 Martina Reutzel
Martina Reutzel Rekha Grewal1
Rekha Grewal1- 1Laboratory for Nutrition in Prevention and Therapy, Biomedical Research Center Seltersberg (BFS), Institute of Nutritional Sciences, Justus-Liebig-University Giessen, Giessen, Germany
- 2Department of Biological Sciences & Cluster of Excellence Macromolecular Complexes, Institute for Molecular Biosciences, Johann Wolfgang Goethe-University Frankfurt, Frankfurt am Main, Germany
Aging is the main risk factor for sporadic Alzheimer’s disease (AD), which is characterized by the cerebral deposition of β-amyloid peptides (Aβ) and cognitive decline. Mitochondrial dysfunction is also characteristic of the disease and represents a hallmark of both, aging and neurodegeneration. We longitudinally followed Aβ levels, cognition, and mitochondrial function in the same cohort of Thy1-APP751SL mice representing a murine model of AD. In the course of time, changes were most prominent at an age of 13 months including the latency time in the passive avoidance test, the activity of complexes I and IV of the mitochondrial respiration chain, and expression of genes related to mitochondrial biogenesis and synaptic plasticity including Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α), CAMP responsive element binding protein 1 (CREB1), and Synaptophysin 1 (SYP1). These changes occurred in parallel with massively increasing cerebral Aβ levels. Other parameters were changed in younger mice including the alteration rate in the Y-maze test and the nesting score when Aβ levels were not changed yet. The results are consistent in the cohort described. However, previous, non-longitudinal studies reported divergent time points for the occurrence of the parameters studied. These findings are discussed in light of the current results.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative, complex disease. Aging is one major risk factor for sporadic AD. On the molecular level, neurotoxic β-amyloid peptides (Aβ) are deposited in the brains of AD patients after β- and γ-secretase cleavage of the amyloid precursor protein (APP) (Pohland et al., 2018; Hampel et al., 2021). An additional characteristic are the intraneuronal neurofibrillary tangles of hyperphosphorylated tau protein, which leads to dysfunction of synapses and disturbed glucose metabolism in brains of AD patients (Naseri et al., 2019; Zhao and Xu, 2021). It has been shown that a particularly early event in the development of AD and the physiological aging process seems to be the decrease of mitochondrial function. Since mitochondria are the powerhouses of the cell and essential for the synthesis of adenosine triphosphate (ATP) during the oxidative phosphorylation (OXPHOS), mitochondrial dysfunction (MD) is proposed to be an early event in both, the physiological aging process and AD (Hauptmann et al., 2009; Zia et al., 2021). MD is characterized by a decrease in cellular mitochondrial membrane potential (MMP) and ATP levels, a reduced capacity of mitochondrial respiration complexes and impairments during mitochondrial biogenesis. In particular, PGC1-α, a transcription factor responsible for inducing mitochondrial biogenesis, has been shown to be decreased both in AD and during aging. PGC1-α can be activated by AMPK-activated kinase, Sirt1, or creb-1 via acetylation or increasing cellular NAD + levels (Li et al., 2017). An increase in PGC1-α was able to reduce the generation of Aβ (Pohland et al., 2018; Mota and Sastre, 2021).
The research of AD and the clarification of exact mechanisms behind the disease is almost inevitable to animal models. Until now, mouse models are most commonly used in AD research. Transgenic AD models are often based on different single or multiple mutations resulting in overexpression of human APP, Aβ, Presenilin 1 or 2 or/and tau proteins (Hall and Roberson, 2012). A variety of promoters are used to overexpress different isoforms of the human amyloid precursor protein. These models are described as a solid basis for visualizing and tracking AD pathology (Hall and Roberson, 2012). However, all models face the major problem that the complexity of AD disease cannot be represented in all its aspects and that they do mainly reflect autosomal dominant rather than sporadic AD. However, they are still an indispensable model for the study of relevant targets and pathways affected in AD. In the current study, mice expressing the human form of APP bearing both, the Swedish (KM670/671NL) and London mutation (V717L) under a murine Thy-1 promoter were used. Pronounced mitochondrial dysfunction in adult Thy1-APP751SL mice, already appeared at 3 months (Hauptmann et al., 2009) when elevated intracellular but not extracellular Aβ deposits are present (Blanchard et al., 2003). We recently confirmed forced α- and β-secretase processing of APP leading to enhanced Aβ1–40 levels in brains of 3 months old Thy1-APP751SL mice and reduced activity of complex IV (C-IV) of the mitochondrial respiration chain (Pohland et al., 2018). In both studies, mitochondrial membrane potential (MMP) and ATP levels were significantly reduced. However, in a more recent study ATP levels were unchanged in brains of 3 months old Thy1-APP751SL mice, although C-IV activity and MMP were significantly reduced (Eckert et al., 2020). Previous studies mostly compared only two study time points. To further elucidate the relationship between aging, mitochondrial dysfunction, Aβ formation, and cognition in mice, we initiated a longitudinal study in which we examined the same cohort at different time points. Cognitive performance, Aβ1–40 levels, mitochondrial parameters, and mRNA levels of relevant genes were examined at 3, 7, and 13 months of age compared to non-transgenic control mice.
Materials and Methods
Animals
Thy1-APP751SL Mice
Male and female C57Bl/6 mice bearing the human Swedish (S:KM670/671NL) and the London (L:V717I) mutations in the 751 amino acid form of human amyloid-beta precursor protein (AβPP) under control of a murine Thy1 promotor were used. All mice were genotyped by tail biopsies and polymerase chain reaction before and after the experiments as previously described (Pohland et al., 2018). Mice were housed in the institute of pharmacology in Frankfurt a. M. according to the German guidelines for animal care with access to water and food ad libitum until they reached the age of 3, 7, and 13 months. Wild-type mice of the same age were used as controls. Experiments were approved by the regional authority (V54-19 c 20/15 – FU/113).
Nestbuilding Behavior
Approximately 1 h before the dark phase, the mouse was moved to a single cage if the mouse was previously housed in a group. If the mouse was housed singly, the test could be performed in the “home” cage. All furnishings except litter were removed from the cage and a nestlet was placed in the cage. After 18 h, the test ended. The nests were photographed and scored by two independent observers. Nests were rated from 1 to 5, with 5 representing a perfect nest and 1 representing an unbuilt nest. The evaluation of the nests was analogous to the protocol described in Deacon (2006).
Passive Avoidance Test
The test was carried out using a passive avoidance step through system (cat. no. 40533/mice Ugo Basile, Germonio, Italy) and a protocol similar to the protocol published by Shiga et al. (2016). The setup of the passive-avoidance-learning test consisted of two chambers, a light and a dark chamber. The mouse was placed in the light chamber (light intensity 75%). The mouse was acclimated to the light chamber for 30 s before the door opened into the dark chamber. The test was stopped after 180 s or if the mouse entered the dark chamber. In the chamber, the mouse received an electric shock (0.2 mA) for 2 s. After 24 h, the experiment was repeated, with the difference that the acclimation time changed to 5 s and the test was aborted after 300 s. In addition, the electric shock was turned off.
One-Trial Y-Maze Test
The mouse was put in one arm of a custom-made Y-shaped maze (material: polyvinyl chloride, length of arms: 36 cm, height of arms: 7 cm, width of arms: 5 cm). Afterward, the mouse was able to explore the maze for 5 min. At the end of the experiment, the number of entries was determined as well as the number of alternations. One alternation was defined as the mouse entered all three arms before it entered an already visited arm. The alternation rate was calculated using the formula [(number of alternations/total number of possible alternations) × 100] (Wolf et al., 2016).
Measurement of Soluble Aβ1–40
Brain samples were homogenized in 10 times the amount of phosphate-buffered saline containing a protease inhibitor cocktail (Roche cOmplete, Mini Protease Inhibitor Cocktail). Afterward, samples were centrifuged (15,000 × g, 30 min, 4°C) and the supernatants were put into a fresh reaction vessel and stored at −80°C until analysis. Aβ1–40 amounts were measured using a specific solid phase sandwich enzyme-linked immunosorbent assay (ELISA; Life Technologies, Carlsbad, CA, United States).
Preparation of Dissociated Brain Cells for ex vivo Studies
Dissociated Brain Cells (DBC) were freshly prepared using one hemisphere of mouse brain. The method is described in detail in Reutzel et al. (2020). Briefly, the brain was washed in medium 1 (138 mM NaCl, 5.4 mM KCl, 0.17 mM Na2HPO4, 0.22 mM KH2PO4, 5.5 mM Glucose × H20, 58.4 mM Sucrose, pH = 7.35), cut into small pieces in 2 ml of medium 1 and was pressed through a 200 μm nylon mesh. Afterward, the brain suspension was filtered through a 102 μm nylon mesh. The cell homogenate was centrifuged (2,000 rpm, 5 min, 4°C) and the resulting pellet was dissolved in 20 ml medium 2 (110 mM NaCl, 5.3 mM KCl, 1.8 mM CaCl2 × 2 137 H20, 1 mM MgCl2 × 6 H20, 25 mM Glucose × H20, 70 mM Sucrose, 20 mM HEPES). The centrifugation step was repeated twice and the pellet dissolved in 4.5 ml Dulbecco’s Modified Eagle Medium (DMEM) without supplements. For ATP measurements twelve replicates of 50 μl cell suspension were seeded into a 96 well plate. Respectively 6 wells were incubated with sodium nitroprusside (0.5 mM). For determination of the mitochondrial membrane potential (MMP) 250 μl were seeded into 24 well plate and 6 wells were incubated with 2 mM sodium nitroprusside. Afterward, the plates were incubated for 3 h in a humified incubator (5% CO2) before measurement of mitochondrial membrane potential or ATP. The remaining suspension was stored for protein determination at −80°C).
Measurement of Mitochondrial Membrane Potential
For the measurement of mitochondrial membrane potential (MMP) DBC’s were incubated for 15 min in a humified incubator (5% CO2) with 0.4 μM of the fluorescence dye Rhodamine-123. The reaction was stopped by adding 250 μl of Hank’s balanced salt solution (HBSS) into each well. The plate was centrifuged (914 g, 5 min, room temperature), the medium aspirated and new HBSS was added into the wells. DBC were triturated and MMP was measured by reading the R123 fluorescence at an excitation wavelength of 490 nm and an emission wavelength of 535 nm Victor X3 multilabel counter). The measurement was repeated four times and the values normalized to protein content of the sample.
Measurement of Adenosine Triphosphate Levels
Adenosine Triphosphate Levels (ATP) concentrations were determined in DBC’s with the ViaLight Plus bioluminescence kit (Lonza, Walkersville, MD, United States). After the incubation, the plate was removed from the incubator and allowed to cool to room temperature for 10 min. All wells were incubated for 10 min with 25 μl lysis buffer in the dark. Next, wells were incubated with 50 μl monitoring reagent. The measurement was conducted according to the manufactures instructions. The emitted light (bioluminescence) was recorded using a Victor X3 multilabel counter and is linearly related to ATP content.
Preparation of Isolated Brain Mitochondria
Half a brain hemisphere (the frontal part) was used for the isolation of brain mitochondria for mitochondrial respiration as previously described (Hagl et al., 2013). Briefly, the sample was homogenized using a potter equipped with a Teflon® pistil in 2 ml mitochondrial respiration medium (MiR05) containing a protease inhibitor cocktail (PI, complete; Roche, Mannheim, Germany). Afterward, the homogenate was centrifuged (1,400 g, 7 min, 4°C). A second centrifugation was conducted with the supernatant for 3 min followed by a centrifugation at 100,000 g for 5 min at 4°C. The resulting pellet containing the mitochondria was dissolved in 250 μl MIRO5 + PI and 80 μl of the solution was used for the measurement of mitochondrial respiration using an Oxygrapph-2k respirometer (Oroboros, Innsbruck, Austria). The remaining mitochondria solution was frozen in liquid nitrogen for citrate synthase activity (120 μl) and protein determination (50 μl).
High-Resolution Respirometry
After injecting 80 μl of the mitochondria suspension into the Oxygraph-2k chamber a protocol elaborated by Prof. Dr. Erich Gnaiger was used including several inhibitors, uncouplers and substrates of the respiratory chain system (Krumschnabel et al., 2015). A detailed description of the different steps of the protocol is described in Hagl et al. (2016). Briefly, the capacity of the oxidative phosphorylation (OXPHOS) was determined using complex-I related substrates pyruvate (5 mM) and malate (2 mM) and ADP (2 mM) followed by the addition of succinate (10 mM). Mitochondrial integrity was measured by addition of cytochrome c (10 μM). Oligomycin (2 μg/ml) was added to determine leak respiration [leak (omy)] and afterward uncoupling was achieved by carbonyl cyanide p-(trifluoromethoxy) phenyl-hydrazone (FCCP, injected stepwise up to 1–1.5 μM). Complex II respiration was measured after the addition of rotenone (0.5 μM). Complex III inhibition was achieved by the addition of antimycin A (2.5 μM) and was subtracted from all respiratory parameters. COX activity was measured after ROX determination by applying 0.5 mM tetramethylphenylenediamine (TMPD) as an artificial substrate of complex IV and 2 mM ascorbate to keep TMPD in the reduced state. Autoxidation rate was determined after the addition of sodium azide (>100 mM), and COX respiration was additionally corrected for autoxidation.
Transcription Analysis by Quantitative Real-Time PCR
RNA isolation was conducted with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions using ∼ 20 mg RNAlater stabilized brain tissue samples (Qiagen, Hilden, Germany). RNA was quantified measuring the absorbance at 260 and 280 nm using NanoDrop™ 2000c spectrometer (Thermo Fisher Scientific, Waltham, MA, United States). Complementary DNA was synthesized from 250 ng total RNA using the iScript cDNA Synthesis Kit (BioRad, Munich, Germany) according to the manufactuer’s instructions and was stored at −80°C. qRT-PCR was conducted using a CfX 96 Connect™ system (BioRad, Munich Germany) (Reutzel et al., 2020). All primers were received from biomol (Hamburg, Germany) or biomers (Ulm, Germany). cDNA for qRT-PCR was diluted 1:5 with RNase free water (Qiagen, Hilden, Germany) and all samples were performed in triplicates. PCR cycling conditions were an initial denaturation at 95°C for 3 min, followed by 45 cycles of 95°C for 10 s, 58°C for 45 s and 72°C for 29 s. Gene expression was analyzed using the –(2ΔΔ Cq) method using BioRad CfX manager software and were normalized to the expression levels of beta 2 microglobulin (B2M) and phosphoglycerate kinase 1 (PGK1). All primer sequences, product sizes and primer concentrations used for quantitative real-time PCR (QRT-PCR) are listed in Table 1.

Table 1. Oligonucleotide primer sequences, product sizes and primer concentrations for quantitative real-time pcr; bp: base pair.
Citrate Synthase Activity
Citrate synthase (CS) activity in isolated mitochondria was determined photometrically as previously published (Pohland et al., 2018; Reutzel et al., 2020). Briefly, a sample of the isolated mitochondria in MiR05 was thawed on ice and the reaction medium [100 μl of 0.1 mM 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB), 25 μl of 10% Triton X-100, 50 μl of 10 mM oxaloacetate, 25 μl of 12.2 mM acetyl coenzyme A, and 790 μl purified water] was mixed and warmed to 30°C for 5 min. Afterward, 10 μl isolated mitochondria were added to the reaction mixture and transferred into a 10 mm quartz cuvette (Hellma® Analytics, Müllheim, Germany). CS activity was measured at 412 nm using a GENESYS 5 spectrophotometer (Spectronic via Thermo Fisher Scientific, Waltham, MA, United States). All samples were measured in duplicates.
Protein Quantification
Protein content was measured with the Pierce™ Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States) according to the Manufacturer’s instructions.
Statistical Analysis
Unless otherwise stated, values are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed by applying a two sided, unpaired Student’s t-test and for multiple comparisons a one-way ANOVA with Tukey’s multiple comparison post test (Prism 8.0 GraphPad Software, San Diego, CA, United States). Statistical significance was defined for p*-values of <0.05, p** < 0.01, p*** < 0.001 and p**** < 0.0001.
Results
To assess the relationship between Aβ formation, cognitive performance and mitochondrial brain function, we used an amyloid-based mouse strain and exclusively followed these parameters longitudinally from one cohort over 13 months. Thy1-APP751SL mice were examined at the ages of 3, 7, and 13 months in comparison to their non-transgenic controls.
Measurement of Soluble Amyloid-β1–40 Brain Levels
Thy1-APP751SL mice aged 13 months showed significantly increased levels of soluble human Amyloid β1–40 brain levels (206 pg/mg protein) compared to mice aged 7 (24 pg/mg protein) and 3 months (9 pg/mg protein) (Figure 1).
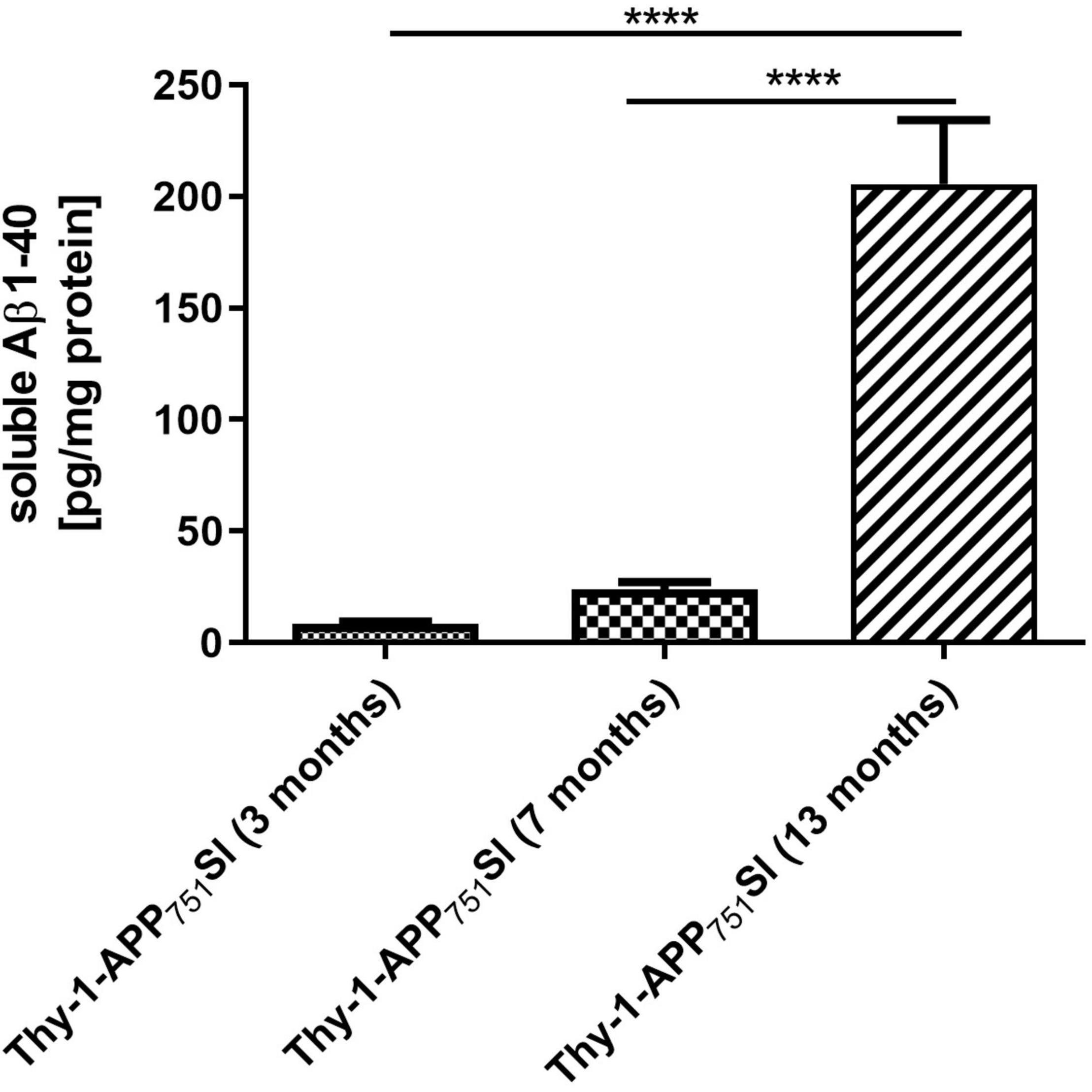
Figure 1. Soluble human Aβ1–40 levels in homogenates isolated from brains of 3, 7, and 13 months old Thy1-APP751SL mice determined using ELISA. Data represent means ± SEM. n = 10; One-Way ANOVA with Tukey’s post-hoc test with ****p < 0.0001.
Effect on Cognition and Nest-Building Behavior During Aging
Thy1-APP751SL mice aged 3, 7, and 13 months were tested over a time period of 5 minutes in the one-trial-Y-Maze test compared to wild-type mice of the same age. The Y-Maze test provides insight into motor skills and spatial learning memory (Wolf et al., 2016). Thy1-APP751SL aged 3 months showed a significant reduced alternation rate compared to control animals of the same age (p < 0.05). At ages 7 and 13 months, there was a numerical decrease in the ratio of alternation compared with controls of the same age (Figure 2A). The results suggest early impairments of the spatial learning memory in 3 months old Thy1-APP751SL mice.
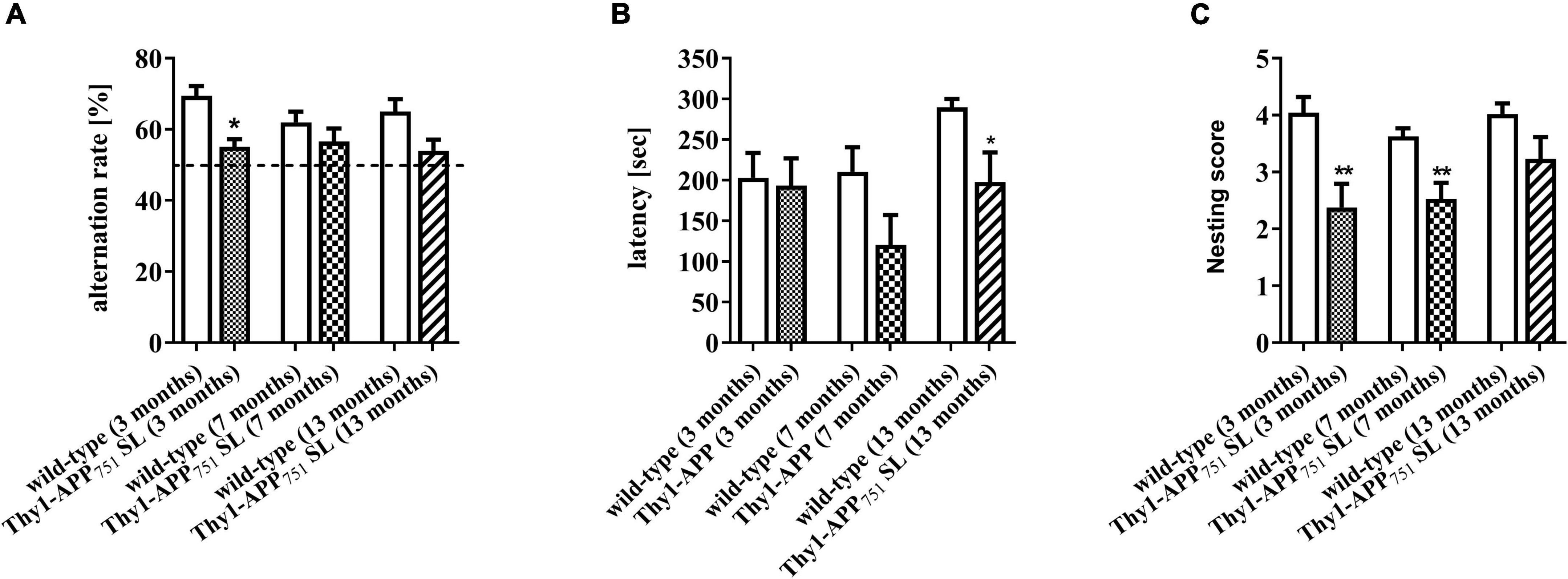
Figure 2. Alternation rate in the Y-Maze spontaneous alternation test (A), latency time day 2 in the Passive Avoidance test (B), and nesting scores (C) of 3, 7, and 13 months old Thy1-APP751SL mice compared to wild-type mice of the same age. n = 13 t-test against wild-type mice of the same age with *p < 0.05 and **p < 0.01.
Thy1-APP751SL mice at the age of 13 months showed a significant reduced latency time in the Passive Avoidance test compared to wild-type mice (Figure 2B). The passive avoidance test gives information on how a mice can remember a negative stimulus and may be an indicator for the function of the long-term memory (Shiga et al., 2016). Results indicate deficits in the long-term memory of 13 months old Thy-1-APP751 mice. At the ages of 3 and 7 months, Thy1-APP751SL mice had a significant deficit in their nest building behavior compared to wild-type mice (Figure 2C). This test is used to assess thermoregulatory behavior, positive motivational states and welfare in mice (Deacon, 2006; Gaskill et al., 2013). Results may indicate decreased motivated behavior and increased stress levels of those mice.
Effects on Mitochondrial Bioenergetics During Aging in Thy1-APP751SL Mice
The mitochondrial respiratory chain in the inner mitochondrial membrane is the main site of ATP formation. The respiratory chain builds up a proton gradient via a total of four respiratory chain complexes [complex I, NADH: ubiquinone oxidoreductase (CI); complex II, succinate-coenzyme Q reductase (CII); complex III, cytochrome-c oxidoreductase (CIII); complex IV, cytochrome-c oxidase (CIV)] the so-called membrane potential, which is the driving force for ATP synthase [F1/F0-ATPase (CV)] for the synthesis of ATP from ADP and inorganic phosphate (Larosa and Remacle, 2018).
Additionally, mitochondrial respiration was measured in isolated brain mitochondria (Figures 3A–C). Thy1-APP751SL mice at the age of 13 months showed significant deficits in CI, CI + CII, and CIV respiration compared to wild-type mice of the same age. However, no significant changes in citrate synthase activity, a mitochondrial mass marker (Rabøl et al., 2010), were observed at any measurement time point (data not shown). MMP-levels showed a numerically increase at the age of 13 months (Table 2). ATP- levels in brains of 3, 7, and 13 months old Thy1-APP751SL were measured compared to wild-type mice of same age. Basal ATP-levels were significant elevated in brains of 7 months old Thy1-APP751SL mice (p < 0.05).
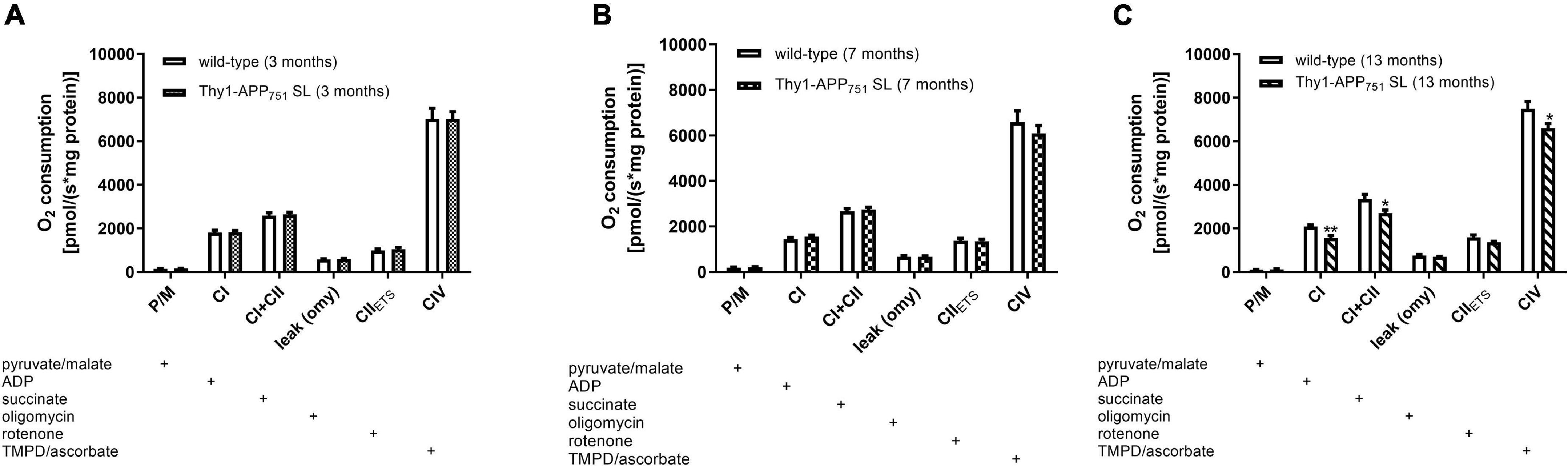
Figure 3. Mitochondrial Respiration in isolated brain mitochondria from 3 (A), 7 (B), and 13 (C) months old Thy1-APP751SL mice compared to wild-type control animals of the same age; the addition of a substance into the oxygraph chamber is indicated with a plus sign; n = 10; mean ± SEM and t-test against wild-type mice of the same age with *p < 0.05 and **p < 0.01.

Table 2. Basal ATP- and MMP-levels in dissociated brain cells of 3, 7, and 13 months old Thy1-APP751SL mice compared to wild-type (wt) control animals of the same age.
Effect on Gene Expression During Aging
Expression of genes involved in mitochondrial biogenesis and mitochondrial respiration, as well as genes which are involved in synaptic plasticity and neurogenesis were measured in brains of 3, 7, and 13 months old Thy1-APP751SL mice compared to wild-type mice of the same age. Expression of β-AMPK and Mitochondrial transcription factor A (TFAM) were significant reduced starting at an age of 7 months indicating impaired mitochondrial biogenesis. All other genes involved in the formation of new mitochondria (CREB1, NRF1, PGC1-α, and SIRT-1) showed a significant decrease at the age of 13 months compared to wild-type mice of the same age. Genes involved in synaptic plasticity, learning behavior and neuronal remodelling (GAP43, SYP1, and BDNF) showed a significant decline in 13 months old Thy1-APP751SL mice compared to wild-type animals. The mRNA expression of complex IV of the respiratory chain showed an increase in brains of 3 months old transgenic mice. As age progresses, mRNA expression of complex I and IV drops significantly at the age 13 months in Thy1-APP751SL (Table 3). Results of mRNA expression patterns of wild-type mice during aging are shown in the supplements.
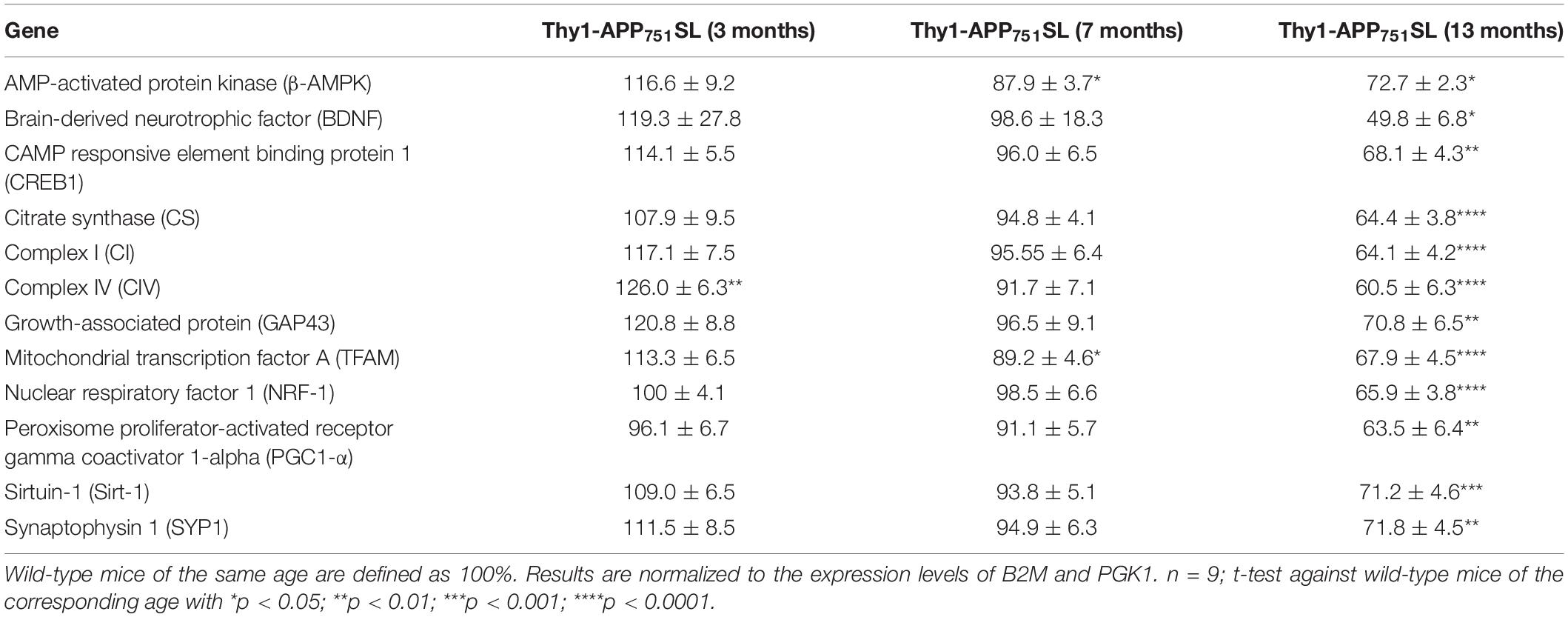
Table 3. Relative normalized mRNA Expression of 3, 7, and 13 months old Thy1-APP751SL mice compared to wild-type mice of the same age.
Discussion
As Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease of aging, we followed the longitudinal brain aging process in Thy1-APP751SL mice as an model of early AD (Hauptmann et al., 2009). Additionally, on the molecular level, it appears that there is a decline in mitochondrial function in both, the aging process and AD (Cadonic et al., 2016; Perez Ortiz and Swerdlow, 2019). Research into AD makes it essential to work with animal models that reflect as many characteristics of this multifactorial disease as possible. However, no animal model exists that can represent all of the extensive molecular changes as well as cognitive impairments in its full extent (Hall and Roberson, 2012; Puzzo et al., 2015). We therefore examined our Aβ-based mouse model, which has not been studied longitudinally before, over a period of 13 months. Neuron-specific expression of the human APP gene depends on the promoter used. While the platelet-derived growth factor B-chain (PDGF-B) and thymocyte differentiation antigen 1 (THY-1) promoters are very specific in leading to APP overexpression in neurons, the prion protein (PrP) promoter is much more active than the first two, but less specific and leads to increased APP expression in liver, kidney, and other tissues (Asante et al., 2002). Cognitive deficits measured by the Y-Maze, passive avoidance, and nesting tests were observed as early as 3 months of age. In the current study, most biochemical changes, including increased Aβ1–40 levels, manifested at 13 months of age in the brains of Thy1-APP751SL mice.
Cognitive Function and Aβ1–40 Levels
Previous studies reported elevated levels of human Aβ starting at an age of 3 months in brains of Thy1-APP751SL mice (Blanchard et al., 2003; Hauptmann et al., 2009; Pohland et al., 2018; Eckert et al., 2020). In the current study soluble Aβ1–40 levels were relatively low at the age of 3 months (9 pg/mg protein), at age of 7 months, only 24 pg/mg soluble Aβ1–40 were determined. At the age of 13 months, significantly elevated Aβ1–40 levels of 206 pg/mg protein were formed in the brain of Thy1-APP751SL mice. These results are in contrast to previous studies including our owns (see Table 4), in which almost three times higher Aβ values were reported at 3 months of age in the same mouse strain (Schuessel et al., 2005; Pohland et al., 2018; Eckert et al., 2020). We can only speculate about the reasons, which may lie in the housing conditions or in the handling of the mice which, however, have not deviated from our standard protocols. In any case, the overall results of the cohort appear to be consistent over time. The most pronounced changes in biochemical parameters were associated with the highest Aβ levels in the oldest mice (see below). Interestingly, cognitive functions were already altered in younger mice of the same cohort.
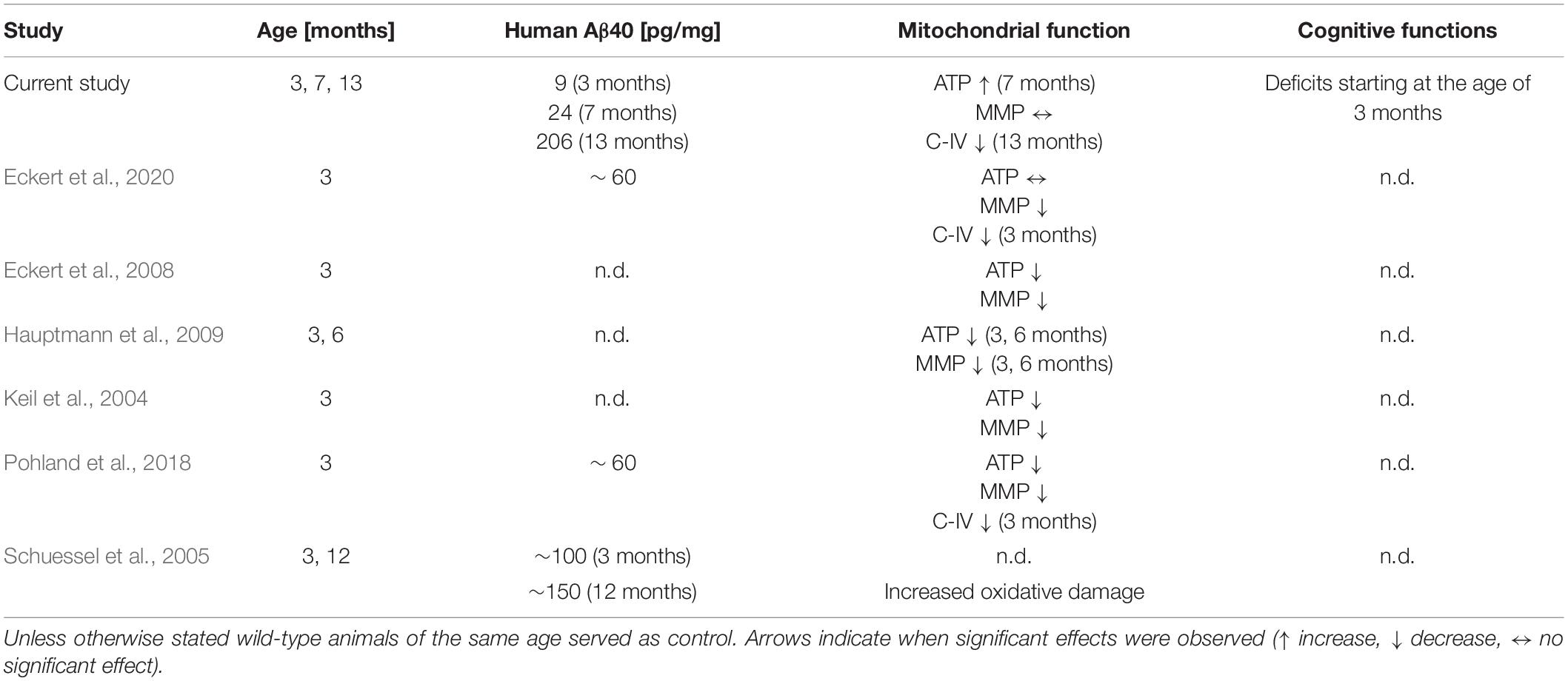
Table 4. Results of studies that determined mitochondrial function in Thy1-APP751SL mice compared with the current longitudinal study focusing on amyloid levels in the brain; n.d. not determined.
Cognitive functions and behavior were assessed using three different behavioral tests. Deficits in nest-building behavior and spatial learning memory of transgenic animals were already observed at the age of 3 months. Deterioration in long-term memory was noted at 7 months of age and manifested at 13 months of age. Most studies in transgenic AD mouse models suggesting cognitive dysfunction due to increasing soluble Aβ levels as well as Aβ plaque deposition (Huber et al., 2018; He et al., 2020; Johnson et al., 2020). Since cerebral Aβ levels of 7-months old animals were very low, one could conclude that the onset of cognitive dysfunction may not be directly related to elevated APP processing. Accordingly, for 18-month-old APP/PS1 mice, Vartak et al. (2019) did not find a correlation between cognitive function and soluble/insoluble Aβ1–40 and Aβ1–42 content in the brain. Eckert et al. (2008) observed a marked increase in soluble intracellular Aβ levels starting at 3 months of age in Thy1-APP751SL mice and a parallel correlation to reduced mitochondrial function, although cognitive functions were not determined in this study.
Longitudinal Mitochondrial Parameters in Thy1-APP751SL Mice
In agreement with previous studies, a significantly reduced activity of mitochondrial C-IV activity was measured (Table 4). However, this deficit again did not occur until 13 months of age, at relatively high Aβ levels of 203 pg/mg protein. Previous studies reported changes in C-IV activity as early as 3 months of age, when Aβ levels were around 60 pg/mg protein (Pohland et al., 2018; Eckert et al., 2020). At 7 months of age, our animals showed only a small and non-significant decrease in C-IV activity. However, at this time point, ATP levels were significantly increased, possibly as a result of a compensatory response.
Interestingly, the improvement in C-IV activity and mitochondrial membrane potential in 3-month-old Thy1-APP751SL mice treated with olesoxime was accompanied by an increase in Aβ levels (Eckert et al., 2020). In this study, the ATP levels in the brains of the 3 month old animals, as in the present one, did not show any change.
Longitudinal Gene Expression Patterns in Thy1-APP751SL Mice
Again, the changes in the brains of Thy1-APP751SL mice were greatest at 13 months of age. The expression levels of all genes studied were significantly reduced at this age. In addition to the plasticity markers SYP1 and GAP43, this also affected all mitochondria-related genes. In 7 months old Thy1-APP751SL mice there is a decrease in the mitochondrial transcription factor A (TFAM) and AMPK-activated protein kinase (β-AMPK), which is one of the key activator for mitochondrial transcription (Li et al., 2017). In accordance with the literature, it is described that in 3xTG-AD mice the mRNA expression of TFAM and other genes involved in mitochondrial biogenesis seem to be already decreased from 1 month of age. This observation was made before a significant deposition of Aβ oligomers took place in brains of transgenic mice (Singulani et al., 2020). Furthermore, the literature describes that in 12-month-old APP mice and hippocampal neurons significant deficits in mitochondrial biogenesis and mitochondrial dynamics take place, which are linked to deficits in the cognitive function of the mice (Manczak et al., 2018). Additionally, in hippocampal neurons Aβ25–35 incubation for 24 h leads to a significant decrease in AMPK and SIRT1 expression (Dong et al., 2016). New mitochondria formation, which is indispensable for maintaining energy supply, is significantly reduced in brains of AD patients, as well as in cellular models of AD. For example, Sheng et al. detected reduced mRNA and protein levels of PGC1-α, NRF-1, NRF2-2, and TFAM in postmortem AD brains and cellular models of AD (Sheng et al., 2012). Directly relevant to our results reduced levels of genes involved in mitochondrial biogenesis were determined in brains of 6- and 12 months old APP transgenic mice (Manczak et al., 2016, 2018; Song et al., 2018).
Limitations
There are limitations to our study. Based on our previous findings, we aimed to investigate the deterioration of mitochondrial and cognitive function with increasing age. However, contrary to expectations, the parameters showed changes only at older ages. Although the cause of this phenomenon cannot be substantiated with evidence, the data nevertheless show that mitochondrial parameters and cognition deteriorate with elevated Aβ levels with increasing age.
Conclusion
A strength of our investigation is the observation of a cohort of Thy1-APP751SL mice over a relatively long period of time. The data suggest a strong relationship between the Aβ formation in the brain of the animals and the measured parameters. This underlines the importance of aging processes for a possible therapy or prevention of the disease.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by V54-19 c 20/15 – FU/113.
Author Contributions
MR generated the figures and involved in most of the experiments during the study and guided AJ. AJ was responsible for the measurement of ELISA and qPCR. MR, RG, and AJ were involved in the animal studies and measured mitochondrial function. GE supervised the work. MR and GE wrote the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Erich Gnaiger for the development of the protocol for the respiration measurements.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.875989/full#supplementary-material
References
Asante, E. A., Gowland, I., Linehan, J. M., Mahal, S. P., and Collinge, J. (2002). Expression pattern of a mini human PrP gene promoter in transgenic mice. Neurobiol. Dis. 10, 1–7. doi: 10.1006/nbdi.2002.0486
Blanchard, V., Moussaoui, S., Czech, C., Touchet, N., Bonici, B., Planche, M., et al. (2003). Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp. Neurol. 184, 247–263.
Cadonic, C., Sabbir, M. G., and Albensi, B. C. (2016). Mechanisms of mitochondrial dysfunction in Alzheimer’s disease. Mol. Neurobiol. 53, 6078–6090.
Deacon, R. M. J. (2006). Assessing nest building in mice. Nat. Protoc. 1, 1117–1119. doi: 10.1038/nprot.2006.170
Dong, W., Wang, F., Guo, W., Zheng, X., Chen, Y., Zhang, W., et al. (2016). Aβ25-35 suppresses mitochondrial biogenesis in primary hippocampal neurons. Cell Mol. Neurobiol. 36, 83–91. doi: 10.1007/s10571-015-0222-6
Eckert, A., Hauptmann, S., Scherping, I., Rhein, V., Müller-Spahn, F., Götz, J., et al. (2008). Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener. Dis. 5, 157–159. doi: 10.1159/000113689
Eckert, G. P., Eckert, S. H., Eckmann, J., Hagl, S., Muller, W. E., and Friedland, K. (2020). Olesoxime improves cerebral mitochondrial dysfunction and enhances Aβ levels in preclinical models of Alzheimer’s disease. Exp. Neurol. 329, 113286. doi: 10.1016/j.expneurol.2020.113286
Gaskill, B. N., Karas, A. Z., Garner, J. P., and Pritchett-Corning, K. R. (2013). Nest building as an indicator of health and welfare in laboratory mice. J. Vis. Exp. 51012. doi: 10.3791/51012
Hagl, S., Berressem, D., Grewal, R., Sus, N., Frank, J., and Eckert, G. P. (2016). Rice bran extract improves mitochondrial dysfunction in brains of aged NMRI mice. Nutr. Neurosci. 19, 1–10. doi: 10.1179/1476830515Y.0000000040
Hagl, S., Kocher, A., Schiborr, C., Eckert, S. H., Ciobanu, I., Birringer, M., et al. (2013). Rice bran extract protects from mitochondrial dysfunction in guinea pig brains. Pharmacol. Res. 76, 17–27. doi: 10.1016/j.phrs.2013.06.008
Hall, A. M., and Roberson, E. D. (2012). Mouse models of Alzheimer’s disease. Brain Res. Bull. 88, 3–12. doi: 10.1016/j.brainresbull.2011.11.017
Hampel, H., Hardy, J., Blennow, K., Chen, C., Perry, G., Kim, S. H., et al. (2021). The Amyloid-β pathway in Alzheimer’s disease. Mol. Psychiatry 26, 5481–5503. doi: 10.1038/s41380-021-01249-0
Hauptmann, S., Scherping, I., Dröse, S., Brandt, U., Schulz, K. L., Jendrach, M., et al. (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 30, 1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005
He, Z., Yang, Y., Xing, Z., Zuo, Z., Wang, R., Gu, H., et al. (2020). Intraperitoneal injection of IFN-γ restores microglial autophagy, promotes amyloid-β clearance and improves cognition in APP/PS1 mice. Cell Death Dis. 11:440. doi: 10.1038/s41419-020-2644-4
Huber, C. M., Yee, C., May, T., Dhanala, A., and Mitchell, C. S. (2018). Cognitive decline in preclinical Alzheimer’s disease: amyloid-beta versus tauopathy. J. Alzheimers Dis. 61, 265–281. doi: 10.3233/JAD-170490
Johnson, E. C. B., Ho, K., Yu, G.-Q., Das, M., Sanchez, P. E., Djukic, B., et al. (2020). Behavioral and neural network abnormalities in human APP transgenic mice resemble those of App knock-in mice and are modulated by familial Alzheimer’s disease mutations but not by inhibition of BACE1. Mol. Neurodegener. 15:53.
Keil, U., Bonert, A., Marques, C. A., Scherping, I., Weyermann, J., Strosznajder, J. B., et al. (2004). Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J. Biol. Chem. 279, 50310–50320. doi: 10.1074/jbc.M405600200
Krumschnabel, G., Fontana-Ayoub, M., Sumbalova, Z., Heidler, J., Gauper, K., Fasching, M., et al. (2015). Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol. Biol. 1264, 245–261. doi: 10.1007/978-1-4939-2257-4_22
Larosa, V., and Remacle, C. (2018). Insights into the respiratory chain and oxidative stress. Biosci. Rep. 38, 1–14. doi: 10.1042/BSR20171492
Li, P. A., Hou, X., and Hao, S. (2017). Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 95, 2025–2029. doi: 10.1002/jnr.24042
Manczak, M., Kandimalla, R., Fry, D., Sesaki, H., and Reddy, P. H. (2016). Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 25, 5148–5166. doi: 10.1093/hmg/ddw330
Manczak, M., Kandimalla, R., Yin, X., and Reddy, P. H. (2018). Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 1332–1342. doi: 10.1093/hmg/ddy042
Mota, B. C., and Sastre, M. (2021). The role of PGC1α in Alzheimer’s disease and therapeutic interventions. Int. J. Mol. Sci. 22:5769. doi: 10.3390/ijms22115769
Naseri, N. N., Wang, H., Guo, J., Sharma, M., and Luo, W. (2019). The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 705, 183–194. doi: 10.1016/j.neulet.2019.04.022
Perez Ortiz, J. M., and Swerdlow, R. H. (2019). Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 176, 3489–3507. doi: 10.1111/bph.14585
Pohland, M., Pellowska, M., Asseburg, H., Hagl, S., Reutzel, M., Joppe, A., et al. (2018). MH84 improves mitochondrial dysfunction in a mouse model of early Alzheimer’s disease. Alzheimers Res. Ther. 10:18. doi: 10.1186/s13195-018-0342-6
Puzzo, D., Gulisano, W., Palmeri, A., and Arancio, O. (2015). Rodent models for Alzheimer’s disease drug discovery. Expert Opin. Drug Discov. 10, 703–711. doi: 10.1517/17460441.2015.1041913
Rabøl, R., Larsen, S., Højberg, P. M. V., Almdal, T., Boushel, R., Haugaard, S. B., et al. (2010). Regional anatomic differences in skeletal muscle mitochondrial respiration in type 2 diabetes and obesity. J. Clin. Endocrinol. Metab. 95, 857–863. doi: 10.1210/jc.2009-1844
Reutzel, M., Grewal, R., Dilberger, B., Silaidos, C., Joppe, A., and Eckert, G. P. (2020). Cerebral mitochondrial function and cognitive performance during aging: a longitudinal study in NMRI mice. Oxid. Med. Cell Longev. 2020:4060769. doi: 10.1155/2020/4060769
Schuessel, K., Schäfer, S., Bayer, T. A., Czech, C., Pradier, L., Müller-Spahn, F., et al. (2005). Impaired Cu/Zn-SOD activity contributes to increased oxidative damage in APP transgenic mice. Neurobiol. Dis. 18, 89–99. doi: 10.1016/j.nbd.2004.09.003
Sheng, B., Wang, X., Su, B., Lee, H.-G., Casadesus, G., Perry, G., et al. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 120, 419–429. doi: 10.1111/j.1471-4159.2011.07581.x
Shiga, T., Nakamura, T. J., Komine, C., Goto, Y., Mizoguchi, Y., Yoshida, M., et al. (2016). A single neonatal injection of ethinyl estradiol impairs passive avoidance learning and reduces expression of estrogen receptor α in the hippocampus and cortex of adult female rats. PLoS One 11:e0146136. doi: 10.1371/journal.pone.0146136
Singulani, M. P., Pereira, C. P. M., Ferreira, A. F. F., Garcia, P. C., Ferrari, G. D., Alberici, L. C., et al. (2020). Impairment of PGC-1α-mediated mitochondrial biogenesis precedes mitochondrial dysfunction and Alzheimer’s pathology in the 3xTg mouse model of Alzheimer’s disease. Exp. Gerontol. 133:110882. doi: 10.1016/j.exger.2020.110882
Song, C., Li, M., Xu, L., Shen, Y., Yang, H., Ding, M., et al. (2018). Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 29, 1517–1524. doi: 10.1097/WNR.0000000000001139
Vartak, R. S., Rodin, A., and Oddo, S. (2019). Differential activation of the mTOR/autophagy pathway predicts cognitive performance in APP/PS1 mice. Neurobiol. Aging 83, 105–113. doi: 10.1016/j.neurobiolaging.2019.08.018
Wolf, A., Bauer, B., Abner, E. L., Ashkenazy-Frolinger, T., and Hartz, A. M. S. A. (2016). Comprehensive behavioral test battery to assess learning and memory in 129S6/Tg2576 Mice. PLoS One 11:e0147733. doi: 10.1371/journal.pone.0147733
Zhao, N., and Xu, B. (2021). The beneficial effect of exercise against Alzheimer’s disease may result from improved brain glucose metabolism. Neurosci. Lett. 763:136182. doi: 10.1016/j.neulet.2021.136182
Keywords: mitochondrial dysfunction, amyloid-beta, Alzheimers’s disease, transgenic mouse models, longitudinal studies
Citation: Reutzel M, Grewal R, Joppe A and Eckert GP (2022) Age-Dependent Alterations of Cognition, Mitochondrial Function, and Beta-Amyloid Deposition in a Murine Model of Alzheimer’s Disease—A Longitudinal Study. Front. Aging Neurosci. 14:875989. doi: 10.3389/fnagi.2022.875989
Received: 14 February 2022; Accepted: 30 March 2022;
Published: 02 May 2022.
Edited by:
Anne Eckert, University Psychiatric Clinic Basel, SwitzerlandReviewed by:
Heather M. Wilkins, University of Kansas Medical Center Research Institute, United StatesKaren Schmitt, Central Institute of Mental Health (ZI), Germany
Copyright © 2022 Reutzel, Grewal, Joppe and Eckert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gunter P. Eckert, ZWNrZXJ0QHVuaS1naWVzc2VuLmRl