
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci., 24 May 2022
Sec. Alzheimer's Disease and Related Dementias
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.890855
This article is part of the Research TopicMolecular and Cellular Mechanisms of Synaptopathies: Emerging Synaptic aging-related molecular pathways in Neurological DisordersView all 11 articles
 Francesco Bellanti1†
Francesco Bellanti1† Vidyasagar Naik Bukke1†
Vidyasagar Naik Bukke1† Archana Moola1
Archana Moola1 Rosanna Villani1
Rosanna Villani1 Caterina Scuderi2
Caterina Scuderi2 Luca Steardo2Gianmauro Palombelli3
Luca Steardo2Gianmauro Palombelli3 Rossella Canese4
Rossella Canese4 Sarah Beggiato4Mario Altamura5Gianluigi Vendemiale1Gaetano Serviddio1
Sarah Beggiato4Mario Altamura5Gianluigi Vendemiale1Gaetano Serviddio1 Tommaso Cassano1*
Tommaso Cassano1*The therapeutic potential of ultramicronized palmitoylethanolamide (um-PEA) was investigated in young (6-month-old) and adult (12-month-old) 3×Tg-AD mice, which received um-PEA for 3 months via a subcutaneous delivery system. Mitochondrial bioenergetics, ATP homeostasis, and magnetic resonance imaging/magnetic resonance spectroscopy were evaluated in the frontal cortex (FC) and hippocampus (HIPP) at the end of um-PEA treatment. Glutamate release was investigated by in vivo microdialysis in the ventral HIPP (vHIPP). We demonstrated that chronic um-PEA treatment ameliorates the decrease in the complex-I respiration rate and the FoF1-ATPase (complex V) activity, as well as ATP content depletion in the cortical mitochondria. Otherwise, the impairment in mitochondrial bioenergetics and the release of glutamate after depolarization was not ameliorated by um-PEA treatment in the HIPP of both young and adult 3×Tg-AD mice. Moreover, progressive age- and pathology-related changes were observed in the cortical and hippocampal metabolism that closely mimic the alterations observed in the human AD brain; these metabolic alterations were not affected by chronic um-PEA treatment. These findings confirm that the HIPP is the most affected area by AD-like pathology and demonstrate that um-PEA counteracts mitochondrial dysfunctions and helps rescue brain energy metabolism in the FC, but not in the HIPP.
Alzheimer's disease (AD) is a consequence of several detrimental processes, such as protein aggregation, oxidative stress, mitochondrial malfunction, and neuroinflammation, finally resulting in the loss of neuronal functions (Blass and Gibson, 1991; Sullivan and Brown, 2005; Yao et al., 2009; Querfurth and Laferla, 2010; Serviddio et al., 2011; Cassano et al., 2016, 2019; Carapelle et al., 2020). Starting from this evidence, multitarget drugs have been increasingly sought after over the last decades (Barone et al., 2019; Cassano et al., 2019). In this regard, palmitoylethanolamide (PEA) seems to exert neuroprotective effects by modulating more therapeutic targets at the same time (Valenza et al., 2021). In fact, it has been demonstrated that PEA, the naturally occurring amide of ethanolamine and palmitic acid, is an endogenous lipid that exerts its beneficial effects through the peroxisome proliferator-activated receptors (PPARs) involvement, transient receptor potential vanilloid type 1 channel, orphan G-protein-coupled receptor 55, and the so-called entourage effect on the endocannabinoid system (Devchand et al., 1996; Delerive et al., 2001; LoVerme et al., 2005, 2006; Scuderi et al., 2011; Tomasini et al., 2015; Bronzuoli et al., 2019; Beggiato et al., 2020a,b). In addition to its known anti-inflammatory activity, PEA protects cultured mouse cerebellar granule cells from glutamate toxicity and reduces histamine-induced cell death in hippocampal cultures (Skaper et al., 1996).
Furthermore, it has been demonstrated that PEA exerts in vitro and in vivo a combination of neuroprotective and anti-inflammatory effects in amyloid-β (Aβ)-induced toxicity by interacting at the PPAR-α nuclear site (D'Agostino et al., 2012; Scuderi et al., 2012, 2014). The neuroprotective action of PEA has been established also in transgenic animal models of AD (D'Agostino et al., 2012; Bronzuoli et al., 2018; Scuderi et al., 2018). In fact, we have previously demonstrated that ultramicronized PEA (um-PEA), a formulation endowed with better bioavailability, can rescue learning and memory impairments in a triple transgenic mouse model of AD (3×Tg-AD) by exerting anti-inflammatory properties, dampening reactive astrogliosis, and promoting the glial neurosupportive function. Moreover, um-PEA strongly suppresses Aβ(1–42) expression and reduces the abnormal phosphorylation of tau in 3×Tg-AD mice (Scuderi et al., 2018).
Mitochondrial dysfunctions are related to inflammation and other energy-dependent disturbances, where the generation of reactive oxygen species (ROS) exceeds the physiological antioxidant activity, causing cellular oxidative damage (Lin and Beal, 2006; Reddy, 2009; Romano et al., 2017). Mitochondrial dysfunction can lead to glutamatergic neurotransmission alterations (Vos et al., 2010) and excitotoxicity (Schinder et al., 1996). In fact, excessive glutamate concentration can cause excitotoxicity, leading to calcium influx, mitochondrial dysfunction, and subsequent cell death (Mahmoud et al., 2019). Therefore, considering the tight link between mitochondria and glutamate, it could be crucial to investigate their alterations in the context of AD pathology.
In the last decade, neuroimaging has been used to complement clinical assessments in the early detection of AD, showing that morphological change (such as measurement of the hippocampal volume) might be an important hallmark for the diagnosis of AD. More recently, the development of magnetic resonance spectroscopy (MRS) in the field of AD revealed that tremendous metabolic changes occur during the progression of AD correlating with cognitive abnormalities (Meyerhoff et al., 1994; Shonk et al., 1995; Frederick et al., 1997; Rose et al., 1999; Kantarci et al., 2000; Huang et al., 2001). Nevertheless, few studies with relatively small sample sizes investigated MR spectral profile alteration as a biomarker for treatment response in AD, and some of them correlated this alteration with psychiatric symptoms in patients with AD (Sweet et al., 2002; Bartha et al., 2008). Although metabolite abnormalities in AD were demonstrated in different samples and pathologically confirmed cases, the pathological significance of these changes is not fully understood. Therefore, in our study, we exploited the availability of the MRS to investigate whether it can provide complementary predictive information regarding (i) progression of the AD-like pathology and (ii) um-PEA treatment effects.
In this article, we used the 3×Tg-AD mice harboring three mutant human genes (betaAPPSwe, PS1M146V, and tauP301L) to directly test the hypothesis that (i) progressive accumulation of Aβ is closely related to mitochondrial, metabolic, and glutamatergic alterations, and (ii) chronic um-PEA administration may ameliorate such alterations. To test these hypotheses, we used an integrated approach, based on ex vivo studies of several mitochondrial bioenergetic parameters, in vivo studies of magnetic resonance imaging (MRI) and spectroscopy (MRS) to evaluate brain metabolites, as well as neurochemical analysis of extracellular levels of glutamate by microdialysis in 3×Tg-AD vs. non-Tg control mice tested at two different stages (mild and severe) of AD-like pathology and subcutaneously treated for 3 months with um-PEA.
A total of 3×Tg-AD male mice and their sex- and age-matched wild-type littermates (non-Tg) (C57BL6/129SvJ) were maintained in controlled conditions (12 h light/12 h dark cycle, temperature 22°C, humidity 50–60%, fresh food, and water ad libitum). All procedures were conducted in accordance with the guidelines of the Italian Ministry of Health (D.L. 26/2014) and the European Parliamentary directive 2010/63/EU. All efforts were made to minimize the number of animals used and their suffering. In addition, 3- and 9-month-old mice were subcutaneously implanted with a 90-day-release pellet containing either 28 mg of um-PEA (Innovative Research of America, Sarasota, Florida; cat# NX-999) or vehicle (cat# NC-111), as previously described (Scuderi et al., 2018). Briefly, an um-PEA or a vehicle pellet was surgically positioned in a subcutaneous pocket created with a blunt probe between the shoulder blades. um-PEA (EPT2110/1) was obtained from the Epitech group (Saccolongo, Italy), and both dosage and administration routes were chosen according to previous data (Costa et al., 2002; Grillo et al., 2013; Scuderi et al., 2018). Both non-Tg and 3×Tg-AD mice were randomly assigned to either vehicle or um-PEA group. At the end of the 90-day treatment, mitochondrial bioenergetics, MRI/MRS experiments, and microdialysis/high-performance liquid chromatography (HPLC) analysis were performed as previously described (Cassano et al., 2012; Scuderi et al., 2018).
On the day of the experiment, 6- and 12-month-old mice were decapitated and brains were rapidly removed and placed on a cold surface to dissect the hippocampus (HIPP), frontal cortex (FC), and cerebellum (Cassano et al., 2012). Experiments were conducted on four samples per group per each brain area. To obtain an optimal sample amount, tissues from three animals were pooled (a total of 12 mice per group). Mitochondria were freshly isolated by using a gradient of Percoll, as previously described (Cassano et al., 2012). Briefly, brain areas were weighted (100 mg) and immersed in ice-cold mitochondrial isolation buffer (MIB) containing 0.25 M sucrose, 0.5 mM K-EDTA, and 10 mM Tris–HCl (pH 7.4). Each sample was homogenized in 3.8 ml of 12% Percoll in MIB (5% w/v) using Dounce homogenizers with glass pestles. In addition, 3 ml of homogenate was then layered onto a previously poured, 3.5 ml 26% Percoll, on 3.5 ml 40% Percoll density gradient. The gradient was centrifuged at 19,000 rpm (30,000 × g) in a Sorval RC-5B type rotor for 5 min. The resulting top layer containing myelin and other cellular debris was carefully removed using a Pasteur pipette and discarded. Fraction 2 containing the mitochondria was removed by pipetting with a 200 ml gel loading tip and diluted 1:4 in cold MIB and centrifuged at 14,000 (15,000 × g) rpm for 10 min. The resulting pellet was then resuspended in 1 ml of MIB and centrifuged at 14,000 rpm (15,000 × g) for 5 min. The final pellet was resuspended in 100 ml of MIB containing 10% 10 mg/ml bovine serum albumin. After isolation, mitochondria were assayed for oxygen consumption at 37°C in a thermostatically controlled oxygraph apparatus equipped with Clark's electrode and a rapid mixing device (Hansatech Instruments, Ltd., Norfolk, England, UK). Mitochondrial respiration was triggered by glutamate and malate as complex I-linked substrates, or succinate (in the presence of rotenone as complex I inhibitor) as complex II-linked substrate. Oxygen uptake in states 3 and 4 and respiratory control index (RCI) was calculated as previously reported (Cassano et al., 2012). Respiratory activity was calculated as oxygen nmol per minute per mg of protein.
The FOF1-ATPase activity was measured following ATP hydrolysis with an ATP-regenerating system coupled to nicotinamide adenine dinucleotide phosphate (NADPH) oxidation, as previously reported (Cassano et al., 2012).
Measurement of ATP concentration in the total homogenates and mitochondrial fractions from brain areas was performed by using a commercial bioluminescent assay kit (Sigma-Aldrich, St. Louis, MO, USA).
The 6- and 12-month-old mice (n = 5–7) underwent MRI/MRS scanning to evaluate genotype- and treatment-induced differences in brain metabolites of FC and HIPP. MRI/MRS analyses were conducted at 4.7 T on a Varian/Agilent Inova horizontal bore system (Agilent, Palo Alto, USA) using a combination of volume and surface coil (RAPID Biomedical, Rimpar, Germany) according to a protocol described in Scuderi et al. (2018). Briefly, mice were anesthetized with isoflurane (IsoFlo, Abbott SpA, Berkshire, UK) 1.5–2.5% in O2 1 L/min. Anatomical T2-weighted sagittal MRIs were acquired for the positioning of the voxels for MRS. Localized 1H-MRS (PRESS TR/TE = 4,000/23 ms) were collected from HIPP and FC (volume 9.5 and 9.1 μl, respectively) as shown in Figure 4A, according to a quantitative protocol (Canese et al., 2012).
The following six metabolites were considered: N-acetyl-aspartate (NAA), myo-inositol (mINS), the sum of creatine and phosphocreatine (Cr + PCr), glutamate (Glu), glutamine (Gln), and total choline (tCho). Metabolite concentrations are expressed in mmol/L (mM). Moreover, we evaluated the NAA/Cr ratio and mINS/Cr.
In vivo microdialysis was performed in awake, freely moving mice, as previously described (Cassano et al., 2012; Romano et al., 2014). Briefly, 6- and 12-month-old anesthetized mice (n = 5–6) were stereotaxically implanted with a CMA/7 guide cannula with a stylet (CMA Microdialysis, Stockholm, Sweden) into the vHIPP (anterior-posterior, −3.0 mm; lateral, +3.0 mm; ventral, −1.8 mm from bregma, Figure 5A), according to the stereotaxis atlas of Franklin and Paxinos (Franklin and Paxinos, 1997). Following a 2-day recovery period, the CMA/7 probe (6-kDa cutoff; 2 mm membrane length) was inserted and dialyses were carried out, in awake, freely moving mice, perfusing each probe with Krebs-Ringer phosphate (KRP) buffer at a flow rate of 1 μl/min. The constituents of the KRP buffer were (in mM) NaCl 145, KCl 2.7, MgCl2 1, CaCl2 2.4, and Na2HPO4 2, buffered at pH 7.4. After a 2-h stabilization period, four baseline samples were collected every 20 min. Thereafter, the probes were perfused for 20 min with KCl (50 mM)-enriched KRP buffer, and then six samples were further collected with the previous KRP buffer. After completion of the microdialysis, the probe position was verified histologically, glutamate was quantified by HPLC coupled to fluorescence detection as previously described (Tomasini et al., 2002; Beggiato et al., 2020a,b).
The sample size was determined based on our previous experiments and using the free software G*Power version 3.1.9.2. All data are expressed as mean ± standard error of the mean (SEM). Within-group variability was analyzed through the Levene test for homogeneity of variances.
Data from mitochondrial bioenergetics and MRI/MRS scanning were analyzed using a 2-way analysis of variance (ANOVA) with genotype and treatment as the between variables. Dunnett's and Tukey's post-hoc tests were used where appropriate to perform multiple comparisons.
Regarding the microdialysis data, the overall basal glutamate levels were calculated as marginal means of the first five dialysate samples (from time −80 up to time 0) and were analyzed by two-way ANOVA with genotype and treatment as the between variables. The neurotransmitter release in response to depolarization (K+-stimulation) (from time 0 up to time 140) was analyzed using three-way ANOVA for repeated measures with genotype (3×Tg-AD vs. non-Tg) and treatment (um-PEA vs. vehicle) as the between variable and time as the within the variable. Dunnett's and Tukey's post-hoc tests were used where appropriate to perform multiple comparisons.
The threshold for statistical significance was set at p < 0.05. The SPSS Statistics version 19 (IBM, Armonk, NY, USA) and GraphPad Prism 6 for Windows (GraphPad Software Inc., San Diego, CA, USA) were used to perform all the statistical analyses and represent graph data, respectively.
Oxygen consumption was detected in mitochondria freshly isolated from FC and HIPP of 6- and 12-month-old non-Tg and 3×Tg-AD mice chronically treated with either vehicle or um-PEA. Statistical analyses are shown in Tables 1, 2.
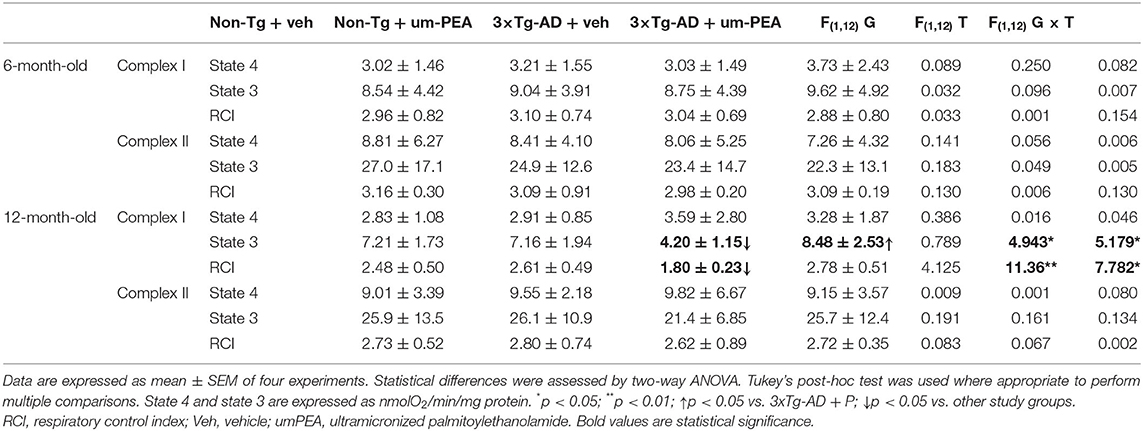
Table 1. Statistical analyses of respiratory activity in mitochondria isolated from the frontal cortex.
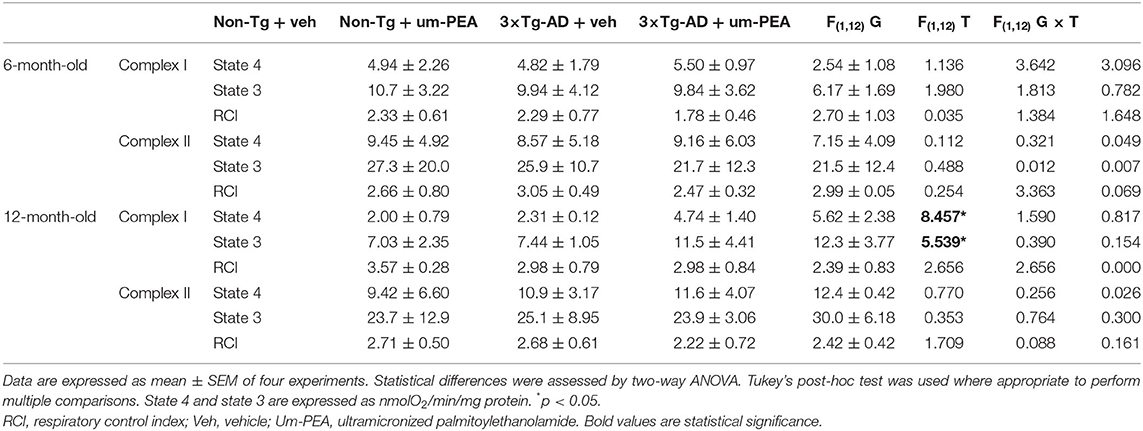
Table 2. Statistical analyses of respiratory activity in mitochondria isolated from the hippocampus.
In 6-month-old mice, no significant difference was observed among experimental groups within the different brain regions (Figures 1A,C). Otherwise, when the NADH-generating substrates glutamate and malate were added to the mitochondrial preparations, a lower respiration activity in state 3 was detected in the FC of vehicle-treated 12-month-old 3×Tg-AD compared to non-Tg mice (Figure 1B), whereas a significant increase in both state 4 and state 3 was detected in the HIPP compared to non-Tg mice (Figure 1D). The ratio between state 3 and state 4, named RCI, was decreased in the FC of vehicle-treated 3×Tg-AD compared to non-Tg mice (Figure 1B), whereas it was unaffected in the HIPP of both genotypes, regardless of treatment (Figure 1D). um-PEA treatment significantly increased the state 3 respiration activity and the RCI in the FC of 3×Tg-AD mice compared to the respective vehicle (Figure 1B).
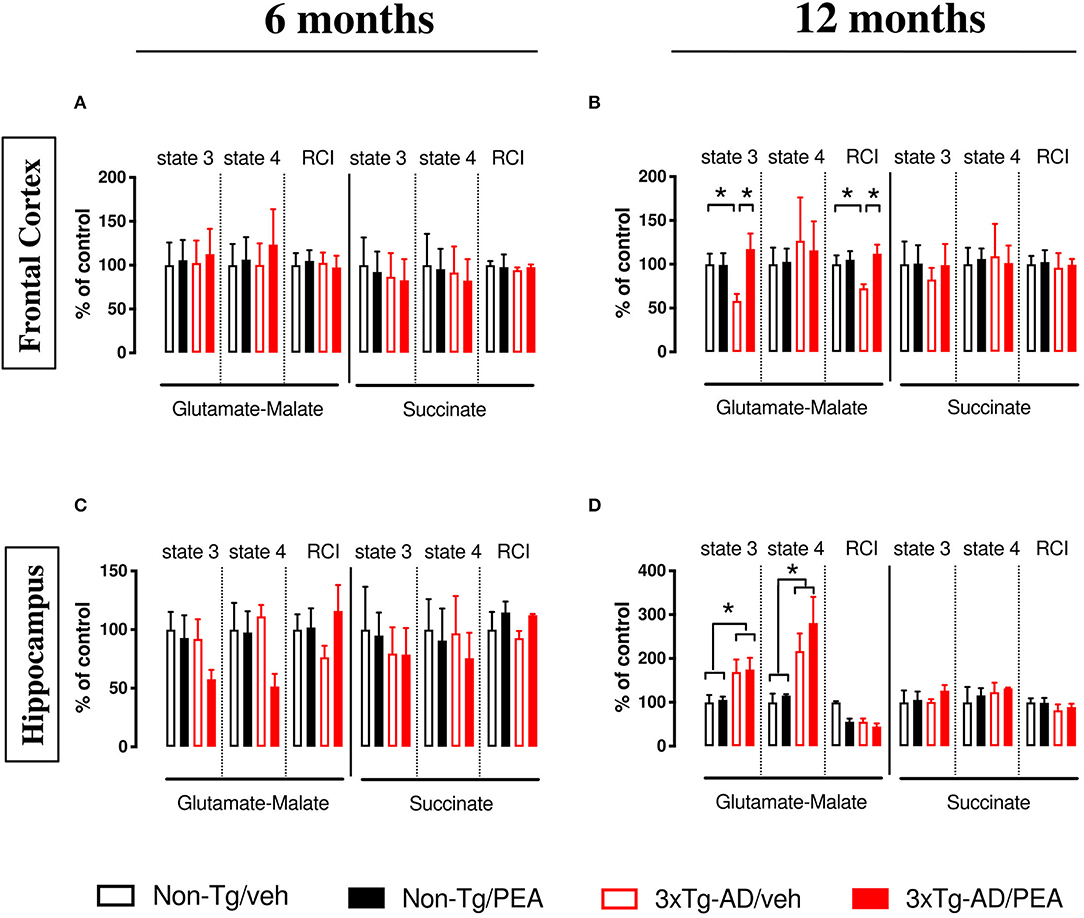
Figure 1. State 4 and state 3 mitochondrial respiratory activity and respiratory control index (RCI) measured in the frontal cortex (A,B), and hippocampus (C,D) of non-Tg (black bars) and 3×Tg-AD (red bars) 6-month-old (A,C) and 12-month-old (B,D) mice treated with vehicle (veh) (open bars) or um-PEA (filled bars), using complex I (glutamate-malate) and complex II (succinate) oxidative substrates. Data are expressed as a percentage of control ± SEM (n = 4). Statistical differences were assessed by two-way ANOVA and Tukey's post-hoc multiple comparisons test. *p < 0.05.
The analysis of complex II respiration, using succinate as a substrate in the presence of rotenone, revealed that cortical (Figure 1B) and hippocampal (Figure 1D) mitochondria were unaffected by both genotype and treatment.
Finally, neither genotype nor treatment affected the respiration activity of the cerebellum when its mitochondria were incubated with either the complex I-linked substrates or complex II-linked substrates (data not shown).
To investigate the effects of chronic um-PEA treatment on ATP homeostasis, FoF1-ATPase (complex V) activity, ADP/O ratio (which expresses the coupling between the phosphorylation activity and mitochondrial respiration), and ATP content were measured in both non-Tg and 3×Tg-AD mice at two progressive stages of the disease (6 and 12 months of age).
Both the FoF1-ATPase activity and the ADP/O ratio were unaffected in mitochondria from FC (Figures 2A,E) and HIPP (Figures 2C,G) of both non-Tg and 3×Tg-AD mice at 6 months of age. Different results were obtained for FoF1-ATPase activity and ADP/O ratio in the presence of glutamate-malate at 12 months of age, where a significant effect of genotype (F(1, 12) = 15.59, p < 0.01; F(1, 12) = 7.98, p < 0.05, respectively) and treatment (F(1, 12) = 5.728, p < 0.05; F(1, 12) = 4.956, p < 0.05, respectively) was observed in the FC (Figures 2B,F). Multiple post-hoc comparison showed a significantly lower FoF1-ATPase activity and ADP/O ratio for vehicle-treated 3×Tg-AD mice compared to non-Tg mice that was counteracted by um-PEA treatment (Figures 2B,F). Regarding the HIPP, statistical analysis revealed only a significant main effect of genotype (F(1, 12) = 6.001, p < 0.05; F(1, 12) = 4.914, p < 0.05, respectively) (Figures 2D,H).
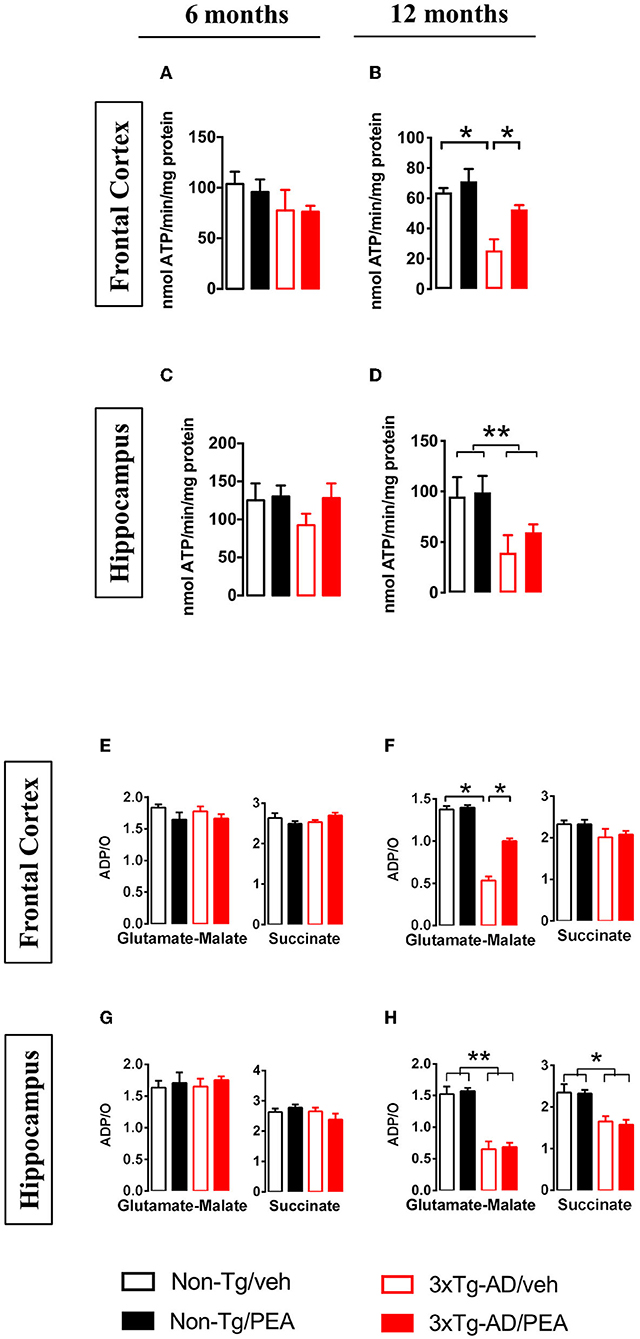
Figure 2. FoF1-ATPase activity (A–D) and ADP/O ratio (E–H) measured in mitochondria isolated from the frontal cortex (A,B,E,F), and hippocampus (C,D,G,H) of non-Tg (black bars) and 3×Tg-AD (red bars) 6-month-old (A,C,E,G) and 12-month-old (B,D,F,H) mice treated with vehicle (open bars) or um-PEA (filled bars). Data are expressed as mean ± SEM (n = 4). Statistical differences were assessed by two-way ANOVA and Tukey's post-hoc test. *p < 0.05; **p < 0.01.
To further evaluate the effects of chronic treatment on ATP homeostasis, we measured ATP levels in tissue homogenates and mitochondrial fractions from both brain regions. Statistical analysis showed a significant main effect of genotype [6 months: (F(1, 12) = 9.738, p < 0.01); 12 months: (F(1, 12) = 34.61, p < 0.001)] and treatment [6 months: (F(1, 12) = 5.372, p < 0.05); 12 months: (F(1, 12) = 6.152, p < 0.001] in the FC at both ages (Figures 3A,B,E,F). In particular, multiple post-hoc comparison showed a significant lower ATP content for vehicle-treated 3×Tg-AD mice compared to non-Tg mice that was counteracted by um-PEA treatment (Figures 3A,B,E,F).
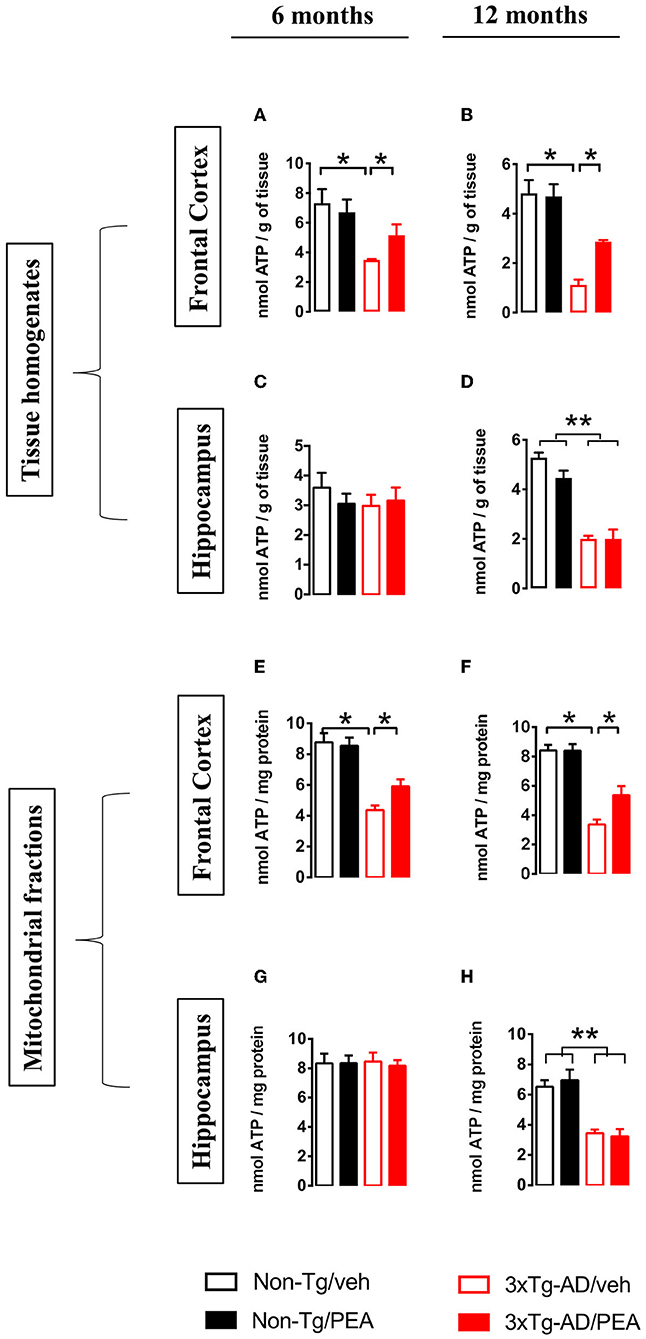
Figure 3. Total homogenate ATP content measured in tissue homogenates and mitochondria isolated from frontal cortex (A,B,E,F) and hippocampus (C,D,G,H) of non-Tg (black bars) and 3×Tg-AD (red bars) 6-month-old (A,C,E,G) and 12-month-old (B,D,F,H) mice treated with vehicle (open bars) or um-PEA (filled bars). Data are expressed as mean ± SEM (n = 4). Statistical differences were assessed by two-way ANOVA and Tukey's post-hoc test. *p < 0.05; **p < 0.01.
Regarding the ATP content in the HIPP, no difference was observed at 6 months of age, whereas a significant main effect of genotype (F(1, 12) = 33.11, p < 0.001) was observed at 12 months of age (Figures 3C,D,G,H).
Magnetic resonance imaging/spectroscopy was used to assess metabolic profiles of treated vs. untreated mice at 6 and 12 months of age. The quantitative results of all metabolite concentrations from FC and HIPP are reported in Figure 4. In the FC, two-way ANOVA analysis revealed no significant differences among groups in the NAA levels at both ages, whereas in the HIPP a main effect of genotype was observed at 6 (F(1, 19) = 10.098, p < 0.01) and 12 months (F(1, 22) = 5.820, p < 0.05).
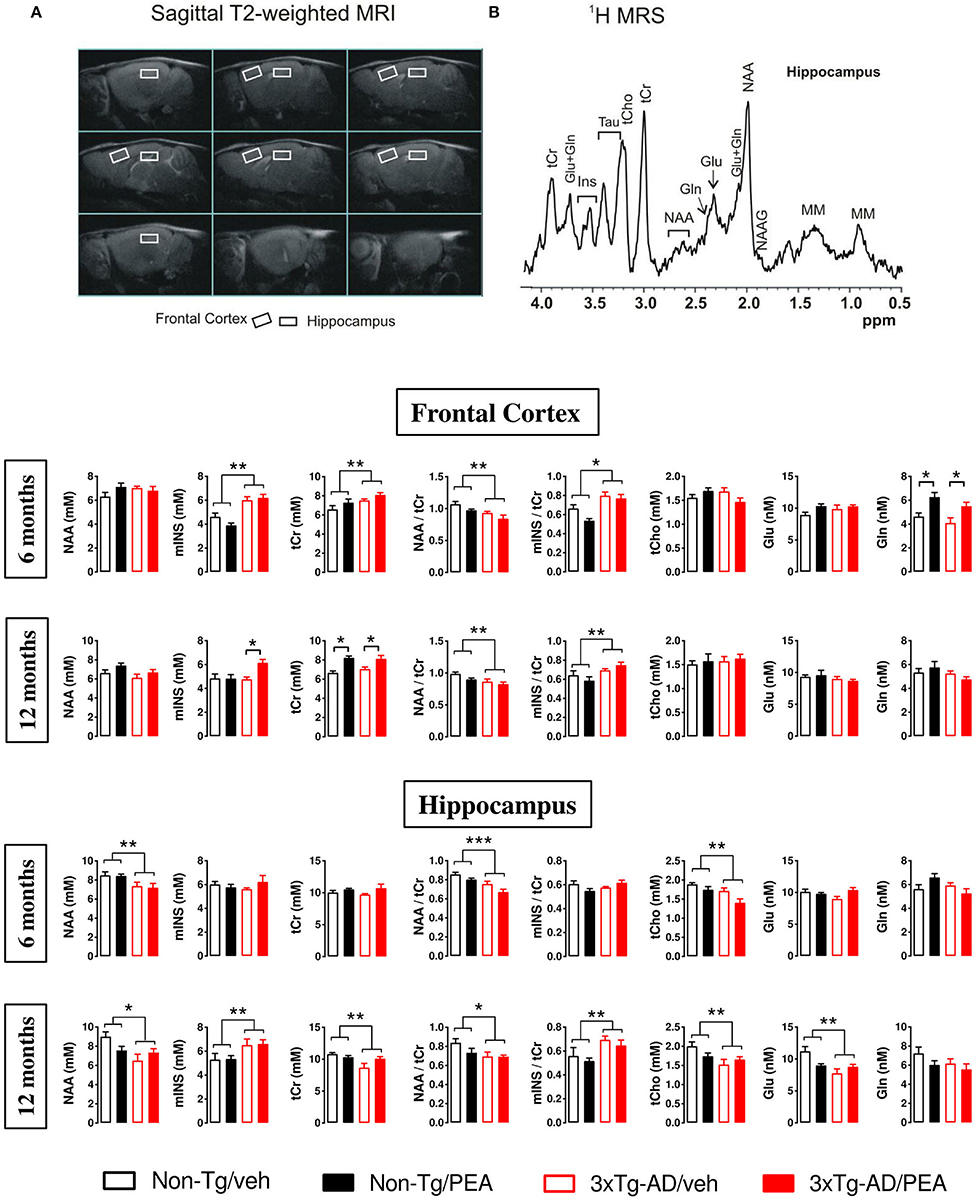
Figure 4. (A) MRI panel: example of in vivo fast spin-echo sagittal anatomical images [repetition time (TR)/echo time (TE) = 3,200/60 ms, consecutive slices]. Voxels localized in the frontal cortex and hippocampus are indicated by white rectangles. (B) MRS panel: examples of in vivo 1 H spectra (PRESS, TR/TE = 4,000/23 ms, NS = 256). Metabolite assignments: NAA, N-acetyl-aspartate; mINS, myo-inositol; tCr, total creatine; tCho, total choline; Glu, glutamate; Gln, glutamine. Histograms showing metabolites measured from the frontal cortex (upper panel) and hippocampus (lower panel) of non-Tg (black bars) and 3×Tg-AD (red bars) 6-month-old and 12-month-old mice treated with vehicle (open bars) or um-PEA (filled bars). Data are expressed as mean ± SEM (n = 5–7). Statistical differences were assessed by two-way ANOVA and Tukey's post-hoc test. *p < 0.05; **p < 0.01.
Statistical analysis for mINS showed a significant main effect of genotype in the FC at 6 months of age (F(1, 19) = 22.072, p < 0.01), whereas a significant main effect of treatment and genotype-by-treatment interaction effect was found at 12 months of age [(Ftreatment(1, 25) = 5.040, p < 0.05); (Fgenotype × treatment(1, 25) = 5.286, p < 0.05)]. Post-hoc comparisons showed a significantly higher mINS level in the um-PEA-treated compared to vehicle-treated 3×Tg-AD mice. Hippocampal mINS levels showed no significant differences among groups at 6 months, whereas a significant main effect of genotype was observed at 12 months of age (F(1, 23) = 6.502, p < 0.05).
Regarding the sum of creatine and phosphocreatine (Cr + PCr), named total creatine (tCr), two-way ANOVA analysis revealed in the FC a significant main effect of genotype at 6 months of age (F(1, 20) = 5.616, p < 0.05), whereas a significant main effect of treatment was observed at 12 months (F(1, 25) = 28.220, p < 0.01). Post-hoc comparisons within genotype showed that um-PEA treatment induced a significant increase in tCr levels compared to the vehicle-treated group. Moreover, hippocampal tCr levels showed no significant differences among groups at 6 months, whereas a significant main effect of genotype was observed at 12 months of age (F(1, 20) = 5.843, p < 0.01).
Statistical analysis for NAA/tCr ratio revealed a significant main effect of genotype at both ages in the FC [6 months: (F(1, 19) = 9.525, p < 0.01); 12 months: (F(1, 25) = 9.111, p < 0.01)], as well as in the HIPP [6 months: (F(1, 19) = 22.910, p < 0.01); 12 months: (F(1, 18) = 5.101, p < 0.05)]. Regarding the mINS/tCr ratio, two-way ANOVA analysis showed a significant main effect of genotype at both ages in the FC [6 months: (F(1, 17) = 18.179, p < 0.05); 12 months: (F(1, 21) = 10.820, p < 0.01)], as well as at 12 months of age in the HIPP (F(1, 18) = 9.217, p < 0.01), whereas no significant differences among groups were found at 6 months in the HIPP.
Statistical analysis for tCho showed no significant differences among groups in the cortical levels at both ages, whereas in the HIPP a main effect of genotype was observed at 6 (F(1, 19) = 14.329, p < 0.01) and 12 months of age (F(1, 24) = 6.007, p < 0.05).
Glu concentrations showed no differences among groups at both ages in the FC, as well as at 6 months in the HIPP; a significant main effect of genotype was observed at 12 months of age in the HIPP (F(1, 22) = 5.619, p < 0.05). Statistical analysis for Gln revealed a significant main effect of treatment at 6 months of age in the FC (F(1, 18) = 14.586, p < 0.01). Post-hoc comparisons within genotype showed that um-PEA treatment induced a significant increase in Gln levels compared to the vehicle-treated group. Moreover, no significant differences among groups were observed at 12 months in the FC, as well as in the HIPP at both ages.
As we have previously published (Scuderi et al., 2018), the basal extracellular glutamate levels in the vHIPP of 6-month-old 3×Tg-AD mice were significantly higher compared to age-matched non-Tg mice, whereas no significant genotype-related difference in basal glutamate levels was observed in 12-month-old mice. Moreover, chronic um-PEA treatment did not affect the basal output of glutamate at both ages (Scuderi et al., 2018). The overall basal glutamate levels are recapitulated in the insets of Figure 5.
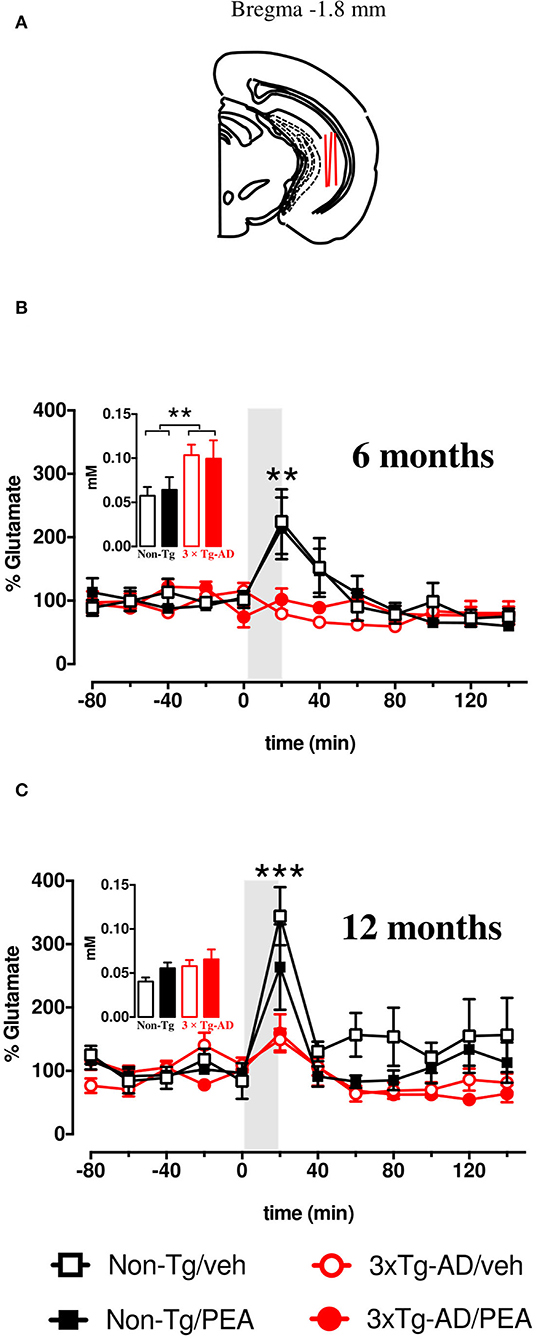
Figure 5. (A) Brain diagrams illustrating the average sites (red lines) where microdialysis probes were implanted and where the representative microphotographs were taken. K+-evoked release of glutamate in the ventral hippocampus of 6- (B) and 12-month-old (C) freely moving non-Tg (squares) and 3×Tg-AD (circles) mice chronically treated with vehicle (empty squares and empty circles, respectively) or um-PEA (black squares and red circles, respectively); gray areas indicate the perfusion with KCl-enriched (50 mM) Krebs-Ringer phosphate (KRP) buffer. Histograms represent average baseline levels (marginal means of five consecutive dialysates: from time −80 up to time 0) observed in non-Tg (black bars) and 3×Tg-AD (red bars) mice treated with vehicle (open bars) and um-PEA (filled bars). Data are expressed as a percentage of control ± SEM (n = 5–6). Statistical differences were assessed by three-way ANOVA and Dunnett's multiple comparison test. **p < 0.01; ***p < 0.001 vs. last baseline within the same group.
After five consecutive samples, the basal extracellular levels of glutamate reached a steady state in the vHIPP of all mice. To address the impact of chronic um-PEA treatment on the impulse-driven glutamate release in the vHIPP of non-Tg and 3×Tg-AD mice, at both mild (6 months of age) and severe (12 months of age) stages of pathology, the probes were perfused with K+-enriched KRP buffer (containing KCl 50 mM) for 20 min (stimulated condition). Under the stimulated condition, the glutamate extracellular levels of non-Tg mice were increased due to the ability of neurons to increase their synaptic activity in response to depolarization. In this study, we observed the total lack of response to K+-stimulation in the vHIPP of 3×Tg-AD mice compared to the non-Tg group, at both 6 and 12 months of age. Moreover, chronic treatment of PEA did not ameliorate the impaired K+-evoked output of glutamate neurotransmission in the 6- and 12-month-old 3×Tg-AD mice (Figures 5A,B). The results of the statistical analyses performed by three-way ANOVA are reported in Table 3.
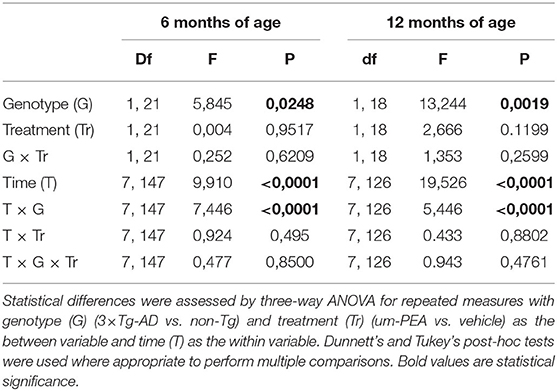
Table 3. Statistical analyses of K+-evoked release of glutamate in the ventral hippocampus of 3×Tg-AD mice and non-Tg mice.
The most novel findings of our study are the demonstration that chronic um-PEA treatment can ameliorate the complex-I respiration rate, the FoF1-ATPase (complex V) activity, as well as ATP content in the cortical mitochondria from 3×Tg-AD mice. Otherwise, mitochondrial bioenergetics, as well as the release of glutamate after depolarization were not ameliorated by um-PEA treatment in the HIPP of both young and adult 3×Tg-AD mice. More interestingly, progressive age- and pathology-related changes were observed in the cortical and hippocampal metabolism that closely mimic the alterations observed in the human AD brain; these metabolic alterations were not affected by chronic um-PEA treatment.
We revealed for the first time that (i) AD-like pathology differently affects mitochondrial bioenergetics in the FC and HIPP and (ii) um-PEA treatment does not promote significant ameliorations simultaneously in those brain regions of 3×Tg-AD mice. This is in line with the temporal- and regional-specific development of AD neuropathology in the brain of 3×Tg-AD mice, which closely mimics their development in the human AD brain (Oddo et al., 2003a,b; Hirata-Fukae et al., 2008).
Compared with many studies, we adopted a longitudinal study design with a sufficiently long observation period that allowed us to assess early alterations and changes over time in both non-transgenic and 3×Tg-AD mice. This study further extends the knowledge of the molecular modification during both normal aging and the progression of AD-like pathology in our murine model of AD (Oddo et al., 2003a,b). Moreover, we analyzed the modulatory effect of PEA on pathways and factors consistently associated with AD-like pathology and symptoms. In fact, we selected two different subsets of 3×Tg-AD mice, which allowed us to investigate the therapeutic potential of um-PEA in restraining the development of AD-like pathology at mild and severe stages, focusing on the mitochondrial bioenergetics, cerebral metabolism, and glutamatergic transmission. Regarding mitochondria and glutamate, we have previously demonstrated that deficits of glutamatergic transmission and mitochondrial dysfunction coexist in the FC and HIPP of 18-month-old 3×Tg-AD mice, which show a substantial number of amyloid plaques and tau pathology; the HIPP was the most affected area at that age (Cassano et al., 2012).
Despite accumulating evidence supporting the link between inflammation, mitochondria, and metabolism, several important questions remain unanswered. Furthermore, there are no studies addressing the effects of PEA on mitochondria bioenergetics in in vivo experimental models of AD, and only one study, to our knowledge, has assessed in vivo the effects of PEA on mitochondrial dysfunction observed in central nervous system pathological conditions (Cristiano et al., 2018). In this regard, Cristiano and colleagues found that PEA treatment improves hippocampal mitochondrial function and reduces oxidative stress in a genetic murine model (BTBR T + tf/J), which exhibits a behavioral phenotype of autism spectrum disorder. In particular, 10-day treatment with um-PEA was able to (i) restore hippocampal mitochondrial state 3 respiration using succinate as substrate, (ii) increase superoxide dismutase (SOD) activity, (iii) counteract ROS, and (iv) decline the energy efficiency, as evidenced by the decreased degree of coupling (Cristiano et al., 2018). Together, these findings demonstrate that PEA counteracts mitochondrial dysfunction and helps rescue brain energy metabolism during pathological states, balancing ROS production/antioxidant defenses, and limiting oxidative stress. Moreover, PEA-treated BTRB mice showed a low activation of pro-inflammatory cytokines at hippocampal and serum levels (Cristiano et al., 2018). Likewise, we have previously observed that 3-month treatment of um-PEA almost completely abolishes the increase in inflammatory markers observed in 6-month-old 3×Tg-AD mice and suppresses the expression of proinflammatory mediators, including interleukin (IL)-1β, IL-16, IL-5, monocyte chemotactic protein 5 (MCP-5), and macrophage colony-stimulating factor (M-CSF), but not inducible nitric oxide synthase (iNOS) and tumor necrosis factor (TNF), while enhancing the anti-inflammatory IL-10 (Scuderi et al., 2018).
In this study, 6-month-old 3×Tg-AD mice with a mild AD-like pathology showed a significant reduction of ATP content in the FC that was counteracted by um-PEA treatment. Otherwise, at 12 months of age, together with severe hallmarks of AD-like pathology, mitochondrial alterations were more evident and regional-specific. In fact, cortical mitochondria of 3×Tg-AD mice exhibited a reduced respiratory capacity, which might partially block electron flow within the respiratory chain and consequently increase oxidative stress; this effect was counteracted by um-PEA. The reduced respiratory activity was not observed after incubation with a complex II-linked substrate. The same pattern of alterations was observed in the cortical mitochondria of 18-month-old 3×Tg-AD mice, which present diffuse extracellular Aβ deposits and extensive human tau immunoreactivity (Cassano et al., 2012). Different from the latter age, at 12 months the reduced oxygen consumption observed in the transgenic cortical preparations was accompanied by alterations of FoF1-ATPase activity and ATP content that were counteracted by um-PEA treatment.
Regarding the HIPP, 12-month-old 3×Tg-AD mice showed the same mitochondrial dysfunctions previously observed with mice at 18 months of age (Cassano et al., 2012). In fact, here we confirmed a significant increase in state 3 and state 4 respiration rates with complex I-linked substrates and a significant impairment of ATP homeostasis. Different from FC, the pharmacological treatment with um-PEA did not ameliorate the hippocampal mitochondrial bioenergetics. Taken together, these findings suggest that um-PEA counteracts mitochondrial dysfunction, helps rescue brain energy metabolism during pathological states only in the FC, and such discrepancy might be due to the difference in Aβ/tau-linked alterations observed between these brain regions. In fact, it has been documented that Aβ and tau pathology differently impact brain regions, with HIPP showing more severe alterations compared to FC (Oddo et al., 2003a,b; Cassano et al., 2012; Bellanti et al., 2017). However, other mechanisms could underlie the regional-specific effect of um-PEA on mitochondrial bioenergetics and other experiments will be necessary to explain this phenomenon.
Brain structures undergo drastic modifications during aging, becoming progressively less interconnected and undergoing several metabolic and structural changes. A clinical scanner by MRS may serve to identify patients with AD before clinical symptom onset to help distinguish AD from other neurodegenerative disorders, as well as evaluate treatment effects.
In this regard, clinical studies demonstrated that NAA, a marker of neuronal integrity, can be detected by MRS and used to differentiate normal aging and pathological dementia (Ackl et al., 2005; Jessen et al., 2005; den Heijer et al., 2006). Besides decreased levels of NAA, several clinical studies demonstrated that mINS, a marker of glial activation, was increased in patients with AD (Glanville et al., 1989; Miller et al., 1993; Bitsch et al., 1999; Jessen et al., 2000; Kantarci et al., 2000; Huang et al., 2001).
Herein, we performed for the first time a comprehensive evaluation of six brain metabolites in the areas most involved in AD-like pathology. Interestingly, HIPP of 3×Tg-AD mice was characterized by lower concentrations of NAA and increased mINS, as seen in human patients with AD (Glanville et al., 1989; Klunk et al., 1992; Miller et al., 1993; Bitsch et al., 1999; Jessen et al., 2000; Kantarci et al., 2000, 2007; Huang et al., 2001; Cheng et al., 2002; Schott et al., 2010). In particular, NAA reduction was observed at both ages (6 and 12 months), whereas mINS increased gradually with mouse age. Similar results were observed with APP/PS1 double transgenic mice, which is characterized by the early appearance of Aβ plaques, neuronal degeneration, and synaptic loss in the brain (Marjanska et al., 2005; Chen et al., 2009). The authors found that NAA was significantly reduced both in the FC and HIPP of 5-month-old APP/PS1 transgenic mice when pathology showed the formation of sparse Aβ plaques in these areas, and the number of neurons decreased. As the age of the mice increased, the NAA decrease was continuously accompanied by the increase in both mINS and the number of Aβ plaques (Marjanska et al., 2005; Chen et al., 2009). In our 3×Tg-AD mice, we found an increase in mINS by age only in the HIPP, but not in the FC, where a significantly higher concentration was observed only at 6 months of age compared to the non-Tg group.
Other results were obtained in the FC with different transgenic AD mice. In fact, no increase in mINS in the FC was observed in the transgenic mouse line PS2APP [PS2N141I × APP(swe)], which develops an age-related cognitive decline associated with severe amyloidosis (Von et al., 2005). Therefore, further studies need to be performed to justify the discrepancy existing in the literature, which may be attributed to the differences in the experimental parameters and animal models of AD-like pathology.
Our longitudinal study revealed a significant decrease in tCr only in the HIPP of 12-month-old 3×Tg-AD mice. This metabolic profile, together with lower NAA, is a clear indicator of the age-related hypometabolism in the HIPP of 3×Tg-AD mice, further suggesting that HIPP is the most affected area at that age (Cassano et al., 2012; Scuderi et al., 2018).
A decrease in NAA/Cr ratio accompanied by an increased mINS/Cr ratio occurs in patients with AD; the latter temporal progression of metabolite abnormalities in patients with AD correlates with the progression of the Aβ/tau-linked alterations (Glanville et al., 1989; Klunk et al., 1992; Miller et al., 1993; Bitsch et al., 1999; Jessen et al., 2000; Kantarci et al., 2000, 2007; Huang et al., 2001; Cheng et al., 2002; Schott et al., 2010). Interestingly, we found a similar pattern of changes in the metabolite ratio both in the FC and HIPP, as seen in human patients with AD.
The role of Cho metabolite in AD is more controversial since several clinical studies demonstrated an increase (Meyerhoff et al., 1994; Pfefferbaum et al., 1999; Kantarci et al., 2007), others no change (Moats et al., 1994; Schuff et al., 1997; Rose et al., 1999; Krishnan et al., 2003), whereas a decrease was reported in one study (Chantal et al., 2002). In our study, we observed a significant decrease of tCho only in the HIPP of both 6- and 12-month-old mice. The Cho cytosolic concentration derives from the breakdown products of phosphatidylcholine (Schuff et al., 1997). Therefore, it has been proposed that catabolism of the phospholipid membrane bilayer allows AD subjects to produce choline to compensate for declining acetylcholine (Wurtman et al., 1985).
Although Glu is the principal excitatory neurotransmitter involved in learning and memory, it has been less well investigated by MRS/MRI. In this regard, few clinical studies revealed a reduction of Glu levels in different brain regions of patients with AD (Antuono et al., 2001; Hattori et al., 2002; Rupsingh et al., 2011) that correlates with the severe reduction of the cerebral metabolic rate for glucose (Mosconi et al., 2008; Bedse et al., 2015; Barone et al., 2016). In fact, glutamate neurotransmission is carried out by a glial-neuronal process that includes the oxidation of glucose and the ATP-dependent glutamine-glutamate cycle. A total of 80–90% of total cerebral glucose usage is attributable to the energy requirements of glutamatergic neurotransmission (Magistretti et al., 1999; Gatta et al., 2016; Pardeshi et al., 2017; Tramutola et al., 2018; Sharma et al., 2019; Bukke et al., 2020a,b). The neurochemical changes obtained from in vivo human MRS studies agree with the results observed in preclinical studies. In fact, the analysis of cortex and HIPP in APP-PS1 mice showed an age-dependent reduction of Glu levels, which correlates with increasing brain amyloidosis (Marjanska et al., 2005). Similar findings were observed in the cortex of APP (Dedeoglu et al., 2004) and PS2APP mice (Von et al., 2005). This is in line with the Glu spectroscopy findings between human AD subjects and the three transgenic mouse models of AD.
In our study, we demonstrated a temporal- and regional-specific alteration of Glu in 3×Tg-AD mice with the HIPP the only affected area. In particular, at 12 months of age, we found a significant reduction (−38%) of Glu in the HIPP of vehicle-treated 3×Tg-AD vs. vehicle-treated non-Tg mice. These results, observed in the vehicle-treated 3×Tg-AD mice, together with lower hippocampal NAA (−45%), tCr (−24%), and tCho (−31%), further support the evidence of an age-related hypometabolism in the HIPP. MRS analysis might be of interest also to depict how neural correlates do change along with pharmacological treatment. In this regard, in our experimental condition, we did not observe any gross treatment-dependent differences between genotypes.
To investigate whether the decrease in hippocampal glutamate levels in the metabolic pool was paralleled by alterations in the glutamatergic nerve terminals, the pattern of glutamate release in basal condition, and in response to K+-stimulation was explored by microdialysis sampling in the vHIPP of 6- and 12-month-old mice. We have previously demonstrated that the basal release of glutamate was significantly increased (+80%) at 6 months of age whereas a trend toward an increase (+43%) was observed in 12-month-old 3×Tg-AD compared to non-Tg mice (Scuderi et al., 2018). Here, we demonstrated that at both ages the depolarization-evoked glutamate release resulted completely disrupted in 3×Tg-AD compared to non-Tg mice, thus suggesting that the deficit of glutamate metabolic pool observed by MRS might sustain, at least in part (at 12 months of age), the decrease of extracellular glutamate concentration in mutant mice.
Our results not only elucidate the temporal correlation among mitochondrion, metabolic alterations, and glutamatergic dysfunction but also, in future studies, allow the assessment of putative therapeutic strategies. However, this study presents some limitations. For instance, additional measurements would allow performing a tight correlation between molecular markers of bioenergetics dysfunction, for instance, hydrogen peroxide production, proton leak or membrane potential, and metabolic alterations. Finally, this study sheds additional light on the effects of PEA treatment, displaying selective bioenergetic effects. At present, the use of a PEA treatment may represent a possible novel frontier for the future clinical treatment of AD.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Ethics Committee of the University of Foggia.
TC, CS, LS, and GS designed the research. FB, VB, MA, RV, GP, RC, and SB performed the research. FB, RC, GV, GS, and TC analyzed data and interpreted the results. FB and TC wrote the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by the Italian Ministry for Education, University and Research (PRIN to LS and GV), and the Department of Clinical and Experimental Medicine (Dipartimenti di eccellenza – Legge 232/2016). This paper/manuscript/study has been published with the financial support of the Department of Medical and Surgical Sciences of the University of Foggia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ackl, N., Ising, M., Schreiber, Y. A., Atiya, M., Sonntag, A., and Auer, D. P. (2005). Hippocampal metabolic abnormalities in mild cognitive impairment and Alzheimer's disease. Neurosci. Lett. 384, 23–28. doi: 10.1016/j.neulet.2005.04.035
Antuono, P. G., Jones, J. L., Wang, Y., and Li, S. J. (2001). Decreased glutamate + glutamine in Alzheimer's disease detected in vivo with (1)H-MRS at 0.5 T. Neurology 56, 737–742. doi: 10.1212/WNL.56.6.737
Barone, E., Di, D. F., Cassano, T., Arena, A., Tramutola, A., Lavecchia, M. A., et al. (2016). Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: a new paradigm. Free Radic. Biol. Med. 91, 127–142. doi: 10.1016/j.freeradbiomed.2015.12.012
Barone, E., Tramutola, A., Triani, F., Calcagnini, S., Di, D. F., Ripoli, C., et al. (2019). Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 56, 2922–2943. doi: 10.1007/s12035-018-1231-5
Bartha, R., Smith, M., Rupsingh, R., Rylett, J., Wells, J. L., and Borrie, M. J. (2008). High field (1)H MRS of the hippocampus after donepezil treatment in Alzheimer disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 786–793. doi: 10.1016/j.pnpbp.2007.12.011
Bedse, G., Di, D. F., Serviddio, G., and Cassano, T. (2015). Aberrant insulin signaling in Alzheimer's disease: current knowledge. Front Neurosci. 9, 204. doi: 10.3389/fnins.2015.00204
Beggiato, S., Cassano, T., Ferraro, L., and Tomasini, M. C. (2020a). Astrocytic palmitoylethanolamide pre-exposure exerts neuroprotective effects in astrocyte-neuron co-cultures from a triple transgenic mouse model of Alzheimer's disease. Life Sci. 257, 118037. doi: 10.1016/j.lfs.2020.118037
Beggiato, S., Tomasini, M. C., Cassano, T., and Ferraro, L. (2020b). Chronic oral palmitoylethanolamide administration rescues cognitive deficit and reduces neuroinflammation, oxidative stress, and glutamate levels in a transgenic murine model of Alzheimer's disease. J. Clin. Med. 9, jcm9020428 doi: 10.3390/jcm9020428
Bellanti, F., Iannelli, G., Blonda, M., Tamborra, R., Villani, R., Romano, A., et al. (2017). Alterations of Clock Gene RNA Expression in Brain Regions of a Triple Transgenic Model of Alzheimer's Disease. J. Alzheimers. Dis. 59, 615–631. doi: 10.3233/JAD-160942
Bitsch, A., Bruhn, H., Vougioukas, V., Stringaris, A., Lassmann, H., Frahm, J., et al. (1999). Inflammatory CNS demyelination: histopathologic correlation with in vivo quantitative proton MR spectroscopy. AJNR Am. J. Neuroradiol. 20, 1619–1627.
Blass, J. P., and Gibson, G. E. (1991). The role of oxidative abnormalities in the pathophysiology of Alzheimer's disease. Rev. Neurol. (Paris) 147 513–525.
Bronzuoli, M. R., Facchinetti, R., Steardo, L. Jr., Romano, A., Stecca, C., Passarella, S., et al. (2018). Palmitoylethanolamide dampens reactive astrogliosis and improves neuronal trophic support in a triple transgenic model of Alzheimer's disease: in vitro and in vivo evidence. Oxid. Med. Cell Longev. 2018, 4720532. doi: 10.1155/2018/4720532
Bronzuoli, M. R., Facchinetti, R., Valenza, M., Cassano, T., Steardo, L., and Scuderi, C. (2019). Astrocyte function is affected by aging and not Alzheimer's disease: a preliminary investigation in hippocampi of 3xTg-AD mice. Front. Pharmacol. 10, 644. doi: 10.3389/fphar.2019.00644
Bukke, V. N., Archana, M., Villani, R., Romano, A. D., Wawrzyniak, A., Balawender, K., et al. (2020a). The dual role of glutamatergic neurotransmission in Alzheimer's disease: from pathophysiology to pharmacotherapy. Int. J. Mol. Sci. 21, ijms21207452 doi: 10.3390/ijms21207452
Bukke, V. N., Villani, R., Archana, M., Wawrzyniak, A., Balawender, K., Orkisz, S., et al. (2020b). The glucose metabolic pathway as a potential target for therapeutics: crucial role of glycosylation in Alzheimer's disease. Int. J. Mol. Sci. 21, ijms21207739 doi: 10.3390/ijms21207739
Canese, R., Pisanu, M. E., Mezzanzanica, D., Ricci, A., Paris, L., Bagnoli, M., et al. (2012). Characterisation of in vivo ovarian cancer models by quantitative 1H magnetic resonance spectroscopy and diffusion-weighted imaging. NMR Biomed. 25, 632–642. doi: 10.1002/nbm.1779
Carapelle, E., Mundi, C., Cassano, T., and Avolio, C. (2020). Interaction between cognitive reserve and biomarkers in Alzheimer disease. Int. J. Mol. Sci. 21, ijms21176279 doi: 10.3390/ijms21176279
Cassano, T., Calcagnini, S., Carbone, A., Bukke, V. N., Orkisz, S., Villani, R., et al. (2019). Pharmacological treatment of depression in Alzheimer's disease: a challenging task. Front Pharmacol. 10, 1067. doi: 10.3389/fphar.2019.01067
Cassano, T., Pace, L., Bedse, G., Lavecchia, A. M., De, M. F., Gaetani, S., et al. (2016). Glutamate and mitochondria: two prominent players in the oxidative stress-induced neurodegeneration. Curr. Alzheimer Res. 13, 185–197. doi: 10.2174/1567205013666151218132725
Cassano, T., Serviddio, G., Gaetani, S., Romano, A., Dipasquale, P., Cianci, S., et al. (2012). Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging 33, 1121–1112. doi: 10.1016/j.neurobiolaging.2011.09.021
Chantal, S., Labelle, M., Bouchard, R. W., Braun, C. M., and Boulanger, Y. (2002). Correlation of regional proton magnetic resonance spectroscopic metabolic changes with cognitive deficits in mild Alzheimer disease. Arch. Neurol. 59, 955–962. doi: 10.1001/archneur.59.6.955
Chen, T. F., Chen, Y. F., Cheng, T. W., Hua, M. S., Liu, H. M., and Chiu, M. J. (2009). Executive dysfunction and periventricular diffusion tensor changes in amnesic mild cognitive impairment and early Alzheimer's disease. Hum. Brain Mapp. 30, 3826–3836. doi: 10.1002/hbm.20810
Cheng, L. L., Newell, K., Mallory, A. E., Hyman, B. T., and Gonzalez, R. G. (2002). Quantification of neurons in Alzheimer and control brains with ex vivo high resolution magic angle spinning proton magnetic resonance spectroscopy and stereology. Magn Reson. Imaging 20, 527–533. doi: 10.1016/S0730-725X(02)00512-X
Costa, B., Conti, S., Giagnoni, G., and Colleoni, M. (2002). Therapeutic effect of the endogenous fatty acid amide, palmitoylethanolamide, in rat acute inflammation: inhibition of nitric oxide and cyclo-oxygenase systems. Br. J. Pharmacol. 137, 413–420. doi: 10.1038/sj.bjp.0704900
Cristiano, C., Pirozzi, C., Coretti, L., Cavaliere, G., Lama, A., Russo, R., et al. (2018). Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T+tf/J mice: contribution of central and peripheral mechanisms. Brain Behav. Immun. 74, 166–175. doi: 10.1016/j.bbi.2018.09.003
D'Agostino, G., Russo, R., Avagliano, C., Cristiano, C., Meli, R., and Calignano, A. (2012). Palmitoylethanolamide protects against the amyloid-beta25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 37, 1784–1792. doi: 10.1038/npp.2012.25
Dedeoglu, A., Choi, J. K., Cormier, K., Kowall, N. W., and Jenkins, B. G. (2004). Magnetic resonance spectroscopic analysis of Alzheimer's disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 1012, 60–65. doi: 10.1016/j.brainres.2004.02.079
Delerive, P., Fruchart, J. C., and Staels, B. (2001). Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 169, 453–459. doi: 10.1677/joe.0.1690453
den Heijer, T., Sijens, P. E., Prins, N. D., Hofman, A., Koudstaal, P. J., Oudkerk, M., et al. (2006). MR spectroscopy of brain white matter in the prediction of dementia. Neurology 66, 540–544. doi: 10.1212/01.wnl.0000198256.54809.0e
Devchand, P. R., Keller, H., Peters, J. M., Vazquez, M., Gonzalez, F. J., and Wahli, W. (1996). The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 384, 39–43. doi: 10.1038/384039a0
Franklin, K. B. J., and Paxinos, G. (1997). The Mouse Brain in Stereotaxic Coordinates, San Diego: Academic Press.
Frederick, B. B., Satlin, A., Yurgelun-Todd, D. A., and Renshaw, P. F. (1997). In vivo proton magnetic resonance spectroscopy of Alzheimer's disease in the parietal and temporal lobes. Biol. Psychiatry 42, 147–150. doi: 10.1016/S0006-3223(97)00242-4
Gatta, E., Lefebvre, T., Gaetani, S., dos, S. M., Marrocco, J., Mir, A. M., et al. (2016). Evidence for an imbalance between tau O-GlcNAcylation and phosphorylation in the hippocampus of a mouse model of Alzheimer's disease. Pharmacol. Res. 105, 186–197. doi: 10.1016/j.phrs.2016.01.006
Glanville, N. T., Byers, D. M., Cook, H. W., Spence, M. W., and Palmer, F. B. (1989). Differences in the metabolism of inositol and phosphoinositides by cultured cells of neuronal and glial origin. Biochim. Biophys. Acta 1004, 169–179. doi: 10.1016/0005-2760(89)90265-8
Grillo, S. L., Duggett, N. A., Ennaceur, A., and Chazot, P. L. (2013). Non-invasive infra-red therapy (1072 nm) reduces beta-amyloid protein levels in the brain of an Alzheimer's disease mouse model, TASTPM. J. Photochem. Photobiol. B 123, 13–22. doi: 10.1016/j.jphotobiol.2013.02.015
Hattori, N., Abe, K., Sakoda, S., and Sawada, T. (2002). Proton MR spectroscopic study at 3 Tesla on glutamate/glutamine in Alzheimer's disease. Neuroreport 13, 183–186. doi: 10.1097/00001756-200201210-00041
Hirata-Fukae, C., Li, H. F., Hoe, H. S., Gray, A. J., Minami, S. S., Hamada, K., et al. (2008). Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 1216, 92–103. doi: 10.1016/j.brainres.2008.03.079
Huang, W., Alexander, G. E., Chang, L., Shetty, H. U., Krasuski, J. S., Rapoport, S. I., et al. (2001). Brain metabolite concentration and dementia severity in Alzheimer's disease: a (1)H MRS study. Neurology 57, 626–632. doi: 10.1212/WNL.57.4.626
Jessen, F., Block, W., Traber, F., Keller, E., Flacke, S., Papassotiropoulos, A., et al. (2000). Proton MR spectroscopy detects a relative decrease of N-acetylaspartate in the medial temporal lobe of patients with AD. Neurology 55, 684–688. doi: 10.1212/WNL.55.5.684
Jessen, F., Traeber, F., Freymann, N., Maier, W., Schild, H. H., Heun, R., et al. (2005). A comparative study of the different N-acetylaspartate measures of the medial temporal lobe in Alzheimer's disease. Dement. Geriatr. Cogn. Disord. 20, 178–183. doi: 10.1159/000087095
Kantarci, K., Jack, C. R. Jr., Xu, Y. C., Campeau, N. G., O'Brien, P. C., Smith, G. E., et al. (2000). Regional metabolic patterns in mild cognitive impairment and Alzheimer's disease: A 1H MRS study. Neurology 55, 210–217. doi: 10.1212/WNL.55.2.210
Kantarci, K., Weigand, S. D., Petersen, R. C., Boeve, B. F., Knopman, D. S., Gunter, J., et al. (2007). Longitudinal 1H MRS changes in mild cognitive impairment and Alzheimer's disease. Neurobiol. Aging 28, 1330–1339. doi: 10.1016/j.neurobiolaging.2006.06.018
Klunk, W. E., Panchalingam, K., Moossy, J., McClure, R. J., and Pettegrew, J. W. (1992). N-acetyl-L-aspartate and other amino acid metabolites in Alzheimer's disease brain: a preliminary proton nuclear magnetic resonance study. Neurology 42, 1578–1585. doi: 10.1212/WNL.42.8.1578
Krishnan, K. R., Charles, H. C., Doraiswamy, P. M., Mintzer, J., Weisler, R., Yu, X., et al. (2003). Randomized, placebo-controlled trial of the effects of donepezil on neuronal markers and hippocampal volumes in Alzheimer's disease. Am. J. Psychiatry 160, 2003–2011. doi: 10.1176/appi.ajp.160.11.2003
Lin, M. T., and Beal, M. F. (2006). Alzheimer's APP mangles mitochondria. Nat. Med. 12, 1241–1243. doi: 10.1038/nm1106-1241
LoVerme, J., La, R. G., Russo, R., Calignano, A., and Piomelli, D. (2005). The search for the palmitoylethanolamide receptor. Life Sci. 77, 1685–1698. doi: 10.1016/j.lfs.2005.05.012
LoVerme, J., Russo, R., La, R. G., Fu, J., Farthing, J., Mattace-Raso, G., et al. (2006). Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J. Pharmacol. Exp. Ther. 319, 1051–1061. doi: 10.1124/jpet.106.111385
Magistretti, P. J., Pellerin, L., Rothman, D. L., and Shulman, R. G. (1999). Energy on demand. Science 283, 496–497. doi: 10.1126/science.283.5401.496
Mahmoud, S., Gharagozloo, M., Simard, C., and Gris, D. (2019). Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 8, 184. doi: 10.3390/cells8020184
Marjanska, M., Curran, G. L., Wengenack, T. M., Henry, P. G., Bliss, R. L., Poduslo, J. F., et al. (2005). Monitoring disease progression in transgenic mouse models of Alzheimer's disease with proton magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. U. S. A 102, 11906–11910. doi: 10.1073/pnas.0505513102
Meyerhoff, D. J., MacKay, S., Constans, J. M., Norman, D., Van, D. C., Fein, G., et al. (1994). Axonal injury and membrane alterations in Alzheimer's disease suggested by in vivo proton magnetic resonance spectroscopic imaging. Ann. Neurol. 36, 40–47. doi: 10.1002/ana.410360110
Miller, B. L., Moats, R. A., Shonk, T., Ernst, T., Woolley, S., and Ross, B. D. (1993). Alzheimer disease: depiction of increased cerebral myo-inositol with proton MR spectroscopy. Radiology 187, 433–437. doi: 10.1148/radiology.187.2.8475286
Moats, R. A., Ernst, T., Shonk, T. K., and Ross, B. D. (1994). Abnormal cerebral metabolite concentrations in patients with probable Alzheimer disease. Magn. Reson. Med. 32, 110–115. doi: 10.1002/mrm.1910320115
Mosconi, L., Tsui, W. H., Herholz, K., Pupi, A., Drzezga, A., Lucignani, G., et al. (2008). Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer's disease, and other dementias. J. Nucl. Med. 49, 390–398. doi: 10.2967/jnumed.107.045385
Oddo, S., Caccamo, A., Kitazawa, M., Tseng, B. P., and Laferla, F. M. (2003a). Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol. Aging 24, 1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003b). Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/S0896-6273(03)00434-3
Pardeshi, R., Bolshette, N., Gadhave, K., Ahire, A., Ahmed, S., Cassano, T., et al. (2017). Insulin signaling: an opportunistic target to minify the risk of Alzheimer's disease. Psychoneuroendocrinology 83, 159–171. doi: 10.1016/j.psyneuen.2017.05.004
Pfefferbaum, A., Adalsteinsson, E., Spielman, D., Sullivan, E. V., and Lim, K. O. (1999). In vivo brain concentrations of N-acetyl compounds, creatine, and choline in Alzheimer disease. Arch. Gen. Psychiatry 56, 185–192. doi: 10.1001/archpsyc.56.2.185
Querfurth, H. W., and Laferla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Reddy, P. H. (2009). Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer's disease. CNS. Spectr. 14, 8–13. doi: 10.1017/S1092852900024901
Romano, A., Pace, L., Tempesta, B., Lavecchia, A. M., Macheda, T., Bedse, G., et al. (2014). Depressive-like behavior is paired to monoaminergic alteration in a murine model of Alzheimer's disease. Int. J. Neuropsychopharmacol. 18, pyu020. doi: 10.1093/ijnp/pyu020
Romano, A., Serviddio, G., Calcagnini, S., Villani, R., Giudetti, A. M., Cassano, T., et al. (2017). Linking lipid peroxidation and neuropsychiatric disorders: focus on 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 111, 281–293. doi: 10.1016/j.freeradbiomed.2016.12.046
Rose, S. E., de Zubicaray, G. I., Wang, D., Galloway, G. J., Chalk, J. B., Eagle, S. C., et al. (1999). A 1H MRS study of probable Alzheimer's disease and normal aging: implications for longitudinal monitoring of dementia progression. Magn. Reson. Imaging 17, 291–299. doi: 10.1016/S0730-725X(98)00168-4
Rupsingh, R., Borrie, M., Smith, M., Wells, J. L., and Bartha, R. (2011). Reduced hippocampal glutamate in Alzheimer disease. Neurobiol. Aging 32, 802–810. doi: 10.1016/j.neurobiolaging.2009.05.002
Schinder, A. F., Olson, E. C., Spitzer, N. C., and Montal, M. (1996). Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16, 6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996
Schott, J. M., Frost, C., MacManus, D. G., Ibrahim, F., Waldman, A. D., and Fox, N. C. (2010). Short echo time proton magnetic resonance spectroscopy in Alzheimer's disease: a longitudinal multiple time point study. Brain 133, 3315–3322. doi: 10.1093/brain/awq208
Schuff, N., Amend, D., Ezekiel, F., Steinman, S. K., Tanabe, J., Norman, D., et al. (1997). Changes of hippocampal N-acetyl aspartate and volume in Alzheimer's disease. A proton MR spectroscopic imaging and MRI study. Neurology 49, 1513–1521. doi: 10.1212/WNL.49.6.1513
Scuderi, C., Bronzuoli, M. R., Facchinetti, R., Pace, L., Ferraro, L., Broad, K. D., et al. (2018). Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer's disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 8, 32. doi: 10.1038/s41398-017-0076-4
Scuderi, C., Esposito, G., Blasio, A., Valenza, M., Arietti, P., Steardo, L. Jr., et al. (2011). Palmitoylethanolamide counteracts reactive astrogliosis induced by beta-amyloid peptide. J. Cell Mol. Med. 15, 2664–2674. doi: 10.1111/j.1582-4934.2011.01267.x
Scuderi, C., Stecca, C., Valenza, M., Ratano, P., Bronzuoli, M. R., Bartoli, S., et al. (2014). Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer's disease. Cell Death. Dis. 5, e1419. doi: 10.1038/cddis.2014.376
Scuderi, C., Valenza, M., Stecca, C., Esposito, G., Carratu, M. R., and Steardo, L. (2012). Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-alpha. J. Neuroinflammation. 9, 49. doi: 10.1186/1742-2094-9-49
Serviddio, G., Romano, A. D., Cassano, T., Bellanti, F., Altomare, E., and Vendemiale, G. (2011). Principles and therapeutic relevance for targeting mitochondria in aging and neurodegenerative diseases. Curr. Pharm. Des. 17, 2036–2055. doi: 10.2174/138161211796904740
Sharma, N., Tramutola, A., Lanzillotta, C., Arena, A., Blarzino, C., Cassano, T., et al. (2019). Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer's disease. Neurobiol. Dis. 125, 176–189. doi: 10.1016/j.nbd.2019.02.003
Shonk, T. K., Moats, R. A., Gifford, P., Michaelis, T., Mandigo, J. C., Izumi, J., et al. (1995). Probable Alzheimer disease: diagnosis with proton MR spectroscopy. Radiology 195, 65–72. doi: 10.1148/radiology.195.1.7892497
Skaper, S. D., Buriani, A., Dal, T. R., Petrelli, L., Romanello, S., Facci, L., et al. (1996). The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc. Natl. Acad. Sci. U. S. A. 93, 3984–3989. doi: 10.1073/pnas.93.9.3984
Sullivan, P. G., and Brown, M. R. (2005). Mitochondrial aging and dysfunction in Alzheimer's disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 407–410. doi: 10.1016/j.pnpbp.2004.12.007
Sweet, R. A., Panchalingam, K., Pettegrew, J. W., McClure, R. J., Hamilton, R. L., Lopez, O. L., et al. (2002). Psychosis in Alzheimer disease: postmortem magnetic resonance spectroscopy evidence of excess neuronal and membrane phospholipid pathology. Neurobiol. Aging 23, 547–553. doi: 10.1016/S0197-4580(02)00009-X
Tomasini, M. C., Borelli, A. C., Beggiato, S., Ferraro, L., Cassano, T., Tanganelli, S., et al. (2015). Differential effects of palmitoylethanolamide against amyloid-beta induced toxicity in cortical neuronal and astrocytic primary cultures from wild-type and 3xTg-AD Mice. J. Alzheimers Dis. 46, 407–421. doi: 10.3233/JAD-143039
Tomasini, M. C., Ferraro, L., Bebe, B. W., Tanganelli, S., Cassano, T., Cuomo, V., et al. (2002). Delta(9)-tetrahydrocannabinol increases endogenous extracellular glutamate levels in primary cultures of rat cerebral cortex neurons: involvement of CB(1) receptors. J. Neurosci. Res. 68, 449–453. doi: 10.1002/jnr.10242
Tramutola, A., Sharma, N., Barone, E., Lanzillotta, C., Castellani, A., Iavarone, F., et al. (2018). Proteomic identification of altered protein O-GlcNAcylation in a triple transgenic mouse model of Alzheimer's disease. Biochim. Biophys. Acta Mol. Basis. Dis. 1864, 3309–3321. doi: 10.1016/j.bbadis.2018.07.017
Valenza, M., Facchinetti, R., Menegoni, G., Steardo, L., and Scuderi, C. (2021). Alternative targets to fight Alzheimer's disease: focus on astrocytes. Biomolecules. 11. doi: 10.3390/biom11040600
Von, K. M., Kunnecke, B., Metzger, F., Steiner, G., Richards, J. G., Ozmen, L., et al. (2005). Altered metabolic profile in the frontal cortex of PS2APP transgenic mice, monitored throughout their life span. Neurobiol. Dis. 18, 32–39. doi: 10.1016/j.nbd.2004.09.005
Vos, M., Lauwers, E., and Verstreken, P. (2010). Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic. Neurosci. 2, 139. doi: 10.3389/fnsyn.2010.00139
Wurtman, R. J., Blusztajn, J. K., and Maire, J. C. (1985). “Autocannibalism” of choline-containing membrane phospholipids in the pathogenesis of Alzheimer's disease-A hypothesis. Neurochem. Int. 7, 369–372. doi: 10.1016/0197-0186(85)90127-5
Keywords: glutamate, mitochondria, hippocampus, frontal cortex, microdialysis, Alzheimer's disease
Citation: Bellanti F, Bukke VN, Moola A, Villani R, Scuderi C, Steardo L, Palombelli G, Canese R, Beggiato S, Altamura M, Vendemiale G, Serviddio G and Cassano T (2022) Effects of Ultramicronized Palmitoylethanolamide on Mitochondrial Bioenergetics, Cerebral Metabolism, and Glutamatergic Transmission: An Integrated Approach in a Triple Transgenic Mouse Model of Alzheimer's Disease. Front. Aging Neurosci. 14:890855. doi: 10.3389/fnagi.2022.890855
Received: 06 March 2022; Accepted: 19 April 2022;
Published: 24 May 2022.
Edited by:
Ashok K. Shetty, Texas A&M University College of Medicine, United StatesReviewed by:
Sandra Milena Sanabria Barrera, Fundación Cardiovascular de Colombia, ColombiaCopyright © 2022 Bellanti, Bukke, Moola, Villani, Scuderi, Steardo, Palombelli, Canese, Beggiato, Altamura, Vendemiale, Serviddio and Cassano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tommaso Cassano, dG9tbWFzby5jYXNzYW5vQHVuaWZnLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.