 Liqian Zhou
Liqian Zhou Yuzhuang Wang
Yuzhuang Wang Lihong Peng
Lihong Peng Zejun Li
Zejun Li Xueming Luo1*
Xueming Luo1*- 1School of Computer Science, Hunan University of Technology, Zhuzhou, China
- 2School of Computer Science, Hunan Institute of Technology, Hengyang, China
Introduction: Drug-target interaction prediction is one important step in drug research and development. Experimental methods are time consuming and laborious.
Methods: In this study, we developed a novel DTI prediction method called EnGDD by combining initial feature acquisition, dimensional reduction, and DTI classification based on Gradient boosting neural network, Deep neural network, and Deep Forest.
Results: EnGDD was compared with seven stat-of-the-art DTI prediction methods (BLM-NII, NRLMF, WNNGIP, NEDTP, DTi2Vec, RoFDT, and MolTrans) on the nuclear receptor, GPCR, ion channel, and enzyme datasets under cross validations on drugs, targets, and drug-target pairs, respectively. EnGDD computed the best recall, accuracy, F1-score, AUC, and AUPR under the majority of conditions, demonstrating its powerful DTI identification performance. EnGDD predicted that D00182 and hsa2099, D07871 and hsa1813, DB00599 and hsa2562, D00002 and hsa10935 have a higher interaction probabilities among unknown drug-target pairs and may be potential DTIs on the four datasets, respectively. In particular, D00002 (Nadide) was identified to interact with hsa10935 (Mitochondrial peroxiredoxin3) whose up-regulation might be used to treat neurodegenerative diseases. Finally, EnGDD was used to find possible drug targets for Parkinson's disease and Alzheimer's disease after confirming its DTI identification performance. The results show that D01277, D04641, and D08969 may be applied to the treatment of Parkinson's disease through targeting hsa1813 (dopamine receptor D2) and D02173, D02558, and D03822 may be the clues of treatment for patients with Alzheimer's disease through targeting hsa5743 (prostaglandinendoperoxide synthase 2). The above prediction results need further biomedical validation.
Discussion: We anticipate that our proposed EnGDD model can help discover potential therapeutic clues for various diseases including neurodegenerative diseases.
1. Introduction
Identification of Drug-Target Interactions (DTIs) for various diseases is one key step in drug research and development (Peng et al., 2015; Zhang et al., 2019; Chu et al., 2022; Zhou et al., 2022; Liu et al., 2023), however, it is time-consuming, costly, and low-success rate (Dickson and Gagnon, 2004; Kola and Landis, 2017; Peng et al., 2022b; Shen et al., 2022). Drug repositioning (Wang and Zeng, 2013; Liang et al., 2022a; Tian et al., 2022b; Sun et al., 2023) can find new indications from existing drugs and expand their scopes and uses. Drug repositioning demonstrates several advantages compared to an entirely new drug design. First, it has less risk related to subsequent efficacy trial failure. Second, drug research and development time can be shortened because most of the preclinical testing and safety assessment have already been done. Finally, less investment is required because drug repositioning can still provide significant data in preclinical, clinical phase I, and clinical phase II stages. In summary, drug repositioning has been widely applied to DTI inference (Tian et al., 2022a; Zhang et al., 2022a).
To date, various drug repositioning methods (Chen et al., 2012, 2016) have been used to identify potential DTIs. These methods can be roughly divided into three categories: docking simulation (Guo et al., 2022; Peng et al., 2022a; Zhao et al., 2023), network-based methods, and machine learning-based methods. Docking simulation first obtains 3D structures of drugs and proteins and then runs molecular simulations to compute the binding ability for each drug-target pair (Li et al., 2006; Pujadas et al., 2008). However, 3D structures of a few proteins are unknown (for example, membrane proteins), thus it is not possible to detect potential drugs interacting with these proteins (Opella, 2013). Furthermore, docking simulation-based DTI identification can be challenging.
Network-based methods (Lotfi Shahreza et al., 2018) provide an efficient way for DTI prediction. Network-based methods integrate protein-protein similarity, drug-drug similarity, and known DTIs in to a heterogeneous network and develop network algorithms to find new DTIs (Chen et al., 2012). For example, Chen et al. (2012) proposed a random walk with a restart-based method. Mei et al. (2013) developed a bipartite local model BLM-NII. Van Laarhoven and Marchiori (2013) designed a computational model WNN-GIP by combining weighted nearest neighbor with Gaussian interaction profiles.
Machine learning-based methods use machine learning models to capture relationships between drugs and targets. For example, Precup et al. (2012), Buza and Peška (2017) designed K-nearest neighbor models with hubness-aware regression technique to alleviate the detrimental effect of bad hubs in a DTI network. In particular, deep learning has obtained wide application in DTI prediction. For example, Zong et al. (2017) developed a deep learning model based on the topology of a multipartite DTI network, Wang et al. (2018) used a deep ensemble learning model with a stacked autoencoder, Öztürk et al. (2018) designed a deep learning model with character representations, You et al. (2019) exploited a deep ensemble learning method with LASSO regression, Cheng et al. (2021) combined multi-head self-attention and graph attention network, Lee and Nam (2022) explored a sequence-based approach, Li et al. (2022) designed a dual-stream graph neural network, Mukherjee et al. (2022) used a deep graph convolutional network and LSTM, Zhang et al. (2022b) exploited a graph neural network, and Tayebi et al. (2022) designed a deep ensemble-balanced learning model.
The above three types of methods effectively identify potential DTIs. Network-based methods predict possible DTIs by combining topological information and node features in a DTI network. However, network-based methods cannot identify potential DTIs for new drugs or targets. Machine learning-based methods utilized feature information involved in drugs and targets and can significantly improve DTI prediction performance. However, machine learning-based methods are susceptive to data quality and feature selection and need huge amounts of data. In this study, we developed a novel DTI prediction method called EnGDD by combining initial feature acquisition, dimensional reduction, and DTI classification based on Gradient boosting neural network (Grownet) (Badirli et al., 2020), Deep Neural Network (DNN) and Deep Forest (DeepForest) (Zhou and Feng, 2019).
Parkinson's Disease (PD) and Alzheimer's Disease (AD) are two common neurodegenerative diseases. PD is mainly characterized by movement disorders, muscle stiffness, tremor, and other symptoms. Its pathogenesis involves many aspects including environment, genetics, and neurochemistry (Poewe et al., 2017). AD has cognitive impairment, memory loss, language impairment, and other symptoms. Its pathogenesis is still unclear. Although a few drugs have been applied to their therapies, new therapeutic clues are still essential to the two diseases (Iraji et al., 2020; Yiannopoulou and Papageorgiou, 2020; Liang et al., 2022b; Lin et al., 2022). Thus, we used the proposed EnGDD method and found new therapeutic clues for PD and AD.
2. Materials and methods
2.1. Data preparation
Yamanishi_08 has been widely used as a gold standard dataset in the field of DTI prediction. It was collected from the KEGG BRITE (Kanehisa et al., 2017), BRENDA (Schomburg et al., 2004), SuperTarget (Günther et al., 2007), and DrugBank (Wishart et al., 2008) databases. It was categorized into four DTI datasets based on different target proteins, that is, nuclear receptors (NR), G protein-coupled receptors (GPCR), ion channels (IC), and enzymes (E). Chu et al. (2021) collected new drugs, new targets, and new DTIs and further updated the four DTI datasets. On the four datasets, there are 886 DTIs between 541 drugs and 33 targets, 5383 DTIs between 1680 drugs and 156 targets, 6385 DTIs between 765 drugs and 238 targets, and 7371 DTIs between 1777 drugs and 1411 targets after an update, respectively. We used the four datasets to capture potential DTIs.
2.2. Initial feature acquisition
ChemDes (Dong et al., 2015) and BioTriangle (Dong et al., 2016) were used to extract the initial features of drugs and targets. ChemDes (Dong et al., 2015) is a freely available tool for molecular description and fingerprint calculation. A drug can be first represented by a Simplified Molecular Input Line Entry System (SMILES) string. SMILES string is then converted into fingerprints via ChemDes. In this study, we use ChemDes and describe each drug as a 1,538-dimensional vector.
BioTriangle (Dong et al., 2016) provides 14 types of biological features to represent each target protein. These features include amino acid composition, dipeptide composition, tripeptide composition, CTD composition, CTD transition, CTD distribution, M-B autocorrelation, Moran autocorrelation, Geary autocorrelation, conjoint triad features, quasi-sequence order descriptors, sequence order coupling number, pseudo amino acid composition 1, and pseudo amino acid composition 2. In this study, we use BioTriangle and describe each target as a 10029-dimensional vector.
2.3. Dimensional reduction
The dimensions of the extracted drug and target features are high and there is a large amount of robust information. We reduce the feature dimensions using Principal Component Analysis (PCA). PCA is a common machine learning algorithm mainly used for dimensionality reduction and feature extraction. It can map raw high-dimensional data into low-dimensional space while preserving main information and structure of data, thus making the prediction model more efficient and accurate. Finally, drugs and targets can be denoted as two d-dimensional vectors. In addition the two vectors are concatenated and each drug-target pair is represented using a 2d-dimensional vector x.
2.4. DTI classification
We first compute interaction probability for each drug-target pair using Grownet, DNN, and DeepForest, respectively. The probability is then integrated by the soft voting and each drug-target can be classified. The details are shown in Figure 1.
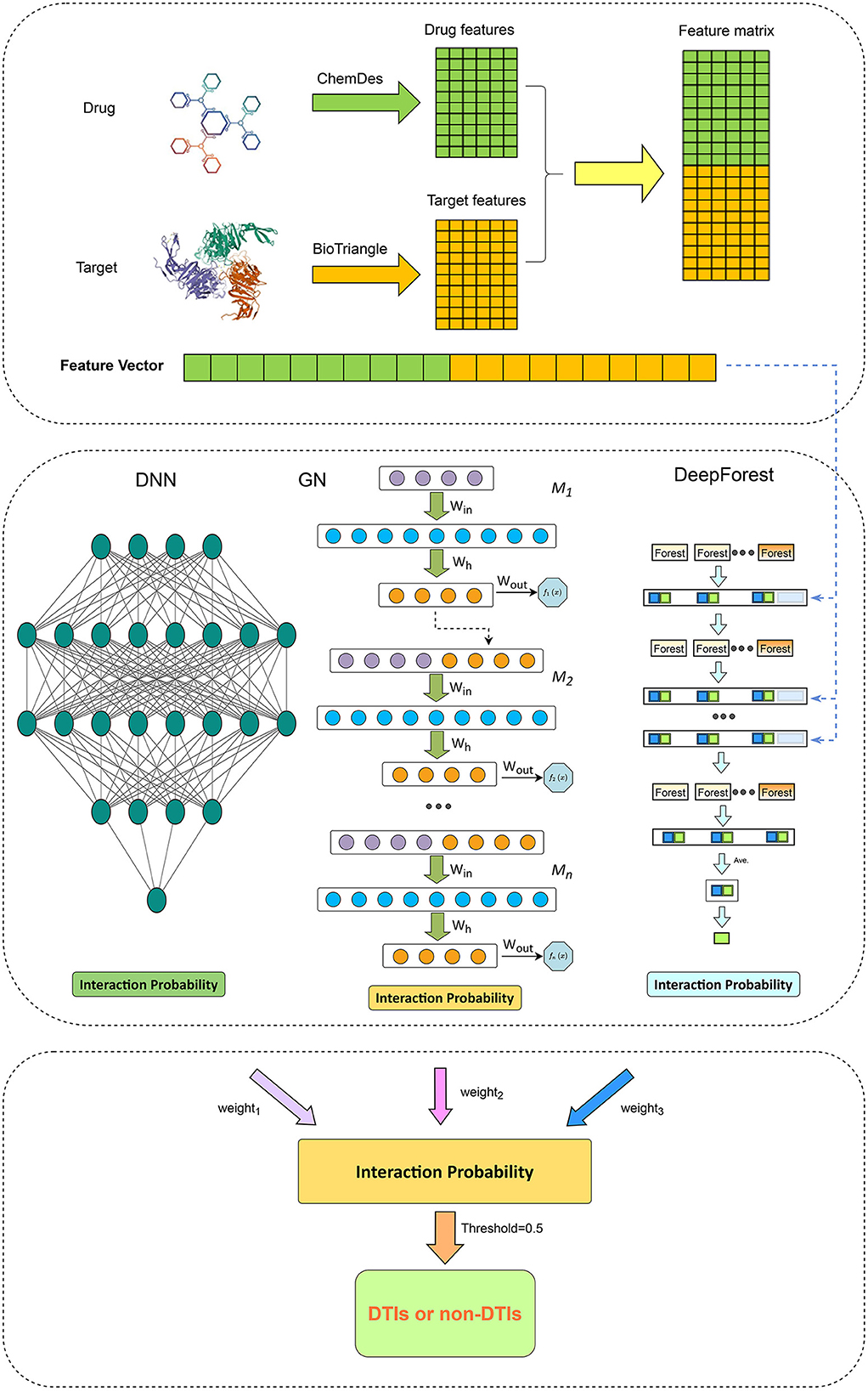
Figure 1. The pipeline for drug-target interaction prediction based on Grownet, DNN, and DeepForest.
2.4.1. Gradient boosting neural network
Gradient boosting machine (Friedman, 2001; ZhouZhou et al., 2021) is a function estimation approach based on numerical optimization and obtains wide application (Peng et al., 2021). Grownet (Badirli et al., 2020) is a gradient boosting framework with shallow neural networks. As shown in Figure 1, Grownet uses shallow neural networks as basic learners and propagates information from the previous classifier to the next one.
At each layer, the learner is trained based on DTI features. The final output is one weighted sum of outputs from all learners: where K denotes the number of learners.
For a drug-target pair xi with a 2d-dimensional feature in a DTI dataset , Grownet obtains its label through K additive functions by Equation (1):
where and αk denote the space of multilayer perceptrons and the step size, respectively. fk is a shallow neural network with linear activation function in the output layer.
Let denote the output at the (t−1)th layer for xi, we minimize the following loss function by Equation (2):
Thus, the objective function is simplified by Equation (3):
where ŷi = −gi/hi, gi and hi denote the first-order and second-order gradients of the objective function at xi, respectively.
2.4.2. Deep neural network
Deep neural networks have been broadly applied in the field of bioinformatics. In this section, we designed a DNN to classify unknown drug-target pairs. The constructed DNN comprises an input layer, multiple hidden layers, and a larger output layer. For a drug-target pair xi with 2d-dimensional features, the input layer feeds xi to the network.
We minimize the following binary cross-entropy function to quantify how many of the predicted labels differ from the real ones by Equation (4):
where yi and denote true labels and the predicted interaction probability of xi. We use the Adam algorithm (Kingma and Ba, 2014) to train the DNN. The training is implemented with 100 epochs and each epoch has a mini-batch with the size of 64.
The final output layer with a single neuron and the sigmoid function is used to output an interaction probability for xi by Equation (5):
2.4.3. Deep forest
To solve complex tasks, learning models increasingly go deep (Cai et al., 2021; Li et al., 2022). Non-neural network style-based deep models demonstrate powerful learning abilities when they can go deep. DeepForest (Zhou and Feng, 2019) is a non-neural network style deep learning model and is constructed upon multi-grained cascade framework. It demonstrates the powerful classification performance and less training time.
In this study, we used DeepForest with no more than 20 layers to classify unobserved drug-target pairs. We choose random forests (Qi, 2012; Biau and Scornet, 2016) and extra trees (Geurts et al., 2006) as basic classifiers in DeepForest. Random forest (Qi, 2012; Biau and Scornet, 2016) is a non-parametric and interpretable classification model. It is an ensemble of many random decision trees and has a better performance in classification tasks with complex data structure, small sample size, and high-dimensional feature space. An extra tree (Geurts et al., 2006) is an ensemble of unpruned decision trees. It can better reduce variance by completely randomly selecting cut-points and minimizing classification bias by using whole learning samples.
As shown in Figure 1, each cascade layer in DeepForest comprises five random forests and five extra trees. Each predictor consists of 100 decision trees. In each layer, each predictor computes a ratio of a given DTI feature belonging to a positive or negative class. The predicted probabilities from all learners produce a class vector. The vector in addition to the raw DTI feature vector is used as input in the next layer.
In particular, similar to DNN, deep forest utilizes a cascade structure. In the structure, each level receives features from its preceding level, and outputs the results to the next level. Therefore, although the proportion of a 20-dimensional class vector in the input layer may be relatively smaller, its proportion in a DTI feature vector will increase with the deepening of the number of layers. Therefore, the 20-dimensional class vector cannot be drowned out in DeepForest.
2.4.4. Ensemble learning
Ensemble learning demonstrates better classification performance than a single classifier. Thus, we combined Grownet, DNN, and DeepForest and developed a hybrid model for DTI identification based on the soft voting approach by Equation (6):
where CGrownet, CDNN, and Cdeepforest represent DTI prediction results from Grownet, DNN, and DeepForest, respectively. α, β, and γ denote the corresponding weights. In particular, one drug-target pair is labeled as positive if its interaction score is greater than 0.5; otherwise, the pair is classified as negative.
3. Results
3.1. Evaluation metrics
In the experiments, precision, recall, accuracy, F1 score, AUC, and AUPR were used to measure the classification performance of our proposed EnGDD method. Higher values indicate better prediction ability for the above metrics. The experiments were repeated 20 times and their average values were selected as the final results. The former four metrics are defined by Equations (7)–(10):
where TP, FP, FN, and TN indicate true positives, false positives, false negatives, and true negatives.
AUC denotes the area under the receiver operating characteristic (ROC) curve and AUPR denotes the area under the Precision-Recall (PR) curve.
3.2. Experimental settings
PaDEL in ChemDes was used to extract drug features. The number of drug features obtained from PaDEL were as follows: 120 constitutional descriptors, 346 autocorrelation descriptors, 42 basak descriptors, 6 BCUT descriptors, 96 burden descriptors, 56 connectivity descriptors, 489 E-state descriptors, 3 Kappa descriptors, 15 molecular property descriptors, 6 quantum chemical descriptors, and 265 topological descriptors. All features in BioTriangle were applied to depict target proteins. Finally, we obtained one 100-dimensional feature for drugs and targets after dimensional reduction, respectively.
In addition, to obtain more accurate and stable prediction results, we used grid search to set the final parameters in the ensemble model. Grid search is a common hyperparameter optimization method and can be used to determine the final parameters in a model. We used it to traverse the parameter space and try all possible hyperparameter combinations. The optimal parameter combination is then selected as the final parameters of the ensemble model. Experimental settings in DNN were the same as Zhou et al. (2021). For Deepforest, we set max_layers = 20, n_estimators = 5, n_trees = 100, predictor = “forest”, and max_depth = None. For Grownet, we set lr = 0.05, num_nets = 20, batch_size = 64, boost_rate = 1.0, epochs_per_stage = 1, correct_epoch = 1, and L2 = 0.001.
Three 5-fold cross validations (CVs) were performed to assess the DTI prediction performance of EnGDD:
1. Five-fold CV on drugs (CVd, DTI prediction for new drugs): 80% of drugs were randomly selected as training data and the remaining 20% was taken as test data in each round.
2. Five-fold CV on targets (CVt, DTI prediction for new targets): 80% of targets were randomly selected as training data and the remaining 20% is taken as test data in each round.
3. Five-fold CV on drug-target pairs (CVdt, DTI prediction for drug-target pairs): 80% of drug-target pairs were randomly selected as training data and the remaining 20% is taken as test data in each round.
There are a few positive DTIs and it is a lack of negative DTIs on the four DTI datasets. If negative DTIs are not reasonably selected, it is easy to cause overfitting. Undersampling is an approach that deals with the data imbalance problem, and has been used to address situations where the number of samples in one category of data is far less than that in the other categories. Consequently, we used an undersampling approach to balance the datasets. That is, the ratio of positive drug-target pairs to negative drug-target pairs is set to 1 to solve the data imbalance problem.
3.3. Comparison with seven state-of-the-art DTI prediction methods
We compared the proposed EnGDD algorithm with seven state-of-the-art DTI prediction models to measure the classification ability of EnGDD, i.e., BLM-NII, NRLMF, WNNGIP, NEDTP, DTi2Vec, RoFDT, and MolTrans. To identify potential DTIs, BLM-NII (Mei et al., 2013) used a bipartite local model and neighbor interaction profiles, NRLMF (Liu et al., 2016) designed a neighborhood regularized logistic matrix factorization method, WNNGIP (Van Laarhoven and Marchiori, 2013) combined a weighted nearest neighbor profile and Gaussian interaction profile, NEDTP (An and Yu, 2021) is a heterogeneous network embedding framework, DTi2Vec (Thafar et al., 2021) integrated network embedding and ensemble learning, RoFDT (Wang et al., 2022) proposed a rotation forest model. Huang et al. (2021) proposed a molecular interaction transformer (MolTrans) to capture possible DTIs. MolTrans first designed an augmented transformer encoder to extract the semantic relationships among sub-structures from unlabeled biomedical data and then used a knowledge inspired sub-structural pattern detection method for more accurate DTI prediction.
Table 1 illustrates the DTI prediction performance of EnGDD and the other seven DTI prediction models under CVd. From Table 1, we observed that although EnGDD computed smaller precisions on GPCRs and ion channels than MolTrans. It computed better accuracy, F1-score, AUC, and AUPR than the other seven DTI prediction models. In particular, EnGDD obtained the best AUCs and AUPRs on nuclear receptors, GPCR, ion channels, and enzymes among the eight DTI prediction models. It computed AUCs of 0.9351, 0.9634, 0.9025, and 0.8697 on the four datasets, outperforming 1.85%, 0.88%, 0.63%, and 2.58% than the second-best approach, respectively. It calculated AUPRs of 0.9367, 0.9636, 0.9200, and 0.8855 on the four datasets, better 2.58%, 1.06%, 0.97%, and 2.60% than the second-best approach, respectively. Figure 2 shows the ROC and PR curves of the eight DTI prediction models and corresponding AUCs and AUPRs on four DTI datasets. The above results demonstrate that EnGDD obtained powerful DTI prediction performance under CVd and can be applied to effectively find potential targets for new drugs.
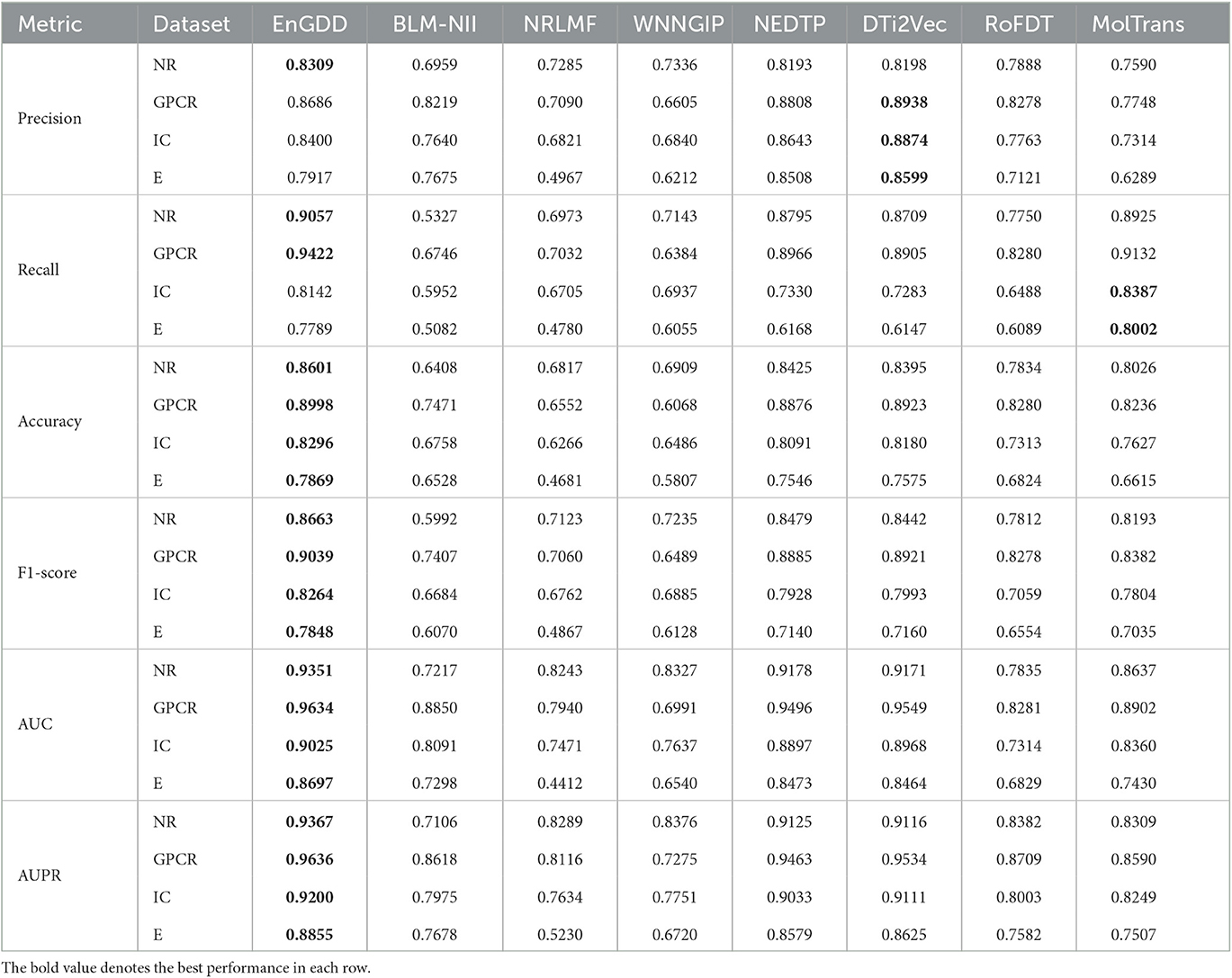
Table 1. Performance of eight DTI prediction methods on CVd.
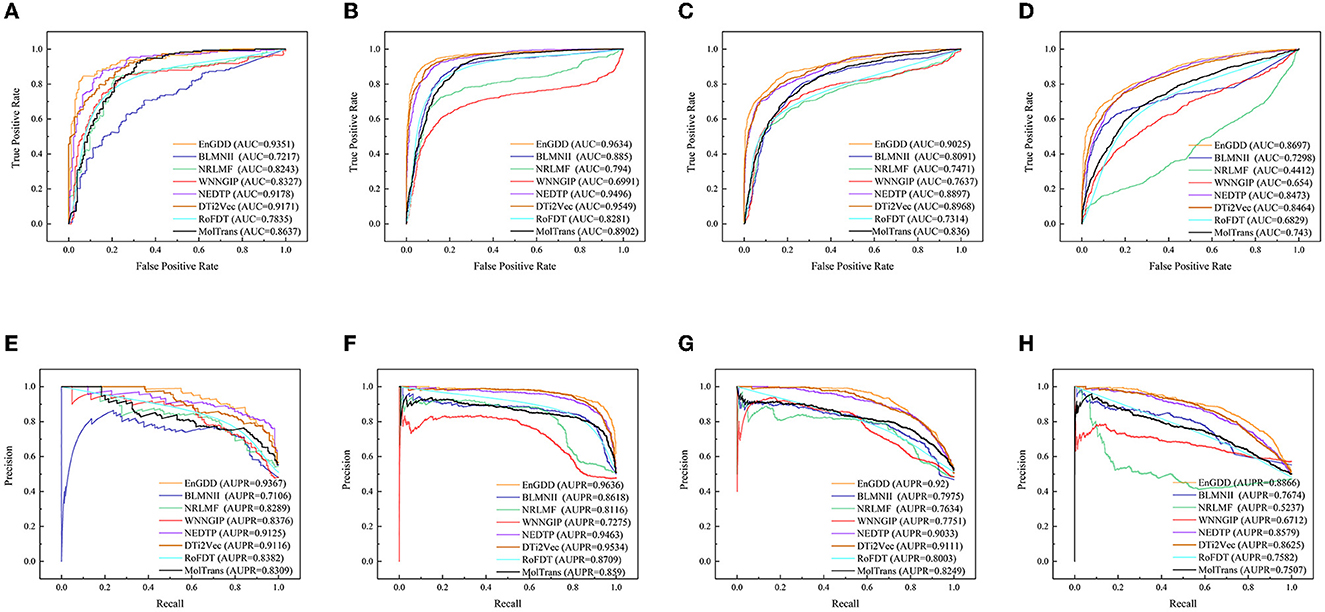
Figure 2. The ROC and PR curves of eight models under CVd. Subfigures (A–D) denote the ROC curves of all methods on the nuclear receptor, GPCR, ion channel, and enzyme datasets. Subfigures (E–H) denote the precision-recall curves of all methods on the nuclear receptor, GPCR, ion channel, and enzyme datasets under CVd.
Table 2 shows the DTI inference performance of the eight DTI prediction models under CVt. The results from Table 2 shows that EnGDD computed lower performance on nuclear receptors than WNNGIP. It may be caused by a small sample size on nuclear receptors. It significantly outperformed WNNGIP on GPCRs, ion channels, and enzymes, which contain larger samples. In addition, although DTi2Vec computed better precision, AUC, and AUPR than EnGDD on ion channels, the differences are very small. It may be caused by its different data structure. Figure 3 gives the ROC and PR curves of the eight models and corresponding AUCs and AUPRs on the four DTI datasets. In summary, EnGDD can be used to screen potential drugs for new targets.
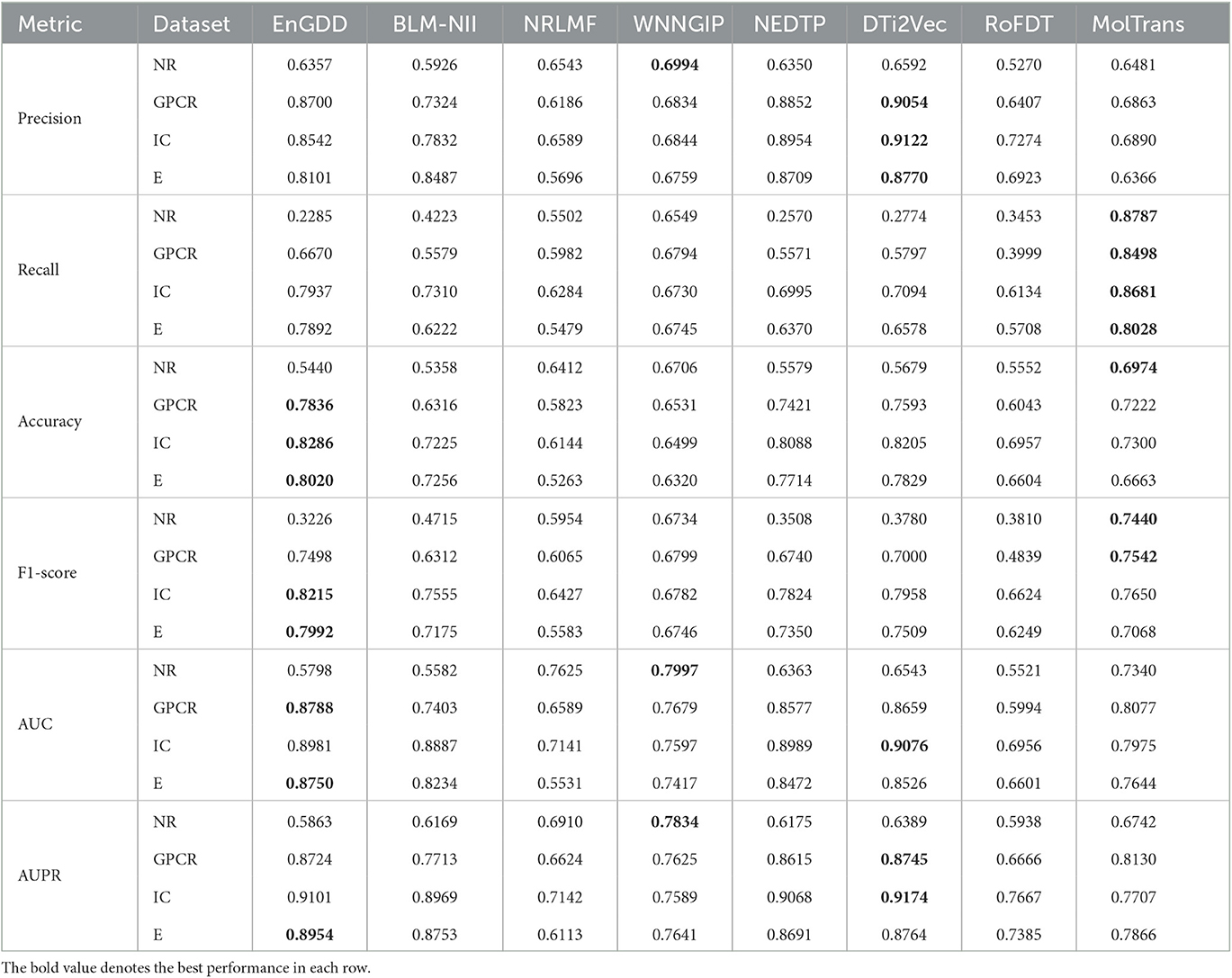
Table 2. Performance of eight DTI prediction methods on CVt.
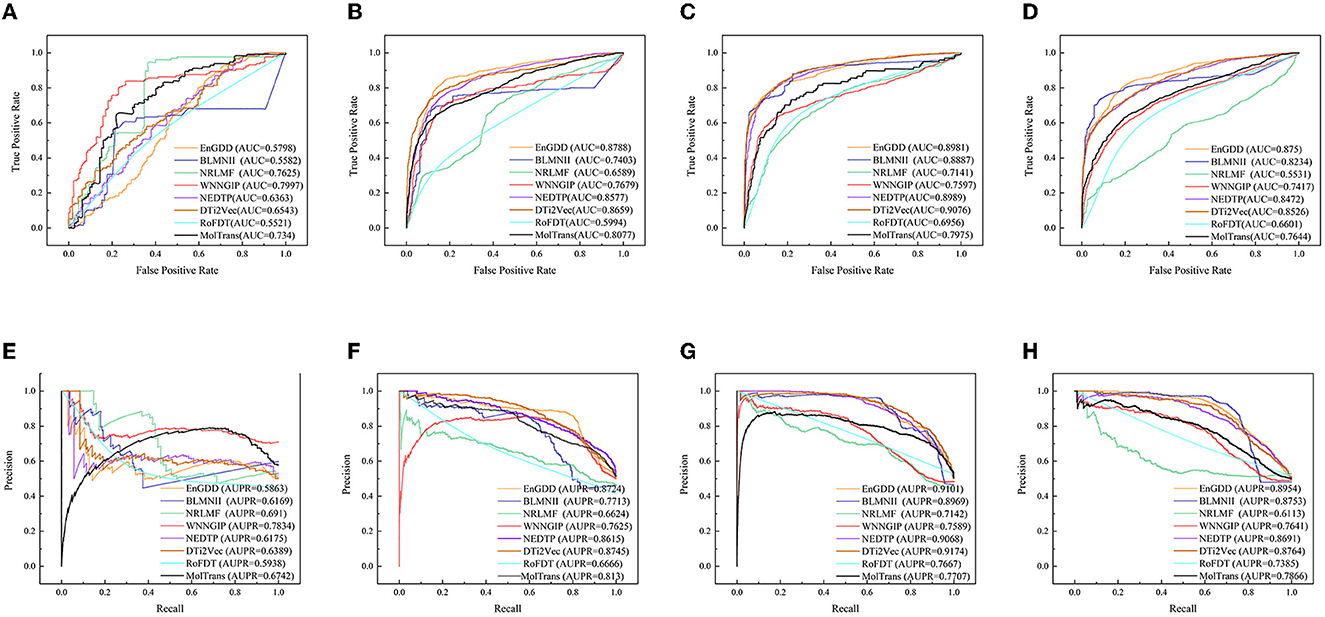
Figure 3. The ROC and PR curves of eight models under CVt. Subfigures (A–D) denote the ROC curves of all methods on the nuclear receptor, GPCR, ion channel, and enzyme datasets. Subfigures (E–H) denote the precision-recall curves of all methods on the nuclear receptor, GPCR, ion channel, and enzyme datasets under CVt.
Table 3 gives DTI prediction results of the eight models under CVdt. The results from Table 3 demonstrate that although EnGDD computed sightly lower precisions, accuracies, and F1-scores than DTi2Vec, it obtained the best recalls, AUCs, and AUPRs on the four DTI datasets. AUC and AUPR are two more important evaluation metrics. EnGDD computed the best AUCs of 0.9457, 0.9753, 0.9786, and 0.9559, outperforming 3.53%, 0.83%, 0.07%, and 1.16% than the second-best approach on the four datasets, respectively. It achieved the best AUPRs of 0.9451, 0.9748, 0.9811, and 0.9618, better 3.31%, 0.91%, 0.23%, and 1.38% than the second-best approach, respectively. Figure 4 shows the ROC and PR curves of the eight models and corresponding AUCs and AUPRs on the datasets. The results suggest that EnGDD can better capture possible DTIs from unknown drug-target pairs.
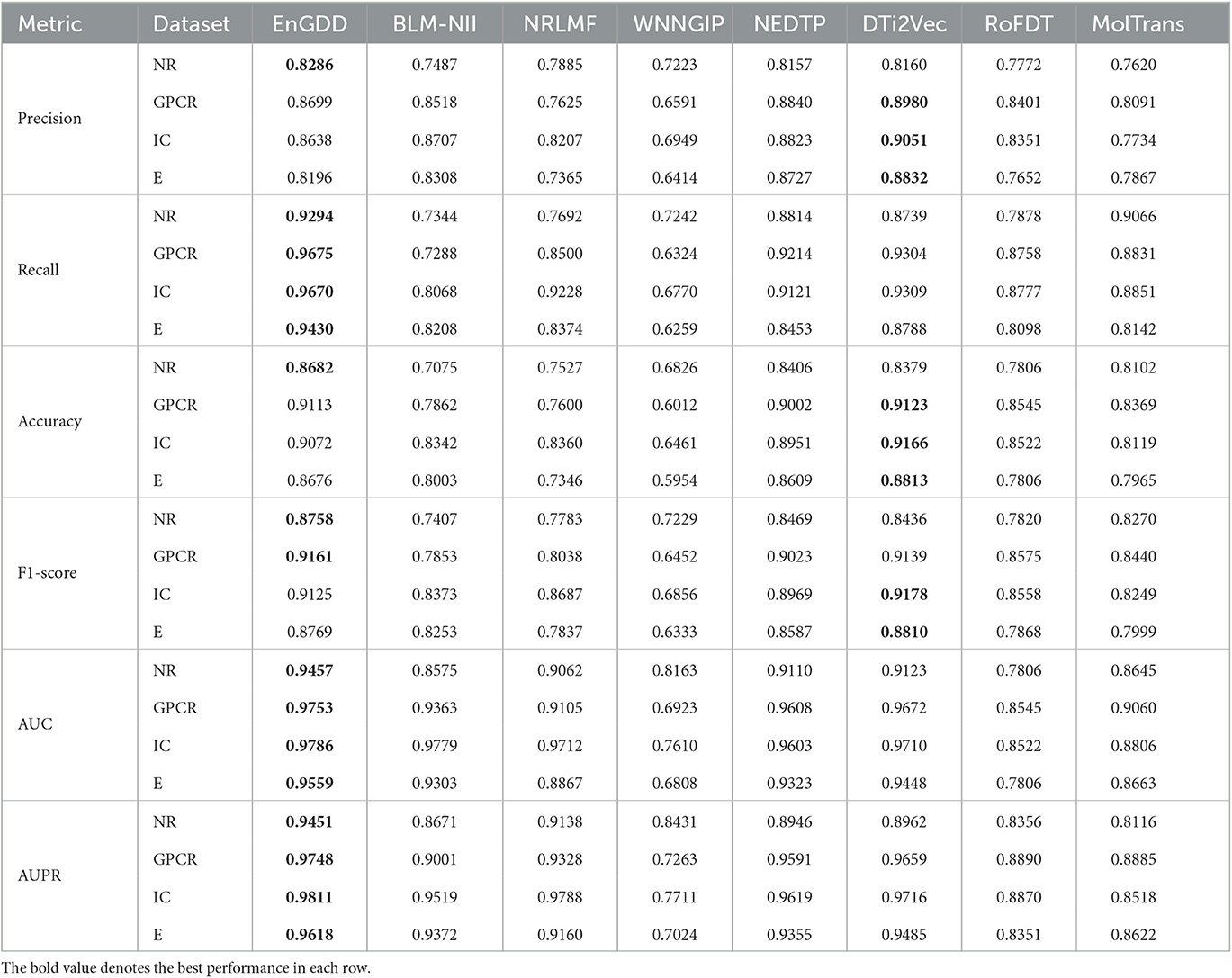
Table 3. Performance of eight DTI prediction methods on CVdt.

Figure 4. The ROC and PR curves of eight models under CVdt. Subfigures (A–D) denote the ROC curves of all methods on the nuclear receptor, GPCR, ion channel, and enzyme datasets. Subfigures (E–H) denote the precision-recall curves of all methods on the nuclear receptor, GPCR, ion channel, and enzyme datasets under CVdt.
3.4. Comparison of EnGDD with three individual models
Our proposed EnGDD method combined three individual deep learning models, that is, Grownet, DNN, and DeepForest. In order to measure the performance of ensemble learning on DTI prediction, we compared EnGDD with the three models under three different cross validations (CVd, CVt, and CVdt). Tables 4–6 show the comparison results under the three cross validations.
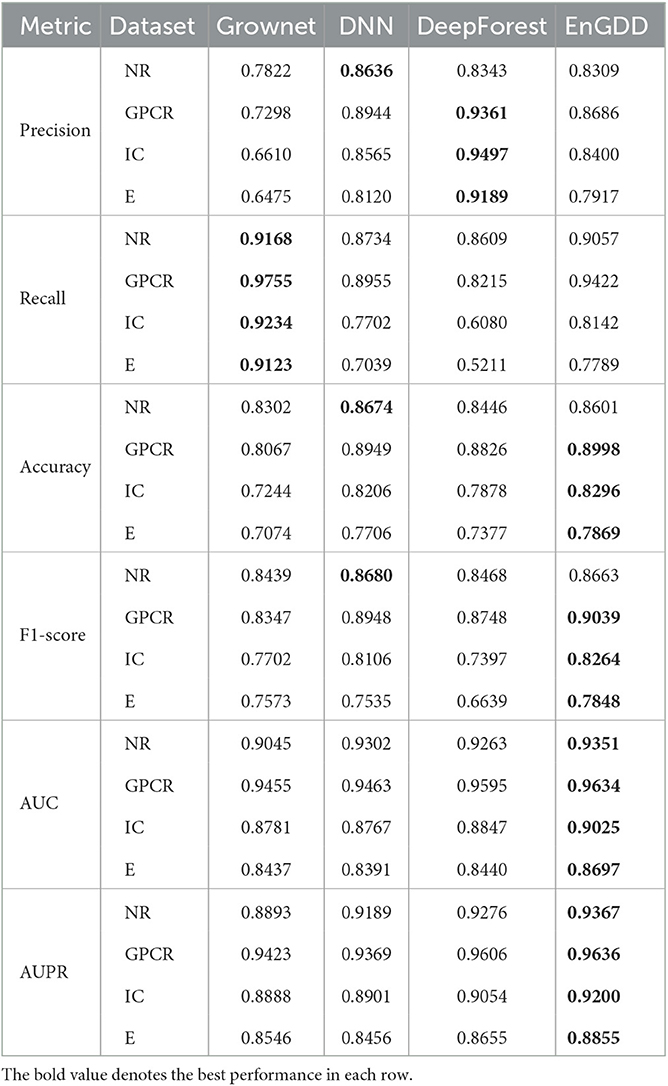
Table 4. Performance of EnGDD and three individual models on CVd.
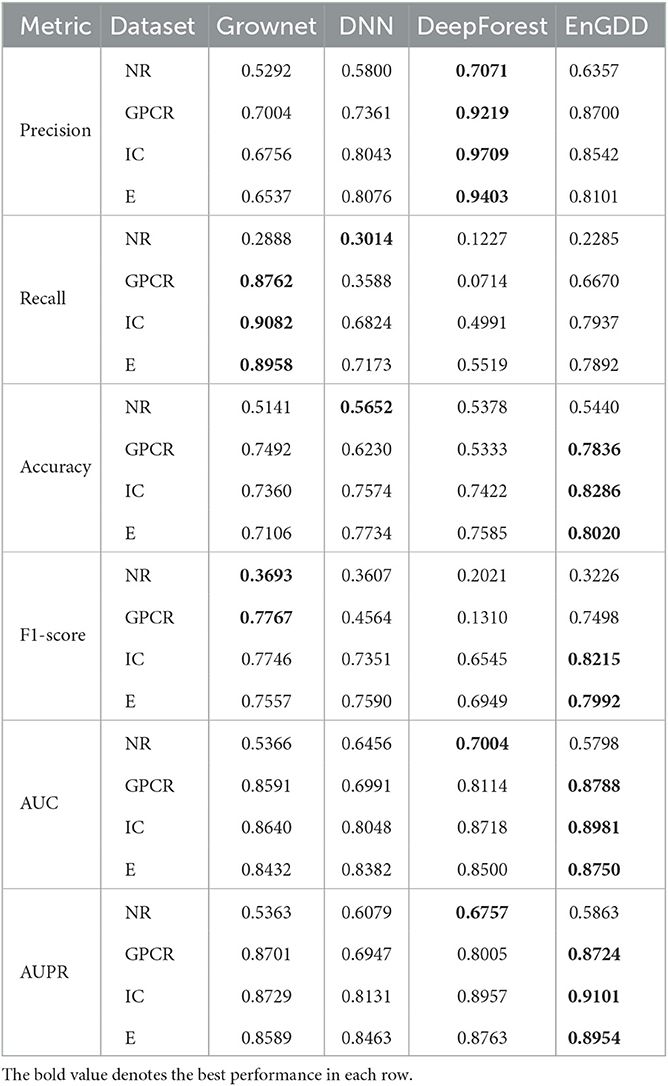
Table 5. Performance of EnGDD and three individual models on CVt.
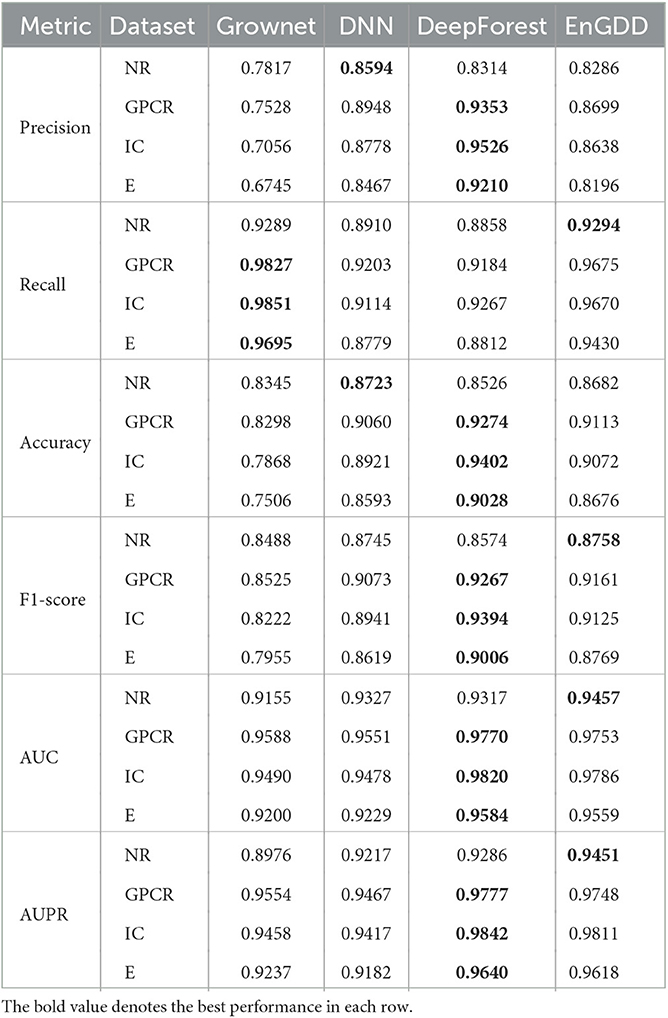
Table 6. Performance of EnGDD and three individual models on CVdt.
As shown in Table 4, EnGDD obtained the best accuracy, F1-score, AUC, and AUPR on GPCR, ion channel, and enzyme under CVd. Although EnGDD computed slightly lower accuracy and F1 score than DNN on the nuclear receptor, the differences are only 0.0073 and 0.0017. Although EnGDD calculated relatively lower precision and recall than DeepForest and Grownet, respectively, it computed a better F1 score than them. The results show that EnGDD can be appropriate to screen possible targets for a new drug.
As shown in Table 5, EnGDD obtained better accuracy, AUC, and AUPR on GPCR, ion channel, and enzyme under CVt. In particular, all methods computed very low recall and F1 score and relatively low precision, accuracy, AUC, and AUPR on nuclear receptors under CVt. There are only 33 targets for nuclear receptor. When conducting cross validation on targets, the testing set only contains about 6 targets. Thus, all methods calculated lower performance on nuclear receptors.
As shown in Table 6, although EnGDD computed slightly lower performance than DeepForest under CVdt, the values are very tiny and can be neglected especially compared with the ones under CVd and CVt. Thus, we used an ensemble of Grownet, DNN, and DeepForest to identify potential DTIs.
4. Case study
4.1. Possible DTI prediction from unknown drug-target pairs
We used EnGDD to find possible DTIs from unknown drug-target pairs on the four DTI datasets after confirming its performance. We first computed interaction probability for each drug-target pair based on EnGDD and ranked each drug-target pair in descending order based on the computed probabilities. Figures 5–8 list the top 100 drug-target pairs with the highest interaction probabilities on the four DTI datasets, respectively. In the figures, black solid lines and black dotted lines indicate known and unknown DTIs obtained from EnGDD, respectively. Deep sky blue diamonds and yellow ellipses denote drugs and targets.
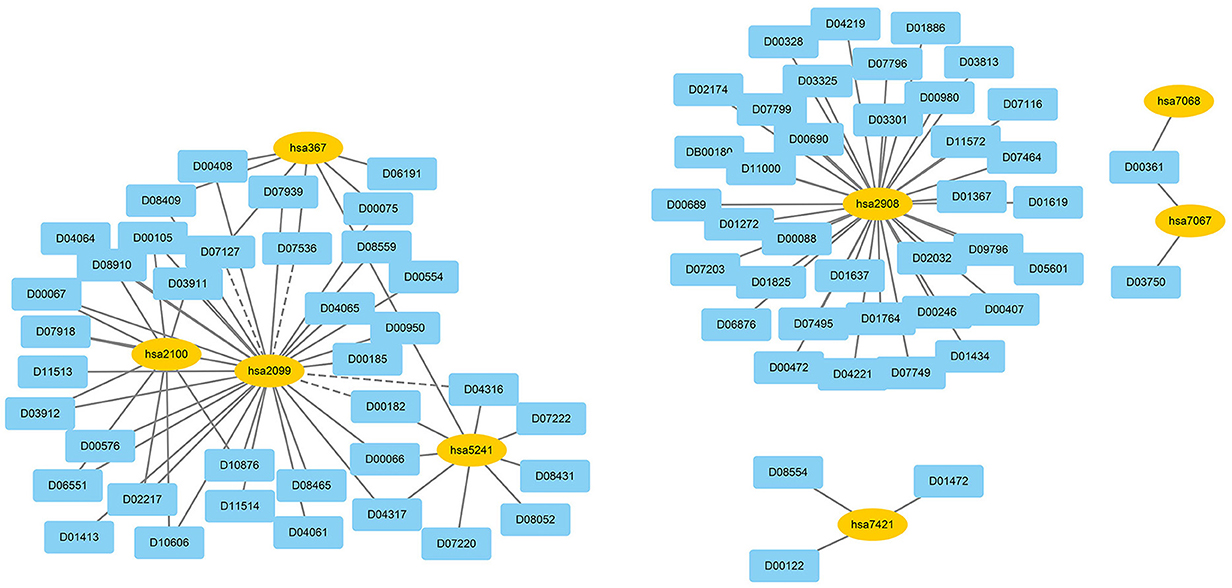
Figure 5. The predicted top 100 DTIs on nuclear receptors.
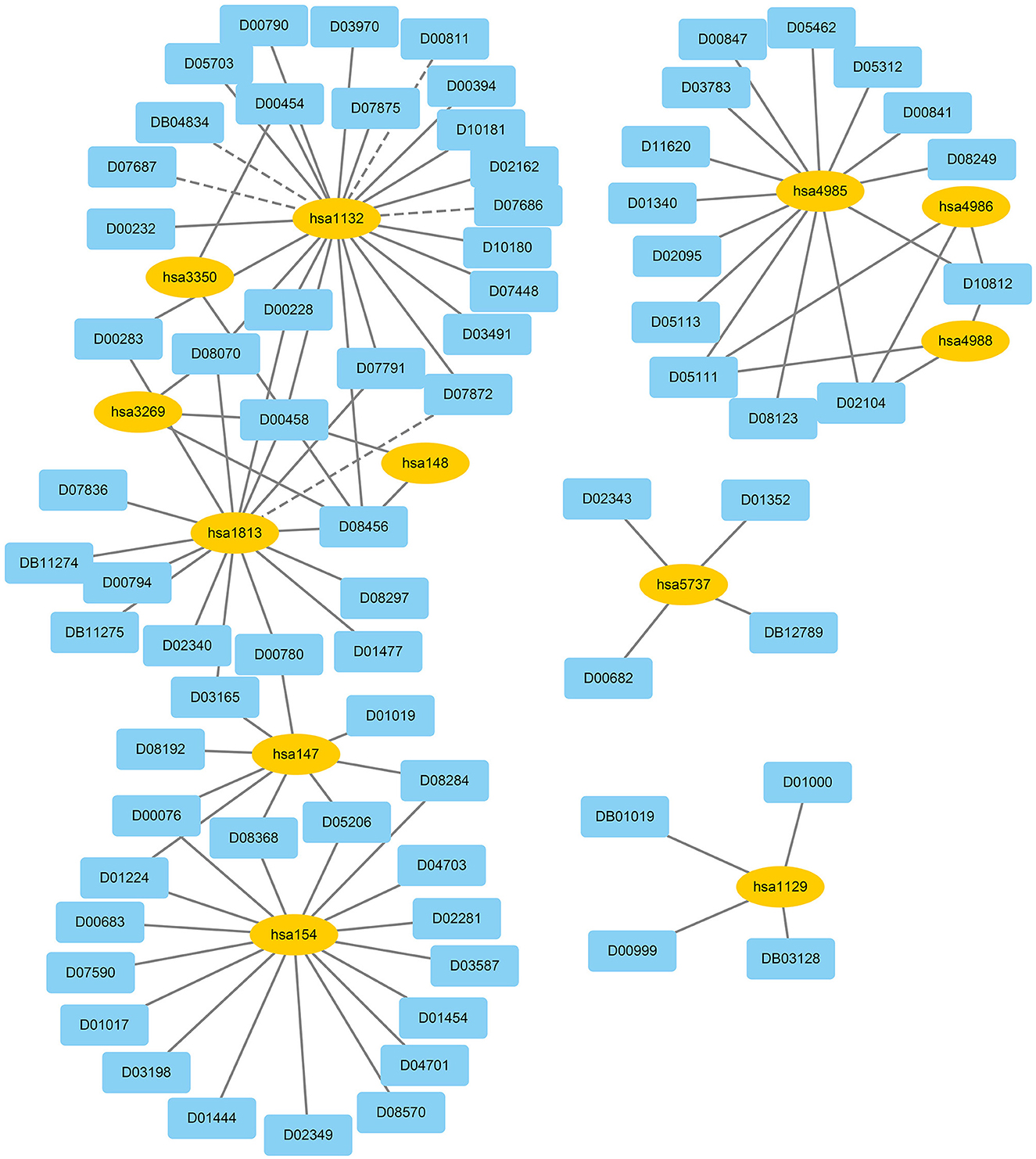
Figure 6. The predicted top 100 DTIs on GPCRs.
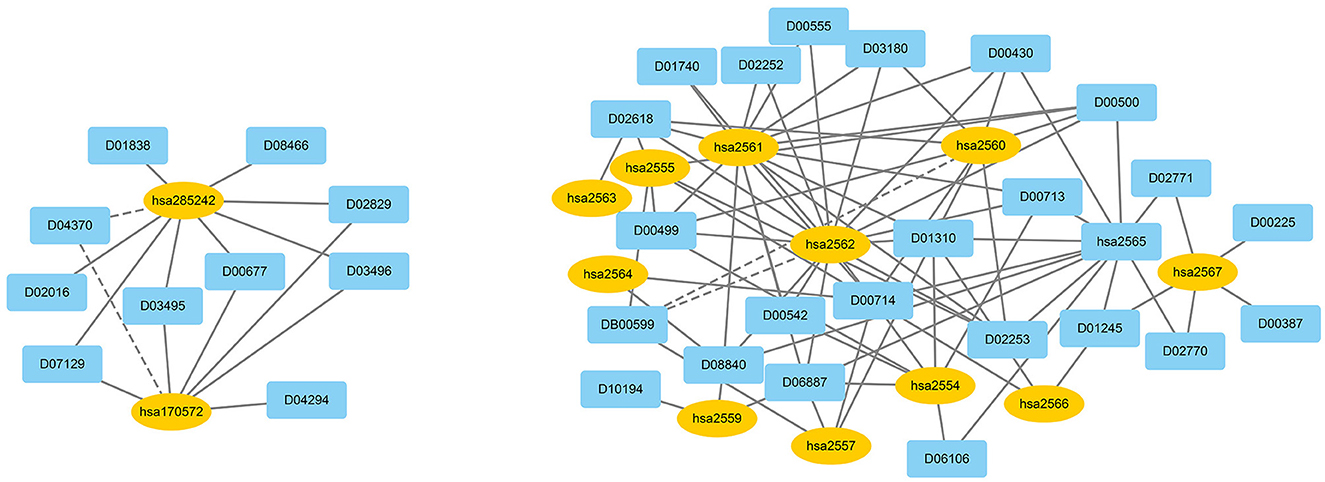
Figure 7. The predicted top 100 DTIs on ion channels.
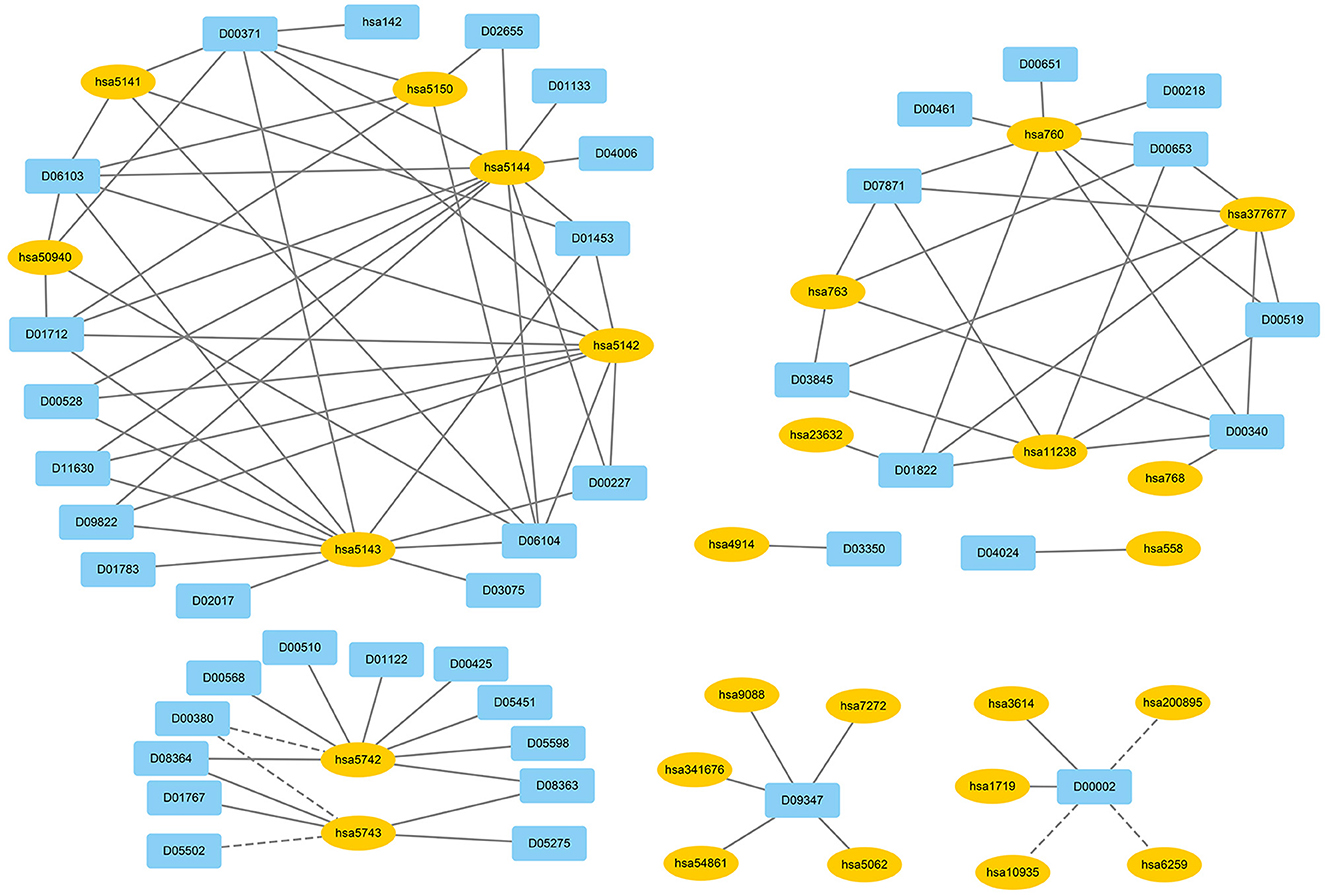
Figure 8. The predicted top 100 DTIs on enzymes.
On the nuclear receptor dataset, we predicted that D00182 and hsa2099 have a higher interaction probability among unknown drug-target pairs. D00182 (Norethisterone) is a synthetic second-generation progestin and used to protect cortical bone. Norethindrone with daily 5 or 10 mg can produce the same functions on biochemical markers of bone turnover as estrogen (DeCherney, 1993; Ferrero et al., 2010; Syed, 2022). hsa2099 (Estrogen receptor) is a nuclear hormone receptor binding to estrogen response elements with high affinity. The steroid hormones and their receptors are densely linked to cellular proliferation and differentiation in tissues (Klinge, 2001). In addition, progestins affect the bone that may be caused by stimulation of translocation relevant to the estrogen receptor and part of norethindrone is transformed to ethinyl estradiol in the rat liver (DeCherney, 1993), which further verified that D00182 and hsa2099 may interact with each other.
On the GPCR dataset, D07872 and hsa1813 were predicted to interact with a higher interaction probability. D07872 (Dosulepin) is a tricyclic antidepressant and weak inhibitor of dopamine reuptake. It interacts with many receptors (Nakagawa et al., 2000; Lepping and Menkes, 2007). It is used in patients suffering from ineffective alternative therapies because of its toxicity potential. hsa1813 is a dopamine receptor. Its activity is usually mediated by G proteins that can inhibit adenylyl cyclase (Nakagawa et al., 2000). It can be antagonized by anticancer small molecule ONC201 in clinical trials for high-grade gliomas and other cancers (Prabhu et al., 2019).
On the ion channel dataset, DB00599 and hsa2562 demonstrated a higher interaction probability. DB00599 (Thiopental) is a barbiturate. The drug can produce general anesthesia, treat convulsions, and reduce intracranial pressure (Dickinson et al., 2002). It can induce general anesthesia or complete anesthesia with short duration by intravenous administration. It is also utilized to control convulsive states for hypnosis and reduce increased intracranial pressure for neurosurgical patients (Wishart et al., 2008). hsa2562 (gamma-aminobutyric acid type A receptor subunit beta3) is a heteropentameric receptor for GABA that mainly inhibit neurotransmitter in the vertebrate brain and is also a receptor of diazepines and various anesthetics (Khair and Salvucci, 2021).
On the enzyme dataset, EnGDD predicted that D00002 may interact with hsa10935 with a higher interaction probability. D00002 (Nadide) is an important metabolic intermediate and can be used as an enzyme cofactor in redox reactions. It is involved in various enzymatic reactions as an electron carrier. It regulates various cellular functions including energy metabolism and DNA repair (Navarro et al., 2022; Pencina et al., 2023). hsa10935 (Mitochondrial peroxiredoxin 3) help to scavenge reactive oxygen species (Wang et al., 2021). It can also protect hippocampal neurons against excitotoxic injury in vivo. The up-regulation of peroxiredoxin-3 might be used to treat neurodegenerative diseases.
4.2. Target prediction for Parkinson's disease and Alzheimer's disease
In the KEGG database, D00777, D00059, D00780, D00784, D01277, D02004, D04641, D05768, D08969, D00558, D00781, D00785, D00786, and D02562 are known to be the therapeutic clues of PD (Kanehisa et al., 2017).
D00777 (Amantadine hydrochloride) is a drug only in oral formulations. It can be used to treat the PD patients. It has good absorption and little drug is present in the circulation. Associations between amantadine therapeutic effects and plasma concentrations have been confirmed by different studies (Aoki and Sitar, 1988). D00059 (Levodopa) has been validated for its “miraculous” effect in PD patients in 1961. L-dopa decarboxylase was an enzyme that can generate dopamine from levodopa. Patients with PD have a severe striatal dopamine deficit. Now, levodopa has been a “gold standard” of PD drug treatment (Hornykiewicz, 2010).
D00780 (Bromocriptine mesylate) is a dopamine receptor with an antioxidant effect (Ashhar et al., 2021). It has dopaminergic and antidyskinetic activities and is utilized to treat PD patients. Bromocriptine can selectively bind to postsynaptic dopamine D2 receptors in the central nervous system to implement the inhibition of neurotransmission and the effect of antidyskinetic (Kim et al., 2023). D00784 (Ropinirole hydrochloride) is a non-ergoline dopamine receptor agonist. It can efficiently control motor symptoms in early PD patients and has good toleration to PD (Sethi et al., 1998).
D01277 (Droxidopa) is an orally active synthetic amino acid. Neurogenic orthostatic hypotension is a fall in blood pressure on standing and notably affects PD. Droxidopa has been applied to the treatment of neurogenic orthostatic hypotension by FDA. Kaufmann et al. (2015). D02004 (Apomorphine hydrochloride) is an effective D1 and D2 dopamine agonist. It has a rapid antiparkinsonian function after subcutaneous administration and the effect is comparable with one of levodopa. Many studies suggest that Apomorphine is an effective therapeutic strategy for motor symptoms in PD (Unti et al., 2015).
D04641 (Istradefylline) is an adenosine A2A receptor antagonist. Kondo et al. (2015) detected the safety and effective of Istradefylline after administration once daily for 52 weeks in PD patients who experience wearing-off symptoms on levodopa treatment. They found that Istradefylline therapy was well-tolerated in levodopa-treated PD patients. D05768 (Rotigotine) is a non-ergolinic dopamine D3/D2/D1 receptor agonist. It administrates via a transdermal system and has been evaluated for the therapy of idiopathic PD (Reynolds et al., 2005). D08969 (Pimavanserin tartrate) is used to treat L-dopa-induced psychosis in PD. It is safe, well-tolerated and efficacious in the treatment of L-dopa-induced psychosis and doesn't worsen motor symptoms (Abbas and Roth, 2008).
Levodopa-D00558 (carbidopa) intestinal gel is used to treat advanced Levodopa-responsive PD with severe motor fluctuations and dyskinesia when other therapies fail to give satisfactory results in several countries (Wirdefeldt et al., 2016). D00781 (Entacapone) is a Chocolate-O-methyltransferase inhibitor. The addition of Entacapone in PD patients who have motor fluctuations can improve motor fluctuations (Schrag, 2005). (Mishra et al., 2019) found that D00785 (Selegiline hydrochloride) loaded nano lipid carrier administered through the nasal route has the potential for PD management therapy. D00786 (Tolcapone) may be beneficial to the PD patients who have not still developed motor fluctuations (Waters et al., 1998). D02562 (Rasagiline mesylate) is a potent and non-reversible MAO-B inhibitor. It has neuroprotective activities and a good safety and a helpful clinical effect in fluctuating PD patients who have been given an add-on to chronic levodopa therapy (Rabey et al., 2000).
The above 14 drugs are known to be therapy strategies for PD patients Table 7 gives the top 10 targets interacting with these drugs with the highest probabilities. In Table 7, each row lists a drug associated with PD and its predicted top 10 targets. The bold fonts denote that corresponding targets have no interaction with the query drug but are predicted to interact with the drug. The normal fonts denote that the query drug is known to interact with corresponding targets and the interactions are also predicted. In particular, D00059, D00780, D00784, D02004, and D05768 have been validated to interact with hsa1813 on the GPCR dataset. EnGDD predicted that D01277, D04641, and D08969 may interact with hsa1813 with the ranking of 5, 7, and 2, respectively. hsa1813 is a dopamine receptor D2. Dopamine is a neurotransmitter in the brain. Its concentration is directly associated with PD. Its low concentration in the substantia nigra can inhibit the transmission of nerve impulses and makes the brain fail to transduct signals in the proper way, which causes the brain and other body parts to lose connection (Latif et al., 2021). The above results show that D01277, D04641, and D08969 can be applied to the PD treatment by targeting hsa1813.
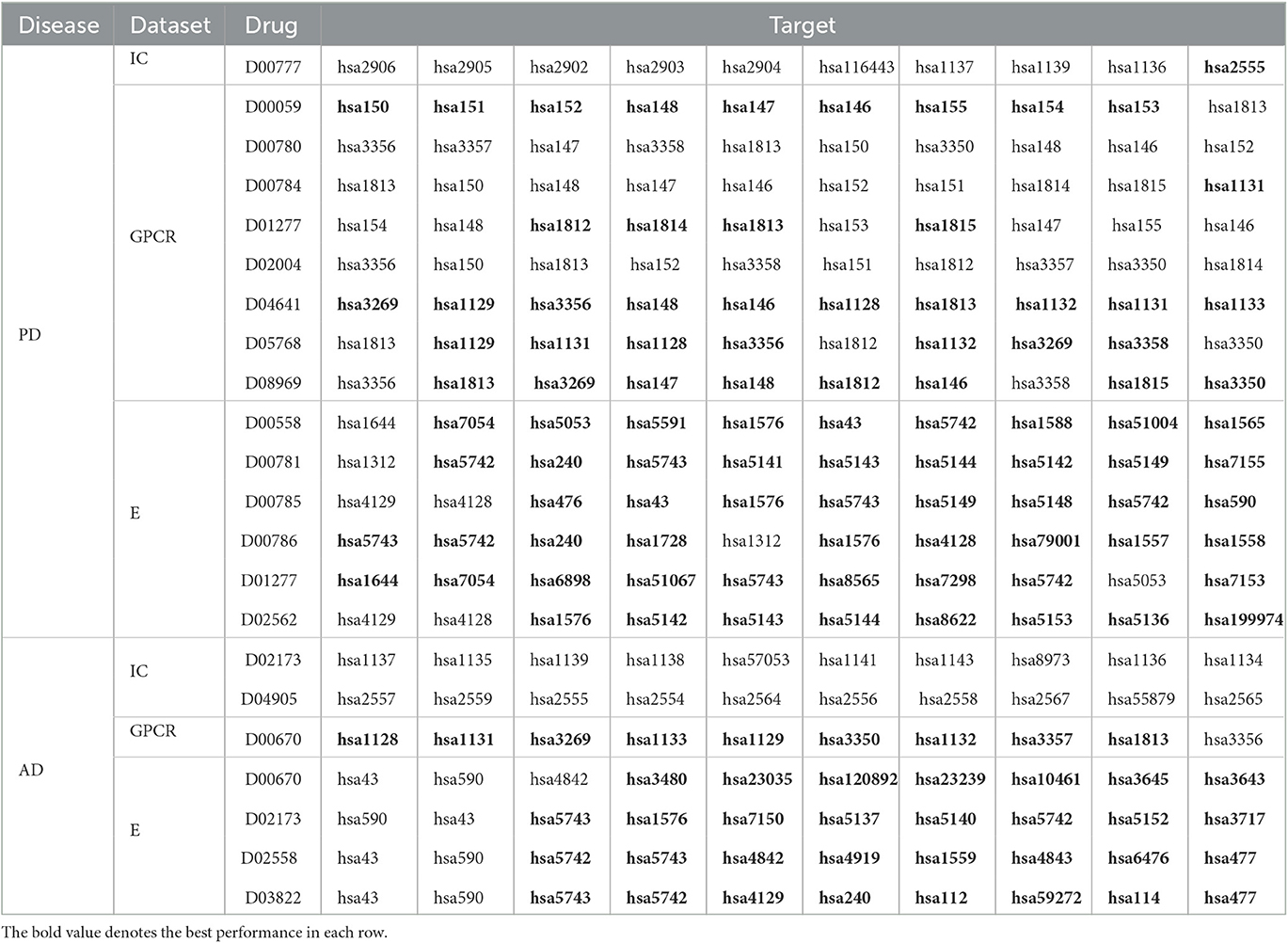
Table 7. Target prediction for Parkinson's disease and Alzheimer's disease.
In the KEGG database, D02173, D04905, D00670, D02558, and D03822 are the therapeutic clues of AD. Table 7 gives the top 10 targets interacting with these drugs with the highest probabilities.
D02173 (Galantamine hydrobromide) is a tertiary alkaloid extracted from plants. It is now synthesized and used to treat mild to moderate AD and provides one choice of an acetylcholinesterase inhibitor for the treatment of AD (Zarotsky et al., 2003). D04905 (Memantine hydrochloride) is the first drug approved by the US FDA and used to treat moderate to severe AD (Witt et al., 2004). D00670 (Donepezil hydrochloride) is one class of AChE inhibitors that can be used for the therapy of AD. It has longer and more selective function and manageable adverse effects (Sugimoto, 2001).
Cholinesterase inhibitors are “first-line” agents used for the treatment of AD. D02558 (Rivastigmine tartrate) and donepezil (cholinesterase inhibitors) have a dose-response association. Rivastigmine tartrate is as a carbamate inhibitor of acetylcholinesterase and is used for the treatment of mild to moderate AD under the trade name of Exelon (Shamsi et al., 2020). D03822 (Rivastigmine) has been reported to improve or maintain AD patients' performance including cognitive function, global function, and behavior. Its efficacy and tolerability have been confirmed by many clinical trials (Williams et al., 2003).
The above 5 drugs are known to be the therapeutic clues of AD. In Table 7, each row lists a drug associated with AD and its predicted top 10 targets. The bold fonts denote that corresponding targets have no interaction with the query drug but are predicted to interact with the drug. The normal fonts denote that the query drug interacts with corresponding targets and the interactions are also predicted. Particularly, D02173, D02558, and D03822 are three agents used in the treatment of AD. EnGDD predicted that the three drugs may interact with hsa5743 with the ranking of 3, 4, and 3, respectively. hsa5743 is prostaglandin-endoperoxide synthase 2 (PTGS2). Xie et al. (2022) have reported that baicalein may influence the progression of AD through regulating the expression of PTGS2. Thus, D02173, D02558, and D03822 may be the clues of treatment for AD patients through targeting PTGS2.
5. Conclusion
In this study, we developed a computational method EnGDD for possible DTI identification. EnGDD combined feature extraction, dimensional reduction, and DTI classification with an ensemble of Grownet, DNN, and DeepForest. EnGDD obtained better performance than the other seven DTI prediction models. Parkinson's disease and Alzheimer's disease are two neurodegenerative diseases. The results from the case studies by EnGDD show that D00002 (Nadide) may be a potential drug for neurodegenerative diseases. In addition, hsa1813 and hsa5743 may be possible targets of Parkinson's disease and Alzheimer's disease, respectively.
In the future, we will design a novel deep learning model to improve DTI prediction performance and find potential drugs and targets for neurodegenerative diseases (Chen et al., 2019; Sun et al., 2022; Wang et al., 2023; Zhang et al., 2023). In addition, with the rapid development of artificial intelligence technologies, novel drug research, and development for the two diseases can be performed by molecular generation and retrosynthesis (Sridharan et al., 2022; Yu et al., 2023).
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
LZ, YW, ZL, and XL: conceptualization and validation. YW and LP: methodology and data curation. LZ and YW: software. LZ and LP: investigation. YW: writing—original draft preparation and visualization. LZ, YW, and LP: writing—review and editing. LZ, ZL, and XL: supervision. LZ and XL: project administration. LZ, LP, and ZL: funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
LZ was funded by the National Natural Science Foundation of China under Grant No. 62072172 and the Natural Science Foundation of Hunan province under Grant No. 2021JJ30219. ZL was supported by the National Natural Science Foundation of China under Grant No. 62172158. LP was supported by the National Natural Science Foundation of China under Grant No. 61803151.
Acknowledgments
We really appreciate reviewer for the valuable comments. We would like to thank all authors of the cited references.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbas, A., and Roth, B. L. (2008). Pimavanserin tartrate: a 5-ht2a inverse agonist with potential for treating various neuropsychiatric disorders. Expert Opin. Pharmacother. 9, 3251–3259. doi: 10.1517/14656560802532707
An, Q., and Yu, L. (2021). A heterogeneous network embedding framework for predicting similarity-based drug-target interactions. Brief. Bioinform. 22, bbab275.
Aoki, F. Y., and Sitar, D. S. (1988). Clinical pharmacokinetics of amantadine hydrochloride. Clin. Pharmacokinet. 14, 35–51.
Ashhar, M. U., Kumar, S., Ali, J., and Baboota, S. (2021). CCRD based development of bromocriptine and glutathione nanoemulsion tailored ultrasonically for the combined anti-Parkinson effect. Chem. Phys. Lipids 235, 105035. doi: 10.1016/j.chemphyslip.2020.105035
Badirli, S., Liu, X., Xing, Z., Bhowmik, A., Doan, K., and Keerthi, S. S. (2020). Gradient boosting neural networks: growNet. arXiv preprint arXiv:2002.07971. doi: 10.48550/arXiv.2002.07971
Biau, G., and Scornet, E. (2016). A random forest guided tour. Test 25, 197–227. doi: 10.1007/s11749-016-0481-7
Buza, K., and Peška, L. (2017). Drug-target interaction prediction with bipartite local models and hubness-aware regression. Neurocomputing 260, 284–293. doi: 10.1016/j.neucom.2017.04.055
Cai, L., Lu, C., Xu, J., Meng, Y., Wang, P., Fu, X., et al. (2021). Drug repositioning based on the heterogeneous information fusion graph convolutional network. Brief. Bioinform. 22, bbab319. doi: 10.1093/bib/bbab319
Chen, X., Liu, M.-X., and Yan, G.-Y. (2012). Drug-target interaction prediction by random walk on the heterogeneous network. Mol. Biosyst. 8, 1970–1978. doi: 10.1039/c2mb00002d
Chen, X., Yan, C. C., Zhang, X., Zhang, X., Dai, F., Yin, J., et al. (2016). Drug-target interaction prediction: databases, web servers and computational models. Brief. Bioinform. 17, 696–712. doi: 10.1093/bib/bbv066
Chen, X., Zhu, C.-C., and Yin, J. (2019). Ensemble of decision tree reveals potential mirna-disease associations. PLoS Comput. Biol. 15, e1007209. doi: 10.1371/journal.pcbi.1007209
Cheng, Z., Yan, C., Wu, F.-X., and Wang, J. (2021). Drug-target interaction prediction using multi-head self-attention and graph attention network. IEEE/ACM Trans. Comput. Biol. Bioinform. 19, 2208–2218. doi: 10.1109/TCBB.2021.3077905
Chu, Y., Kaushik, A. C., Wang, X., Wang, W., Zhang, Y., Shan, X., et al. (2021). Dti-cdf: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features. Brief. Bioinform. 22, 451–462. doi: 10.1093/bib/bbz152
Chu, Y., Zhang, Y., Wang, Q., Zhang, L., Wang, X., Wang, Y., et al. (2022). A transformer-based model to predict peptide–hla class i binding and optimize mutated peptides for vaccine design. Nat. Mach. Intell. 4, 300–311. doi: 10.1038/s42256-022-00459-7
DeCherney, A. (1993). Physiologic and pharmacologic effects of estrogen and progestins on bone. J. Reproduct. Med. 38(12 Suppl), 1007–1014.
Dickinson, R., M. de Sousa, S. L., Lieb, W. R., and Franks, N. P. (2002). Selective synaptic actions of thiopental and its enantiomers. J. Am. Soc. Anesthesiol. 96, 884–892. doi: 10.1097/00000542-200204000-00016
Dickson, M., and Gagnon, J. P. (2004). Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 3, 417–429. doi: 10.1038/nrd1382
Dong, J., Cao, D.-S., Miao, H.-Y., Liu, S., Deng, B.-C., Yun, Y.-H., et al. (2015). Chemdes: an integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 7, 1–10. doi: 10.1186/s13321-015-0109-z
Dong, J., Yao, Z.-J., Wen, M., Zhu, M.-F., Wang, N.-N., Miao, H.-Y., et al. (2016). Biotriangle: a web-accessible platform for generating various molecular representations for chemicals, proteins, DNAs/RNAs and their interactions. J. Cheminform. 8, 1–13. doi: 10.1186/s13321-016-0146-2
Ferrero, S., Camerini, G., Ragni, N., Venturini, P., Biscaldi, E., and Remorgida, V. (2010). Norethisterone acetate in the treatment of colorectal endometriosis: a pilot study. Human Reproduct. 25, 94–100. doi: 10.1093/humrep/dep361
Friedman, J. H. (2001). Greedy function approximation: a gradient boosting machine. Ann. Stat. 29, 1189–1232. doi: 10.1214/aos/1013203451
Geurts, P., Ernst, D., and Wehenkel, L. (2006). Extremely randomized trees. Mach. Learn. 63, 3–42. doi: 10.1007/s10994-006-6226-1
Günther, S., Kuhn, M., Dunkel, M., Campillos, M., Senger, C., Petsalaki, E., et al. (2007). Supertarget and matador: resources for exploring drug-target relationships. Nucleic Acids Res. 36(Suppl_1), D919–D922. doi: 10.1093/nar/gkm862
Guo, S.-S., Liu, J., Zhou, X.-G., and Zhang, G.-J. (2022). Deepumqa: ultrafast shape recognition-based protein model quality assessment using deep learning. Bioinformatics 38, 1895–1903. doi: 10.1093/bioinformatics/btac056
Hornykiewicz, O. (2010). A brief history of levodopa. J. Neurol. 257(Suppl 2), 249–252. doi: 10.1007/s00415-010-5741-y
Huang, K., Xiao, C., Glass, L. M., and Sun, J. (2021). Moltrans: molecular interaction transformer for drug–target interaction prediction. Bioinformatics 37, 830–836. doi: 10.1093/bioinformatics/btaa880
Iraji, A., Khoshneviszadeh, M., Firuzi, O., Khoshneviszadeh, M., and Edraki, N. (2020). Novel small molecule therapeutic agents for Alzheimer disease: focusing on bace1 and multi-target directed ligands. Bioorgan. Chem. 97, 103649. doi: 10.1016/j.bioorg.2020.103649
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kaufmann, H., Norcliffe-Kaufmann, L., and Palma, J.-A. (2015). Droxidopa in neurogenic orthostatic hypotension. Expert Rev. Cardiovasc. Ther. 13, 875–891. doi: 10.1586/14779072.2015.1057504
Khair, A. M., and Salvucci, A. E. (2021). Phenotype expression variability in children with gabrb3 heterozygous mutations. Oman Med. J. 36, e240. doi: 10.5001/omj.2021.27
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., et al. (2023). Pubchem 2023 update. Nucleic Acids Res. 51, D1373–D1380. doi: 10.1093/nar/gkac956
Kingma, D. P., and Ba, J. (2014). Adam: a method for stochastic optimization. arXiv preprint arXiv:1412.6980. doi: 10.48550/arXiv.1412.6980
Klinge, C. M. (2001). Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 29, 2905–2919. doi: 10.1093/nar/29.14.2905
Kola, I., and Landis, J. (2017). Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 3, 711–715. doi: 10.1038/nrd1470
Kondo, T., Mizuno, Y., Group, J. I. S., et al. (2015). A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin. Neuropharmacol. 38, 41–46. doi: 10.1097/WNF.0000000000000073
Latif, S., Jahangeer, M., Maknoon Razia, D., Ashiq, M., Ghaffar, A., Akram, M., et al. (2021). Dopamine in Parkinson's disease. Clin. Chim. Acta 522, 114–126. doi: 10.1016/j.cca.2021.08.009
Lee, I., and Nam, H. (2022). Sequence-based prediction of protein binding regions and drug–target interactions. J. Cheminform. 14, 1–15. doi: 10.1186/s13321-022-00584-w
Lepping, P., and Menkes, D. B. (2007). Abuse of dosulepin to induce mania. Addiction 102, 1166–1167. doi: 10.1111/j.1360-0443.2007.01828.x
Li, H., Gao, Z., Kang, L., Zhang, H., Yang, K., Yu, K., et al. (2006). Tarfisdock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 34(Suppl_2), W219–W224. doi: 10.1093/nar/gkl114
Li, Y., Liang, W., Peng, L., Zhang, D., Yang, C., and Li, K.-C. (2022). Predicting drug-target interactions via dual-stream graph neural network. IEEE/ACM Trans. Comput. Biol. Bioinform. 1–11. doi: 10.1109/TCBB.2022.3204188
Liang, Y., Wu, Y., Zhang, Z., Liu, N., Peng, J., and Tang, J. (2022a). Hyb4mC: a hybrid DNA2vec-based model for DNA n4-methylcytosine sites prediction. BMC Bioinformatics 23, 258. doi: 10.1186/s12859-022-04789-6
Liang, Y., Zhang, Z.-Q., Liu, N.-N., Wu, Y.-N., Gu, C.-L., and Wang, Y.-L. (2022b). Magcnse: predicting lncRNA-disease associations using multi-view attention graph convolutional network and stacking ensemble model. BMC Bioinformatics 23, 189. doi: 10.1186/s12859-022-04715-w
Lin, S., Zhang, G., Wei, D.-Q., and Xiong, Y. (2022). Deeppse: prediction of polypharmacy side effects by fusing deep representation of drug pairs and attention mechanism. Comput. Biol. Med. 149, 105984. doi: 10.1016/j.compbiomed.2022.105984
Liu, J., Zhao, K., and Zhang, G. (2023). Improved model quality assessment using sequence and structural information by enhanced deep neural networks. Brief. Bioinform. 24, bbac507. doi: 10.1093/bib/bbac507
Liu, Y., Wu, M., Miao, C., Zhao, P., and Li, X.-L. (2016). Neighborhood regularized logistic matrix factorization for drug-target interaction prediction. PLoS Comput. Biol. 12, e1004760. doi: 10.1371/journal.pcbi.1004760
Lotfi Shahreza, M., Ghadiri, N., Mousavi, S. R., Varshosaz, J., and Green, J. R. (2018). A review of network-based approaches to drug repositioning. Brief. Bioinform. 19, 878–892. doi: 10.1093/bib/bbx017
Mei, J.-P., Kwoh, C.-K., Yang, P., Li, X.-L., and Zheng, J. (2013). Drug-target interaction prediction by learning from local information and neighbors. Bioinformatics 29, 238–245. doi: 10.1093/bioinformatics/bts670
Mishra, N., Sharma, S., Deshmukh, R., Kumar, A., and Sharma, R. (2019). Development and characterization of nasal delivery of selegiline hydrochloride loaded nanolipid carriers for the management of Parkinson's disease. Central Nerv. Syst. Agents Med. Chem. 19, 46–56. doi: 10.2174/1871524919666181126124846
Mukherjee, S., Ghosh, M., and Basuchowdhuri, P. (2022). “DeepGLSTM: deep graph convolutional network and LSTM based approach for predicting drug-target binding affinity,” in Proceedings of the 2022 SIAM International Conference on Data Mining (SDM) (Arlington, VA: SIAM), 729–737.
Nakagawa, T., Ukai, K., Ohyama, T., Gomita, Y., and Okamura, H. (2000). Effect of dopaminergic drugs on the reserpine-induced lowering of hippocampal theta wave frequency in rats. Jpn. J. Psychopharmacol. 20, 71–76.
Navarro, M. N., Gomez de las Heras, M. M., and Mittelbrunn, M. (2022). Nicotinamide adenine dinucleotide metabolism in the immune response, autoimmunity and inflammageing. Brit. J. Pharmacol. 179, 1839–1856. doi: 10.1111/bph.15477
Opella, S. J. (2013). Structure determination of membrane proteins by nuclear magnetic resonance spectroscopy. Annu. Rev. Anal. Chem. 6, 305. doi: 10.1146/annurev-anchem-062012-092631
Öztürk, H., Özgür, A., and Ozkirimli, E. (2018). Deepdta: deep drug-target binding affinity prediction. Bioinformatics 34, i821–i829. doi: 10.1093/bioinformatics/bty593
Pencina, K. M., Valderrabano, R., Wipper, B., Orkaby, A. R., Reid, K. F., Storer, T., et al. (2023). Nicotinamide adenine dinucleotide augmentation in overweight or obese middle-aged and older adults: a physiologic study. J. Clin. Endocrinol. Metab. dgad027. doi: 10.1210/clinem/dgad027
Peng, C.-X., Zhou, X.-G., Xia, Y.-H., Liu, J., Hou, M.-H., and Zhang, G.-J. (2022a). Structural analogue-based protein structure domain assembly assisted by deep learning. Bioinformatics 38, 4513–4521. doi: 10.1093/bioinformatics/btac553
Peng, L., Liao, B., Zhu, W., Li, Z., and Li, K. (2015). Predicting drug–target interactions with multi-information fusion. IEEE J. Biomed. Health Inform. 21, 561–572. doi: 10.1109/JBHI.2015.2513200
Peng, L., Wang, C., Tian, X., Zhou, L., and Li, K. (2021). Finding lncRNA-protein interactions based on deep learning with dual-net neural architecture. IEEE/ACM Trans. Comput. Biol. Bioinform. 19, 3456–3468. doi: 10.1109/TCBB.2021.3116232
Peng, L., Wang, F., Wang, Z., Tan, J., Huang, L., Tian, X., et al. (2022b). Cell–cell communication inference and analysis in the tumour microenvironments from single-cell transcriptomics: data resources and computational strategies. Brief. Bioinform. 23, bbac234. doi: 10.1093/bib/bbac234
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3, 1–21. doi: 10.1038/nrdp.2017.13
Prabhu, V. V., Madhukar, N. S., Gilvary, C., Kline, C. L. B., Oster, S., El-Deiry, W. S., et al. (2019). Dopamine receptor d5 is a modulator of tumor response to dopamine receptor d2 antagonism. Clin. Cancer Res. 25, 2305–2313. doi: 10.1158/1078-0432.CCR-18-2572
Precup, R.-E., Kovács, S., Preitl, S., and Petriu, E. M. (2012). Applied Computational Intelligence in Engineering and Information Technology: Revised and Selected Papers from the 6th IEEE International Symposium on Applied Computational Intelligence and Informatics SACI 2011, volume 1. Timisoara: Springer Science & Business Media.
Pujadas, G., Vaque, M., Ardevol, A., Blade, C., Salvado, M., Blay, M., et al. (2008). Protein-ligand docking: a review of recent advances and future perspectives. Curr. Pharmaceut. Anal. 4, 1–19. doi: 10.2174/157341208783497597
Qi, Y. (2012). “Random forest for bioinformatics,” in Ensemble Machine Learning (Springer), 307–323.
Rabey, J., Sagi, I., Huberman, M., Melamed, E., Korczyn, A., Giladi, N., et al. (2000). Rasagiline mesylate, a new mao-b inhibitor for the treatment of Parkinson's disease: a double-blind study as adjunctive therapy to levodopa. Clin. Neuropharmacol. 23, 324–330. doi: 10.1097/00002826-200011000-00005
Reynolds, N. A., Wellington, K., and Easthope, S. E. (2005). Rotigotine: in Parkinson's disease. CNS Drugs 19, 973–981. doi: 10.2165/00023210-200519110-00006
Schomburg, I., Chang, A., Ebeling, C., Gremse, M., Heldt, C., Huhn, G., et al. (2004). Brenda, the enzyme database: updates and major new developments. Nucleic Acids Res. 32(Suppl_1), D431–D433. doi: 10.1093/nar/gkh081
Schrag, A. (2005). Entacapone in the treatment of Parkinson's disease. Lancet Neurol, 4, 366–370. doi: 10.1016/S1474-4422(05)70098-3
Sethi, K. D., O'Brien, C., Hammerstad, J., Adler, C., Davis, T., Taylor, R., et al. (1998). Ropinirole for the treatment of early parkinson disease: a 12-month experience. Arch. Neurol. 55, 1211–1216.
Shamsi, A., Mohammad, T., Anwar, S., Alajmi, M. F., Hussain, A., Hassan, M. I., et al. (2020). Probing the interaction of rivastigmine tartrate, an important alzheimer's drug, with serum albumin: attempting treatment of Alzheimer's disease. Int. J. Biol. Macromol. 148, 533–542. doi: 10.1016/j.ijbiomac.2020.01.134
Shen, L., Liu, F., Huang, L., Liu, G., Zhou, L., and Peng, L. (2022). VDA-RWLRLS: an anti-SARS-CoV-2 drug prioritizing framework combining an unbalanced bi-random walk and laplacian regularized least squares. Comput. Biol. Med. 140, 105119. doi: 10.1016/j.compbiomed.2021.105119
Sridharan, B., Goel, M., and Priyakumar, U. D. (2022). Modern machine learning for tackling inverse problems in chemistry: molecular design to realization. Chem. Commun. 58, 5316–5331. doi: 10.1039/D1CC07035E
Sugimoto, H. (2001). Donepezil hydrochloride: a treatment drug for Alzheimer's disease. Chem. Rec. 1, 63–73. doi: 10.1002/1528-0691(2001)1:1<63::AID-TCR9>3.0.CO;2-J
Sun, F., Sun, J., and Zhao, Q. (2022). A deep learning method for predicting metabolite–disease associations via graph neural network. Brief. Bioinform. 23, bbac266. doi: 10.1093/bib/bbac266
Sun, X., Zhang, Y., Li, H., Zhou, Y., Shi, S., Chen, Z., et al. (2023). DRESIS: the first comprehensive landscape of drug resistance information. Nucleic Acids Res. 51, D1263–D1275. doi: 10.1093/nar/gkac812
Syed, Y. Y. (2022). Relugolix/estradiol/norethisterone (norethindrone) acetate: a review in symptomatic uterine fibroids. Drugs 1–8. doi: 10.1007/s40265-022-01790-4
Tayebi, A., Yousefi, N., Yazdani-Jahromi, M., Kolanthai, E., Neal, C. J., Seal, S., et al. (2022). Unbiaseddti: mitigating real-world bias of drug-target interaction prediction by using deep ensemble-balanced learning. Molecules 27, 2980. doi: 10.3390/molecules27092980
Thafar, M. A., Olayan, R. S., Albaradei, S., Bajic, V. B., Gojobori, T., et al. (2021). DTI2VEC: drug–target interaction prediction using network embedding and ensemble learning. J. Cheminform. 13, 1–18. doi: 10.1186/s13321-021-00552-w
Tian, G., Wang, Z., Wang, C., Chen, J., Liu, G., Xu, H., et al. (2022a). A deep ensemble learning-based automated detection of COVID-19 using lung CT images and vision transformer and convnext. Front. Microbiol. 13, 1024104. doi: 10.3389/fmicb.2022.1024104
Tian, X., Shen, L., Gao, P., Huang, L., Liu, G., Zhou, L., et al. (2022b). Discovery of potential therapeutic drugs for COVID-19 through logistic matrix factorization with kernel diffusion. Front. Microbiol. 13, 740382. doi: 10.3389/fmicb.2022.740382
Unti, E., Ceravolo, R., and Bonuccelli, U. (2015). Apomorphine hydrochloride for the treatment of Parkinson's disease. Expert Rev. Neurotherapeut. 15, 723–732. doi: 10.1586/14737175.2015.1051468
Van Laarhoven, T., and Marchiori, E. (2013). Predicting drug-target interactions for new drug compounds using a weighted nearest neighbor profile. PLoS ONE 8, e66952. doi: 10.1371/journal.pone.0066952
Wang, L., You, Z.-H., Chen, X., Xia, S.-X., Liu, F., Yan, X., et al. (2018). A computational-based method for predicting drug-target interactions by using stacked autoencoder deep neural network. J. Comput. Biol. 25, 361–373. doi: 10.1089/cmb.2017.0135
Wang, T., Sun, J., and Zhao, Q. (2023). Investigating cardiotoxicity related with herg channel blockers using molecular fingerprints and graph attention mechanism. Comput. Biol. Med. 153, 106464. doi: 10.1016/j.compbiomed.2022.106464
Wang, Y., Wang, L., Wong, L., Zhao, B., Su, X., Li, Y., et al. (2022). ROFDT: Identification of drug–target interactions from protein sequence and drug molecular structure using rotation forest. Biology 11, 741. doi: 10.3390/biology11050741
Wang, Y., and Zeng, J. (2013). Predicting drug-target interactions using restricted boltzmann machines. Bioinformatics 29, i126–i134. doi: 10.1093/bioinformatics/btt234
Wang, Y., Zhao, Y., Wang, Z., Sun, R., Zou, B., Li, R., et al. (2021). Peroxiredoxin 3 inhibits acetaminophen-induced liver pyroptosis through the regulation of mitochondrial ROS. Front. Immunol. 12, 652782. doi: 10.3389/fimmu.2021.652782
Waters, C., Kurth, M., Bailey, P., Shulman, L., LeWitt, P., Dorflinger, E., et al. (1998). Tolcapone in stable Parkinson's disease: efficacy and safety of long-term treatment. Neurology 50(5 Suppl 5), S39–S45. doi: 10.1212/WNL.50.5_Suppl_5.S39
Williams, B. R., Nazarians, A., and Gill, M. A. (2003). A review of rivastigmine: a reversible cholinesterase inhibitor. Clin. Therapeut. 25, 1634–1653. doi: 10.1016/S0149-2918(03)80160-1
Wirdefeldt, K., Odin, P., and Nyholm, D. (2016). Levodopa–carbidopa intestinal gel in patients with Parkinson's disease: a systematic review. CNS Drugs 30, 381–404. doi: 10.1007/s40263-016-0336-5
Wishart, D. S., Knox, C., Guo, A. C., Cheng, D., Shrivastava, S., Tzur, D., et al. (2008). Drugbank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36(Suppl_1), D901–D906. doi: 10.1093/nar/gkm958
Witt, A., MacDonald, N., and Kirkpatrick, P. (2004). Memantine hydrochloride. Nat. Rev. Drug Discov. 3, 109–110. doi: 10.1038/nrd1311
Xie, T., Pei, Y., Shan, P., Xiao, Q., Zhou, F., Huang, L., and Wang, S. (2022). Identification of miRNA-mRNA pairs in the Alzheimer's disease expression profile and explore the effect of mir-26a-5p/PTGS2 on amyloid-β induced neurotoxicity in Alzheimer's disease cell model. Front. Aging Neurosci. 14, 909222. doi: 10.3389/fnagi.2022.909222
Yiannopoulou, K. G., and Papageorgiou, S. G. (2020). Current and future treatments in Alzheimer disease: an update. J. Central Nerv. Syst. Dis. 12, 1179573520907397. doi: 10.1177/1179573520907397
You, J., McLeod, R. D., and Hu, P. (2019). Predicting drug-target interaction network using deep learning model. Comput. Biol. Chem. 80, 90–101. doi: 10.1016/j.compbiolchem.2019.03.016
Yu, T., Boob, A. G., Volk, M. J., Liu, X., Cui, H., and Zhao, H. (2023). Machine learning-enabled retrobiosynthesis of molecules. Nat. Catal. 6, 1–15. doi: 10.1038/s41929-022-00909-w
Zarotsky, V., Sramek, J. J., and Cutler, N. R. (2003). Galantamine hydrobromide: an agent for Alzheimer's disease. Am. J. Health Syst. Pharm. 60, 446–452. doi: 10.1093/ajhp/60.5.446
Zhang, C., Mou, M., Zhou, Y., Zhang, W., Lian, X., Shi, S., et al. (2022a). Biological activities of drug inactive ingredients. Brief. Bioinform. 23, bbac160. doi: 10.1093/bib/bbac160
Zhang, W., Lin, W., Zhang, D., Wang, S., Shi, J., and Niu, Y. (2019). Recent advances in the machine learning-based drug-target interaction prediction. Curr. Drug Metab. 20, 194–202. doi: 10.2174/1389200219666180821094047
Zhang, Z., Chen, L., Zhong, F., Wang, D., Jiang, J., Zhang, S., et al. (2022b). Graph neural network approaches for drug-target interactions. Curr. Opin. Struct. Biol. 73, 102327. doi: 10.1016/j.sbi.2021.102327
Zhang, Z., Xu, J., Wu, Y., Liu, N., Wang, Y., and Liang, Y. (2023). CAPSNet-LDA: predicting lncRNA-disease associations using attention mechanism and capsule network based on multi-view data. Brief. Bioinform. 24, bbac531. doi: 10.1093/bib/bbac531
Zhao, K., Xia, Y., Zhang, F., Zhou, X., Li, S. Z., and Zhang, G. (2023). Protein structure and folding pathway prediction based on remote homologs recognition using pathreader. Commun. Biol. 6, 243. doi: 10.1038/s42003-023-04605-8
Zhou, L., Duan, Q., Tian, X., Xu, H., Tang, J., and Peng, L. (2021). LPI-hyADBS: a hybrid framework for lncRNA-protein interaction prediction integrating feature selection and classification. BMC Bioinformatics 22, 568. doi: 10.1186/s12859-021-04485-x
Zhou, Y., Zhang, Y., Lian, X., Li, F., Wang, C., Zhu, F., et al. (2022). Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 50, D1398–D1407. doi: 10.1093/nar/gkab953
ZhouZhou, L., Wang, Z., Tian, X., and Peng, L. (2021). LPI-deepGBDT: a multiple-layer deep framework based on gradient boosting decision trees for lncRNA-protein interaction identification. BMC Bioinformatics 22, 479. doi: 10.21203/rs.3.rs-477640/v1
Keywords: drug-target interaction, gradient boosting neural network, deep neural network, deep forest, Parkinson's disease, Alzheimer's disease
Citation: Zhou L, Wang Y, Peng L, Li Z and Luo X (2023) Identifying potential drug-target interactions based on ensemble deep learning. Front. Aging Neurosci. 15:1176400. doi: 10.3389/fnagi.2023.1176400
Received: 28 February 2023; Accepted: 10 May 2023;
Published: 15 June 2023.
Edited by:
Ailan Wang, Geneis (Beijing) Co., Ltd., ChinaReviewed by:
Guohua Huang, Shaoyang University, ChinaYing Liang, Jiangxi Agricultural University, China
Han Wang, Northeast Normal University, China
Copyright © 2023 Zhou, Wang, Peng, Li and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zejun Li, bHpqZm94QGhuaXQuZWR1LmNu; Xueming Luo, bGlvbnZlckBodXQuZWR1LmNu