
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci., 25 August 2022
Sec. Alzheimer's Disease and Related Dementias
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.933979
This article is part of the Research TopicMolecular and Cellular Mechanisms of Synaptopathies: Emerging Synaptic aging-related molecular pathways in Neurological DisordersView all 11 articles
 Sara Cazzaro1,2
Sara Cazzaro1,2 Cenxiao Fang2
Cenxiao Fang2 Hirah Khan2
Hirah Khan2 Richard Witas2
Richard Witas2 Teresa R. Kee1,2
Teresa R. Kee1,2 Jung-A. A. Woo1*
Jung-A. A. Woo1* David E. Kang1,3*
David E. Kang1,3*Increasing evidence indicates that the accumulation misfolded proteins in Alzheimer’s disease (AD) arises from clearance defects in the autophagy-lysosome pathway. Misfolded proteins such as Aβ and tau are secreted in small extracellular vesicles (i.e., exosomes) and are propagated from cell to cell in part through secreted small extracellular vesicles (sEVs). Recent studies suggest that autophagic activity and exosome secretion are coregulated events, and multiple autophagy-related proteins are found in sEVs, including the cargo receptors Sqstm1/p62 and optineurin. However, whether and how autophagy cargo receptors per se regulate the secretion of sEVs is unknown. Moreover, despite the prominent role of actin dynamics in secretory vesicle release, its role in EV secretion is unknown. In this study, we leveraged the dual axes of Slingshot Homolog-1 (SSH1), which inhibits Sqstm1/p62-mediated autophagy and activates cofilin-mediated actin dynamics, to study the regulation of sEV secretion. Here we show that cargo receptors Sqstm1/p62 and optineurin inhibit sEV secretion, an activity that requires their ability to bind ubiquitinated cargo. Conversely, SSH1 increases sEV secretion by dephosphorylating Sqstm1/p62 at pSer403, the phospho-residue that allows Sqstm1/p62 to bind ubiquitinated cargo. In addition, increasing actin dynamics through the SSH1-cofilin activation pathway also increases sEV secretion, which is mimicked by latrunculin B treatment. Finally, Aβ42 oligomers and mutant tau increase sEV secretion and are physically associated with secreted sEVs. These findings suggest that increasing cargo receptor engagement with autophagic cargo and reducing actin dynamics (i.e., SSH1 inhibition) represents an attractive strategy to promote misfolded protein degradation while reducing sEV-mediated cell to cell spread of pathology.
Emerging evidence indicates that the release and uptake of extracellular vesicles (EVs) represents an important form of intercellular communication that could transmit beneficial or pathogenic signals across different cells (Rajendran et al., 2014). Normal EVs have been shown to regulate neuronal neurite outgrowth, survival, and synaptic activity (Wang et al., 2011; Antonucci et al., 2012) as well as microglial activation (Verderio et al., 2012; Prada et al., 2013). EVs are secreted by all cell types and contain specific sets of RNA and proteins (membrane and cytosolic) (Valadi et al., 2007; Skog et al., 2008). Large microvesicles (MVs) (150–1000 nm) are derived from evagination and excision of the plasma membrane, whereas the smaller exosomes (30–150 nm) are intraluminal vesicles (ILVs) formed by inward budding of the limiting membrane of late endosomes or multivesicular bodies (MVBs), and are secreted by fusion with the plasma membrane (Raposo and Stoorvogel, 2013). Despite their ubiquitous presence in the brain, the role of EVs in brain cells and their potential effects on neurodegenerative processes are relatively unknown. In pathological settings, proteins prone to aggregation (i.e., Aβ, Tau, α-synuclein, and prion) are secreted in EVs, potentially providing an avenue for spreading such misfolded proteins to neighboring healthy cells (Rajendran et al., 2006; Vella et al., 2007; Danzer et al., 2012; Saman et al., 2012). For example, the spread of tau requires neighboring microglia and exosomes (Asai et al., 2015), and tau overexpression also abnormally recruits other proteins to exosomes (Saman et al., 2014). Alzheimer’s disease (AD) brain-derived exosomes contain tau and Aβ oligomers that can seed and propagate pathology (Sardar Sinha et al., 2018; Miyoshi et al., 2021). Other studies have shown beneficial effects of certain exosomes in reducing amyloid load and protecting against synaptic plasticity deficits (An et al., 2013; Yuyama et al., 2014).
Recent studies suggest that EV secretion of pathological proteins and autophagy pathways may be linked. For example, autophagy activation with the mTOR inhibitor rapamycin inhibits exosomal prion release, whereas inhibition of autophagy increases exosomal prion secretion (Abdulrahman et al., 2018). In a similar vein, inhibition of lysosome function by bafilomycin A1 increases TDP-43 and α-synuclein release in EVs (Iguchi et al., 2016; Minakaki et al., 2018), suggesting that failure of the autophagy-lysosome pathway (ALP) directs autophagosomes containing misfolded proteins for secretion in association with EVs. This secretory pathway has been proposed to involve the fusion of autophagosomes with MVBs to form amphisomes, which are then secreted by fusion with the plasma membrane (Gordon et al., 1992; Liou et al., 1997; Fader et al., 2008; Ganesan and Cai, 2021). Interestingly, autophagy cargo receptors, such as Sqstm1/p62 and optineurin, are often found in secreted exosomes (Gudbergsson and Johnsen, 2019), while they play vital roles in the clearance of misfolded proteins such as tau in AD (Xu et al., 2019; Woo et al., 2020; Fang et al., 2021; Roca-Agujetas et al., 2021a,b). However, whether prototypic autophagy cargo receptors regulate exosome secretion is unknown. Moreover, despite the crucial role of actin dynamics in secretory vesicle docking and secretion (Li et al., 2018), how actin dynamics regulate exosome secretion is also unknown. We and others previously showed that the protein phosphatase Slingshot Homolog-1 (SSH1) contains two major separable activities in the control of actin dynamics through cofilin activation through dephosphorylation at Ser3 (Niwa et al., 2002) and autophagy inhibition through p62 dephosphorylation at pSer403 (Fang et al., 2021). In this study, using a combination of DiI labeling of EVs and differential centrifugation, we explored the role of autophagy cargo receptors and actin dynamics regulated by SSH1 in small EV (sEV, aka exosome) secretion. Our findings highlight the roles of autophagy cargo receptors and actin dynamics in regulating sEV secretion through mechanisms impacting AD pathogenesis.
Mouse hippocampus-derived neuroblastoma cells HT22, mouse embryonic fibroblast-derived NIH3T3, and tetracycline-inducible human embryonic kidney 293 cells overexpressing tau P301L (iHEK P301L) were cultured in Dulbecco’s modified Eagle’s medium (DMEM 1X) (Gibco, 11965-092) supplemented with 10% fetal bovine serum (FBS) (Sigma, 12306C) and 1% penicillin-streptomycin (P/S) (Gibco, 15140-122), and BM cyclin (Roche, 10799050001). iHEKP301L cells were induced with 1 μg/mL tetracycline when seeded, and cells were grown for 48 h before performing experimental assays. Cells were maintained at 37°C with 5% CO2 levels.
Cortical primary neurons were cultured from C57BL6 or tauP301S P0 mouse pups as previously described (Woo et al., 2015b; Fang et al., 2021). Briefly, the cortex was dissected in ice-cold HBSS and digested with 0.25% Trypsin-EDTA (1X) (Gibco, 25200-056), then plated in culture-treated plates with neurobasal medium (Invitrogen, 21103049) supplemented with 2% GlutaMAX (Invitrogen, 35050061) and 2% B27 supplement (Invitrogen, 17504044). Cells were maintained at 37°C with 5% CO2 levels.
pMXs-puro GFP-p62 (Addgene, 38277) (Itakura and Mizushima, 2011), pMXs-puro GFP-p62ΔC (addgene, 38282) (Itakura and Mizushima, 2011), ECFP-SSH1ΔC (Kurita et al., 2008), pEGFP-N1 human cofilin WT (addgene, 50859) (Garvalov et al., 2007), pEGFP-N1 human cofilin S3A (addgene, 50860) (Garvalov et al., 2007), pEGFP-N1 human cofilin S3E (addgene, 50861) (Garvalov et al., 2007), pOPTN-EGFP (addgene, 27052) (Park et al., 2006), pOPTN E478G-EGFP (addgene, 68848) (Turturro et al., 2014) were obtained from corresponding sources. Plasmids p3xFlag-SSH1 and p3xFlag-SSH1ΔN, and GFP-p62 403E were generated in the Kang lab as previously documented (Fang et al., 2021).
DNA plasmids were transiently transfected using Lipofectamine 2000 (Invitrogen, 11668-019) and reduced serum media Opti-MEM I (Gibco, 31985-070). After 4–6 h post-transfection, the media was replaced with new complete medium. Cells were grown for 48 h after transfection.
Small extracellular vesicles within the size range of exosomes were isolated by serial centrifugation as previously described (Witas et al., 2017). Briefly, 48 h after transfection, cells were washed twice with PBS (Gibco, 10010-023), and media was replaced with DMEM 1X supplemented with 10% exosome-depleted fetal bovine serum (FBS) (Gibco, A27208-03) and 1% P/S. Twenty-four hours later, media was collected for EV isolation. Samples were centrifuged at 4°C at 800 × g for 10 min to remove cells and large bodies, and supernatants were centrifuged at 4°C at 2,500 × g for 15 min to remove cell debris. Supernatants were then centrifuged at 4°C at 10,000 × g for 30 min to separate pellet microvesicles. Finally, the resulting supernatants were labeled with DiI and centrifuged 4°C at 100,000 × g for 1 h to pellet small EVs. Small EV pellets were washed with PBS and used for analysis.
Vesicles were stained with DiI as previously described (Witas et al., 2017). Briefly, post-microvesicle supernatants were rocked for 15 min in 5 μM DiI (Thermo Fisher, D3911) solution at 37°C. After EVs isolation and washing, pellets were resuspended in 5 μl of fluorochrome mounting solution (Thermo scientific, TA-030-FM), and the entire volume was placed on a glass slide and covered with a glass coverslip for imaging and analysis.
Isolated EVs were resuspended in 500 μl of PBS and ran on NanoSight™ LM10 with nanoparticle tracking analysis (Malvern). Using the NanoSight software, three videos of 60 s each were taken and analyzed for particle size distribution.
For cell protein extraction, cells were washed with 1× PBS and then resuspended in RIPA lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100 [Amresco, 0694–1 L], 0.1% SDS) with protease inhibitor (GeneDEPOT, P3100-010) and phosphatase inhibitor (GeneDEPOT, P3200-005). Sample concentrations were determined and equalized using Pierce BCA Protein Assay Kit (Thermo Scientific, 23225), and then used for immunoblotting. For EV protein extraction, EVs isolated from the media of two 10 cm plates were resuspended in RIPA buffer with protease and phosphatase inhibitors. The samples were equalized based on corresponding protein concentrations in cell lysates.
Equal protein amounts were loaded and ran in SDS-PAGE gels, then transferred to nitrocellulose membranes (GE Healthcare, 10600002). Blots were probed with primary antibodies overnight at 4°C (1:1000 dilution in TBS-T) and with HRP-conjugated secondary antibodies overnight at 4°C (1:1000 dilution in TBS-T) before detection with ECL western blot reagents (Pierce, 34578) and imaged with Fuji LAS-4000 imager (LAS-4000, Pittsburgh, PA, United States). All images were quantified using NIH ImageJ software.
Antibodies were purchased from the following commercial sources: Aβ (Cell Signaling Technology, 8243S), Tau (Santa Cruz Biotech, sc-390476), Hsc70 (Enzo Life Sciences, ADI-SPA-815-F), TSG101 (Thermo Fisher, PA5-31260), GFP (Cell Signaling Technology, 2956S), β-Actin (Santa Cruz Biotech, sc-47778), p62 (Cell Signaling Technology, 5114), Flag (Sigma Aldrich, F1804), SSH1 (ECM Biosciences, SP1711) (Cell Signaling Technology, 13578), Cofilin (Cell Signaling Technology, 5175s), Peroxidase-Conjugated AffiniPure Goat anti-mouse IgG (Jackson ImmunoResearch, 115-035-033), Peroxidase-conjugated AffiniPure Goat anti-rabbit IgG (Jackson ImmunoResearch, 111-035-033), donkey anti-rat IgG-HRP (Southern Biotech, 6430-05).
Amyloid-β 1-42 (GenicBio, A-42-T-1), Rapamycin (Sigma Aldrich, R0395), Bafilomycin A1 (Sigma Aldrich, B1793), Latrunculin B (Sigma Aldrich, L5288), Jasplakinolide (AdipoGen Life Sciences, 102396-24-7), p62 siRNA (Cell Signaling Technology, 6399S), SSH1 siRNA (Dharmacon GE Healthcare, 5′-GAG GAG CUG UCC CGA UGA C-3′), Cofilin siRNA (Dharmacon GE Healthcare, 5′-GGA GGA CCU GGU GUU CAU C-3′).
Confocal images were captured with the Olympus FV10i confocal microscope (Tokyo, Japan). All images were quantified using NIH ImageJ software.
To measure the secretion of exosomes, hereafter referred to as small EVs (sEVs), we used medium containing exosome-depleted (exo-free) FBS to collect conditioned medium from cells. Post-microvesicle (10,000 × g) supernatant was labeled with the fluorescent lipophilic dye DiI and ultracentrifuged at 100,000 × g to pellet sEVs (Figure 1A). We then visualized and quantified isolated sEVs on microscopic slides by fluorescence confocal microscopy. A similar method of DiI-labeling of EVs has previously been documented (Lehmann et al., 2008; Beer et al., 2015; Witas et al., 2017). As earlier studies showed that the autophagy activator and mTOR inhibitor rapamycin significantly inhibits sEV secretion (Fader et al., 2008; Abdulrahman et al., 2018; Bhat et al., 2021), we first tested the effects of rapamycin on sEV secretion in NIH3T3 cells. Indeed, rapamycin treatment for 24 h significantly reduced sEV secretion by ∼30% (Figures 1B,C). Western blotting for exosome markers Hsc70 and TSG101 showed corresponding reductions of both proteins in isolated sEVs but not in whole-cell lysates (Figure 1D). Nanoparticle tracking analysis (NTA) showed that the isolated sEVs are within the expected size range, the vast majority of which were under ∼150 nm in diameter with a minor proportion in the 150–250 nm range (Figure 1E). By contrast, inhibition of lysosomes with bafilomycin A1 treatment for 6 h significantly increased sEV release into the conditioned medium by ∼45% (Figures 1F,G), in agreement with previous studies (Iguchi et al., 2016; Minakaki et al., 2018).
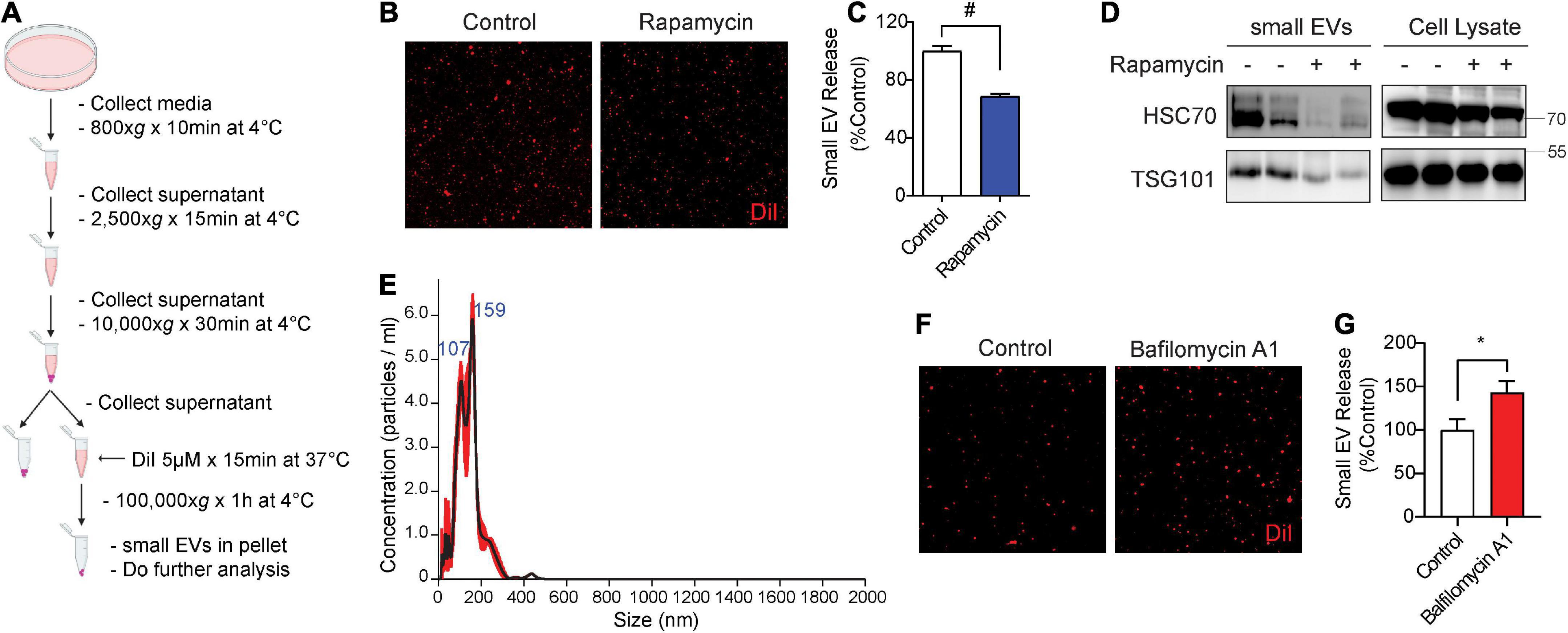
Figure 1. Rapamycin decreases and bafilomycin A1 increases small EV secretion. (A) Schematic of small EV isolation method. (B) Representative images of DiI-stained small EVs secreted from NIH3T3 cells with or without 200 nM rapamycin treatment for 24 h. (C) Quantification of secreted small EVs for (B). Data are presented as means ± SEM. n = 3 independent experiments, t-test, #P < 0.0001. (D) Representative immunoblots for the indicated proteins in secreted small EVs and cell lysates from NIH3T3 cells with or without 200 nM rapamycin treatment for 24 h. (E) Representative NTA analysis of small EVs isolated from NIH3T3 cells. The black line indicates concentration and size of particles, and the red line indicates standard error. (F) Representative images of DiI-stained small EVs secreted from NIH3T3 cells with or without 200 nM bafilomycin A1 treatment for 6 h. (G) Quantification of secreted small EVs for (F). Data are presented as means ± SEM. n = 4 independent experiments, t-test, *P = 0.02.
While previous studies have observed autophagy cargo receptors such as Sqstm1/p62 and optineurin in isolated sEVs (Gudbergsson and Johnsen, 2019), whether such cargo receptors regulate sEV secretion is unknown. Hence, we tested if overexpression of Sqstm1/p62 or optineurin alters sEV secretion. Hereafter, we use p62 in reference to Sqstm1/p62. The C-terminal region of p62 contains the UBA and LIR domains essential for binding to ubiquitinated cargo and LC3, respectively (Figure 2A; Katsuragi et al., 2015). The UBA region of p62 is subject to phosphorylation at Ser403 by ULK1, CK2, and/or TBK1, allowing p62 to bind ubiquitinated cargo (Pilli et al., 2012; Katsuragi et al., 2015; Lim et al., 2015; Matsumoto et al., 2015; Sanchez-Martin and Komatsu, 2018). GFP-p62 transfection significantly decreased sEV secretion by ∼50% compared to control GFP transfection (Figures 2B–D). However, transfection of p62 lacking the UBA and LIR domains (p62ΔC) failed to alter sEV secretion (Figures 2B–D), suggesting that p62 engagement with ubiquitinated cargo and/or LC3 binding are necessary to inhibit sEV release. Like p62, transfection of optineurin also suppressed sEV secretion. However, the ALS-linked optineurin E478G mutant, which fails to bind ubiquitin (Nakazawa et al., 2016), also failed to alter sEV secretion (Figures 2E–G), indicating that ubiquitin binding is a necessary step in suppressing sEV secretion by optineurin. To determine if endogenous p62 normally inhibits sEV secretion, we transfected cells with p62 siRNA. As expected, p62 knockdown significantly increased sEV secretion by ∼40% (Figures 2H–J). Hence, these observations indicate that autophagy cargo receptors, p62 and optineurin, inhibit sEV secretion by engaging ubiquitinated cargo.
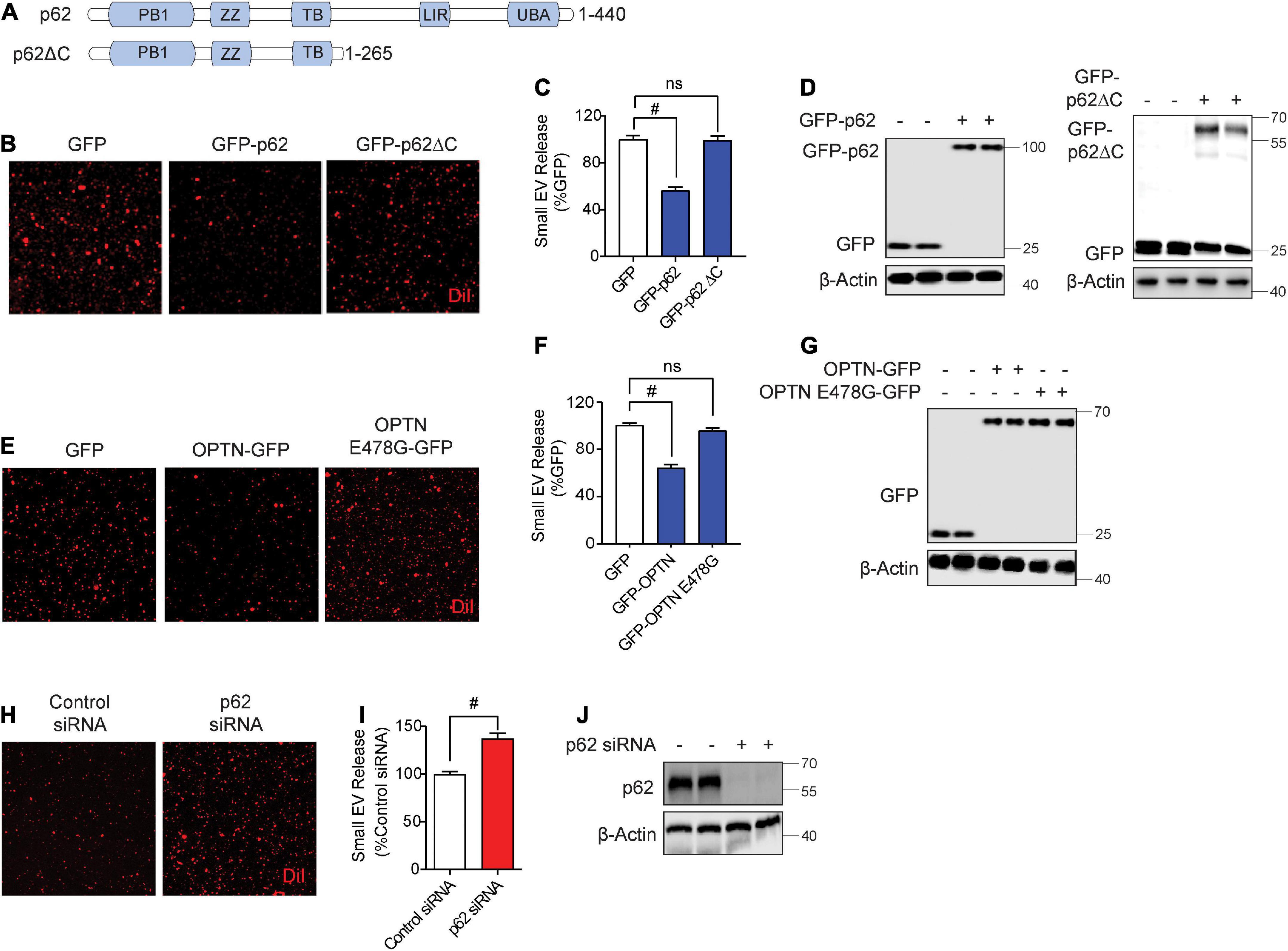
Figure 2. Autophagy cargo receptors, p62 and optineurin, inhibit small EV secretion through the ubiquitin association domain. (A) Schematic of p62 full length and p62ΔC proteins with their respective domains. (B) Representative images of DiI-stained small EVs secreted from NIH3T3 cells expressing GFP, GFP-p62, or GFP-p62ΔC. (C) Quantification of secreted small EVs for (B). Data are presented as means ± SEM. n = 3 independent experiments, 1-way ANOVA, followed by Dunnett’s post-hoc, #P < 0.0001. (D) Representative immunoblots of GFP-p62, GFP-p62ΔC, GFP, and β-Actin protein levels in lysates from cells for (B). (E) Representative images of DiI-stained small EVs secreted from NIH3T3 cells expressing GFP, GFP-OPTN, or GFP-OPTN E478G. (F) Quantification of secreted small EVs for (E). Data are presented as means ± SEM. n = 3 independent experiments, 1-way ANOVA, followed by Dunnett’s post-hoc, #P < 0.0001. n.s. = not significant. (G) Representative immunoblots for GFP, GFP-OPTN, and β-Actin protein levels in cell lysates for (E). (H) Representative images of DiI-stained small EVs secreted from NIH3T3 cells transfected with control siRNA or p62 siRNA. (I) Quantification of secreted small EVs for (H). Data are presented as means ± SEM. n = 4 independent experiments, t-test, #P < 0.0001. (J) Representative immunoblots for p62 and β-Actin protein levels in cell lysates for (H).
We recently showed that SSH1 contains a modular activity in inhibiting p62 autophagy flux by dephosphorylating p62 at pSer403, the phospho-residue that allows p62 binding to ubiquitinated cargo (Fang et al., 2021). SSH1 siRNA significantly decreased sEV secretion by nearly ∼40% (Figures 3A–C), indicating that endogenous SSH1 promotes sEV secretion. Conversely, forced expression of SSH1 significantly increased sEV secretion (Figures 3D–F). In p62 depleted cells, which exhibited significantly elevated sEV secretion (Figures 3G–I), SSH1 failed to increase sEV secretion (Figures 3G–I), indicating that SSH1-induced sEV secretion requires p62. To determine if SSH1-induced sEV secretion is specifically through p62 dephosphorylation at pSer403, we co-transfected cells with or without SSH1 and GFP control, GFP-p62, or GFP-p62-S403E, the latter mutant which mimics constitutive p62 phosphorylation at Ser403 (Fang et al., 2021). As expected, SSH1 overexpression significantly increased sEV secretion in the setting of GFP transfection and significantly reversed the decline in sEV secretion induced by GFP-p62 (Figures 3J–L). However, SSH1 failed to change the reduction in sEV secretion caused by GFP-p62-S403E (Figures 3J–L), indicating that SSH1 increases sEV secretion by dephosphorylating p62 at pSer403, a step that renders p62 inactive for binding to ubiquitinated cargo. Hence, these results indicate that the ability of p62 to engage ubiquitinated cargo is critical to SSH1-regulated sEV secretion. We did not investigate SSH1-mediated effects on optineurin-induced inhibition of sEV secretion, as SSH1 does not alter optineurin-mediated autophagy (Fang et al., 2021).
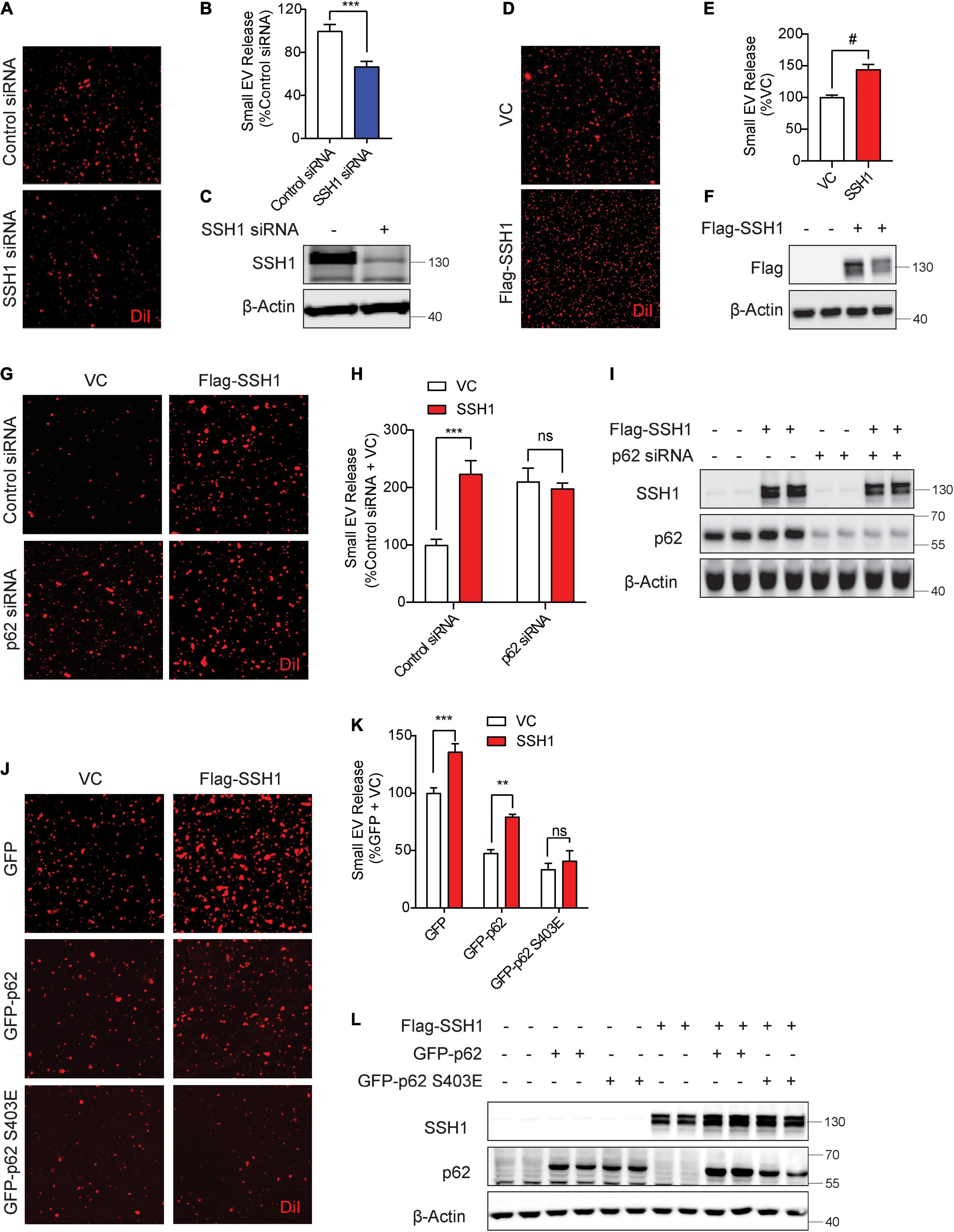
Figure 3. Slingshot homolog-1 (SSH1)-mediated p62 inhibition at pSer403 increases small EV secretion. (A) Representative images of DiI-stained small EVs secreted from NIH3T3 cells transfected with control siRNA or SSH1 siRNA. (B) Quantification of secreted small EVs for (A). Data are presented as means ± SEM. n = 3 independent experiments, t-test, ***P = 0.0005. (C) Representative immunoblots for SSH1 and β-Actin protein levels in cell lysates for (A). (D) Representative images of DiI-stained small EVs secreted from NIH3T3 cells expressing VC or Flag-SSH1 (E) Quantification of secreted small EVs for (D). Data are presented as means ± SEM. n = 3 independent experiments, t-test, #P < 0.0001 (F) Representative immunoblots for Flag and β-Actin protein levels in cell lysates for (D). (G) Representative images of DiI-stained small EVs secreted from NIH3T3 cells co-transfected with VC or Flag-SSH1 and control siRNA or p62 siRNA. (H) Quantification of secreted small EVs for (G). Data are presented as means ± SEM. n = 3 independent experiments, 2-way ANOVA, followed by Sidak’s post-hoc, ***P = 0.0004. n.s. = not significant. (I) Representative immunoblots showing SSH1, p62, and β-Actin protein levels in cell lysates for (G). (J) Representative images of DiI-stained small EVs secreted from NIH3T3 cells co-transfected with VC or Flag-SSH1 and GFP, GFP-p62, or GFP-p62 S403E. (K) Quantification of secreted small EVs for (J). Data are presented as means ± SEM. n = 3 independent experiments, 2-way ANOVA, followed by Sidak’s post-hoc, ***P = 0.0007, **P = 0.001. n.s. = not significant. (L) Representative immunoblots for SSH1, p62, and β-Actin protein levels in cell lysates for (J).
Slingshot homolog-1 was initially discovered as the major phosphatase that activates the actin severing and depolymerizing protein cofilin by its dephosphorylation at pSer3, increasing actin dynamics (Niwa et al., 2002; Kurita et al., 2007, 2008). SSH1-mediated dephosphorylation of cofilin and p62 are separable activities, as SSH1ΔN lacking the cofilin binding site dephosphorylates p62 but not cofilin, while the SSH1ΔC lacking the p62 binding site dephosphorylates cofilin but not p62 (Fang et al., 2021; Figure 4A). Hence, we tested if SSH1ΔN and SSH1ΔC increase sEV secretion compared to vector control and full-length SSH1. Surprisingly, SSH1ΔN or SSH1ΔC overexpression significantly increased sEV secretion compared to vector control, albeit less effectively than full-length SSH1 (Figures 4B–D). Given that SSH1ΔC increases cofilin activation, we next tested whether cofilin per se increases sEV secretion. Indeed, cofilin overexpression alone significantly increased sEV secretion by ∼30% (Figures 4E–G). The constitutively active cofilin-S3A similarly increased sEV secretion, whereas the inactive and dominant-negative cofilin-S3E (Liu et al., 2017; Shaw and Bamburg, 2017; Woo et al., 2019), which mimics the phosphorylated state, significantly decreased sEV secretion by ∼40% (Figures 4H–J). Moreover, cofilin siRNA significantly reduced sEV secretion by ∼35% (Figures 4K–M), indicating that endogenous cofilin promotes sEV secretion.
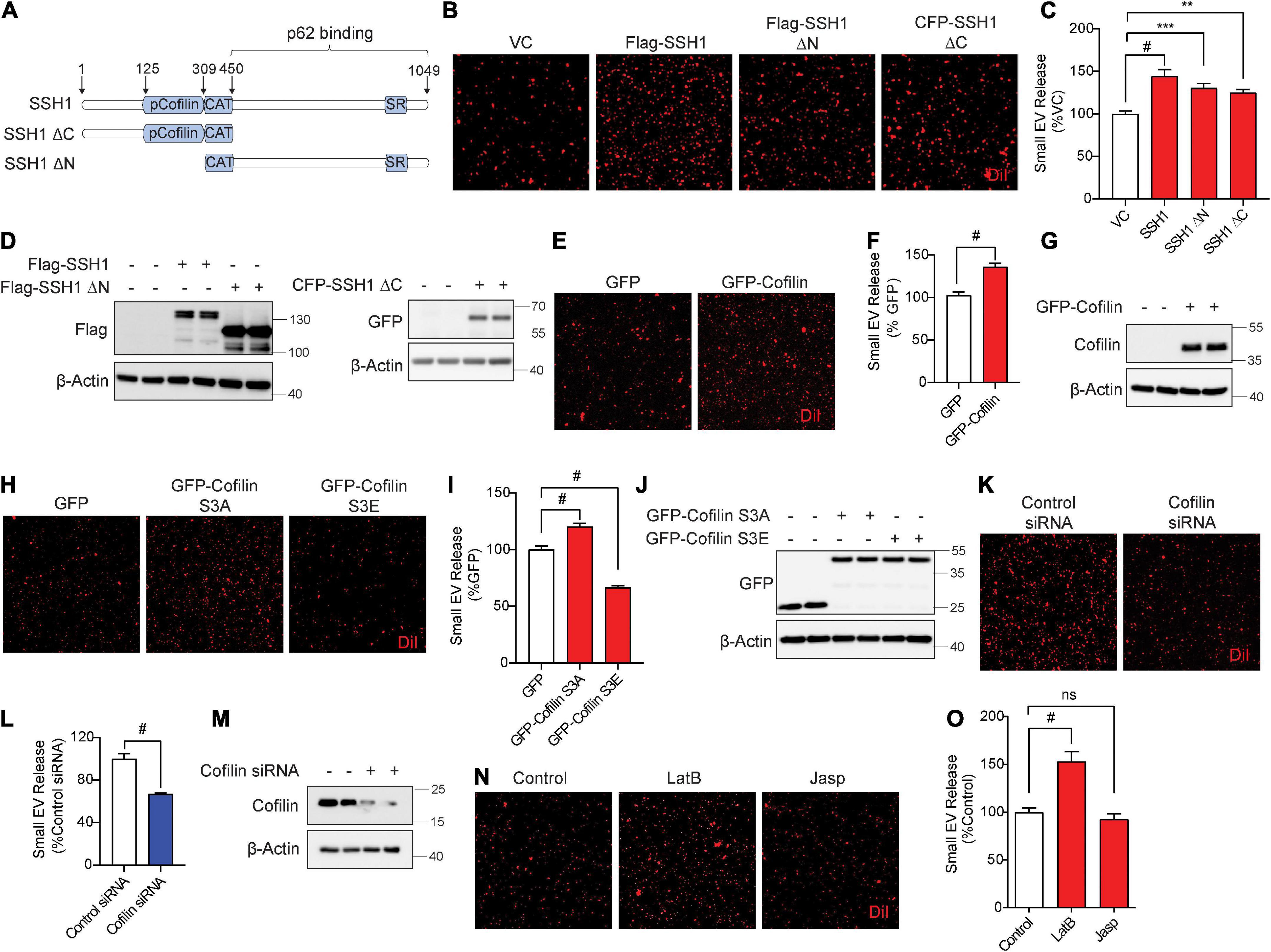
Figure 4. Slingshot homolog-1 (SSH1) increases small EV secretion partially through cofilin activation and F-actin disruption. (A) Schematic of SSH1 full length, SSH1ΔC, and SSH1ΔN proteins. (B) Representative images of DiI-stained small EVs secreted from NIH3T3 cells expressing VC, Flag-SSH1, Flag-SSH1ΔN, or CFP-SSH1ΔC. (C) Quantification of secreted small EVs for (B). Data are presented as means ± SEM. n = 3 independent experiments, 1-way ANOVA, followed by Dunnett’s post-hoc, #P < 0.0001, ***P = 0.001, **P = 0.0069. (D) Representative immunoblots for Flag, GFP, and β-Actin protein levels in cell lysates for (B). (E) Representative images of DiI-stained small EVs secreted from NIH3T3 cells expressing GFP or GFP-Cofilin. (F) Quantification of secreted small EVs for (E). Data are presented as means ± SEM. n = 3 independent experiments, t-test, #P < 0.0001. (G) Representative immunoblots for cofilin and β-Actin protein levels in cell lysates for (E). (H) Representative images of DiI-stained small EVs secreted from NIH3T3 cells expressing GFP, GFP-cofilin-S3A, or GFP-cofilin-S3E. (I) Quantification of secreted small EVs for (H). Data are presented as means ± SEM. n = 3 independent experiments, 1-way ANOVA, followed by Dunnett’s post-hoc, #P < 0.0001. (J) Representative immunoblots for GFP and β-Actin protein levels in cell lysates for (H). (K) Representative images of DiI-stained small EVs secreted from NIH3T3 cells transfected with control siRNA or cofilin siRNA. (L) Quantification of secreted small EVs for (K). Data are presented as means ± SEM. n = 3 independent experiments, t-test, #P < 0.0001. (M) Representative immunoblots for cofilin and β-Actin protein levels in cell lysates for (K). (N) Representative images of DiI-stained small EVs secreted from NIH3T3 cells with or without 100 nM Latrunculin B (LatB) or 100 nM Jasplakinolide (Jasp) treatment for 8 h. (O) Quantification of secreted small EVs for (N). Data are presented as means ± SEM. n = 3 independent experiments, 1-way ANOVA, followed by Dunnett’s post-hoc, #P < 0.0001. n.s. = not significant.
As actin filament severing and depolymerization activity of cofilin promoted sEV secretion, we tested if latrunculin B and jasplakinolide, drugs known to inhibit actin polymerization (Wakatsuki et al., 2001) and nucleate actin (Bubb et al., 2000), respectively, alter sEV secretion. Compared to the vehicle control, preventing actin polymerization by latrunculin B significantly increased sEV secretion by ∼50%, whereas promoting actin nucleation by jasplakinolide had no significant effect on sEV secretion (Figures 4N,O). These results are consistent with the observation that activated cofilin, which severs and depolymerizes F-actin, increases sEV release.
Aβ42 oligomers (Aβ42o) increase cofilin activation via SSH1, disrupt F-actin (Woo et al., 2015b) and enhance mTOR signaling (Caccamo et al., 2010). Moreover, misfolded mutant tau inhibits p62-mediated autophagy flux (Fang et al., 2021) and promotes F-actin bundling (Fulga et al., 2007; Cabrales Fontela et al., 2017). We tested if these AD signature pathologies alter sEV secretion and are in turn associated with sEVs. For treatment with Aβ42o, we used mouse hippocampus-derived HT22 neuroblastoma cells, as HT22 cells are responsive to Aβ42o-induced cofilin activation (Woo et al., 2015b). Aβ42o were prepared as previously described (Woo et al., 2015b). Aβ42o (250 nM) treatment to HT22 cells for 24 h significantly increased sEV secretion by nearly ∼3-fold (Figures 5A,B). Likewise, Western blotting for Hsc70 and TSG101 also demonstrated significant increases in these exosome markers by Aβ42o treatment in isolated sEVs but not in cell lysates (Figures 5C,D). The treated Aβ42o were readily detected in isolated sEVs (Figure 5C). Like in NIH3T3 cells, isolation of DiI-labeled sEVs in HT22 cells yielded the vast majority of nanovesicles <150 nm in diameter by NTA (Figure 5E), consistent with the range for exosomes. To determine whether misfolded tau alters sEV secretion, we utilized Frontotemporal dementia with Parkinsonism-17 (FTDP-17) tauP301S mutant and littermate WT neurons from P0 pups grown for 9 days in vitro (DIV9). On DIV9, the media was replaced with fresh media and collected after 24 h. TauP301S neurons exhibited significantly increased sEV secretion compared to littermate WT neurons (Figures 5F,G). To confirm this finding in a different way using another FTDP-17-linked tau mutation, we utilized tetracycline-inducible HEK293 expressing the tauP301L mutation. Tetracycline-inducible expression of tauP301L significantly increased exosome markers Hsc70 and TSG101 in isolated sEVs but not in cell lysates (Figures 5H,I), indicating that misfolded tau increases sEV secretion in neurons and HEK293 cells. TauP301L was also readily detected in association with isolated sEVs (Figure 5H), indicating that misfolded tau not only drives sEV release but is also a content of sEVs.
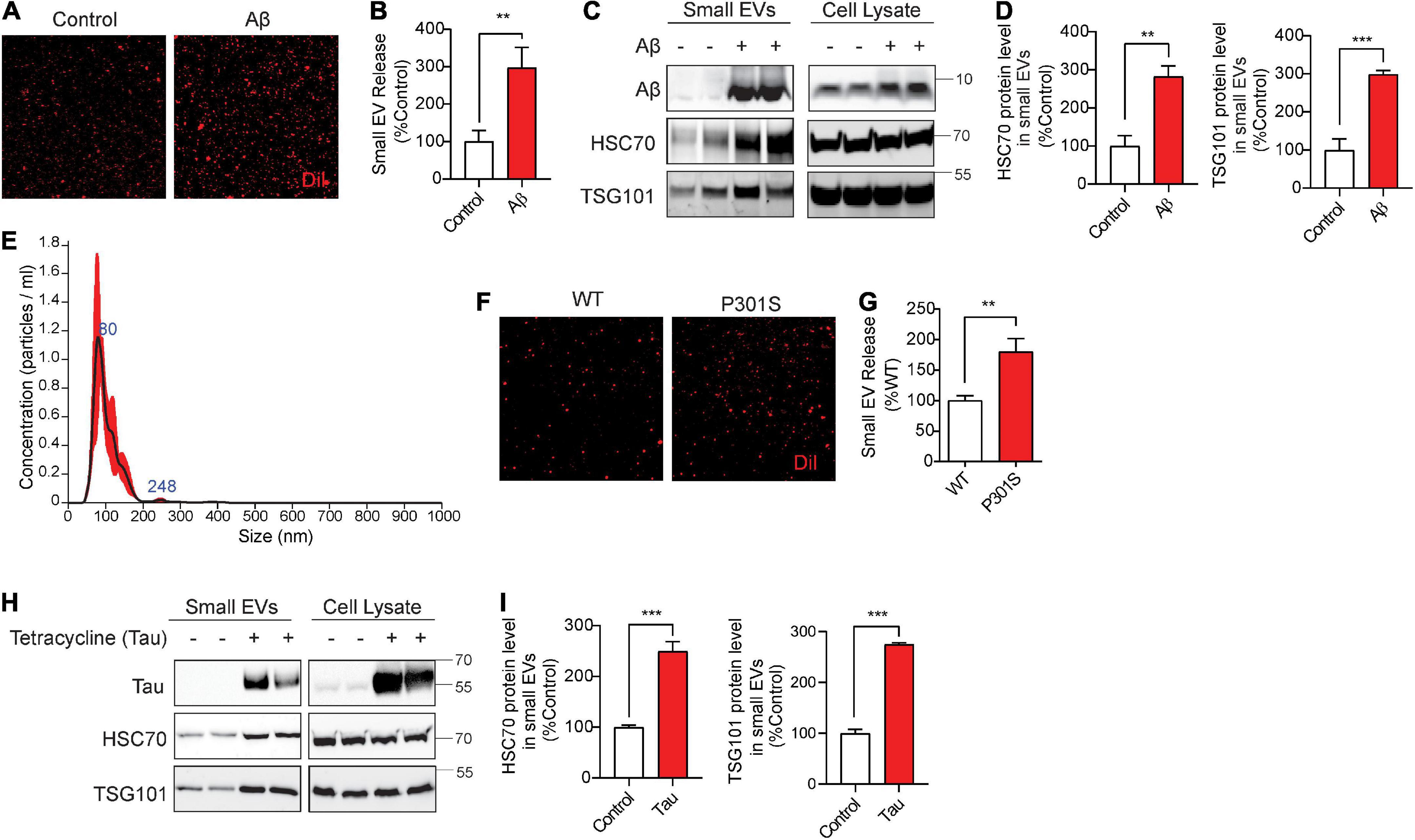
Figure 5. Aβ42 oligomers (Aβ42) oligomers and misfolded tau increase small EV secretion and are associated with secreted small EVs. (A) Representative images of DiI-stained small EVs secreted from HT22 cells with or without 250 nM Aβ treatment for 24 h. (B) Quantification of secreted small EVs for (A). Data are presented as means ± SEM. n = 3 independent experiments, t-test, **P < 0.005. (C) Representative immunoblots for the indicated proteins in secreted small EVs and cell lysates from HT22 cells with or without 250 nM Aβ treatment for 24 h. (D) Quantification of Hsc70 and TSG101 (small EV markers) for (C). Data are presented as means ± SEM. n = 4 independent experiments, t-test, ***P = 0.0007, **P < 0.004. (E) Representative NTA analysis of vesicle size distribution of small EVs isolated from HT22 cells. The black line indicates concentration and size of particles, and the red line indicates standard error. (F) Representative images of DiI-stained small EVs secreted by DIV10 cortical primary neurons derived from C57BL6 (WT) or tauP301S (P301S) mice. (G) Quantification of secreted small EVs for (F). Data are presented as means ± SEM. n = 3 independent experiments, t-test, **P = 0.002. (H) Representative immunoblots for the indicated proteins in secreted small EVs and cell lysates from tet-inducible iHEK-P301L cells expressing tauP301L, with or without tetracycline. (I) Quantification of Hsc70 and TSG101 (small EV markers) for (H). Data are presented as means ± SEM. n = 4 independent experiments, t-test, ***P < 0.0003.
Multiple studies have implicated the involvement of the SSH1-cofilin pathway in Aβ-induced dendritic spine shrinkage and F-actin loss (Zhao et al., 2006; Shankar et al., 2007) as well as the accumulation of multiple pathologies, including cofilin-actin pathology (Minamide et al., 2000; Rahman et al., 2014), Aβ pathology (Liu et al., 2019), and tau pathology (Woo et al., 2019). Cofilin activity is increased in the brains of AD patients (Zhao et al., 2006; Kim et al., 2013) and the APP/PS1 mouse model (Woo et al., 2015b). At the same time, the autophagy-lysosome pathway is overwhelmed and impaired in AD (Lin et al., 2003; Nixon et al., 2005; Salminen et al., 2012; Hebron et al., 2014; Rea et al., 2014; Tanji et al., 2014; Feng et al., 2019; Roca-Agujetas et al., 2021a,b), which contributes to the accumulation of misfolded proteins and dysfunctional mitochondria (Ramesh Babu et al., 2008; Xu et al., 2019; Woo et al., 2020; Fang et al., 2021; Roca-Agujetas et al., 2021a,b). Meanwhile, AD pathological proteins Aβ and tau are secreted in exosomes and are propagated from cell to cell in part via exosomes (Asai et al., 2015; Sardar Sinha et al., 2018; Miyoshi et al., 2021). Yet, the pathobiological relationship between these drivers of AD pathogenesis and sEV secretion are unknown. In this study, we took advantage of the dual axes of SSH1 signaling in the inhibition of autophagy (Fang et al., 2021) and the promotion of actin dynamics (Niwa et al., 2002; Kurita et al., 2007, 2008) to study the corresponding pathways in sEV secretion. We used the neutral term sEVs to place no bias on the origin of the EVs; however, sEVs are largely equivalent to exosomes based on their predominant size range. Here we showed for the first time that autophagy cargo receptors, p62 and optineurin, inhibit sEV secretion, an activity that requires their ability to engage ubiquitinated cargo. SSH1, which inhibits p62 binding to cargo (Fang et al., 2021), increased sEV secretion, but not in p62-depleted cells or cells expressing the p62-S403E mutation. However, the N-terminal domain of SSH1 (SSH1ΔC), which activates cofilin but does not inhibit p62 (Fang et al., 2021), increased sEV secretion through the cofilin activation pathway. Accordingly, latrunculin B, which prevents actin polymerization and indirectly promotes F-actin depolymerization (Wakatsuki et al., 2001), mimicked the effect of cofilin in enhancing sEV secretion. Finally, we showed that AD pathological proteins, Aβ oligomers and mutant tau, both of which impact autophagy and F-actin dynamics pathways (Fulga et al., 2007; Caccamo et al., 2010; Woo et al., 2015b; Cabrales Fontela et al., 2017; Fang et al., 2021), increased sEV secretion and are also contents of secreted sEVs. These findings, therefore, implicate autophagy cargo receptor-cargo interactions and actin dynamics in regulating sEV secretion through mechanisms contributing to AD pathogenesis.
Multiple studies have shown that autophagosomes accumulate in AD brains (Lin et al., 2003; Nixon et al., 2005; Hebron et al., 2014; Feng et al., 2019), indicative of impairment in the autophagy-lysosome pathway. Autophagosomes can fuse with multivesicular bodies (MVBs) containing intraluminal vesicles to form amphisomes or fuse directly with lysosomes for degradation (Ganesan and Cai, 2021; Figure 6). Our observations that the ability of p62 and optineurin to inhibit sEV secretion depends on their ability to engage ubiquitinated cargo are intriguing in light of the significantly reduced p62 Ser403 phosphorylation in AD brains (Tanji et al., 2014), a phospho-residue required for p62 to bind ubiquitinated cargo (Pilli et al., 2012; Katsuragi et al., 2015; Lim et al., 2015; Matsumoto et al., 2015; Sanchez-Martin and Komatsu, 2018). We interpret this to indicate that the binding of autophagy cargo receptors to ubiquitinated cargo promotes autophagy flux and fusion of autophagosomes and amphisomes with lysosomes (Fang et al., 2021), which diverts amphisomes away from fusing with the plasma membrane and are instead degraded by lysosomes (Figure 6A). Indeed, cargo binding to p62 promotes autophagosome maturation by enhancing the synthesis of LC3 and its conversion to LC3-II (Cha-Molstad et al., 2017; Figure 6A). In support of this notion, SSH1 increased sEV secretion through its ability to dephosphorylate p62 at pSer403, which reduces cargo binding and inhibits p62 autophagy flux (Fang et al., 2021; Figure 6B). Interestingly, calcium elevation or reactive oxygen species (ROS) induced by Aβ oligomers or other stressors activate SSH1 (Davis et al., 2011; Woo et al., 2015a,b) through calcineurin activation (Wang et al., 2005; Tu et al., 2014) and/or 14-3-3 oxidation (Wang et al., 2005), respectively, the latter which releases SSH1 from 14-3-3 inhibitory control (Figure 6B). Such calcium and reactive oxygen species (ROS)-mediated control of SSH1 activation is consistent with our observation that Aβ42 oligomers also increase sEV secretion (Figure 6B). Like SSH1, inhibition of p62-mediated autophagy flux by misfolded tau (Fang et al., 2021) is also consistent with increased sEV secretion by mutant tau in this study, likely by diverting autophagosomes away from lysosomes toward secretory amphisomes (Figure 6B).
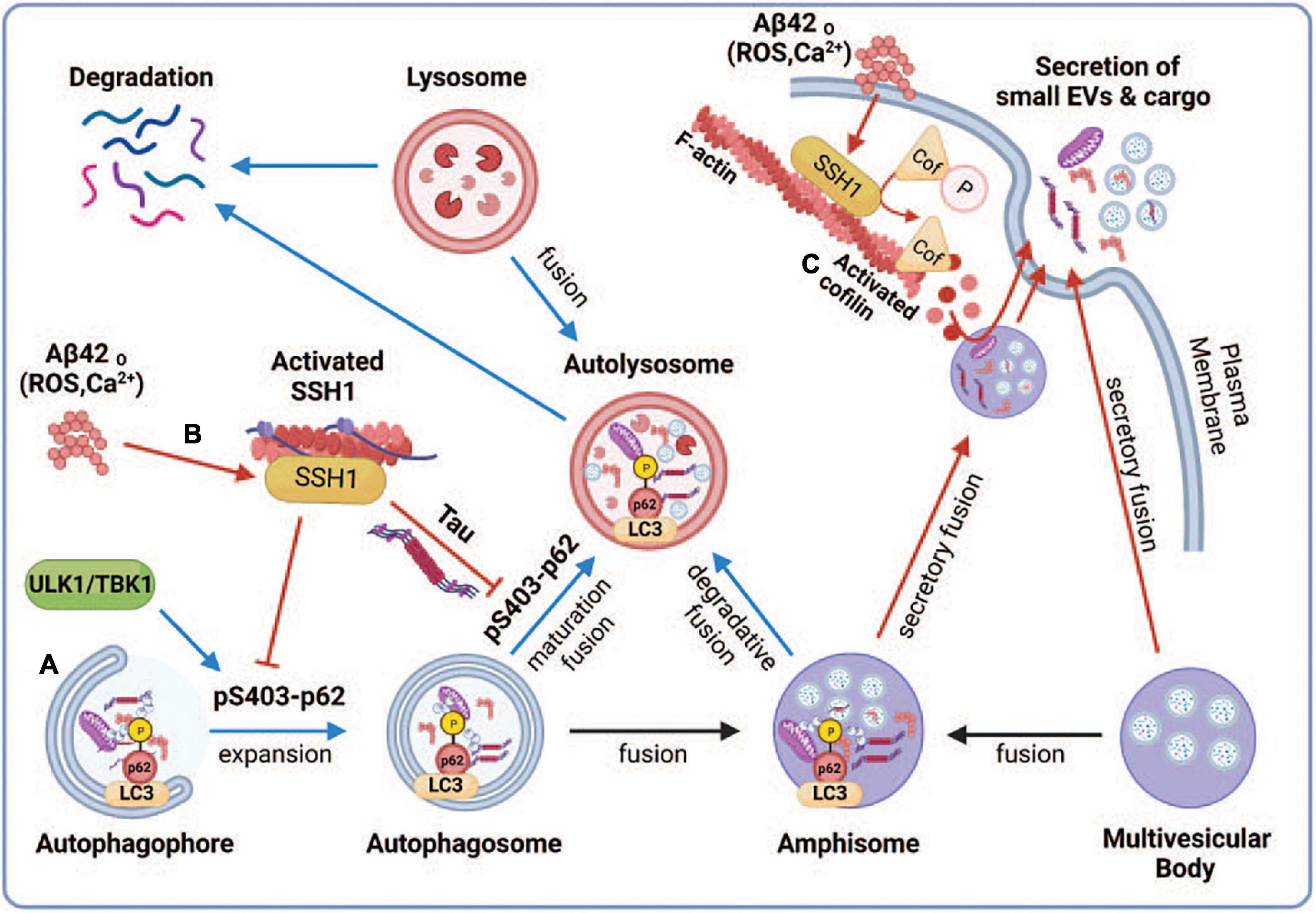
Figure 6. Proposed model depicting the regulation of small EV secretion by p62-mediated autophagy and cofilin-mediated actin dynamics through the SSH1 pathway. Note that blue lines/arrows favor lysosomal degradation, whereas red lines/arrows favor secretory clearance. (A) Accumulation of misfolded proteins or dysfunctional mitochondria activates autophagy through ULK1 and TBK1, which phosphorylate p62 at Ser403, resulting in p62 binding to ubiquitinated cargo and activation of LC3 to drive autophagosome maturation. This promotes autophagy flux and fusion of autophagosomes and amphisomes with lysosomes, resulting in the degradation of ubiquitinated cargo together with intraluminal vesicles from amphisomes, thereby reducing small EV secretion. (B) Oxidative stress (ROS) or calcium elevation induced by Aβ oligomers or otherwise activates SSH1, which dephosphorylates p62 at pSer403 and inhibits p62 autophagy flux. Dephosphorylated p62 is less able to bind ubiquitinated cargo and activate LC3, which slows autophagy flux and fusion of autophagosomes and amphisomes with lysosomes. This process diverts amphisomes toward the secretory fusion pathway and increases small EV secretion. Like SSH1, inhibition of p62 autophagy flux by misfolded tau (Fang et al., 2021) also likely contributes to increased small EV secretion. (C) Actin filaments serve to recruit vesicles and create a diffusion barrier for vesicles to gain access to the plasma membrane. SSH1 activation by ROS or calcium also activates cofilin, which severs and depolymerizes the F-actin network near the plasma membrane. This process allows large secretory amphisomes to access membrane docking sites, facilitating small EV secretion. This illustration was generated at Biorender.com.
Control of actin dynamics is critical to maintaining cell morphology and other cellular processes, including membrane protein trafficking, endocytosis, phagocytosis, and exocytosis (Carlsson, 2018; Li et al., 2018; Liu et al., 2019). However, the role of actin dynamics in sEV secretion is essentially unknown. The actin-binding protein cortactin positively regulates sEV secretion by binding to F-actin and Arp2/3, stabilizing docking sites at the plasma membrane (Sinha et al., 2016). Like exocytosis of secretory vesicles, sEV secretion is increased by calcium elevation (Savina et al., 2003). At the microscale level, the actin-based cytoskeleton serves to both recruit vesicles and function as a diffusion barrier that prevents vesicles from gaining access to docking sites on the plasma membrane (Li et al., 2018). Hence, a dense network of actin filaments would serve as a far more significant barrier to the much larger MVBs or amphisomes. As SSH1-mediated cofilin activation is increased by calcium elevation (Wang et al., 2005), the F-actin severing and depolymerizing cofilin activities would be expected to trim the dense F-actin network, allowing MVBs and amphisomes to gain access to plasma membrane docking sites (Figure 6C). However, excessive depolymerization of F-actin could also hinder sEV secretion by failing to recruit MVBs or amphisomes to the F-actin mesh close to the plasma membrane. Studies on secretory vesicles have shown that it is the oscillation of calcium signals and actin dynamics (depolymerization and polymerization) that coordinately control vesicle access to docking sites and exocytosis (Li et al., 2018). Indeed, cofilin-S3A, which is far more active than wild-type (WT) cofilin, was slightly less effective than WT cofilin in increasing sEV secretion, which may be due to the excessive actin disruptive activity of this mutant. On the other hand, the inactive and dominant-negative cofilin-S3E significantly decreased sEV secretion, likely due to the inhibition of endogenous SSH1 (Liu et al., 2017; Shaw and Bamburg, 2017; Woo et al., 2019). Like cofilin, 100 nM latrunculin B treatment for 8 h, a dose that partially disrupts F-actin (Wakatsuki et al., 2001), had a net positive effect on sEV secretion.
Our studies showed that Aβ oligomers and misfolded tau not only increase sEV secretion but are also secreted in association with sEVs. This coupling appears to be a way for cells to efficiently expel misfolded proteins, which otherwise would impede and congest the autophagy machinery. Excessive secretion of misfolded proteins by “secretory autophagy” in association with sEVs or otherwise could then serve as seeds for the propagation of pathology from cell to cell (Rajendran et al., 2006; Vella et al., 2007; Danzer et al., 2012; Saman et al., 2012, 2014; Asai et al., 2015; Sardar Sinha et al., 2018; Miyoshi et al., 2021).
Our data suggest that increasing cargo receptor engagement with autophagic cargo and reducing actin dynamics (i.e., SSH1 inhibition) represents an attractive strategy to promote misfolded protein degradation while reducing sEV-mediated cell to cell spread of pathology.
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of South Florida (USF) and Case Western Reserve University (CWRU).
SC: investigation, methodology, and writing. CF, HK, and RW: investigation and methodology. TK: data analysis and visualization. JW: investigation, writing, review, and funding acquisition. DK: conceptualization, writing, visualization, and funding acquisition. All authors have read and approved the manuscript.
This work was supported in part by grants from the National Institutes of Health (NIH) (R01AG059721, JW; R01AG067741, DK and JW; R01NS122350, DK; and RF1AG053060, DK) and Veterans Affairs (BX004680, DK).
We thank Dr. Laura Blair and Dr. Chad Dickey for providing the iHEK-tauP301L cells.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aβ42o, Aβ42 oligomers; Arp2/3, actin related protein 2/3 complex; AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; ALP, autophagy-lysosome pathway; CK2, casein kinase 2; EVs, extracellular vesicles; sEVs, small EVs; F-actin, fIlamentous actin; FTDP-17, frontotemporal dementia with parkinsonism-17; Hsc70, heat shock cognate 70 kDa protein; FP, green fluorescent protein; ILVs, intraluminal vesicles; LIR, LC3 interacting region; LC3, microtubule-associated protein light chain 3; MVs, microvesicles; MVBs, multivesicular bodies; mTOR, mammalian target of rapamycin; NTA, nanoparticle tracking analysis; SSH1, slingshot homolog-1; sequestosome-1, Sqstm1/p62; TBK1, tank binding kinase 1; TSG101, tumor susceptibility gene 101 protein; UBA, ubiquitin association; ULK1, Unc-51 like autophagy activating kinase 1.
Abdulrahman, B. A., Abdelaziz, D. H., and Schatzl, H. M. (2018). Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem. 293, 8956–8968. doi: 10.1074/jbc.RA117.000713
An, K., Klyubin, I., Kim, Y., Jung, J. H., Mably, A. J., O’Dowd, S. T., et al. (2013). Exosomes neutralize synaptic-plasticity-disrupting activity of Abeta assemblies in vivo. Mol. Brain 6:47. doi: 10.1186/1756-6606-6-47
Antonucci, F., Turola, E., Riganti, L., Caleo, M., Gabrielli, M., Perrotta, C., et al. (2012). Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 31, 1231–1240. doi: 10.1038/emboj.2011.489
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Beer, L., Zimmermann, M., Mitterbauer, A., Ellinger, A., Gruber, F., Narzt, M. S., et al. (2015). Analysis of the Secretome of Apoptotic Peripheral Blood Mononuclear Cells: Impact of Released Proteins and Exosomes for Tissue Regeneration. Sci. Rep. 5:16662. doi: 10.1038/srep16662
Bhat, O. M., Yuan, X., Kukreja, R. C., and Li, P. L. (2021). Regulatory role of mammalian target of rapamycin signaling in exosome secretion and osteogenic changes in smooth muscle cells lacking acid ceramidase gene. FASEB J. 35:e21732. doi: 10.1096/fj.202100385R
Bubb, M. R., Spector, I., Beyer, B. B., and Fosen, K. M. (2000). Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 275, 5163–5170. doi: 10.1074/jbc.275.7.5163
Cabrales Fontela, Y., Kadavath, H., Biernat, J., Riedel, D., Mandelkow, E., and Zweckstetter, M. (2017). Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nat. Commun. 8:1981. doi: 10.1038/s41467-017-02230-8
Caccamo, A., Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120. doi: 10.1074/jbc.M110.100420
Carlsson, A. E. (2018). Membrane bending by actin polymerization. Curr. Opin. Cell Biol. 50, 1–7. doi: 10.1016/j.ceb.2017.11.007
Cha-Molstad, H., Yu, J. E., Feng, Z., Lee, S. H., Kim, J. G., Yang, P., et al. (2017). p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat. Commun. 8:102. doi: 10.1038/s41467-017-00085-7
Danzer, K. M., Kranich, L. R., Ruf, W. P., Cagsal-Getkin, O., Winslow, A. R., Zhu, L., et al. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegeneration 7:42. doi: 10.1186/1750-1326-7-42
Davis, R. C., Marsden, I. T., Maloney, M. T., Minamide, L. S., Podlisny, M., Selkoe, D. J., et al. (2011). Amyloid beta dimers/trimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin-phosphorylation. Mol. Neurodegeneration. 6:10. doi: 10.1186/1750-1326-6-10
Fader, C. M., Sanchez, D., Furlan, M., and Colombo, M. I. (2008). Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 9, 230–250. doi: 10.1111/j.1600-0854.2007.00677.x
Fang, C. X., Woo, J. A. A., Liu, T., Zhao, X. Y., Cazzaro, S., Yan, Y., et al. (2021). SSH1 impedes SQSTM1/p62 flux and MAPT/Tau clearance independent of CFL (cofilin) activation. Autophagy 17, 2144–2165. doi: 10.1080/15548627.2020.1816663
Feng, Q., Luo, Y., Zhang, X. N., Yang, X. F., Hong, X. Y., Sun, D. S., et al. (2019). MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy 16, 641–658. doi: 10.1080/15548627.2019.1633862
Fulga, T. A., Elson-Schwab, I., Khurana, V., Steinhilb, M. L., Spires, T. L., Hyman, B. T., et al. (2007). Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 9, 139–148. doi: 10.1038/ncb1528
Ganesan, D., and Cai, Q. (2021). Understanding amphisomes. Biochem. J. 478, 1959–1976. doi: 10.1042/BCJ20200917
Garvalov, B. K., Flynn, K. C., Neukirchen, D., Meyn, L., Teusch, N., Wu, X., et al. (2007). Cdc42 regulates cofilin during the establishment of neuronal polarity. J. Neurosci. 27, 13117–13129. doi: 10.1523/JNEUROSCI.3322-07.2007
Gordon, P. B., Hoyvik, H., and Seglen, P. O. (1992). Prelysosomal and lysosomal connections between autophagy and endocytosis. Biochem. J. 283, 361–369. doi: 10.1042/bj2830361
Gudbergsson, J. M., and Johnsen, K. B. (2019). Exosomes and autophagy: rekindling the vesicular waste hypothesis. J. Cell Commun. Signal. 13, 443–450. doi: 10.1007/s12079-019-00524-8
Hebron, M. L., Algarzae, N. K., Lonskaya, I., and Moussa, C. (2014). Fractalkine signaling and Tau hyper-phosphorylation are associated with autophagic alterations in lentiviral Tau and Abeta1-42 gene transfer models. Exp. Neurol. 251, 127–138. doi: 10.1016/j.expneurol.2013.01.009
Iguchi, Y., Eid, L., Parent, M., Soucy, G., Bareil, C., Riku, Y., et al. (2016). Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139, 3187–3201. doi: 10.1093/brain/aww237
Itakura, E., and Mizushima, N. (2011). p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 192, 17–27. doi: 10.1083/jcb.201009067
Katsuragi, Y., Ichimura, Y., and Komatsu, M. (2015). p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282, 4672–4678. doi: 10.1111/febs.13540
Kim, T., Vidal, G. S., Djurisic, M., William, C. M., Birnbaum, M. E., Garcia, K. C., et al. (2013). Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341, 1399–1404. doi: 10.1126/science.1242077
Kurita, S., Gunji, E., Ohashi, K., and Mizuno, K. (2007). Actin filaments-stabilizing and -bundling activities of cofilin-phosphatase Slingshot-1. Genes Cells 12, 663–676. doi: 10.1111/j.1365-2443.2007.01078.x
Kurita, S., Watanabe, Y., Gunji, E., Ohashi, K., and Mizuno, K. (2008). Molecular dissection of the mechanisms of substrate recognition and F-actin-mediated activation of cofilin-phosphatase Slingshot-1. J. Biol. Chem. 283, 32542–32552. doi: 10.1074/jbc.M804627200
Lehmann, B. D., Paine, M. S., Brooks, A. M., McCubrey, J. A., Renegar, R. H., Wang, R., et al. (2008). Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 68, 7864–7871. doi: 10.1158/0008-5472.CAN-07-6538
Li, P., Bademosi, A. T., Luo, J., and Meunier, F. A. (2018). Actin Remodeling in Regulated Exocytosis: Toward a Mesoscopic View. Trends Cell Biol. 28, 685–697. doi: 10.1016/j.tcb.2018.04.004
Lim, J., Lachenmayer, M. L., Wu, S., Liu, W., Kundu, M., Wang, R., et al. (2015). Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 11:e1004987. doi: 10.1371/journal.pgen.1004987
Lin, W. L., Lewis, J., Yen, S. H., Hutton, M., and Dickson, D. W. (2003). Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J. Neurocytol. 32, 1091–1105. doi: 10.1023/B:NEUR.0000021904.61387.95
Liou, W., Geuze, H. J., Geelen, M. J., and Slot, J. W. (1997). The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 136, 61–70. doi: 10.1083/jcb.136.1.61
Liu, T., Wang, F., LePochat, P., Woo, J. A., Bukhari, M. Z., Hong, K. W., et al. (2017). Cofilin-mediated Neuronal Apoptosis via p53 Translocation and PLD1 Regulation. Sci. Rep. 7:11532. doi: 10.1038/s41598-017-09996-3
Liu, T., Woo, J. A., Yan, Y., LePochat, P., Bukhari, M. Z., and Kang, D. E. (2019). Dual role of cofilin in APP trafficking and amyloid-beta clearance. FASEB J. 33, 14234–14247. doi: 10.1096/fj.201901268R
Matsumoto, G., Shimogori, T., Hattori, N., and Nukina, N. (2015). TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 24, 4429–4442. doi: 10.1093/hmg/ddv179
Minakaki, G., Menges, S., Kittel, A., Emmanouilidou, E., Schaeffner, I., and Barkovits, K. (2018). Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 14, 98–119. doi: 10.1080/15548627.2017.1395992
Minamide, L. S., Striegl, A. M., Boyle, J. A., Meberg, P. J., and Bamburg, J. R. (2000). Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2, 628–636. doi: 10.1038/35023579
Miyoshi, E., Bilousova, T., Melnik, M., Fakhrutdinov, D., Poon, W. W., and Vinters, H. V. (2021). Exosomal tau with seeding activity is released from Alzheimer’s disease synapses, and seeding potential is associated with amyloid beta. Lab Invest. 101, 1605–1617. doi: 10.1038/s41374-021-00644-z
Nakazawa, S., Oikawa, D., Ishii, R., Ayaki, T., Takahashi, H., and Takeda, H. (2016). Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat. Commun. 7:12547. doi: 10.1038/ncomms12547
Niwa, R., Nagata-Ohashi, K., Takeichi, M., Mizuno, K., and Uemura, T. (2002). Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108, 233–246. doi: 10.1016/s0092-8674(01)00638-9
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Park, B. C., Shen, X., Samaraweera, M., and Yue, B. Y. (2006). Studies of optineurin, a glaucoma gene: Golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am. J. Pathol. 169, 1976–1989. doi: 10.2353/ajpath.2006.060400
Pilli, M., Arko-Mensah, J., Ponpuak, M., Roberts, E., Master, S., and Mandell, M. A. (2012). TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234. doi: 10.1016/j.immuni.2012.04.015
Prada, I., Furlan, R., Matteoli, M., and Verderio, C. (2013). Classical and unconventional pathways of vesicular release in microglia. Glia 61, 1003–1017. doi: 10.1002/glia.22497
Rahman, T., Davies, D. S., Tannenberg, R. K., Fok, S., Shepherd, C., Dodd, P. R., et al. (2014). Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer’s disease. J. Alzheimer’s Dis. 42, 1443–1460. doi: 10.3233/JAD-140393
Rajendran, L., Bali, J., Barr, M. M., Court, F. A., Kramer-Albers, E. M., Picou, F., et al. (2014). Emerging roles of extracellular vesicles in the nervous system. J. Neurosci. 34, 15482–15489. doi: 10.1523/JNEUROSCI.3258-14.2014
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Ramesh Babu, J., Lamar Seibenhener, M., Peng, J., Strom, A. L., Kemppainen, R., Cox, N., et al. (2008). Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 106, 107–120. doi: 10.1111/j.1471-4159.2008.05340.x
Raposo, G., and Stoorvogel, W. (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383. doi: 10.1083/jcb.201211138
Rea, S. L., Majcher, V., Searle, M. S., and Layfield, R. (2014). SQSTM1 mutations–bridging Paget disease of bone and ALS/FTLD. Exp. Cell Res. 325, 27–37. doi: 10.1016/j.yexcr.2014.01.020
Roca-Agujetas, V., Barbero-Camps, E., de Dios, C., Podlesniy, P., Abadin, X., Morales, A., et al. (2021a). Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegeneration 16:15. doi: 10.1186/s13024-021-00435-6
Roca-Agujetas, V., de Dios, C., Abadin, X., and Colell, A. (2021b). Upregulation of brain cholesterol levels inhibits mitophagy in Alzheimer disease. Autophagy 17, 1555–1557. doi: 10.1080/15548627.2021.1920814
Salminen, A., Kaarniranta, K., Haapasalo, A., Hiltunen, M., Soininen, H., and Alafuzoff, I. (2012). Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Progress Neurobiol. 96, 87–95. doi: 10.1016/j.pneurobio.2011.11.005
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Saman, S., Lee, N. C., Inoyo, I., Jin, J., Li, Z., Doyle, T., et al. (2014). Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer’s disease. J. Alzheimer’s Dis. 40:S47–S70. doi: 10.3233/JAD-132135
Sanchez-Martin, P., and Komatsu, M. (2018). p62/SQSTM1 - steering the cell through health and disease. J. Cell Sci. 131:jcs222836. doi: 10.1242/jcs.222836
Sardar Sinha, M., Ansell-Schultz, A., Civitelli, L., Hildesjo, C., Larsson, M., Lannfelt, L., et al. (2018). Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 136, 41–56. doi: 10.1007/s00401-018-1868-1
Savina, A., Furlan, M., Vidal, M., and Colombo, M. I. (2003). Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J. Biol. Chem. 278, 20083–20090. doi: 10.1074/jbc.M301642200
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Shaw, A. E., and Bamburg, J. R. (2017). Peptide regulation of cofilin activity in the CNS: A novel therapeutic approach for treatment of multiple neurological disorders. Pharmacol. Ther. 175, 17–27. doi: 10.1016/j.pharmthera.2017.02.031
Sinha, S., Hoshino, D., Hong, N. H., Kirkbride, K. C., Grega-Larson, N. E., Seiki, M., et al. (2016). Cortactin promotes exosome secretion by controlling branched actin dynamics. J. Cell Biol. 214, 197–213. doi: 10.1083/jcb.201601025
Skog, J., Wurdinger, T., van Rijn, S., Meijer, D. H., Gainche, L., Sena-Esteves, M., et al. (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476. doi: 10.1038/ncb1800
Tanji, K., Miki, Y., Ozaki, T., Maruyama, A., Yoshida, H., Mimura, J., et al. (2014). Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2:50. doi: 10.1186/2051-5960-2-50
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegeneration 9:48. doi: 10.1186/1750-1326-9-48
Turturro, S., Shen, X., Shyam, R., Yue, B. Y., and Ying, H. (2014). Effects of mutations and deletions in the human optineurin gene. Springerplus 3:99. doi: 10.1186/2193-1801-3-99
Valadi, H., Ekstrom, K., Bossios, A., Sjostrand, M., Lee, J. J., and Lotvall, J. O. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659. doi: 10.1038/ncb1596
Vella, L. J., Sharples, R. A., Lawson, V. A., Masters, C. L., Cappai, R., and Hill, A. F. (2007). Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211, 582–590. doi: 10.1002/path.2145
Verderio, C., Muzio, L., Turola, E., Bergami, A., Novellino, L., Ruffini, F., et al. (2012). Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 72, 610–624. doi: 10.1002/ana.23627
Wakatsuki, T., Schwab, B., Thompson, N. C., and Elson, E. L. (2001). Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J. Cell Sci. 114, 1025–1036. doi: 10.1242/jcs.114.5.1025
Wang, S., Cesca, F., Loers, G., Schweizer, M., Buck, F., and Benfenati, F. (2011). Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J. Neurosci. 31, 7275–7290. doi: 10.1523/JNEUROSCI.6476-10.2011
Wang, Y., Shibasaki, F., and Mizuno, K. (2005). Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J. Biol. Chem. 280, 12683–12689. doi: 10.1074/jbc.M411494200
Witas, R., Chaput, D., Khan, H., Stevens, S. M. Jr., and Kang, D. (2017). Isolation and Proteomic Analysis of Microvesicles and Exosomes from HT22 Cells and Primary Neurons. Methods Mol. Biol. 1598, 255–267. doi: 10.1007/978-1-4939-6952-4_12
Woo, J. A., Boggess, T., Uhlar, C., Wang, X., Khan, H., and Cappos, G. (2015a). RanBP9 at the intersection between cofilin and Abeta pathologies: rescue of neurodegenerative changes by RanBP9 reduction. Cell Death Dis. 6:1676. doi: 10.1038/cddis.2015.37
Woo, J. A., Liu, T., Fang, C. C., Castano, M. A., Kee, T., Yrigoin, K., et al. (2020). beta-Arrestin2 oligomers impair the clearance of pathological tau and increase tau aggregates. Proc. Natl. Acad. Sci. U.S. A. 117, 5006–5015. doi: 10.1073/pnas.1917194117
Woo, J. A., Liu, T., Fang, C. C., Cazzaro, S., Kee, T., LePochat, P., et al. (2019). Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun. Biol. 2:112. doi: 10.1038/s42003-019-0359-9
Woo, J. A., Zhao, X., Khan, H., Penn, C., Wang, X., Joly-Amado, A., et al. (2015b). Slingshot-Cofilin activation mediates mitochondrial and synaptic dysfunction via Abeta ligation to beta1-integrin conformers. Cell Death Differ. 22, 1069–1070. doi: 10.1038/cdd.2015.41
Xu, Y., Zhang, S., and Zheng, H. (2019). The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy 15, 583–598. doi: 10.1080/15548627.2018.1532258
Yuyama, K., Sun, H., Sakai, S., Mitsutake, S., Okada, M., Tahara, H., et al. (2014). Decreased amyloid-beta pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 289, 24488–24498. doi: 10.1074/jbc.M114.577213
Keywords: SSH1, amyloid, tau, exosome, p62, optineurin, actin dynamics, autophagy
Citation: Cazzaro S, Fang C, Khan H, Witas R, Kee TR, Woo J-AA and Kang DE (2022) Slingshot homolog-1 mediates the secretion of small extracellular vesicles containing misfolded proteins by regulating autophagy cargo receptors and actin dynamics. Front. Aging Neurosci. 14:933979. doi: 10.3389/fnagi.2022.933979
Received: 01 May 2022; Accepted: 08 August 2022;
Published: 25 August 2022.
Edited by:
Rodrigo Morales, University of Texas Health Science Center at Houston, United StatesReviewed by:
Melissa Calegaro Nassif, Universidad Mayor, ChileCopyright © 2022 Cazzaro, Fang, Khan, Witas, Kee, Woo and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David E. Kang, ZGVrOTRAY2FzZS5lZHU=; Jung-A. A. Woo, amF3MzMwQGNhc2UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.