 Jiajie Li1
Jiajie Li1 Yi Li
Yi Li Gang Chen
Gang Chen Jingying Pan
Jingying Pan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 06 July 2023
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1190563
Pain is common and frequent in many neurodegenerative diseases, although it has not received much attention. In Huntington’s disease (HD), pain is often ignored and under-researched because attention is more focused on motor and cognitive decline than psychiatric symptoms. In HD progression, pain symptoms are complex and involved in multiple etiologies, particularly mental issues such as apathy, anxiety and irritability. Because of psychiatric issues, HD patients rarely complain of pain, although their bodies show severe pain symptoms, ultimately resulting in insufficient awareness and lack of research. In HD, few studies have focused on pain and pain-related features. A detailed and systemic pain history is crucial to assess and explore pain pathophysiology in HD. This review provides an overview concentrating on pain-related factors in HD, including neuropathology, frequency, features, affecting factors and mechanisms. More attention and studies are still needed in this interesting field in the future.
Neurodegenerative diseases are critical problems that seriously affect the social economy and patients’ quality of life. Huntington’s disease (HD) is a genetic neurodegenerative disease represented by progressive dysfunction, including motor damage and psychiatric impairments (Dale and van Duijn, 2015; Maiuri et al., 2019), and has a 50% risk of being inherited by children (McColgan and Tabrizi, 2018). The average lifespan of HD is approximately 20 years from initial symptoms to death. To date, the main studies and treatments of HD focus on motor and cognitive problems, thereby ignoring pain issues, even though some patients complain about painful indications.
Pain is an unpleasant feeling or emotional injury, often identified through oral description, questionnaires or pain evaluation (de Tommaso et al., 2016a; Buhmann et al., 2020). Pain is a troublesome problem and has been extensively studied in Alzheimer’s disease (AD), Parkinson’s disease (PD) and motor neuron diseases (MND), with prevalence of 38–75%, 68–95% and 19–85%, respectively (de Tommaso et al., 2016a). Surprisingly, few articles about pain in HD have been reported, as pain is not considered a major theme because HD patients prefer to consult about motor issues rather than sensory problems. It is well-known that pain occurs in patients with normal sensory systems once they receive direct or indirect damage (Yam et al., 2018; Tsuda, 2021). Because of neurodegeneration, HD patients show sensory dysfunctions and abnormal pain behaviors that are rarely mentioned (de Tommaso et al., 2016a). In addition, chorea and cognitive symptoms deteriorate significantly as HD progresses, and patients with mental problems, such as issues with memory, emotion, and social cognition, find it difficult to describe their painful suffering, resulting in pain issues being greatly ignored and overlooked (Snowden, 2017; Achterberg et al., 2021).
This review intends to discuss research on pain in HD, including neuropathology, prevalence, symptoms, impacting factors and possible mechanisms. Overall, more studies and explorations of pain in HD are urgently required in the future.
It is difficult to obtain an exact prevalence estimation because HD is a rare disease with a great diversity of influencing factors. Because of geographical differences, epidemiological investigations often show under- or over-assessment of the total population (Snowden, 2017). Present data indicate that the average HD prevalence is approximately 5–10/100,000 in the UK but much higher in Scotland and England (Evans et al., 2013). The highest reported in North America is 13.7/100,000 (Fisher and Hayden, 2014; McColgan and Tabrizi, 2018). HD prevalence is higher in Europe than in Asia, at approximately 0.1–0.7/100,000 (Pringsheim et al., 2012; Sipila et al., 2015; Xu and Wu, 2015). In addition, lower prevalence is observed in black species rather than white and mixed species (McColgan and Tabrizi, 2018). The prevalence difference between ethnic groups might be closely associated with different HTT genotypes (Xu and Wu, 2015; Baig et al., 2016).
Huntingtin protein (Htt, 350kD) is abundantly expressed in the central nervous system, and its mutation can trigger HD. In general, there are approximately 17–20 CAG repeats in the Htt gene on chromosome 4 (Wyant et al., 2017). Abnormal CAG length is considered to be responsible for genic mutation and HD occurrence. Due to CAG expansion, the causative gene is considered to have neuronal toxicity and cause HD disease (McColgan and Tabrizi, 2018). There is a great correlation between CGA length, morbidity and progression: longer CGA repeats are usually linked with earlier onset and a higher rate. HD cases were reported to have more than 35 repeats, and people with a CAG size of 30–35 are deemed to gain little HD (Rubinsztein et al., 1996). Nevertheless, mutant gene carriers with more than 39 CAG repeats are predicted to develop disease and exhibit symptoms within several years (Rubinsztein et al., 1996; McColgan and Tabrizi, 2018; Figure 1).
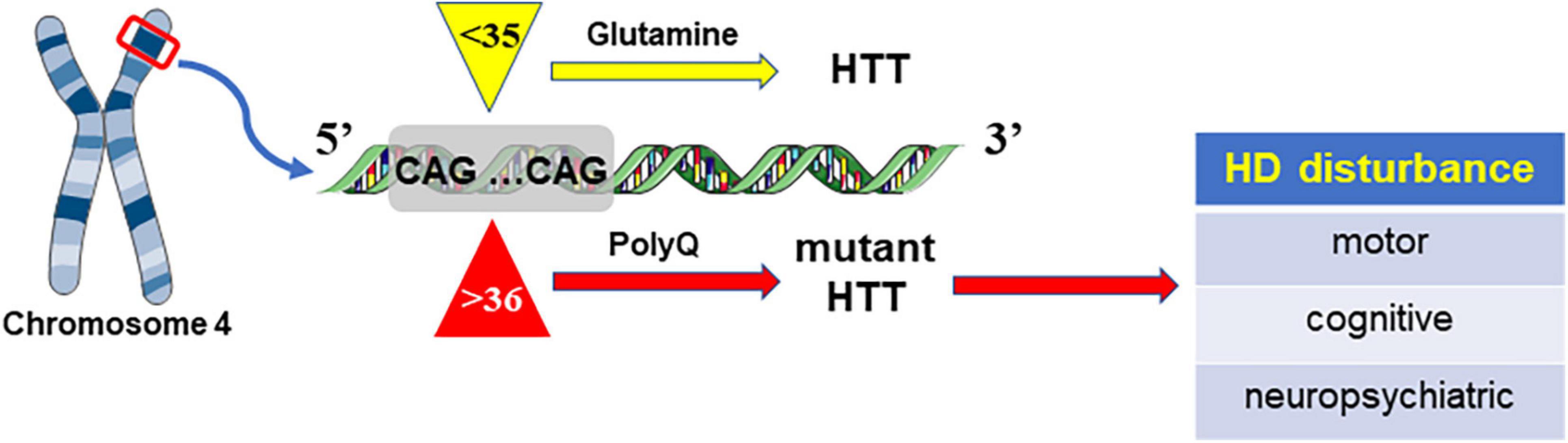
Figure 1. HD is caused by mutant Htt due to the increase in CAG repeats, which can lead to abnormal extension of polyglutamine (polyQ).
HD is a brain-derived degenerative disease with widespread and progressive brain shrinkage, structural decline and functional impairment, especially in the striatum (Reiner et al., 1988; Fjodorova et al., 2015; Reiner and Deng, 2018). Although the striatum is the initial pathological site, there is still progressive damage in different cortical layers of the HD brain, from posterior to anterior, that generates symptoms during the whole lifespan (Rosas et al., 2002; Nana et al., 2014; Jimenez-Sanchez et al., 2017). Other areas, including the hippocampus, thalamus, and cerebellum, also undergo various levels of neuronal loss on the basis of HD stages. In addition, HD patients also display other symptoms, such as weight loss, muscle defects, and cardiac damage (Arenas et al., 1998; Gilbert, 2009). In general, males and females are impacted equally, and the average age of HD onset with symptoms is approximately 20–65 years, lasting about 20 years and then developing severe intellectual disability and eventually leading to death (Snowden, 2017; Wyant et al., 2017).
CAG length can expand when passed down from parents to children, especially when delivered through males, therefore leading to a high probability of HD symptoms appearing in childhood and adolescence prior to age 21, which is identified as Juvenile Huntington’s disease (JHD)(Reyes Molon et al., 2010; Quigley, 2017). JHD develops and progresses more rapidly than adult HD because of the extensive CAG repeats with a high mutation rate and longer length compared with their parents (Quarrell et al., 2012). Motor impairments remain the main symptoms in JHD, including chorea, gait changes, speech deficits and so on (Robertson et al., 2012; Snowden, 2017). Cognitive decline and psychiatric factors are also manifested in JHD, such as learning and memory problems, anxiety, aggression and depression, speech and language impairments (Fusilli et al., 2018; Cronin et al., 2019; Lesinskiene et al., 2020). Due to a series of functional deficiencies, suicide frequently occurs in JHD.
Mutant huntingtin (mHtt) can cause severe neuronal dysfunction, especially in medium spiny neurons (MSNs) in the striatum area, which is selectively susceptible to mHtt (Bohanna et al., 2008; Sprenger et al., 2019). Both autopsy reports and magnetic resonance imaging (MRI) have revealed tremendous atrophy in the striatum and cortical white matter (Bohanna et al., 2008; Tabrizi et al., 2011; McColgan and Tabrizi, 2018). Striatal degeneration can appear in the early HD phase, sometimes 10–15 years earlier than clinical detection (Paulsen et al., 2008; Wolf et al., 2013). It is well known that the striatum is a crucial part of the “pain matrix” and deals with various pain-related processes, such as sensorial discrimination, emotional processing, and cognitive evaluation (Fenton et al., 2015; Frediani and Bussone, 2019; Faraj et al., 2021). The striatum is principally responsible for affective- or cognitive- pain dimensions (Thompson and Neugebauer, 2019; Zhou et al., 2019; Tan and Kuner, 2021), which are crucial to determining the level of suffering from uncomfortable pain and then remembering, integrating and responding appropriately (Price, 2000; Nees and Becker, 2018). In addition, unusual sensory activation of the cortex has been discovered in HD (Boecker et al., 1999). Other brain areas associated with the “pain matrix” that are responsible for multiple pain dimensions include cortex, insula, thalamus, and somatosensory cortices (Apkarian et al., 2005; Perrotta et al., 2012; Cardenas Fernandez, 2015). Relevant to previous studies, different levels of atrophy have been discovered in the “pain matrix” regions of HD patient brains. Disease progression is greatly linked with the atrophy level in the HD brain (Bohanna et al., 2008; Coppen et al., 2016, 2018; Wilson et al., 2018; Johnson and Gregory, 2019). The basal ganglia are closely related to acute and chronic pain and show some changes in pain processing. In the mid stage of HD, an obvious delay in pain progression was found at the spinal cord level compared with the general population (De Tommaso et al., 2011; Perrotta et al., 2012; de Tommaso et al., 2016b). Overall, dysfunction at any degree and in any area has the possibility of causing unmodulated, transient or sustained pain.
Pain has been studied and reported in AD, PD and other neurological disorders. However, pain in HD has rarely received attention, with only a few studies concerning pain issues. Several clinical studies have shown that pain in HD patients is greatly ignored and treated insufficiently, hence it is becoming a seriously undervalued issue (Andrich et al., 2009; de Tommaso et al., 2016a).
Pain development is closely influenced by psychological features, including depression, anxiety, apathy and irritability. Psychiatric symptoms occur frequently during all HD periods, especially the late stage, with an onset of approximately 33–76% (van Duijn et al., 2007, 2014). Some early studies reported that HD patients with depression had obviously painful symptoms; the greater the levels of anxiety and depression, the greater the severity of pain (Albin and Young, 1988; Arran et al., 2014). One study with 1474 HD gene carriers found that the pain prevalence ranged from 32% in the early period to 50% in the late period (Underwood et al., 2017), which was similar to another small sample study with a pain prevalence ranging from 11–62% (De Tommaso et al., 2011; Calvert et al., 2013). Another meta-analysis reported that the average pain prevalence in HD was about 41%, ranging from 36 to 46% (Sprenger et al., 2019). Recently, a worldwide pain-HD investigation showed that in HD mutation carriers, 34% had pain intervention, 17% underwent painful conditions, and 13% were treated with analgesics. For HD carriers without mutation, 42% had pain intervention and pain frequency was 12% in the middle period and 15% in the late period (Sprenger et al., 2021). The average prevalence of JHD in total HD cases is approximately 5–10% (Reyes Molon et al., 2010; Quigley, 2017; Lesinskiene et al., 2020).
As an essential non-motor feature, pain has been undervalued and not linked with HD for a long time until a brief report in 1988 said that two HD cases had strong sensory symptoms. One patient described an unusual sensory feeling as intermittent “bee-sting pain”, which occasionally occurred on any body part, lasting several seconds or minutes. The pain frequency and intensity became worse in the following months, and rare therapeutic approaches could reduce his pain (Albin and Young, 1988). Another HD victim also suffered an uncomfortable sensation and intermittent, sharp-tingling pain in her arms and legs. She continued to complain of severe “lancinating pain” and auditory abnormality. Both eventually committed suicide (Albin and Young, 1988). These two cases only represent a small part of the HD population, indicating the essential role of sensory problems in HD, which cannot be overlooked. Some other studies have also shown abnormal sensory-induced potentials in HD patients (Oepen et al., 1981; Ehle et al., 1984). A marathon runner experienced serious muscle pain after running, which continued for several weeks and worsened his performance (Kosinski et al., 2007). In addition, a study with 90 sample, aimed to study pain in premanifest period HD and found that approximately 49% of them used narcotics, but only 14% used narcotics in the general population (Sprenger et al., 2019). The high use of narcotics indicates that pain is indeed a severe but unrecognized symptom in HD.
Studies concentrating on pain in HD display controversial results. In a study of HD with 19 cases, 11 patients estimated a high pain score, but only 3 of them obtained analgesia treatment (McColgan and Tabrizi, 2018). Another study with 28 HD samples reported that HD patients display a slowing pain development, and only 3 mild-stage patients expressed pain responses after laser stimuli (De Tommaso et al., 2011). Cognitive deficiency includes damage to language, memory, attention and decision-making, which could result in abnormal behavior in HD patients (Singh and Agrawal, 2021). Slow pain progression might disturb sensory information integration and then lead to insufficient or inaccurate behavior in response to painful issues. An increasing number of studies have shown that HD patients experience pain but show a deficit in pain recognition. Because of motor and cognitive deficits, later-stage HD patients rarely complain of pain even with serious pain symptoms, suggesting the possibility of pain signal transfer delay or dysfunction (Albin and Young, 1988; Andrich et al., 2009; Baez et al., 2015). In addition to cognitive deficits, there are other behavioral abnormalities in HD, including emotional damage (psychosis, depression, anxiety) and empathy deficits, which cause negative pain recognition (Baez et al., 2015). The painful stimuli may interrupt sensorimotor information integration (Fil-Balkan et al., 2018; Millian-Morell et al., 2018) and lead to global deterioration of HD (De Tommaso et al., 2011; de Tommaso et al., 2016a).
There are still some other interesting findings concerning specific pain in HD. It has been reported that a series of abnormal pain events occur and change as the disease develops, including back pain, headache, limb pain, abdominal pain (Sprenger et al., 2021). In an elderly study, the relationship between gastroesophageal inflammation and HD was explored. Many HD patients have gastritis or esophagitis symptoms but without complaints, which has an obvious relationship with the duration and severity of HD (Andrich et al., 2009). The increase in abdominal pain might be highly related to the gastroesophageal disturbances in the end HD stage. Additionally, fractures seem to increase as HD progresses, which might be linked with bone density alteration (Carroll et al., 2015).
The correlation between pain and HD is interesting, involving a series of affecting factors. First, HD itself may cause pain and is rarely treated, partly due to the insufficient awareness of pain from physicians, which is supported by the incoherence between painful conditions and analgesic use (Sprenger et al., 2021). Second, neuronal loss or dysfunction could also cause a lack of self-awareness in HD, making it difficult to gather pain-related data through self-evaluation (Madariaga et al., 2021). Third, the incoherence between pain and HD might be clarified by basal ganglia disturbance, which could increase pain severity but with less or no pain behavior, probably owing to disorders in sensation, motor function and/or cognition (Borsook et al., 2010). Fourth, emotional factors are critical and closely linked with pain behavior in the HD population. A high frequency of emotional issues, such as depression and anxiety, are popular in the HD manifest periods (Sprenger et al., 2021). Finally, different pain criteria could cause different pain assessment data, which might underestimate the pain prevalence in the HD population. For example, specific dystonia is not considered a painful symptom, but it frequently appears and triggers pain in HD progression (Buhmann et al., 2020).
As HD progresses, an increasing number of affecting factors appear and are present, such as sex, illness stage, daily exercise, medicine and so on, and these factors interact with one another. Therefore, systematic studies are required to explore the detailed relationship between pain and HD in the future.
As a typical genetic disease, HD is primarily characterized by motor and psychiatric dysfunction, and rarely focuses on pain abnormalities. Here, we outline some potential mechanisms associated with pain in HD, summarized in Figure 2.
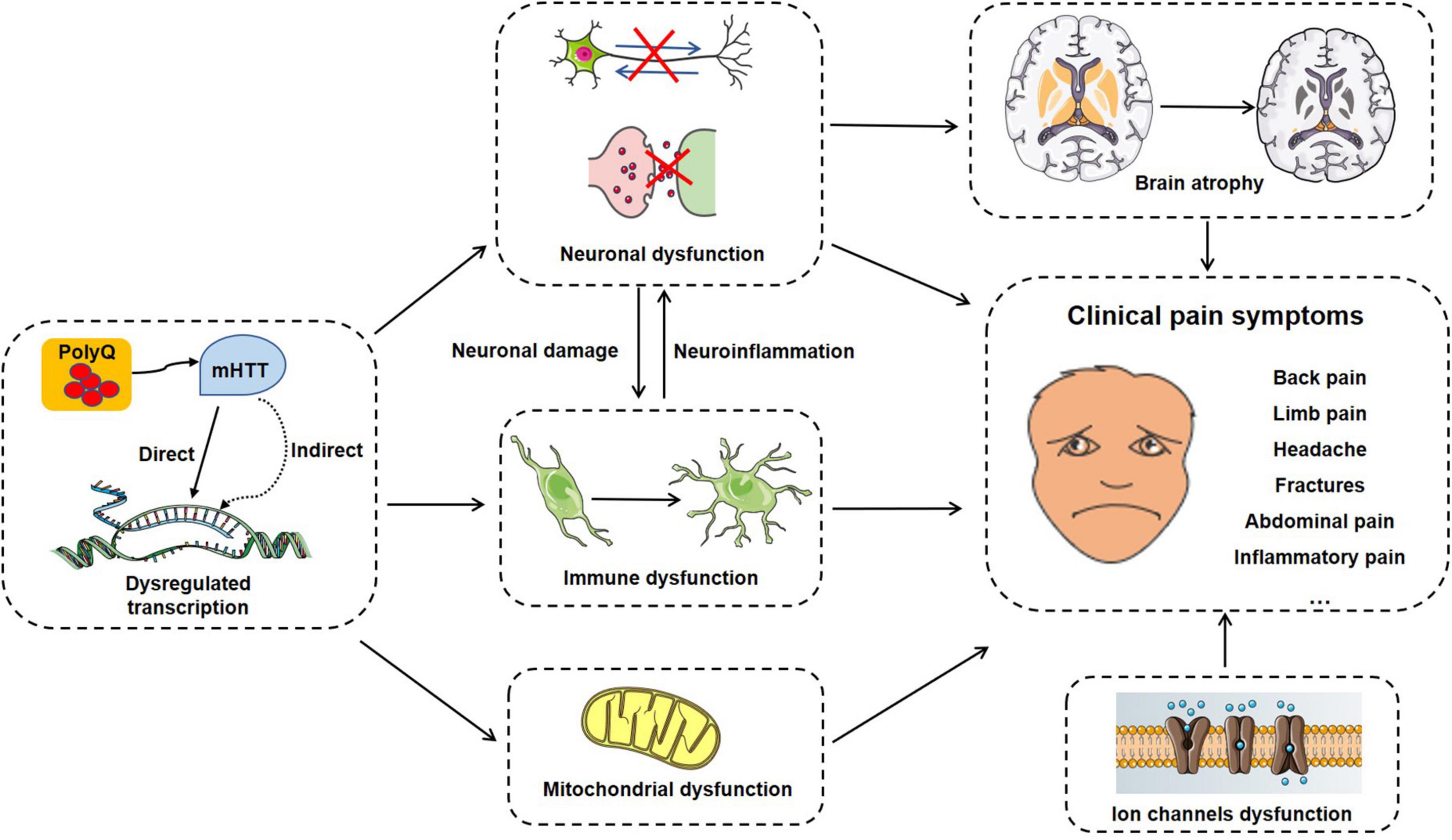
Figure 2. A summary of potential pain-related molecular mechanisms, cellular processes and clinical symptoms in HD progress.
The well-known characteristics of HD is progressive brain and basal ganglia atrophy, involving many functional regions and leading to motor and cognitive dysfunction as well as dementia and psychosis. Studies have reported that many regions of the brain are influenced in HD, including gray matter volume decrease, cortical thinning, and striatum deterioration.
Brain structures, such as the cortex, thalamus, cerebellum, and hippocampus, are essential areas related to pain sensory, function and functionally integrate with each other to modulate pain processing. However, the particular parts of brain atrophy associated with pain in HD are seldom considered. The brain regions involved in pain and HD are summarized and listed below (Table 1).
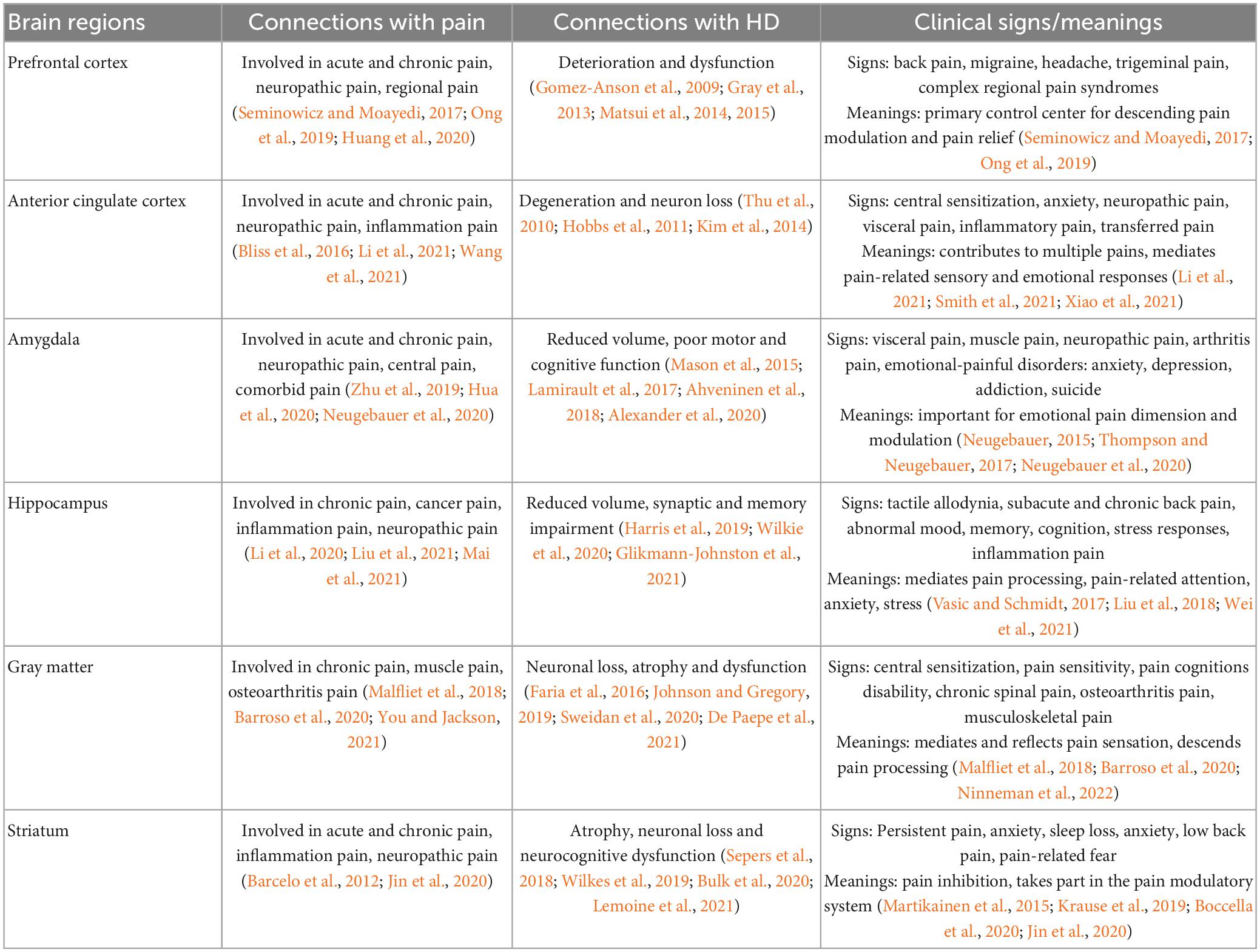
Table 1. The brain regions involved in pain and HD.
As major pain regulation areas, spinal cord (SC) and peripheral nervous systems (PNS) play vital roles in pain processing and transduction, also showed morphological and functional disorders in HD. SC gradual atrophy were detected and confirmed in manifest HD patients, as well as in the early stage (Muhlau et al., 2014; Wilhelms et al., 2017). One postmortem study detected the expression and location of mHTT in the SC of HD patients and found that mHTT preferred to express in the spinal gray matter (Sciacca and Cicchetti, 2017). The deterioration of gray matter and white matter in SC were also observed in HD mice. The expression of mHTT in oligodendrocytes resulted in myelination abnormalities, which can be rescued by mHTT reduction (Ferrari Bardile et al., 2021). The SC impairments in HD inevitably brought many sensory issues, such as pain. Abnormal pain signals in SC were observed along with a dysfunction of pain signal transportation, which finally induced sensory alternation in HD patients (De Tommaso et al., 2011; Perrotta et al., 2012). At SC level, mHTT could result in significant changes of pain behavior and pain-related cytokine in HD mice (Lin et al., 2018).
In SC and PNS, it was reported that glia cells participated in pain responses mainly through regulating pain signals transmission, neuroinflammation and neuron-glia interactions. Glial cells were highly involved in the development of HD processing. The special expression of mHTT in glial cells contributed to normal function lose and neuropathic pathology (Wilhelms et al., 2017; Wilton and Stevens, 2020). Both in HD patients and animal models, the cell morphology, metabolism and functions of astrocyte were greatly changed as well as their interactions with neurons (Skotte et al., 2018; Osipovitch et al., 2019). In addition, the abnormal expression of mHTT in astrocytes is greatly related with HD pathology motivation (Diaz-Castro et al., 2019; Wood et al., 2019). The cell size and structural changes of microglial were explored in HD, along with motility and migration disorders. Meanwhile, microglia cells in HD showed upregulated levels of inflammatory cytokines, contributed to pain processing and abnormity (Diaz-Castro et al., 2019; Liu et al., 2019). In HD mice, phenotypic and molecular studies revealed that the structural and functional deficits of oligodendrocyte appeared in HD early stage, causing severe oligodendrocytes’ dysfunctions, including thinner myelin sheaths, remyelination impairments and less response to demyelinating injury (Teo et al., 2016, 2019).
One of the main pathological mechanisms in HD is transcription disorder. Many genes showed expression changes in HD (Lee et al., 2013; Sipione et al., 2016). Mutant Htt can interact with transcription factors, such as p53, CREB and PGC-1a, and disrupt cell survival, energy metabolism and protein expression (Jiang et al., 2006; Chaturvedi et al., 2012; Kim et al., 2016; Aravindan et al., 2020). As a key neurotrophic factor, the transcription, expression and transport of BDNF, which is essential for striatal neuron survival, are badly impaired in HD (Hong et al., 2016). Therefore, the stratum becomes the most vulnerable region due to a low level of BDNF (Park, 2018). In addition, synaptic dysfunction of the cortical- striatum is due to deficiency in the BDNF pathway via p75 (postsynaptic receptor), which is also thought to act with TrkB in HD (Pan et al., 2018).
Huntington-associated protein 1 (HAP1) is the earliest protein found to interact with Htt and contributes to cargo (vesicle, receptor, and neurotrophic factor) trafficking, the function of which is interfered with by mHtt (Jimenez-Sanchez et al., 2017), including the synthesis and release of BDNF in the cortex and the retrograde transport of TrkB in the striatum (Park, 2018). HAP1 is highly involved not only in HD but also in pain progression, and enriched in the spinal dorsal horn and dorsal root ganglia (Islam et al., 2017, 2020), which are considered the “primary sensory center”. In our study, we found that HAP1 could regulate Cav1.2 surface expression, which in turn influenced neuronal excitability, BDNF secretion, and inflammatory responses and ultimately modulated pain progression (Pan et al., 2023). HAP1 deficient mice exhibited mechanical allodynia and hyperalgesia inhibition in acute and chronic pain models (Gloor et al., 2022).
Neuronal abnormality is one of the initial pathological changes in HD, along with synaptic plasticity alteration, involving many factors, such as gene transcription, protein expression and transmission (Jimenez-Sanchez et al., 2017). As a scaffold protein, Htt contributes to vesicle transport through microtubules and motor proteins, while mHtt can inhibit the axonal delivery of cargoes and organelles, reduce neurotransmission and hinder signaling (Jimenez-Sanchez et al., 2017; Snowden, 2017; McColgan and Tabrizi, 2018). Axonal delivery is important for cargo transfer to membranes and expression. However, the transport of many aborted receptors, including GABAA (γ-aminobutyric acid type A), AMPA (a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid), and NMDA (N-methyl-d-aspartate) receptors, is found in HD, disrupting synaptic plasticity and excitability. Some studies have found that GABAergic neurons are reduced in neuropathic pain, and that elevated levels of GABAergic mediators and GABAA receptors might reverse and attenuate pain (Knabl et al., 2008; St John Smith, 2018). In HD, redundant neurotransmission induced by NMDA receptors could cause neuronal death, specifically in striatal neurons. The function of NMDA receptors has been studied in multiple pain models, playing a pivotal role in attenuating central sensitization and pain hypersensitivity after stimulation (Kreutzwiser and Tawfic, 2019; Shin et al., 2020). Stimulated NMDA receptors can lead to calcium influx, which is critical for synaptic plasticity. AMPA receptors can regulate synaptic strength and plasticity and alleviate different types of pain hypersensitivity, including neuropathic (Wang et al., 2021), neuroinflammatory (Kopach et al., 2018), chronic (Bliss et al., 2016) and postoperative (Kopach et al., 2018; Khan et al., 2019; Kopach and Voitenko, 2021) pain. However, the balance between presynaptic, postsynaptic and extra-synaptic activities is altered in HD, termed excitotoxicity (Jimenez-Sanchez et al., 2017).
Activated glial cells interact with neurons and affect pain processing by releasing neurotransmitters, molecules, inflammatory cytokines and chemokines. Immune dysfunction is an important factor in neurodegenerative disease. In HD, changes in immune cells and inflammatory responses have been discovered in the brain and peripheral system, indicating a potential role in pathogenesis (Andre et al., 2016). Increasing evidence has shown increased levels of proinflammatory mediators in early and late HD patients (Trager et al., 2014; Andre et al., 2016). When mHtt was chosen to be expressed in astrocytes, no notable changes appeared in glia or neurons, but mice showed pathological symptoms in the late stage. HD astrocytes present defects in the secretion of chemokines CCL5, BDNF and low levels of K+ channels, which can regulate neuronal excitability (Hong et al., 2016; Saba et al., 2020). In HD, nuclear factor-κB (NF-κB) acts as an inflammatory indicator and regulates the expression of various inflammatory cytokines, including IL-1 and TNF-α (Yusuf et al., 2021). NF-kB signals increase and evoke inflammatory responses in HD myeloid cells (Trager et al., 2014). In PNS, mutant huntingtin could influence inflammatory responses through NF-kB signaling inhibition. Downregulation of NF-κB attenuates inflammatory gene expression and suppresses inflammatory pain responses (Xiang et al., 2019; Yang et al., 2020; Kato et al., 2021). Microglial activation was discovered in the brains of HD patients and mice and is involved in neuronal degeneration. In addition, microglia expressing mHtt also exhibit decreased migration (Kwan et al., 2012; Crotti et al., 2014). Moreover, bone marrow transplantation could inhibit pathology by increasing the levels of cytokines and chemokines in HD mouse models (Jimenez-Sanchez et al., 2017).
Mitochondria are involved in ATP production, calcium balance, and protein synthesis, which influence neuronal survival, growth and apoptosis (Dai et al., 2020; Doyle and Salvemini, 2021). Some studies have suggested that mitochondrial abnormalities are found in HD and are probably involved in the pain process (Jimenez-Sanchez et al., 2017). Some studies found that huntingtin mutation could cause mitochondrial calcium imbalance by binding with the outer membrane, interrupting the axonal transport of mitochondria and ultimately reducing ATP production (Shirendeb et al., 2011; Reddy and Shirendeb, 2012). Ca2+ is an essential signaling molecular, the abnormal activities of which play complex roles in HD (Jimenez-Sanchez et al., 2017), thereby leading to abnormal pain responses indirectly or directly. The fusion-fission cycles of mitochondria are regulated by dynamin-related protein 1 (Drp1), the deficiency of which can inhibit mHtt-evoked mitochondrial toxicity and delay HD development (Zhan et al., 2018; Dai et al., 2020). Drp1 and its associated mitochondrial malfunction have been reported to influence the pathological process of neuropathic pain (Jimenez-Sanchez et al., 2017; Zhan et al., 2018; Kun et al., 2019). Because of mitochondrial dysfunction, HD patients and models exhibit reactive oxygen species (ROS) (Jimenez-Sanchez et al., 2017), which have been observed and function in neuropathic pain (Flippo and Strack, 2017; Dai et al., 2020).
Ion channels are considered as major targets for regulating pain sensation, due to their notable distribution in sensory neurons and key tissues. A lot of pain-related ion channels have been discovered and identified as potential pathogenic factors in HD, including transient receptor potential (TRP) channels, calcium (Ca+) channels, potassium (K+) channels, acid sensing ion channels (ASICs).
TRP channels are ubiquitously diffused in CNS, PNS and SC as well as dorsal root ganglia (DRG). TRP channels are mainly expressed in membranes of neurons and non-neuronal cells, mediating ion homeostasis and emerging as crucial pain transducers (Nilius and Owsianik, 2011; Moore et al., 2018; Lee et al., 2021). It had reported that TRP subfamilies TRPC (canonical) and TRPV (vanilloid) were associated with HD. Decreased level of endogenous TRPC1 was explored in HD striatal neurons, together with increased level of TRPC5 glutathionylation, leading to neurons apoptosis in HD mice (Hong et al., 2015, 2020). Furthermore, pharmacological experiments demonstrated the potential effects of TRPV1 channel in HD models, suggesting the possible contribution of TRPV1 in HD dysfunctions (Lastres-Becker et al., 2003).
Calcium channels have fundamental roles in neuronal health and functions and have a series of subtypes according different structures, pharmacological and physiological properties. L-type channels are abundantly expressed in CNS and SC, regulating Ca2+ influx and neuronal sensitization (Roca-Lapirot et al., 2018; Hopp, 2021). Its subtype Cav1.2 was highly involved in HD, whose expression and functions were interfered by mHTT (Miranda et al., 2019). The store-operated Ca2+ entry (SOCE), a major Ca2+ entry pathway, was greatly increased in HD and attributed to Ca2+ homeostasis disorder by activating mHTT indirectly (Czeredys, 2020). Besides, the interaction between mHTT and inositol-1,4,5-triphosphate receptors (IP3Rs) can lead to abnormally Ca2+ levels in R6/2 HD mouse, containing high intracellular Ca2+ level and low mitochondrial Ca2+ level (Mackay et al., 2018; Schrank et al., 2020). Furthermore, ryanodine receptor (RyR) associated Ca2+ abnormity had been investigated and documented in HD (Dridi et al., 2020).
Potassium (K+) channels are functional for pain processes by regulating pain transmission and modulation, whose dysfunction can induce a series of pain problems (Tsantoulas, 2015; Busserolles et al., 2016). In HD, K+ channels also have important roles in physiological and pathological conditions. Glia cells express many rectifying K + channel (Kir) subfamilies and function in HD processes (Zhang et al., 2018). In R6/2 and Q175 HD mouse models, Kir4.1 decrease can cause disturbances of astrocyte–mediated K+ homeostasis (Tong et al., 2014; Nwaobi et al., 2016). Evidence indicated that both HD patients and models showed obvious decrease of Kir4.1 level in astrocytes, leading to a high K+ extracellular concentration and neuronal damages (Nwaobi et al., 2016; Wang et al., 2022). Furthermore, KATP channels had been indicated to associate with abnormal neuronal firing in HD mice (Atherton et al., 2016; Rallapalle et al., 2021).
Additionally, acid sensing ion channels (ASICs) are highly expressed in nervous system, and greatly involved in both pain processing and HD development (Deval and Lingueglia, 2015; Sluka and Gregory, 2015; Zhou et al., 2016; Zhu et al., 2022).
HD, as a classic neurodegenerative disorder, provides scientists with ideas for many interesting studies on social, financial and medical issues. Pain is one of the major features, a subjective and direct way to express discomfort and suffering. As HD progresses, pain can be produced directly by many elements, such as neuron loss, atrophy, muscle decline, and bone density reduction; however, subjective pain behavior might be limited or concealed owing to motor and sensory dysfunction and cognitive deficiency. Because of cognitive problems, HD patients suffer from pain but rarely complain, resulting in pain that is limited and neglected, therefore leading to underestimation and no treatments. As a result of insufficient attention, special evaluations and therapeutic guidelines are unavailable, and clinical treatments for pain are limited to analgesics and anti-inflammatory agents without a full speculation of potential mechanisms. However, in many other diseases, particularly AD and PD, specific scales and treatments are used and applied to pain issues, which are considered fully as a major symptom instead of a secondary problem.
This review notes that the major issues related to pain are medical neglect and carelessness, as attention and treatment are mostly focused on motor-related symptoms. Hence, health doctors and caregivers should pay more attention to pain problems during HD progression, especially in the later stage. The correlation between HD and pain should be further investigated. Current studies and evidence associated with pain in HD suggest a huge challenge and opportunity for scientists in the neuronal-pain field. Future studies should focus on pain-related factors and intrinsic mechanisms, pay more attention to specific scales, equipment, treatments and identify effective therapeutic methods for HD patients undergoing pain conditions.
GC and JP designed and drafted the manuscript. All authors contributed to the manuscript revision and approved the final version.
This work was supported by the National Natural Science Foundation of China (82001168, 32070998, and 82101455), the Overseas Study Foundation of Jiangsu Province Education Department, and the Students’ Practice Innovation Training Programs of Jiangsu Province Higher School.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Achterberg, W., Lautenbacher, S., Husebo, B., Erdal, A., and Herr, K. (2021). Pain in dementia. Schmerz 35, 130–138.
Ahveninen, L., Stout, J., Georgiou-Karistianis, N., Lorenzetti, V., and Glikmann-Johnston, Y. (2018). Reduced amygdala volumes are related to motor and cognitive signs in Huntington’s disease: The IMAGE-HD study. NeuroImage Clin. 18, 881–887. doi: 10.1016/j.nicl.2018.03.027
Albin, R., and Young, A. (1988). Somatosensory phenomena in Huntington’s disease. Mov. Disord. 3, 343–346.
Alexander, B., Georgiou-Karistianis, N., Beare, R., Ahveninen, L., Lorenzetti, V., Stout, J., et al. (2020). Accuracy of automated amygdala MRI segmentation approaches in Huntington’s disease in the IMAGE-HD cohort. Hum. Brain Mapp. 41, 1875–1888. doi: 10.1002/hbm.24918
Andre, R., Carty, L., and Tabrizi, S. (2016). Disruption of immune cell function by mutant huntingtin in Huntington’s disease pathogenesis. Curr. Opin. Pharmacol. 26, 33–38.
Andrich, J., Wobben, M., Klotz, P., Goetze, O., and Saft, C. (2009). Upper gastrointestinal findings in Huntington’s disease: Patients suffer but do not complain. J. Neural Transm. 116, 1607–1611.
Apkarian, A., Bushnell, M., Treede, R., and Zubieta, J. (2005). Human brain mechanisms of pain perception and regulation in health and disease. Eur. J. Pain 9, 463–484.
Aravindan, S., Chen, S., Choudhry, H., Molfetta, C., Chen, K., and Liu, A. (2020). Osmolytes dynamically regulate mutant huntingtin aggregation and CREB function in Huntington’s disease cell models. Sci. Rep. 10:15511. doi: 10.1038/s41598-020-72613-3
Arenas, J., Campos, Y., Ribacoba, R., Martin, M., Rubio, J., Ablanedo, P., et al. (1998). Complex I defect in muscle from patients with Huntington’s disease. Ann. Neurol. 43, 397–400.
Arran, N., Craufurd, D., and Simpson, J. (2014). Illness perceptions, coping styles and psychological distress in adults with Huntington’s disease. Psychol. Health Med. 19, 169–179. doi: 10.1080/13548506.2013.802355
Atherton, J., McIver, E., Mullen, M., Wokosin, D., Surmeier, D., and Bevan, M. (2016). Early dysfunction and progressive degeneration of the subthalamic nucleus in mouse models of Huntington’s disease. Elife 5:e21616. doi: 10.7554/eLife.21616
Baez, S., Herrera, E., Gershanik, O., Garcia, A., Bocanegra, Y., Kargieman, L., et al. (2015). Impairments in negative emotion recognition and empathy for pain in Huntington’s disease families. Neuropsychologia 68, 158–167. doi: 10.1016/j.neuropsychologia.2015.01.012
Baig, S., Strong, M., and Quarrell, O. (2016). The global prevalence of Huntington’s disease: A systematic review and discussion. Neurodegener. Dis. Manag. 6, 331–343.
Barcelo, A., Filippini, B., and Pazo, J. (2012). The striatum and pain modulation. Cell. Mol. Neurobiol. 32, 1–12.
Barroso, J., Vigotsky, A., Branco, P., Reis, A., Schnitzer, T., Galhardo, V., et al. (2020). Brain gray matter abnormalities in osteoarthritis pain: A cross-sectional evaluation. Pain 161, 2167–2178. doi: 10.1097/j.pain.0000000000001904
Bliss, T., Collingridge, G., Kaang, B., and Zhuo, M. (2016). Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci. 17, 485–496.
Boccella, S., Marabese, I., Guida, F., Luongo, L., Maione, S., and Palazzo, E. (2020). The modulation of pain by metabotropic glutamate receptors 7 and 8 in the dorsal striatum. Curr. Neuropharmacol. 18, 34–50.
Boecker, H., Ceballos-Baumann, A., Bartenstein, P., Weindl, A., Siebner, H., Fassbender, T., et al. (1999). Sensory processing in Parkinson’s and Huntington’s disease: Investigations with 3D H(2)(15)O-PET. Brain 122, 1651–1665.
Bohanna, I., Georgiou-Karistianis, N., Hannan, A., and Egan, G. (2008). Magnetic resonance imaging as an approach towards identifying neuropathological biomarkers for Huntington’s disease. Brain Res. Rev. 58, 209–225. doi: 10.1016/j.brainresrev.2008.04.001
Borsook, D., Upadhyay, J., Chudler, E., and Becerra, L. (2010). A key role of the basal ganglia in pain and analgesia–insights gained through human functional imaging. Mol. Pain 6:27. doi: 10.1186/1744-8069-6-27
Buhmann, C., Kassubek, J., and Jost, W. (2020). Management of pain in Parkinson’s disease. J. Parkinsons Dis. 10, S37–S48.
Bulk, M., Hegeman-Kleinn, I., Kenkhuis, B., Suidgeest, E., van Roon-Mom, W., Lewerenz, J., et al. (2020). Pathological characterization of T2*-weighted MRI contrast in the striatum of Huntington’s disease patients. NeuroImage Clin. 28:102498. doi: 10.1016/j.nicl.2020.102498
Busserolles, J., Tsantoulas, C., Eschalier, A., and Lopez Garcia, J. (2016). Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 157, S7–S14. doi: 10.1097/j.pain.0000000000000368
Calvert, M., Pall, H., Hoppitt, T., Eaton, B., Savill, E., and Sackley, C. (2013). Health-related quality of life and supportive care in patients with rare long-term neurological conditions. Qual. Life Res. 22, 1231–1238.
Cardenas Fernandez, R. (2015). [The neuromatrix and its importance in pain neurobiology]. Investig. Clin. 56, 109–110.
Carroll, J., Bates, G., Steffan, J., Saft, C., and Tabrizi, S. (2015). Treating the whole body in Huntington’s disease. Lancet Neurol. 14, 1135–1142.
Chaturvedi, R., Hennessey, T., Johri, A., Tiwari, S., Mishra, D., Agarwal, S., et al. (2012). Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington’s disease. Hum. Mol. Genet. 21, 3474–3488. doi: 10.1093/hmg/dds178
Coppen, E., Jacobs, M., van den Berg-Huysmans, A., van der Grond, J., and Roos, R. (2018). Grey matter volume loss is associated with specific clinical motor signs in Huntington’s disease. Parkinsonism Relat. Disord. 46, 56–61.
Coppen, E., van der Grond, J., Hafkemeijer, A., Rombouts, S., and Roos, R. (2016). Early grey matter changes in structural covariance networks in Huntington’s disease. NeuroImage Clin. 12, 806–814.
Cronin, T., Rosser, A., and Massey, T. (2019). Clinical presentation and features of juvenile-onset Huntington’s disease: A systematic review. J. Huntingtons Dis. 8, 171–179.
Crotti, A., Benner, C., Kerman, B., Gosselin, D., Lagier-Tourenne, C., Zuccato, C., et al. (2014). Mutant huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 17, 513–521. doi: 10.1038/nn.3668
Czeredys, M. (2020). Dysregulation of neuronal calcium signaling via store-operated channels in Huntington’s disease. Front. Cell Dev. Biol. 8:611735. doi: 10.3389/fcell.2020.611735
Dai, C., Guo, Y., and Chu, X. (2020). Neuropathic Pain: The dysfunction of Drp1, mitochondria, and ROS homeostasis. Neurotoxicity Res. 38, 553–563. doi: 10.1007/s12640-020-00257-2
Dale, M., and van Duijn, E. (2015). Anxiety in Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 27, 262–271.
De Paepe, A., Ara, A., Garcia-Gorro, C., Martinez-Horta, S., Perez-Perez, J., Kulisevsky, J., et al. (2021). Gray matter vulnerabilities predict longitudinal development of apathy in Huntington’s disease. Mov. Disord. 36, 2162–2172. doi: 10.1002/mds.28638
de Tommaso, M., Arendt-Nielsen, L., Defrin, R., Kunz, M., Pickering, G., and Valeriani, M. (2016a). Pain in neurodegenerative disease: Current knowledge and future perspectives. Behav. Neurol. 2016:7576292.
de Tommaso, M., Franco, G., Ricci, K., Montemurno, A., and Sciruicchio, V. (2016b). Laser evoked potentials in early and presymptomatic Huntington’s disease. Behav. Neurol. 2016:8613729.
De Tommaso, M., Serpino, C., Difruscolo, O., Cormio, C., Sciruicchio, V., Franco, G., et al. (2011). Nociceptive inputs transmission in Huntington’s disease: A study by laser evoked potentials. Acta Neurol. Belgica 111, 33–40.
Deval, E., and Lingueglia, E. (2015). Acid-sensing ion channels and nociception in the peripheral and central nervous systems. Neuropharmacology 94, 49–57.
Diaz-Castro, B., Gangwani, M., Yu, X., Coppola, G., and Khakh, B. (2019). Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 11:eaaw8546.
Doyle, T., and Salvemini, D. (2021). Mini-review: Mitochondrial dysfunction and chemotherapy-induced neuropathic pain. Neurosci. Lett. 760:136087. doi: 10.1016/j.neulet.2021.136087
Dridi, H., Liu, X., Yuan, Q., Reiken, S., Yehia, M., Sittenfeld, L., et al. (2020). Role of defective calcium regulation in cardiorespiratory dysfunction in Huntington’s disease. JCI Insight 5:e140614.
Ehle, A., Stewart, R., Lellelid, N., and Leventhal, N. (1984). Evoked potentials in Huntington’s disease. A comparative and longitudinal study. Arch. Neurol. 41, 379–382.
Evans, S., Douglas, I., Rawlins, M., Wexler, N., Tabrizi, S., and Smeeth, L. (2013). Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 84, 1156–1160.
Faraj, M., Lipanski, N., Morales, A., Goldberg, E., Bluth, M., Marusak, H., et al. (2021). A virtual reality meditative intervention modulates pain and the pain neuromatrix in patients with opioid use disorder. Pain Med. 22, 2739–2753. doi: 10.1093/pm/pnab162
Faria, A., Ratnanather, J., Tward, D., Lee, D., van den Noort, F., Wu, D., et al. (2016). Linking white matter and deep gray matter alterations in premanifest Huntington disease. NeuroImage Clin. 11, 450–460.
Fenton, B., Shih, E., and Zolton, J. (2015). The neurobiology of pain perception in normal and persistent pain. Pain Manag. 5, 297–317.
Ferrari Bardile, C., Sidik, H., Quek, R., Yusof, N., Garcia-Miralles, M., and Pouladi, M. (2021). Abnormal spinal cord myelination due to oligodendrocyte dysfunction in a model of Huntington’s disease. J. Huntingtons Dis. 10, 377–384. doi: 10.3233/JHD-210495
Fil-Balkan, A., Salci, Y., Keklicek, H., Armutlu, K., Aksoy, S., Kayihan, H., et al. (2018). Sensorimotor integration training in Parkinson‘s disease. Neurosciences 23, 208–215.
Fisher, E., and Hayden, M. (2014). Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 29, 105–114. doi: 10.1002/mds.25717
Fjodorova, M., Noakes, Z., and Li, M. (2015). How to make striatal projection neurons. Neurogenesis 2:e1100227.
Flippo, K., and Strack, S. (2017). Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 130, 671–681.
Fusilli, C., Migliore, S., Mazza, T., Consoli, F., De Luca, A., Barbagallo, G., et al. (2018). Biological and clinical manifestations of juvenile Huntington’s disease: A retrospective analysis. Lancet Neurol. 17, 986–993.
Gilbert, G. (2009). Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 73, 1506–13.
Glikmann-Johnston, Y., Mercieca, E., Carmichael, A., Alexander, B., Harding, I., and Stout, J. (2021). Hippocampal and striatal volumes correlate with spatial memory impairment in Huntington’s disease. J. Neurosci. Res. 99, 2948–2963. doi: 10.1002/jnr.24966
Gloor, Y., Matthey, A., Sobo, K., Mouterde, M., Kosek, E., Pickering, G., et al. (2022). Uncovering a genetic polymorphism located in huntingtin associated protein 1 in modulation of central pain sensitization signaling pathways. Front. Neurosci. 16:807773. doi: 10.3389/fnins.2022.807773
Gomez-Anson, B., Alegret, M., Munoz, E., Monte, G., Alayrach, E., Sanchez, A., et al. (2009). Prefrontal cortex volume reduction on MRI in preclinical Huntington’s disease relates to visuomotor performance and CAG number. Parkinsonism Relat. Disord. 15, 213–219. doi: 10.1016/j.parkreldis.2008.05.010
Gray, M., Egan, G., Ando, A., Churchyard, A., Chua, P., Stout, J., et al. (2013). Prefrontal activity in Huntington’s disease reflects cognitive and neuropsychiatric disturbances: The IMAGE-HD study. Exp. Neurol. 239, 218–228. doi: 10.1016/j.expneurol.2012.10.020
Harris, K., Armstrong, M., Swain, R., Erzinclioglu, S., Das, T., Burgess, N., et al. (2019). Huntington’s disease patients display progressive deficits in hippocampal-dependent cognition during a task of spatial memory. Cortex 119, 417–427. doi: 10.1016/j.cortex.2019.07.014
Hobbs, N., Pedrick, A., Say, M., Frost, C., Dar Santos, R., Coleman, A., et al. (2011). The structural involvement of the cingulate cortex in premanifest and early Huntington’s disease. Mov. Disord. 26, 1684–1690.
Hong, C., Choi, S., Kwak, M., Jeong, B., Ko, J., Park, H., et al. (2020). TRPC5 channel instability induced by depalmitoylation protects striatal neurons against oxidative stress in Huntington’s disease. Biochim. Biophys. Acta Mol. Cell Res. 1867:118620. doi: 10.1016/j.bbamcr.2019.118620
Hong, C., Seo, H., Kwak, M., Jeon, J., Jang, J., Jeong, E., et al. (2015). Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 138, 3030–3047. doi: 10.1093/brain/awv188
Hong, Y., Zhao, T., Li, X., and Li, S. (2016). Mutant huntingtin impairs BDNF release from astrocytes by disrupting conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 36, 8790–8801. doi: 10.1523/JNEUROSCI.0168-16.2016
Hopp, S. (2021). Targeting microglia L-type voltage-dependent calcium channels for the treatment of central nervous system disorders. J. Neurosci. Res. 99, 141–162.
Hua, T., Chen, B., Lu, D., Sakurai, K., Zhao, S., Han, B., et al. (2020). General anesthetics activate a potent central pain-suppression circuit in the amygdala. Nat. Neurosci. 23, 854–868.
Huang, S., Zhang, Z., Gambeta, E., Xu, S., Thomas, C., Godfrey, N., et al. (2020). Dopamine inputs from the ventral tegmental area into the medial prefrontal cortex modulate neuropathic pain-associated behaviors in mice. Cell Rep. 31:107812.
Islam, M., Maeda, N., Miyasato, E., Jahan, M., Tarif, A., Ishino, T., et al. (2020). Expression of huntingtin-associated protein 1 in adult mouse dorsal root ganglia and its neurochemical characterization in reference to sensory neuron subpopulations. IBRO Rep. 9, 258–269. doi: 10.1016/j.ibror.2020.10.001
Islam, M., Takeshita, Y., Yanai, A., Imagawa, A., Jahan, M., Wroblewski, G., et al. (2017). Immunohistochemical analysis of huntingtin-associated protein 1 in adult rat spinal cord and its regional relationship with androgen receptor. Neuroscience 340, 201–217.
Jiang, H., Poirier, M., Liang, Y., Pei, Z., Weiskittel, C., Smith, W., et al. (2006). Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 23, 543–551.
Jimenez-Sanchez, M., Licitra, F., Underwood, B., and Rubinsztein, D. (2017). Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harbor Perspect. Med. 7:a024240.
Jin, Y., Meng, Q., Mei, L., Zhou, W., Zhu, X., Mao, Y., et al. (2020). A somatosensory cortex input to the caudal dorsolateral striatum controls comorbid anxiety in persistent pain. Pain 161, 416–428.
Johnson, E., and Gregory, S. (2019). Huntington’s disease: Brain imaging in Huntington’s disease. Progr. Mol. Biol. Transl. Sci. 165, 321–369.
Kato, K., Akeda, K., Miyazaki, S., Yamada, J., Muehleman, C., Miyamoto, K., et al. (2021). NF-kB decoy oligodeoxynucleotide preserves disc height in a rabbit anular-puncture model and reduces pain induction in a rat xenograft-radiculopathy model. Eur. Cells Mater. 42, 90–109. doi: 10.22203/eCM.v042a07
Khan, A., Khan, S., and Kim, Y. (2019). Insight into pain modulation: Nociceptors sensitization and therapeutic targets. Curr. Drug Targets 20, 775–788.
Kim, E., Thu, D., Tippett, L., Oorschot, D., Hogg, V., Roxburgh, R., et al. (2014). Cortical interneuron loss and symptom heterogeneity in Huntington disease. Ann. Neurol. 75, 717–727.
Kim, S., Shahani, N., Bae, B., Sbodio, J., Chung, Y., Nakaso, K., et al. (2016). Allele-specific regulation of mutant huntingtin by Wig1, a downstream target of p53. Hum. Mol. Genet. 25, 2514–2524.
Knabl, J., Witschi, R., Hosl, K., Reinold, H., Zeilhofer, U., Ahmadi, S., et al. (2008). Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 451, 330–334.
Kopach, O., and Voitenko, N. (2021). Spinal AMPA receptors: Amenable players in central sensitization for chronic pain therapy? Channels 15, 284–297. doi: 10.1080/19336950.2021.1885836
Kopach, O., Krotov, V., Shysh, A., Sotnic, A., Viatchenko-Karpinski, V., Dosenko, V., et al. (2018). Spinal PKCalpha inhibition and gene-silencing for pain relief: AMPAR trafficking at the synapses between primary afferents and sensory interneurons. Sci. Rep. 8:10285. doi: 10.1038/s41598-018-28512-9
Kosinski, C., Schlangen, C., Gellerich, F., Gizatullina, Z., Deschauer, M., Schiefer, J., et al. (2007). Myopathy as a first symptom of Huntington’s disease in a marathon runner. Mov. Disord. 22, 1637–1640.
Krause, A., Prather, A., Wager, T., Lindquist, M., and Walker, M. (2019). The pain of sleep loss: A brain characterization in humans. J. Neurosci. 39, 2291–2300.
Kreutzwiser, D., and Tawfic, Q. (2019). Expanding role of NMDA receptor antagonists in the management of pain. CNS Drugs 33, 347–374. doi: 10.1007/s40263-019-00618-2
Kun, L., Lu, L., Yongda, L., Xingyue, L., and Guang, H. (2019). Hyperbaric oxygen promotes mitophagy by activating CaMKKbeta/AMPK signal pathway in rats of neuropathic pain. Mol. Pain 15:1744806919871381. doi: 10.1177/1744806919871381
Kwan, W., Trager, U., Davalos, D., Chou, A., Bouchard, J., Andre, R., et al. (2012). Mutant huntingtin impairs immune cell migration in Huntington disease. J. Clin. Investig. 122, 4737–4747.
Lamirault, C., Yu-Taeger, L., Doyere, V., Riess, O., Nguyen, H., and El Massioui, N. (2017). Altered reactivity of central amygdala to GABAAR antagonist in the BACHD rat model of Huntington disease. Neuropharmacology 123, 136–147. doi: 10.1016/j.neuropharm.2017.05.032
Lastres-Becker, I., de Miguel, R., De Petrocellis, L., Makriyannis, A., Di Marzo, V., and Fernandez-Ruiz, J. (2003). Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington’s disease. J. Neurochem. 84, 1097–1109.
Lee, C., Cantle, J., and Yang, X. (2013). Genetic manipulations of mutant huntingtin in mice: New insights into Huntington’s disease pathogenesis. FEBS J. 280, 4382–4394. doi: 10.1111/febs.12418
Lee, K., Jo, Y., Chung, G., Jung, J., Kim, Y., and Park, C. (2021). Functional importance of transient receptor potential (TRP) channels in neurological disorders. Front. Cell Dev. Biol. 9:611773. doi: 10.3389/fcell.2021.611773
Lemoine, L., Lunven, M., Bapst, B., Cleret de Langavant, L., de Gardelle, V., and Bachoud-Levi, A. (2021). The specific role of the striatum in interval timing: The Huntington’s disease model. NeuroImage Clin. 32:102865.
Lesinskiene, S., Rojaka, D., Praninskiene, R., Morkuniene, A., Matuleviciene, A., and Utkus, A. (2020). Juvenile Huntington’s disease: Two case reports and a review of the literature. J. Med. Case Rep. 14:173.
Li, L., Zou, Y., Liu, B., Yang, R., Yang, J., Sun, M., et al. (2020). Contribution of the P2X4 receptor in rat hippocampus to the comorbidity of chronic pain and depression. ACS Chem. Neurosci. 11, 4387–4397.
Li, X., Matsuura, T., Xue, M., Chen, Q., Liu, R., Lu, J., et al. (2021). Oxytocin in the anterior cingulate cortex attenuates neuropathic pain and emotional anxiety by inhibiting presynaptic long-term potentiation. Cell Rep. 36:109411. doi: 10.1016/j.celrep.2021.109411
Lin, Y., Hsiao, H., Wu, S., and Liu, Y. (2018). Huntington mice demonstrate diminished pain response in inflammatory pain model. Anesth. Analg. 126, 661–669.
Liu, H., Chou, K., and Chen, W. (2018). Migraine and the hippocampus. Curr. Pain Headache Rep. 22:13.
Liu, X., Xie, Z., Li, S., He, J., Cao, S., and Xiao, Z. (2021). PRG-1 relieves pain and depressive-like behaviors in rats of bone cancer pain by regulation of dendritic spine in hippocampus. Int. J. Biol. Sci. 17, 4005–4020. doi: 10.7150/ijbs.59032
Liu, Y., Ying, Y., Li, Y., Eyo, U., Chen, T., Zheng, J., et al. (2019). Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci. 22, 1771–1781. doi: 10.1038/s41593-019-0511-3
Mackay, J., Nassrallah, W., and Raymond, L. (2018). Cause or compensation?-Altered neuronal Ca(2+) handling in Huntington’s disease. CNS Neurosci. Ther. 24, 301–310.
Madariaga, V., Overdorp, E., Claassen, J., Brazil, I., and Oosterman, J. (2021). Association between self-reported pain, cognition, and neuropathology in older adults admitted to an outpatient memory clinic-a cross-sectional study. Brain Sci. 11:1156. doi: 10.3390/brainsci11091156
Mai, C., Tan, Z., Xu, Y., Zhang, J., Huang, Z., Wang, D., et al. (2021). CXCL12-mediated monocyte transmigration into brain perivascular space leads to neuroinflammation and memory deficit in neuropathic pain. Theranostics 11, 1059–1078. doi: 10.7150/thno.44364
Maiuri, T., Suart, C., Hung, C., Graham, K., Barba Bazan, C., and Truant, R. (2019). DNA damage repair in Huntington’s disease and other neurodegenerative diseases. Neurotherapeutics 16, 948–956.
Malfliet, A., Kregel, J., Coppieters, I., De Pauw, R., Meeus, M., Roussel, N., et al. (2018). Effect of pain neuroscience education combined with cognition-targeted motor control training on chronic spinal pain: A randomized clinical trial. JAMA Neurol. 75, 808–817.
Martikainen, I., Nuechterlein, E., Pecina, M., Love, T., Cummiford, C., Green, C., et al. (2015). Chronic back pain is associated with alterations in dopamine neurotransmission in the ventral striatum. J. Neurosci. 35, 9957–9965.
Mason, S., Zhang, J., Begeti, F., Guzman, N., Lazar, A., Rowe, J., et al. (2015). The role of the amygdala during emotional processing in Huntington’s disease: From pre-manifest to late stage disease. Neuropsychologia 70, 80–89. doi: 10.1016/j.neuropsychologia.2015.02.017
Matsui, J., Vaidya, J., Johnson, H., Magnotta, V., Long, J., Mills, J., et al. (2014). Diffusion weighted imaging of prefrontal cortex in prodromal Huntington’s disease. Hum. Brain Mapp. 35, 1562–1573.
Matsui, J., Vaidya, J., Wassermann, D., Kim, R., Magnotta, V., Johnson, H., et al. (2015). Prefrontal cortex white matter tracts in prodromal Huntington disease. Hum. Brain Mapp. 36, 3717–3732.
McColgan, P., and Tabrizi, S. (2018). Huntington’s disease: A clinical review. Eur. J. Neurol. 25, 24–34.
Millian-Morell, L., Lopez-Alburquerque, T., Rodriguez-Rodriguez, A., Gomez-Nieto, R., Carro, J., Meilan, J., et al. (2018). Relations between sensorimotor integration and speech disorders in Parkinson’s disease. Curr. Alzheimer Res. 15, 149–156.
Miranda, A., Cardozo, P., Silva, F., de Souza, J., Olmo, I., Cruz, J., et al. (2019). Alterations of calcium channels in a mouse model of Huntington’s disease and neuroprotection by blockage of Ca(V)1 channels. ASN Neuro 11:1759091419856811.
Moore, C., Gupta, R., Jordt, S., Chen, Y., and Liedtke, W. (2018). Regulation of pain and itch by TRP channels. Neurosci. Bull. 34, 120–142.
Muhlau, M., Engl, C., Boucard, C., Schmidt, P., Biberacher, V., Gorsch, I., et al. (2014). Spinal cord atrophy in early Huntington’s disease. Ann. Clin. Transl. Neurol. 1, 302–306.
Nana, A., Kim, E., Thu, D., Oorschot, D., Tippett, L., Hogg, V., et al. (2014). Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington’s disease. J. Huntingtons Dis. 3, 45–64. doi: 10.3233/JHD-140092
Nees, F., and Becker, S. (2018). Psychological processes in chronic pain: Influences of reward and fear learning as key mechanisms - behavioral evidence, neural circuits, and maladaptive changes. Neuroscience 387, 72–84. doi: 10.1016/j.neuroscience.2017.08.051
Neugebauer, V., Mazzitelli, M., Cragg, B., Ji, G., Navratilova, E., and Porreca, F. (2020). Amygdala, neuropeptides, and chronic pain-related affective behaviors. Neuropharmacology 170:108052.
Nilius, B., and Owsianik, G. (2011). The transient receptor potential family of ion channels. Genome Biol. 12:218.
Ninneman, J., Gretzon, N., Stegner, A., Lindheimer, J., Falvo, M., Wylie, G., et al. (2022). Pain, but not physical activity, is associated with gray matter volume differences in gulf war veterans with chronic pain. J. Neurosci. 42, 5605–5616.
Nwaobi, S., Cuddapah, V., Patterson, K., Randolph, A., and Olsen, M. (2016). The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 132, 1–21.
Oepen, G., Doerr, M., and Thoden, U. (1981). Visual (VEP). and somatosensory (SSEP). evoked potentials in Huntington’s chorea. Electroencephalogr. Clin. Neurophysiol. 51, 666–670. doi: 10.1016/0013-4694(81)90211-x
Ong, W., Stohler, C., and Herr, D. (2019). Role of the prefrontal cortex in pain processing. Mol. Neurobiol. 56, 1137–1166.
Osipovitch, M., Asenjo Martinez, A., Mariani, J., Cornwell, A., Dhaliwal, S., Zou, L., et al. (2019). Human ESC-derived chimeric mouse models of Huntington’s disease reveal cell-intrinsic defects in glial progenitor cell differentiation. Cell Stem Cell 24:e107. doi: 10.1016/j.stem.2018.11.010
Pan, J., Zhao, Y., Sang, R., Yang, R., Bao, J., Wu, Y., et al. (2023). Huntington-associated protein 1 inhibition contributes to neuropathic pain by suppressing Cav1.2 activity and attenuating inflammation. Pain 164, e286–e302. doi: 10.1097/j.pain.0000000000002837
Pan, Y., Zhu, Y., Yang, W., Tycksen, E., Liu, S., Palucki, J., et al. (2018). The role of Twist1 in mutant huntingtin-induced transcriptional alterations and neurotoxicity. J. Biol. Chem. 293, 11850–11866.
Park, H. (2018). Cortical axonal secretion of BDNF in the striatum is disrupted in the mutant-huntingtin knock-in mouse model of Huntington’s disease. Exp. Neurobiol. 27, 217–225.
Paulsen, J., Langbehn, D., Stout, J., Aylward, E., Ross, C., Nance, M., et al. (2008). Detection of Huntington’s disease decades before diagnosis: The Predict-HD study. J. Neurol. Neurosurg. Psychiatry 79, 874–880.
Perrotta, A., Serpino, C., Cormio, C., Serrao, M., Sandrini, G., Pierelli, F., et al. (2012). Abnormal spinal cord pain processing in Huntington’s disease. The role of the diffuse noxious inhibitory control. Clin. Neurophysiol. 123, 1624–1630.
Price, D. (2000). Psychological and neural mechanisms of the affective dimension of pain. Science 288, 1769–1772.
Pringsheim, T., Wiltshire, K., Day, L., Dykeman, J., Steeves, T., and Jette, N. (2012). The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 27, 1083–1091.
Quarrell, O., O’Donovan, K., Bandmann, O., and Strong, M. (2012). The prevalence of juvenile Huntington’s disease: A review of the literature and meta-analysis. PLoS Curr. 4:e4f8606b742ef3. doi: 10.1371/4f8606b742ef3
Quigley, J. (2017). Juvenile Huntington’s disease: Diagnostic and treatment considerations for the psychiatrist. Curr. Psychiatry Rep. 19:9. doi: 10.1007/s11920-017-0759-9
Rallapalle, V., King, A., and Gray, M. (2021). BACHD mice recapitulate the striatal parvalbuminergic interneuron loss found in Huntington’s disease. Front. Neuroanat. 15:673177. doi: 10.3389/fnana.2021.673177
Reddy, P., and Shirendeb, U. (2012). Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim. Biophys. Acta 1822, 101–110.
Reiner, A., Albin, R., Anderson, K., D’Amato, C., Penney, J., and Young, A. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 85, 5733–5737.
Reiner, A., and Deng, Y. (2018). Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci. Ther. 24, 250–280.
Reyes Molon, L., Yanez Saez, R., and Lopez-Ibor Alcocer, M. (2010). Juvenile Huntington’s disease: A case report and literature review. Actas Espanolas Psiquiatria 38, 285–294.
Robertson, L., Santini, H., O’Donovan, K., Squitieri, F., Barker, R., Rakowicz, M., et al. (2012). Current pharmacological management in juvenile Huntington’s disease. PLoS Curr. 4:RRN1304. doi: 10.1371/currents.RRN1304
Roca-Lapirot, O., Radwani, H., Aby, F., Nagy, F., Landry, M., and Fossat, P. (2018). Calcium signalling through L-type calcium channels: Role in pathophysiology of spinal nociceptive transmission. Br. J. Pharmacol. 175, 2362–2374. doi: 10.1111/bph.13747
Rosas, H., Liu, A., Hersch, S., Glessner, M., Ferrante, R., Salat, D., et al. (2002). Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology 58, 695–701. doi: 10.1212/wnl.58.5.695
Rubinsztein, D., Leggo, J., Coles, R., Almqvist, E., Biancalana, V., Cassiman, J., et al. (1996). Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am. J. Hum. Genet. 59, 16–22.
Saba, J., Lopez Couselo, F., Turati, J., Carniglia, L., Durand, D., de Laurentiis, A., et al. (2020). Astrocytes from cortex and striatum show differential responses to mitochondrial toxin and BDNF: Implications for protection of striatal neurons expressing mutant huntingtin. J. Neuroinflammation 17:290. doi: 10.1186/s12974-020-01965-4
Schrank, S., Barrington, N., and Stutzmann, G. (2020). Calcium-handling defects and neurodegenerative disease. Cold Spring Harbor Perspect. Biol. 12:a035212.
Sciacca, G., and Cicchetti, F. (2017). Mutant huntingtin protein expression and blood-spinal cord barrier dysfunction in huntington disease. Ann. Neurol. 82, 981–994.
Seminowicz, D., and Moayedi, M. (2017). The dorsolateral prefrontal cortex in acute and chronic pain. J. Pain 18, 1027–1035.
Sepers, M., Smith-Dijak, A., LeDue, J., Kolodziejczyk, K., Mackie, K., and Raymond, L. (2018). Endocannabinoid-specific impairment in synaptic plasticity in striatum of Huntington’s disease mouse model. J. Neurosci. 38, 544–554.
Shirendeb, U., Reddy, A., Manczak, M., Calkins, M., Mao, P., Tagle, D., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455.
Singh, A., and Agrawal, N. (2021). Metabolism in Huntington’s disease: A major contributor to pathology. Metab. Brain Dis. 37, 1757–1771. doi: 10.1007/s11011-021-00844-y
Sipila, J., Hietala, M., Siitonen, A., Paivarinta, M., and Majamaa, K. (2015). Epidemiology of Huntington’s disease in Finland. Parkinsonism Relat. Disord. 21, 46–49.
Sipione, S., Rigamonti, D., Valenza, M., Zuccato, C., Conti, L., Pritchard, J., et al. (2016). Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum. Mol. Genet. 11, 1953–65.
Skotte, N., Andersen, J., Santos, A., Aldana, B., Willert, C., Norremolle, A., et al. (2018). Integrative characterization of the R6/2 mouse model of Huntington’s disease reveals dysfunctional astrocyte metabolism. Cell Rep. 23, 2211–2224.
Sluka, K., and Gregory, N. (2015). The dichotomized role for acid sensing ion channels in musculoskeletal pain and inflammation. Neuropharmacology 94, 58–63. doi: 10.1016/j.neuropharm.2014.12.013
Smith, M., Asada, N., and Malenka, R. (2021). Anterior cingulate inputs to nucleus accumbens control the social transfer of pain and analgesia. Science 371, 153–159. doi: 10.1126/science.abe3040
Snowden, J. (2017). The Neuropsychology of Huntington’s Disease. Arch. Clin. Neuropsychol. 32, 876–887.
Sprenger, G., Roos, R., van Zwet, E., Reijntjes, R., Achterberg, W., and de Bot, S. (2021). The prevalence of pain in Huntington’s disease in a large worldwide cohort. Parkinsonism Relat. Disord. 89, 73–78.
Sprenger, G., van der Zwaan, K., Roos, R., and Achterberg, W. (2019). The prevalence and the burden of pain in patients with Huntington disease: A systematic review and meta-analysis. Pain 160, 773–783. doi: 10.1097/j.pain.0000000000001472
St John Smith, E. (2018). Advances in understanding nociception and neuropathic pain. J. Neurol. 265, 231–238.
Sweidan, W., Bao, F., Bozorgzad, N., and George, E. (2020). White and gray matter abnormalities in manifest Huntington’s disease: Cross-sectional and longitudinal analysis. J. Neuroimaging 30, 351–358.
Tabrizi, S., Scahill, R., Durr, A., Roos, R., Leavitt, B., Jones, R., et al. (2011). Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: The 12-month longitudinal analysis. Lancet Neurol. 10, 31–42.
Tan, L., and Kuner, R. (2021). Neocortical circuits in pain and pain relief. Nat. Rev. Neurosci. 22, 458–471. doi: 10.1038/s41583-021-00468-2
Teo, R., Ferrari Bardile, C., Tay, Y., Yusof, N., Kreidy, C., Tan, L., et al. (2019). Impaired remyelination in a mouse model of Huntington disease. Mol. Neurobiol. 56, 6873–6882. doi: 10.1007/s12035-019-1579-1
Teo, R., Hong, X., Yu-Taeger, L., Huang, Y., Tan, L., Xie, Y., et al. (2016). Structural and molecular myelination deficits occur prior to neuronal loss in the YAC128 and BACHD models of Huntington disease. Hum. Mol. Genet. 25, 2621–2632. doi: 10.1093/hmg/ddw122
Thompson, J., and Neugebauer, V. (2017). Amygdala plasticity and pain. Pain Res. Manag. 2017:8296501.
Thompson, J., and Neugebauer, V. (2019). Cortico-limbic pain mechanisms. Neurosci. Lett. 702, 15–23.
Thu, D., Oorschot, D., Tippett, L., Nana, A., Hogg, V., Synek, B., et al. (2010). Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain 133, 1094–1110. doi: 10.1093/brain/awq047
Tong, X., Ao, Y., Faas, G., Nwaobi, S., Xu, J., Haustein, M., et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17, 694–703.
Trager, U., Andre, R., Lahiri, N., Magnusson-Lind, A., Weiss, A., Grueninger, S., et al. (2014). HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain 137, 819–833.
Tsantoulas, C. (2015). Emerging potassium channel targets for the treatment of pain. Curr. Opin. Support. Palliat. Care 9, 147–154.
Tsuda, M. (2021). [Pain signal processing in the spinal dorsal horn and glial cells]. Brain Nerve 73, 803–810.
Underwood, M., Bonas, S., Dale, M., and REGISTRY Investigators of the European Huntington’s Disease Network (2017). Huntington’s disease: Prevalence and psychological indicators of pain. Mov. Disord. Clin. Pract. 4, 198–204. doi: 10.1002/mdc3.12376
van Duijn, E., Craufurd, D., Hubers, A., Giltay, E., Bonelli, R., Rickards, H., et al. (2014). Neuropsychiatric symptoms in a European Huntington’s disease cohort (REGISTRY). J. Neurol. Neurosurg. Psychiatry 85, 1411–1418.
van Duijn, E., Kingma, E., and van der Mast, R. (2007). Psychopathology in verified Huntington’s disease gene carriers. J. Neuropsychiatry Clin. Neurosci. 19, 441–448.
Vasic, V., and Schmidt, M. (2017). Resilience and vulnerability to pain and inflammation in the hippocampus. Int. J. Mol. Sci. 18:739.
Wang, S., Wang, B., Shang, D., Zhang, K., Yan, X., and Zhang, X. (2022). Ion channel dysfunction in astrocytes in neurodegenerative diseases. Front. Physiol. 13:814285. doi: 10.3389/fphys.2022.814285
Wang, Y., Wang, J., Xia, S., Gutstein, H., Huang, Y., Schluter, O., et al. (2021). Neuropathic pain generates silent synapses in thalamic projection to anterior cingulate cortex. Pain 162, 1322–1333. doi: 10.1097/j.pain.0000000000002149
Wei, X., Centeno, M., Ren, W., Borruto, A., Procissi, D., Xu, T., et al. (2021). Activation of the dorsal, but not the ventral, hippocampus relieves neuropathic pain in rodents. Pain 162, 2865–2880.
Wilhelms, W., Bellenberg, B., Koster, O., Weiler, F., Hoffmann, R., Gold, R., et al. (2017). Progressive spinal cord atrophy in manifest and premanifest Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 88, 614–616.
Wilkes, F., Abaryan, Z., Ching, C., Gutman, B., Madsen, S., Walterfang, M., et al. (2019). Striatal morphology and neurocognitive dysfunction in Huntington disease: The IMAGE-HD study. Psychiatry Res. Neuroimaging 291, 1–8.
Wilkie, C., Barnes, J., Benson, C., Brymer, K., Nafar, F., and Parsons, M. (2020). Hippocampal synaptic dysfunction in a mouse model of Huntington disease is not alleviated by ceftriaxone treatment. eNeuro 7:ENEURO.0440-19.2020. doi: 10.1523/ENEURO.0440-19.2020
Wilson, H., Dervenoulas, G., and Politis, M. (2018). Structural magnetic resonance imaging in Huntington’s disease. Int. Rev. Neurobiol. 142, 335–380.
Wilton, D., and Stevens, B. (2020). The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol. Dis. 143:104963.
Wolf, R., Thomann, P., Thomann, A., Vasic, N., Wolf, N., Landwehrmeyer, G., et al. (2013). Brain structure in preclinical Huntington’s disease: A multi-method approach. Neurodegener. Dis. 12, 13–22.
Wood, T., Barry, J., Yang, Z., Cepeda, C., Levine, M., and Gray, M. (2019). Mutant Huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 28, 487–500.
Wyant, K., Ridder, A., and Dayalu, P. (2017). Huntington’s disease-update on treatments. Curr. Neurol. Neurosci. Rep. 17:33.
Xiang, H., Lin, L., Hu, X., Zhu, H., Li, H., Zhang, R., et al. (2019). AMPK activation attenuates inflammatory pain through inhibiting NF-kappaB activation and IL-1beta expression. J. Neuroinflammation 16:34.
Xiao, X., Ding, M., and Zhang, Y. (2021). Role of the anterior cingulate cortex in translational pain research. Neurosci. Bull. 37, 405–422.
Yam, M., Loh, Y., Tan, C., Khadijah Adam, S., Abdul Manan, N., and Basir, R. (2018). General pathways of pain sensation and the major neurotransmitters involved in pain regulation. Int. J. Mol. Sci. 19:2164. doi: 10.3390/ijms19082164
Yang, H., Wu, L., Deng, H., Chen, Y., Zhou, H., Liu, M., et al. (2020). Anti-inflammatory protein TSG-6 secreted by bone marrow mesenchymal stem cells attenuates neuropathic pain by inhibiting the TLR2/MyD88/NF-kappaB signaling pathway in spinal microglia. J. Neuroinflammation 17:154.
You, B., and Jackson, T. (2021). Gray matter volume differences between more versus less resilient adults with chronic musculoskeletal pain: A voxel-based morphology study. Neuroscience 457, 155–164.
Yusuf, I., Chen, H., Cheng, P., Chang, C., Tsai, S., Chuang, J., et al. (2021). Fibroblast growth factor 9 stimulates neuronal length through NF-kB signaling in striatal cell Huntington’s disease models. Mol. Neurobiol. 58, 2396–2406.
Zhan, L., Li, R., Sun, Y., Dou, M., Yang, W., He, S., et al. (2018). Effect of mito-TEMPO, a mitochondria-targeted antioxidant, in rats with neuropathic pain. Neuroreport 29, 1275–1281. doi: 10.1097/WNR.0000000000001105
Zhang, X., Wan, J., and Tong, X. (2018). Potassium channel dysfunction in neurons and astrocytes in Huntington’s disease. CNS Neurosci. Ther. 24, 311–318.
Zhou, R., Wu, X., Wang, Z., Xie, Y., Ge, J., and Chen, F. (2016). Novel insights into acid-sensing ion channels: Implications for degenerative diseases. Aging Dis. 7, 491–501. doi: 10.14336/AD.2015.1213
Zhou, W., Jin, Y., Meng, Q., Zhu, X., Bai, T., Tian, Y., et al. (2019). A neural circuit for comorbid depressive symptoms in chronic pain. Nat. Neurosci. 22, 1649–1658.
Zhu, X., Zhou, W., Jin, Y., Tang, H., Cao, P., Mao, Y., et al. (2019). A central amygdala input to the parafascicular nucleus controls comorbid pain in depression. Cell Rep. 29, 3847–3858.e5.
Keywords: Huntington disease, pain, prevalence, neuropathology, pain symptoms
Citation: Li J, Wang Y, Yang R, Ma W, Yan J, Li Y, Chen G and Pan J (2023) Pain in Huntington’s disease and its potential mechanisms. Front. Aging Neurosci. 15:1190563. doi: 10.3389/fnagi.2023.1190563
Received: 21 March 2023; Accepted: 19 June 2023;
Published: 06 July 2023.
Edited by:
Yong Ho Kim, Gachon University, Republic of KoreaReviewed by:
Md. Mahbubur Rahman, Gachon University College of Medicine, Republic of KoreaCopyright © 2023 Li, Wang, Yang, Ma, Yan, Li, Chen and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingying Pan, cGFuamluZ3lpbmcwNTA1QDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.