 Lihua Yu†
Lihua Yu† Guoping Peng†
Guoping Peng† Yuan Yuan
Yuan Yuan Min Tang
Min Tang Ping Liu
Ping Liu Xiaoyan LiuJie NiYi Li
Xiaoyan LiuJie NiYi Li Caihong Ji
Caihong Ji Ziqi FanWenli Zhu
Ziqi FanWenli Zhu Benyan Luo*
Benyan Luo* Qing Ke*
Qing Ke*- Department of Neurology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Background: Rapid-onset dystonia parkinsonism (RDP) is a rare disease caused by ATP1A3 mutation with considerable clinical heterogeneity. Increased knowledge of RDP could be beneficial in its early diagnosis and treatment.
Objective: This study aimed to summarize the gene mutation spectrum of ATP1A3 associated with RDP, and to explore the correlation of ATP1A3 variants with RDP clinical phenotypes.
Methods: In this study, we reported two RDP patients from a family with a novel inherited ATP1A3 variant. Then, we reviewed and analyzed the available literature in English focused on ATP1A3-causative RDP. A total of 35 articles covering 15 families (59 patients) and 36 sporadic RDP cases were included in our analysis.
Results: The variant A813V (2438C>T) in ATP1A3 found in our cases was a novel mutant. Delays in diagnosis were common, with a mean delay time of 14 years. ATP1A3 had distinct RDP-related mutation hotspots, which consisted of exon8, 14, 17, and 18, and the most frequently occurring variants were T613M and I578S. Approximately 74.5% of patients have specific triggers before disease onset, and 82.1% of RDPs have stable symptoms within 1 month. The incidence rates of dystonia and bradykinesia are 100 and 88.1%, respectively. The onset site varied and exhibited a rostrocaudal gradient distribution pattern in 45% of patients with RDP. Approximately 63.6% of patients had mild improvement after receiving comprehensive interventions, especially in gait disturbance amelioration.
Conclusion: In patients with acute and unexplained dystonia or bradykinesia, gene screening on ATP1A3 should be timely performed. When a diagnosis has been made, treatments that may be effective are to be attempted. Our study would be helpful for the early diagnosis and treatment of ATP1T3-related RDP.
Introduction
Rapid-onset dystonia-parkinsonism (RDP, DYT12) identified a quarter century ago is a rare disorder characterized by the abrupt onset of asymmetric dystonia and parkinsonism, with bradykinesia, gait instability, and prominent bulbar symptoms (Brashear et al., 1996; Heinzen et al., 2014). RDP episodes typically occur within hours of a triggering event, such as fevers, alcohol consumption, exercise, emotional stress, childbirth, or infections, and progress in a few hours to a week (Tarsy et al., 2010; Barbano et al., 2012). Subsequently, the clinical symptoms of most patients maintain relatively stable, and levodopa treatment shows only no or only a minimal benefit (Heinzen et al., 2014). Disease onset within age ranges from 9 months to 55 years has been previously reported (Brashear et al., 2007). The clinical manifestations of RDP are heterogeneous, and family members can have different clinical symptoms. Thus, RDP diagnostic delay is very common (Tan et al., 2015; Haq et al., 2019). Understanding the RDP phenotype profile is of great significance for its early diagnosis and treatment.
ATP1A3, as an autosomal dominant pathogenic gene, is the only causative gene for RDP (Brashear et al., 1996, 2007). It encodes a neuron-specific P-type Na+/K+ ATPase that is closely related to the sodium-coupled transport of a variety of organic and inorganic molecules, osmoregulation, and electrical excitability of nerves and muscles (Heinzen et al., 2014). ATP1A3 variation is also the primary cause of alternating hemiplegia of childhood (AHC), Relapsing encephalopathy with cerebellar ataxia (RECA) (Dard et al., 2015; Biela et al., 2021), early onset epileptic encephalopathy (Paciorkowski et al., 2015; Schirinzi et al., 2018), and cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) (Heinzen et al., 2012). The clinical phenotype of AHC is characterized by recurrent episodes of hemiplegia and dystonia alternating in laterality, and the onset age is usually before 18 months (Rosewich et al., 2012), which is quite different from RDP. In addition, there is a specific AHC/RDP intermediate phenotype, with clinical symptoms overlapping with those of AHC and RDP. This type often presents symptoms of AHC in the early stage of the disease, and as the disease progresses, the clinical manifestations and outcomes are similar to those of RDP (Termsarasab et al., 2015; Pereira et al., 2016). Until now, nearly 30 mutations of ATP1A3 related to RDP and the AHC/RDP intermediate type have been identified.
In this study, we reported cases of two RDP patients from a family carrying a novel ATP1A3 mutation. We reviewed all ATP1A3-related RDP and AHC/RDP intermediate patients reported in the previously published English literature. We then performed a genotype-phenotype correlation analysis using the collected data.
Materials and methods
Patient cohorts
Two patients, including the proband and her subject mother, were enrolled in our study. Both fulfill the clinical diagnostic criteria (Rosewich et al., 2017), received detailed medical history collection and physical examination, performed head MRI scanning, cardiac color Doppler ultrasound, lumbar puncture examinations, and collected peripheral blood for whole exome sequencing.
In addition, we searched all published English literature with the keywords “Rapid-onset dystonia parkinsonism”, or “RDP” and “ATP1A3” in Pubmed and Web of Science database, and then enrolled all clinically diagnosed as RDP or intermediate AHC/RDP. All patients included in this study met the clinical diagnostic criteria for RDP or intermediate AHC/RDP (Pereira et al., 2016; Haq et al., 2019). Through manual screening, we included a total of 35 English literature and our two cases, including 15 RDP families (total of 59 patients) and 36 sporadic patients, were performed secondary analysis. Among these 95 patients, 23 of them only had ATP1A3 mutation information, for which clinical data were not available, and further genotype–phenotype correlation analysis was not included. Ultimately, a total of 60 typical RDP patients and 12 intermediated RDP/AHC patients were included in our genotype-phenotype analysis.
This study was approved by the Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University, China. Both of the patients in this study agreed to participate and provided written informed consent.
Whole exome sequencing and Sanger sequencing
The proband was subjected to whole exome sequencing (WES) analysis to test point mutation or small deletion/insertion. The genetic testing was carried out at Running Gene Medical Laboratory (Beijing, China). Genomic DNA was extracted and tested according to the manufacturer's standard procedure as reported in our previous study (Yu et al., 2021). The potential variant found in WES was validated by the Sanger sequencing method. Then the variant found in the proband was tested in the subjected mother and asymptomatic father using the same method (Yu et al., 2021).
Computational prediction of variant pathogenicity
We used PolyPhen-2 (https://genetics.bwh.harvard.edu/pph2/), MutPreds (http://mutpred.mutdb.org/#qform), PROVEAN (https://provean.jcvi.org/protein_batch_submit.php?species=human), programs to predict the effect of missense variants. Then, the Clinvar database, Pubmed database, and the Human Gene Mutation Database (HGMD) were used to screen mutations reported in previous studies. The variant detected in our study was compared in the gene database of normal healthy people, including the Chinese Millionome Database (CMDB) (https://cmdb.bgi.com/cmdb/), 1000 Genomes Project (https://www.internationalgenome.org/) (Sampson et al., 2016; Yu et al., 2021).
Statistical analysis
We used the Shapiro–Wilk to test whether the age of the patient fits a Normal distribution and the Mann–Whitney U test was used to detect the age differences between subgroups. The Chi-square test was used to compare differences in rates. All the tests were performed with SPSS 26 software.
Results
Identification of a novel ATP1A3 mutation in a familial RDP
An 18-year-old female (III-1, the proband) Chinese patient presented abruptly with dysphagia and speech difficulty after a headache. Within 2 days, she gradually developed right-upper limb bradykinesia and dystonia, and lower limb stiffness. One year later, after a cough, the dystonia and bradykinesia of the limbs were significantly aggravated. She had an abnormal gait and could not walk independently. Her symptoms did not progress and remained stable. She took levodopa/carbidopa and trihexyphenidyl for 6.5 months, but with no obvious effect. She had a positive family history, and her mother had similar symptoms. The patient reported no exposure to drugs, toxins, or an uneventful medical history of neurological disorders. At the age of 20, she visited our department. Given her symptoms, we refined a series of tests. Psychological assessments using HAMD and HAMA scales revealed that she had significant anxiety and mild depression. Cognitive function assessments with MMSE and MoCA scales showed normal scores, and 3T-cranial MRI findings were normal. Cerebrospinal fluid examination (CSF) showed decreased homovanillic acid level. Using whole exome sequencing (WES), an ATP1A3 missense heterozygous mutation was detected and verified by Sanger sequencing. ATP1A3 variant c.2438C>T, located in exon18, results in alanine at position 813 being replaced by valine (p.Ala813Val, A813V). This novel variant was not found in Pubmed, HGMD, and 1000 Genomes projects. Therefore, the patient was diagnosed with RDP. After 1 month of treatments with baclofen, coenzyme Q10, and rehabilitation exercises, partial gait improvement was noted.
Mother of the proband (II-2) showed the symptoms after she abruptly adopted an abnormal posture and unsteady gait following a strenuous exercise. Sanger sequencing confirmed that she carried the same ATP1A3 c.2438C>T missense mutation. She did not receive any medication due to mild symptoms. Father of the proband (II-1) was completely normal and did not carry this variant (Figure 1).
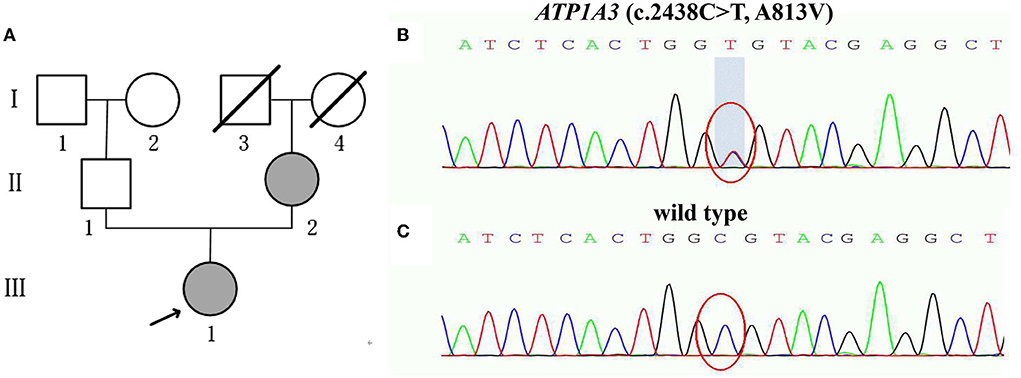
Figure 1. (A) Pedigree of the family with RDP showing the affected cases (fully shaded: indicate the ATP1A3 (c.2438C>T, A813V) mutation; (B,C) Sanger sequencing analysis of the mutation A813V; (B) Heterozygous p.A813V mutation carrier (the proband III-1 and the mother of the proband II-2); (C) Normal control (the father of the proband II-1).
ATP1A3 A813V was a potentially pathogenic mutant for RDP
The proband's manifestations met the clinical diagnostic criteria for typical RDP. The results suggested that ATP1A3 variant alleles were co-segregated in the family. The mutation site is located in exon18 of ATP1A3, which constituted the mutation hotspot region. In the ATP1A3 A813V variant, the original alanine was replaced by valine. As observed by crystallography, the side chain of valine was much larger than alanine, which changed the conformation of the ATP1A3 protein, thereby impairing its biological activity. The nature of the variant (A813V) was predicted to be highly disruptive, as indicated by a PolyPhen2 score of 0.663 (possibly damaging) and a SIFT-PROVEAN finding of deleterious (−2.846). In addition, the predicted result of MutPred2 was damage (0.575) and the predicted molecular mechanism was the loss of Helix. According to the criteria recommended by the ACMG grading guidelines criteria, the ATP1A3 variant A813V was rated as a likely pathogenic mutant (Richards et al., 2015) (Supplementary figure 1). Taken altogether, variant A813V of ATP1A3 potentially altered its function.
Clinical and genetic spectrum of patients with ATP1A3-related RDP and intermediated AHC/RDP
A total of 95 patients with RDP and intermediate AHC/RDP, including 15 family and 36 sporadic patients with ATP1A3 mutations, were enrolled in our study. The manifestations and genetic information of all enrolled patients are summarized in Supplementary table 1 (Pittock et al., 2000; Wunderlich et al., 2002; de Carvalho Aguiar et al., 2004; Zaremba et al., 2004; Kamphuis et al., 2006; Brashear et al., 2007, 2012; Lee et al., 2007; Kamm et al., 2008; Zanotti-Fregonara et al., 2008; Anselm et al., 2009; Blanco-Arias et al., 2009; Svetel et al., 2010; Tarsy et al., 2010; Barbano et al., 2012; Roubergue et al., 2013; Oblak et al., 2014; Rosewich et al., 2014a,b; Sasaki et al., 2014; Tan et al., 2015; Termsarasab et al., 2015; Wilcox et al., 2015; Liu et al., 2016; Nicita et al., 2016; Pereira et al., 2016; Sampson et al., 2016; Sweadner et al., 2016; Sousa et al., 2017; Wenzel et al., 2017; Marrodan et al., 2018; Haq et al., 2019; Boonsimma et al., 2020; Yuan et al., 2020; Hoshino et al., 2021; Nomura et al., 2021). Due to the lack of clinical data of some patients, a total of 60 patients with typical RDP and 12 patients with intermediate AHC/RDP were finally included for further analysis (Tables 1, 2) (Anselm et al., 2009; Brashear et al., 2012; Rosewich et al., 2014a; Sasaki et al., 2014; Termsarasab et al., 2015; Nicita et al., 2016; Pereira et al., 2016; Sousa et al., 2017; Boonsimma et al., 2020).
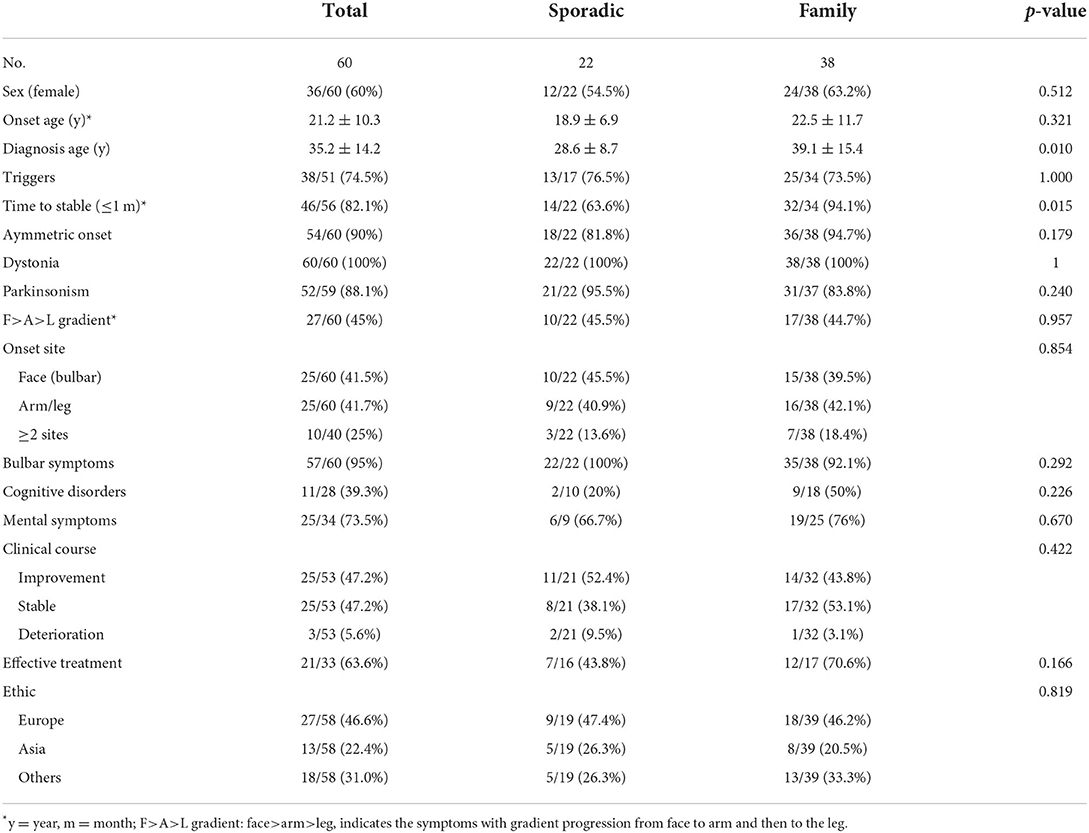
Table 1. Clinical characteristics of ATP1A3-related RDPs.
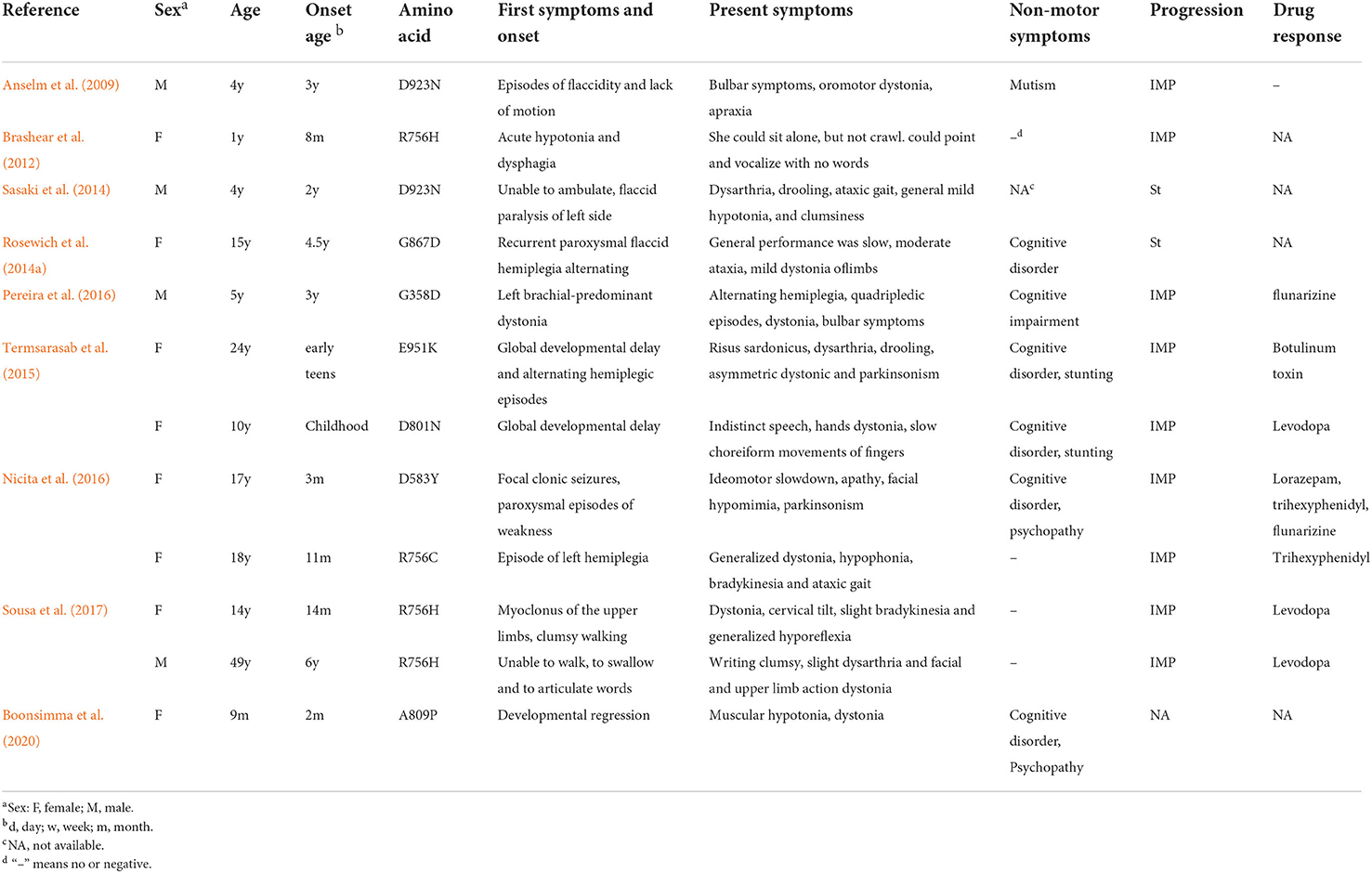
Table 2. Summary of the genetic and clinical features of the intermediate AHC/RDP patients.
Systematic review of clinical features of RDP patients
A systematic review of the available published clinical data of RDP patients with ATP1A3 mutations was conducted in this study. A total of 60 typical RDP, composed of 38 familial RDPs from 15 pedigrees and 22 unrelated sporadic RDPs, were included in our analysis (Table 1). Overall, the proportion of female patients (60%) was slightly higher than that of male patients (40%). The age at RDP onset was 21.2 ± 10.3 years (range 4–55 years), the diagnosis age was 35.2 ± 14.2 years, and the average diagnostic delay time was 14 years. About 74.5% (38/51) of the patients had definite triggers before the onset of RDP or before their symptoms significantly worsened. The triggers vary, and even the same patient met with different triggers. The most common triggering factors (over 80% of the reported triggers) included psychological stress, fever (heat), alcohol, exercise, head trauma, and childbirth. Other less frequently seen triggering factors were also reported, such as medicine (prochlorperazine) intake, mock air defense drill, anesthetic application, respiratory tract infection, travel sickness, and operation. Most patients experienced a rapid onset process, with 82.1% reaching a stable state within 30 days.
In terms of clinical symptoms, all patients had dystonia; 88.1% of them also presented with parkinsonism (mainly bradykinesia and muscle rigidity), and 90% of the patients experienced an asymmetric onset. A common rostrocaudal gradient [Face (Bulbar) > Arm > Leg] was presented, along with the characteristic manifestations that were found in 45% of RDP patients. The proportion of the patients with a limb (arm/leg) onset and face (bulbar) onset accounted for 41.5 and 41.7%, respectively. Bulbar symptoms were very common, appearing in 95% of the RDPs. Non-motor symptoms, such as cognitive disorders and mental symptoms, were not uncommon, especially the latter appearing in ~73.5% of the patients, whereas cognitive disorders occurred in 22.7% of patients. There was a lack of effective drugs for treating RDP, and RDP patients had poor responses to dopamine/carbidopa (Pittock et al., 2000; Rosewich et al., 2014b). In this study, comprehensive treatments showed partial efficacy in 63.6% of patients, with the most significantly improved symptom being gait disturbance. In terms of auxiliary examination, blood test and CT/MRI examination did not reveal specific changes, but severe brain atrophy was detected in one patient using cranial MRI (Sweadner et al., 2016). Molecular imaging tests, including PET-CT and SPECT, indicated some specific changes (Table 3) (Kamphuis et al., 2006; Zanotti-Fregonara et al., 2008; Anselm et al., 2009; Svetel et al., 2010; Tarsy et al., 2010). Decreased expression of homovanillic acid (HAV) in cerebrospinal fluid was found in RDP patients; Among eight patients who underwent the HAV test, three had a decreased HAV level. The potentially effective treatments included benzodiazepines (diazepam, or lorazepam), anti-parkinsonian drugs (levodopa/carbidopa, trihexyphenidyl, or amantadine), and other drugs such as baclofen, propranolol, a vitamin compound, and botulinum toxin injection. Of these, baclofen and trihexyphenidyl were primarily reported (Supplementary table 1). However, the current evidence regarding the efficacy of deep brain stimulation (DBS) treatment was very limited and controversial. In a previous study, the symptoms of one sporadic patient were not alleviated with DBS of bilateral globus pollidus internus (Gpi) (Deutschlander et al., 2005), whereas a modest benefit was achieved in another patient using DBS of the GPi (Kamm et al., 2008). Considering the clinical course of RDP, both improvement and stable state rates were 47.2%, and the deteriorating case was rare (5.6%). The results of previous functional studies on ATP1A3-related RDP, mainly involving the decreased activity of Na+-K+ ATPase and reduced affinity to cytoplasm Na+, were summarized in Table 4 (Rodacker et al., 2006; Blanco-Arias et al., 2009; Heinzen et al., 2012; Sweadner et al., 2016).
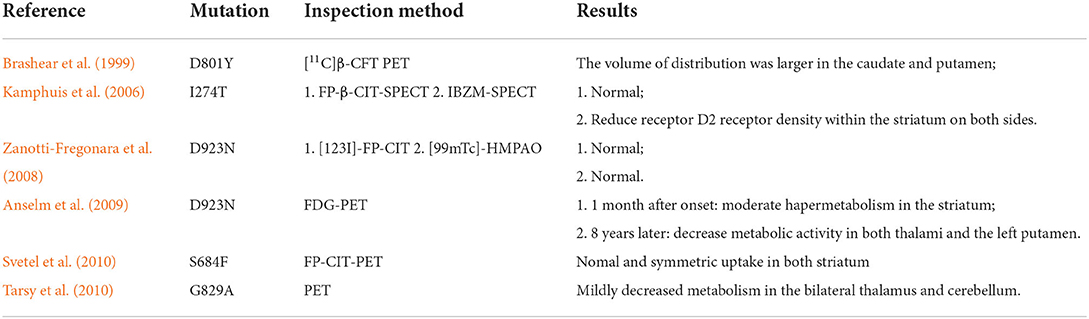
Table 3. Summary of molecular imaging findings on RDP and intermediate AHC/RDP.

Table 4. The functional findings of ATP1A3 mutations on RDP and intermediate AHC/RDP.
Based on family history, we divided all RDP cases into two subgroups, sporadic and familial RDP. We compared their data and analyzed the difference in the clinical symptoms between the two groups. Of all clinical symptoms, only the diagnosis age showed a significant difference between the two groups (p = 0.010).
Systematic review of clinical features of patients with intermediate AHC/RDP
A total of 12 patients diagnosed with intermediate AHC/RDP were included in our analysis, including 10 sporadic and 1 pedigree case. The male-to-female sex ratio was 1:2. The ages of these patients at onset were usually <5 years, with a mean diagnosis age of 13.49 ± 13.44 years, and a delay in diagnosis age was still very common. The majority of patients had definite triggers before disease onset, which was similar to RDP occurrence, and the most common triggering factors included fever, mental disorder, and exercise.
The onset symptoms and intermediate AHC/RDP vary greatly, manifesting as episodes of flaccidity, recurrent paroxysmal flaccid hemiplegia, limb dystonia or hypotonia of the limbs, myoclonus, and other non-motor symptoms such as seizure and global developmental delay. As the disease progressed, motor symptoms, including bulbar symptoms, dystonia, parkinsonism, and ataxic gait became increasingly common. In the early stage of intermediate AHC/RDP, the patients' clinical symptoms were similar to those of patients with AHC. With the progression of the disease, the phenotype of the intermediate AHC/RDP mimicked that of RDP. With regard to drug treatment, there was also a lack of effective treatments for intermediate AHC/RDP patients. Similarly with RDP treatment, partial effects might be achieved by administrating levodopa, trihexyphenidyl, lorazepam, and botulinum toxin injection. In addition, flunarizine was also reported to be potentially effective in two previous studies (Nicita et al., 2016; Pereira et al., 2016). In terms of outcome, the prognosis of patients with intermediate AHC/RDP was better than that of RDP patients, with ~9 of 12 patients showing improved condition (Table 2).
Genotype-phenotype correlation analysis
A total of 32 ATP1A3 mutations have been reported in the literature, including 29 missense mutations and 3 nonsense mutations. Among them, 23 genetic mutations were only associated with the RDP phenotype, 7 were associated with the intermediate AHC/RDP phenotype, and the other 2 variants D923N and R756H were linked with RDP and intermediate AHC/RDP. Of all ATP1A3 variants, T613M and I758S were predominant, with 25 and 15 RDP patients carrying these two variants, respectively. The mutational clusters were obvious, most of the mutations (87.5%) located in exons 8, 14, 17, and 18, with the corresponding areas being domain transmembrane regions (T3 and T5), and cytoplasmic regions (between T4 and T5). Approximately 58.3% of the mutations in the intermediate AHC/RDP mutational clusters are located in exons 9 and 17 (Figure 2). Most ATP1A3 mutants were associated with a single disease phenotype, including RDP, AHC, or intermediate AHC/RDP, while a small portion of the mutants was correlated with different phenotypes. For example, ATP1A3 D923N and R756H could present RDP or intermediate AHC/RDP phenotype (Zanotti-Fregonara et al., 2008; Anselm et al., 2009; Brashear et al., 2012; Tan et al., 2015), while ATP1A3 variants 1013Ydul, E277K, D923N were linked with the phenotype of RDP or AHC (Termsarasab et al., 2015).
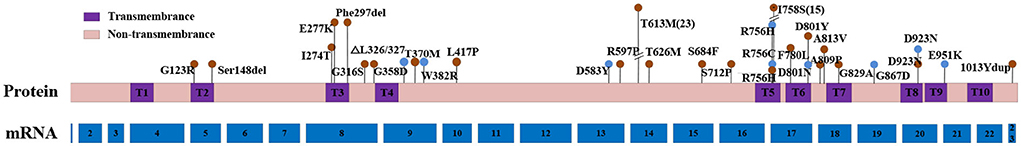
Figure 2. Schematic diagram of the location of RDP or intermediate AHC/RDP-causing mutations in ATP1A3 mRNA and protein. Red dots show RDP-causing mutations and blue dots dedicate intermediate AHC/RDP- causing mutations. The height of the dot represents the number of reported cases for this mutation, the shortest dot represents only one reported case for this mutation. The number of mRNA represent each corresponding exon. RDP, rapid-onset dystonia parkinsonism; AHC, alternating hemiplegia of childhood.
In previously reported RDP families, patients carrying the same mutation usually showed similar clinical manifestations and pathological changes. Oblak et al. (2014) reported three RDP patients from the same family with ATP1A3 I758S mutant who had analogous clinical manifestations, including similar triggers, cognitive disorder, and severe psychotic disorder, the pathological changes of them were also comparable. In another RDP family with ATP1A3 Ser148del variant, all nine patients were female, and the only male carrier did not show any symptoms (Wilcox et al., 2015). Barbano et al. (2012) reported an RDP family with ATP1A3 T613M mutant, four subjects not only had the same onset and symptoms, but also show similar non-motor symptoms, including cognitive disorder with predominant verbal fluency impairment, and mood disorder.
Discussion
In this study, we reported a Chinese RDP family carrying a novel ATP1A3 variant 2438C>T (A813V), which was predicted to be a potential pathogenic mutation. Our case provided a genetic marker for the diagnosis and treatment of RDP, the discovery of this variant also expanded the existing knowledge on the genetic spectrum of RDP. We included all English literature on RDP and intermediate AHC/RDP, and a systematic review and genotype-phenotype correlation analysis were performed on the clinical and genetic findings of 60 RPD patients and 12 patients with intermediate AHC/RDP (Tables 1, 2). The following major results were found: (1) RDP diagnosis delay was common; (2) There was evidence supporting a genotype-phenotype correlation in RDP; (3) ATP1A3-related RDP and intermediate AHC/RDP might be different phenotypes of the same disease, also called a spectrum disorder; (4) The recognition of onset symptoms and timely genetic screening on ATP1A3 mutations facilitated early diagnosis of RDP; (5) There were no significant differences exist in the clinical manifestations between family and sporadic patients.
Diagnosis delay is very common for ATP1A3-related RDP and intermediate AHC/RDP. We found that the diagnosis age was delayed by an average of about 14 years compared with the onset age. This phenomenon was more pronounced in the RDP family, where the diagnosis age and diagnosis delays were 39.1 ± 15.4 years and 16.6 years, respectively, much higher than those of sporadic RDP patients. Thus, patients can lose their opportunity for early treatment, and the disease burden on patients and their families would be increased. Several reasons may contribute to this finding: (1) Incomplete penetrance of ATP1A3 is very common, the symptoms of the parents of the proband may be milder, more atypical, and even asymptomatic, resulting in some patients being diagnosed at an older age; (2) In the early period when the disease of RDP was recognized for family patients, the genetic diagnosis technologies at that time were not advanced, which also might lead to misdiagnosis or diagnosis delay. (3) As a rare disease, the clinical manifestations of RDP are heterogeneous. Some neurologists may not have sufficient knowledge about this disease. (4) The most important reason may be that the previous clinical diagnosis criteria were too strict, such as seeking a clear rostrocaudal gradient (F>A>L) or features of Parkinsonism (Brashear et al., 2007). In our statistical analysis, rostrocaldal gradient accounted for only 45%, whereas the rate of parkinsonism was 88%. Therefore, based on the development of genetic diagnostic technologies, we suggest the following changes in the diagnosis criteria: (I) unexplained acute onset; (II) dystonia (especially with prominent bulbar findings); (III) with or without bradykinesia; (IV) with ATP1A3 variant. Some other symptoms such as clear trigger presence before onset, a clear rostrocaudal gradient, onset age of more than 18 months, the onset-to-stable time within 1 month, no or minimal responses to levodopa, and non-motor symptoms (psychiatric symptoms and cognitive dysfunction) support the diagnosis of RDP (Haq et al., 2019).
Genotype-phenotype correlation constitutes an important aspect of RDP pathogenesis. The predominant ATP1A3 mutation locations in RDP patients were in exons 8, 14, 17, and 18 (a rate of 87.5%), the corresponding functional areas were domain transmembrane regions (T3 and T5) and the cytoplasmic region between T4 and T5, which was different from the reported common mutation regions in exon 5, 7, 8, 9, 16, 20, 21, and 22 of ATP1A3 in AHC (Rosewich et al., 2014b). About 58.3% of the mutations in intermediate AHC/RDP mutational clusters are located in exons 9 and 17. The most common ATP1A3 mutants were T613M and I758S, which were found in 42.1% of all RDP patients. The majority of patients with T613M mutant came from Europe, and the founder effect may be a plausible explanation. Patients carrying the same ATP1A3 mutation tend to exhibit similar clinical phenotypes, including not only similar motor symptoms, but also non-motor symptoms, such as cognitive impairment, psychotic disorder, and verbal fluency impairment (Barbano et al., 2012; Oblak et al., 2014; Wilcox et al., 2015). We expect this correlation to become stronger as more ATP1A3-related RDP cases are reported. Meanwhile, the clinical heterogeneity of ATP1A3-related RDPS was significant, and the same mutation could be associated with different clinical phenotypes. The phenotypes of the patients with D923N mutant were presented as AHC, RDP, or intermediate AHC/RDP (Zanotti-Fregonara et al., 2008; Anselm et al., 2009; Roubergue et al., 2013), patients with R756H mutant could manifest as RDP or intermediate AHC/RDP phenotype (Brashear et al., 2012; Tan et al., 2015; Nicita et al., 2016). Additionally, ATP1A3 E277K and 1013Ydul mutants were reported to be associated with RDP or AHC phenotype (Blanco-Arias et al., 2009; Brashear et al., 2012; Termsarasab et al., 2015). The reason for this phenomenon may be that ATP1A3-related diseases may be a spectrum of diseases. In addition to mutations in ATP1A3, other factors such as environmental factors and the interaction with other genes may also contribute to the pathogenesis of RDP (Oblak et al., 2014).
Currently, extensive research has been conducted on ATP1A3 mutations that may be a pathogenic cause for RDP or intermediate AHC/RDP. The reported variants included ATP1A3 T613M, 1013Ydup, D293N, G867D, and D801N (Table 4). The mutations of ATP1A3 don't result in reduced expression of the ATP1A3 mRNA and protein, mainly causing decreased activity of Na+-K+ ATPase, lowering its affinity to cytoplasmic Na+ (Rodacker et al., 2006; Blanco-Arias et al., 2009; Sweadner et al., 2016). Similar findings are shown for the effects of ATP1A3 mutations in AHC (Heinzen et al., 2012). Based on this evidence, improving ATPase activity and affinity for Na+ may be an important direction for the treatment of ATP1A3-related disease.
Until now, ATP1A3 is the only discovered pathogenic gene for RDP, AHC, intermediate AHC/RDP and CAPOS (Brashear et al., 2007; Heinzen et al., 2012). If a patient has suspicious clinical manifestations, timely ATP1A3 screening is conducive to the early diagnosis of ATP1A3-related diseases. In addition, the identification of gene mutation is also the basis for genetic counseling and future gene modification therapy. In addition to genetic screening, some other auxiliary examinations may also facilitate the early and effective diagnosis of RDP. Downregulated expression of homovanillic acid (HAV) in CSF was found in 37.5% (3/8) of reported RDP patients. In addition, some studies have established that RDP patients also show specific PET and SPECT molecular imaging findings (Table 3). Therefore, when the clinical symptoms of patients are atypical or genetic screening cannot be performed, the HAV test of CSF and molecular imaging can provide more evidence for RDP diagnosis.
There is a lack of effective treatments for RDP and intermediate RDP/AHC, and patients exhibit almost no responses to levodopa. However, other treatments may be minimally effective, such as trihexyphenidyl, lorazepam, baclofen, flunarizine, Botulinum toxin injection, and rehabilitation therapy (Supplementary table 1). Therefore, for patients diagnosed with RDP and intermediate AHC/RDP, the above treatments should be recommended. Nevertheless, in our study, ~47.2% of RDP and 75% of AHC/RDP patients achieved partial improvements, especially showing ameliorated gait impairment (Table 1). Deep brain stimulation (DBS) therapy is a promising but controversial treatment for RDP. In the past, two RDP patients were reported to undergo DBS surgery in GPi. The therapy was completely ineffective for one patient, while the other patient showed partial improvements in the motor symptoms (Deutschlander et al., 2005; Kamm et al., 2008). However, future advancements in surgical methods and changes in intervention targets may enable breakthroughs in the current treatment status in this field.
Based on our analysis, we make the following recommendations for the diagnosis and treatment of patients with RDP. For patients with acute and unexplained dystonia or bradykinesia, gene screening on ATP1A3 should be timely performed. When a diagnosis has been made, treatments that may be effective are to be attempted. We believe that the most potentially effective treatments in the future will be the following: (1) genetic modification therapy of ATP1A3; (2) drugs that improve the NA+-K+ ATPase activity; and (3) DBS surgery. The current understanding of RDP is still very limited. Further research on the clinical manifestations, the functions of the mutations in ATP1A3, and the discovery and development of effective treatments are necessary.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University, China. The patients/participants provided their written informed consent to participate in this study.
Author contributions
LY: design, analysis, and writing. GP: statistical analysis. YY: diagnosis of the cases. MT: collect case information. PL: literature query. XL: document analysis. JN: reference retrieval. CJ: drawing of the tables. ZF: drawing of the figures. WZ: literature screening. BL: editing of final version of the manuscript. QK: design and editing of final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Zhejiang Province (LY20H170003, LY), the National Natural Science Foundation (81471300, LY); the National Key R&D Program of China (2020YFB1313501, QK), the Natural Science Foundation of Zhejiang Province (LY21H090010, QK), the National Natural Science Foundation (82001287, WZ), and the National Natural Science Foundation of China (82101251, PL).
Acknowledgments
We thank our patients for participating in this study. We thank Medsci (https://www.medsci.cn) for the English language editing of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.933893/full#supplementary-material
References
Anselm, I. A., Sweadner, K. J., Gollamudi, S., Ozelius, L. J., and Darras, B. T. (2009). Rapid-onset dystonia-parkinsonism in a child with a novel atp1a3 gene mutation. Neurology 73, 400–401. doi: 10.1212/WNL.0b013e3181b04acd
Barbano, R. L., Hill, D. F., Snively, B. M., Light, L. S., Boggs, N., McCall, W. V., et al. (2012). New triggers and non-motor findings in a family with rapid-onset dystonia-parkinsonism. Parkinsonism Relat. Disord. 18, 737–741. doi: 10.1016/j.parkreldis.2012.03.020
Biela, M., Rydzanicz, M., Szymanska, K., Pieniawska-Smiech, K., Lewandowicz-Uszynska, A., Chruszcz, J., et al. (2021). Variants of ATP1A3 in residue 756 cause a separate phenotype of relapsing encephalopathy with cerebellar ataxia (RECA)-Report of two cases and literature review. Mol. Genet. Genomic Med. 9, e1722. doi: 10.1002/mgg3.1772
Blanco-Arias, P., Einholm, A. P., Mamsa, H., Concheiro, C., Gutierrez-de-Teran, H., Romero, J., et al. (2009). A C-terminal mutation of ATP1A3 underscores the crucial role of sodium affinity in the pathophysiology of rapid-onset dystonia-parkinsonism. Hum. Mol. Genet. 18, 2370–2377. doi: 10.1093/hmg/ddp170
Boonsimma, P., Michael Gasser, M., Netbaramee, W., Wechapinan, T., Srichomthong, C., Ittiwut, C., et al. (2020). Mutational and phenotypic expansion of ATP1A3-related disorders: report of nine cases. Gene 749, 144709. doi: 10.1016/j.gene.2020.144709
Brashear, A., Dobyns, W. B., de Carvalho Aguiar, P., Borg, M., Frijns, C. J., Gollamudi, S., et al. (2007). The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 130, 828–835. doi: 10.1093/brain/awl340
Brashear, A., Farlow, M. R., Butler, I. J., Kasarskis, E. J., and Dobyns, W. B. (1996). Variable phenotype of rapid-onset dystonia-parkinsonism. Mov. Disord. 11,151–156. doi: 10.1002/mds.870110206
Brashear, A., Mink, J. W., Hill, D. F., Boggs, N., McCall, W. V., Stacy, M. A., et al. (2012). ATP1A3 mutations in infants: a new rapid-onset dystonia-Parkinsonism phenotype characterized by motor delay and ataxia. Dev. Med. Child Neurol. 54, 1065–1067. doi: 10.1111/j.1469-8749.2012.04421.x
Brashear, A., Mulholland, G. K., Zheng, Q. H., Farlow, M. R., Siemers, E. R., and Hutchins, G. D. (1999). PET imaging of the pre-aynaptic dopamine uptake sites in rapid-onset dystonia-parkinsonism (RDP). Mov. Disord. 14, 132–137. doi: 10.1002/1531-8257(199901)14:1<132::AID-MDS1022>3.0.CO;2-J
Dard, R., Mignot, C., Durr, A., Lesca, G., Sanlaville, D., Roze, E., et al. (2015). Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Dev. Med. Child Neurol. 57, 1183–1186. doi: 10.1111/dmcn.12927
de Carvalho Aguiar, P., Sweadner, K. J., Penniston, J. T., Zaremba, J., Liu, L., Caton, M., et al. (2004). Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175. doi: 10.1016/j.neuron.2004.06.028
Deutschlander, A., Asmus, F., Gasser, T., Steude, U., and Botzel, K. (2005). Sporadic rapid-onset dystonia-parkinsonism syndrome: failure of bilateral pallidal stimulation. Mov. Disord. 20, 254–257. doi: 10.1002/mds.20296
Haq, I. U., Snively, B. M., Sweadner, K. J., Suerken, C. K., Cook, J. F., Ozelius, L. J., et al. (2019). Revising rapid-onset dystonia-parkinsonism: Broadening indications for ATP1A3 testing. Mov. Disord. 34, 1528–1536. doi: 10.1002/mds.27801
Heinzen, E. L., Arzimanoglou, A., Brashear, A., Clapcote, S. J., Gurrieri, F., Goldstein, D. B., et al. (2014). Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 13, 503–514. doi: 10.1016/S1474-4422(14)70011-0
Heinzen, E. L., Swoboda, K. J., Hitomi, Y., Gurrieri, F., Nicole, S., de Vries, B., et al. (2012). De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat. Genet. 44, 1030–1034. doi: 10.1038/ng.2358
Hoshino, K., Sweadner, K. J., Kawarai, T., Saute, J. A., Freitas, J., Damasio, J., et al. (2021). Rapid-onset dystonia-parkinsonism phenotype consistency for a novel variant of ATP1A3 in patients across 3 global populations. Neurol. Genet. 7, e562. doi: 10.1212/NXG.0000000000000562
Kamm, C., Fogel, W., Wächter, T., Schweitzer, K., Berg, D., Kruger, R., et al. (2008). Novel ATP1A3 mutation in a sporadic RDP patient with minimal benefit from deep brain stimulation. Neurology 70, 1501–1503. doi: 10.1212/01.wnl.0000310431.41036.e0
Kamphuis, D. J., Koelman, H., Lees, A. J., and Tijssen, M. A. (2006). Sporadic rapid-onset dystonia-parkinsonism presenting as Parkinson's disease. Mov. Disord. 21, 118–119. doi: 10.1002/mds.20695
Lee, J. Y., Gollamudi, S., Ozelius, L. J., Kim, J. Y., and Jeon, B. S. (2007). ATP1A3 mutation in the first asian case of rapid-onset dystonia-parkinsonism. Mov. Disord. 22, 1808–1809. doi: 10.1002/mds.21638
Liu, Y., Lu, Y., Zhang, X., Xie, S., Wang, T., Wu, T., et al. (2016). A case of rapid-onset dystonia-parkinsonism accompanied by pyramidal tract impairment. BMC Neurol. 16, 218. doi: 10.1186/s12883-016-0743-8
Marrodan, M., Rossi, M., and Merello, M. (2018). Rapid-onset dystonia-parkinsonism preceded by a single episode of subacute persisting hemiparesis: expanding the ATP1A3-related disorders phenotype. J. Neurol. Sci. 392, 44–45. doi: 10.1016/j.jns.2018.07.002
Nicita, F., Travaglini, L., Sabatini, S., Garavaglia, B., Panteghini, C., Valeriani, M., et al. (2016). Childhood-onset ATP1A3-related conditions: report of two new cases of phenotypic spectrum. Parkinsonism Relat. Disord. 30, 81–82. doi: 10.1016/j.parkreldis.2016.05.029
Nomura, S., Kashiwagi, M., Tanabe, T., Oba, C., Yanagi, K., Kaname, T., et al. (2021). Rapid-onset dystonia-parkinsonism with ATP1A3 mutation and left lower limb paroxysmal dystonia. Brain Dev. 43, 566–570. doi: 10.1016/j.braindev.2020.12.009
Oblak, A. L., Hagen, M. C., Sweadner, K. J., Haq, I., Whitlow, C. T., Maldjian, J. A., et al. (2014). Rapid-onset dystonia-parkinsonism associated with the I758S mutation of the ATP1A3 gene: a neuropathologic and neuroanatomical study of four siblings. Acta Neuropathol. 128, 81–98. doi: 10.1007/s00401-014-1279-x
Paciorkowski, A. R., McDaniel, S. S., Jansen, L. A., Tully, H., Tuttle, E., Ghoneim, D. H., et al. (2015). Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 56, 422–430. doi: 10.1111/epi.12914
Pereira, P., Guerreiro, A., Fonseca, M., Halpern, C., Pinto-Basto, J., and Monteiro, J. P. (2016). A distinct phenotype in a novel ATP1A3 mutation: connecting the two ends of a spectrum. Mov. Disord. Clin. Pract. 3, 398–401. doi: 10.1002/mdc3.12263
Pittock, S. J., Joyce, C., O'Keane, V., Hugle, B., Hardiman, M. O., Brett, F., et al. (2000). Rapid-onset dystonia-parkinsonism: a clinical and genetic analysis of a new kindred. Neurology 55, 991–995. doi: 10.1212/WNL.55.7.991
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rodacker, V., Toustrup-Jensen, M., and Vilsen, B. (2006). Mutations Phe785Leu and Thr618Met in Na+,K+-ATPase, associated with familial rapid-onset dystonia parkinsonism, interfere with Na+ interaction by distinct mechanisms. J. Biol. Chem. 281, 18539–18548. doi: 10.1074/jbc.M601780200
Rosewich, H., Baethmann, M., Ohlenbusch, A., Gartner, J., and Brockmann, K. (2014a). A novel ATP1A3 mutation with unique clinical presentation. J. Neurol. Sci. 341, 133–135. doi: 10.1016/j.jns.2014.03.034
Rosewich, H., Ohlenbusch, A., Huppke, P., Schlotawa, L., Baethmann, M., Carrilho, I., et al. (2014b). The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology 82, 945–955. doi: 10.1212/WNL.0000000000000212
Rosewich, H., Sweney, M. T., DeBrosse, S., Ess, K., Ozelius, L., Andermann, E., et al. (2017). Research conference summary from the 2014 international task force on ATP1A3-related disorders. Neurol. Genet. 3, e139. doi: 10.1212/NXG.0000000000000139
Rosewich, H., Thiele, H., Ohlenbusch, A., Maschke, U., Altmüller, J., Frommolt, P., et al. (2012). Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol. 11, 764–773. doi: 10.1016/S1474-4422(12)70182-5
Roubergue, A., Roze, E., Vuillaumier-Barrot, S., Fontenille, M. J., Meneret, A., Vidailhet, M., et al. (2013). The multiple faces of the ATP1A3-related dystonic movement disorder. Mov. Disord. 28, 1457–1459. doi: 10.1002/mds.25396
Sampson, J. B., Michaeli, T. H., Wright, B. A., Goldman, J. E., Vonsattel, J. P., and Fahn, S. (2016). Basal ganglia gliosis in a case of rapid-onset dystonia-parkinsonism (DYT12) with a novel mutation in ATPase 1A3 (ATP1A3). Mov. Disord. Clin. Pract. 3, 618–620. doi: 10.1002/mdc3.12354
Sasaki, M., Ishii, A., Saito, Y., and Hirose, S. (2014). Intermediate form between alternating hemiplegia of childhood and rapid-onset dystonia-parkinsonism. Mov. Disord. 29, 153–154. doi: 10.1002/mds.25659
Schirinzi, T., Graziola, F., Nicita, F., Travaglini, L., Stregapede, F., Valeriani, M., et al. (2018). Childhood rapid-onset ataxia: expanding the phenotypic spectrum of ATP1A3 mutations. Cerebellum 17:489–493. doi: 10.1007/s12311-018-0920-y
Sousa, A. L., Alonso, I., and Magalhaes, M. (2017). A Portuguese rapid-onset dystonia-parkinsonism case with atypical features. Neurol. Sci. 38, 1713–1714. doi: 10.1007/s10072-017-2996-4
Svetel, M., Ozelius, L. J., Buckley, A., Lohmann, K., Brajkovic, L., Klein, C., et al. (2010). Rapid-onset dystonia-parkinsonism: case report. J. Neurol. 257, 472–474. doi: 10.1007/s00415-009-5385-y
Sweadner, K. J., Toro, C., Whitlow, C. T., Snively, B. M., Cook, J. F., Ozelius, L. J., et al. (2016). ATP1A3 mutation in adult rapid-onset ataxia. PLoS ONE 11, e0151429. doi: 10.1371/journal.pone.0151429
Tan, A. H., Ozelius, L. J., Brashear, A., Lang, A. E., Ahmad-Annuar, A., Tan, C. T., et al. (2015). Rapid-onset dystonia-parkinsonism in a chinese girl with a de novo ATP1A3 c.2267G>A (p.R756H) genetic mutation. Mov. Disord. Clin. Pract. 2, 74–75. doi: 10.1002/mdc3.12122
Tarsy, D., Sweadner, K. J., and Song, P. C. (2010). Case records of the Massachusetts General Hospital. Case 17-2010 - a 29-year-old woman with flexion of the left hand and foot and difficulty speaking. N. Engl. J. Med. 362, 2213–2219. doi: 10.1056/NEJMcpc1002112
Termsarasab, P., Yang, A. C., and Frucht, S. J. (2015). Intermediate phenotypes of ATP1A3 mutations: phenotype-genotype correlations. Tremor. Other Hyperkinet. Mov. 5, 336. doi: 10.5334/tohm.255
Wenzel, G. R., Lohmann, K., and Kuhn, A. A. (2017). A novel de-novo mutation in the ATP1A3 gene causing rapid-onset dystonia parkinsonism. Parkinsonism Relat. Disord. 37, 120–122. doi: 10.1016/j.parkreldis.2017.02.009
Wilcox, R., Braenne, I., Bruggemann, N., Winkler, S., Wiegers, K., Bertram, L., et al. (2015). Genome sequencing identifies a novel mutation in ATP1A3 in a family with dystonia in females only. J. Neurol. 262, 187–193. doi: 10.1007/s00415-014-7547-9
Wunderlich, S., Csoti, I., Reiners, K., Gunthner-Lengsfeld, T., Schneider, C., Becker, G., et al. (2002). Camptocormia in Parkinson's disease mimicked by focal myositis of the paraspinal muscles. Mov. Disord. 17, 598–600. doi: 10.1002/mds.10110
Yu, L. H., Peng, G. P., Yuan, Y., Liu, X. Y., Ji, F., Li, Y., et al. (2021). Novel compound heterozygous of PARKIN causes early-onset Parkinson's disease. Neurosci. Lett. 744, 135597. doi: 10.1016/j.neulet.2020.135597
Yuan, Y., Ran, L., Lei, L., Zhu, H., Zhu, X., and Chen, H. (2020). The expanding phenotypic spectrums associated with ATP1A3 mutation in a family with rapid-onset dystonia Parkinsonism. Neurodegener. Dis. 20, 84–89. doi: 10.1159/000511733
Zanotti-Fregonara, P., Vidailhet, M., Kas, A., Ozelius, L. J., Clot, F., Hindie, E., et al. (2008). [123I]-FP-CIT and [99mTc]-HMPAO single photon emission computed tomography in a new sporadic case of rapid-onset dystonia-parkinsonism. J. Neurol. Sci. 273, 148–151. doi: 10.1016/j.jns.2008.06.033
Keywords: ATP1A3, gene mutation, rapid-onset dystonia parkinsonism (RDP), genotype-phenotype correlation analysis, early diagnosis of RDP
Citation: Yu L, Peng G, Yuan Y, Tang M, Liu P, Liu X, Ni J, Li Y, Ji C, Fan Z, Zhu W, Luo B and Ke Q (2022) ATP1A3 mutation in rapid-onset dystonia parkinsonism: New data and genotype-phenotype correlation analysis. Front. Aging Neurosci. 14:933893. doi: 10.3389/fnagi.2022.933893
Received: 01 May 2022; Accepted: 04 July 2022;
Published: 01 August 2022.
Edited by:
Azlina Ahmad-Annuar, University of Malaya, MalaysiaReviewed by:
Federica Graziola, IRCCS Carlo Besta Neurological Institute Foundation, ItalyAlessandro Capuano, Azienda Sanitaria Locale di Viterbo, Italy
Naoko Ishihara, Fujita Health University, Japan
Copyright © 2022 Yu, Peng, Yuan, Tang, Liu, Liu, Ni, Li, Ji, Fan, Zhu, Luo and Ke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Ke, S2VxaW5nMjAwM0B6anUuZWR1LmNu; Benyan Luo, bHVvYmVueWFuQHpqdS5lZHUuY24=
†These authors share first authorship