: 10.1016/j.nbd.2018.11.010
Andrews-Zwilling, Y., Bien-Ly, N., Xu, Q., Li, G., Bernardo, A., Yoon, S. Y., et al. (2010). Apolipoprotein E4 causes age-and tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 30, 13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010
Arends, Y. M., Duyckaerts, C., Rozemuller, J. M., Eikelenboom, P., and Hauw, J. J. (2000). Microglia, amyloid and dementia in alzheimer disease. A correlative study. Neurobiol Aging 21, 39–47. doi: 10.1016/s0197-4580(00)00094-4
Babcock, K. R., Page, J. S., Fallon, J. R., and Webb, A. E. (2021). Adult hippocampal neurogenesis in aging and Alzheimer’s disease. Stem Cell Reports 16, 681–693. doi: 10.1016/j.stemcr.2021.01.019
Bac, B., Hicheri, C., Weiss, C., Buell, A., Vilcek, N., Spaeni, C., et al. (2023). The TgF344-AD rat: behavioral and proteomic changes associated with aging and protein expression in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 123, 98–110. doi: 10.1016/j.neurobiolaging.2022.12.015
Baglietto-Vargas, D., Moreno-Gonzalez, I., Sanchez-Varo, R., Jimenez, S., Trujillo-Estrada, L., Sanchez-Mejias, E., et al. (2010). Calretinin interneurons are early targets of extracellular amyloid-beta pathology in PS1/Abeta PP Alzheimer mice hippocampus. J. Alzheimers Dis. 21, 119–132. doi: 10.3233/JAD-2010-100066
Bazzigaluppi, P., Beckett, T. L., Koletar, M. M., Lai, A. Y., Joo, I. L., Brown, M. E., et al. (2018). Early-stage attenuation of phase-amplitude coupling in the hippocampus and medial prefrontal cortex in a transgenic rat model of Alzheimer’s disease. J. Neurochem. 144, 669–679. doi: 10.1111/jnc.14136
Beal, M. F., Mazurek, M. F., Svendsen, C. N., Bird, E. D., and Martin, J. B. (1986). Widespread reduction of somatostatin-like immunoreactivity in the cerebral cortex in Alzheimer’s disease. Ann. Neurol. 20, 489–495. doi: 10.1002/ana.410200408
Berkowitz, L. E., Harvey, R. E., Drake, E., Thompson, S. M., and Clark, B. J. (2018). Progressive impairment of directional and spatially precise trajectories by TgF344-Alzheimer’s disease rats in the Morris water task. Sci. Rep. 8:16153. doi: 10.1038/s41598-018-34368-w
Bernaud, V. E., Bulen, H. L., Peña, V. L., Koebele, S. V., Northup-Smith, S. N., Manzo, A. A., et al. (2022). Task-dependent learning and memory deficits in the TgF344-AD rat model of Alzheimer’s disease: three key timepoints through middle-age in females. Sci. Rep. 12:14596. doi: 10.1038/s41598-022-18415-1
Bi, D., Wen, L., Wu, Z., and Shen, Y. (2020). GABAergic dysfunction in excitatory and inhibitory (E/I) imbalance drives the pathogenesis of Alzheimer’s disease. Alzheimers Dement. 16, 1312–1329. doi: 10.1002/alz.12088
Boissonneault, V., Filali, M., Lessard, M., Relton, J., Wong, G., and Rivest, S. (2009). Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer’s disease. Brain 132, 1078–1092. doi: 10.1093/brain/awn331
Boldrini, M., Fulmore, C. A., Tartt, A. N., Simeon, L. R., Pavlova, I., Poposka, V., et al. (2018). Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell 22, 589–599.e5. doi: 10.1016/j.stem.2018.03.015
Brady, D. R., and Mufson, E. J. (1997). Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s disease brain. Neuroscience 80, 1113–1125. doi: 10.1016/s0306-4522(97)00068-7
Cai, Z., Hussain, M. D., and Yan, L. J. (2014). Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 124, 307–321. doi: 10.3109/00207454.2013.833510
Calvo-Flores Guzmán, B., Vinnakota, C., Govindpani, K., Waldvogel, H. J., Faull, R. L. M., and Kwakowsky, A. (2018). The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 146, 649–669. doi: 10.1111/jnc.14345
Candy, J. M., Gascoigne, A. D., Biggins, J. A., Smith, A. I., Perry, R. H., Perry, E. K., et al. (1985). Somatostatin immunoreactivity in cortical and some subcortical regions in Alzheimer’s disease. J. Neurol. Sci. 71, 315–323. doi: 10.1016/0022-510x(85)90070-x
Carello-Collar, G., Bellaver, B., Ferreira, P. C. L., Ferrari-Souza, J. P., Ramos, V. G., Therriault, J., et al. (2023). The GABAergic system in Alzheimer’s disease: a systematic review with meta-analysis. Mol. Psychiatry 28, 5025–5036. doi: 10.1038/s41380-023-02140-w
Ceyzériat, K., Zilli, T., Fall, A. B., Millet, P., Koutsouvelis, N., Dipasquale, G., et al. (2021). Treatment by low-dose brain radiation therapy improves memory performances without changes of the amyloid load in the TgF344-AD rat model. Neurobiol. Aging 103, 117–127. doi: 10.1016/j.neurobiolaging.2021.03.008
Chaney, A. M., Lopez-Picon, F. R., Serrière, S., Wang, R., Bochicchio, D., Webb, S. D., et al. (2021). Prodromal neuroinflammatory, cholinergic and metabolite dysfunction detected by PET and MRS in the TgF344-AD transgenic rat model of AD: a collaborative multi-modal study. Theranostics 11, 6644–6667. doi: 10.7150/thno.56059
Chaudry, O., Ndukwe, K., Xie, L., Figueiredo-Pereira, M., Serrano, P., and Rockwell, P. (2022). Females exhibit higher Glu A2 levels and outperform males in active place avoidance despite increased amyloid plaques in TgF344-Alzheimer’s rats. Sci. Rep. 12:19129. doi: 10.1038/s41598-022-23801-w
Cheng, A., Wang, J., Ghena, N., Zhao, Q., Perone, I., King, T. M., et al. (2020). SIRT3 Haploinsufficiency aggravates loss of GABAergic interneurons and neuronal network Hyperexcitability in an Alzheimer’s disease model. J. Neurosci. 40, 694–709. doi: 10.1523/JNEUROSCI.1446-19.2019
Chun, H., and Lee, C. J. (2018). Reactive astrocytes in Alzheimer’s disease: a double-edged sword. Neurosci. Res. 126, 44–52. doi: 10.1016/j.neures.2017.11.012
Cohen, R. M., Rezai-Zadeh, K., Weitz, T. M., Rentsendorj, A., Gate, D., Spivak, I., et al. (2013). A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J. Neurosci. 33, 6245–6256. doi: 10.1523/JNEUROSCI.3672-12.2013
Couillard-Despres, S., Winner, B., Schaubeck, S., Aigner, R., Vroemen, M., Weidner, N., et al. (2005). Doublecortin expression levels in adult brain reflect neurogenesis. Eur. J. Neurosci. 21, 1–14. doi: 10.1111/j.1460-9568.2004.03813.x
Culig, L., Chu, X., and Bohr, V. A. (2022). Neurogenesis in aging and age-related neurodegenerative diseases. Ageing Res. Rev. 78:101636. doi: 10.1016/j.arr.2022.101636
Czéh, B., Simon, M., Schmelting, B., Hiemke, C., and Fuchs, E. (2006). Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 31, 1616–1626. doi: 10.1038/sj.npp.1300982
Czéh, B., Vardya, I., Varga, Z., Febbraro, F., Csabai, D., Martis, L. S., et al. (2018). Long-term stress disrupts the structural and functional integrity of GABAergic neuronal networks in the medial prefrontal cortex of rats. Front. Cell. Neurosci. 12:148. doi: 10.3389/fncel.2018.00148
Czéh, B., Varga, Z. K., Henningsen, K., Kovács, G. L., Miseta, A., and Wiborg, O. (2015). Chronic stress reduces the number of GABAergic interneurons in the adult rat hippocampus, dorsal-ventral and region-specific differences. Hippocampus 25, 393–405. doi: 10.1002/hipo.22382
Dani, M., Wood, M., Mizoguchi, R., Fan, Z., Walker, Z., Morgan, R., et al. (2018). Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754. doi: 10.1093/brain/awy188
DaRocha-Souto, B., Scotton, T. C., Coma, M., Serrano-Pozo, A., Hashimoto, T., Serenó, L., et al. (2011). Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J. Neuropathol. Exp. Neurol. 70, 360–376. doi: 10.1097/NEN.0b013e318217a118
Davies, P., Katzman, R., and Terry, R. D. (1980). Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementia. Nature 288, 279–280. doi: 10.1038/288279a0
Demars, M., Hu, Y. S., Gadadhar, A., and Lazarov, O. (2010). Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 88, 2103–2117. doi: 10.1002/jnr.22387
Deuschl, G., Beghi, E., Fazekas, F., Varga, T., Christoforidi, K. A., Sipido, E., et al. (2020). The burden of neurological diseases in Europe: an analysis for the global burden of disease study 2017. Lancet Public Health 5, e551–e567. doi: 10.1016/S2468-2667(20)30190-0
Do Carmo, S., and Cuello, A. C. (2013). Modeling Alzheimer’s disease in transgenic rats. Mol. Neurodegener. 8:37. doi: 10.1186/1750-1326-8-37
Drummond, E., and Wisniewski, T. (2017). Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133, 155–175. doi: 10.1007/s00401-016-1662-x
Fang, X., Tang, C., Zhang, H., Border, J. J., Liu, Y., Shin, S. M., et al. (2023). Longitudinal characterization of cerebral hemodynamics in the TgF344-AD rat model of Alzheimer’s disease. Geroscience. 45, 1471–1490. doi: 10.1007/s11357-023-00773-x
Ferrari-Souza, J. P., Ferreira, P. C. L., Bellaver, B., Tissot, C., Wang, Y. T., Leffa, D. T., et al. (2022). Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 27, 4781–4789. doi: 10.1038/s41380-022-01716-2
Fowler, C. F., Goerzen, D., Devenyi, G. A., Madularu, D., Chakravarty, M. M., and Near, J. (2022). Neurochemical and cognitive changes precede structural abnormalities in the TgF344-AD rat model. Brain Commun. 4:fcac 072. doi: 10.1093/braincomms/fcac072
Fu, H., Liu, B., Frost, J. L., Hong, S., Jin, M., Ostaszewski, B., et al. (2012). Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia 60, 993–1003. doi: 10.1002/glia.22331
Gerrits, E., Brouwer, N., Kooistra, S. M., Woodbury, M. E., Vermeiren, Y., Lambourne, M., et al. (2021). Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 141, 681–696. doi: 10.1007/s00401-021-02263-w
Giesers, N. K., and Wirths, O. (2020). Loss of hippocampal Calretinin and Parvalbumin interneurons in the 5XFAD mouse model of Alzheimer’s disease. ASN Neuro 12:1759091420925356. doi: 10.1177/1759091420925356
Giusti, V., Kaur, G., Giusto, E., and Civiero, L. (2024). Brain clearance of protein aggregates: a close-up on astrocytes. Mol. Neurodegener. 19:5. doi: 10.1186/s13024-024-00703-1
Götz, J., and Ittner, L. M. (2008). Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 9, 532–544. doi: 10.1038/nrn2420
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Gundersen, H. J., Bagger, P., Bendtsen, T. F., Evans, S. M., Korbo, L., Marcussen, N., et al. (1988). The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS 96, 857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x
Gundersen, H. J., Jensen, E. B., Kiêu, K., and Nielsen, J. (1999). The efficiency of systematic sampling in stereology--reconsidered. J. Microsc. 193, 199–211. doi: 10.1046/j.1365-2818.1999.00457.x
Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., et al. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933. doi: 10.1093/brain/awy132
Hao, L., Shi, M., Ma, J., Shao, S., Yuan, Y., Liu, J., et al. (2024). A cholecystokinin analogue ameliorates cognitive deficits and regulates mitochondrial dynamics via the AMPK/Drp 1 pathway in APP/PS1 mice. J. Prev Alzheimers Dis. 11, 382–401. doi: 10.14283/jpad.2024.6
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388. doi: 10.1016/0165-6147(91)90609-v
Harro, J., and Oreland, L. (1992). Age-related differences of cholecystokinin receptor binding in the rat brain. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 16, 369–375. doi: 10.1016/0278-5846(92)90088-v
Hayes, A., Thaker, U., Iwatsubo, T., Pickering-Brown, S. M., and Mann, D. M. (2002). Pathological relationships between microglial cell activity and tau and amyloid beta protein in patients with Alzheimer’s disease. Neurosci. Lett. 331, 171–174. doi: 10.1016/s0304-3940(02)00888-1
Hernandez, C. M., McCuiston, M. A., Davis, K., Halls, Y., Carcamo Dal Zotto, J. P., Jackson, N. L., et al. (2024). In a circuit necessary for cognition and emotional affect, Alzheimer’s-like pathology associates with neuroinflammation, cognitive and motivational deficits in the young adult TgF344-AD rat. Brain Behav Immun. Health 39:100798. doi: 10.1016/j.bbih.2024.100798
Hijazi, S., Smit, A. B., and van Kesteren, R. E. (2023). Fast-spiking parvalbumin-positive interneurons in brain physiology and Alzheimer’s disease. Mol. Psychiatry 28, 4954–4967. doi: 10.1038/s41380-023-02168-y
Huh, S., Baek, S. J., Lee, K. H., Whitcomb, D. J., Jo, J., Choi, S. M., et al. (2016). The reemergence of long-term potentiation in aged Alzheimer’s disease mouse model. Sci. Rep. 6:29152. doi: 10.1038/srep29152
Jankowsky, J. L., Slunt, H. H., Ratovitski, T., Jenkins, N. A., Copeland, N. G., and Borchelt, D. R. (2001). Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17, 157–165. doi: 10.1016/s1389-0344(01)00067-3
Jin, K., Peel, A. L., Mao, X. O., Xie, L., Cottrell, B. A., Henshall, D. C., et al. (2004). Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 101, 343–347. doi: 10.1073/pnas.2634794100
Kelberman, M. A., Anderson, C. R., Chlan, E., Rorabaugh, J. M., McCann, K. E., and Weinshenker, D. (2022). Consequences of Hyperphosphorylated tau in the locus Coeruleus on behavior and cognition in a rat model of Alzheimer’s disease. J. Alzheimers Dis. 86, 1037–1059. doi: 10.3233/JAD-215546
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement 4, 575–590. doi: 10.1016/j.trci.2018.06.014
Kwon, H. S., and Koh, S. H. (2020). Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9:42. doi: 10.1186/s40035-020-00221-2
Leon, W. C., Canneva, F., Partridge, V., Allard, S., Ferretti, M. T., DeWilde, A., et al. (2010). A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J. Alzheimers Dis. 20, 113–126. doi: 10.3233/JAD-2010-1349
Li, J., Liu, Y., Yin, C., Zeng, Y., and Mei, Y. (2024). Structural and functional remodeling of neural networks in β-amyloid driven hippocampal hyperactivity. Ageing Res. Rev. 101:102468. doi: 10.1016/j.arr.2024.102468
Liu, Y. J., Liu, T. T., Jiang, L. H., Liu, Q., Ma, Z. L., Xia, T. J., et al. (2021). Identification of hub genes associated with cognition in the hippocampus of Alzheimer’s disease. Bioengineered 12, 9598–9609. doi: 10.1080/21655979.2021.1999549
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D. M., and Landreth, G. E. (2009). Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009
Mazurek, M. F., and Beal, M. F. (1991). Cholecystokinin and somatostatin in Alzheimer’s disease postmortem cerebral cortex. Neurology 41, 716–719. doi: 10.1212/wnl.41.5.716
Melgosa-Ecenarro, L., Doostdar, N., Radulescu, C. I., Jackson, J. S., and Barnes, S. J. (2023). Pinpointing the locus of GABAergic vulnerability in Alzheimer’s disease. Semin. Cell Dev. Biol. 139, 35–54. doi: 10.1016/j.semcdb.2022.06.017
Moon, M., Cha, M. Y., and Mook-Jung, I. (2014). Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J. Alzheimers Dis. 41, 233–241. doi: 10.3233/JAD-132417
Moreno-Jiménez, E. P., Flor-García, M., Terreros-Roncal, J., Rábano, A., Cafini, F., Pallas-Bazarra, N., et al. (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 25, 554–560. doi: 10.1038/s41591-019-0375-9
Moreno-Jiménez, E. P., Terreros-Roncal, J., Flor-García, M., Rábano, A., and Llorens-Martín, M. (2021). Evidences for adult hippocampal neurogenesis in humans. J. Neurosci. 41, 2541–2553. doi: 10.1523/JNEUROSCI.0675-20.2020
Morrone, C. D., Bazzigaluppi, P., Beckett, T. L., Hill, M. E., Koletar, M. M., Stefanovic, B., et al. (2020). Regional differences in Alzheimer’s disease pathology confound behavioural rescue after amyloid-β attenuation. Brain 143, 359–373. doi: 10.1093/brain/awz371.Erratum in: Brain. 2020 Mar 1; 143(3): e23. doi:10.1093/brain/awz409
Nagele, R. G., Wegiel, J., Venkataraman, V., Imaki, H., Wang, K. C., and Wegiel, J. (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674. doi: 10.1016/j.neurobiolaging.2004.01.007
Novati, A., Singer-Mikosch, E., Yu-Taeger, L., Clemensson, E., and Nguyen, H. P. (2022). Rat models of major neurodegenerative disorders. Ageing Neur Dis. 2:17. doi: 10.20517/and.2022.19
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Palop, J. J., and Mucke, L. (2016). Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17, 777–792. doi: 10.1038/nrn.2016.141
Pang, K., Jiang, R., Zhang, W., Yang, Z., Li, L. L., Shimozawa, M., et al. (2022). An app knock-in rat model for Alzheimer’s disease exhibiting Aβ and tau pathologies, neuronal death and cognitive impairments. Cell Res. 32, 157–175. doi: 10.1038/s41422-021-00582-x
Paul, C. M., Magda, G., and Abel, S. (2009). Spatial memory: theoretical basis and comparative review on experimental methods in rodents. Behav. Brain Res. 203, 151–164. doi: 10.1016/j.bbr.2009.05.022
Paxinos, G., and Watson, C. (1998). The rat brain in stereotaxic coordinates. San Diego: Academic Press.
Pentkowski, N. S., Berkowitz, L. E., Thompson, S. M., Drake, E. N., Olguin, C. R., and Clark, B. J. (2018). Anxiety-like behavior as an early endophenotype in the TgF344-AD rat model of Alzheimer’s disease. Neurobiol. Aging 61, 169–176. doi: 10.1016/j.neurobiolaging.2017.09.024
Perry, E. K., Johnson, M., Ekonomou, A., Perry, R. H., Ballard, C., and Attems, J. (2012). Neurogenic abnormalities in Alzheimer’s disease differ between stages of neurogenesis and are partly related to cholinergic pathology. Neurobiol. Dis. 47, 155–162. doi: 10.1016/j.nbd.2012.03.033
Plagman, A., Hoscheidt, S., McLimans, K. E., Klinedinst, B., Pappas, C., Anantharam, V., et al. (2019). Alzheimer’s disease neuroimaging initiative. Cholecystokinin and Alzheimer’s disease: a biomarker of metabolic function, neural integrity, and cognitive performance. Neurobiol. Aging 76, 201–207. doi: 10.1016/j.neurobiolaging.2019.01.002
Popović, M., Caballero-Bleda, M., Kadish, I., and Van Groen, T. (2008). Subfield and layer-specific depletion in calbindin-D28K, calretinin and parvalbumin immunoreactivity in the dentate gyrus of amyloid precursor protein/presenilin 1 transgenic mice. Neuroscience 155, 182–191. doi: 10.1016/j.neuroscience.2008.05.023
Proskauer Pena, S. L., Mallouppas, K., Oliveira, A. M. G., Zitricky, F., Nataraj, A., and Jezek, K. (2021). Early spatial memory impairment in a double transgenic model of Alzheimer’s disease TgF-344 AD. Brain Sci. 11:1300. doi: 10.3390/brainsci11101300
Ramos, B., Baglietto-Vargas, D., del Rio, J. C., Moreno-Gonzalez, I., Santa-Maria, C., Jimenez, S., et al. (2006). Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging 27, 1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022
Reich, N., and Hölscher, C. (2024). Cholecystokinin (CCK): a neuromodulator with therapeutic potential in Alzheimer’s and Parkinson’s disease. Front. Neuroendocrinol. 73:101122. doi: 10.1016/j.yfrne.2024.101122
Reid, H. M. O., Chen-Mack, N., Snowden, T., and Christie, B. R. (2021). Understanding changes in hippocampal interneurons subtypes in the pathogenesis of Alzheimer’s disease: a systematic review. Brain Connect 11, 159–179. doi: 10.1089/brain.2020.0879
Rice, H. C., Marcassa, G., Chrysidou, I., Horré, K., Young-Pearse, T. L., Müller, U. C., et al. (2020). Contribution of GABAergic interneurons to amyloid-β plaque pathology in an APP knock-in mouse model. Mol. Neurodegener. 15:3. doi: 10.1186/s13024-019-0356-y
Ries, M., and Sastre, M. (2016). Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 8:160. doi: 10.3389/fnagi.2016.00160
Rorabaugh, J. M., Chalermpalanupap, T., Botz-Zapp, C. A., Fu, V. M., Lembeck, N. A., Cohen, R. M., et al. (2017). Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 140, 3023–3038. doi: 10.1093/brain/awx232
Rubio, S. E., Vega-Flores, G., Martínez, A., Bosch, C., Pérez-Mediavilla, A., del Río, J., et al. (2012). Accelerated aging of the GABAergic septohippocampal pathway and decreased hippocampal rhythms in a mouse model of Alzheimer’s disease. FASEB J. 26, 4458–4467. doi: 10.1096/fj.12-208413
Rusznák, K., Horváth, Á. I., Pohli-Tóth, K., Futácsi, A., Kemény, Á., Kiss, G., et al. (2022). Experimental arthritis inhibits adult hippocampal neurogenesis in mice. Cells 11:791. doi: 10.3390/cells11050791
Saré, R. M., Cooke, S. K., Krych, L., Zerfas, P. M., Cohen, R. M., and Smith, C. B. (2020). Behavioral phenotype in the TgF344-AD rat model of Alzheimer’s disease. Front. Neurosci. 14:601. doi: 10.3389/fnins.2020.00601
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chételat, G., Teunissen, C. E., et al. (2021). Alzheimer’s disease. Lancet 397, 1577–1590. doi: 10.1016/S0140-6736(20)32205-4
Schmid, L. C., Mittag, M., Poll, S., Steffen, J., Wagner, J., Geis, H. R., et al. (2016). Dysfunction of somatostatin-positive interneurons associated with memory deficits in an Alzheimer’s disease model. Neuron 92, 114–125. doi: 10.1016/j.neuron.2016.08.034
Scopa, C., Marrocco, F., Latina, V., Ruggeri, F., Corvaglia, V., La Regina, F., et al. (2020). Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 27, 934–948. doi: 10.1038/s41418-019-0409-3.Erratum in: Cell Death Differ. 2020 Jun; 27(6): 2035. doi:10.1038/s41418-019-0478-3
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Serrano-Pozo, A., Muzikansky, A., Gómez-Isla, T., Growdon, J. H., Betensky, R. A., Frosch, M. P., et al. (2013). Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol. Exp. Neurol. 72, 462–471. doi: 10.1097/NEN.0b013e3182933788
Shi, A., Petrache, A. L., Shi, J., and Ali, A. B. (2020). Preserved Calretinin interneurons in an app model of Alzheimer’s disease disrupt hippocampal inhibition via upregulated P2Y1 Purinoreceptors. Cereb. Cortex 30, 1272–1290. doi: 10.1093/cercor/bhz165
Shu, S., Xu, S. Y., Ye, L., Liu, Y., Cao, X., Jia, J. Q., et al. (2023). Prefrontal parvalbumin interneurons deficits mediate early emotional dysfunction in Alzheimer’s disease. Neuropsychopharmacology 48, 391–401. doi: 10.1038/s41386-022-01435-w
Shu, S., Zhu, H., Tang, N., Chen, W., Li, X., Li, H., et al. (2016). Selective degeneration of entorhinal-CA1 synapses in Alzheimer’s disease via activation of DAPK1. J. Neurosci. 36, 10843–10852. doi: 10.1523/JNEUROSCI.2258-16.2016
Soler, H., Dorca-Arévalo, J., González, M., Rubio, S. E., Ávila, J., Soriano, E., et al. (2017). The GABAergic septohippocampal connection is impaired in a mouse model of tauopathy. Neurobiol. Aging 49, 40–51. doi: 10.1016/j.neurobiolaging.2016.09.006
Sos, K. E., Mayer, M. I., Takács, V. T., Major, A., Bardóczi, Z., Beres, B. M., et al. (2020). Amyloid β induces interneuron-specific changes in the hippocampus of APPNL-F mice. PLoS One 15:e0233700. doi: 10.1371/journal.pone.0233700
Srivastava, H., Lasher, A. T., Nagarajan, A., and Sun, L. Y. (2023). Sexual dimorphism in the peripheral metabolic homeostasis and behavior in the TgF344-AD rat model of Alzheimer’s disease. Aging Cell 22:e13854. doi: 10.1111/acel.13854
Stanley, E. M., Fadel, J. R., and Mott, D. D. (2012). Interneuron loss reduces dendritic inhibition and GABA release in hippocampus of aged rats. Neurobiol. Aging 33, 431.e1–431.e13. doi: 10.1016/j.neurobiolaging.2010.12.014
Stoiljkovic, M., Kelley, C., Stutz, B., Horvath, T. L., and Hajós, M. (2019). Altered cortical and hippocampal excitability in TgF344-AD rats modeling Alzheimer’s disease pathology. Cereb. Cortex 29, 2716–2727. doi: 10.1093/cercor/bhy140
Takahashi, H., Brasnjevic, I., Rutten, B. P., Van Der Kolk, N., Perl, D. P., Bouras, C., et al. (2010). Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer’s disease. Brain Struct. Funct. 214, 145–160. doi: 10.1007/s00429-010-0242-4
Tang, Y., Yan, Y., Mao, J., Ni, J., and Qing, H. (2023). The hippocampus associated GABAergic neural network impairment in early-stage of Alzheimer’s disease. Ageing Res. Rev. 86:101865. doi: 10.1016/j.arr.2023.101865
Tobin, M. K., Musaraca, K., Disouky, A., Shetti, A., Bheri, A., Honer, W. G., et al. (2019). Human hippocampal neurogenesis persists in aged adults and Alzheimer’s disease patients. Cell Stem Cell 24, 974–982.e3. doi: 10.1016/j.stem.2019.05.003
Van Dam, D., and De Deyn, P. P. (2011). Animal models in the drug discovery pipeline for Alzheimer’s disease. Br. J. Pharmacol. 164, 1285–1300. doi: 10.1111/j.1476-5381.2011.01299.x
van den Berg, M., Toen, D., Verhoye, M., and Keliris, G. A. (2023). Alterations in theta-gamma coupling and sharp wave-ripple, signs of prodromal hippocampal network impairment in the TgF344-AD rat model. Front. Aging Neurosci. 15:1081058. doi: 10.3389/fnagi.2023.1081058
Varga, Z., Csabai, D., Miseta, A., Wiborg, O., and Czéh, B. (2017). Chronic stress affects the number of GABAergic neurons in the orbitofrontal cortex of rats. Behav. Brain Res. 316, 104–114. doi: 10.1016/j.bbr.2016.08.030
von Bohlen Und Halbach, O. (2007). Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 329, 409–420. doi: 10.1007/s00441-007-0432-4
Waller, R., Mandeya, M., Viney, E., Simpson, J. E., and Wharton, S. B. (2020). Histological characterization of interneurons in Alzheimer’s disease reveals a loss of somatostatin interneurons in the temporal cortex. Neuropathology 40, 336–346. doi: 10.1111/neup.12649
Wang, R., Dineley, K. T., Sweatt, J. D., and Zheng, H. (2004). Presenilin 1 familial Alzheimer’s disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience 126, 305–312. doi: 10.1016/j.neuroscience.2004.03.048
Wang, R., and Reddy, P. H. (2017). Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers Dis. 57, 1041–1048. doi: 10.3233/JAD-160763
Wen, P. H., Hof, P. R., Chen, X., Gluck, K., Austin, G., Younkin, S. G., et al. (2004). The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp. Neurol. 188, 224–237. doi: 10.1016/j.expneurol.2004.04.002
Wu, Y., and Eisel, U. L. M. (2023). Microglia-astrocyte communication in Alzheimer’s disease. J. Alzheimers Dis. 95, 785–803. doi: 10.3233/JAD-230199
Xin, S. H., Tan, L., Cao, X., Yu, J. T., and Tan, L. (2018). Clearance of amyloid Beta and tau in Alzheimer’s disease: from mechanisms to therapy. Neurotox. Res. 34, 733–748. doi: 10.1007/s12640-018-9895-1
Xu, Y., Zhao, M., Han, Y., and Zhang, H. (2020). GABAergic inhibitory interneuron deficits in Alzheimer’s disease: implications for treatment. Front. Neurosci. 14:660. doi: 10.3389/fnins.2020.00660
Yang, X., Yao, C., Tian, T., Li, X., Yan, H., Wu, J., et al. (2018). A novel mechanism of memory loss in Alzheimer’s disease mice via the degeneration of entorhinal-CA1 synapses. Mol. Psychiatry 23, 199–210. doi: 10.1038/mp.2016.151
Zallo, F., Gardenal, E., Verkhratsky, A., and Rodríguez, J. J. (2018). Loss of calretinin and parvalbumin positive interneurones in the hippocampal CA1 of aged Alzheimer’s disease mice. Neurosci. Lett. 681, 19–25. doi: 10.1016/j.neulet.2018.05.027
Zeng, Q., Zheng, M., Zhang, T., and He, G. (2016). Hippocampal neurogenesis in the APP/PS1/nestin-GFP triple transgenic mouse model of Alzheimer’s disease. Neuroscience 314, 64–74. doi: 10.1016/j.neuroscience.2015.11.054
Keywords: astrocyte, Barnes maze, cell number, cholecystokinin, hippocampus, microglia, medial prefrontal cortex, CCK+ interneurons
Citation: Futácsi A, Rusznák K, Szarka G, Völgyi B, Wiborg O and Czéh B (2025) Quantification and correlation of amyloid-β plaque load, glial activation, GABAergic interneuron numbers, and cognitive decline in the young TgF344-AD rat model of Alzheimer’s disease. Front. Aging Neurosci. 17:1542229. doi: 10.3389/fnagi.2025.1542229
Edited by:
Md. Golam Sharoar, Oakland University William Beaumont School of Medicine, United StatesReviewed by:
Sumit Sarkar, National Center for Toxicological Research (FDA), United StatesSonia Do Carmo, McGill University, Canada
Md. Imamul Islam, University of Manitoba, Canada
Copyright © 2025 Futácsi, Rusznák, Szarka, Völgyi, Wiborg and Czéh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boldizsár Czéh, Y3plaC5ib2xkaXpzYXJAcHRlLmh1