 Olayemi Joseph Olajide
Olayemi Joseph Olajide Claudia La Rue
Claudia La Rue Andreas Bergdahl
Andreas Bergdahl Clifton Andrew Chapman
Clifton Andrew Chapman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 06 October 2022
Sec. Alzheimer's Disease and Related Dementias
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.960314
This article is part of the Research Topic Molecular and Cellular Mechanisms of Synaptopathies: Emerging Synaptic aging-related molecular pathways in Neurological Disorders View all 11 articles
Increasing evidence suggests that mitochondrial dysfunction and aberrant release of mitochondrial reactive oxygen species (ROS) play crucial roles in early synaptic perturbations and neuropathology that drive memory deficits in Alzheimer’s disease (AD). We recently showed that solubilized human amyloid beta peptide 1–42 (hAβ1–42) causes rapid alterations at glutamatergic synapses in the entorhinal cortex (EC) through the activation of both GluN2A- and GluN2B-containing NMDA receptors. However, whether disruption of mitochondrial dynamics and increased ROS contributes to mechanisms mediating hAβ1–42-induced synaptic perturbations in the EC is unknown. Here we assessed the impact of hAβ1–42 on mitochondrial respiratory functions, and the expression of key mitochondrial and synaptic proteins in the EC. Measurements of mitochondrial respiratory function in wild-type EC slices exposed to 1 μM hAβ1–42 revealed marked reductions in tissue oxygen consumption and energy production efficiency relative to control. hAβ1–42 also markedly reduced the immunoexpression of both mitochondrial superoxide dismutase (SOD2) and mitochondrial-cytochrome c protein but had no significant impact on cytosolic-cytochrome c expression, voltage-dependent anion channel protein (a marker for mitochondrial density/integrity), and the immunoexpression of protein markers for all five mitochondrial complexes. The rapid impairments in mitochondrial functions induced by hAβ1–42 were accompanied by reductions in the presynaptic marker synaptophysin, postsynaptic density protein (PSD95), and the vesicular acetylcholine transporter, with no significant changes in the degradative enzyme acetylcholinesterase. We then assessed whether reducing hAβ1–42-induced increases in ROS could prevent dysregulation of entorhinal synaptic proteins, and found that synaptic impairments induced by hAβ1–42 were prevented by the mitochondria-targeted antioxidant drug mitoquinone mesylate, and by the SOD and catalase mimetic EUK134. These findings indicate that hAβ1–2 can rapidly disrupt mitochondrial functions and increase ROS in the entorhinal, and that this may contribute to synaptic dysfunctions that may promote early AD-related neuropathology.
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder that results from the progressive loss of neurons in selective brain areas, and is the primary cause of dementia worldwide (Fan et al., 2020; Guo et al., 2020; Olajide et al., 2021). Neuropathology in AD is hallmarked by abnormal accumulation of amyloid beta peptide (Aβ), a highly neurotoxic derivative of the amyloid precursor protein (APP) which is a transmembrane protein that is particularly concentrated in synapses (Haass and Selkoe, 2007; Masters and Selkoe, 2012; Mucke and Selkoe, 2012; Hampel et al., 2021). Aβ is thought to interact with multiple biological mechanisms to incite AD-related modifications in neurons, and several lines of experimental evidence, including our own, have shown that Aβ can rapidly dysregulate proteins mediating excitatory synaptic transmission (Selkoe, 2002; Coleman and Yao, 2003; Shankar and Walsh, 2009; Overk and Masliah, 2014; Tu et al., 2014; Olajide and Chapman, 2021; Olajide et al., 2021). These synaptic changes are thought to lead to cognitive deficits and memory failure early in AD (Arendt, 2009; Mucke and Selkoe, 2012; Marsh and Alifragis, 2018; Guo et al., 2020), but the molecular mechanisms through which Aβ induces excitotoxicity and neurodegeneration are still poorly understood.
Synaptic transmission relies heavily on mitochondria that generate energy through ATP and nicotinamide adenine dinucleotide (NAD +), maintain calcium homeostasis and buffering, and regulate cell signaling (Calkins et al., 2011; Akhter et al., 2017; Pickett et al., 2018). Perturbation of mitochondrial functions may contribute directly to impaired synaptic transmission in early AD (Cavallucci et al., 2013; Akhter et al., 2017; Olajide et al., 2017a; Xiao et al., 2017; Pickett et al., 2018; Khosravi and Harner, 2020), and Aβ can disrupt mitochondrial energy production and lead to increases in the synthesis of reactive oxygen species (ROS) and oxidative damage (Calkins et al., 2011; Hampel et al., 2021; Olajide et al., 2021; Ashleigh et al., 2022). Cellular ROS are natural by-products of mitochondrial aerobic respiration that result predominantly from leakage of electrons at complexes I and III of the electron transport chain. While ROS serve essential cellular functions, excess ROS can induce oxidative damage and AD-related neuropathology (Murphy, 2009; Patten et al., 2010; Wang et al., 2020). Under physiological conditions, oxidative damage by the major ROS, superoxide (O2−), is prevented by mitochondrial superoxide dismutase (SOD2), an antioxidant enzyme that catalyzes the dismutation of O2− to hydrogen peroxide (Murphy, 2009). However, Aβ can induce mitochondrial dysfunction both by increasing O2− production and by depleting the cellular antioxidant defense system, which causes degeneration of synaptic elements and alters neurotransmission processes in neurons (Tönnies and Trushina, 2017; Wang et al., 2020; Ionescu-Tucker and Cotman, 2021; Misrani et al., 2021). The overexpression of SOD2 in AD mouse models reportedly reduces Aβ deposition and prevents memory deficits (Dumont et al., 2009; Massaad et al., 2009), whereas mutant AD mice with depleted SOD2 expression show increased Aβ levels and accelerated synaptic dysfunction and cognitive decline (Li et al., 2004; Esposito et al., 2006).
The entorhinal cortex (EC) is important for cognitive functions including memory and is among the first cortical regions to be affected by AD pathology (van Hoesen et al., 1991; Velayudhan et al., 2013; Khan et al., 2014; Zhou et al., 2016; Grubman et al., 2019; Olajide et al., 2021). Subpopulations of neurons are differentially vulnerable to the toxicity of oxidative stress (Wang and Michaelis, 2010), and the EC is one of the earliest temporal lobe structures to show both oxidative impairment and Aβ accumulation (Nunomura et al., 2001; Terni et al., 2010; Olajide et al., 2021). Armand-Ugon et al. (2017) reported reduced expression of mitochondrial complexes I, II, IV, and V in the EC, but not in the frontal cortex, during the initial stages of AD, and this mitochondrial dysfunction and oxidative impairment may drive early synaptic failure in AD (Tönnies and Trushina, 2017). We have shown recently that human Aβ1–42 (hAβ1–42) rapidly alters elements of glutamatergic synapses in the EC through activation of both GluN2A and GluN2B subunit-containing NMDA receptors (Olajide and Chapman, 2021). This may drive the selective vulnerability of the EC to AD-type neurodegeneration (Olajide et al., 2021), but whether increased ROS and mitochondrial dysfunction contribute to the susceptibility of EC to early AD-related synaptic degeneration has not been explored.
Here, we investigated how mitochondrial proteins, respiratory function, and key synaptic proteins are affected in the EC of brain slices from wild-type rats following exposure to soluble hAβ1–42. We then assessed the ability of two novel and specific pharmacological ROS scavengers and inhibitors, the mitochondria-targeted antioxidant mitoquinone mesylate (MitoQ) and the SOD and catalase mimetic EUK134, to block the degradation of synaptic proteins induced by hAβ1–42 in the EC. Our results support the idea that mitochondrial alterations induced by hAβ1–42 are a central factor in early synaptic degeneration in the EC, and contribute to a more detailed understanding of the molecular mechanisms driving neurodegeneration in the EC during early AD.
Experiments were conducted according to the guidelines of the Canadian Council on Animal Care, and experimental procedures were approved by the Concordia University Animal Research Ethics Committee (Permit Number: 30000253). Six to ten-week-old male Long-Evans rats (Charles River) were housed and placed on a reverse 12 h: 12 h light-dark cycle, with free access to rat chow and water. Acute brain slices were obtained following isoflurane anesthesia and decapitation as previously described (Glovaci and Chapman, 2019). Brains were rapidly removed and cooled (4°C) in high-sucrose ACSF containing (in mM) 250 sucrose, 2 KCl, 1.25 NaH2PO4, 7 MgCl2, 26 NaHCO3, 0.5 CaCl2 and 10 dextrose, saturated with 95% O2 and 5% CO2. Horizontal slices (400 μm thick) were obtained throughout the ventral to the dorsal extent of the brain in cooled high-sucrose ACSF using a vibratome (Leica, VT1200). The EC was carefully excised from each slice, using a flat blade in contact with the vibratome blade (Paxinos and Watson, 1997; Olajide and Chapman, 2021). Obtained tissue was placed in normal ACSF consisting (in mM) of 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 dextrose saturated with 95% O2 and 5% CO2 at 32°C for 30 min. Assignment of EC slices taken from the right or left hemispheres were alternated between treatment groups at each consecutive level, so that tissue in the control and treated conditions was obtained from both hemispheres across the dorso-ventral extent of the EC.
Drugs were prepared as stock solutions and diluted to final concentrations just before use. Tissue was incubated for 3 h in submersion chambers containing ACSF at 22–24°C, saturated with 95% O2 and 5% CO2. EC slices were incubated in 1 μM hAβ1–42 (MW 4514.08; Abcam, AB120301) with a final concentration of DMSO of 0.1%, while control EC slices were exposed to 0.1% DSMO in ACSF. Solubilization and preparation of hAβ1–42 were done as previously described (Olajide and Chapman, 2021). hAβ1–42 was first solubilized in DMSO at 500 μM, sonicated for 15 min at room temperature, and then centrifuged at 15,000 × g at 4°C for 20 min. The supernatant was stored at −80°C in 10 μL aliquots and diluted in ACSF just before use. This method is known to result in low molecular weight β-oligomers including monomers to tetramers (Bitan et al., 2003; Stine et al., 2003) which are the most neurotoxic (Masters and Selkoe, 2012; Guo et al., 2020). It is possible for some protofibrils to develop following this preparation, but significant fibrillary aggregation requires longer incubation times and higher concentrations (>10 μM; O’Nuallain et al., 2004; Wogulis et al., 2005).
The role of mitochondrial ROS and oxidative stress was assessed by applying the mitochondria-targeted antioxidant drug mitoquinone mesylate (MitoQ, 500 nM; Toronto Research Chemicals; M372215), and SOD/catalase mimetic drug EUK134 (250 nM; Cayman Chemical; 10006329) during incubation of slices in hAβ1–42 or DMSO. Each treatment group included slices from at least 6 animals.
A sequential substrate addition protocol was conducted to assess mitochondrial coupled and uncoupled oxygen consumption, LEAK respiration, and membrane integrity using a two-chamber polarographic sensor (Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria). hAβ1–42-incubated and control EC samples were assessed simultaneously in both chambers under similar reaction conditions (n = 5 or 6). The measurements of oxygen consumption were performed in MiR05 at 37°C. MiR05 contains (in mM) 0.5 EGTA, 3.0 MgCl2⋅6H2O, 60 K-lactobionate, 20 taurine, 10 KH2PO4, 20 HEPES, 110 sucrose, and 1 g/L BSA (pH 7.1). The oxygen flux was registered and analyzed by the DatLab 7.0 software. Between 2 and 3 mg (wet weight) of treated entorhinal tissue was placed in the Oxygraph and oxygen levels were increased to approximately 480 pmol. Saponin (50 μg/mL) was added to the chambers to permeabilize the tissue before the experiment was begun. In the protocol, non-phosphorylating LEAK-respiration was induced by adding the Complex I-linked substrates malate (2 mM), pyruvate (5 mM), and glutamate (5 mM). Subsequently, the OXPHOS-capacity of Complex I-linked activity was measured after the addition of a saturating concentration of ADP (5 mM). Cytochrome c (10 μM) was then added to assess mitochondrial membrane damage. OXPHOS-capacity with combined Complex I and II-linked substrates was assessed by the addition of succinate (10 mM). This was followed by FCCP (carbonylcyanide-4 (trifluoromethoxy) phenylhydrazone, 1 μM) to test for uncoupling. The acceptor control ratio (ACR), which measures the degree of coupling between oxidation and phosphorylation, was calculated as the rate of oxygen consumption during ADP phosphorylation divided by the rate of non-phosphorylating LEAK-respiration induced by glutamate administration.
To perform Western immunoblotting, treated EC tissue was collected into microfuge tubes and snap-frozen. Tissues were disrupted with a tissue sonicator (QSonica: Q55) in homogenization buffer (10 mM Tris, pH 7.4, 1 mM EGTA, 1 mM EDTA, 0.5 DTT, 0.1 μM okadaic acid, 1 mM Na3VO4, 1 mM PMSF and 10 μg/mL leupeptin). Mitochondrial protein purification was done following extraction in mitochondrial homogenization buffer (10 mM Tris-HCl, pH 6.7, 10 mM KCl, 0.15 mM MgCl2, 1 mM PMSF, 1 mM DTT) followed by resuspension in mitochondrial suspension buffer (10 mM Tris HCl-pH 6.7, 0.15 mM MgCl2, 0.25 mM sucrose, 1 mM PMSF, 1 mM DTT). The quantity of protein in each sample was determined using BCA Protein Assay (Thermo Fisher: 23,227) and an ELISA Fluorostar Analysis System Plate Reader. Bovine serum albumin (BSA) was used as the standard for protein quantification. Protein samples (20–30 μg) were resolved on Tris-glycine 8–12% SDS-PAGE gels. The resolved proteins were transferred from gels to nitrocellulose membrane (Bio-Rad: 1620112) and blocked for 1–2 h in either 5% milk or 5% BSA (as indicated below for each specific antibody) in Tris-buffered saline (TBS) containing 0.2% Tween-20 (TBST). All antibodies used were solubilized (0.5–2.0 mg/mL) by manufacturers in buffered saline or culture supernatant containing 0.02–0.1% sodium azide (pH 7.2–7.6). Primary antibodies were diluted for overnight incubation of membranes at 4°C, and included total oxidative phosphorylation (OXPHOS) rodent antibody cocktail (1:2,000, 5% milk in TBST, MitoSciences, MS604), rabbit anti-SOD2 antibody (1:4,000, 5% milk in TBST; Proteintech, 24127-1-AP), rabbit anti-Cytochrome c antibody (1:2,000, 5% milk in TBST; Abcam, AB133504), mouse anti-VDAC-1/Porin antibody (1:2,000, 5% BSA in TBST, Abcam, AB14734), mouse anti-synaptophysin (1:3,000, 5% BSA in TBST; Sigma-Aldrich, MAB5258), rabbit anti-PSD95 (1:3,000, 5% milk in TBST; Abcam, AB18258), rabbit anti-choline acetylcholinesterase (1:1,000, 5% milk in TBST; Abcam, AB183591), rabbit anti-vesicular acetylcholine transporter (1:2,000, 5% milk in TBST; Abcam, AB235201), mouse anti-vinculin (1:4,000, 5% milk in TBST; Abcam, AB130007), and mouse anti-β-Actin (1:5,000, 5% BSA in TBST; Abcam, AB8226). Membranes were then washed 3 times for 5 min each in TBST and incubated at room temperature with either peroxidase-conjugated goat anti-mouse secondary antibody (1:4,000; Millipore Sigma, AP124P) or peroxidase-conjugated goat anti-rabbit secondary antibody (1:5,000; Millipore Sigma, AP132P) for 1–2 h. Immunoreactivity was detected using ECL Western blotting substrate (Thermo Fisher Scientific, 32106) and visualized using a CDP-STAR chemiluminescence system (Amersham hyperfilm ECL). All antibody signals were normalized against loading control (β-Actin or vinculin) immunoreactivity. Western blot data were compiled from six animals, and bands were quantified by densitometric analysis using Image-J software (version 1.41).
Results obtained from mitochondrial respiratory measurements were tested statistically using a two-tailed Student’s t-test. Data from Western blot densitometry were analyzed using either two-tailed Student’s t-tests or one-way analyses of variance (ANOVA) with Sidak’s multiple comparisons tests. All data analyses were done using GraphPad Prism software version 8.0.1 with significance set at p < 0.05. Bar graphs indicate the mean and standard deviation, normalized to the largest control value in percentage (for protein analysis), and include plots showing values obtained from individual animals.
During the pathogenesis of AD, the EC selectively accumulates neurotoxic Aβ and shows the earliest signs of oxidative impairment and neurodegeneration (Terni et al., 2010; Olajide et al., 2021), but the mechanisms involved are still unclear. We therefore examined the rapid effects of hAβ1–42 on mitochondrial functions in entorhinal tissue using high-resolution respirometry. Utilization of respiration substrates in entorhinal samples was significantly reduced by exposing brain slices to 1 μM hAβ1–42 for a period of 3 h (Figure 1A1). Relative to control, hAβ1–42 markedly reduced respiration in EC slices (in pmol/s/mg; n = 5–6) following the addition of glutamate (2.9 ± 1.2 vs. 5.6 ± 1.0 in controls; p = 0.0211), ADP (3.8 ± 3.3 vs. 20.2 ± 2.4; p = 0.0022), cytochrome c (4.2 ± 3.7 vs. 19.4 ± 2.7; p = 0.0036), succinate (13.7 ± 5.9 vs. 36.0 ± 4.7; p = 0.0045), and FCCP (11.7 ± 5.0 vs. 27.0 ± 4.0; p = 0.0070). However, incubation of EC slices with hAβ1–42 did not significantly alter the consumption of malate (0.7 ± 0.2 vs. 1.1 ± 0.5 in controls; p = 0.2427), and pyruvate (1.8 ± 1.2 vs. 3.5 ± 0.6; p = 0.0570). hAβ1–42 also markedly reduced the ratio between oxidation and phosphorylation which measures the efficiency of the mitochondria in generating ATP per given amount of oxygen, and is shown by assessments of the ACR (1.1 ± 0.6 vs. 3.6 ± 0.4 in controls; p < 0.0001; Figure 1A2).
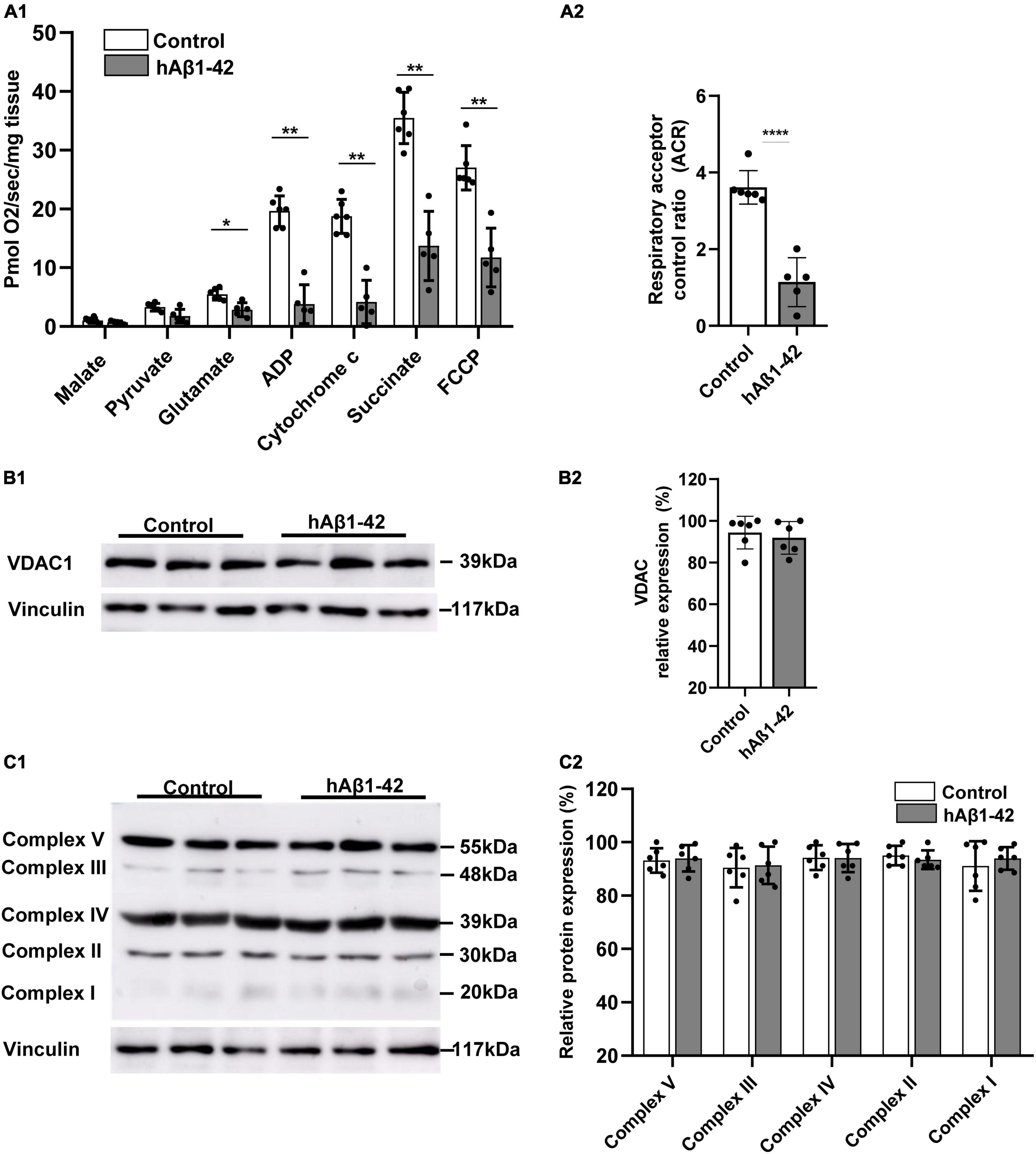
Figure 1. Impact of hAβ1–42 on mitochondrial respiration and function in the entorhinal cortex. (A1) Bar graphs represent mitochondrial substrate utilization in permeabilized entorhinal tissue previously treated with 1 μM hAβ1–42 for 3 h vs. control tissue, assessed through high-resolution respirometry (n = 5–6). (A2) Graph showing the acceptor control ratio (ACR) which measures the relative efficiency of phosphorylation and is determined by dividing ADP by glutamate average rates of respiration. hAβ1–42 treatment significantly reduced the ACR relative to control. (B1) Representative immunoblots of mitochondrial membrane and gatekeeper protein voltage-dependent anion channel 1 (VDAC1) and vinculin (loading control) in entorhinal lysates following incubation in control medium or hAβ1–42. (B2) Bar graphs showing the normalized protein expression of VDAC1 (n = 6). (C1) Representative immunoblots of mitochondrial complex I to V with total OXPHOS rodent antibody cocktail with vinculin serving as a loading control. (C2) Graphs showing normalized data for all five mitochondrial subunits in hAβ1–42-treated slices vs. the largest expression in the control group (n = 6) (*p < 0.05; **p < 0.01; ****p < 0.0001).
We next assessed the impact of hAβ1–42 on immunoexpression of markers for the integrity of the mitochondrial membrane and for the five mitochondrial protein complexes that make up the electron transport chain. Compared to control, hAβ1–42 treatment did not alter immunoblot expression of the mitochondrial membrane and gatekeeper protein voltage-dependent anion channel 1 (VDAC1; 91.9 ± 7.3 vs. 94.3 ± 7.8 in controls; p = 0.6125) (Figure 1B). Similarly, immunoblotting of complexes I to V with total OXPHOS rodent antibody cocktail (Figure 1C) revealed no significant changes (n = 6) in protein expression between control and hAβ1–42-treated entorhinal slices. When compared to control, hAβ1–42 (1 μM) treatment for 3 h did not alter the normalized protein expression of Complex V (93.2 ± 4.6 and 94.0 ± 5.0; p = 0.785), Complex III (90.5 ± 7.4 and 91.4 ± 7.0; p = 0.833), Complex IV (94.2 ± 4.6 and 94.1 ± 5.3; p = 0.961), Complex II (95.1 ± 3.6 and 93.4 ± 3.5; p = 0.560), and Complex I (91.1 ± 9.3 and 94.0 ± 4.3; p = 0.589) in entorhinal lysates. Therefore, although hAβ1–42 treatment reduces mitochondrial function as reflected in respirometry analysis, it did not significantly affect the expression of protein markers for mitochondrial integrity and for respiratory complexes.
Our finding that hAβ1–42 rapidly impairs mitochondrial respiration in the EC suggests that a resulting oxidative redox imbalance, due to increased leakage of mitochondrial electrons and ROS formation, could have deleterious effects on the mitochondrial antioxidant system. We therefore characterized the expression of the mitochondrial oxidative scavenger enzyme SOD2, and both mitochondrial and cytosolic cytochrome c protein. Representative immunoblots and quantification data show that, compared to control, hAβ1–42 markedly reduced the relative expression of SOD2 (53.3 ± 14.0 vs. 90.4 ± 8.1 in controls; p = 0.0002) (Figure 2A). Mitochondrial cytochrome c expression was also reduced by application of hAβ1–42 (33.6 ± 17.2 vs. 86.0 ± 12.3 in controls; p = 0.0001), and this effect was specific to mitochondria because immunoexpression of cytosolic cytochrome c was not significantly affected (81.7 ± 13.6 vs. 89.6 ± 11.3 p = 0.298).
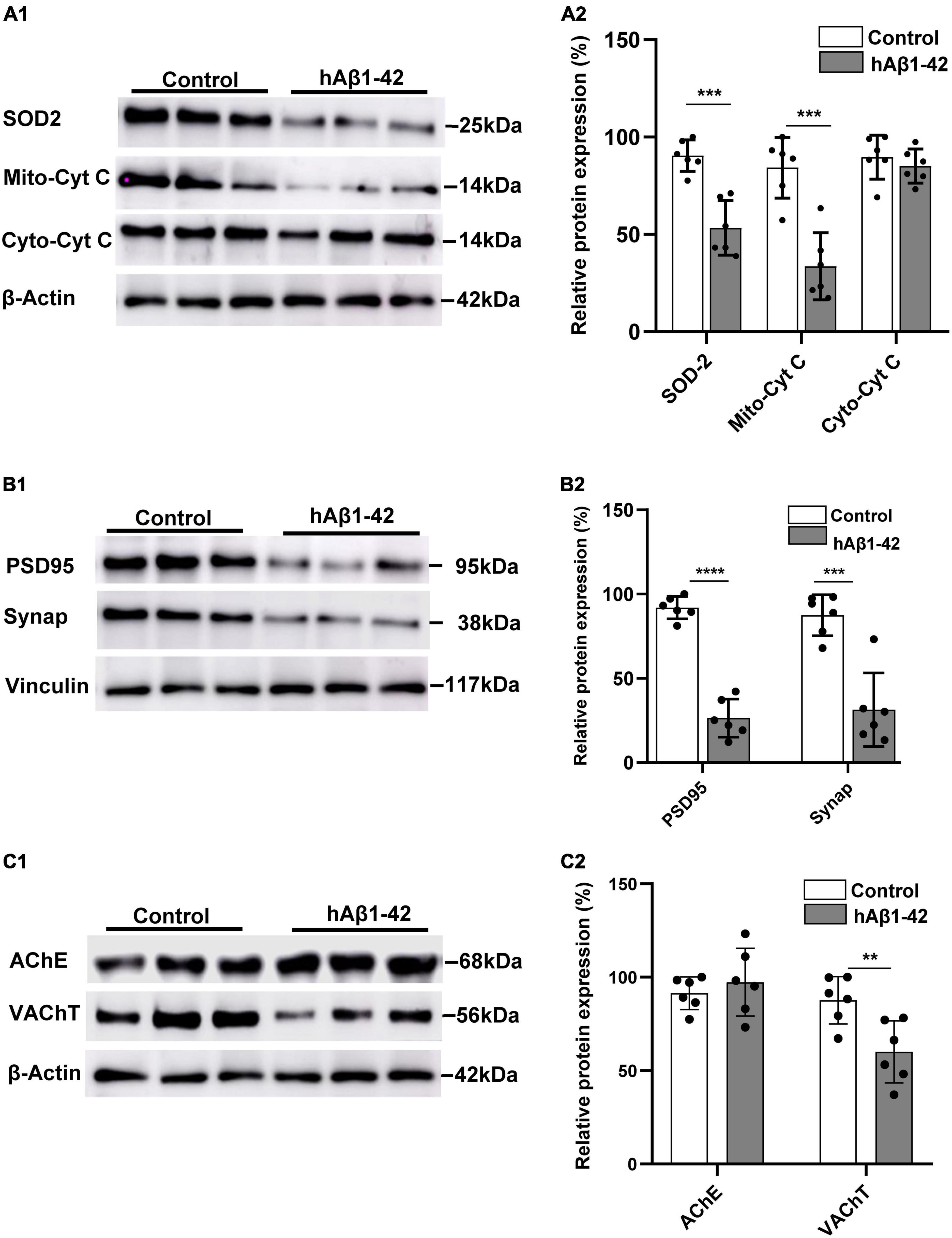
Figure 2. Effects of hAβ1–42 on key mitochondrial elements, synaptic proteins, and cholinergic markers in the entorhinal cortex. (A1) Representative immunoblots of mitochondrial superoxide dismutase 2 (SOD2), mitochondrial cytochrome c (Mito-cyt C), cytosolic cytochrome c (Cyto-cyt C), and β-actin loading control in entorhinal lysates treated with hAβ1–42 and control medium. (A2) Normalized expression of SOD2, Mito-cyt C, and Cyto-cyt C in hAβ1–42-treated entorhinal samples compared to control (n = 6). (B1) Representative immunoblots of postsynaptic density protein (PSD95), presynaptic marker synaptophysin (Synap), and vinculin loading control in entorhinal lysates. (B2) Quantification data showing the normalized expression of both PSD95 and Synap in slices incubated with hAβ1–42 vs. control (n = 6). (C1) Representative immunoblots of cholinergic markers acetylcholinesterase (AChE), vesicular acetylcholine transporter (VAChT), and β-actin (loading control). (C) Bar graphs showing normalized expression of AChE and VAChT in hAβ1–42–treated slices and control (n = 6) (**p < 0.01; ***p < 0.005; ****p < 0.0001).
A reduction in the efficiency and energy production capacity of the mitochondrial electron transport chain, increased mitochondrial ROS, and oxidative stress could impact both pre- and post-synaptic elements, and we therefore evaluated the effects of hAβ1–42 on the presynaptic marker synaptophysin, and the postsynaptic density protein 95 (PSD95). We found that hAβ1–42-induced mitochondrial dysregulation was accompanied by a marked reduction in relative immunoexpression of both PSD95 (91.9 ± 6.6 and 26.4 ± 11.3; p < 0.0001) and synaptophysin (87.5 ± 12.2 and 31.3 ± 21.8; p = 0.0003) (Figure 2B).
Reductions in cholinergic function in the EC is one of the earliest manifestations of AD (Hampel et al., 2018), and we therefore assessed the effects of hAβ1–42 on the expression of the vesicular acetylcholine transporter (VAChT) and the degradative enzyme acetylcholinesterase (AChE). Although hAβ1–42 did not cause a significant change in AChE immunoexpression when compared to control (97.4 ± 18.2 vs. 91.5 ± 8.7 in controls; p = 0.5433), hAβ1–42 markedly reduced the expression of VAChT in entorhinal lysates vs. control (60.0 ± 16.6 vs. 87.7 ± 12.7; p = 0.0312) (Figure 2C), suggesting a disruption in the function of cholinergic terminals in the EC.
To determine if increased ROS associated with mitochondrial dysregulation is related to the reductions in mitochondrial enzymes and synaptic proteins induced by hAβ1–42, we tested the ability of two novel, structurally distinct ROS scavengers (MitoQ and EUK134) to block protein reductions induced by hAβ1–42. The mitochondria-targeted antioxidant MitoQ administered alone had no significant effect on the expression of mitochondrial enzymes, but was found to block changes in both SOD2 and mitochondrial cytochrome c induced by hAβ1–42 (compare Figure 3A and Figure 2A). There was no significant main effect of treatment on the relative expression of SOD2 between control slices (92.2 ± 6.4) and slices incubated with MitoQ alone (90.4 ± 15.6) or MitoQ with hAβ1–42 (88.7 ± 8.9) [F(2, 15) = 0.15, p = 0.8646]. Similarly, the expression of mitochondrial cytochrome c protein was not significantly different between control slices (90.7.0 ± 8.3) and slices treated with either MitoQ (94.5 ± 9.8) or MitoQ and hAβ1–42 (88.0 ± 11.4) [F(2, 15) = 0.65, p = 0.5355] (Figure 3A).
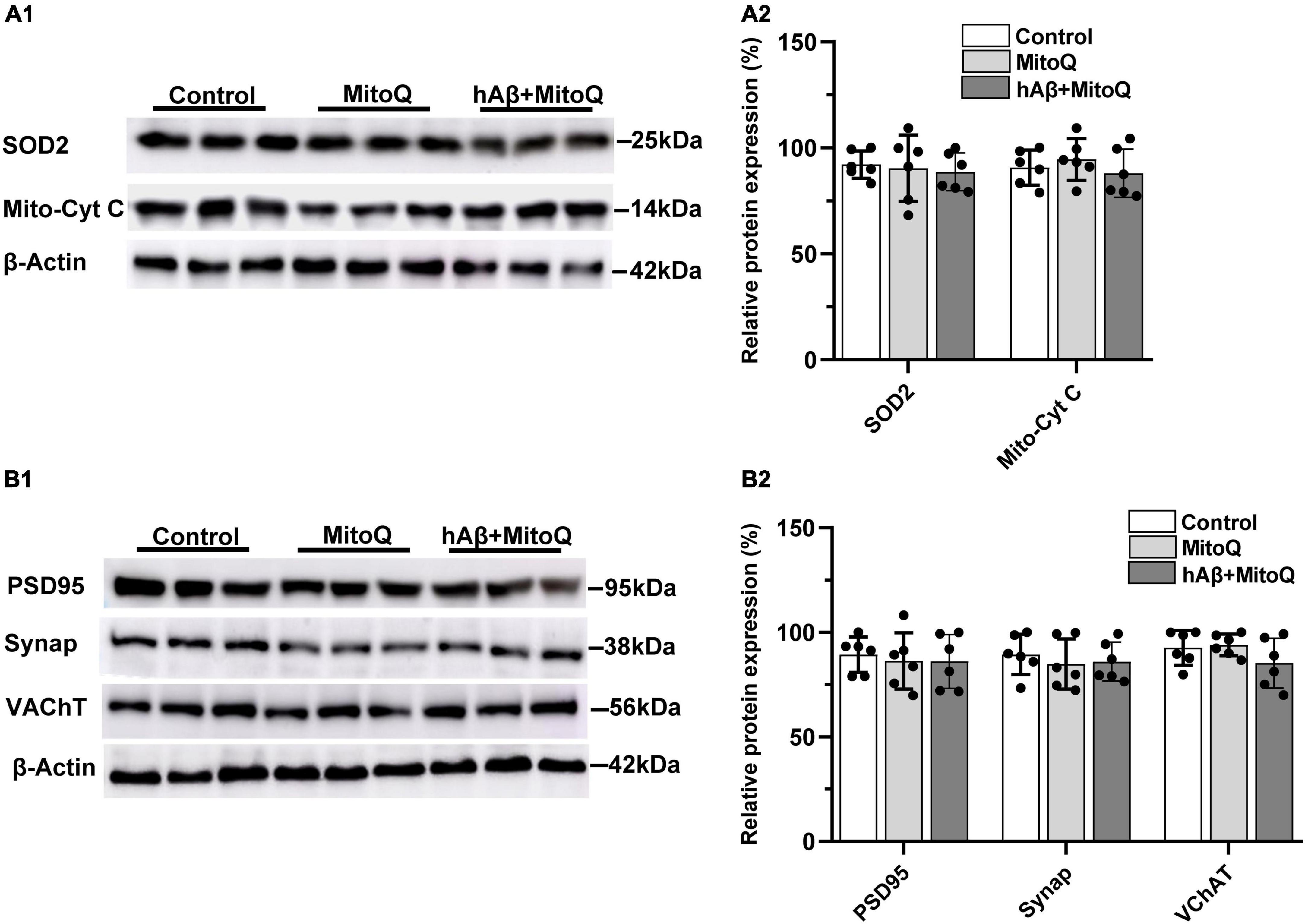
Figure 3. Inhibiting increases in reactive oxygen species with MitoQ prevents hAβ1–42-induced changes in mitochondrial and synaptic proteins. (A1) Representative immunoblots of superoxide dismutase 2 (SOD2), mitochondrial cytochrome c (Mito-cyt C), and the loading control β-Actin, are shown in slices treated with MitoQ, hAβ1–42 with MitoQ, and control. (A2) Normalized relative expression of SOD2 and Mito-cyt C proteins (n = 6). (B1) Representative immunoblots of postsynaptic density protein PSD95, presynaptic marker synaptophysin (Synap.), vesicular acetylcholine transporter (VAChT), and the loading control β-Actin are shown in tissue treated with MitoQ, hAβ1–42 with MitoQ, and control. Bar graphs indicate normalized relative expression of PSD95, Synap., and VAChT (B2).
We then assessed if blocking excess ROS induced by hAβ1–42 using MitoQ would also block reductions in the immunoexpression of PSD95, synaptophysin, and the vesicular acetylcholine transporter (VChAT). The application of MitoQ had no effect alone, but it prevented reductions in all three proteins induced by hAβ1–42 (compare Figure 3B and Figures 2B,C). There were no significant main effects of treatment (n = 6) on the expression of PSD95 [F(2, 15) = 0.14, p = 0.8742], synaptophysin [F(2, 15) = 0.31, p = 0.7386], and VChAT [F(2, 15) = 1.66, p = 0.2226]. Preventing increases in ROS induced by hAβ1–42 can therefore prevent degradation of key synaptic proteins.
The idea that increases in mitochondrial ROS induced by hAβ1–42 mediate synaptic degeneration in entorhinal slices was further tested by determining if the synthetic SOD and catalase mimetic EUK134 could block the reductions in synaptic and mitochondrial proteins induced by hAβ1–42. Application of EUK134 blocked reductions in both SOD2 and mitochondrial cytochrome c induced by hAβ1–42 (compare Figure 4A and Figure 2A), and there were no significant main effects of treatment with EUK134, or with EUK134 and hAβ1–42, (n = 6), on the relative expression of SOD2 [F(2, 15) = 0.95, p = 0.4084] or mitochondrial cytochrome c [F(2, 15) = 0.61, p = 0.5578].
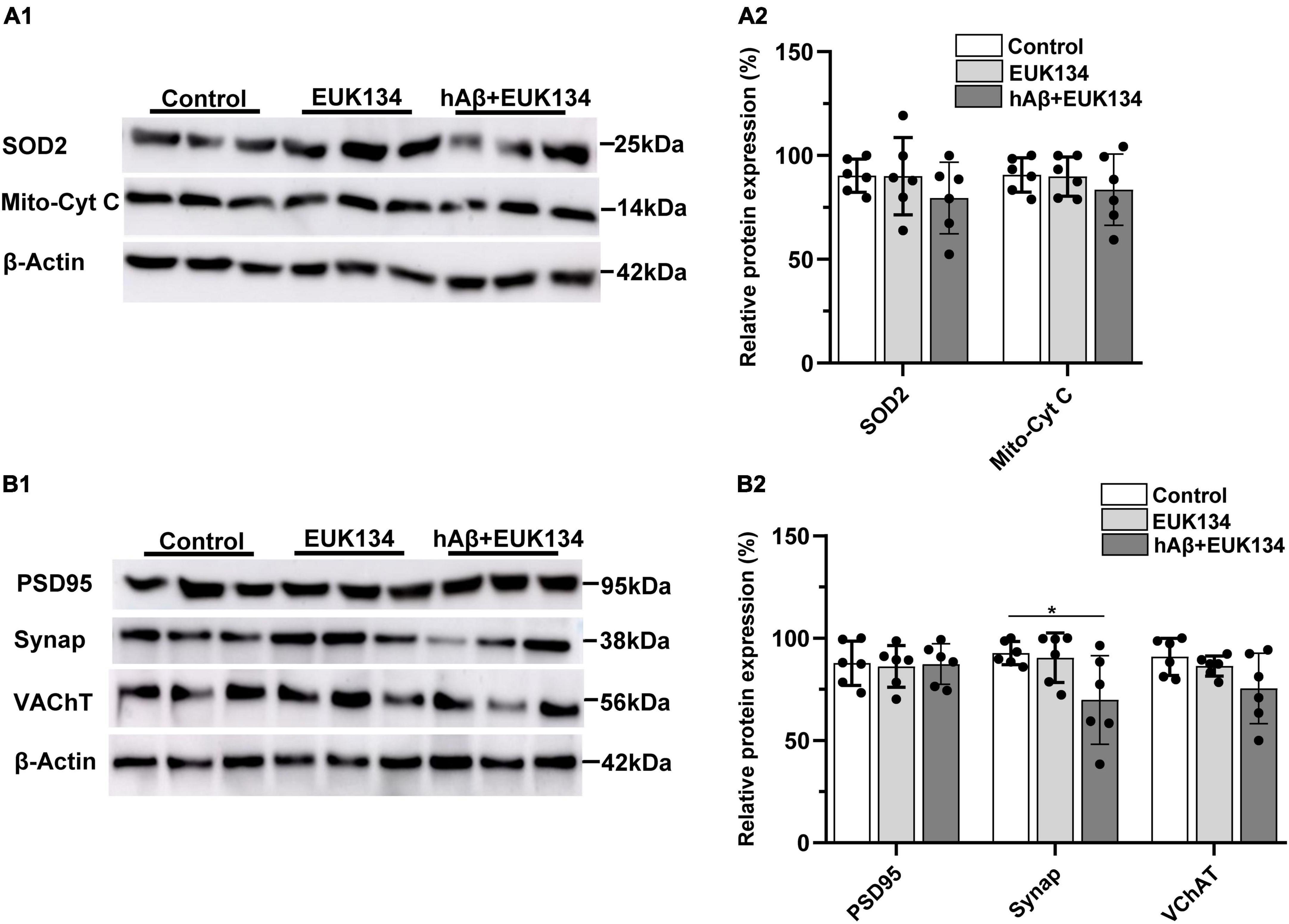
Figure 4. Inhibiting increases in reactive oxygen species with EUK134 prevents hAβ1–42-induced changes in mitochondrial and synaptic proteins. (A1) Representative immunoblots of superoxide dismutase 2 (SOD2), mitochondrial cytochrome c (Mito-cyt C), and the loading control β-Actin, are shown in slices treated with EUK134, hAβ1–42 with EUK134, and control. (A2) Normalized relative expression of SOD2 and Mito-cyt C proteins (n = 6). (B1) Representative immunoblots of postsynaptic density protein PSD95, presynaptic marker synaptophysin (Synap.), vesicular acetylcholine transporter (VAChT), and the loading control β-Actin are shown for tissue treated with EUK134, hAβ1–42 with EUK134, and control. Bar graphs indicate normalized relative expression of PSD95, Synap., and VAChT (B2) (*p < 0.05).
We also found that EUK134 rescued hAβ1–42-induced reductions in immunoexpression of PSD95 and VChAT, and partially rescued hAβ1–42-induced reductions in the presynaptic protein synaptophysin (compare Figure 4B and Figures 2B,C). There was no significant effect of treatment with EUK134 or EUK134 and hAβ1–42 on the relative expression of PSD95 [F(2, 15) = 0.04, p = 0.9650] and VChAT [F(2, 15) = 2.83, p = 0.0905]. There was, however, a significant main effect of treatment on synaptophysin immunoexpression [F(2, 15) = 4.44, p = 0.0305] in which the expression of synaptophysin was significantly reduced in slices treated with EUK134 + hAβ1–42 relative to control slices (69.8 ± 21.6 vs. 92.7 ± 5.7 in controls; p = 0.0480), but not relative to slices treated with EUK134 alone (90.4 ± 12.1; p = 0.0811). The mean expression of synaptophysin following treatment with hAβ1–42 in the presence of EUK134 (69.8 ± 21.6) was greater than that following treatment with hAβ1–42 alone (31.3 ± 21.8; Figure 2B), however, suggesting that EUK134 was partially protective in preventing reductions in synaptophysin. These findings suggest that hAβ1–42-induced rapid synaptic impairments in entorhinal tissue can be significantly rescued by inhibiting mitochondrial ROS levels.
A previous report from our lab has shown that hAβ1–42 causes rapid degeneration of presynaptic and postsynaptic elements in the EC through activation of GluN2A- and GluN2B-containing NMDA glutamate receptors (Olajide and Chapman, 2021). The selective vulnerability of the EC to early AD-related synaptic damage and neurodegeneration (Du et al., 2004; Velayudhan et al., 2013; Zhou et al., 2016), may reflect the dysregulation of neural mechanisms including NMDA glutamate receptors that contribute to learning and memory (Olajide et al., 2021). Aβ can accumulate in mitochondria where it impairs mitochondrial dynamics and upregulates oxidative stress by impairing mitochondrial respiratory function and the production of adenosine triphosphate (ATP) (Wang et al., 2014, 2020; Ashleigh et al., 2022). Further, activation of NMDA receptors by hAβ1–42 can lead to increases in calcium influx, the rapid facilitation of AMPA glutamate receptor responses, and hyperexcitability that increases metabolic demands on mitochondria, ultimately culminating in oxidative stress and synaptic dysfunction (Findley et al., 2019; Guo et al., 2020; Wang et al., 2020). We have found here that exposure to 1 μM hAβ1–42 for a period of 3 h markedly reduces mitochondrial respiratory function and integrity in the EC. Further, the resulting increase in ROS appears to be a major driver of the rapid degeneration of both pre- and post-synaptic proteins in the EC because the ROS scavengers Mito-Q and EUK134 strongly inhibited reductions in synaptophysin and PSD-95. The reductions in synaptic proteins observed here imply a reduction in number of synapses, but it is not clear if this would be expressed following this relatively brief application of hAβ1–42. However, these results point to a major role of mitochondrial dysregulation in the synaptic degeneration induced by hAβ1–42, which may contribute to the early progression of AD in the EC (Olajide et al., 2021). The present results may reflect mitochondrial dysfunction in glia as well as neurons, and mitochondrial dysfunction in glia is also thought to be a major contributor to the progression of AD (Mulica et al., 2021).
In our experimental model we have applied moderate concentrations of Aβ to wildtype slices for 3 h, but the progression of AD involves circulation of lower concentrations of Aβ over greatly extended periods of time. Although our model may reflect degenerative mechanisms that are expressed early in the progression of AD, and reflect the early selective vulnerability of the EC to Aβ toxicity (Olajide et al., 2021), the changes in mitochondrial function and synaptic proteins that we have observed might also occur in later stages of AD after prolonged exposure to lower concentrations of Aβ.
We have found here that hAβ1–42 markedly reduces mitochondrial-coupled oxygen consumption, and functional membrane integrity as reflected by oxygen use in the presence of cytochrome c. High-resolution respirometry showed no significant changes in non-phosphorylating LEAK respiration during the addition of the Complex I-linked substrates malate, pyruvate or glutamate, but the OXPHOS capacity of Complex I-linked activity was markedly reduced by hAβ1–42, as shown by the approximately fivefold reduction in oxygen utilization during the addition of ADP. Exposure to hAβ1–42 also impaired the function of Complex II, because the addition of succinate, which reflects the combined OXPHOS capacity of Complex I and Complex II, was also significantly reduced. The rapid defects in mitochondrial-related metabolic pathways provide direct evidence that impaired bioenergetic machinery in mitochondria is an early driver of AD-related pathology in the EC. Similarly, OXPHOS and associated pathways are significantly downregulated within the hippocampus during AD, and reductions in Complex I, III, and IV of OXPHOS have been reported in both early and definite AD brains (Manczak et al., 2004; Brooks et al., 2007).
Further, the indication that hAβ1–42-induced impairments in entorhinal electron transport chain complexes can dysregulate bioenergetics is corroborated by our findings that the efficiency of the link between oxidation and the production of ATP by phosphorylation of ADP was reduced by hAβ1–42, as reflected by both reductions in the uncoupling capacity of the mitochondria during the addition of FCCP, and by a reduction in the ACR. The ACR expresses the rate of oxygen consumption during ADP phosphorylation relative to the rate of non-phosphorylating LEAK-respiration induced by glutamate. Our data corresponds with several reports indicating a large and consistent decline in mitochondrial substrate utilization and energy production in the neocortex during the prodromal stages of AD (Croteau et al., 2018; Wang et al., 2020), suggesting that mitochondrial bioenergetics dysfunction plays an early role in the pathogenesis of AD in the EC.
Mitochondrial cytochrome c transfers electrons to complex IV in the electron transport chain which is the primary site of cellular oxygen consumption (Timón-Gómez et al., 2018). We found that hAβ1–42 caused about a 2.5-fold reduction in mitochondrial cytochrome c function, suggesting alterations to the mitochondrial membrane in entorhinal tissue. We further found that, although total cytoplasmic cytochrome c protein expression was unaffected, the expression of mitochondrial cytochrome c was greatly reduced by treatment with hAβ1–42. An increase in cytosolic cytochrome c could be expected from previous work showing that Aβ can activate intrinsic apoptotic pathways and the release of mitochondrial cytochrome c into the cytosol (Kim et al., 2014), but the lack of an increase in cytosolic cytochrome c in the present study may be due to the relatively brief duration of exposure to Aβ. Deficiency of cytochrome c oxidase (Complex IV) is the most common defect in the mitochondrial electron transport chain in AD, leading to an increase in ROS production, a decrease in energy stores, and a disruption of energy metabolism (Rak et al., 2016). We found a marked reduction in the expression of SOD2, a key antioxidant enzyme that scavenges superoxide in mitochondria, and this is consistent with a rapid increase in mitochondrial ROS production induced by hAβ1–42 that depletes the energy metabolism machinery in the EC.
Although hAβ1–42 significantly reduced respiration and bioenergetics, it did not alter the expression of protein markers for the five mitochondrial Complexes or the expression of the mitochondrial voltage-dependent anion channel 1 (VDAC1). VDAC1 is the most abundant protein on the outer membrane of mitochondria, and it serves as a gatekeeper for the passage of metabolites, and is crucial for the metabolic functions of mitochondria (Camara et al., 2017). It is well established that the expression of mitochondrial proteins and genes are among the most prominent changes in the cortex during AD (Cottrell et al., 2001; Valla et al., 2001; Minjarez et al., 2016; Adav et al., 2019), and the lack of a significant effect of hAβ1–42 on the expression of mitochondrial complex proteins observed here may reflect the resistance of these proteins to rapid degradation over the relativity short incubation time.
Reductions in synaptic proteins induced by hAβ1–42 are likely to have resulted from increased ROS and oxidative stress in entorhinal neurons. Incubation in hAβ1–42 resulted in a marked reduction in SOD2, which is the primary antioxidant enzyme that scavenges superoxide in mitochondria, suggesting that SOD2 may have been depleted due to increases in ROS and overwhelming activity-dependent utilization of SOD2. The reduction in SOD2 may have further impaired ROS scavenging in entorhinal neurons thereby exacerbating oxidative damage and a loss of mitochondrial cytochrome c function. Excessive ROS and oxidative stress have been extensively linked to synaptic loss associated with aging and AD (Patten et al., 2010; Tönnies and Trushina, 2017; Guo et al., 2020), and increases in ROS associated with mitochondrial dysfunction that exceed the scavenging ability of entorhinal neurons is likely to have contributed to the observed loss of synaptic proteins. Consistent with our previous findings in the EC (Olajide and Chapman, 2021), both the presynaptic marker synaptophysin and the postsynaptic marker PSD-95, were reduced by hAβ1–42.
The degeneration of cholinergic terminals in the EC is one of the earliest neuropathologies in AD (Francis et al., 1999; Gil-Bea et al., 2005; Hamam et al., 2007; Schaeffer and Gattaz, 2008). Although we found that the degradative enzyme acetylcholinesterase was not affected by hAβ1–42, we did observe a reduction in the vesicular acetylcholine transporter (VAChT), suggesting a reduced function in cholinergic terminals. Several other reports have also shown marked reductions in the expression of VAChT following Aβ infusion in rats and in AD models (Ikeda et al., 2000; Pákáski and Kálmán, 2008; Schliebs and Arendt, 2011; Hampel et al., 2018).
ROS-mediated oxidative stress is thought to have a major role in synaptic dysregulation and neurodegeneration in AD, and various antioxidants, and the genetic overexpression of SOD2, have been explored as therapeutic strategies for AD (Dumont et al., 2009; Massaad et al., 2009; Bonda et al., 2010; Olajide et al., 2017a,b, 2018; Tönnies and Trushina, 2017; Misrani et al., 2021). We co-incubated slices with hAβ1–42 and the ROS inhibitors MitoQ or EUK134 to determine if reducing oxidative stress could prevent the degeneration of synaptic proteins in the EC. Both MitoQ and EUK134 inhibited hAβ1–42-induced reductions in both SOD2 and mitochondrial cytochrome c, likely by maintaining a sufficient ROS scavenging capacity in entorhinal neurons and preventing overwhelming demands on SOD2. In addition, co-incubation with MitoQ blocked hAβ1–42-induced decreases in synaptophysin, PSD-95, and VAChT, indicating that oxidative stress arising from insufficient mitochondrial capacity can drive degeneration of these synaptic proteins. Similarly, the ROS inhibitor EUK134 prevented reductions in PSD-95 and VAChT, and greatly attenuated reductions in synaptophysin induced by hAβ1–42. Others have also found that decreasing ROS using MitoQ and EUK134 can prevent synaptic degeneration induced by oxidative stress in cortical and hippocampal neurons (Ma et al., 2011; Mcmanus et al., 2011; Reddy et al., 2012; Sanmartín et al., 2017; Yu et al., 2018; Cenini and Voos, 2019). These studies suggest that the protective effect of ROS inhibition that we have observed following acute application of hAβ1–42 to wildtype slices might be similarly protective for the advancement of neurodegeneration in more chronic models of AD.
Mitochondrial dysfunction resulting in increased ROS production and oxidative stress precedes the formation of neuritic plaques and neurofibrillary tangles, and is thought to contribute substantially to the earliest stages of AD and the onset of cognitive decline and memory loss (Uttara et al., 2009; Wang et al., 2014; Tönnies and Trushina, 2017; Ashleigh et al., 2022). We have shown here that short-term exposure of wild-type EC slices to hAβ1–42 results in deficits in mitochondrial respiration, and have provided evidence that oxidative stress associated with increased ROS is a major factor in the rapid degeneration of key pre- and post-synaptic proteins in the EC. Further, these mechanisms may be a major contributor to the reduction in cholinergic transmission in the EC, which is an early pathology thought to contribute to cognitive decline.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Animal Research Ethics Committee, Concordia University.
OO, AB, and CC contributed to conception and design of the study. OO, AB, and CL acquired and analyzed the data. OO wrote the first draft of the manuscript. All authors contributed to manuscript revision and approved the submitted version.
This research was funded by the Natural Sciences and Engineering Research Council of Canada to CC and AB and the Merit Scholarship to OO from the Fonds de Recherché du Quebec–Nature et Technologies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adav, S. S., Park, J. E., and Sze, S. K. (2019). Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 12:8. doi: 10.1186/S13041-019-0430-Y/FIGURES/6
Akhter, F., Chen, D., Yan, S. F., and Yan, S. S. (2017). Mitochondrial perturbation in Alzheimer’s disease and diabetes. Prog. Mol. Biol. Transl. Sci. 146, 341–361. doi: 10.1016/bs.pmbts.2016.12.019
Armand-Ugon, M., Ansoleaga, B., Berjaoui, S., and Ferrer, I. (2017). Reduced mitochondrial activity is early and steady in the entorhinal cortex but it is mainly unmodified in the frontal cortex in Alzheimer’s disease. Curr. Alzheimer Res. 14, 1327–1334. doi: 10.2174/1567205014666170505095921
Arendt, T. (2009). Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 118, 167–179. doi: 10.1007/s00401-009-0536-x
Ashleigh, T., Swerdlow, R. H., and Beal, M. F. (2022). The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. doi: 10.1002/alz.12683
Bitan, G., Vollers, S. S., and Teplow, D. B. (2003). Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 278, 34882–34889. doi: 10.1074/jbc.M300825200
Bonda, D. J., Wang, X., Perry, G., Nunomura, A., Tabaton, M., Zhu, X., et al. (2010). Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 59, 290–294. doi: 10.1016/j.neuropharm.2010.04.005
Brooks, W. M., Lynch, P. J., Ingle, C. C., Hatton, A., Emson, P. C., Faull, R. L. M., et al. (2007). Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 1127, 127–135. doi: 10.1016/J.BRAINRES.2006.09.106
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Camara, A. K. S., Zhou, Y. F., Wen, P. C., Tajkhorshid, E., and Kwok, W. M. (2017). Mitochondrial VDAC1: A key gatekeeper as potential therapeutic target. Front. Physiol. 8:460. doi: 10.3389/FPHYS.2017.00460/BIBTEX
Cavallucci, V., Ferraina, C., and D’Amelio, M. (2013). Key role of mitochondria in Alzheimer’s disease synaptic dysfunction. Curr. Pharm. Des. 19, 6440–6450. doi: 10.2174/1381612811319360005
Cenini, G., and Voos, W. (2019). Mitochondria as potential targets in Alzheimer disease therapy: An update. Front. Pharmacol. 10:902. doi: 10.3389/FPHAR.2019.00902/BIBTEX
Coleman, P. D., and Yao, P. J. (2003). Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging 24, 1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001
Cottrell, D. A., Blakely, E. L., Johnson, M. A., Ince, P. G., and Turnbull, D. M. (2001). Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 57, 260–264. doi: 10.1212/WNL.57.2.260
Croteau, E., Castellano, C. A., Fortier, M., Bocti, C., Fulop, T., Paquet, N., et al. (2018). A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 107, 18–26. doi: 10.1016/J.EXGER.2017.07.004
Du, A. T., Schuff, N., Kramer, J. H., Ganzer, S., Zhu, X. P., Jagust, W. J., et al. (2004). Higher atrophy rate of entorhinal cortex than Hippocampus in AD. Neurology 62, 422–427. doi: 10.1212/01.WNL.0000106462.72282.90
Dumont, M., Wille, E., Stack, C., Calingasan, N. Y., Beal, M. F., and Lin, M. T. (2009). Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J. 23, 2459–2466. doi: 10.1096/FJ.09-132928
Esposito, L., Raber, J., Kekonius, L., Yan, F., Yu, G. Q., Bien-Ly, N., et al. (2006). Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 26, 5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006
Fan, L., Mao, C., Hu, X., Zhang, S., Yang, Z., Hu, Z., et al. (2020). New insights into the pathogenesis of Alzheimer’s disease. Front. Neurol. 10:1312. doi: 10.3389/fneur.2019.01312
Findley, C. A., Bartke, A., Hascup, K. N., and Hascup, E. R. (2019). Amyloid beta-related alterations to glutamate signaling dynamics during Alzheimer’s disease progression. ASN Neuro 11:1759091419855541. doi: 10.1177/1759091419855541
Francis, P. T., Palmer, A. M., Snape, M., and Wilcock, G. K. (1999). The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 66, 137–147. doi: 10.1136/jnnp.66.2.137
Gil-Bea, F. J., García-Alloza, M., Domínguez, J., Marcos, B., and Ramírez, M. J. (2005). Evaluation of cholinergic markers in Alzheimer’s disease and in a model of cholinergic deficit. Neurosci. Lett. 375, 37–41. doi: 10.1016/j.neulet.2004.10.062
Glovaci, I., and Chapman, C. A. (2019). Dopamine induces release of calcium from internal stores in layer II lateral entorhinal cortex fan cells. Cell Calcium 80, 103–111. doi: 10.1016/j.ceca.2019.04.003
Grubman, A., Chew, G., Ouyang, J. F., Sun, G., Choo, X. Y., McLean, C., et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097. doi: 10.1038/s41593-019-0539-4
Guo, T., Zhang, D., Zeng, Y., Huang, T. Y., Xu, H., and Zhao, Y. (2020). Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 15:40. doi: 10.1186/s13024-020-00391-7
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hamam, B. N., Sinai, M., Poirier, G., and Chapman, C. A. (2007). Cholinergic suppression of excitatory synaptic responses in layer II of the medial entorhinal cortex. Hippocampus 17, 103–113. doi: 10.1002/hipo.20249
Hampel, H., Hardy, J., Blennow, K., Chen, C., Perry, G., Kim, S. H., et al. (2021). The amyloid-β pathway in Alzheimer’s disease. Mol. Psychiatry 26, 5481–5503. doi: 10.1038/s41380-021-01249-0
Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., et al. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933. doi: 10.1093/brain/awy132
Ikeda, E., Shiba, K., Mori, H., Ichikawa, A., Sumiya, H., Kuji, I., et al. (2000). Reduction of vesicular acetylcholine transporter in beta-amyloid protein-infused rats with memory impairment. Nucl. Med. Commun. 21, 933–937. doi: 10.1097/00006231-200010000-00007
Ionescu-Tucker, A., and Cotman, C. W. (2021). Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 107, 86–95. doi: 10.1016/J.NEUROBIOLAGING.2021.07.014
Khan, U. A., Liu, L., Provenzano, F. A., Berman, D. E., Profaci, C. P., Sloan, R., et al. (2014). Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 17, 304–311. doi: 10.1038/nn.3606
Khosravi, S., and Harner, M. E. (2020). The MICOS complex, a structural element of mitochondria with versatile functions. Biol. Chem. 401, 765–778. doi: 10.1515/hsz-2020-0103
Kim, J., Yang, Y., Song, S. S., Na, J. H., Oh, K. J., Jeong, C., et al. (2014). Beta-amyloid oligomers activate apoptotic BAK pore for cytochrome c release. Biophys. J. 107, 1601–1608. doi: 10.1016/j.bpj.2014.07.074
Li, F., Calingasan, N. Y., Yu, F., Mauck, W. M., Toidze, M., Almeida, C. G., et al. (2004). Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 89, 1308–1312. doi: 10.1111/J.1471-4159.2004.02455.X
Ma, T., Hoeffer, C. A., Wong, H., Massaad, C. A., Zhou, P., Iadecola, C., et al. (2011). Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J. Neurosci. 31, 5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011
Manczak, M., Park, B. S., Jung, Y., and Reddy, P. H. (2004). Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 5, 147–162. doi: 10.1385/NMM
Marsh, J., and Alifragis, P. (2018). Synaptic dysfunction in Alzheimer’s disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 13, 616–623. doi: 10.4103/1673-5374.230276
Massaad, C. A., Washington, T. M., Pautler, R. G., and Klann, E. (2009). Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106:13576. doi: 10.1073/PNAS.0902714106
Masters, C. L., and Selkoe, D. J. (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006262. doi: 10.1101/cshperspect.a006262
Mcmanus, M. J., Murphy, M. P., and Franklin, J. L. (2011). The mitochondria-targeted antioxidant mitoq prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 31:15703. doi: 10.1523/JNEUROSCI.0552-11.2011
Minjarez, B., Calderón-González, K. G., Rustarazo, M. L. V., Herrera-Aguirre, M. E., Labra-Barrios, M. L., Rincon-Limas, D. E., et al. (2016). Identification of proteins that are differentially expressed in brains with Alzheimer’s disease using iTRAQ labeling and tandem mass spectrometry. J. Proteom. 139, 103–121. doi: 10.1016/J.JPROT.2016.03.022
Misrani, A., Tabassum, S., and Yang, L. (2021). Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front. Aging Neurosci. 13:57. doi: 10.3389/FNAGI.2021.617588/BIBTEX
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006262. doi: 10.1101/cshperspect.a006338
Mulica, P., Grünewald, A., and Pereira, S. L. (2021). Astrocyte-neuron metabolic crosstalk in neurodegeneration: A mitochondrial perspective. Front. Endocrinol. 12:668517. doi: 10.3389/fendo.2021.668517
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem J. 417, 1–13. doi: 10.1042/BJ20081386
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/JNEN/60.8.759
Olajide, O. J., and Chapman, C. A. (2021). Amyloid-β (1-42) peptide induces rapid NMDA receptor-dependent alterations at glutamatergic synapses in the entorhinal cortex. Neurobiol. Aging 105, 296–309. doi: 10.1016/J.NEUROBIOLAGING.2021.05.006
Olajide, O. J., Asogwa, N. T., Moses, B. O., and Oyegbola, C. B. (2017a). Multidirectional inhibition of cortico-hippocampal neurodegeneration by kolaviron treatment in rats. Metab. Brain Dis. 32, 1147–1161. doi: 10.1007/s11011-017-0012-6
Olajide, O. J., Fatoye, J. O., Idowu, O. F., Ilekoya, D., Gbadamosi, I. T., Gbadamosi, M. T., et al. (2018). Reversal of behavioral decline and neuropathology by a complex vitamin supplement involves modulation of key neurochemical stressors. Environ. Toxicol. Pharmacol. 62, 120–131. doi: 10.1016/j.etap.2018.07.005
Olajide, O. J., Suvanto, M. E., and Chapman, C. A. (2021). Molecular mechanisms of neurodegeneration in the entorhinal cortex that underlie its selective vulnerability during the pathogenesis of Alzheimer’s disease. Biol. Open 10:bio056796. doi: 10.1242/bio.056796
Olajide, O. J., Yawson, E. O., Gbadamosi, I. T., Arogundade, T. T., Lambe, E., Obasi, K., et al. (2017b). Ascorbic acid ameliorates behavioural deficits and neuropathological alterations in rat model of Alzheimer’s disease. Environ. Toxicol. Pharmacol. 50, 200–211. doi: 10.1016/j.etap.2017.02.010
O’Nuallain, B., Williams, A. D., Westermark, P., and Wetzel, R. (2004). Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 279, 17490–17499. doi: 10.1074/jbc.M311300200
Overk, C. R., and Masliah, E. (2014). Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem. Pharmacol. 88, 508–516. doi: 10.1016/j.bcp.2014.01.015
Pákáski, M., and Kálmán, J. (2008). Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem. Int. 53, 103–111. doi: 10.1016/j.neuint.2008.06.005
Patten, D. A., Germain, M., Kelly, M. A., and Slack, R. S. (2010). Reactive oxygen species: Stuck in the middle of neurodegeneration. J. Alzheimers Dis. 20, S357–S367. doi: 10.3233/JAD-2010-100498
Paxinos, G., and Watson, C. (1997). The rat brain in stereotaxic coordinates, 3rd Edn. San Diego, CA: Academic Press.
Pickett, E. K., Rose, J., McCrory, C., McKenzie, C. A., King, D., Smith, C., et al. (2018). Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 136, 747–757. doi: 10.1007/s00401-018-1903-2
Rak, M., Bénit, P. P., Chrétien, D., Bouchereau, J., Schiff, M., El-Khoury, R., et al. (2016). Mitochondrial cytochrome c oxidase deficiency. Clin. Sci. 130, 393–407. doi: 10.1042/CS20150707
Reddy, P. H., Tripathi, R., Troung, Q., Tirumala, K., Reddy, T. P., Anekonda, V., et al. (2012). abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 1822:639. doi: 10.1016/J.BBADIS.2011.10.011
Sanmartín, C. D., Veloso, P., Adasme, T., Lobos, P., Bruna, B., Galaz, J., et al. (2017). RyR2-mediated Ca2+ release and mitochondrial ROS generation partake in the synaptic dysfunction caused by amyloid β peptide oligomers. Front. Mol. Neurosci. 10:115. doi: 10.3389/FNMOL.2017.00115/BIBTEX
Schaeffer, E. L., and Gattaz, W. F. (2008). Cholinergic and glutamatergic alterations beginning at the early stages of Alzheimer disease: Participation of the phospholipase A2 enzyme. Psychopharmacology 198, 1–27. doi: 10.1007/s00213-008-1092-0
Schliebs, R., and Arendt, T. (2011). The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563. doi: 10.1016/j.bbr.2010.11.058
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Shankar, G. M., and Walsh, D. M. (2009). Alzheimer’s disease: Synaptic dysfunction and Aβ. Mol. Neurodegener. 4:48. doi: 10.1186/1750-1326-4-48
Stine, W. B., Dahlgren, K. N., Krafft, G. A., and LaDu, M. J. (2003). In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622. doi: 10.1074/jbc.M210207200
Terni, B., Boada, J., Portero-Otin, M., Pamplona, R., and Ferrer, I. (2010). Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of alzheimer’s disease pathology. Brain Pathol. 20, 222–233. doi: 10.1111/j.1750-3639.2009.00266.x
Timón-Gómez, A., Nývltová, E., Abriata, L. A., Vila, A. J., Hosler, J., and Barrientos, A. (2018). Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 76:163. doi: 10.1016/J.SEMCDB.2017.08.055
Tönnies, E., and Trushina, E. (2017). Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 57:1105. doi: 10.3233/JAD-161088
Tu, S., Okamoto, S.-I., Lipton, S. A., and Xu, H. (2014). Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9:48. doi: 10.1186/1750-1326-9-48
Uttara, B., Singh, A., Zamboni, P., and Mahajan, R. (2009). Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 7, 65–74. doi: 10.2174/157015909787602823
van Hoesen, G. W., Hyman, B. T., and Damasio, A. R. (1991). Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus 1, 1–8. doi: 10.1002/hipo.450010102
Valla, J., Berndt, J. D., and Gonzalez-Lima, F. (2001). Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: Superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci. 21:4923. doi: 10.1523/JNEUROSCI.21-13-04923.2001
Velayudhan, L., Proitsi, P., Westman, E., Muehlboeck, J. S., Mecocci, P., Vellas, B., et al. (2013). Entorhinal cortex thickness predicts cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 33, 755–766. doi: 10.3233/JAD-2012-121408
Wang, X., and Michaelis, E. K. (2010). Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2:12. doi: 10.3389/fnagi.2010.00012
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 15:30 doi: 10.1186/S13024-020-00376-6
Wang, X., Wang, W., Li, L., Perry, G., Lee, H. G., and Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1240–1247. doi: 10.1016/j.bbadis.2013.10.015
Wogulis, M., Wright, S., Cunningham, D., Chilcote, T., Powell, K., and Rydel, R. E. (2005). Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J. Neurosci. 25, 1071–1080. doi: 10.1523/JNEUROSCI.2381-04.2005
Xiao, T., Jiao, B., Zhang, W., Pan, C., Wei, J., Liu, X., et al. (2017). Identification of CHCHD10 mutation in chinese patients with Alzheimer disease. Mol. Neurobiol. 54, 5243–5247. doi: 10.1007/s12035-016-0056-3
Yu, Q., Wang, Y., Du, F., Yan, S., Hu, G., Origlia, N., et al. (2018). Overexpression of endophilin A1 exacerbates synaptic alterations in a mouse model of Alzheimer’s disease. Nat. Commun. 9:2968. doi: 10.1038/S41467-018-04389-0
Keywords: Alzheimer’s disease, mitochondria, acetylcholine, entorhinal cortex, oxidative stress, reactive oxygen species, synaptic proteins
Citation: Olajide OJ, La Rue C, Bergdahl A and Chapman CA (2022) Inhibiting amyloid beta (1–42) peptide-induced mitochondrial dysfunction prevents the degradation of synaptic proteins in the entorhinal cortex. Front. Aging Neurosci. 14:960314. doi: 10.3389/fnagi.2022.960314
Received: 02 June 2022; Accepted: 20 September 2022;
Published: 06 October 2022.
Edited by:
Panteleimon Giannakopoulos, Université de Genève, SwitzerlandReviewed by:
Natali Lujan Chanaday Ricagni, University of Pennsylvania, United StatesCopyright © 2022 Olajide, La Rue, Bergdahl and Chapman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clifton Andrew Chapman, YW5kcmV3LmNoYXBtYW5AY29uY29yZGlhLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.