 Xiangyang Xiong
Xiangyang Xiong Tianpeng Hu1
Tianpeng Hu1 Ping Lei
Ping Lei- 1Department of Geriatrics, Tianjin Medical University General Hospital, Tianjin, China
- 2Tianjin Neurological Institute, Tianjin, China
Although there are still no satisfactory answers to the question of why we need to sleep, a better understanding of its function will help to improve societal attitudes toward sleep. Sleep disorders are very common in neurodegenerative diseases and are a key factor in the quality of life of patients and their families. Alzheimer’s disease (AD) is an insidious and irreversible neurodegenerative disease. Along with progressive cognitive impairment, sleep disorders and disturbances in circadian rhythms play a key role in the progression of AD. Sleep and circadian rhythm disturbances are more common in patients with AD than in the general population and can appear early in the course of the disease. Therefore, this review discusses the bidirectional relationships among circadian rhythm disturbances, sleep disorders, and AD. In addition, pharmacological and non-pharmacological treatment options for patients with AD and sleep disorders are outlined.
Sleep disorders, circadian rhythm disturbances and Alzheimer’s disease
Sleep is induced by neurons, therefore, it is believed that death due to sleep deprivation is the result of impaired brain function. In a very early study, puppies that were completely deprived of sleep died within days, with the most serious lesions occurring in the brain. Through a histological study of the dead puppies, degenerative changes in spinal ganglion neurons, cerebellar Purkinje cells and frontal cortex neurons were observed (Bentivoglio and Grassi-Zucconi, 1997). In a recent study, the movement of chromosomes (chromosome dynamics) in the neurons of zebrafish was photographed at regular intervals; sleep was found to triple the chromosome dynamics. In general, sleep increases chromosome dynamics, which changes the structure of chromosomes and reduces DNA damage, thus reducing death (Zada et al., 2019). Additionally, a human clinical study found a decrease in temporal lobe nerve activity after sleep deprivation (Nir et al., 2017). Sleep is an active process: specific brain structures are activated during specific stages of sleep and deactivated during other stages. Normal sleep stages includes non-rapid eye movement (non-REM) sleep and rapid eye movement (REM) sleep, which have different EEG characteristics (Tamaki et al., 2020). A sleep cycle lasts approximately every 90 min, and these cycles occur four to five times a night (Tamaki et al., 2020). Sleep is typically necessary for brain activity. By promoting the clearance of brain metabolites, sleep plays a key role in maintaining the dynamic balance of metabolism (Xie et al., 2013). Sleep can also save energy and help animals escape the notice of predators. Furthermore, sleep is important in consolidating memory (Girardeau and Lopes-Dos-Santos, 2021). The average person needs approximately 8 h of sleep per night, but many otherwise healthy people deprive themselves of adequate sleep, resulting in fatigue, poor decision-making and a higher risk of accidents (Bandyopadhyay and Sigua, 2019). Various studies have shown that chronic sleep deprivation leads to poor concentration, emotional instability (Yoo et al., 2007), increased sensitivity to pain (Alexandre et al., 2017), metabolic and cardiovascular diseases (Broussard et al., 2012; Huang et al., 2020), immune dysfunction and even, in extreme cases, death. A long-term mismatch between circadian rhythms and lifestyle may be associated with a high risk of several diseases, such as neurodegenerative diseases, inflammation, cancer, and metabolic disorders (Hirota and Kay, 2015; Figure 1). These negative effects of sleep deprivation elucidate the significance of sleep. A balanced and adequate sleep cycle is one of the main factors determining human quality of life. Many factors contribute to sleep deprivation, including environmental, psychological, and physical factors. Changes in the environment affect many aspects of our daily lives, and sleep disorders may occur due to increased ambient noise and fluctuations in light and temperature (LeGates et al., 2012; Kroller-Schon et al., 2018). Mental health also plays a crucial role in maintaining regular sleep patterns, and mental illnesses, such as stress, anxiety, and depression, interfere with regular sleep cycles, which may translate into insomnia (O’Loughlin et al., 2021). Additionally, psychiatric disorders such as schizophrenia or neurodegenerative diseases such as Alzheimer’s are often associated with sleep deprivation, such as shortened rapid eye movement sleep cycles and reduced sleep spindle activity (Fernandez and Luthi, 2020). Modern lifestyle changes such as unbalanced work hours, excessive caffeine intake, and increased screen time potentially affect the quality and quantity of sleep. However, the dynamic balance of normal biological circadian rhythms is necessary to achieve better cognitive and task performance. For example, night shift workers experience varying degrees of cognitive impairment. Shift work can forcibly disrupt the normal sleep-wake cycle, resulting in insufficient sleep and excessive fatigue, which, in turn, affects long-term health and safety (Kecklund and Axelsson, 2016). Substance abuse and alcohol intake have also been implicated as the causes of an increasing number of sleep disorders (Guo et al., 2016; Hasler and McClung, 2021). For instance, alcohol consumption inhibits REM sleep, which in turn impairs the performance of routine tasks (Chan et al., 2015). Additionally, sleep deprivation can affect the balance of important neurotransmitters in the brain; one important example is dopamine (DA), the disruption of which has been shown to increase susceptibility to substance use (Hasler and McClung, 2021). The negative effects of sleep deprivation described above encompass only a small portion of these effects, but can help convey the importance of sleep.
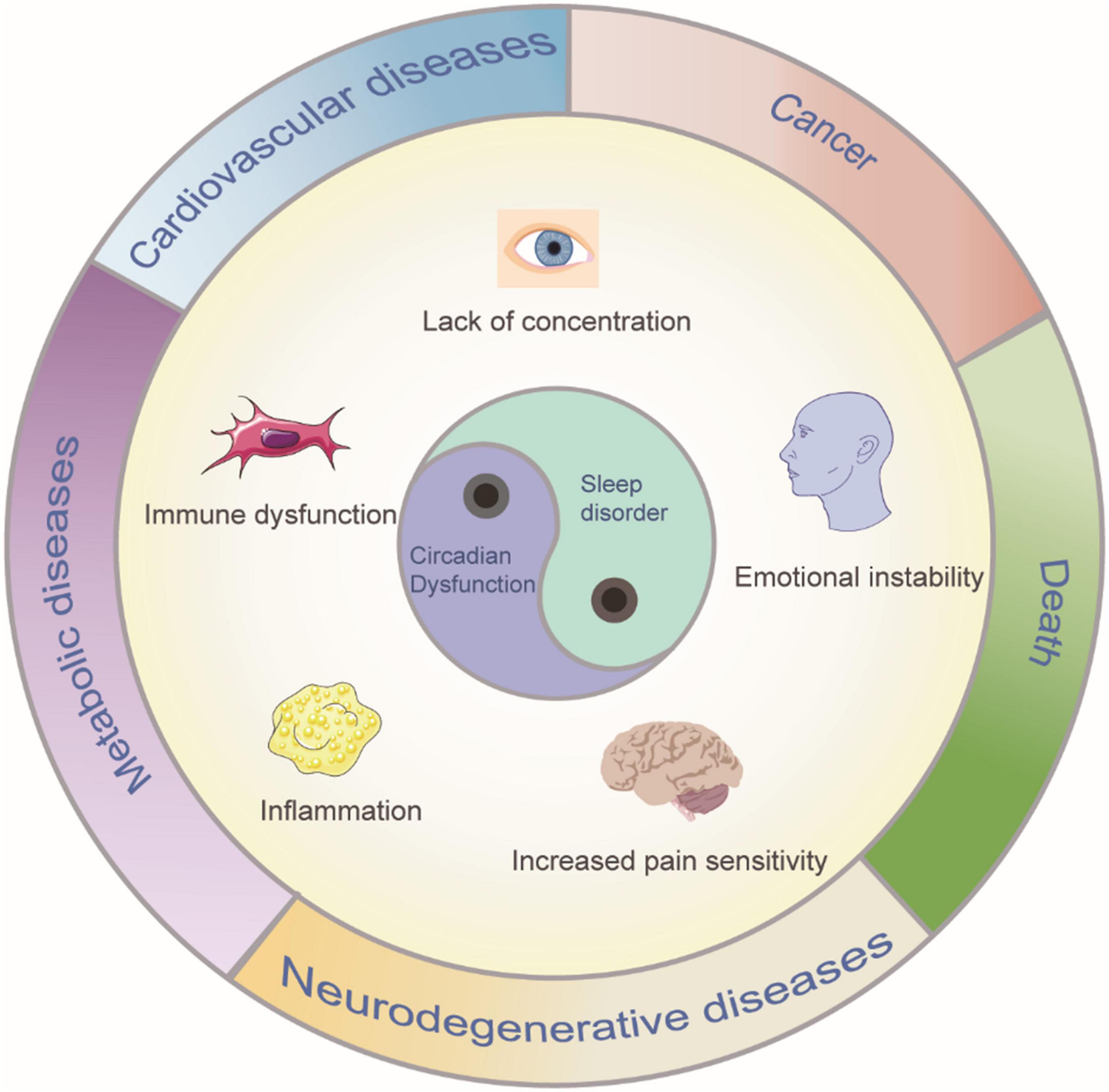
Figure 1. Sleep disorder and circadian dysfunction may lead to clinical symptoms and diseases. Such consequences can include poor concentration, emotional instability, increased sensitivity to pain, metabolic, and cardiovascular diseases, immune dysfunction and even, in extreme cases, death.
Alzheimer’s disease (AD) was first identified in 1907 when the German physician Alois Alzheimer reported a case in a 56-year-old woman, subsequently, the disease was named after him (Alzheimer et al., 1995). Currently, approximately 44 million people worldwide have AD or related dementias, and that number is expected to reach approximately 135 million by 2050 (Hossain et al., 2019). Typical features of AD are the gradual deterioration of memory, language and cognitive function. Sleep and circadian rhythm disturbances are very common in patients with AD, and up to 45% of patients experience sleep problems. The clinical presentation of AD is characterized by memory loss and cognitive dysfunction. With the increase of age, changes in the brain as well as in other cells of the body are evident. Examples include changes in the genome and epigenome (Schumacher et al., 2021), loss of protein balance (Hetz, 2021), a decline in cellular and subcellular function (Bulati et al., 2017), and deregulation of signaling systems (Martinez-Jimenez et al., 2017). Most people gradually experience difficulties such as losing the ability to think and/or remember certain things and begin to notice these changes. However, difficulty remembering new information is the most common early feature of AD because in the initial stages, changes occur in areas of the brain involved in learning. In the later stages of AD, changes in the brain lead to increasingly severe symptoms, including disorientation, behavioral and mood changes, increasing uncertainty about events, places and times, unfounded doubts about family, friends and professional caregivers, severe memory loss, and difficulties in everyday tasks such as swallowing, speaking and walking. The prevalence of AD has progressively increased since the 1950s, this disease has a duration of 1–20 years or more and shortens life expectancy (Selkoe, 2013).
Growing evidence for the role of sleep disorder in the pathophysiology of AD has resulted in the proposal of a bidirectional relationship (Ju et al., 2014; Guarnieri and Sorbi, 2015; Villa et al., 2015). The identification of sleep disorders and circadian rhythm disturbances can serve as initial markers for AD diagnosis (Saeed and Abbott, 2017). The pathogenesis of AD is still unclear, though the most influential factors appear to be extracellular plaque deposits of β-amyloid (Aβ) and tau-containing intracellular neurofibrillary tangles (NFTs) (Knopman et al., 2021; Figure 2). Aβ is derived from amyloid precursor protein (APP), which is cleaved by β-secretase and γ-secretase, and NFTs, which are aggregates of hyperphosphorylated tau proteins. However, autopsy of these classic pathological features only explain the manifestation of dementia to a limited extent (Matthews et al., 2009). The risk of AD attributable to heredity is approximately 70%. Earlier studies have identified genetic causes of AD, which include dominant mutations in the genes encoding APP, Presennilin-1 and Presennilin-2 (Goate et al., 1991; Schellenberg et al., 1992; Levy-Lahad et al., 1995). These genes are critical to our understanding of the pathogenesis of AD. Sortilin-related receptor 1 (SORL1) is also thought to be an important genetic cause of late-onset AD (Rogaeva et al., 2007). In addition, several genes that are potential risk factors for AD have been identified. The most strongly associated risk genes were apolipoprotein E (ApoE) (Corder et al., 1993; Uddin et al., 2019), glycogen synthase kinase 3β (GSK3β) (Hernandez et al., 2009), dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) (Kimura et al., 2007), translocase of outer mitochondrial membrane 40 (TOMM40), clusterin (CLU) (Thambisetty et al., 2010) and phosphatidylinositol-binding clathrin assembly (PICALM). These genes may play an important role in identifying pathways and potential drug targets associated with AD. However, recent studies have shown that certain individuals do not need at least 7 h of sleep per night due to their genetics. This group of short sleepers need only 4 or 5 h of sleep each night to be energized and refreshed and thus exhibit a reduction in required sleep throughout their lifetime. More importantly, this reduction in sleep does not impact their cognitive function or induce AD. The short sleep ability is associated with mutations in the hereditary natural short sleep gene, in which the development of tau pathology is slowed according to findings in mice carrying mutations in the DEC2-P384R and Npsr1-Y206H short sleep genes (Dong et al., 2022).
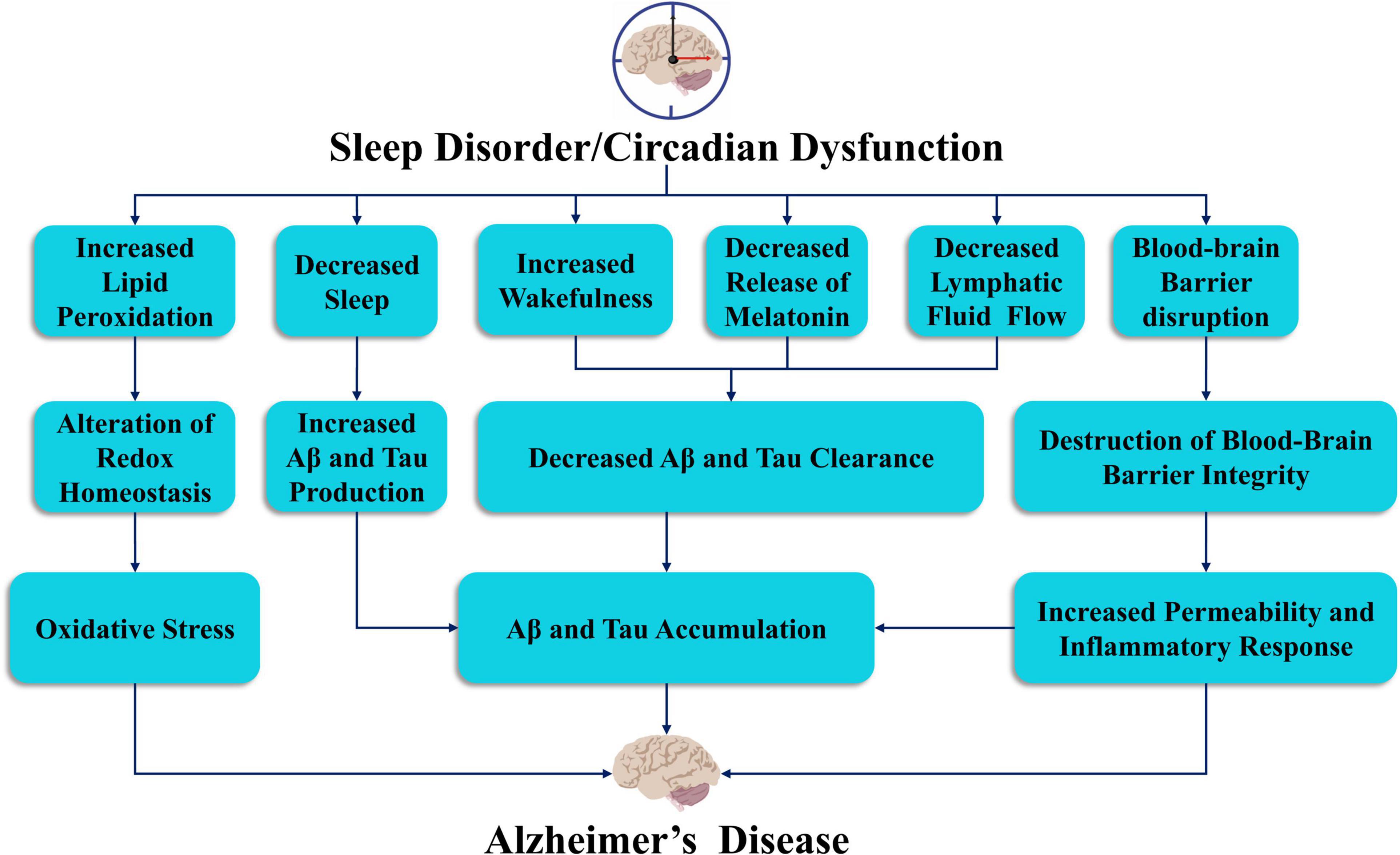
Figure 2. The link between sleep deprivation and circadian dysfunction in the pathogenesis of Alzheimer’s disease. Sleep and circadian rhythm disorders may lead to oxidative stress, accumulation of Aβ and tau proteins, decreased release of melatonin, reduced lymphatic flow, and damage to the blood-brain barrier, which may lead to the development of AD.
In this review, we explore the evidence linking sleep disorders, circadian rhythm disorders and AD, focusing on the complexity of the main mechanisms underlying the relationship between sleep disorders and AD. In addition, this review highlights potential therapeutic strategies for reducing sleep disorders in patients with AD. We review evidence that sleep affects the pathogenesis of AD and summarize three key research areas: (1) the potential risks of sleep disorders for Alzheimer’s disease, (2) the mechanism by which sleep disorders lead to Alzheimer’s disease, and (3) the treatment of sleep disorders in patients with AD. On the basis of these findings, we recommend highly individualized early interventions. We aim to provide policy makers, researchers and clinicians with necessary background information to fully understand this review’s clinical relevance, the potential pathophysiology of AD, and potential methods of early diagnosis and prevention to establish better treatment in terms of personalized care and protection of elderly individuals.
Sleep disorders are a potential risk factor for Alzheimer’s disease
Various studies have shown that sleep is a natural and important biological activity and that sleep disorders can lead to neurodegeneration that is not limited to progressive disease but occurs before cognitive impairment, and thus, they may be considered a marker of neurodegeneration (Lim et al., 2013; Hahn et al., 2014).
In the brain, many areas, neurotransmitters and trigger switches strictly control the sleep-wake cycle. Sleep regulatory areas are distributed throughout the brain, including the brainstem, midbrain, thalamus, hypothalamus, and basal forebrain (Saper et al., 2005). These regions regulate arousal, promote and inhibit NREM and REM sleep, and regulate circadian rhythms. A study reported that slowing circadian rhythms in healthy, community-dwelling older women increases the odds of developing mild cognitive impairment (MCI) and dementia (Tranah et al., 2011). This conclusion was confirmed in a recent study (Posner et al., 2021). In fact, weaker circadian rhythms are also associated with lower cognitive function, especially in older women without dementia (Walsh et al., 2014). Additionally, sleep disorders are the leading cause of AD, and most people with AD exhibit some type of sleep disorder (Cui and Li, 2013; Cui et al., 2015). A previous study clearly emphasized the importance of sleep, as sleep deprivation in rats led to death within 2–3 weeks (Rechtschaffen, 1998).
In recent years, the interaction between sleep disorders and AD has attracted increasing attention. Accumulating evidence indicates that many diseases, including AD, multiple sclerosis, and Parkinson’s disease (PD), are directly associated with sleep disorders (Barun, 2013; Mendelsohn and Larrick, 2013; Ondo, 2014). Moreover, sleep disorders are not only an important clinical feature of AD but also an important risk factor. Targeted sleep regulation has become an important strategy and therapeutic target for AD prevention and treatment.
Study on the mechanism of sleep disorders leading to Alzheimer’s disease
Sleep disorders affect Aβ aggregation, leading to the onset of Alzheimer’s disease
Sleep-wake cycles and circadian rhythms play a critical role in controlling Aβ levels (Figure 2). Both mouse and human sleep disorders increase Aβ levels in the brain and potentially increase plaque deposition (Kang et al., 2009; Ooms et al., 2014; Ju et al., 2017). Moreover, Aβ levels exhibit a significant circadian rhythm in both mouse and human models, and Aβ in the interstitial fluid (ISF) of the brain is positively correlated with wakefulness, negatively correlated with sleep duration, and significantly negatively correlated with the amount of NREM sleep (Holth et al., 2019). In addition, levels of Aβ in human cerebrospinal fluid (CSF) samples were also found to be highest during wakefulness and lowest during sleep, with a difference of up to 30% between the two levels (Lucey et al., 2018). Previously, the central nervous system was widely believed to lack lymphatic circulation, but this view has been updated upon the discovery of a network of meningeal lymphatic vessels in rodents, non-human primates, and human dura mater (Aspelund et al., 2015; Louveau et al., 2015; Absinta et al., 2017). The lymphatic system of mice has also been shown to participate in the removal of solutes, including Aβ, from the ISF of mice (Iliff et al., 2012). Apparently, sleep, whether normal or induced by anesthetics, increases ISF space in the brain by 60%, which is essential for the clearance of toxic metabolites such as Aβ (Xie et al., 2013). If animal models experience persistent insomnia, they die within days or weeks (Kress et al., 2014). This finding was also observed in patients with fatal familial insomnia (Kress et al., 2014). In addition, sleep deprivation increases the accumulation of Aβ in transgenic mice, conversely, proper sleep reduces Aβ levels and enhances clearance (Xie et al., 2013). These findings suggest that sleep disorders increase the risk of developing AD by increasing the production of Aβ. The relationship between AD and Aβ has been recognized in preclinical and clinical studies, but Aβ accumulation can also be found in some cognitively healthy people (Davis and Chisholm, 1999; Price et al., 2009). Conversely, neurodegeneration can also occur without plaque accumulation (Chetelat, 2013). Indeed, Aβ plaques may not be the only cause of AD, and there may be no obvious causal relationship between them (Murphy and LeVine, 2010; Figure 2). Clinical drug development for Aβ as a therapeutic target has failed, which urges us to further study the mechanism of AD and whether Aβ is a final metabolite rather than the initiator of the pathophysiological process.
Sleep disorders affect tau levels, leading to Alzheimer’s disease onset
Although substantial evidence suggests that Aβ aggregation initiates the pathogenesis of AD, such as by driving tau protein aggregation and diffusion, tau aggregation appears to promote neurodegenerative lesions (Simic et al., 2016; Figure 2). The researchers measured tau protein levels in the ISF and CSF to examine whether tau levels were affected by the sleep-wake cycle and sleep deprivation. The results showed that the tau level in the ISF in mice increased by approximately 90% in the normal waking state and by 100% in the sleep deprivation state. In humans, tau levels in the CSF also increased by more than 50% during sleep deprivation. Thus, the sleep-wake cycle regulates tau levels in the ISF, and sleep deprivation increases tau levels in the ISF and CSF as well as pathological diffusion (Holth et al., 2019). In addition, dysfunctional tau protein metabolism has been observed in circadian rhythm disturbances and sleep disorders (Barthelemy et al., 2020). Many brain regions involved in sleep regulation, such as the parabrachial nucleus, locus coeruleus, midbrain periaqueductal gray, dorsal raphe nucleus, lateral hypothalamic nucleus, tuberomammillary nucleus, and basal forebrain, are characterized by different tau-related pathologies. Unlike Aβ levels, total and phosphorylated tau levels in the CSF predict cognitive decline in preclinical and clinical AD (Fagan et al., 2007; Mattsson et al., 2009). Furthermore, tau pathology has recently been shown to be the earliest observable AD-like change in the human brain, with abnormal tau phosphorylation and aggregation in the locus coeruleus beginning in early adulthood (Braak et al., 2011) and extending to other connected regions even before amyloid is detected (Stratmann et al., 2016). Recent studies have also found that the norepinephrine metabolite DOPEGAL covalently modifies the K353 site of the tau protein in the locus coeruleus, promoting tau aggregation and the spread of lesions (Kang et al., 2022). Despite these observations, little is known about the impact of tau pathology on sleep in preclinical or clinical AD. These areas highlight the important role of tau in AD sleep disorders (Figure 2).
Other factors potentially leading from sleep disorders to Alzheimer’s disease
One study induced chronic REM sleep disorders in mice by chronic sleep fragmentation (SF), which enhanced neural activity in the medial habenula (MHB), a brain region increasingly implicated in negative effects. Cholinergic neurons (CHNs) in the MHB exhibited higher spontaneous firing rates and enhanced regularity of brain slice firing after 5 days of chronic SF. When glutamate, GABA, acetylcholine, and histamine receptors were inhibited, the changes in SF-induced firing were unchanged, indicating that this autonomic mechanism was independent of synaptic transmission. In addition, the SF-induced hyperactivity of CHNs in the medial habenular nucleus was not due to the enhancement of the excitability of the lamina propria but was accompanied by depolarization of the resting membrane potential. SF impaired the function of TASK-3 in CHNs in the medial habenular nucleus, which may have led to the depolarization of the resting membrane potential and spontaneous firing. These results not only proved that REM sleep disorder leads to hyperactivity of CHNs in the MHB but also identified the key molecular substrates affected by REM sleep disorder. Chronic REM SF induces CHN hyperactivity in the MHB by regulating the K2P channel TASK-3. Sleep disorders may thus affect mood regulation through this molecular mechanism, which may induce neurodegenerative diseases (Ge et al., 2021).
With increasing age, the interruption of the sleep-wake cycle is accompanied by increasing oxidative stress, which indicates that the accumulation of age-related oxidative damage may lead to the interruption of the sleep-wake cycle (Koh et al., 2006; Figure 2). The latest research has also shown that mortality caused by severe sleep deprivation may be due to oxidative stress, particularly in the intestinal tract. Sleep deprivation can lead to death through the accumulation of reactive oxygen species (ROS) in the intestinal tract, but this accumulation can be reversed by oral antioxidant compounds or targeted intestinal antioxidant enzymes (Vaccaro et al., 2020).
Melatonin released from the pineal gland can regulate circadian rhythms and prevent additional Aβ accumulation (Figure 2). Previous studies have shown that melatonin levels in the CSF in patients with AD are decreased, even during preclinical AD and disease progression. Recent studies have shown that melatonin directly binds to and inhibits the function of death-associated protein kinase 1 (DAPK1) in AD, thereby reducing the accumulation and phosphorylation of tau protein and promoting synaptic growth and microtubule assembly (Chen et al., 2020). This finding indicates that the loss of melatonin may enhance the development of AD.
The blood–brain barrier (BBB) is a large regulatory and exchange interface between the brain and the peripheral circulation. Sleep deprivation impairs the function of the BBB. Chronic sleep deprivation not only reduced the expression of inducible nitric oxide synthase, endothelin-1, and glucose transporter in BBB microvascular endothelial cells but also reduced brain uptake of 2-deoxy-D-arabino-hexose. Increased inflammation associated with COX-2 altered vascular reactivity, decreased glucose transporters and impaired BBB permeability, which led to the occurrence of AD (He et al., 2014; Figure 2).
Treatment of sleep disorders in patients with Alzheimer’s disease
The treatment of sleep disorders in patients with AD aims to improve the quality of life of patients and caregivers. Because of the impact of sleep disorders on AD, systematic clinical evaluation is essential for selecting the most appropriate treatment for each patient (Figure 3).
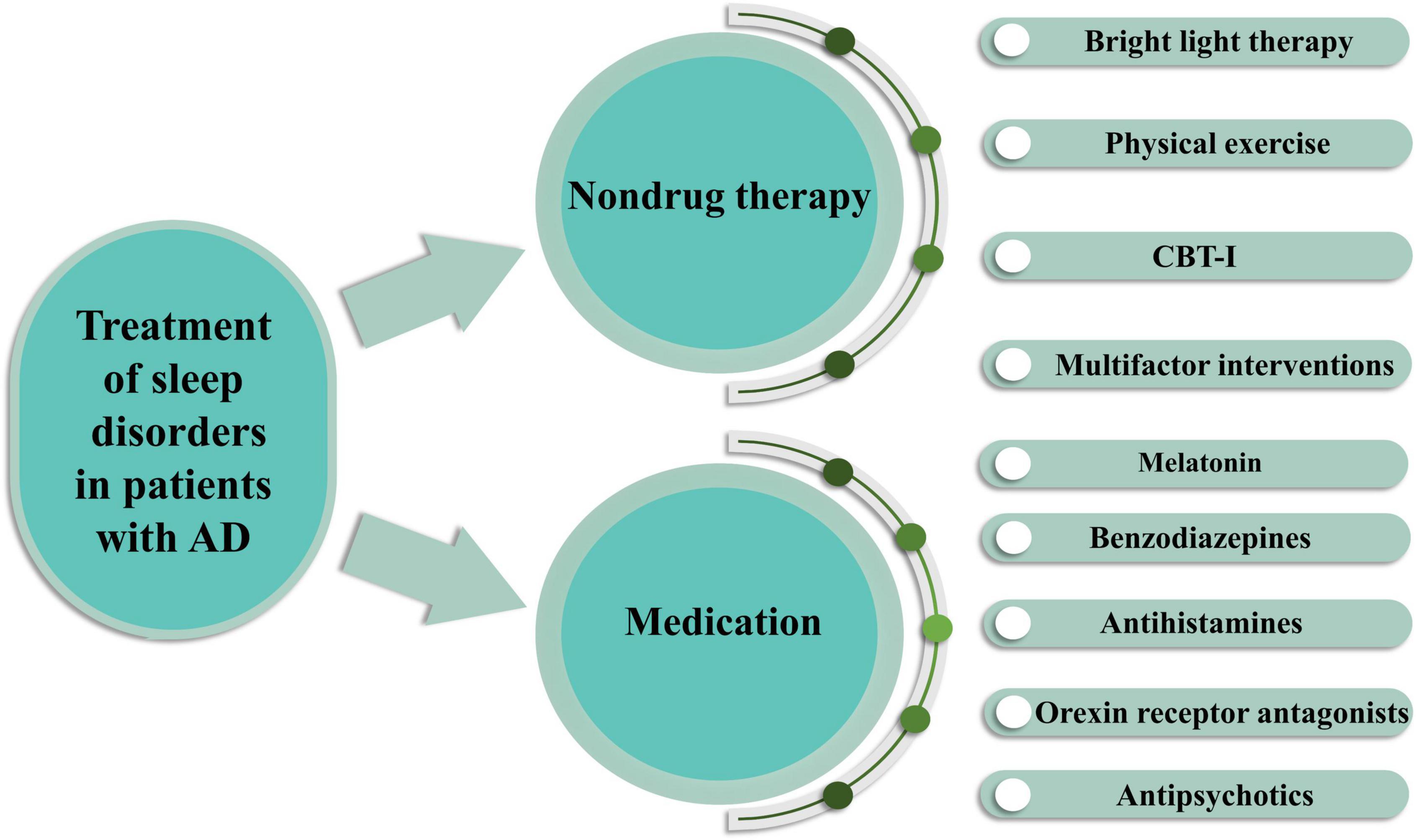
Figure 3. Treatment of sleep disorders in AD patients by pharmacological and non-pharmacological means. Non-drug therapy includes bright light therapy, physical exercise, cognitive behavioral therapy for insomnia (CBT-I) and multifactor interventions. Drug treatments include melatonin, benzodiazepines, antihistamines, orexin receptor antagonists, antipsychotics.
Non-drug therapy
Bright light therapy
Non-drug therapy is typically considered a first-line treatment for patients with AD, although there is limited scientific evidence as to its effectiveness. However, in a structured literature review, BLT was shown to be an effective treatment (Brown et al., 2013; Figure 3). Currently, the mechanism by which bright light therapy (BLT) treats sleep disorders in patients with AD is believed to be through retinal neurons; these retinal neurons contain a photosensitive pigment that can transmit light signals to the suprachiasmatic nucleus (SCN), which is the epicenter of circadian rhythms and regulates the secretion of melatonin (Wen et al., 2020). Melatonin synchronizes endogenous circadian rhythms, and its synthesis and secretion are affected by the photoperiod (Shirani and St, 2009). After stimulation from a light source, photosensory ganglion cells in the retina alter the neurotransmitter (e.g., 5-hydroxytryptamine, dopamine, and norepinephrine) and neuroendocrine levels (e.g., sex hormones, thyroid hormones, and melatonin) as well as the neuroimmune system (e.g., IL, TNF-α, and IFN) through visual and non-visual pathways; these changes further influence the mental state and behavior of patients with AD (Maynard et al., 2017). Among these pathways, the visual pathway involves photoreceptors that receive light stimuli, which act on intrinsically photosensitive retinal ganglion cells (ipRGCs); these signals are transmitted to the hypothalamic suprachiasmatic nucleus through the retinal hypothalamic tract, thus regulating a variety of light-related physiological processes, such as stimulating the expression of circadian-related genes and proteins, affecting spontaneous action potential, and regulating pineal secretion of melatonin (Shirani and St, 2009; Sonoda et al., 2020). Therefore, BLT influences biological circadian rhythms, mood and sleep in ways that improve the sleep quality of patients with AD. The wavelength, dose and optimal timing of light therapy are closely related to its therapeutic effect (Shirani and St, 2009). In a 24-week cluster randomized trial, the effect of BLT on the emotion of dementia patients was greater than that of the control group after 16 weeks, suggesting that BLT may be beneficial to dementia patients (Kolberg et al., 2021). However, a new systematic review of the application of BLT in elderly patients with cognitive impairment shows that while BLT improves the management of sleep, behavioral and emotional disorders in patients with cognitive impairment, its impact on cognition, quality of life and functional status is limited (Cibeira et al., 2020). The American Academy of Sleep Medicine published practical parameters for the use of bright light to treat sleep and circadian rhythm disturbances 10–20 years ago (Chesson et al., 1999; Morgenthaler et al., 2007), however, there is no recognized gold standard for bright light exposure, including when to implement it, the duration, the safest and most effective wavelengths of light, or the best mode of exposure. There is increasing interest in the efficacy of BLT alone or in combination with other therapies for treating sleep disorders in patients with AD. However, the systematic review reported great deal of heterogeneity. Therefore, future studies should aim to determine the optimal intensity, frequency and duration of BLT interventions and assess their effectiveness.
Physical exercise
Regular exercise is an important measure to prevent the development of circadian rhythm disturbances and stimulates resynchronization of circadian rhythms (Figure 3). The potential mechanism underlying the relationship between exercise and sleep is a complex physiological process involving multiple systems. However, acute and chronic exercise exert different effects on sleep according to the mode, intensity, duration, and dosage of exercise as well as patient age and sex (Driver and Taylor, 2000). Exercise promotes sleep by regulating the central nervous system, endocrine system, metabolism, and body temperature during sleep (Uchida et al., 2012). Exercise can lead to a significant increase in the duration of stages 3 and 4 of slow wave sleep (SWS) (Hori et al., 2001). Habitual exercise enhances vagus nerve regulation, leading to bradycardia, which reflects enhanced parasympathetic nerve control (Sandercock et al., 2005) and may improve sleep and mood through monoamine activation, hypothalamus-pituitary-adrenal axis activation or expression of brain-derived neurotrophic factor (BDNF) (Brosse et al., 2002). Exercise affects thermoregulation (McGinty and Szymusiak, 1990) and body recovery (Adam and Oswald, 1983), according to the energy conservation hypothesis (Berger and Phillips, 1988), promoting sleep. Timely exposure to circadian phase-shifting stimulation can not only normalize circadian rhythms but also improve sleep (Campbell et al., 1993). Therefore, exercise in well-lit conditions may improve the sleep of shift workers, air travelers, patients with atypical depression and older people with abnormal circadian rhythms (Driver and Taylor, 2000). The latest research shows that skeletal muscle, an endocrine organ, can release various muscle cytokines during exercise and exert an anti-inflammatory effect (Hoffmann and Weigert, 2017). Aerobic exercise can inhibit neuroinflammation and activation of microglia (Mee-Inta et al., 2019). This form of exercise can also prevent inflammatory reactions and learning and memory impairments caused by acute sleep deprivation and reverse the decline in cognitive function caused by sleep deprivation (Kojima et al., 2020). A previous randomized controlled study showed that elderly people with moderate sleep problems improved their sleep quality with regular moderate-intensity exercise (King et al., 1997). Studies have also shown that participating in non-strenuous daytime activities can have a beneficial effect on sleep in individuals with dementia. In one study, compared with the control group, dementia patients with severe sleep disorders randomly received 15–30 min of personalized social activities every day for 21 consecutive days, which significantly reduced their daytime sleepiness, sleep latency and nocturnal awakenings (Richards et al., 2005). Another study showed that 30 min of collective activity twice a week for 12 weeks improved sleep and increased the total sleep duration compared with the control group by significantly reducing the number of nighttime arousals after falling asleep. In addition, moderate physical activity has been suggested to improve the sleep of patients with AD (Joranson et al., 2021). Although there is plenty of evidence that increases in physical activity can significantly improve sleep and cognitive abilities in patients with AD, health care professionals rarely systematically advise physical activity to prevent or treat diseases that affect the brain. Physical activity decreases with age, especially among women, low-income people, and in some ethnic groups (August and Sorkin, 2011). Notably, these people are disproportionately affected by AD, so clinicians should discuss the benefits of physical activity for maintaining brain health.
Cognitive behavioral therapy for insomnia
The American College of Physicians (ACP) recommends Cognitive behavioral therapy for insomnia (CBT-I) as the initial treatment for chronic insomnia (Qaseem et al., 2016). The ACP also recommends that clinicians and patients use a standardized decision-making approach, including discussion of benefits, hazards, and costs, to decide whether to use drugs when CBT-I alone does not work (Qaseem et al., 2016). CBT-I is a multimodal therapy that combines several behavioral and cognitive interventions. Specifically, this therapy includes education, stimulus control instructions, bed rest time limits and relaxation training. CBT-I produces reliable, lasting benefits in 70–80% of patients and may reduce the use of sedatives (Trauer et al., 2015). This therapy can be provided by trained therapists or through self-directed, fully automated online programs. Overall, CBT-I has moderate to significant effects on sleep-related outcomes, including time to fall asleep, continuity of sleep, feeling rested, and sleep duration (Trauer et al., 2015). Brief behavioral treatment for insomnia (BBTI) is an evidence-based, easy-to-manage method derived from CBT-I and can also be used in a variety of treatment environments (Buysse et al., 2011). BBTI includes four behavioral interventions to improve sleep consolidation by increasing the “driving force” of sleep, improving sleep regularity, reducing arousal, and increasing the link between being in bed and sleeping as follows: (1) reducing bedtime to match actual sleep onset, (2) getting up at the same time every day, regardless of sleep duration, (3) not going to bed unless sleepy, and (4) not lying in bed unless attempting to sleep (Buysse et al., 2017). The patient’s progress is monitored through daily sleep diaries and weekly phone calls or electronic communications. As sleep becomes more consolidated, patients can gradually increase their bed rest time to achieve the best balance between sleep continuity and sleep duration.
Multifactor interventions
Because of the risk of drug side effects, behavioral strategies are often recommended as first-line treatments for patients with dementia and sleep disorders (Figure 3). Standard recommendations include setting a regular bedtime and wake-up time, limiting daytime naps and time spent resting in bed, establishing a regular mealtime, avoiding alcohol, nicotine and caffeine, and emptying the bladder before going to bed. Additionally, the sleep environment should be maintained at an appropriate temperature (not be too hot or too cold) and light and noise should be minimized (McCurry and Ancoli-Israel, 2003). In a randomized controlled trial of a nocturnal insomnia treatment for patients with AD, a combination of sleep health education, moderate physical activity and increased light significantly improved the quality of sleep compared with that of the control group, and this therapeutic effect was maintained until the 6-month follow-up (McCurry et al., 2005). Diet and exercise affect cognitive decline and the risk of dementia through endogenous factors triggered by food metabolism and exercise (e.g., changes in apoptosis and decreased integrity of hippocampal progenitor cells), according to a new study. These endogenous factors travel to the brain through the bloodstream and may interact with the hippocampus to cause cognitive decline and dementia (Du Preez et al., 2022). Therefore, the combination of exercise and diet is a feasible and promising method to reduce or prevent sleep disorders in AD patients.
Medication
Melatonin
Melatonin, a neurohormone secreted by the pineal gland, has received considerable research interest as an alternative drug for the treatment of sleep dysfunction in neurodegenerative diseases (Figure 3). Initially, using neuroblastoma cell lines exposed to Aβ, researchers found that melatonin had antioxidant and anti-apoptotic effects in AD (Pappolla et al., 1997). Further research found that melatonin can inhibit Aβ (Pappolla et al., 1998). These early studies reflect the increasing interest in melatonin as a potential modification therapy to improve the symptoms of patients with AD and sleep disorders. Subsequent studies have shown that melatonin improves behavioral defects in AD-model mice through antioxidant mechanisms (Raghavendra and Kulkarni, 2001) and that exogenous melatonin has anti-tau activity (Liu and Wang, 2002). Moreover, in AD-model animals, melatonin reduced Aβ levels (Matsubara et al., 2003), reduced neuroinflammation (Gunasingh et al., 2008), and reduced damage to hippocampal neuroplasticity (Stefanova et al., 2015). A recent study showed that melatonin plays an important role in regulating circadian rhythm disturbances by controlling clock genes, it also attenuates Aβ accumulation and tau hyperphosphorylation by regulating the GSK3 and CDK5 signaling pathways (Hossain et al., 2019). Similarly, a previous randomized, double-blind, placebo-controlled, crossover study showed that short-term use of melatonin improved sleep, mood and cognition in elderly individuals with mild cognitive impairment (Jean-Louis et al., 1998). However, in a randomized clinical trial, exogenous melatonin had no effect on objective sleep outcomes in patients with AD, though it did have a positive effect on sleep therapy (Zhang et al., 2016). Other studies have failed to find any benefits of melatonin for sleep quality in patients with AD (Serfaty et al., 2002; Gehrman et al., 2009). These different results may be explained by the limited duration of the trial and the inclusion of different stages of dementia and AD patients. Therefore, the relationship between melatonin and AD-related sleep disorders needs to be further evaluated in future studies.
Benzodiazepines
Benzodiazepines are derivatives of 1,4-benzodiazepine and act mainly on the reticular formation of the brainstem and on the limbic system. These drugs reduce sleep latency by activating γ-aminobutyric acid (GABA) receptors, the main inhibitory neurotransmitter receptors in the brain, thus improving sleep quality. Benzodiazepines have become the most widely used sedative and hypnotic drugs in clinical practice. Several early studies have shown that benzodiazepines reduce REM sleep and/or slow wave sleep, with reliable improvements demonstrated in patients with chronic sleep disorders (Nowell et al., 1997), later studies showed that these two sleep stages are essential for memory consolidation (Rasch and Born, 2013; Todorova and Zugaro, 2019) and the clearance of neurotoxic waste (Rasmussen et al., 2018; Figure 3). Conversely, one study found that taking benzodiazepines increased the risk of AD, especially with long-term use (Billioti et al., 2014): there was a 43–51% increase in the risk of developing AD in patients who took benzodiazepines. Further analysis indicated that long-term use of benzodiazepines was associated with a greater risk of developing AD than short-term use, supporting a causal relationship (Yaffe and Boustani, 2014). However, the results from a large cohort study suggest that increased benzodiazepine use is not associated with faster cognitive decline. Those with the lowest exposure to benzodiazepines had a slightly higher risk of developing dementia, but those with the highest level of benzodiazepine exposure did not have a higher risk of developing dementia (Gray et al., 2016). These results do not support a causal relationship between benzodiazepines and dementia. However, given the numerous adverse effects of benzodiazepines, the use of such drugs in the neurodegenerative disease population should be approached with caution.
Treatment of sleep disorders in patients with Alzheimer’s disease with other drugs
Antihistamines, especially diphenhydramine, are over-the-counter drugs that are widely prescribed to the elderly. These drugs have significant side effects, including sedation, cognitive impairment, increased daytime drowsiness, and anticholinergic reactions. Because of the risk of side effects, the use of antihistamines should not be the first choice of treatment for elderly individuals. Some studies have found that antidepressants have a significant effect on sleep disorders in patients with AD and they may delay the decline in cognitive function. Orexin receptor antagonists can also improve the total sleep duration of patients with mild to moderate AD-related insomnia (Herring et al., 2020). Antipsychotics are another class of drugs used to treat sleep disorders in neurodegenerative diseases. Studies have shown that the low-dose atypical antipsychotic risperidone improves the 5-year prognosis of AD patients with sleep disorders (Yin et al., 2015; Figure 3).
Because of the complexity of sleep disorders in patients with AD, it is impossible to treat them with only one drug or a combination of drugs. The key strategy is to help patients maintain high quality sleep, delay the decline in cognitive, and behavioral performance, and alleviate specific concerns. Therefore, it is highly important for researchers to establish new treatments for the special genetic, molecular and cellular mechanisms of the disease.
Summary and prospects
Sleep disorders and circadian rhythm disturbances are very common in patients with AD, the etiology is complex, involving many factors, and these disorders appear early in the course of the disease. This series of problems seriously affects the quality of life of patients, family members and caregivers. To accurately diagnose neurological diseases and develop care plans to improve sleep quality and reduce the burden on caregivers, it is important to provide a comprehensive review of current drugs and the possible causes of sleep changes in patients. Through the combined use of drug and non-drug treatments, early interventions can delay the course of sleep disorders in patients with AD.
Although substantial progress has been made in understanding the basic mechanisms that control the biological clock and the neural circuits involved in sleep, little is known about how these systems are affected in the brain in neurodegenerative diseases, which can differ based on sex, APOE4 status, depression and drug use. A more detailed understanding is needed of the mechanisms by which specific neurodegenerative diseases and pathogenic proteins affect the circadian rhythm and sleep system, as well as the interaction between sleep and the circadian rhythm system in AD. Sleep disorders may appear as an early marker of disease pathology that may exist in the brain years to decades before clinical symptoms appear. The extent to which sleep disorders are a risk factor for the occurrence or progression of neurodegenerative disease is unclear. Other prospective studies using biomarkers such as cerebrospinal fluid and PET Aβ concentrations are needed to help document biological changes associated with sleep disorders. In addition, sleep disorders in the elderly need to be assessed in clinical trials to determine sleep indicators and evaluate cognitive results to indicate whether improved sleep can help prevent or delay the occurrence or progression of dementia. The role of the lymphatic system in neurodegeneration should be another priority. Although the comprehensive study of the circadian rhythm system, sleep and neurodegeneration in the lymphatic system is still in its infancy, there is considerable potential to use these powerful systems to control many key aspects of brain function to prevent neurodegeneration.
Author contributions
XX, PL, and FC conceived, designed, and drafted the manuscript. XX wrote the original draft preparation. TH, ZY, and YZ contributed to the review and edit of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 81772060 and 82072166) and the Key projects of Science and Technology support of Tianjin key R&D plan (grant no. 20YFZCSY00030). This work was funded by the Tianjin Key Medical Discipline (Specialty) Construction Project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Absinta, M., Ha, S. K., Nair, G., Sati, P., Luciano, N. J., Palisoc, M., et al. (2017). Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. eLife 6:29738. doi: 10.7554/eLife.29738
Adam, K., and Oswald, I. (1983). Protein synthesis, bodily renewal and the sleep-wake cycle. Clin. Sci. 65, 561–567. doi: 10.1042/cs0650561
Alexandre, C., Latremoliere, A., Ferreira, A., Miracca, G., Yamamoto, M., Scammell, T. E., et al. (2017). Decreased alertness due to sleep loss increases pain sensitivity in mice. Nat. Med. 23, 768–774. doi: 10.1038/nm.4329
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Aspelund, A., Antila, S., Proulx, S. T., Karlsen, T. V., Karaman, S., Detmar, M., et al. (2015). A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 212, 991–999. doi: 10.1084/jem.20142290
August, K. J., and Sorkin, D. H. (2011). Racial/ethnic disparities in exercise and dietary behaviors of middle-aged and older adults. J. Gen. Intern. Med. 26, 245–250. doi: 10.1007/s11606-010-1514-7
Bandyopadhyay, A., and Sigua, N. L. (2019). What is sleep deprivation? Am. J. Respir. Crit. Care Med. 199, 11–12. doi: 10.1164/rccm.1996P11
Barthelemy, N. R., Liu, H., Lu, W., Kotzbauer, P. T., Bateman, R. J., and Lucey, B. P. (2020). Sleep deprivation affects tau phosphorylation in human cerebrospinal fluid. Ann. Neurol. 87, 700–709. doi: 10.1002/ana.25702
Barun, B. (2013). Pathophysiological background and clinical characteristics of sleep disorders in multiple sclerosis. Clin. Neurol. Neurosurg. 115(Suppl. 1), S82–S85. doi: 10.1016/j.clineuro.2013.09.028
Bentivoglio, M., and Grassi-Zucconi, G. (1997). The pioneering experimental studies on sleep deprivation. Sleep 20, 570–576. doi: 10.1093/sleep/20.7.570
Berger, R. J., and Phillips, N. H. (1988). Comparative aspects of energy metabolism, body temperature and sleep. Acta Physiol. Scand. Suppl. 574, 21–27.
Billioti, D. G. S., Moride, Y., Ducruet, T., Kurth, T., Verdoux, H., Tournier, M., et al. (2014). Benzodiazepine use and risk of Alzheimer’s disease: case-control study. BMJ 349:g5205. doi: 10.1136/bmj.g5205
Braak, H., Thal, D. R., Ghebremedhin, E., and Del, T. K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Brosse, A. L., Sheets, E. S., Lett, H. S., and Blumenthal, J. A. (2002). Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med. 32, 741–760. doi: 10.2165/00007256-200232120-00001
Broussard, J. L., Ehrmann, D. A., Van Cauter, E., Tasali, E., and Brady, M. J. (2012). Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Ann. Intern. Med. 157, 549–557. doi: 10.7326/0003-4819-157-8-201210160-00005
Brown, C. A., Berry, R., Tan, M. C., Khoshia, A., Turlapati, L., and Swedlove, F. (2013). A critique of the evidence base for non-pharmacological sleep interventions for persons with dementia. Dementia 12, 210–237. doi: 10.1177/1471301211426909
Bulati, M., Caruso, C., and Colonna-Romano, G. (2017). From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing”. Ageing Res. Rev. 36, 125–136. doi: 10.1016/j.arr.2017.04.001
Buysse, D. J., Germain, A., Moul, D. E., Franzen, P. L., Brar, L. K., Fletcher, M. E., et al. (2011). Efficacy of brief behavioral treatment for chronic insomnia in older adults. Arch. Intern. Med. 171, 887–895. doi: 10.1001/archinternmed.2010.535
Buysse, D. J., Rush, A. J., and Reynolds, C. R. (2017). Clinical management of insomnia disorder. JAMA 318, 1973–1974. doi: 10.1001/jama.2017.15683
Campbell, S. S., Dawson, D., and Anderson, M. W. (1993). Alleviation of sleep maintenance insomnia with timed exposure to bright light. J. Am. Geriatr. Soc. 41, 829–836. doi: 10.1111/j.1532-5415.1993.tb06179.x
Chan, J. K., Trinder, J., Colrain, I. M., and Nicholas, C. L. (2015). The acute effects of alcohol on sleep electroencephalogram power spectra in late adolescence. Alcohol Clin. Exp. Res. 39, 291–299. doi: 10.1111/acer.12621
Chen, D., Mei, Y., Kim, N., Lan, G., Gan, C. L., Fan, F., et al. (2020). Melatonin directly binds and inhibits death-associated protein kinase 1 function in Alzheimer’s disease. J. Pineal Res. 69:e12665. doi: 10.1111/jpi.12665
Chesson, A. J., Littner, M., Davila, D., Anderson, W. M., Grigg-Damberger, M., Hartse, K., et al. (1999). Practice parameters for the use of light therapy in the treatment of sleep disorders. Standards of Practice Committee, American Academy of Sleep Medicine. Sleep 22, 641–660. doi: 10.1093/sleep/22.5.641
Chetelat, G. (2013). Alzheimer disease: abeta-independent processes-rethinking preclinical AD. Nat. Rev. Neurol. 9, 123–124. doi: 10.1038/nrneurol.2013.21
Cibeira, N., Maseda, A., Lorenzo-Lopez, L., Rodriguez-Villamil, J. L., Lopez-Lopez, R., and Millan-Calenti, J. C. (2020). Application of light therapy in older adults with cognitive impairment: a systematic review. Geriatr. Nurs. 41, 970–983. doi: 10.1016/j.gerinurse.2020.07.005
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Cui, B., and Li, K. (2013). Chronic noise exposure and Alzheimer disease: is there an etiological association? Med. Hypotheses. 81, 623–626. doi: 10.1016/j.mehy.2013.07.017
Cui, B., Li, K., Gai, Z., She, X., Zhang, N., Xu, C., et al. (2015). Chronic noise exposure acts cumulatively to exacerbate Alzheimer’s disease-like amyloid-beta pathology and neuroinflammation in the rat hippocampus. Sci. Rep. 5:12943. doi: 10.1038/srep12943
Davis, J. N., and Chisholm, J. C. (1999). Alois Alzheimer and the amyloid debate. Nature 400:810. doi: 10.1038/23571
Dong, Q., Gentry, N. W., McMahon, T., Yamazaki, M., Benitez-Rivera, L., Wang, T., et al. (2022). Familial natural short sleep mutations reduce Alzheimer pathology in mice. iScience 25:103964. doi: 10.1016/j.isci.2022.103964
Driver, H. S., and Taylor, S. R. (2000). Exercise and sleep. Sleep Med. Rev. 4, 387–402. doi: 10.1053/smrv.2000.0110
Du Preez, A., Lefevre-Arbogast, S., Houghton, V., de Lucia, C., Low, D. Y., Helmer, C., et al. (2022). The serum metabolome mediates the concert of diet, exercise, and neurogenesis, determining the risk for cognitive decline and dementia. Alzheimers Dement. 18, 654–675. doi: 10.1002/alz.12428
Fagan, A. M., Roe, C. M., Xiong, C., Mintun, M. A., Morris, J. C., and Holtzman, D. M. (2007). Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch. Neurol. 64, 343–349. doi: 10.1001/archneur.64.3.noc60123
Fernandez, L., and Luthi, A. (2020). Sleep spindles: mechanisms and functions. Physiol. Rev. 100, 805–868. doi: 10.1152/physrev.00042.2018
Ge, F., Mu, P., Guo, R., Cai, L., Liu, Z., Dong, Y., et al. (2021). Chronic sleep fragmentation enhances habenula cholinergic neural activity. Mol. Psychiatry 26, 941–954. doi: 10.1038/s41380-019-0419-z
Gehrman, P. R., Connor, D. J., Martin, J. L., Shochat, T., Corey-Bloom, J., and Ancoli-Israel, S. (2009). Melatonin fails to improve sleep or agitation in double-blind randomized placebo-controlled trial of institutionalized patients with Alzheimer disease. Am. J. Geriatr. Psychiatry 17, 166–169. doi: 10.1097/JGP.0b013e318187de18
Girardeau, G., and Lopes-Dos-Santos, V. (2021). Brain neural patterns and the memory function of sleep. Science 374, 560–564. doi: 10.1126/science.abi8370
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Gray, S. L., Dublin, S., Yu, O., Walker, R., Anderson, M., Hubbard, R. A., et al. (2016). Benzodiazepine use and risk of incident dementia or cognitive decline: prospective population based study. BMJ 352;i90. doi: 10.1136/bmj.i90
Guarnieri, B., and Sorbi, S. (2015). Sleep and cognitive decline: a strong bidirectional relationship. It is time for specific recommendations on routine assessment and the management of sleep disorders in patients with mild cognitive impairment and dementia. Eur. Neurol. 74, 43–48. doi: 10.1159/000434629
Gunasingh, M. J., Philip, J. E., Ashok, B. S., Kirubagaran, R., Jebaraj, W. C., Davis, G. D., et al. (2008). Melatonin prevents amyloid protofibrillar induced oxidative imbalance and biogenic amine catabolism. Life Sci. 83, 96–102. doi: 10.1016/j.lfs.2008.05.011
Guo, R., Simasko, S. M., and Jansen, H. T. (2016). Chronic alcohol consumption in rats leads to desynchrony in diurnal rhythms and molecular clocks. Alcohol Clin. Exp. Res. 40, 291–300. doi: 10.1111/acer.12944
Hahn, E. A., Wang, H. X., Andel, R., and Fratiglioni, L. (2014). A change in sleep pattern may predict Alzheimer disease. Am. J. Geriatr. Psychiatry 22, 1262–1271. doi: 10.1016/j.jagp.2013.04.015
Hasler, B. P., and McClung, C. A. (2021). Delayed circadian rhythms and substance abuse: dopamine transmission’s time has come. J. Clin. Investig. 131:e152832. doi: 10.1172/JCI152832
He, J., Hsuchou, H., He, Y., Kastin, A. J., Wang, Y., and Pan, W. (2014). Sleep restriction impairs blood-brain barrier function. J. Neurosci. 34, 14697–14706. doi: 10.1523/JNEUROSCI.2111-14.2014
Hernandez, F., de Barreda, E. G., Fuster-Matanzo, A., Goni-Oliver, P., Lucas, J. J., and Avila, J. (2009). The role of GSK3 in Alzheimer disease. Brain Res. Bull. 80, 248–250. doi: 10.1016/j.brainresbull.2009.05.017
Herring, W. J., Ceesay, P., Snyder, E., Bliwise, D., Budd, K., Hutzelmann, J., et al. (2020). Polysomnographic assessment of suvorexant in patients with probable Alzheimer’s disease dementia and insomnia: a randomized trial. Alzheimers Dement. 16, 541–551. doi: 10.1002/alz.12035
Hetz, C. (2021). Adapting the proteostasis capacity to sustain brain healthspan. Cell 184, 1545–1560. doi: 10.1016/j.cell.2021.02.007
Hirota, T., and Kay, S. A. (2015). Identification of small-molecule modulators of the circadian clock. Methods Enzymol. 551, 267–282. doi: 10.1016/bs.mie.2014.10.015
Hoffmann, C., and Weigert, C. (2017). Skeletal muscle as an endocrine organ: the role of myokines in exercise adaptations. Cold Spring Harb. Perspect. Med. 7:a029793. doi: 10.1101/cshperspect.a029793
Holth, J. K., Fritschi, S. K., Wang, C., Pedersen, N. P., Cirrito, J. R., Mahan, T. E., et al. (2019). The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884. doi: 10.1126/science.aav2546
Hori, T., Sugita, Y., Koga, E., Shirakawa, S., Inoue, K., Uchida, S., et al. (2001). Proposed supplements and amendments to ‘a manual of standardized terminology, techniques and scoring system for sleep stages of human subjects’, the Rechtschaffen & Kales (1968) standard. Psychiatry Clin. Neurosci. 55, 305–310. doi: 10.1046/j.1440-1819.2001.00810.x
Hossain, M. F., Uddin, M. S., Uddin, G., Sumsuzzman, D. M., Islam, M. S., Barreto, G. E., et al. (2019). Melatonin in Alzheimer’s disease: a latent endogenous regulator of neurogenesis to mitigate alzheimer’s neuropathology. Mol. Neurobiol. 56, 8255–8276. doi: 10.1007/s12035-019-01660-3
Huang, T., Mariani, S., and Redline, S. (2020). Sleep irregularity and risk of cardiovascular events: the multi-ethnic study of atherosclerosis. J. Am. Coll. Cardiol. 75, 991–999. doi: 10.1016/j.jacc.2019.12.054
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 4, 111r–147r. doi: 10.1126/scitranslmed.3003748
Jean-Louis, G., von Gizycki, H., and Zizi, F. (1998). Melatonin effects on sleep, mood, and cognition in elderly with mild cognitive impairment. J. Pineal Res. 25, 177–183. doi: 10.1111/j.1600-079x.1998.tb00557.x
Joranson, N., Olsen, C., Calogiuri, G., Ihlebaek, C., and Pedersen, I. (2021). Effects on sleep from group activity with a robotic seal for nursing home residents with dementia: a cluster randomized controlled trial. Int. Psychogeriatr. 33, 1045–1056. doi: 10.1017/S1041610220001787
Ju, Y. E., Lucey, B. P., and Holtzman, D. M. (2014). Sleep and Alzheimer disease pathology–a bidirectional relationship. Nat. Rev. Neurol. 10, 115–119. doi: 10.1038/nrneurol.2013.269
Ju, Y. S., Ooms, S. J., Sutphen, C., Macauley, S. L., Zangrilli, M. A., Jerome, G., et al. (2017). Slow wave sleep disruption increases cerebrospinal fluid amyloid-beta levels. Brain 140, 2104–2111. doi: 10.1093/brain/awx148
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kang, S. S., Meng, L., Zhang, X., Wu, Z., Mancieri, A., Xie, B., et al. (2022). Tau modification by the norepinephrine metabolite DOPEGAL stimulates its pathology and propagation. Nat. Struct. Mol. Biol. 29, 292–305. doi: 10.1038/s41594-022-00745-3
Kecklund, G., and Axelsson, J. (2016). Health consequences of shift work and insufficient sleep. BMJ 355:i5210. doi: 10.1136/bmj.i5210
Kimura, R., Kamino, K., Yamamoto, M., Nuripa, A., Kida, T., Kazui, H., et al. (2007). The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 16, 15–23. doi: 10.1093/hmg/ddl437
King, A. C., Oman, R. F., Brassington, G. S., Bliwise, D. L., and Haskell, W. L. (1997). Moderate-intensity exercise and self-rated quality of sleep in older adults. A randomized controlled trial. JAMA 277, 32–37.
Knopman, D. S., Amieva, H., Petersen, R. C., Chetelat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Primers 7:33. doi: 10.1038/s41572-021-00269-y
Koh, K., Evans, J. M., Hendricks, J. C., and Sehgal, A. (2006). A Drosophila model for age-associated changes in sleep:wake cycles. Proc. Natl. Acad. Sci. U.S.A. 103, 13843–13847. doi: 10.1073/pnas.0605903103
Kojima, S., Abe, T., Morishita, S., Inagaki, Y., Qin, W., Hotta, K., et al. (2020). Acute moderate-intensity exercise improves 24-h sleep deprivation-induced cognitive decline and cerebral oxygenation: a near-infrared spectroscopy study. Respir. Physiol. Neurobiol. 274:103354. doi: 10.1016/j.resp.2019.103354
Kolberg, E., Hjetland, G. J., Thun, E., Pallesen, S., Nordhus, I. H., Husebo, B. S., et al. (2021). The effects of bright light treatment on affective symptoms in people with dementia: a 24-week cluster randomized controlled trial. BMC Psychiatry 21:377. doi: 10.1186/s12888-021-03376-y
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, H. S., Zeppenfeld, D., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
Kroller-Schon, S., Daiber, A., Steven, S., Oelze, M., Frenis, K., Kalinovic, S., et al. (2018). Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur. Heart J. 39, 3528–3539. doi: 10.1093/eurheartj/ehy333
LeGates, T. A., Altimus, C. M., Wang, H., Lee, H. K., Yang, S., Zhao, H., et al. (2012). Aberrant light directly impairs mood and learning through melanopsin-expressing neurons. Nature 491, 594–598. doi: 10.1038/nature11673
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977. doi: 10.1126/science.7638622
Lim, A. S., Kowgier, M., Yu, L., Buchman, A. S., and Bennett, D. A. (2013). Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 36, 1027–1032. doi: 10.5665/sleep.2802
Liu, S. J., and Wang, J. Z. (2002). Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol. Sin. 23, 183–187.
Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., et al. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. doi: 10.1038/nature14432
Lucey, B. P., Hicks, T. J., McLeland, J. S., Toedebusch, C. D., Boyd, J., Elbert, D. L., et al. (2018). Effect of sleep on overnight cerebrospinal fluid amyloid beta kinetics. Ann. Neurol. 83, 197–204. doi: 10.1002/ana.25117
Martinez-Jimenez, C. P., Eling, N., Chen, H. C., Vallejos, C. A., Kolodziejczyk, A. A., Connor, F., et al. (2017). Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355, 1433–1436. doi: 10.1126/science.aah4115
Matsubara, E., Bryant-Thomas, T., Pacheco Quinto, J., Henry, T. L., Poeggeler, B., Herbert, D., et al. (2003). Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 85, 1101–1108. doi: 10.1046/j.1471-4159.2003.01654.x
Matthews, F. E., Brayne, C., Lowe, J., McKeith, I., Wharton, S. B., and Ince, P. (2009). Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med. 6:e1000180. doi: 10.1371/journal.pmed.1000180
Mattsson, N., Zetterberg, H., Hansson, O., Andreasen, N., Parnetti, L., Jonsson, M., et al. (2009). CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302, 385–393. doi: 10.1001/jama.2009.1064
Maynard, M. L., Zele, A. J., Kwan, A. S., and Feigl, B. (2017). Intrinsically photosensitive retinal ganglion cell function, sleep efficiency and depression in advanced age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 58, 990–996. doi: 10.1167/iovs.16-20659
McCurry, S. M., and Ancoli-Israel, S. (2003). Sleep dysfunction in Alzheimer’s disease and other dementias. Curr. Treat. Options Neurol. 5, 261–272. doi: 10.1007/s11940-003-0017-9
McCurry, S. M., Gibbons, L. E., Logsdon, R. G., Vitiello, M. V., and Teri, L. (2005). Nighttime insomnia treatment and education for Alzheimer’s disease: a randomized, controlled trial. J. Am. Geriatr. Soc. 53, 793–802. doi: 10.1111/j.1532-5415.2005.53252.x
McGinty, D., and Szymusiak, R. (1990). Keeping cool: a hypothesis about the mechanisms and functions of slow-wave sleep. Trends Neurosci. 13, 480–487. doi: 10.1016/0166-2236(90)90081-k
Mee-Inta, O., Zhao, Z. W., and Kuo, Y. M. (2019). Physical exercise inhibits inflammation and microglial activation. Cells 8:691. doi: 10.3390/cells8070691
Mendelsohn, A. R., and Larrick, J. W. (2013). Sleep facilitates clearance of metabolites from the brain: glymphatic function in aging and neurodegenerative diseases. Rejuvenation Res. 16, 518–523. doi: 10.1089/rej.2013.1530
Morgenthaler, T. I., Lee-Chiong, T., Alessi, C., Friedman, L., Aurora, R. N., Boehlecke, B., et al. (2007). Practice parameters for the clinical evaluation and treatment of circadian rhythm sleep disorders. An American Academy of Sleep Medicine report. Sleep 30, 1445–1459. doi: 10.1093/sleep/30.11.1445
Murphy, M. P., and LeVine, H. R. (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323. doi: 10.3233/JAD-2010-1221
Nir, Y., Andrillon, T., Marmelshtein, A., Suthana, N., Cirelli, C., Tononi, G., et al. (2017). Selective neuronal lapses precede human cognitive lapses following sleep deprivation. Nat. Med. 23, 1474–1480. doi: 10.1038/nm.4433
Nowell, P. D., Mazumdar, S., Buysse, D. J., Dew, M. A., Reynolds, C. R., and Kupfer, D. J. (1997). Benzodiazepines and zolpidem for chronic insomnia: a meta-analysis of treatment efficacy. JAMA 278, 2170–2177.
O’Loughlin, J., Casanova, F., Jones, S. E., Hagenaars, S. P., Beaumont, R. N., Freathy, R. M., et al. (2021). Using Mendelian Randomisation methods to understand whether diurnal preference is causally related to mental health. Mol. Psychiatry 26, 6305–6316. doi: 10.1038/s41380-021-01157-3
Ondo, W. G. (2014). Sleep/wake problems in Parkinson’s disease: pathophysiology and clinicopathologic correlations. J. Neural Transm. 121(Suppl. 1), S3–S13. doi: 10.1007/s00702-014-1239-6
Ooms, S., Overeem, S., Besse, K., Rikkert, M. O., Verbeek, M., and Claassen, J. A. (2014). Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 71, 971–977. doi: 10.1001/jamaneurol.2014.1173
Pappolla, M., Bozner, P., Soto, C., Shao, H., Robakis, N. K., Zagorski, M., et al. (1998). Inhibition of Alzheimer beta-fibrillogenesis by melatonin. J. Biol. Chem. 273, 7185–7188. doi: 10.1074/jbc.273.13.7185
Pappolla, M. A., Sos, M., Omar, R. A., Bick, R. J., Hickson-Bick, D. L. M., Reiter, R. J., et al. (1997). Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 17:1683. doi: 10.1523/JNEUROSCI.17-05-01683.1997
Posner, A. B., Tranah, G. J., Blackwell, T., Yaffe, K., Ancoli-Israel, S., Redline, S., et al. (2021). Predicting incident dementia and mild cognitive impairment in older women with nonparametric analysis of circadian activity rhythms in the Study of Osteoporotic Fractures. Sleep 44:zsab119. doi: 10.1093/sleep/zsab119
Price, J. L., McKeel, D. J., Buckles, V. D., Roe, C. M., Xiong, C., Grundman, M., et al. (2009). Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 30, 1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002
Qaseem, A., Kansagara, D., Forciea, M. A., Cooke, M., and Denberg, T. D. (2016). Management of chronic insomnia disorder in adults: a clinical practice guideline from the american college of physicians. Ann. Intern. Med. 165, 125–133. doi: 10.7326/M15-2175
Raghavendra, V., and Kulkarni, S. K. (2001). Possible antioxidant mechanism in melatonin reversal of aging and chronic ethanol-induced amnesia in plus-maze and passive avoidance memory tasks. Free Radic. Biol. Med. 30, 595–602. doi: 10.1016/s0891-5849(00)00447-0
Rasch, B., and Born, J. (2013). About sleep’s role in memory. Physiol. Rev. 93, 681–766. doi: 10.1152/physrev.00032.2012
Rasmussen, M. K., Mestre, H., and Nedergaard, M. (2018). The glymphatic pathway in neurological disorders. Lancet Neurol. 17, 1016–1024. doi: 10.1016/S1474-4422(18)30318-1
Rechtschaffen, A. (1998). Current perspectives on the function of sleep. Perspect. Biol. Med. 41, 359–390. doi: 10.1353/pbm.1998.0051
Richards, K. C., Beck, C., O’Sullivan, P. S., and Shue, V. M. (2005). Effect of individualized social activity on sleep in nursing home residents with dementia. J. Am. Geriatr. Soc. 53, 1510–1517. doi: 10.1111/j.1532-5415.2005.53460.x
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T., Zou, F., et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177. doi: 10.1038/ng1943
Saeed, Y., and Abbott, S. M. (2017). Circadian disruption associated with Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 17:29. doi: 10.1007/s11910-017-0745-y
Sandercock, G. R., Bromley, P. D., and Brodie, D. A. (2005). Effects of exercise on heart rate variability: inferences from meta-analysis. Med. Sci. Sports Exerc. 37, 433–439. doi: 10.1249/01.mss.0000155388.39002.9d
Saper, C. B., Scammell, T. E., and Lu, J. (2005). Hypothalamic regulation of sleep and circadian rhythms. Nature 437, 1257–1263. doi: 10.1038/nature04284
Schellenberg, G. D., Bird, T. D., Wijsman, E. M., Orr, H. T., Anderson, L., Nemens, E., et al. (1992). Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 258, 668–671. doi: 10.1126/science.1411576
Schumacher, B., Pothof, J., Vijg, J., and Hoeijmakers, J. (2021). The central role of DNA damage in the ageing process. Nature 592, 695–703. doi: 10.1038/s41586-021-03307-7
Selkoe, D. J. (2013). SnapShot: pathobiology of Alzheimer’s disease. Cell 154, 468. doi: 10.1016/j.cell.2013.07.003
Serfaty, M., Kennell-Webb, S., Warner, J., Blizard, R., and Raven, P. (2002). Double blind randomised placebo controlled trial of low dose melatonin for sleep disorders in dementia. Int. J. Geriatr. Psychiatry 17, 1120–1127. doi: 10.1002/gps.760
Shirani, A., and St, L. E. (2009). Illuminating rationale and uses for light therapy. J. Clin. Sleep Med. 5, 155–163.
Simic, G., Babic, L. M., Wray, S., Harrington, C., Delalle, I., Jovanov-Milosevic, N., et al. (2016). Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6:6. doi: 10.3390/biom6010006
Sonoda, T., Li, J. Y., Hayes, N. W., Chan, J. C., Okabe, Y., Belin, S., et al. (2020). A noncanonical inhibitory circuit dampens behavioral sensitivity to light. Science 368, 527–531. doi: 10.1126/science.aay3152
Stefanova, N. A., Maksimova, K. Y., Kiseleva, E., Rudnitskaya, E. A., Muraleva, N. A., and Kolosova, N. G. (2015). Melatonin attenuates impairments of structural hippocampal neuroplasticity in OXYS rats during active progression of Alzheimer’s disease-like pathology. J. Pineal Res. 59, 163–177. doi: 10.1111/jpi.12248
Stratmann, K., Heinsen, H., Korf, H. W., Del, T. D., Ghebremedhin, E., Seidel, K., et al. (2016). Precortical phase of Alzheimer’s disease (AD)-related tau cytoskeletal pathology. Brain Pathol. 26, 371–386. doi: 10.1111/bpa.12289
Tamaki, M., Wang, Z., Barnes-Diana, T., Guo, D., Berard, A. V., Walsh, E., et al. (2020). Complementary contributions of non-REM and REM sleep to visual learning. Nat. Neurosci. 23, 1150–1156. doi: 10.1038/s41593-020-0666-y
Thambisetty, M., Simmons, A., Velayudhan, L., Hye, A., Campbell, J., Zhang, Y., et al. (2010). Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch. Gen. Psychiatry 67, 739–748. doi: 10.1001/archgenpsychiatry.2010.78
Todorova, R., and Zugaro, M. (2019). Isolated cortical computations during delta waves support memory consolidation. Science 366, 377–381. doi: 10.1126/science.aay0616
Tranah, G. J., Blackwell, T., Stone, K. L., Ancoli-Israel, S., Paudel, M. L., Ensrud, K. E., et al. (2011). Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann. Neurol. 70, 722–732. doi: 10.1002/ana.22468
Trauer, J. M., Qian, M. Y., Doyle, J. S., Rajaratnam, S. M., and Cunnington, D. (2015). Cognitive behavioral therapy for chronic insomnia: a systematic review and meta-analysis. Ann. Intern. Med. 163, 191–204. doi: 10.7326/M14-2841
Uchida, S., Shioda, K., Morita, Y., Kubota, C., Ganeko, M., and Takeda, N. (2012). Exercise effects on sleep physiology. Front. Neurol. 3:48. doi: 10.3389/fneur.2012.00048
Uddin, M. S., Kabir, M. T., Al, M. A., Abdel-Daim, M. M., Barreto, G. E., and Ashraf, G. M. (2019). APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol. Neurobiol. 56, 2450–2465. doi: 10.1007/s12035-018-1237-z
Vaccaro, A., Kaplan, D. Y., Nambara, K., Pollina, E. A., Lin, C., Greenberg, M. E., et al. (2020). Sleep loss can cause death through accumulation of reactive oxygen species in the gut. Cell 181, 1307–1328. doi: 10.1016/j.cell.2020.04.049
Villa, C., Ferini-Strambi, L., and Combi, R. (2015). The synergistic relationship between Alzheimer’s disease and sleep disorders: an update. J. Alzheimers Dis. 46, 571–580. doi: 10.3233/JAD-150138
Walsh, C. M., Blackwell, T., Tranah, G. J., Stone, K. L., Ancoli-Israel, S., Redline, S., et al. (2014). Weaker circadian activity rhythms are associated with poorer executive function in older women. Sleep 37, 2009–2016. doi: 10.5665/sleep.4260
Wen, S., Ma, D., Zhao, M., Xie, L., Wu, Q., Gou, L., et al. (2020). Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat. Neurosci. 23, 456–467. doi: 10.1038/s41593-020-0586-x
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Yaffe, K., and Boustani, M. (2014). Benzodiazepines and risk of Alzheimer’s disease. BMJ 349:g5312. doi: 10.1136/bmj.g5312
Yin, Y., Liu, Y., Zhuang, J., Pan, X., Li, P., Yang, Y., et al. (2015). Low-dose atypical antipsychotic risperidone improves the 5-year outcome in Alzheimer’s disease patients with sleep disturbances. Pharmacology 96, 155–162. doi: 10.1159/000435889
Yoo, S. S., Gujar, N., Hu, P., Jolesz, F. A., and Walker, M. P. (2007). The human emotional brain without sleep–a prefrontal amygdala disconnect. Curr. Biol. 17, R877–R878. doi: 10.1016/j.cub.2007.08.007
Zada, D., Bronshtein, I., Lerer-Goldshtein, T., Garini, Y., and Appelbaum, L. (2019). Sleep increases chromosome dynamics to enable reduction of accumulating DNA damage in single neurons. Nat. Commun. 10:895. doi: 10.1038/s41467-019-08806-w
Keywords: sleep disorders, circadian rhythm disturbances, Alzheimer’s disease, amyloid-β, tau
Citation: Xiong X, Hu T, Yin Z, Zhang Y, Chen F and Lei P (2022) Research advances in the study of sleep disorders, circadian rhythm disturbances and Alzheimer’s disease. Front. Aging Neurosci. 14:944283. doi: 10.3389/fnagi.2022.944283
Received: 15 May 2022; Accepted: 27 July 2022;
Published: 17 August 2022.
Edited by:
Korey Kam, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
David Alcantara-Gonzalez, Nathan Kline Institute for Psychiatric Research, United StatesRyan Falck, University of British Columbia, Canada
Copyright © 2022 Xiong, Hu, Yin, Zhang, Chen and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Lei, bGVpcGluZzE5NzRAMTYzLmNvbQ==