
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 26 September 2023
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1223911
 Jonathan R. Roth1,2
Jonathan R. Roth1,2 Ruan Carlos Macedo de Moraes1Brittney P. Xu1Savannah R. Crawley1
Ruan Carlos Macedo de Moraes1Brittney P. Xu1Savannah R. Crawley1 Malghalara A. Khan1
Malghalara A. Khan1 Girish C. Melkani1*
Girish C. Melkani1*Huntington’s disease (HD) is a neurodegenerative disease characterized by movement and cognitive dysfunction. HD is caused by a CAG expansion in exon 1 of the HTT gene that leads to a polyglutamine (PQ) repeat in the huntingtin protein, which aggregates in the brain and periphery. Previously, we used Drosophila models to determine that Htt-PQ aggregation in the heart causes shortened lifespan and cardiac dysfunction that is ameliorated by promoting chaperonin function or reducing oxidative stress. Here, we further study the role of neuronal mutant huntingtin and how it affects peripheral function. We overexpressed normal (Htt-PQ25) or expanded mutant (Htt-PQ72) exon 1 of huntingtin in Drosophila neurons and found that mutant huntingtin caused age-dependent Htt-PQ aggregation in the brain and could cause a loss of synapsin. To determine if this neuronal dysfunction led to peripheral dysfunction, we performed a negative geotaxis assay to measure locomotor performance and found that neuronal mutant huntingtin caused an age-dependent decrease in locomotor performance. Next, we found that rapamycin reduced Htt-PQ aggregation in the brain. These results demonstrate the role of neuronal Htt-PQ in dysfunction in models of HD, suggest that brain-periphery crosstalk could be important to the pathogenesis of HD, and show that rapamycin reduces mutant huntingtin aggregation in the brain.
Huntington’s disease (HD) is a devastating neurodegenerative disease that is characterized by progressive degeneration of the basal ganglia in the brain. This results in a clinical presentation of chorea, which is the spontaneous arrhythmic movement of limbs, alongside cognitive and psychiatric dysfunction. HD is monogenic and is caused by a CAG repeat expansion in exon 1 of the HTT gene, leading to a polyglutamine (PQ) expansion in the huntingtin protein. Typically, healthy people have 26 or fewer CAG repeats in HTT, while greater than 35 repeats leads to HD, and the age of onset correlates with the CAG repeat number (Duyao et al., 1993; Lee et al., 2012). Currently, there are no effective disease-modifying therapies for HD (Dash and Mestre, 2020).
HD displays prominent dysfunction in both the brain and periphery, especially in muscles. HTT is expressed in multiple tissues throughout the body (Strong et al., 1993) and PQ aggregates are present in the brain (Difiglia et al., 1997) and in skeletal and cardiac muscle (Sathasivam et al., 1999), where it can contribute to dysfunction (Van Der Burg et al., 2009). We previously studied the role of mutant huntingtin in the heart because HD patients are at higher risk of dying from cardiac dysfunction (Lanska et al., 1988) and found that mutant huntingtin expressed in the heart causes PQ aggregation, premature mortality, and cardiac dysfunction in Drosophila (Melkani et al., 2013). The importance of huntingtin in the periphery is supported by the finding that a mouse model expressing mutant huntingtin ubiquitously has severe cardiac dysfunction (Mihm et al., 2007). Beyond the heart, there is significant evidence that HTT is important in skeletal muscle, as movement dysfunction and skeletal muscle weakness are hallmarks of HD (Busse et al., 2008; Zielonka et al., 2014). Additionally, it is possible that brain-muscle crosstalk contributes to dysfunction in HD (Chuang and Demontis, 2021), especially with recent evidence showing that muscle-specific manipulations can affect brain function (Ehlen et al., 2017). However, there is still a gap in knowledge for how brain-muscle crosstalk contributes to dysfunction in HD, and how neuronal HTT affects muscle function.
Understanding the mechanisms underlying HD is important for developing effective therapies. HTT is involved in axonal trafficking, transcriptional regulation, and cell survival (Schulte and Littleton, 2011). Beyond these important endogenous roles for normal huntingtin, we previously found that oxidative stress and protein misfolding stress contribute to mutant huntingtin–induced dysfunction in the heart (Melkani et al., 2013). Additionally, huntingtin has key roles in regulating autophagy (Cortes and La Spada, 2014), and either huntingtin knockdown or overexpressing mutant huntingtin disrupts autophagosome dynamics (Wong and Holzbaur, 2014). Autophagy dysfunction can contribute to abnormal protein aggregation (Tyedmers et al., 2010), impaired autophagy worsens huntingtin aggregation (Ravikumar et al., 2002), and autophagy is disrupted in models of HD (Martinez-Vicente et al., 2010). Thus, improving autophagy could be beneficial for HD, and it has been proposed to do this with rapamycin (Rubinsztein et al., 2007), which stimulates autophagy by inhibiting TOR (Raught et al., 2001; Li et al., 2014) and improves lifespan in flies and mice (Bjedov et al., 2010). This approach has yielded positive results, as rapamycin reduces mutant huntingtin-associated dysfunction in cell culture and animal models of HD (Ravikumar et al., 2004; Berger et al., 2006; King et al., 2008; Sarkar et al., 2008; Bailus et al., 2021). These studies have focused on the effects of rapamycin on molecular and photoreceptor neuron outcomes, however, and its effects on in vivo aggregation and locomotor performance has been understudied in Drosophila.
Here, we determined the effects of mutant huntingtin in neurons using well-characterized Drosophila models of HD that have proven to be powerful tools (Krench and Littleton, 2013; Lewis and Smith, 2016). We measured the age-dependent effects of neuronal mutant huntingtin on PQ aggregation in the brain and on synapsin levels. We next studied brain-muscle crosstalk to determine whether neuronal mutant huntingtin affects locomotor performance, and if neuron-to-muscle signaling could contribute to dysfunction in the context of HD. Finally, we determined the effects of rapamycin on PQ aggregation in the brain and climbing muscle locomotor performance. Our results determine the age-dependent effects of mutant huntingtin in the brain, support the importance of autophagy in HD, and highlight the potential of rapamycin to prevent aggregation of mutant huntingtin.
As before (Melkani et al., 2013; Gill et al., 2015; Villanueva et al., 2019; Livelo et al., 2023), flies were housed together (about 10 flies per 25 mm vial) in an incubator at 25°C with constant humidity under a 12 h light:12 h dark cycle. Flies were maintained on standard Drosophila cornmeal food diet and changed to new food every 4–7 days. We used the following drivers lines: Elav(X)-Gal4 from Bloomington Drosophila Stock Center (BDSC) (BL#458) and Elav(II)-Gal4 (BL#8765). We used the following UAS lines: UAS-eGFP from BDSC (BL#5431), and HD model lines that expressed huntingtin exon 1 with 72 CAG repeats (HTTex1-PQ72-GFP) or 25 CAG repeats (HTTex1-PQ25-GFP) tagged with GFP from Dr. Norbert Perrimon (Zhang et al., 2010). Progeny of Elav drivers crossed with w1118 flies were used as a driver control throughout because the lines were on a w1118 background.
Fly heads were removed under a dissecting microscope and fixed with 4% PFA in PBS for 30 min at room temperature with mild agitation, then washed 3 × 10 min in 1× PBS and stored at 4°C in PBS before cryosectioning. Heads were placed in a mold with OTC and sectioned at 20 μm with a Leica CM3050 S cryostat and placed on glass slides (Fisher #15-188-48). After drying for 30 min, slides were washed 3 × 10 min with 1× PBS to remove dried OTC, then permeabilized with 1× TBST for 10 min. We blocked the slides with 3% BSA in TBST for 30 min and incubated with primary antibody for synapsin (1:500, UI Developmental Studies Hybirdoma Bank #3C11) in 1% BSA in TBST overnight at 4°C in a humidity chamber. Slides were then washed 3 × 10 with PBS and incubated for 1 h at room temperature in AlexaFluor-594 anti-mouse fluorescent secondary antibody (1:1000, ThermoFisher # A-11032). Finally, slides were washed 3 × 10 min with 1× PBS and mounted with ProLong Diamond Antifade Mountant with DAPI (ThermoFisher # P36971). After setting overnight, multichannel fluorescence images were taken with an Olympus BX63F fluorescence microscope using CellSens Software at 10× magnification to view one head section per image.
PQ-GFP aggregation was quantified using FIJI software (ImageJ v1.53v) similar to how we have quantified punctate proximity ligation signal before (Rush et al., 2020). The FITC channel images were adjusted in a batch to maximize signal-to-noise, then thresholded to exclude any background signal. ImageJ particle analyzer was run with size and circularity specified to exclude non-punctate signal on a region of interest selected around the brain, and number of puncta per section was reported.
Syanpsin immunofluorescence intensity was quantified using FIJI software (ImageJ v1.53v) similar to how we have quantified BIN1 protein levels before (Voskobiynyk et al., 2020). The TRITC channel images were adjusted in a batch to maximize signal-to-noise, then mean fluorescent intensity for each image was measured in a region of interest selected around the brain. For each experiment, we ran samples in parallel but with no primary antibody to measure background fluorescence. We measured this background fluorescence in the brain and subtracted mean background fluorescence from the fluorescence value for each image. At least two sections per fly were imaged and mean fluorescence from each fly was calculated and reported.
We conducted negative geotaxis assay as before (Ali et al., 2011; Villanueva et al., 2019). Briefly, flies were transferred into clean vials (about 10 per vial) and briefly allowed to acclimate. The vial was sharply tapped onto a pad to stimulate negative geotaxis response, and the proportion of flies the climbed to a 7 cm mark within 10 s was recorded by video for analysis. This was repeated a total of four times for each vial of flies, and the average of the four runs was calculated.
We conducted flight index assay as before (Drummond et al., 1991; Villanueva et al., 2019). Briefly, one fly at a time was released into a large plexiglass box with a light at the top. Flight index was determined by the fly’s ability to fly up (a flight index value of 6), horizontally (4), down (2), or unable to fly (0). The average flight index was calculated by dividing the sum of individual flight index values by the number of flies for each group.
Rapamycin (LC Laboratories # R-5000) was dissolved to 50 mM in ethanol, then added to food at 1:250 before pouring into vials for a final concentration of 200 μM, which is sufficient to extend fly lifespan (Bjedov et al., 2010). For vehicle control, ethanol alone was added at 1:250. To avoid developmental effects of rapamycin (Zhang et al., 2000), flies were kept on standard Drosophila cornmeal food for 1 week post-eclosion, then split into two groups and put on either rapamycin or vehicle control. Flies were kept on rapamycin or vehicle control for 3 weeks before conducting experiments at 4 weeks of age.
About 10 flies per group per cohort were flash-frozen together and stored at −80°C until use. Heads were removed and put in 150 μL Zymo Lysis Buffer and homogenized at room temperature using the Zymo Quick-RNA MicroPrep Kit (Zymo Research #R1051). RNA was purified using minicolumns and digested with DNAse I for 15 min. Next, the RNA was suspended in RNAse/DNAse free water and quantified with an Aligent Bio Tek Synergy LX Multi-mode and stored at −80°C until use. Next, we used 250 ng of RNA and 4 μL iScript RT Supermax (Bio-Rad #1708840) to synthesize cDNA. Reverse transcription was performed with 5 min of priming at 25°C, 20 min of reverse transcriptase at 46°C, then 1 min of inactivation at 95°C. We used 5 ng of cDNA for qPCR with 200 ng primers per reaction, using Ssa Advanced Universal SYBR Green Supermix (Bio-Rad #1725275) in a CFX Opus Real-Time PCR System (Bio-Rad). Expression was normalized to Rpl11 (60S ribosomal protein) and Tbp (TATA-box binding protein). PCR specificity was verified by comparing the melting curve to a predicted melting curve for the specific sequence generated by the software μMELT Quartz.1 Results are presented as 2 − ΔΔCt values normalized to the expression of Rpl11 and Tbp and control samples. All reactions were performed in triplicate.
The following primers were used: Atg1-F: CGTCAGCCTGGTCATGGAGTA; Atg1-R: TAACGGTATCCTCGCTGAG; Hid-F: CACCGACCAAGTGCTATACG; Hid-R: GGCGGATACTGGAAGATTTGC; Pten-F: GTGCAAACGCAAACAGCCTA; Pten-R: ATCCAGTTCTGGTGGCTTCG; Ilp5-F: GCGGATTTGGATAGCTCCGA; Ilp5-R: AAAGGAACACGATTTGCGGC; Rpl11-F: CGATCTGGGCATCAAGTACGA; Rpl11-R: TTGCGCTTCCTGTGGTTCAC; Tbp-F: ATGCCCTGAGCAACATCCAC; Tbp-R: GGATCAGCGGAACCTGGTG.
Statistics and test values are described in each figure legend. 1-way or 2-way ANOVA were used, and which effect was tested is reported in each legend along with sample sizes. We used Dunnett’s post hoc to compare means to GFP mean. Data values were organized in Microsoft Excel and plotted using Graphpad Prism 9, which was also used for statistical analyses. α < 0.05 was defined as our significance threshold. Data were presented as mean ± SEM.
First, we used the UAS-Gal4 system in Drosophila (Osterwalder et al., 2001) to express different huntingtin constructs in neurons. We used the Elav-Gal4 driver on the 2nd chromosome (Elav(II)) to target expression to mature neurons (Robinow and White, 1988), and, along with a driver control crossed with wildtype W1118 flies (/+), expressed three UAS constructs in neurons: GFP as an expression control, HTT exon 1 with 72 CAG repeats fused to GFP (which we refer to as Htt-PQ72), and HTT exon 1 with 25 CAG repeats fused to GFP (Htt-PQ25) (Zhang et al., 2010). We confirmed with fluorescent imaging that the GFP constructs expressed a similar amount (Supplementary Figure S1). While we did not observe any aggregation at eclosion (P0) or 1 week post-eclosion (P7), we found significant PQ-GFP aggregation in the Elav(II) > Htt-PQ72 fly brains at beginning at 2 weeks pose-eclosion (P14) and at 4 weeks post-eclosion (Figures 1A–C). We next verified whether these results were reproducible using a different Elav-Gal4 line, so we crossed the same UAS constructs with an Elav-Gal4 driver on the X chromosome (Elav(X)). In contrast to when driven by Elav(II), 45% of Elav(X) > Htt-PQ72 died before P28. To avoid survivorship bias, we measured PQ-GFP aggregation at an earlier timepoint at P21. PQ-GFP aggregated much faster in the Elav(X) > Htt-PQ72 flies than the Elav(II) > Htt-PQ72 flies, beginning by P7, and having much higher aggregation by 3 weeks (P21) than then Elav(II) > Htt-PQ72 flies had at P28 (Figures 1D–F). We interpreted that this could be due to higher expression with the Elav(X) driver than with the Elav(II) driver, which could also explain the differences in premature mortality between the drivers. Together, these results show that overexpressing mutant huntingtin in neurons causes an age- and driver-dependent aggregation of PQ-GFP in the brain.
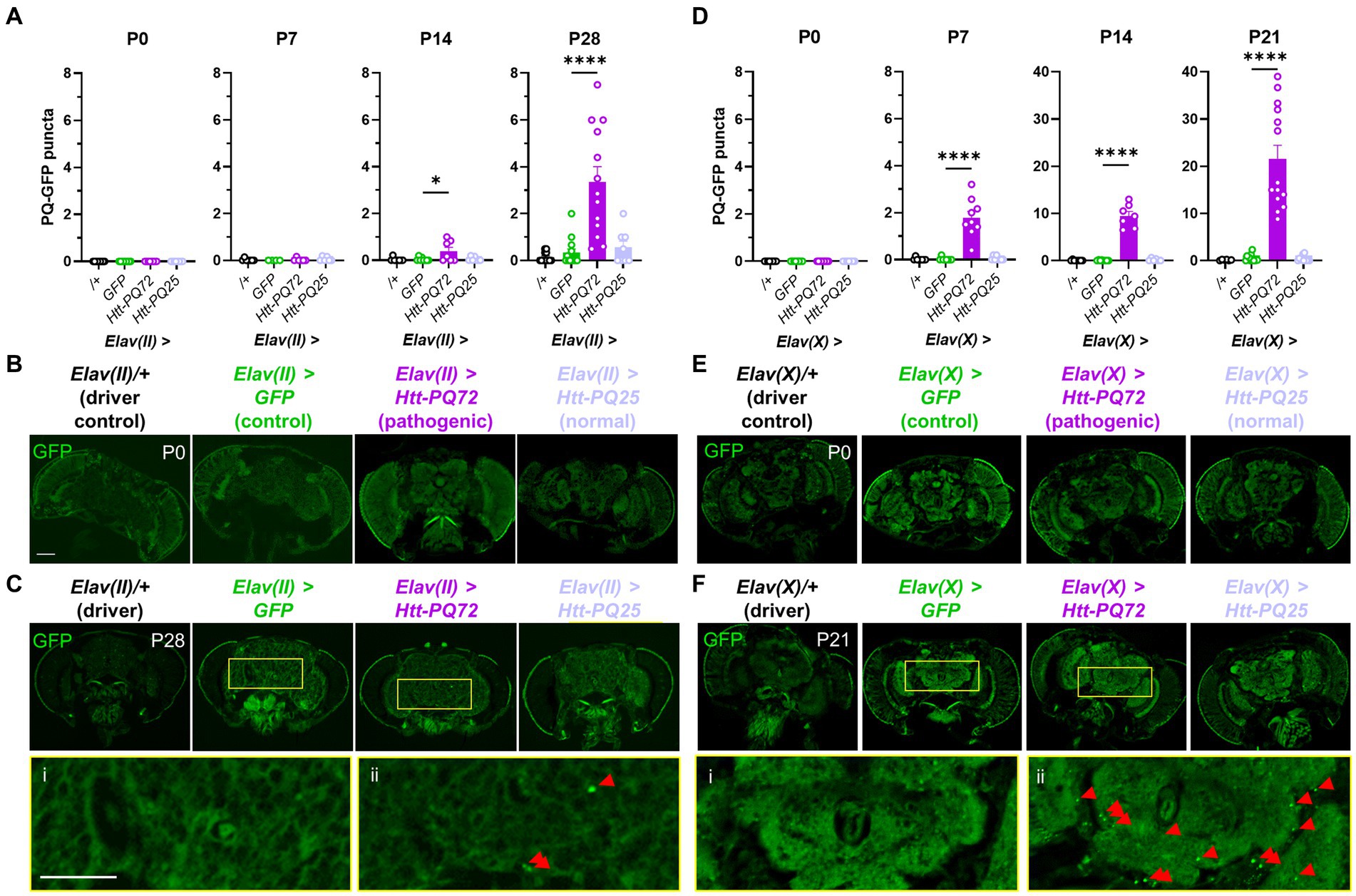
Figure 1. Neuronal Htt-PQ72 aggregates in the brain. (A) Quantification of aggregates in the brain show that PQ-GFP aggregates only in the brains of Elav(II) > Htt-PQ72 flies with age starting at 2 weeks of age (P14), but not in driver control, GFP, or Htt-PQ25 flies (ANOVA for all, P0: p = 1.0 because no variability, N = 8–9 flies per group; P7: F(3,19) = 0.74, p = 0.54, N = 4–7 flies per group; P14: F(3,26) = 4.3, p = 0.01, N = 7–8 flies per group; P28: F(3,42) = 16.6, p < 0.0001, N = 8–13 flies per group). (B) Representative images of flies at eclosion (P0). Scale bar = 100 μm. (C) Representative images of heads at 4 weeks of age (P28) showing aggregation. Yellow boxes show inset displayed below of brains of Elav(II) > GFP flies (i) without aggregates and Elav(II) > Htt-PQ72 flies (ii) with aggregates. Scale bar = 100 μm. (D) Quantification of aggregates in the brain show that PQ-GFP aggregates only in the brains of Elav(X) > Htt-PQ72 flies with age starting by 1 week of age (ANOVA for all, P0: p = 1.0 because no variability, N = 7–9 flies per group; P7: F(3,29) = 35.5, p < 0.0001, N = 8–9 flies per group; P14: F(3,25) = 107.5, p < 0.0001, N = 6–8 flies per group; P21: F(3,40) = 37.7, p < 0.0001, N = 6–14 flies per group). Note the change in y-axis between P7 and P14. (E) Representative images of heads at eclosion (P0). Scale bar = 100 μm. (F) Representative images of heads at 3 weeks of age (P21) showing aggregation. Yellow boxes show inset displayed below of brains of Elav(X) > GFP flies (i) without aggregates and Elav(X) > Htt-PQ72 flies (ii) with aggregates. Note the higher levels of aggregation when driven by Elav(X) compared to Elav(II). All: Data displayed as mean ± SEM. Dunnett’s post hoc. *p < 0.05 and ****p < 0.0001 compared to GFP. Red arrows point to PQ-GFP aggregates in the insets.
Synaptic dysfunction is a common early feature in neurodegeneration, especially HD (Smith-Dijak et al., 2019), and synapse loss often precedes neuronal loss (Milnerwood and Raymond, 2010). Thus, we used immunohistochemistry to measure synapsin levels to determine whether Htt-PQ72 could induce changes at the synapse that are consistent with synaptic dysfunction. Synapsin is critical for neurotransmitter vesicle release (Cesca et al., 2010) and binds mutant huntingtin in the presynaptic compartment to inhibit its phosphorylation (Xu et al., 2013), which predicts dysfunction in HD models (Liévens et al., 2002). We found that at 3 weeks of age, Elav(X) > Htt-PQ72 flies had reduced synapsin levels in the brain relative to GFP, while Elav(X) > Htt-PQ25 flies did not (Figures 2A,C). However, we did not see a significant change in synapsin staining in any flies using the Elav(II) driver at 4 weeks of age (Figures 2B,D). To determine whether synapsin loss occurs with PQ-GFP aggregation or after significant aggregation, we stained for synapsin in P7 Elav(X) flies, when PQ-GFP aggregation has already occurred (Figure 1A), and found that there was no difference in synapsin staining (Figures 2E,F). Thus, PQ-GFP aggregation precedes synapsin loss in the brain of Elav(X) > Htt-PQ72 flies, which occurs with age.
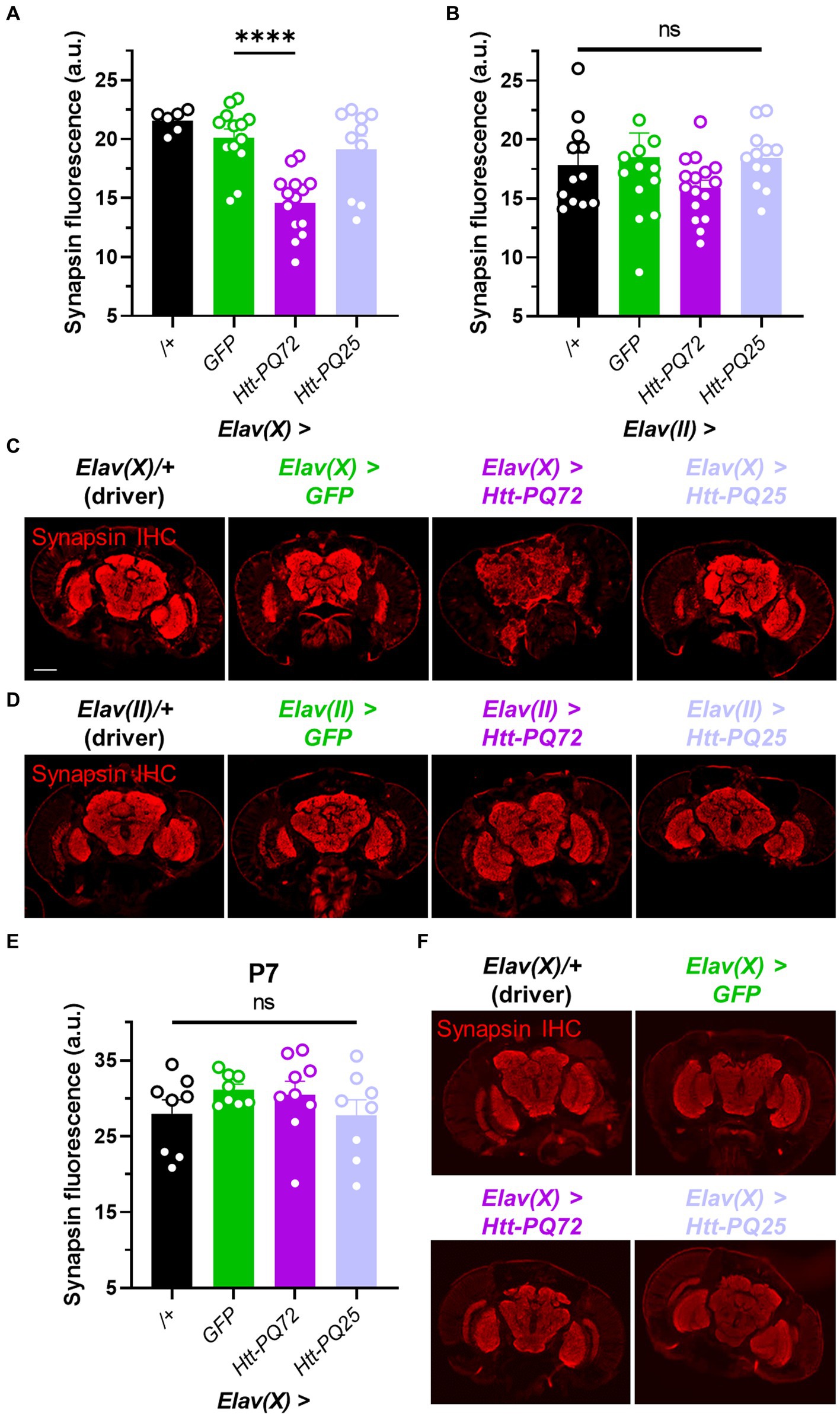
Figure 2. Neuronal Htt-PQ72 can induce synapsin loss in the brain. (A) Quantification of synapsin staining shows a decrease of synapsin in Elav(X) > Htt-PQ72 flies at P21 (ANOVA F(3,39) = 13.6, p < 0.0001, Dunnett’s post hoc ****p < 0.0001 compared to GFP, N = 6–14 flies per group). (B) Quantification of synapsin staining shows no differences in synapsin levels in the brains of flies driven by Elav(II) at P28 (ANOVA F(3,49) = 1.1, p = 0.37, N = 12–16 flies per group). (C) Representative immunofluorescent images stained for presynaptic marker synapsin in the brain of P21 flies driven by Elav(X). Scale bar = 100 μm. (D) Representative images of P28 Elav(II) fly heads stained for synapsin. (E) Quantification of synapsin staining shows no difference between any groups driven by Elav(X) at P7, when aggregates start to form (ANOVA F(3,29) = 1.0, p = 0.39, N = 8–9 flies per group). (F) Representative images of P7 Elav(X) fly heads stained for synapsin. All: Data displayed as mean ± SEM.
HD is characterized by muscle dysfunction, and we have previously found that Htt-PQ72 causes dysfunction in cardiac and skeletal muscle (Melkani et al., 2013; Barwell et al., 2023). It has been proposed that this peripheral muscle dysfunction is primarily caused by peripheral expression of mutant huntingtin, and that peripheral mutant huntingtin could cause neuronal dysfunction in the brain (Chuang and Demontis, 2021). We wanted to complement this idea and test the converse – whether neuronal huntingtin can disrupt locomotor performance independent from expression in the muscle. To do so, we conducted two assays to measure the performance of two types of skeletal muscle: negative geotaxis assay, which measures climbing muscle performance, and flight index assay, which measures flight muscle performance (Villanueva et al., 2019). We chose to use the Elav(II) driver to allow for observing more subtle phenotypes induced by lower aggregation or expression of constructs. While there were no significant changes in climbing muscle performance at P7, before PQ-GFP aggregation, or at P14, when aggregation is beginning (Figure 1A), Elav(II) > Htt-PQ72 flies displayed progressive dysfunction in climbing muscle performance starting at P21, beyond the loss of muscle performance associated with normal aging (Figure 3A). There was also some climbing muscle dysfunction in Elav(II) > Htt-PQ25 flies at P28, which is not surprising since some dysfunction has been reported by overexpression of shorter PQ chains with aging in vivo (Morley et al., 2002). To determine if neuronal mutant huntingtin causes general muscle performance loss, we performed a flight index assay to measure flight muscle performance. To our surprise, we found no change in flight muscle performance at P28 (Figure 3B). As it is possible that higher PQ-GFP in neurons could decrease flight muscle performance, we repeated the flight index assay using the Elav(X) driver. Again, we observed no change in flight muscle performance at 3 weeks of age (Figure 3C). Together, these results show that neuronal Htt-PQ72 can decrease climbing muscle performance in an age-dependent manner without decreasing flight muscle performance.
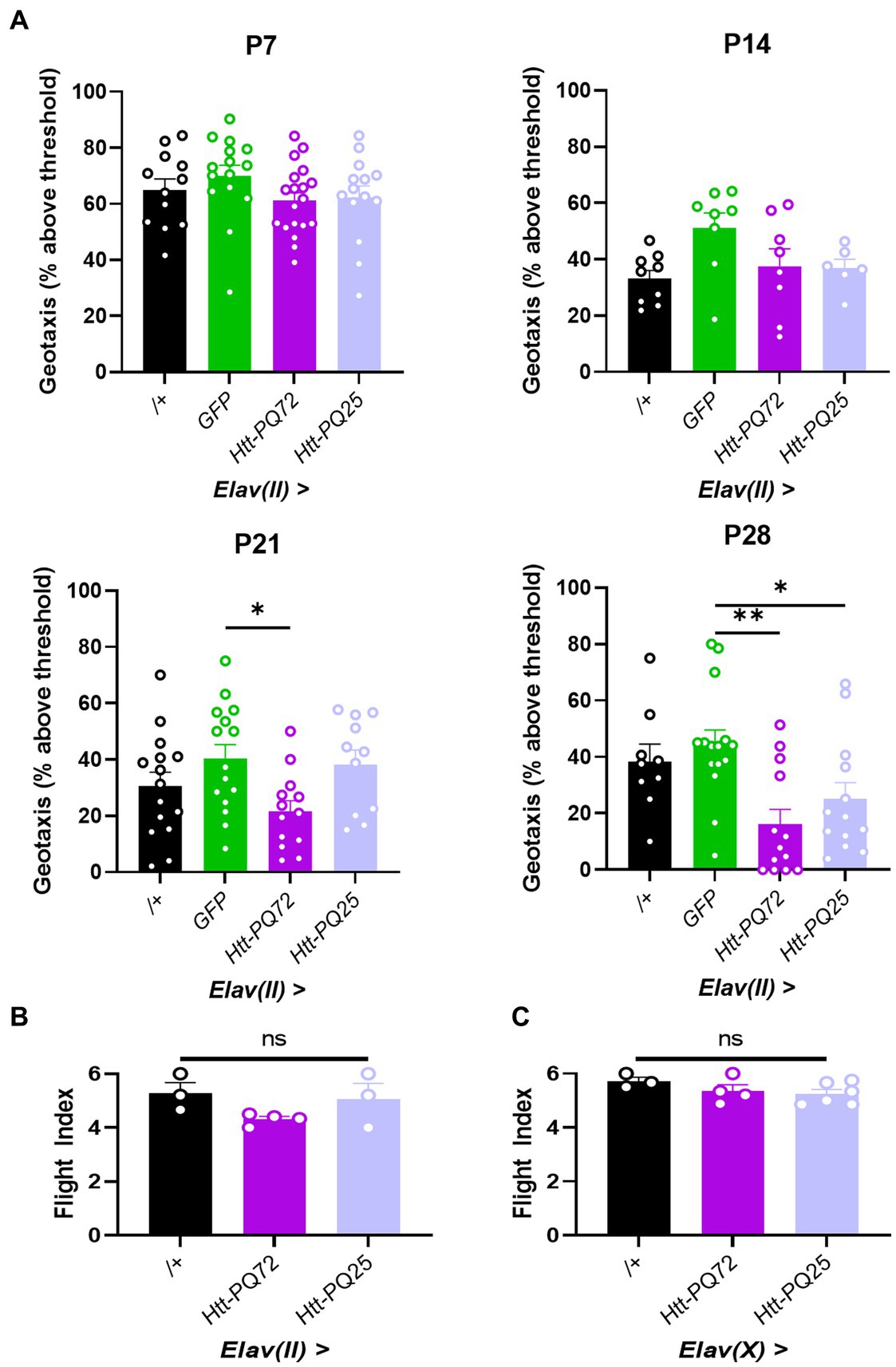
Figure 3. Neuronal Htt-PQ72 decreases climbing muscle performance with age. (A) Negative geotaxis assay demonstrates progressive loss of climbing muscle performance in Elav(II) > Htt-PQ72 flies beginning at P21 (ANOVA for all, P7: F(3,56) = 1.2, p = 0.31, n = 12–19 cohorts per group; P14: F(3,27) = 2.9, p = 0.06, n = 6–9 cohorts per group; P21: F(3,50) = 3.2, p = 0.03, n = 11–15 cohorts per group; P28: F(3,46) = 5.6, p = 0.002, n = 9–15 cohorts per group. All: Dunnett’s post hoc *p < 0.05 and **p < 0.01 compared to GFP). (B) Flight index assay demonstrates no significant difference in flight muscle performance in Elav(II)-driven flies at P28 (ANOVA F(2,7) = 2.2, p = 0.19, n = 3–4 cohorts with N = 14–19 flies per group). (C) Flight index assay demonstrates no significant difference in flight muscle performance in Elav(X)-driven flies at P21 (ANOVA F(2,10) = 1.5, p = 0.29, n = 3–5 cohorts with N = 11–35 flies per group). All: Data displayed as mean ± SEM.
Autophagy plays an important role in aging and metabolism, and has been implicated in many neurodegenerative diseases including HD (Croce and Yamamoto, 2019). Rapamycin stimulates autophagy and prevents some mutant huntingtin-induced dysfunction (Djajadikerta et al., 2020), so we next tested whether it impacted PQ-GFP aggregation in the brain. We placed 1 week-old flies on food supplemented with 200 μM rapamycin or vehicle control food for 3 weeks. First, we verified the mechanism of action of rapamycin by isolating heads and conducting PCR for genes associated with autophagy and TOR signaling (Figure 4A). We found that rapamycin significantly increases expression of Atg1, which is responsible for the formation of autophagosomes (Mizushima, 2010; Stjepanovic et al., 2014). Additionally, we measured expression of Hid, which can induce autophagy (Baehrecke, 2003; Juhász and Sass, 2005), and pten, which is negatively associated with TOR activity (Manning and Cantley, 2007; Das et al., 2012), and found them increased by rapamycin, demonstrating that rapamycin treatment led to inhibition of TOR as expected. As an independent measure of TOR activity, we measured ilp5 expression, which is positively regulated by TOR and is important for nutrient signaling and metabolism (Texada et al., 2022; Ling and Raikhel, 2023). Rapamycin significantly decreased ilp5, again demonstrating that rapamycin inhibited TOR signaling (Figure 4B). Next, we determined whether rapamycin prevented PQ-GFP aggregation and other mHTT-associated dysfunction. We found that rapamycin reduced PQ-GFP aggregation, though it did not prevent a loss of synapsin in aged Elav(X) flies (Figures 4C–E). Next, we determined whether rapamycin was sufficient to prevent PQ-GFP aggregation and locomotor performance loss in Elav(II) flies. We found that rapamycin again reduced PQ-GFP aggregation and also prevented a loss of locomotor performance in Elav(II) > Htt-PQ72 flies (Figures 4F–H). There was a decrease in climbing performance in the control flies relative to prior experiments (Figure 3), likely due to chronic ethanol exposure (Chvilicek et al., 2020; Nuñez et al., 2023) as ethanol was used to dissolve the rapamycin and was included in the control food as a vehicle. Altogether, these results show that rapamycin is sufficient to prevent aggregation of mutant huntingtin in the brains of flies and can prevent the loss of locomotor performance.
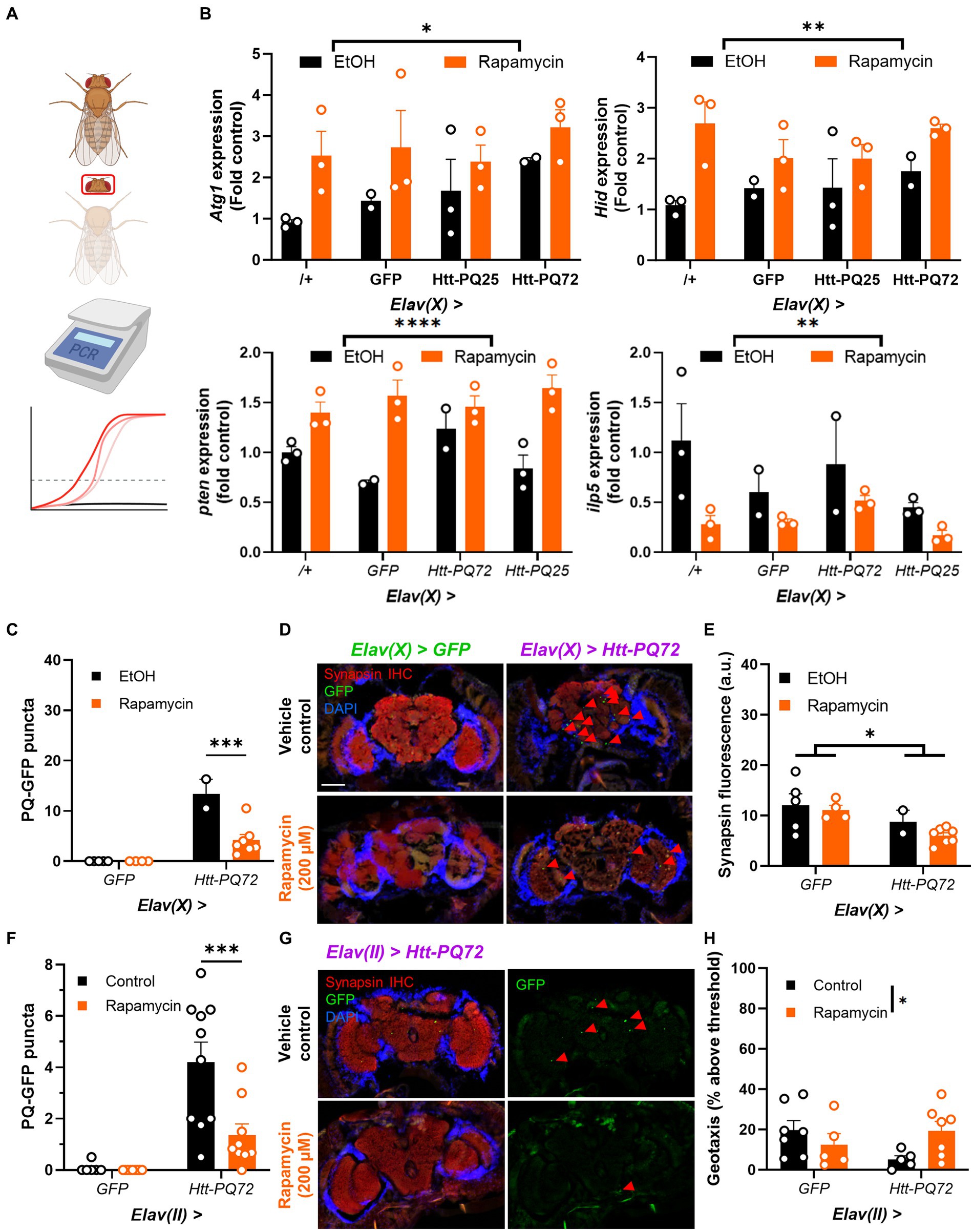
Figure 4. Rapamycin reduces Htt-PQ72 aggregation in the brain and ameliorates flight muscle performance. (A) qPCR workflow. Fly heads were isolated for RNA extraction and qPCR was used to compare transcript levels between groups. This image was created in Biorender. (B) Rapamycin increases Atg1 expression (F(1,14) = 7.218, p = 0.0173), Hid expression (F(1,14) = 13.25, p = 0.0027), and pten expression (2-way ANOVA main effect of rapamycin F(1, 14) = 39.7, p < 0.0001); and decreases ilp5 expression (2-way ANOVA main effect of rapamycin F(1, 14) = 10.0, p = 0.007). N = 2–3 cohorts with n = about 10 flies per group per cohort of fly heads. (C) Quantification of PQ-GFP aggregates in Elav(X)-driven flies treated with 200 μM rapamycin or vehicle control for 3 weeks. Rapamycin decreases aggregation in Elav(X) > Htt-PQ72 fly brains (2-way ANOVA interaction F(1,14) = 15.26, p = 0.002, Dunnett’s post hoc ***p < 0.001 compared to vehicle control, N = 2–7 flies per group). (D) Representative fluorescent images of P28 Elav(X)-driven GFP and Htt-PQ72 flies with vehicle control or Rapamycin for 3 weeks. Scale bar = 100 μm. (E) Quantification of synapsin staining shows a decrease of synapsin in Elav(X) > Htt-PQ72 flies at with no effect of rapamycin (2-way ANOVA main effect of genotype F(1,14) = 6.4, p = 0.02, N = 2–7 flies per group). (F) Quantification of PQ-GFP aggregates in Elav(II)-driven flies treated with 200 μM rapamycin or vehicle control for 3 weeks. Rapamycin decreases aggregation in Elav(II) > Htt-PQ72 fly brains (2-way ANOVA interaction F(1,30) = 6.9, p = 0.014, Dunnett’s post hoc ***p < 0.001 compared to vehicle control, N = 6–10 flies per group). (G) Representative fluorescent images of P28 Elav(II) > Htt-PQ72 flies treated with vehicle control or rapamycin for 3 weeks. (H) Rapamycin modifies the loss of climbing performance in Elav(II) > Htt-PQ72 flies at P28 (2-way ANOVA interaction F(1,20) = 5.2, p = 0.03, n = 5–7 cohorts with N = 34–73 flies per group).
In this work, we describe the effect of expressing Htt-PQ72 in Drosophila neurons. We characterized age-dependent aggregation of PQ-GFP that was greater when driven by Elav(X) than Elav(II), and that it can reduce synapsin in the brain after PQ-GFP aggregation occurs. We next explored the role of neuronal Htt-PQ72 in walking muscle performance and found that it causes age-dependent loss of climbing muscle performance but no effect on flight muscle performance. Finally, we found that rapamycin reduces aggregation of PQ-GFP in the brain and prevents the loss of locomotor performance, supporting the importance of autophagy in HD models and that rapamycin could be beneficial to prevent mutant huntingtin–induced dysfunction.
Brain-periphery crosstalk is likely important in many neurodegenerative diseases, especially HD. Huntingtin is expressed in many tissues throughout the body and mutant huntingtin aggregates have been found in multiple tissues (Van Der Burg et al., 2009). Most preclinical work in HD has studied the role of neuronal huntingtin in the brain and peripheral huntingtin in periphery, though new studies in brain-periphery crosstalk have demonstrated that the periphery can affect the brain, especially skeletal muscles (Ehlen et al., 2017; Matthews et al., 2022). Our results complement these studies and add to the evidence supporting the importance of brain-periphery crosstalk, as neuronal mutant huntingtin is sufficient to reduce locomotor performance without expression of mutant huntingtin in the muscles themselves.
We found it interesting that Htt-PQ72 caused a loss of climbing muscle performance but not flight muscle performance. This suggests that there are specific effects of neuronal mutant huntingtin on specific peripheral tissues, and highlights the importance of studying multiple types of skeletal muscle in models of movement disorders like HD. One potential caveat to these results is that the negative geotaxis assay is dependent on neuronal function (Sun et al., 2018), so it is possible that the climbing muscles themselves were not disrupted, per se, but were impacted by neuronal dysfunction caused by Htt-PQ72. This is supported by the lack of phenotype in the flight muscle performance assay, which is not primarily driven by neuronal function. Additionally, Elav is expressed in peripheral neurons in addition to central nervous system neurons (Robinow and White, 1988), so it is possible that the locomotor dysfunction is due to peripheral neuron dysfunction outside of the brain. Alternatively, it is possible that mHTT toxicity in areas of the brain that control movement could contribute to the locomotor dysfunction, as is hypothesized to occur in humans (Smith et al., 2000). Altogether, it is notable that neuronal mutant huntingtin is sufficient to specifically reduce skeletal muscle performance, likely due to neuronal dysfunction, especially with the prominence of locomotor dysfunction present in HD.
Another potential caveat to our results is the differences seen with using the Elav(II) and Elav(X) drivers, especially regarding the synapsin loss (Figure 2). Two potential explanations for the differences are that they are due to either differences in expression location or different expression levels. Both driver lines are well-established models to express constructs in mature neurons that endogenously express Elav, and there is typically minimal leakage with the UAS-Gal4 system (Osterwalder et al., 2001). Our results were consistent with differences being due to higher expression with the Elav(X) driver, namely the early mortality, higher level of aggregation at an earlier age, and the reduced synapsin. It is notable that PQ-GFP aggregation in P7 Elav(X) > Htt-PQ72 flies and P28 Elav(II) > Htt-PQ72 flies (1.78 vs. 3.36 aggregates per brain section) was closer than differences in aggregation between P21 Elav(X) > Htt-PQ72 flies and P28 Elav(II) > Htt-PQ72 flies (21.57 vs. 3.36 aggregates per brain section), so we speculate that it is possible that a reduction of synapsin levels would occur in the Elav(II) > Htt-PQ72 flies with more age and PQ-GFP aggregation. Thus, we expect that the differences were due to Elav(X) driving higher expression than Elav(II), though we cannot exclude the possibility of different cell-specific expression of the drivers or Gal4 leakage.
The difference between models is also seen in effects of rapamycin, which reduces PQ-GFP aggregation in both models and prevents the loss of locomotor performance in Elav(II) > Htt-PQ72 flies, but does not prevent the loss of synapsin in Elav(X) > Htt-PQ72 flies (Figure 4). There are several possible explanations for this. It is possible that the more severe model (Elav(X)) had too much mHTT-associated toxicity for this dose of rapamycin to prevent synapsin loss, that treating with rapamycin after aggregation occurred was insufficient to prevent it, or that synapsin loss is a downstream effect of mHTT that is independent of its effects of aggregation and locomotor performance. We speculate that it is due to a ceiling effect such that the Elav(X) model has high enough expression that mHTT toxicity has either already occurred or that the dose of rapamycin used was not sufficient to prevent mHTT toxicity.
Both PQ-GFP aggregation and synapsin loss were spread uniformly throughout the brain. This was expected, as Elav-Gal4 drives expression pan-neuronally in all parts of the brain, so Elav > Htt-PQ72 will be expressed in all brain regions and can aggregate throughout. Additionally, since soluble mHTT can be toxic (Leitman et al., 2013), aggregation is not necessary for downstream dysfunction, as is seen with synapsin loss (Figure 2). Altogether, our results support the idea that high levels of non-aggregated mHTT can cause neuronal dysfunction in multiple neuron subtypes throughout the brain, namely a decrease in synapsin.
Currently, there are no highly effective treatments for HD. Rapamycin has been proposed as a potential therapeutic strategy because mutant huntingtin impairs autophagy, leading to neuronal dysfunction (Pircs et al., 2018). Multiple preclinical studies have demonstrated that rapamycin can delay aging (Harrison et al., 2009; Bjedov et al., 2010; Robida-Stubbs et al., 2012), even after just brief treatment during adulthood (Juricic et al., 2022), and that it prevents mutant huntingtin-induced dysfunction (Ravikumar et al., 2004; Sarkar et al., 2008). Our data support this approach as we found that rapamycin reduces mutant huntingtin–induced dysfunction in the brain and periphery. It is important to note that rapamycin can inhibit huntingtin aggregation independent of autophagy (King et al., 2008), and that aggregation may in fact be protective by reducing toxic, soluble forms of mutant huntingtin (Arrasate et al., 2004). Thus, it is important that we found that rapamycin prevented reduced behavioral dysfunction, especially progressive muscle performance dysfunction caused by neuronal mutant huntingtin. HD is rare within neurodegenerative diseases in that it is autosomal dominant and monogenic (Myers, 2004). Thus, it could be possible to identify individuals who will develop HD and treat them before symptom onset if rapamycin is found to prevent development of motor dysfunction in HD. Interestingly, HD is part of a class of neurodegenerative diseases that are characterized by PQ repeat expansions, including several ataxias (Shao and Diamond, 2007), so we speculate that rapamycin could be beneficial in these diseases if there are common mechanisms underlying PQ expansion disorders.
In summary, this study explores the role of mutant huntingtin in neurons. It shows that expressing mutant huntingtin in Drosophila neurons causes an age-dependent increase in mutant huntingtin aggregation in the brain, which can be associated with synapsin loss. Additionally, it supports the importance of brain-muscle crosstalk in HD models, as we found age-dependent loss of climbing muscle performance caused by mutant huntingtin in neurons. Finally, this study adds to the body of work showing the importance of autophagy in HD and the potential therapeutic benefits of rapamycin in HD models.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The manuscript presents research on animals that do not require ethical approval for their study.
JRR, RCM, and GCM contributed to the initial study and experimental design. JRR, RCM, BPX, SRC, and MAK conducted experiments, analysed, and interpreted results. JRR wrote the manuscript with input from RCM, BPX, SRC, MAK, and GCM. All authors contributed to the article and approved the submitted version.
This work was supported by National Institutes of Health (NIH) grants AG065992 and AG068550, and UAB Startup funds 3123226 and 3123227 to GCM.
We would like to thank members of the Melkani lab for input on the project and for technical help with fly stocks. We thank Perrimon and the Bloomington Drosophila Stock Center for the fly stocks.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2023.1223911/full#supplementary-material
Ali, Y. O., Escala, W., Ruan, K., and Zhai, R. G., (2011). Assaying locomotor, learning, and memory deficits in Drosophila models of neurodegeneration. J. Vis. Exp., 11, 2504, doi: 10.3791/2504
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Baehrecke, E. H. (2003). Autophagic programmed cell death in Drosophila. Cell Death Diff. 10, 940–945. doi: 10.1038/sj.cdd.4401280
Bailus, B. J., Scheeler, S. M., Simons, J., Sanchez, M. A., Tshilenge, K.-T., Creus-Muncunill, J., et al. (2021). Modulating Fkbp5/Fkbp51 and autophagy lowers Htt (huntingtin) levels. Autophagy 17, 4119–4140. doi: 10.1080/15548627.2021.1904489
Barwell, T., Raina, S., Page, A., Maccharles, H., and Seroude, L. (2023). Juvenile and adult expression of polyglutamine expanded huntingtin produce distinct aggregate distributions in Drosophila muscle. Hum. Mol. Genet. 32, 2656–2668. doi: 10.1093/hmg/ddad098
Berger, Z., Ravikumar, B., Menzies, F. M., Oroz, L. G., Underwood, B. R., Pangalos, M. N., et al. (2006). Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 15, 433–442. doi: 10.1093/hmg/ddi458
Bjedov, I., Toivonen, J. M., Kerr, F., Slack, C., Jacobson, J., Foley, A., et al. (2010). Mechanisms of life span extension by rapamycin in the fruit Fly Drosophila Melanogaster. Cell Metab. 11, 35–46. doi: 10.1016/j.cmet.2009.11.010
Busse, M. E., Hughes, G., Wiles, C. M., and Rosser, A. E. (2008). Use of hand-held dynamometry in the evaluation of lower limb muscle strength in people with Huntington’s disease. J. Neurol. 255, 1534–1540. doi: 10.1007/s00415-008-0964-x
Cesca, F., Baldelli, P., Valtorta, F., and Benfenati, F. (2010). The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 91, 313–348. doi: 10.1016/j.pneurobio.2010.04.006
Chuang, C. L., and Demontis, F. (2021). Systemic manifestation and contribution of peripheral tissues to Huntington’s disease pathogenesis. Ageing Res. Rev. 69:101358. doi: 10.1016/j.arr.2021.101358
Chvilicek, M. M., Titos, I., and Rothenfluh, A. (2020). The neurotransmitters involved in Drosophila alcohol-induced behaviors. Front. Behav. Neurosci. 14:607700. doi: 10.3389/fnbeh.2020.607700
Cortes, C. J., and La Spada, A. R. (2014). The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov. Today 19, 963–971. doi: 10.1016/j.drudis.2014.02.014
Croce, K. R., and Yamamoto, A. (2019). A role for autophagy in Huntington’s disease. Neurobiol. Dis. 122, 16–22. doi: 10.1016/j.nbd.2018.08.010
Das, F., Ghosh-Choudhury, N., Dey, N., Mandal, C. C., Mahimainathan, L., Kasinath, B. S., et al. (2012). Unrestrained mammalian target of rapamycin complexes 1 and 2 increase expression of phosphatase and tensin homolog deleted on chromosome 10 to regulate phosphorylation of Akt kinase. J. Biol. Chem. 287, 3808–3822. doi: 10.1074/jbc.M111.246397
Dash, D., and Mestre, T. A. (2020). Therapeutic update on Huntington’s disease: symptomatic treatments and emerging disease-modifying therapies. Neurotherapeutics 17, 1645–1659. doi: 10.1007/s13311-020-00891-w
Difiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Djajadikerta, A., Keshri, S., Pavel, M., Prestil, R., Ryan, L., and Rubinsztein, D. C. (2020). Autophagy induction as a therapeutic strategy for neurodegenerative diseases. J. Mol. Biol. 432, 2799–2821. doi: 10.1016/j.jmb.2019.12.035
Drummond, D. R., Hennessey, E. S., and Sparrow, J. C. (1991). Characterisation of missense mutations in the Act88f gene of Drosophila Melanogaster. Mol. Gen. Genet. 226, 70–80. doi: 10.1007/BF00273589
Duyao, M., Ambrose, C., Myers, R., Novelletto, A., Persichetti, F., Frontali, M., et al. (1993). Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 4, 387–392. doi: 10.1038/ng0893-387
Ehlen, J. C., Brager, A. J., Baggs, J., Pinckney, L., Gray, C. L., Debruyne, J. P., et al. (2017). Bmal1 function in skeletal muscle regulates sleep. elife 6:26557. doi: 10.7554/eLife.26557
Gill, S., Le, H. D., Melkani, G. C., and Panda, S. (2015). Time-restricted feeding attenuates age-related cardiac decline in Drosophila. Science 347, 1265–1269. doi: 10.1126/science.1256682
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi: 10.1038/nature08221
Juhász, G., and Sass, M. (2005). Hid can induce, but is not required for autophagy in polyploid larval Drosophila tissues. Eur. J. Cell Biol. 84, 491–502. doi: 10.1016/j.ejcb.2004.11.010
Juricic, P., Lu, Y.-X., Leech, T., Drews, L. F., Paulitz, J., Lu, J., et al. (2022). Long-lasting Geroprotection from brief rapamycin treatment in early adulthood by persistently increased intestinal autophagy. Nat. Aging 2, 824–836. doi: 10.1038/s43587-022-00278-w
King, M. A., Hands, S., Hafiz, F., Mizushima, N., Tolkovsky, A. M., and Wyttenbach, A. (2008). Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol. Pharmacol. 73, 1052–1063. doi: 10.1124/mol.107.043398
Krench, M., and Littleton, J. T. (2013). Modeling Huntington disease in Drosophila: insights into axonal transport defects and modifiers of toxicity. Fly (Austin) 7, 229–236. doi: 10.4161/fly.26279
Lanska, D. J., Lavine, L., Lanska, M. J., and Schoenberg, B. S. (1988). Huntington’s disease mortality in the United States. Neurology 38, 769–772. doi: 10.1212/WNL.38.5.769
Lee, J.-M., Ramos, E. M., Lee, J.-H., Gillis, T., Mysore, J. S., Hayden, M. R., et al. (2012). Cag repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 78, 690–695. doi: 10.1212/WNL.0b013e318249f683
Leitman, J., Ulrich Hartl, F., and Lederkremer, G. Z. (2013). Soluble forms of Polyq-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 4:2753. doi: 10.1038/ncomms3753
Lewis, E. A., and Smith, G. A. (2016). Using Drosophila models of Huntington’s disease as a translatable tool. J. Neurosci. Methods 265, 89–98. doi: 10.1016/j.jneumeth.2015.07.026
Li, J., Kim, S. G., and Blenis, J. (2014). Rapamycin: one drug, many effects. Cell Metab. 19, 373–379. doi: 10.1016/j.cmet.2014.01.001
Liévens, J. C., Woodman, B., Mahal, A., and Bates, G. P. (2002). Abnormal phosphorylation of Synapsin I predicts a neuronal transmission impairment in the R6/2 Huntington’s disease transgenic mice. Mol. Cell. Neurosci. 20, 638–648. doi: 10.1006/mcne.2002.1152
Ling, L., and Raikhel, A. S. (2023). Amino acid-dependent regulation of insulin-like peptide signaling is mediated by Tor and Gata factors in the disease vector mosquito Aedes Aegypti. Proc. Natl. Acad. Sci. U. S. A. 120:E2303234120. doi: 10.1073/pnas.2303234120
Livelo, C., Guo, Y., Abou Daya, F., Rajasekaran, V., Varshney, S., Le, H. D., et al. (2023). Time-restricted feeding promotes muscle function through purine cycle and Ampk signaling in Drosophila obesity models. Nat. Commun. 14:949. doi: 10.1038/s41467-023-36474-4
Manning, B. D., and Cantley, L. C. (2007). Akt/Pkb signaling: navigating downstream. Cells 129, 1261–1274. doi: 10.1016/j.cell.2007.06.009
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 13, 567–576. doi: 10.1038/nn.2528
Matthews, I., Birnbaum, A., Gromova, A., Coutinho, K., Mcgraw, M., Patterson, D., et al. (2022, 2022). Skeletal muscle proteostasis promotes central nervous system rejuvenation and reduces neuroinflammation during aging and neurodegenerative disease. Biorxiv 7:482891. doi: 10.1101/2022.03.07.482891
Melkani, G. C., Trujillo, A. S., Ramos, R., Bodmer, R., Bernstein, S. I., and Ocorr, K. (2013). Huntington’s disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genet. 9:E1004024. doi: 10.1371/journal.pgen.1004024
Mihm, M. J., Amann, D. M., Schanbacher, B. L., Altschuld, R. A., Bauer, J. A., and Hoyt, K. R. (2007). Cardiac dysfunction in the R6/2 mouse model of Huntington's disease. Neurobiol. Dis. 25, 297–308. doi: 10.1016/j.nbd.2006.09.016
Milnerwood, A. J., and Raymond, L. A. (2010). Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 33, 513–523. doi: 10.1016/j.tins.2010.08.002
Mizushima, N. (2010). The role of the Atg1/Ulk1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139. doi: 10.1016/j.ceb.2009.12.004
Morley, J. F., Brignull, H. R., Weyers, J. J., and Morimoto, R. I. (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis Elegans. Proc. Natl. Acad. Sci. U. S. A. 99, 10417–10422. doi: 10.1073/pnas.152161099
Myers, R. H. (2004). Huntington’s disease genetics. NeuroRx 1, 255–262. doi: 10.1602/neurorx.1.2.255
Nuñez, K. M., Catalano, J. L., Scaplen, K. M., and Kaun, K. R. (2023). Ethanol behavioral responses in Drosophila. Cold Spring Harb. Protoc. doi: 10.1101/pdb.top107887
Osterwalder, T., Yoon, K. S., White, B. H., and Keshishian, H. (2001). A conditional tissue-specific transgene expression system using inducible Gal4. Proc. Natl. Acad. Sci. U. S. A. 98, 12596–12601. doi: 10.1073/pnas.221303298
Pircs, K., Petri, R., Madsen, S., Brattås, P. L., Vuono, R., Ottosson, D. R., et al. (2018). Huntingtin aggregation impairs autophagy, leading to Argonaute-2 accumulation and global microrna dysregulation. Cell Rep. 24, 1397–1406. doi: 10.1016/j.celrep.2018.07.017
Raught, B., Gingras, A. C., and Sonenberg, N. (2001). The target of rapamycin (Tor) proteins. Proc. Natl. Acad. Sci. U. S. A. 98, 7037–7044. doi: 10.1073/pnas.121145898
Ravikumar, B., Duden, R., and Rubinsztein, D. C. (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117. doi: 10.1093/hmg/11.9.1107
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of MTor induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Robida-Stubbs, S., Glover-Cutter, K., Lamming, D. W., Mizunuma, M., Narasimhan, S. D., Neumann-Haefelin, E., et al. (2012). Tor signaling and rapamycin influence longevity by regulating Skn-1/Nrf and Daf-16/Foxo. Cell Metab. 15, 713–724. doi: 10.1016/j.cmet.2012.04.007
Robinow, S., and White, K. (1988). The locus Elav of Drosophila Melanogaster is expressed in neurons at all developmental stages. Dev. Biol. 126, 294–303. doi: 10.1016/0012-1606(88)90139-X
Rubinsztein, D. C., Gestwicki, J. E., Murphy, L. O., and Klionsky, D. J. (2007). Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 6, 304–312. doi: 10.1038/nrd2272
Rush, T., Roth, J. R., Thompson, S. J., Aldaher, A. R., Cochran, J. N., and Roberson, E. D. (2020). A peptide inhibitor of tau-Sh3 interactions ameliorates amyloid-Β toxicity. Neurobiol. Dis. 134:104668. doi: 10.1016/j.nbd.2019.104668
Sarkar, S., Krishna, G., Imarisio, S., Saiki, S., O’kane, C. J., and Rubinsztein, D. C. (2008). A rational mechanism for combination treatment of Huntington’s disease using Lithium and rapamycin. Hum. Mol. Genet. 17, 170–178. doi: 10.1093/hmg/ddm294
Sathasivam, K., Hobbs, C., Turmaine, M., Mangiarini, L., Mahal, A., Bertaux, F., et al. (1999). Formation of polyglutamine inclusions in non-Cns tissue. Hum. Mol. Genet. 8, 813–822. doi: 10.1093/hmg/8.5.813
Schulte, J., and Littleton, J. T. (2011). The biological function of the huntingtin protein and its relevance to Huntington’s disease pathology. Curr Trends Neurol 5, 65–78.
Shao, J., and Diamond, M. I. (2007). Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 16, R115–R123. doi: 10.1093/hmg/ddm213
Smith, M. A., Brandt, J., and Shadmehr, R. (2000). Motor disorder in Huntington’s disease begins as a dysfunction in error feedback control. Nature 403, 544–549. doi: 10.1038/35000576
Smith-Dijak, A. I., Sepers, M. D., and Raymond, L. A. (2019). Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 150, 346–365. doi: 10.1111/jnc.14723
Stjepanovic, G., Davies, C. W., Stanley, R. E., Ragusa, M. J., Kim, D. J., and Hurley, J. H. (2014). Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc. Natl. Acad. Sci. 111, 12793–12798. doi: 10.1073/pnas.1407214111
Strong, T. V., Tagle, D. A., Valdes, J. M., Elmer, L. W., Boehm, K., Swaroop, M., et al. (1993). Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet. 5, 259–265. doi: 10.1038/ng1193-259
Sun, J., Xu, A. Q., Giraud, J., Poppinga, H., Riemensperger, T., Fiala, A., et al. (2018). Neural control of startle-induced locomotion by the mushroom bodies and associated neurons in Drosophila. Front. Syst. Neurosci. 12:e00006. doi: 10.3389/fnsys.2018.00006
Texada, M. J., Lassen, M., Pedersen, L. H., Koyama, T., Malita, A., and Rewitz, K. (2022). Insulin signaling couples growth and early maturation to cholesterol intake in Drosophila. Curr. Biol. 32, 1548–1562. doi: 10.1016/j.cub.2022.02.021
Tyedmers, J., Mogk, A., and Bukau, B. (2010). Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788. doi: 10.1038/nrm2993
Van Der Burg, J. M. M., Björkqvist, M., and Brundin, P. (2009). Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 8, 765–774. doi: 10.1016/S1474-4422(09)70178-4
Villanueva, J. E., Livelo, C., Trujillo, A. S., Chandran, S., Woodworth, B., Andrade, L., et al. (2019). Time-restricted feeding restores muscle function in Drosophila models of obesity and circadian-rhythm disruption. Nat. Commun. 10:2700. doi: 10.1038/s41467-019-10563-9
Voskobiynyk, Y., Roth, J. R., Cochran, J. N., Rush, T., Carullo, N. V., Mesina, J. S., et al. (2020). Alzheimer's disease risk gene Bin1 induces tau-dependent network hyperexcitability. elife 9:57354. doi: 10.7554/eLife.57354
Wong, Y. C., and Holzbaur, E. L. F. (2014). The regulation of autophagosome dynamics by huntingtin and Hap1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 34, 1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014
Xu, Q., Huang, S., Song, M., Wang, C. E., Yan, S., Liu, X., et al. (2013). Synaptic mutant huntingtin inhibits synapsin-1 phosphorylation and causes neurological symptoms. J. Cell Biol. 202, 1123–1138. doi: 10.1083/jcb.201303146
Zhang, S., Binari, R., Zhou, R., and Perrimon, N. (2010). A genomewide Rna interference screen for modifiers of aggregates formation by mutant huntingtin in Drosophila. Genetics 184, 1165–1179. doi: 10.1534/genetics.109.112516
Zhang, H., Stallock, J. P., Ng, J. C., Reinhard, C., and Neufeld, T. P. (2000). Regulation of cellular growth by the Drosophila target of rapamycin Dtor. Genes Dev. 14, 2712–2724. doi: 10.1101/gad.835000
Keywords: Huntington’s disease, aggregation, Htt-polyQ, rapamycin, locomotion, neurodegeneration, Drosophila
Citation: Roth JR, Moraes RCMd, Xu BP, Crawley SR, Khan MA and Melkani GC (2023) Rapamycin reduces neuronal mutant huntingtin aggregation and ameliorates locomotor performance in Drosophila. Front. Aging Neurosci. 15:1223911. doi: 10.3389/fnagi.2023.1223911
Edited by:
Bogdan O. Popescu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Xiaolan Fan, Cheng Du, ChinaCopyright © 2023 Roth, Moraes, Xu, Crawley, Khan and Melkani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Girish C. Melkani, Z2lyaXNobWVsa2FuaUB1YWJtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.