
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Aging Neurosci. , 04 July 2022
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.855776
This article is part of the Research Topic Trends in Biomarkers for Neurodegenerative Diseases: Current Research and Future Perspectives View all 18 articles
 Mobina Fathi1†
Mobina Fathi1† Kimia Vakili1†
Kimia Vakili1† Shirin Yaghoobpoor1†
Shirin Yaghoobpoor1† Mohammad Sadegh Qadirifard2,3Mohammadreza Kosari4Navid Naghsh5
Mohammad Sadegh Qadirifard2,3Mohammadreza Kosari4Navid Naghsh5 Afsaneh Asgari taei6
Afsaneh Asgari taei6 Andis Klegeris7Mina Dehghani8Ashkan Bahrami9Hamed Taheri10
Andis Klegeris7Mina Dehghani8Ashkan Bahrami9Hamed Taheri10 Ashraf Mohamadkhani11Ramtin Hajibeygi12
Ashraf Mohamadkhani11Ramtin Hajibeygi12 Mostafa Rezaei Tavirani13*
Mostafa Rezaei Tavirani13* Fatemeh Sayehmiri1*†
Fatemeh Sayehmiri1*†Parkinson's disease (PD), the second most common neurodegenerative disorder, is characterized by neuroinflammation, formation of Lewy bodies, and progressive loss of dopaminergic neurons in the substantia nigra of the brain. In this review, we summarize evidence obtained by animal studies demonstrating neuroinflammation as one of the central pathogenetic mechanisms of PD. We also focus on the protein factors that initiate the development of PD and other neurodegenerative diseases. Our targeted literature search identified 40 pre-clinical in vivo and in vitro studies written in English. Nuclear factor kappa B (NF-kB) pathway is demonstrated as a common mechanism engaged by neurotoxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA), as well as the bacterial lipopolysaccharide (LPS). The α-synuclein protein, which plays a prominent role in PD neuropathology, may also contribute to neuroinflammation by activating mast cells. Meanwhile, 6-OHDA models of PD identify microsomal prostaglandin E synthase-1 (mPGES-1) as one of the contributors to neuroinflammatory processes in this model. Immune responses are used by the central nervous system to fight and remove pathogens; however, hyperactivated and prolonged immune responses can lead to a harmful neuroinflammatory state, which is one of the key mechanisms in the pathogenesis of PD.
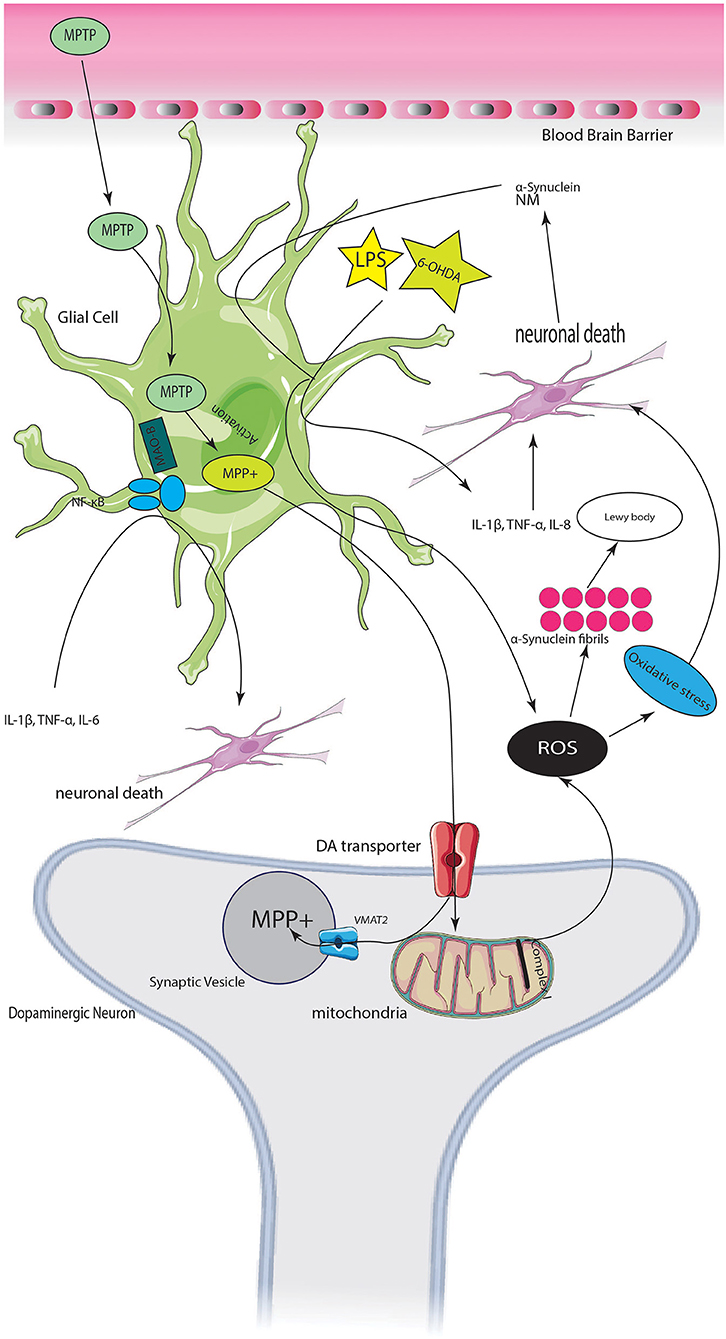
Graphical Abstract. Servier (2022).
Parkinson's disease (PD) is a neurodegenerative disorder defined by dopaminergic neuronal loss in the substantia nigra (SN) pars compacta, aggregation of misfolded α-synuclein (α-syn) as Lewy bodies, and motor dysfunction (Trudler et al., 2015). Degeneration of the nigrostriatal pathway leads to hallmark motor symptoms of this disease, including bradykinesia, rigidity, tremor at rest, and postural instability. Furthermore, most patients exhibit non-motor symptoms, such as sleep disorders, autonomic nervous system dysfunction, and cognitive impairment (Trudler et al., 2015; Kempuraj et al., 2016). Currently, pharmacological and surgical interventions are the main treatment options in PD which provide only symptomatic relief for patients. Among various disease-specific pathogenetic mechanisms, neuroinflammation plays a prominent role in the onset and progression of a broad range of neurodegenerative disorders, including PD, Alzheimer's disease (AD), and multiple sclerosis (MS). It has been demonstrated that the sterile neuroinflammation in neurodegenerative diseases embodies a cascade of events involving abnormal protein aggregates, upregulated inflammatory mediators, and activation of non-neuronal glial cells, which leads to neuronal damage. Neurodegeneration, in turn, induces further glial activation and neuroinflammation in the central nervous system (CNS). Out of the several different non-neuronal cell types of the CNS, microglia are the most prominent contributors to neuroimmune reactions. They express several different pattern recognition receptors (PRRs), such as toll-like receptor (TLR)2, TLR4, TLR9, and receptor for advanced glycation end products (RAGE), that recognize various pathogens and abnormal proteins. Microglial activation leads to the secretion of various pro-inflammatory mediators, including interleukin (IL) 1β, IL-6, and tumor necrosis factor α (TNF-α), to restore tissue hemostasis and also facilitate tissue repair (Ransohoff and Brown, 2012; Ransohoff, 2016; Molteni and Rossetti, 2017). Astrocytes represent another type of glial cells, which are critical for neuronal networks and maintenance of brain tissue homeostasis. Similar to microglia, astrocytes express PRRs and can contribute to neuroimmune responses by releasing a broad range of inflammatory mediators (Cunningham et al., 2019). Although acute inflammatory response can clear abnormal proteins, eliminate cell debris, and promote tissue repair, persistent inflammation is detrimental since it produces harmful inflammatory mediators and cytotoxins, as well as inhibits neural regeneration.
Neuroinflammation contributes to PD pathogenesis throughout the progression of this disease from early α-syn aggregation to causing dopaminergic cell loss and ultimately PD symptoms. For example, the accumulation of misfolded α-syn has been suggested to cause dysregulation of both innate and adaptive immune responses (Harms et al., 2021). Strong support to the neuroinflammatory hypothesis of PD is provided by genome-wide association studies linking sporadic PD to polymorphism in the human leukocyte antigen (HLA) region containing HLA-DR gene (Mohamadkhani et al., 2009; Simón-Sánchez et al., 2009; Hamza et al., 2010; Ahmed et al., 2012). The sustained inflammatory responses have been described in both PD patients and animal models of this disease, which through various mechanisms can cause neuronal dysfunction. In addition to reactive microglia and astrocytes releasing neurotoxic molecules, other mechanisms contributing to neuronal death and neurodegeneration in PD have been discovered; they include brain infiltration and activation of inflammatory mast cells and T lymphocytes, increased oxidative stress, and upregulation of inflammatory signaling molecules (Lyman et al., 2014; Jarrott and Williams, 2016; Kempuraj et al., 2016).
Activated glial cells and infiltrating peripheral immune cells are the main sources of the various pro-inflammatory mediators contributing to the onset and progression of PD. Activated glial cells release a broad range of both pro- and anti-inflammatory cytokines, such as TNF-α, IL-1β/IL-1α, IL-6, IL-8, and IL-10, as well as the brain inflammatory protein called glia maturation factor (GMF), which regulates functions of glial cells and can also induce neurodegeneration in the brain (Lim et al., 2004; Zaheer et al., 2008a,b; Tore and Tuncel, 2009; Kempuraj et al., 2013; Molteni and Rossetti, 2017; Mukai et al., 2018). Activated mast cells release potentially harmful mediators, such as proteases, utilizing the degranulation process (Tore and Tuncel, 2009; Taracanova et al., 2017; Mukai et al., 2018).
Dopaminergic neurotoxins such as 6-OHDA, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and its metabolite 1-methyl-4-phenylpyridinium (MPP+) have been shown to adversely alter neuronal functions in both mature and developing nervous tissue. They have been used to induce PD in animal models where they, in addition to dopaminergic neuronal damage, cause glial activation, oxidative stress, mitochondrial damage, and the release of inflammatory cytokines (Członkowska et al., 1996; Stojkovska et al., 2015; Trudler et al., 2015; Pourasgari and Mohamadkhani, 2020). In vivo and in vitro studies show that the mechanism of action for dopaminergic neurotoxins could involve high-mobility group box 1 (HMGB1) (Huang et al., 2017), which is a nuclear DNA binding non-histone protein that facilitates the assembly of nucleoprotein complexes, but can also be released extracellularly and act as a damage-associated molecular pattern (DAMP) triggering neuroimmune responses (Sims et al., 2010).
The present systematic review aims to identify and evaluate the underlying genes and mechanisms of neuroinflammation in PD pre-clinical studies. Following this, a gene list-independent approach to compare different disease models is used.
Electronic databases PubMed, Scopus, Google Scholar, Web of Science, and EMBASE were searched using the medical subject headings (MeSH) aimed at identifying all research articles related to the topic “The association between neuroinflammation and Parkinson's disease.” Two authors independently conducted the search using search strategies specific for each database and reviewed all relevant peer-reviewed articles published before April 2022. The following search terms were used “[(neuro-inflammation) AND Parkinson's disease],” “[(neuro-inflammation) AND neurodegenerative disease],” “[((inflammation) AND Parkinson's disease) AND brain],” “(adaptive immunity) AND (Parkinson), (immunological biomarkers) AND (Parkinson), (hHumoral immunity) AND (Parkinson),” “[((inflammation) AND neurodegenerative disease) AND brain],” “[(inflammatory markers) AND Parkinson's disease],” and “[(inflammatory markers) AND neurodegenerative disease],” ”[((inflammation) AND Parkinson's disease) AND animal].” A total of 40 articles were included.
This review article considered both quantitative and qualitative data on the association between neuroinflammation and PD, which were obtained by reviewing all available in vivo and in vitro pre-clinical studies relevant to this topic. Review articles and studies with human subjects were excluded. Only articles published in English were included. Duplicate data and low-quality studies, identified by the Systematic Review Center for Laboratory animal Experimentation (SYRCLES) quality assessment checklist (Hooijmans et al., 2014), were excluded. All eligible studied, both with positive and negative findings have been included in this systematic review. SYRCLES quality assessment checklist (Hooijmans et al., 2014) was used to assess selection bias, performance bias, detection bias, attrition bias, reporting bias, and other sources of bias (see Supplementary Table 1).
The relevant studies were selected after the title, abstract, and full-text screening of the articles. In addition, the reference lists of selected studies were reviewed to identify any additional articles, should they have not been identified by the search process. The following information was extracted from each of the identified studies: cell culture and animal model used, the size of the PD model and control groups, inflammatory markers studied, and the key results obtained (see Table 1). This study was approved by the Iranian National Committee for Ethics in Biomedical Sciences (Code of Ethics: IR.SBMU.RETECH.REC.1399.992).
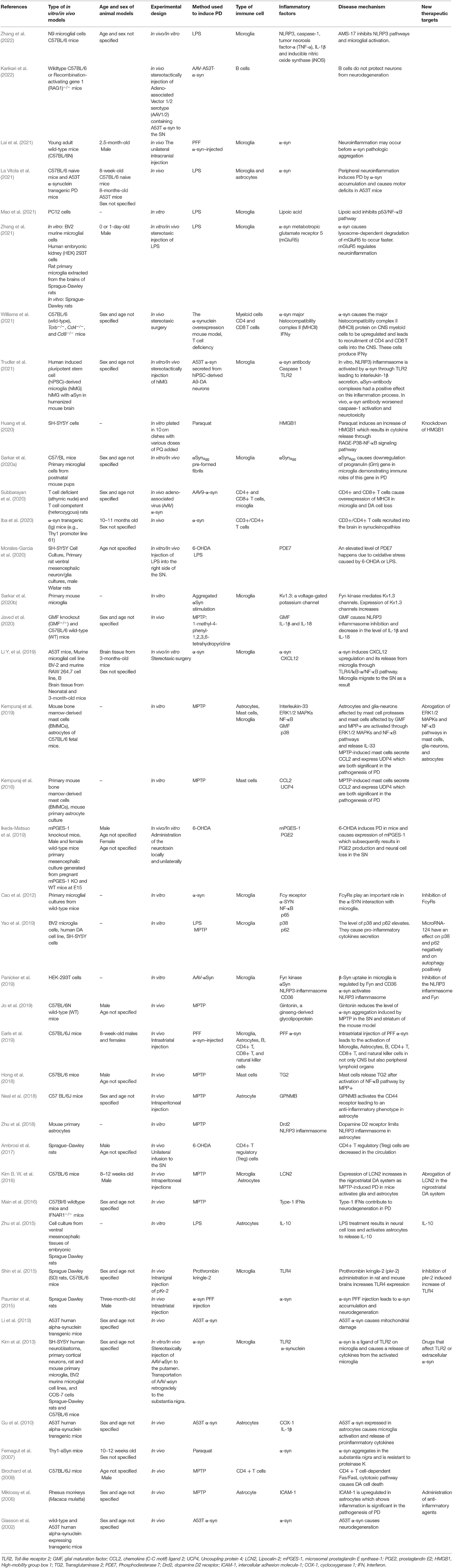
Table 1. A rapid review of articles assessed.
The current study has been run based on PRISMA checklist (Tore and Tuncel, 2009). At first, 2,499 papers were identified through the search on PubMed, and 7,560 were identified through searching other databases (Scopus, Google Scholar, Web of Science, and EMBASE). However, 4,823 of those 10,059 articles were excluded due to duplication in different databases. After screening the abstracts and titles of these obtained studies, 2,121 more papers were excluded due to unavailability of abstracts, or articles written in non-English, or being review articles. Moreover, 3,003 more papers were removed due to being irrelevant to the main subject or human studies. Finally, from the 112 remaining articles, 36 records excluded because of not relevant for the topic of the review. The full texts of the 76 remaining papers were fully assessed, and 36 more studies were excluded due to insufficient or unclear data (n = 19) and low quality (n =17). Finally, 40 articles, published before April 2022, were chosen for systematic review (Figure 1).
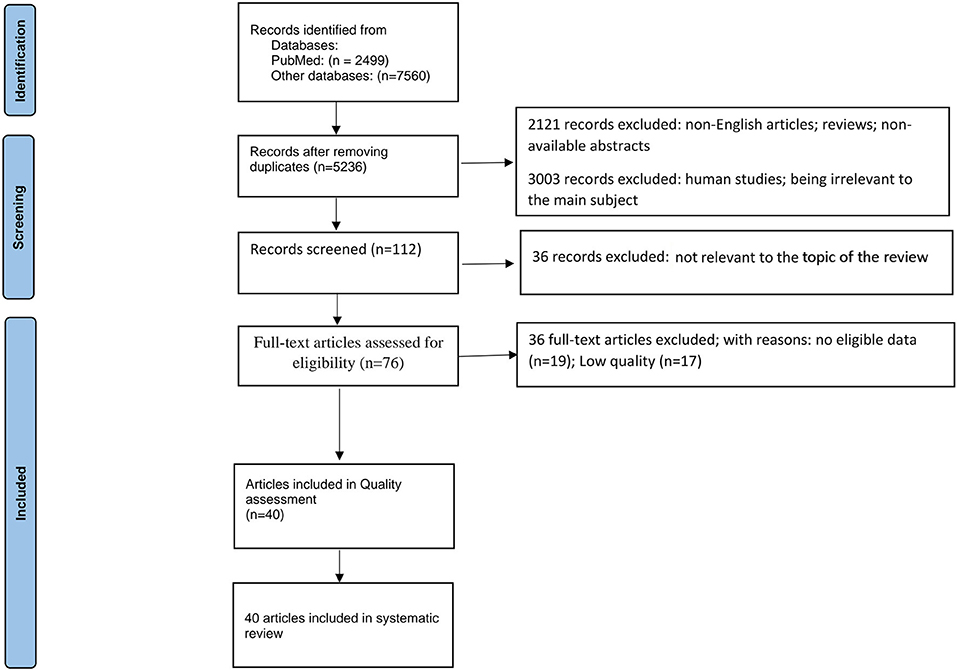
Figure 1. PRISMA 2020 flow diagram for systematic reviews which included searches of databases.
The main results of all the papers included in this review are summarized first based on the neuroinflammatory pathways of PD. The method used to induce PD in animal models might be a key factor which affects the mechanism of neuroinflammation observed in a particular study; therefore, we reviewed the methods in both Table 1 and a separate subsection of the results. The other subsections are introduced based on the most frequently appearing topics and mechanisms discussed in the reviewed articles.
A total of 40 articles are included in this review. Twenty-one out of 40 studies used only in vivo models, 11 were based on in vitro experiments, and 8 studies employed both in vitro and in vivo experiments. A variety of methods were used in different studies to induce PD: 11 studies used MPTP, 10 α-Syn injection, 6 LPS, 5 A53T α-Syn transgenic mice, and 2 paraquat. Two studies used both 6-OHDA and LPS, 1 used both MPTP and LPS, 1 6-OHDA, and 1 used prothrombin kringle 2.
Table 1 illustrates a variety of methods used by the reviewed studies to induce PD in animal models. Each method initiates its own pathway leading to PD-like pathology. Administration of MPTP to different animal species has been used extensively to model PD neuropathology. MPTP mimics the destruction of dopaminergic neurons of the substantia nigra pars compacta observed in PD. The mechanism by which MPTP causes damage to dopaminergic neurons involves a sequence of events including disturbance in the mitochondrial function, oxidative stress and respiratory failure. MPTP leads to inducible nitric oxide synthase (iNOS) overexpression. MPP+ is the active metabolite of MPTP which accumulates in dopaminergic (DA) neurons of the model after treatment with MPTP. MPP+ provokes the production of superoxide radicals which react with nitric oxide and generate peroxynitrite. This substance inhibits the function of many proteins including tyrosine hydroxylase. As a result, the production of dopamine is disturbed leading to damage of DA neurons (Przedborski et al., 2000; Burré et al., 2014, 2018). The loss of DA cells after MPTP administration has also been shown to activate microglia in the SN of rhesus monkeys (McGeer and McGeer, 2007). Therefore, in vivo and in vitro use of MPTP and its metabolite MPP+ can help elucidate the molecular mechanisms of the neuroinflammatory reactions in PD. Injection of lipopolysaccharide (LPS) and the following brain region-specific inflammation in mice provides in vivo models to investigate neurodegenerative diseases such as PD (Noh et al., 2014). LPS treatment in mice causes a decrease in the level of IL-4 and IL-10 but an elevation in the level of TNF-α, interleukin-1β, prostaglandin (PG) E2, and nitric oxide. IL-10 has a neuroprotective effect against LPS intoxication (Tansey and Goldberg, 2010). As shown in Table 1, some studies have used this method to create a PD model. 6-hydroxydopamine (6-OHDA) is another neurotoxin used to create PD models which affects nerve terminals as well as cell bodies and induces death of neurons through inhibition of the mitochondrial respiratory enzymes. In PD models 6-OHDA lesions are made in nigrostriatal dopaminergic pathways (Deumens et al., 2002). One of the studies used paraquat to induce PD in vitro (Trudler et al., 2021). Paraquat is a pesticide exposure to which is epidemiologically known to be a risk factor for PD (Berry et al., 2010). The rotenone-induced PD is another method, which is the best model to study mitochondrial complex I deficiency in PD (Greenamyre et al., 2003).
Synucleins are proteins highly expressed in the brain. There are three members in this protein family: α-, β-, and γ-synuclein. α-Synuclein is a pre-synaptic protein mostly found in nerve terminals (Goedert, 2001). Under physiological conditions, α-Syn interacts with many neuronal proteins to play a variety of functional roles such as inhibiting phospholipase D, regulating microtubules, elevating the rate of tau phosphorylation, etc. It also interacts with soluble NSF attachment protein receptors (SNAREs), which play a role in neurotransmitter release through meditating fusion of vesicles. This indicates that α-Syn is a likely contributor to the release of neurotransmitters (Burré et al., 2010, 2014, 2018). Point mutations in the gene encoding α-Syn (SNCA) are linked to the familial form of PD. A variety of mutations are known to be linked to PD including A53T, A30P, E46K, G51D, and H50Q. We will discuss some of these mutations in transgenic PD models in the following section in more details. Lewy bodies, the hallmark neuronal inclusions observed in neurodegenerative diseases including idiopathic PD, mainly consist of insoluble fibrillar α-syn protein (Goedert, 2001; Marques and Outeiro, 2012; Bendor et al., 2013). In pathological situations, soluble monomeric α-syn generates β-sheet-like oligomers (protofibrils), which form into amyloid fibrils. Amyloid fibrils, then, accumulate in Lewy bodies (Burré et al., 2015). Neurons can also release α-syn, inducing inflammatory responses of microglia (Kim et al., 2013). The fibrillar α-syn, but not its monomeric or oligomeric forms, is the main trigger of neuroinflammation in PD. It interacts with microglial TLR2 as well as another innate immune sensor, the nucleotide oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3), resulting in activation of the nuclear factor κB (NF-κB) and assembly of NLRP3 inflammasomes. This sequence of events ultimately leads to the release of TNF-α and IL-1β by the microglia, two potent pro-inflammatory cytokines that are known to play a role in the pathogenesis of PD (Gustot et al., 2015; Zhou et al., 2016; Panicker et al., 2019; Trudler et al., 2021). AMS-17 is known to inhibit NLRP3 pathways and activation of microglia. Further investigations using PD model mice indicate that microglial endocytosis of α-syn, impairment of lysosomal functions, and the release of lysosomal protease cathepsin B into the cytoplasm are required for α-syn-induced assembly of the NLRP3 inflammasomes in microglia. α-Syn also negatively regulates the AMP-activated protein kinase (AMPK)-mediated autophagy, which leads to the intracellular accumulation of reactive oxygen species (ROS), followed by activation of the NLRP3 inflammasome (Zhou et al., 2016). Furthermore, α-syn binds to the microglial CD36 receptor, resulting in the activation of Fyn kinase and subsequently the activation of the NF-kB pathway. Fyn kinase also participates in the activation of NLRP3 since it is decreased in the fyn−/− mice injected with adeno-associated virus overexpressing α-syn (AAV-αSyn) compared to the wild-type animals (Panicker et al., 2019). In addition, Fcγ receptors (FcγR) located on the surface of microglia can mediate α-syn intracellular trafficking leading to pro-inflammatory activation of these cells (Cao et al., 2012). α-Syn can also promote neuroinflammation by interacting with astrocytes since it has been shown to upregulate secretion of interleukin IL-6 and expression of intercellular adhesion molecule-1 (ICAM-1) by human astrocytes. Notably, the PD-causing mutations of α-syn upregulate its potency as a pro-inflammatory stimulant of astrocytes (Klegeris et al., 2006; Jo et al., 2019). In addition to stimulating glial cells, α-syn facilitates aggregation of other inflammatory proteins. For example, co-localization and co-aggregation of α-syn with S100A9 protein have been observed in Lewy bodies and neuronal cells in the SN and frontal lobe areas of PD patients. S100A9 is a member of a family of structurally homologous calcium-binding S100 proteins, which are involved in many inflammatory and neurodegenerative diseases (Srikrishna, 2012; Markowitz and Carson, 2013; Horvath et al., 2018). Aggregated a-syn leads to downregulation of progranulin (GRN) gene in microglia which affects immune functions of these cells (Sarkar et al., 2020a). Another effect of α-syn is related to metabotropic glutamate receptor 5 (mGluR5). This receptor has a neuroprotective role, but its lysosome-dependent degradation occurs faster as a result of synucleinopathy (Zhang et al., 2021). Even though α-syn has been demonstrated to be an initiator in neuroinflammation processes, some studies indicated that inflammation can occur before synucleinopathy (Lai et al., 2021).
To further study the pathophysiology of α-syn, various transgenic mouse models of PD have been created including α-syn knockout models and models overexpressing wildtype or mutated human α-syn (Fernagut and Chesselet, 2004). α-Syn KO mice are used to identify the role of α-syn in PD pathogenesis. A study on α-syn KO mice demonstrated that these PD models are resistant to acute administration of MPTP, which highlights the key role of α-syn in increasing the vulnerability of DA neurons to neurodegeneration when exposed to environmental neurotoxins (Dauer et al., 2002). As previously mentioned, two missense mutations of α-syn gene are linked to PD: A53T and A30P. Homozygous transgenic A30P*A53T α-syn mice manifest many features of PD phenotype and are useful models to study this disease (Kilpeläinen et al., 2019). Mutant α-syn in transgenic A53T mice does not form aggregates but is distributed in different parts of neurons abnormally leading to motor impairment in transgenic mice followed by further paralysis and death (Giasson et al., 2002; Gispert et al., 2003). In transgenic mice which overexpressed A53T α-syn selectively in astrocytes, microglia became reactive and produced proinflammatory cytokines, such as IL-1β, and upregulated cyclooxygenase (COX)-1 leading to neurodegeneration (Gu et al., 2010). Accumulation of A53T α-syn in transgenic mice leads to peripheral inflammation and motor deficits (La Vitola et al., 2021). A53T α-syn has a destructive effect on mitochondrial function in the DA neurons of transgenic mice. It causes damage to mitochondrial transport and respiratory mechanisms (Li et al., 2013). Some other transgenic mouse models are also created to investigate PD such as Thy1-aSyn and α-syn pre-formed fibril (PFF)-injected models. Intrastriatal injection of PFF α-syn leads to the activation of microglia, astrocytes, B, CD4+ T, CD8+ T, and natural killer cells in not only CNS but also peripheral lymphoid organs (Earls et al., 2019). Thy1-aSyn models overexpress human wildtype α-syn by the murine Thy-1 promoter. In this model, many manifestations of sporadic PD are seen including inflammation, biochemical and molecular changes resembling those observed in PD (Chesselet et al., 2012). To study pre-clinical stages of PD, Thy1-aSyn transgenic mice are helpful as in this model high levels of α-syn cause no DA neuronal death up to 8 months (Fleming et al., 2008).
As described above, the interaction of α-syn with microglia causes activation of NF-kB, which is central to a broad range of neuroinflammatory processes (Tobon-Velasco et al., 2014); therefore, inhibition of this signaling pathway could be a therapeutic target for PD. Panicker et al. treated Fyn−/− mice with LPS to evaluate the role of Fyn in NF-κB activation (Greenamyre et al., 2003). NF-kB signaling in microglia can be activated by the DAMPs released from damaged CNS cells, such as HMGB1, IL-33, ATP, cytochrome C, mitochondrial DNA, and several different heat shock proteins (HSP) (Klegeris, 2021). HMGB1 is a prototypical DAMP present in most nucleated cells, which can be actively secreted or passively released from stimulated and necrotic cells, respectively. a-Syn-induced upregulation of CXCL12 and its release from microglia through TLR4/IκB-α/NF-κB pathway results in microglia migration to the SN (Li Y. et al., 2019). GMF causes NLRP3 inflammasome inhibition and decreases levels of IL-1β and IL-18 (Javed et al., 2020). An in vitro study showed that knockdown of HMGB1 alleviated upregulation of NF-κB signaling and inflammatory responses; therefore, anti-HMGB1 monoclonal antibody therapy should be considered as a potential treatment strategy for PD (Nishibori et al., 2019; Huang et al., 2020). The pro-neuroinflammatory effects of DAMPs in the CNS can be counterbalanced by several different resolution-associated molecular patterns, including HSP10, αB-crystallin, prothymosin α, and binding immunoglobulin protein (BiP), also known as HSP70 (Wenzel et al., 2020). Thus, an in vitro model of PD was used to demonstrate that HSP70 inhibited the mRNA and protein expressions of NF-κB along with another key pro-inflammatory signaling molecule signal transducer and activator of transcription (STAT)-3 (Li et al., 2019). Notably, select miRNAs, such as miR-124 and miR-7, can also inhibit the progression of neuroinflammation in PD models by modulating NF-κB signaling pathways (Zhou et al., 2016; Yao et al., 2019). One study showed the Lipoic acid (LA) has an anti-inflammatory effect by inhibiting the p53/NF-κB pathway in an LPS-induced model of PD (Mao et al., 2021).
The essential role of mast cells in neuroinflammation is supported by several studies. Kempuraj et al. showed that exposure of murine and human mast cells to MPP+ induced release of chemokine c-c motif ligand 2 (CCL2), which in turn has been implicated in the pathogenesis of PD (Kempuraj et al., 2016). MMP+ treatment-induced secretion of IL-33 and a high level of ROS generation by murine mast cells. In addition, mouse mast cell protease (MMCP)-6 and MMCP-7 triggered the release of IL-33 from glia-neuron mixed cultures and primary mouse astrocytes. All these events involved the activation of the NF-kB pathway demonstrating its critical role in neuroinflammation associated with PD (Kempuraj et al., 2019). Furthermore, MPP+ activated NF-κB in mast cells leading to upregulation of transglutaminase 2 (TG2) and subsequent release of the pro-inflammatory TNF-α and IL-1β. This study also demonstrated that TG2-expressing mast cells recruited into SN tissues might contribute to neuroinflammation in PD (Hong et al., 2018).
Nigrostriatal damage correlates with complex alterations in both central and peripheral immunity (Ambrosi et al., 2017). Regarding in vivo studies, some immunological features change in MPTP-treated mice as a result of the damage to central dopaminergic cells (Bieganowska et al., 1993). In 6-OHDA-treated rats, a decrease in the percentage of circulating CD4+ T regulatory (Treg) cells was observed (Ambrosi et al., 2017). Upregulation of a-syn provokes adaptive immune system (Theodore et al., 2008). Upregulation of the major histocompatibility complex II (MHCII) protein in CNS myeloid cells and recruitment of CD4+ and CD8+ T cells into the CNS occur due to α-syn accumulation (Williams et al., 2021). One study demonstrated that CD3+/CD4+ T cells are recruited into the perivascular parenchyma of the neocortex, hippocampus, and striatum in α-syn transgenic mice. This study supports the role of adaptive immune cells in neuroinflammation in synucleinopathies (Iba et al., 2020). Degeneration of DA cells occurs through a CD4+ T cell-dependent Fas/FasL cytotoxic pathway (Brochard et al., 2009). CD4+ and CD8+ T cells, in an α-syn rat model of PD, cause an upregulation of MHCII in microglia and significant loss of DA neurons (Subbarayan et al., 2020). One study on A53T α-syn mice indicated that T cells contribute to the neurodegeneration caused by α-syn, while B cells have no neuroprotective effect against this neurodegeneration (Karikari et al., 2022). Taken together, these studies indicate that the adaptive immune system is associated with neuroinflammation and neurodegeneration in PD.
TLR4 is a cell-surface protein that interacts with bacterial endotoxin and other pathogen-associated molecular patterns (PAMPs) as well as several different DAMPs (Leitner et al., 2019). This receptor is expressed by many different cell types including glial cells. TLR4 activation causes motor impairment in MPTP-treated mice. TLR4 signaling has been suggested to initiate neuroinflammation in PD since microglia express this receptor and α-syn activates it. A decrease in the inflammatory response to α-syn oligomers is observed in murine macrophages derived from bone marrow. In TLR4-deficient mice, the activation of microglia and astrocytes is inhibited by the suppression of NF-κB and the NLRP3 inflammasome signaling pathways (Campolo et al., 2019; Hughes et al., 2019; Shao et al., 2019). An elevated level of microglial TLR4 damages the nigrostriatal DA system. The increase in this receptor occurs after the administration of prothrombin kringle-2 (pKr-2) in rat and mouse brains, which indicates pKr-2 as a new potential TLR4-linked therapeutic target for PD (Shin et al., 2015). Lee et al. report that apoptosis signal-regulating kinase 1 (ASK1) is responsible for the MPTP-induced glial activation and neurotoxicity (Lee et al., 2012). PD model mice were also used to identify glial lipocalin-2 (Lcn-2) as a protein contributing to the disease pathogenesis since its levels were increased in the SN and striatum of the MPTP-treated animals (Kim B. W. et al., 2016).
A microsomal isoform of prostaglandin E synthase-1 (mPGES-1) plays a vital role in the inflammatory processes of peripheral tissues and the CNS by producing the inflammatory PGE2. It contributes to clinical manifestations of inflammation, such as pyrexia and pain, and has been shown to contribute to accelerated neuronal death in PD (Uematsu et al., 2002; Engblom et al., 2003; Kamei et al., 2004). Furthermore, mPGES-1 is upregulated in nigrostriatal DA neurons from postmortem PD brain specimens and the 6-hydroxydopamine (6-OHDA) model of PD. Elevated PGE2 produced by this enzyme is believed to contribute to the DA neuronal death in this model (Ikeda-Matsuo et al., 2019). The type 7 cyclic nucleotide phosphodiesterase 7 (PDE7) is another pro-inflammatory enzyme revealed by several pre-clinical models to contribute to PD pathogenesis. PDE7 inhibitors display anti-inflammatory and neuroprotective activities and can stimulate adult neurogenesis both in vivo and in vitro (Morales-Garcia et al., 2011, 2015, 2017). Recently, small non-coding RNAs, such as microRNAs (miRNAs), have been identified as biomarkers of neurodegenerative processes and shown to be involved in PD pathogenesis (Kim et al., 2016). Specific miRNAs bind to the three prime untranslated regions (3′-UTRs) of target mRNAs regulating gene expression in the post-transcriptional phase (Bartel, 2004). In the MPTP model of PD, type-I IFNs mediate neuroinflammation in its early stages and worsen PD pathology (Main et al., 2016).
As seen in Table 1, BV2 murine microglial cells (four articles) and and SH-SY5Y human neuroblastoma cell line (four articles) are the two most commonly-used cell lines to induce PD in vitro in the articles included in this review. Six in vitro studies used rat/mice primary neuron/glia to model PD rather the previously mentioned cell lines. Primary neuronal cultures are useful PD models obtained from the embryonic rodent brain which resemble the morphology and physiology of human neurons (Lopes et al., 2017a). Experimental variations and difficulty in maintenance are some disadvantages of this model (Slanzi et al., 2020). To study the role of microglial cells in PD pathology, the immortalized murine microglial cell line BV-2 has been widely used instead of primary microglial cells. LPS administration in this model leads to pathological pattern of PD similar to that observed in microglial cells in vitro and in vivo (Henn et al., 2009). SH-SY5Y human neuroblastoma cells is a widely-used PD model which mimics some aspects of DA neuron phenotype, such as the expression of tyrosine hydroxylase, dopamine-β-hydroxylase, and dopamine transporter (Hong-rong et al., 2010). SH-SY5Y neuroblastoma can be differentiated to cells that are similar to cholinergic, dopaminergic, or noradrenergic neurons (Slanzi et al., 2020). However, one disadvantage of this model is that there is no standard differentiation protocol and variation in the source of the cells and their culture maintenance techniques lead to different results. In order to provide a PD model, SH-SY5Y neuroblastoma lineage is manipulated both chemically and genetically. MPP+, 6-OHDA, and rotenone treatment as well as overexpression of mutated α-syn are some examples. SH-SY5Y cells cannot transform MPTP to its metabolite and must be treated by MPP+ itself (Xicoy et al., 2017). Some other cell lines are also mentioned in Table 1. For example, PC12 cells are derived from rat pheochromocytoma of the adrenal medulla. These cells produce and secrete catecholamines. However, due to their tumoral origin, they may manifest altered signaling pathways (Slanzi et al., 2020). HEK 293 cell line (or immortalized human embryonic kidney cells) has been used for large-scale experiments but their non-neuronal origin is the main disadvantage of this model (Falkenburger and Schulz, 2006). Animal models of PD are appropriate for studies on motor deficits in PD as the disease progresses. However, since PD is a human-specific neurodegenerative diseases, its pathology, such as damage to DA neurons, needs to be induced. No animal model has been able to recreate all the PD aspects so far. In vitro models provide a controlled environment to investigate the disease mechanisms. However, these models do not have the complexity of CNS. In addition, the source of cells used, their morphology, physiology, and maintenance under different culture conditions result in different outcomes (Lopes et al., 2017a).
The complex pathological features of PD include the death of dopaminergic neurons in SN as well as α-syn aggregation accompanied by neuroinflammation (Hirsch and Hunot, 2009; Tansey and Goldberg, 2010; Hirsch et al., 2012; Guo et al., 2017; Mohamadkhani, 2018). In the current systematic review, we considered 40 original in vitro and in vivo studies describing the molecular mechanism, signaling pathways, and other factors that potentially contribute to the PD pathophysiology in animal models. Our review highlights the following main findings: (1) the methods applied to create PD animal models include treatment of the animals with MPTP, LPS, 6-OHDA, paraquat, and rotenone, in addition to transgenic animal models (Giasson et al., 2002; Miklossy et al., 2006; Fernagut et al., 2007; Gu et al., 2010; Li et al., 2013; Paumier et al., 2015; Zhu et al., 2015, 2018; Kempuraj et al., 2016, 2019; Kim B. W. et al., 2016; Main et al., 2016; Hong et al., 2018; Neal et al., 2018; Earls et al., 2019; Ikeda-Matsuo et al., 2019; Jo et al., 2019; Li Y. et al., 2019; Panicker et al., 2019; Yao et al., 2019; Huang et al., 2020; Javed et al., 2020; Morales-Garcia et al., 2020; Sarkar et al., 2020a; Lai et al., 2021; La Vitola et al., 2021; Trudler et al., 2021; Williams et al., 2021; Zhang et al., 2021). (2) α-syn is an important regulator of glial immune responses, which can induce neuroinflammation and, thus, lead to PD development (Cao et al., 2012; Kim et al., 2013; Ikeda-Matsuo et al., 2019; Jo et al., 2019; Panicker et al., 2019); (3) the NF-κB pathway, induced by α-syn, contributes to the neuroinflammatory process during PD progression (Zhou et al., 2016; Li et al., 2019; Nishibori et al., 2019; Yao et al., 2019; Huang et al., 2020; Wenzel et al., 2020; Klegeris, 2021); (4) mast cells are important contributors to the disease pathogenesis in PD mouse models (Kempuraj et al., 2016, 2019; Hong et al., 2018); (5) adaptive immune system plays role in neuroinflammation and neurodegeneration in PD pathogenesis (Bieganowska et al., 1993; Brochard et al., 2009; Ambrosi et al., 2017; Iba et al., 2020). (6) additional factors contributing to PD neuroinflammation involve the pKr-2 protein, which can facilitate neuroinflammation and PD progression in rodent models by upregulating microglial TLR4 (Shin et al., 2015); mPGES-1, which is upregulated in the SN of 6-OHDA-induced PD models and could lead to neurotoxicity (Uematsu et al., 2002; Engblom et al., 2003; Kamei et al., 2004; Morales-Garcia et al., 2011, 2015, 2017; Kim et al., 2016; Ikeda-Matsuo et al., 2019); and type-I IFN, which aggravates PD by inducing neuroinflammation in the early stages of this disease (Main et al., 2016).
MPTP is one of the agents used to create animal Parkinson models (Miklossy et al., 2006; Kim B. W. et al., 2016; Main et al., 2016; Hong et al., 2018; Neal et al., 2018; Zhu et al., 2018; Kempuraj et al., 2019; Yao et al., 2019). For performing its toxicity, MPTP is initially transformed to MPP+. In DA neurons, ASK1 conveys the MPP+-induced signals leading to the generation of glial-activating molecules like COX-2. During the MPTP-induced toxicity in mice, ASK1 signaling plays a significant role as a connection between neuroinflammation and oxidative stress (Lee et al., 2012). A review by Guo et al. illustrates that ASK1 signaling is linked to the pathogenesis of several neurodegenerative diseases including PD (Guo et al., 2017). Therefore, ASK1 is important in the neurotoxicity caused by MPTP treatment. Furthermore, it is suggested that ASK1 may be a target for treating or preventing PD and other neurodegenerative diseases (Guo et al., 2017). Treatment of mice with MPTP also increases glial Lcn-2 levels, according to Kim B. W. et al. (2016). This is consistent with previously reported inflammatory functions of this protein potentially playing a role in various age-related CNS diseases. Lcn-2 has been shown to promote cell death and iron dysregulation, in addition to neuroinflammation, leading to cognitive impairments. Therefore, elevated Lcn-2 levels can be considered a risk factor for age-related CNS disorders (Dekens et al., 2021). Additionally, it is indicated that MPP+ causes superoxide radicals to form, which combine with nitric oxide to form peroxynitrite. Many proteins, including tyrosine hydroxylase, are inhibited by this chemical. As a result, dopamine synthesis is disrupted, leading to injury of DA neurons (Liberatore et al., 1999; Przedborski et al., 2000, 2004). McGeer et al. demonstrated that in the SN of rhesus monkeys, the loss of DA cells after MPTP treatment caused microglia activation, which contributes to neuroinflammation (McGeer and McGeer, 2007) (Figure 2).
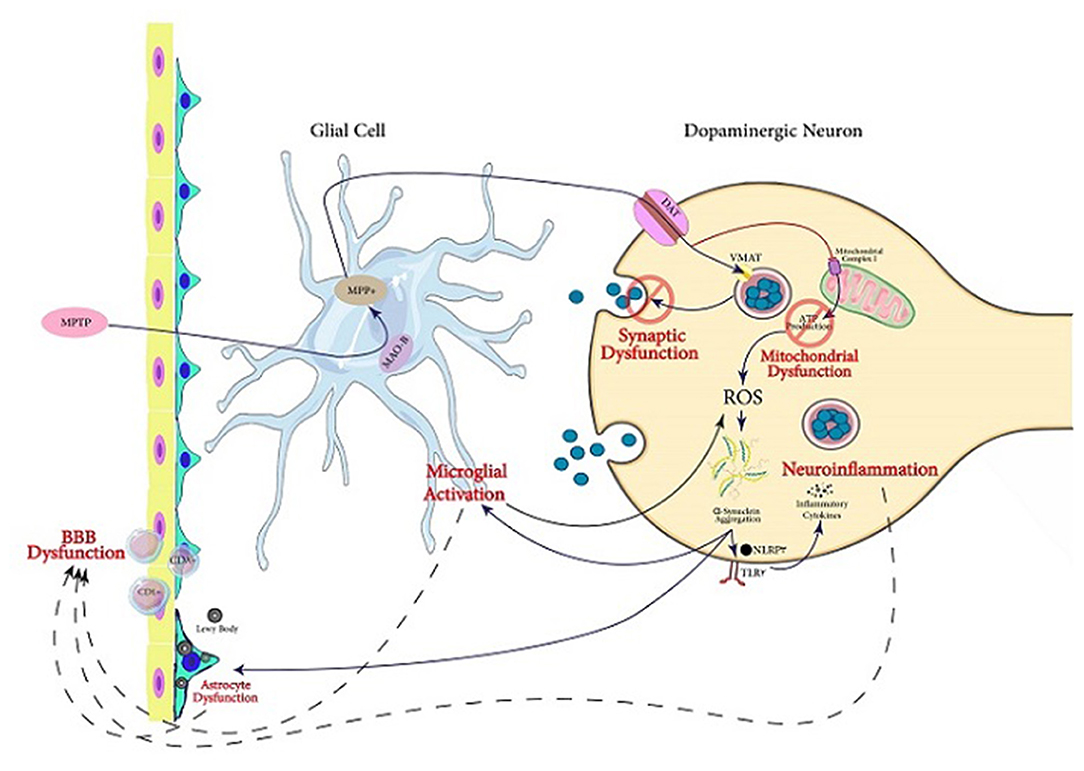
Figure 2. Servier (2022) mechanisms involved in PD pathogenesis. In this figure, the mechanisms following MPTP exposure through which dopaminergic cell death occurs are illustrated. The active metabolite of MPTP, MPP+, is produced in glial cells and transfers into dopaminergic neurons via DA transporter. In the neuron, this activated metabolite causes synaptic dysfunction as well as mitochondrial dysfunction which triggers aggregation of α-Syn. It also can lead to neuroinflammation and microglial activation. Acting together, the mentioned mechanisms can lead to dysfunction of BBB, which is one of the main pathological findings in PD, in addition to Lewy bodies and α-Syn aggregation.
An LPS-induced mouse model of neuroinflammation has been shown to be a useful tool for studying the pathogenic mechanisms behind neurodegeneration and testing possible therapeutic agents (Noh et al., 2014; Zhu et al., 2015; Panicker et al., 2019; Yao et al., 2019; Morales-Garcia et al., 2020; La Vitola et al., 2021; Zhang et al., 2021). The TLR4 and NF-κB signaling pathway is activated by LPS injections, which stimulates microglia. This causes release of IL-6, TNF-α, and insulin-like growth factor 1 (IGF-1), which can be beneficial or harmful to surrounding tissues depending on conditions (Wyss-Coray and Mucke, 2002; Block et al., 2007; Noh et al., 2014). In both the mouse and the rat, LPS injection causes the expression of pro-inflammatory markers such as TNF-α and IL-1b in the entire brain and plasma (Qin et al., 2007; Henry et al., 2009; Nikodemova and Watters, 2011; Oskvig et al., 2012; Molteni et al., 2013). LPS injections produce systemic inflammation and neuroinflammation, which lead to an increase in Aβ levels and neuronal cell death, resulting in cognitive impairment. Thus, systemic inflammation can play a role in the progression of cognitive impairments observed in Alzheimer's disease (AD) and PD (Zhao et al., 2019). According to the study by Morales-Garcia et al. (2020), PDE7 is involved in the progression of neuronal damage in neurodegenerative illnesses, in part due to its role in the regulation of neuroinflammation, suggesting that it could be a key factor in the advancement of PD. In fact, this study detected a strong rise of PDE7 expression, both in vitro and in vivo, primarily in microglial cells, implying that PDE7 expression is critical for the neuroinflammatory response triggered by these cells, which leads to an increase in DA neuron degeneration (Morales-Garcia et al., 2020).
Another agent used for creating PD animal models is 6-OHDA (Ikeda-Matsuo et al., 2019; Morales-Garcia et al., 2020). 6-OHDA was the first chemical agent shown to have selective neurotoxic effects on catecholaminergic pathways (Ungerstedt, 1968; Sachs and Jonsson, 1975). 6-OHDA causes catecholaminergic neuron degeneration by using the same catecholamine transport mechanism as dopamine and norepinephrine (Figure 3) in the substantia nigra, the nigrostriatal tract, or the striatum to specifically target the nigrostriatal DA system (Perese et al., 1989; Przedbroski et al., 1995). DA neurons start degenerating 24 h after 6-OHDA (Przedbroski et al., 1995; Schwarting and Huston, 1996) injections into the SN or the nigrostriatal tract, and striatal dopamine is reduced 2–3 days later (Faull and Laverty, 1969). There is substantial evidence that oxidative stress plays a role in the neurotoxic effects of 6-OHDA. In the presence of iron, 6-OHDA-induced degeneration has been linked to the production of hydrogen peroxide and hydroxyl radicals (Sachs and Jonsson, 1975). The fact that intranigral iron injection has similar neurotoxic effects to 6-OHDA suggests that iron may play a role in 6-OHDA-induced degeneration. Furthermore, investigations have shown that 6-OHDA causes a decrease in glutathione peroxidase (GSH) and superoxide dismutase (SOD) activity as well as an increase in malondialdehyde levels in the striatum (Perumal et al., 1992; Kumar et al., 1995). 6-OHDA is similarly harmful to mitochondrial complex I, resulting in the generation of superoxide free radicals (Hasegawa et al., 1990; Cleeter et al., 1992).
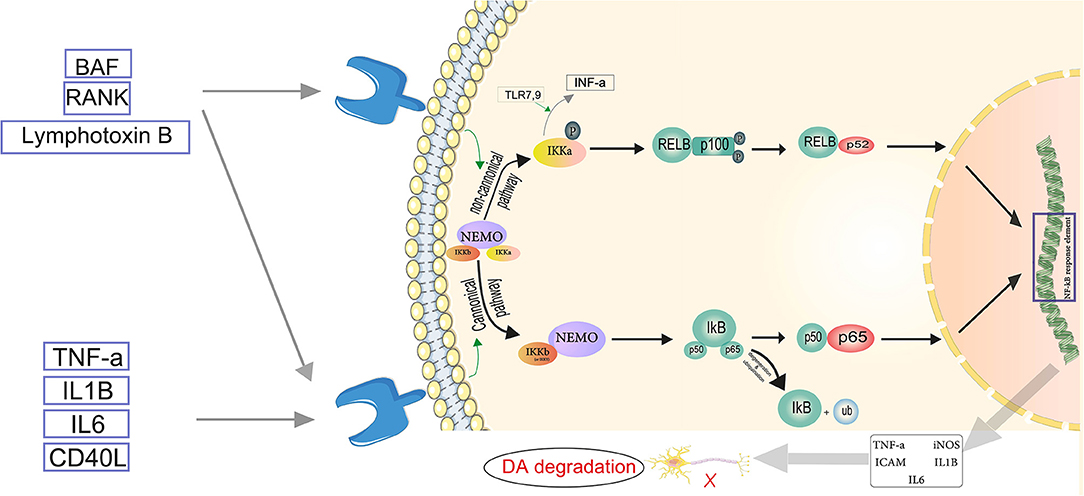
Figure 3. Servier (2022) the neuroinflammatory cascade mediated by NF-κB. TNF-α, IL-1β, IL-6, and CD-40L are proinflammatory substances activating the canonical pathway. In the canonical pathway, an inhibitor of κβ kinase (IKK) β (or IKKγ) is required for NF-κB activation. IKKβ phosphorylates Iκβ. The regulatory subunit of the IKK complex is the NF-κB essential modulator (NEMO). In the cytosol, IκB is degraded by proteasomes, and the phosphorylated heterodimer of NF-κB (p50–p65) is transferred to the nucleus and binds to the NF-κB response element. Thus, pro-inflammatory mediators such as TNF-α, IL-1β, IL-6, iNOS, and ICAM become activated, which play role in the degradation of dopaminergic neurons (DA). In the non-canonical pathway, NEMO phosphorylates IKK-α and induces proteasomal destruction as well as proteasomal processing of p100, a subunit of the NF-B heterodimer, creating the p52-RELB active heterodimer. IKκ induces INF-α production, triggered by TLR7,9. The p52-RELB active heterodimer enters the nucleus and binds to the NF-κB response element, regulating the expression of pro-inflammatory factors. Eventually NF-κB mediated neuroinflammation plays role in Parkinson's disease through DA degradation.
Paraquat, a well-studied neurotoxic agent, is commonly regarded as one of the environmental elements contributing to PD (Fernagut et al., 2007; Huang et al., 2020). Because of its structural similarities to MPP+, Paraquat has presented as a possible risk factor for PD. Paraquat can pass the blood-brain-barrier, but only to a limited degree. It induces a dose-dependent decrease in DA nigral neurons and striatal DA innervation, and subsequent reduced mobility, when administered systemically to mice (Boireau et al., 1995). Paraquat method of action is thought to entail oxidative stress, and its harmful effects could be mediated through the mitochondria because of its structural resemblance to MPP+ (Betarbet et al., 2002). Paraquat has been shown in several studies to cause neuroinflammation and microglial activation. Underlying inflammatory processes greatly increase the sensitivity of DA neurons to toxic damage (Purisai et al., 2007; Mitra et al., 2011). According to a study by Ishola et al. TNF-α levels in the midbrain were considerably elevated by Paraquat, indicating neuroinflammation (Ishola et al., 2018). TNF-α is a cytokine that makes up the acute phase reaction and is a cell signaling protein implicated in inflammatory cascades (Tweedie et al., 2007; Ishola et al., 2013). Xiao et al. reported that Paraquat could activate BV-2 microglia and cause neuroinflammation. This involves inhibition of Akt1 activation, mediated by the increased ROS. Also, Paraquat treatment was followed by a significant increase in the expression of M1 microglia markers including TNF-α, IL-1β, and IL-6. Therefore, Paraquat elevated the M1 phenotype of BV-2 microglia (Xiao et al., 2022). According to Mitra et al. Paraquat induced ROS production and differential α-syn expression, which promoted neuroinflammation. This was characterized by area-specific changes in microglial cell localization and appearance, as well as an increase in TNF-α expression patterns in the substantia nigra, frontal cortex, and hippocampus (Mitra et al., 2011). Paraquat may also act through HGMB1 (Huang et al., 2020). In SH-SY5Y cells, a well-established in vitro model for PD research, Paraquat exposure resulted in a significant increase in HMGB1, which was translocated to cytosol and then released into the extracellular milieu of SH-SY5Y cells in a concentration and time-dependent manner. The activation of the RAGE-P38-NF-kB signaling pathway and the generation of inflammatory cytokines such as TNF-α and IL-6 were both reduced when HMGB1 was knocked out. These findings suggest that HMGB1 plays role in Paraquat-induced cell death by increasing neuroinflammatory responses and activating RAGE signaling pathways (Huang et al., 2020).
Another agent used for inducing PD in animal models is rotenone. This agent is an inhibitor of mitochondrial complex I, which is neurotoxic to non-DA and DA neurons (Chen et al., 2006; Choi et al., 2015). This is to some extent because of its inhibitory impact on mitochondria and also causing elevation of oxidative stress. It is indicated in studies using primary cultured glia or microglia that rotenone triggers oxidative stress and neuroinflammation, leading to elevated secretion of pro-inflammatory cytokines (Ye et al., 2016). According to Main et al. type-I IFNs play critical role in mediating the neuroinflammation caused by rotenone in both primary cultured glia and neurons in vitro. They also observed a neuroprotective effect by attenuating type-I IFNs signaling. This confirms the important role of these cytokines in neuroinflammation, which leads to death of neurons in chronic neuropathologies (Main et al., 2017) as was shown in another study by Main et al. on MPTP-induced mice (Main et al., 2016).
According to this systematic review, α-syn has a potentially significant role in the pathogenesis of PD. α-Syn performs many functions through its interactions with different proteins (Burré et al., 2018). It can inhibit phospholipase D (PLD). Many experimental studies, clearly show that α-syn and PLD have a functional interaction. PLD2 overexpression in the rat substantia nigra pars compacta, for example, induced dopaminergic neuron death due to increased lipase activity, while α-syn co-expression decreased PLD2 toxicity (Mendez-Gomez et al., 2018). Furthermore, PLD1 controls autophagic flux and clearance of α-syn aggregates (Bae et al., 2014), whereas overexpression of wild-type α-syn in human neuroblastoma cells reduces PLD1 expression (Conde et al., 2018). An important interaction of α-syn is with SNARE, which facilitates the formation of synaptic vesicles (Burré et al., 2010, 2014, 2018; Huang et al., 2019). SNARE complex assembly requires monomeric α-syn, and deficiencies in this protein can impair vesicle formation. Aggregated α-syn, on the other hand, prevents SNARE-mediated membrane fusion (Hawk et al., 2019). The findings explain a potential mechanism for SNARE-mediated neuronal dopamine release deficiencies leading to neurodegeneration due to a lack of monomeric α-syn and/or increased insoluble α-syn aggregation. However, since SNARE proteins are required for a variety of membrane fusion processes, changes in α-syn could affect vesicle production in a variety of cell types, particularly during fetal development when α-syn is most widely expressed (Baltic et al., 2004). Because vesicle formation is essential for microglia to phagocytose and transfer extracellular cargo to the lysosome for destruction, the deficiency of endogenous α-syn may have an impact on vesicle formation. During the beginning phase of autophagy, SNARE proteins are necessary for the synthesis of precursor vesicles that convert to the phagophore (Wang et al., 2020). This microglial process aids in the elimination of harmful proteins and protects against neuron-derived α-syn aggregation (Choi et al., 2016). Microglia's ability to remove misfolded α-syn could be harmed if vesicle production during autophagy is impaired and consequently contribute to aggregation of α-syn. The SNARE complex is also involved in vesicle exocytosis during microglia cytokine release (Murray et al., 2005). One probable reason for decreased cytokine release is α-syn interaction with SNAP23, a subunit of SNARE complex. Many microglial activities rely on vesicle production to protect against harmful protein buildup and to enhance inflammatory responses in the brain (Gardai et al., 2013).
α-sSyn induces neuroinflammation directly. This process is initiated by α-syn activating microglial TLR2 and NLRP3, which ultimately increases their secretion of IL-1β, TNF-α, and other pro-inflammatory cytokines (Panicker et al., 2019). For example, Bauernfeind et al. (2009) and Qiao et al. (2012) have demonstrated that α-syn is recognized by microglial TLR2, leading to activation of the NF-κB pathway and subsequent production of IL-1β (Bauernfeind et al., 2009; Qiao et al., 2012). Wang et al. (2019) highlight that NLRP3 inflammasome activation not only induces IL-1β secretion by microglia, but also causes a type of inflammatory cell death known as pyroptosis leading to rupture of microglial plasma membrane and further release of IL-1β (Wang et al., 2019). A meta-analysis by Qin et al. showed that peripheral levels of several inflammatory cytokines including TNF-α and IL-1β are higher in PD patients compared to healthy controls (Qin et al., 2016). IL-1β released from microglia has been suggested to participate in inflammatory responses that cause impairment of DA neurons (Block et al., 2007). Injection of IL-1β into the SN of rats induces the death of DA neurons, which is the pathological feature of PD (Ferrari et al., 2006; Block et al., 2007). This, in turn, increases the release of α-syn and creates a vicious circle that amplifies neuroinflammation and accelerates the pathogenesis of PD. Due to its critical role in neuroinflammation, inhibition of NLRP3 pathways is a recently suggested therapeutic strategy for PD (Wang et al., 2019). FcγR on the surface of microglia can mediate α-syn intracellular trafficking, causing microglia to become pro-inflammatory (Cao et al., 2012). According to a study by Javed et al. GMF inhibits the NLRP3 inflammasome and leads to a decrease in the levels of IL-1β and IL-18 (Javed et al., 2020). Miklossy et al. showed that ICAM-1 is upregulated in astrocytes of PD and MPTP-treated monkeys. Also, lymphocyte function-associated antigen 1 (LFA-1) was increased in the reactive microglia. Therefore, inflammation is a potential factor in PD pathogenesis (Miklossy et al., 2006).
The NF-κB pathway is found to be a significant mechanism driving neuroinflammatory reactions (Kempuraj et al., 2019). The two major routes involved in the activation of NF-κB are the canonical or classical pathway and the non-canonical or alternate pathway. The canonical pathway involves dimers of Rel proteins p50 and p65 forming complexes with inhibitory complex IκBα in the cytosol, where they are activated and regulate the production of pro-inflammatory cytokines (Lawrence, 2009). TNF, LPS, IL-1β, and T cell receptor or B cell receptor, as well as other cell, surfaces receptors such as TLRs, TNF receptor, and IL-1 receptor, activate NF-κB throughout the canonical pathway (Baeuerle and Baltimore, 1996). Members of the TNF receptor superfamily, such as B cell-activating factor (BAF), receptor activator of NF-κB (RANK), lymphotoxin B (LT) receptor, and CD40, activate the non-canonical NF-κB pathway in response to diverse stimuli. These receptors also activate the canonical pathway at the same time. Only IKKα homodimers, not IKKβ or IKKγ implicated in the canonical pathway for IκB phosphorylation, are responsible for non-canonical NF-κB pathway activation (Singh et al., 2020) (Figure 3). Inhibition or capture of HMBG1 can suppress this pathway and has been considered as a therapeutic approach. A review by Nishibori et al. illustrates that administration of anti-HMGB1 monoclonal antibodies inhibits Dneuronal loss in a 6-OHDA rat model of PD by suppressing ROS production and neuroinflammation (Nishibori et al., 2019). The protective effects of miR-124 and miR-7 against inflammation may make them protective in PD as already suggested by Titze-de-Almeida and Titze-de-Almeida, who describe the potential benefits of miR-7 replacement therapy in this disease (Titze-de-Almeida and Titze-de-Almeida, 2018). Microglia migration to the substantia nigra is triggered by α-syn-induced increase of CXCL12 and its release from microglia via the TLR4/IB/NF-κB pathway (Ahmed et al., 2012).α-Syn is also a CD36 agonist (Panicker et al., 2019). It has been shown that CD36 and Fyn kinase facilitate the uptake of α-syn by microglia and initiate the assembly of inflammasomes through a protein kinase Cδ-dependent nuclear translocation of NF-κB-p65. Furthermore, uptake of α-syn is reduced in Fyn-deficient microglia and bone marrow-derived macrophages (BMDM) that lack CD36. Therefore, Fyn plays an important role in promoting neuroinflammation in PD (Panicker et al., 2019). In addition, based on a genome-wide association study (GWAS), the Fyn locus is linked to the increased risk of PD (Nalls et al., 2019). Sarkar et al. showed that in PD models, Kv1.3 is elevated. The downstream mediator of the NF-κB and p38 MAPK pathways, the Fyn/PKC signaling cascade, proximally controlled the Kv1.3 upregulation. They showed that Kv1.3 overexpression contributes significantly to neuroinflammation-mediated neurodegeneration in PD models. These findings also point to a possible Kv1.3-mediated signaling pathway which can modulate microglial inflammation in PD (Sarkar et al., 2020b). Sarkar et al. in another study discovered new molecular pathways for α-syn aggregation-induced neuroinflammation. α-Syn upregulated the expression of RNA binding proteins in mouse microglia, implying higher RNA processing and splicing as well as mitochondrial oxidative stress. They also found evidence for decreased microglial progranulin as a new disease mechanism in PD, suggesting that lysosomal dysfunction and autophagy are involved in the disease pathogenesis (Sarkar et al., 2020a). Zhang et al. proposed another novel mechanism. They discovered that mGluR5 was critical in preventing α-syn-induced neuroinflammation. This effect was dependent on the interaction between mGluR5 and α-syn, as well as mGluR5 degradation via the lysosomal pathway induced by α-syn. According to this study, the separation of the mGluR5—α-syn complex in microglia is induced by increased mGluR5 expression (Zhang et al., 2021).
A large number of αsyn transgenic mice models have been developed to replicate a spectrum of clinical and behavioral characteristics of PD and other synucleinopathies. The form of α-syn expressed (wild type vs. mutant) and its promoter-specific expression pattern are the key differences between the existing mouse lines (Kahle, 2008; Chesselet and Richter, 2011; Magen and Chesselet, 2011). A number of mouse lines with α-syn deficiency have also been employed to help researchers understand the roles of this protein in the brain cellular processes (Abeliovich et al., 2000; Specht and Schoepfer, 2004; Kokhan et al., 2012). Transgenic α-syn KO mice (Abeliovich et al., 2000) were initially reported to be unimpaired in spatial memory learning, as demonstrated by the Morris Water Maze (MWM) challenge (Chen et al., 2002). A further study indicated that there are actually cognitive abnormalities in this transgenic mouse model but at more advanced ages compared to the initial report (Kokhan et al., 2012). A likely confounding element in understanding the potential role of α-syn in cognitive dysfunction seems to be the compensatory function of gamma synuclein (γ-syn) in synaptic regulation during the absence of α-syn, resulting in the alleviation of cognitive abnormalities in α-syn-KO mice (Senior et al., 2008). Therefore, γ-syn could perform a compensatory function by restoring cognitive functions in α-syn-KO mice (Hatami and Chesselet, 2015).
A53T α-syn causes neurodegeneration (Giasson et al., 2002; Gu et al., 2010). Mice with A53T α-syn have significant motor impairments, which can lead to paralysis and death (Giasson et al., 2002). In a study by Gu et al. (2010), inflammation and microglial activation were promoted by the A53T α-syn, which caused astrogliosis, particularly in the midbrain, brainstem, and spinal cord. This study also discovered a significant DA neurons loss in the midbrain and motor neurons of the spinal cord in symptomatic mice, which could explain the paralysis characteristics of mutant mice. Furthermore, COX-1-mediated inflammatory pathways can play a role in neurodegeneration, as indicated by the COX-1 inhibitor's ability to lengthen the lifespan of A53T mice. The A53T α-syn mice also developed age-dependent α-syn inclusions, which mimic the pathology seen in people with PD. Overexpression of A53T α-syn inhibited complex I function in DA neurons of transgenic mice (Chinta et al., 2010), depolarized mitochondrial membrane potential, increased ROS in human neuroblastoma cells (Parihar et al., 2009), and induced mitochondrial autophagy in neurons bearing the A53T mutation (Chinta et al., 2010; Choubey et al., 2011). Furthermore, α-syn has been demonstrated to alter mitochondrial motility (Xie and Chung, 2012). One theory for the mechanism underlying the effect of A53T α-syn on mitochondria is that it raises Ca2+ signal in neurons, which has been demonstrated to limit mitochondrial mobility (Yi et al., 2004; Wang and Schwarz, 2009). Previous investigations have suggested that A53T α-syn can create Ca2+ permeable holes in the plasma membrane (Furukawa et al., 2006) and can control Ca2+ entry pathway (Hettiarachchi et al., 2009). Li et al. discovered that A53T α-syn decreased both overall mitochondrial mobility and the fraction of mobile mitochondria. In other words, in A53T α-syn neurons, the percentage of stationary mitochondria rose (Li et al., 2013); therefore, it is probable that A53T α-syn controls syntaphilin or myosin (Kang et al., 2008; Pathak et al., 2010) to improve anchoring of stationary mitochondria. Mice expressing A30P α-syn have failed to exhibit alterations in locomotor activity, and dopamine levels in spite of the buildup of α-syn in various brain regions (Kahle et al., 2001; Yavich et al., 2004; Freichel et al., 2007). Kilpeläinen et al. characterized homozygous double mutant A30P*A53T α-syn transgenic mice and reported that these animals did show early onset and age-dependent alterations in striatal dopaminergic function and locomotor activity, as well as formation of α-syn oligomers, suggesting that it could be a useful tool for modeling early onset PD associated with familial SNCA mutations (Kilpeläinen et al., 2019). Thy-1 promoter has been used for producing transgenic α-syn overexpressing mice. The α-syn transgene is widely expressed in these mice, and cytoplasmic and nuclear inclusions containing human α-syn appear in various brain areas, including the cortex, hippocampus, olfactory bulb, and to a lesser extent, the substantia nigra (Cenci and Björklund, 2020).
Our review also highlighted the roles of adaptive immune response in PD models. T lymphocyte infiltration and enhanced MHC II immunoreactivity were seen in MPTP-treated mice, but no B lymphocyte infiltration was reported (Kurkowska-Jastrzebska et al., 1999; Karikari et al., 2022). Furthermore, the injection of regulatory T cells reduced the neurotoxicity of MPTP (Reynolds et al., 2007). In contrast to T lymphocytes' obvious participation in human PD, there is no indication of B lymphocytes' presence in the brains of animal models of PD. It is worth mentioning that mice lacking both B and T lymphocytes were resistant to MPTP toxicity in a recent study employing the MPTP mouse model of PD (Benner et al., 2008). In the past, research on neuroinflammation in AD and PD has primarily focused on aberrant innate immune system activation (Benner et al., 2008; Rodrigues et al., 2014; Caplan and Maguire-Zeiss, 2018; Labzin et al., 2018). Recent data suggests that changes in the adaptive immune response may also play a role in inflammation and neurodegeneration in Alzheimer's disease and age-related synucleinopathies (Kannarkat et al., 2013; Allen Reish and Standaert, 2015; Baird et al., 2019). α-Syn oligomers and fibrils increased the ratio of CD8+ to CD4+ T cells in the CNS and decreased the expression of STAT3, CD25, and CD127 in CD3+CD4+ T cells. CD4+ T cell infiltration into the CNS has also been linked to changes in the phenotype of brain microglia (Olesen et al., 2018). Thus, CD3+CD4+ T cells' homing and tolerance capabilities are affected by α-syn aggregates (Olesen et al., 2018). In acute neurotoxic models of PD, such as MPTP-injected mice, substantial T cell infiltration was detected in the substantia nigra at the first day following MPTP challenge, and gradually decreased and normalized by day 30 (Chandra et al., 2017). In addition to the potentially neurotoxic impacts of cytokines secreted by Th1 or Th17 cells (Park et al., 2017; Storelli et al., 2019), Th2 and Treg cells are considered to suppress innate immune activation in the CNS, indicating that an imbalance in T cell types may cause overactivation of glia and chronic inflammation (Gendelman and Appel, 2011; Olson and Gendelman, 2016; von Euler Chelpin and Vorup-Jensen, 2017). Previous research has revealed that α-syn aggregates are released into the extracellular space under pathological situations (Desplats et al., 2009; Lee et al., 2014; Emmanouilidou and Vekrellis, 2016; Steiner et al., 2018), in which they can potentially stimulate T cells. In the context of MHC class II, two types of antigen-presenting cells are reported to display epitopes originating from the α-syn Y39 region. IL-5 from CD4+ T cells and IFN from CD8+ T cells are the main triggers of this response (Sulzer et al., 2017). As a result, α-syn peptides can behave as antigenic epitopes, activating T cell responses, which could explain the link between PD and specific MHC alleles (Sulzer et al., 2017). Recent research has found that extracellular α-syn has a variety of impacts on CD4+ and CD8+ T cell populations in the peripheral and central nervous systems, implying that α-syn variations affect CD4+ T cell homing and tolerance capacity (Olesen et al., 2018). Another study used a combination of human α-syn PFF and AAV-human-α-syn injections into the rat substantia nigra and found both microglia activation and CD4+ and CD8+ T cell infiltration (Thakur et al., 2017). Furthermore, extracellular α-syn aggregates have been reported to inhibit CD25 expression, which could explain why recently activated T cells in PD have a lower survival potential (Olesen et al., 2018).
It is reported that Fas-deficient animals have less MPTP-induced DA neuron loss (Hayley et al., 2004). While the Fas/FasL pathway has been linked to the removal of activated macrophages and therefore to the resolution of inflammation in the setting of antigen presentation (Ashany et al., 1995), new evidence reveals that this pathway may alternatively generate proinflammatory cytokines in tissue macrophages (Park et al., 2003). As a result, CD4+ Th FasL-mediated activation of microglial cells may play a role in the inflammatory response and degeneration of DA neurons. FasL produced by T cells may potentially play a role in inflammatory responses in astrocytes, which are known to be highly resistant to Fas-mediated cell death and to produce proinflammatory cytokines and chemokines in response to Fas ligation (Choi and Benveniste, 2004). Fas expression has been found to be elevated on these glial cells in the MPTP model (Ferrer et al., 2000; Hayley et al., 2004). Alternatively, cell-to-cell interaction between infiltrating CD4+ T cells and DA neurons may cause neuronal death (Giuliani et al., 2003).
Our review unveils the critical role of mast cells in the pathogenesis of PD. As stated above, MPP+ treatment induces mast cell activation and a subsequent increase in levels of CCL2, IL-33, and ROS generation (Kempuraj et al., 2016, 2019). This is in line with a review by Sandhu and Kulka (2021) that reported MPP + is an active metabolite of MPTP that causes activation of mouse bone marrow-derived mast cells (BMMC) and increased release of CCL-2 and MMP-3. Thus, IL-33 secretion by mast cells is increased after MPP+ treatment and plays its role through a heterodimeric receptor complex consisting of suppression of tumorigenicity 2 (ST2) and the accessory IL-1 receptor protein (IL-1RAP). The IL-33/ST2 pathway is involved in CNS homeostasis and its pathologies, including neurodegenerative diseases (Sun et al., 2021). Mast cells are a population of IL-33 targeting cells, recognizing it by IL-33 receptor, ST2 (Lunderius-Andersson et al., 2012). Mast cells activation is observed in PD brains and may play role in neuroinflammation in this disease (Kempuraj et al., 2015, 2019).
TG2 is expressed by mast cells in MPTP-treated mice, which can stimulate inflammatory cytokines and neuroinflammation (Hong et al., 2018). A review by Kim et al. (2013) confirms the roles of TG2 in PD and other neurodegenerative diseases. This gene has been reported to encode an enzyme with four different activities, including protein disulfide isomerase, transamidase, protein kinase, and GTPase. Its transamination function can cause a toxic and insoluble aggregation of amyloid and other proteins. In the Lewy bodies, large numbers of isopeptide bonds produced by TG2 were found. The SH-SY5Y neuroblastoma cell line was treated with MPP+, which greatly elevated TG2 activity (Beck et al., 2006; Verhaar et al., 2011). It is established that α-syn is one of TG2's substrates (Junn et al., 2003). TG2 catalyzes the cross-linking of α-synuclein, resulting in the formation of insoluble, high-molecular-weight aggregates. TG2 was recently discovered to be a substrate of PINK1, a PD-associated Ser/Thr protein kinase. PINK1 phosphorylates TG2 directly, increasing protein stability by preventing proteasomal breakdown (Min et al., 2015). As a result, PINK1 regulates TG2 activity, which may be linked to the production of aggresomes in neural cells (Min et al., 2015). According to recent studies, endoplasmic reticulum (ER) dysfunction is a key component of PD development. As a result, proper TG2 function is intimately linked to ER function. In MPP-treated SH-SY5Y cells, for example, biochemical contact and colocalization between TG2 and ER were detected (Verhaar et al., 2012). In a separate investigation, it was discovered that the localization of TG2 to the granular ER compartment in the PD brain is highly selective for stressed and melanized neurons (Wilhelmus et al., 2011). In this regard, TG2 inhibitors may be a promising therapy for alleviating the brain diseases which TG2 plays role in (Min and Chung, 2018). Additionally, inhibition of pKr-2 is identified as a potential therapeutic strategy in PD since pKr-2 treatment of rats causes an increase in microglial TLR4, which is essential for their immune activation (Shin et al., 2015). Furthermore, TLR4 agonists can cause necroptosis, which leads to cell death and release of their intracellular contents triggering innate immune responses and neuroinflammation (Yu et al., 2021); therefore, TLR4 antagonists could have therapeutic potential in PD and other neuroinflammatory disorders as already summarized by Leitner et al. (2019). Suppression of PGE2 is another potential treatment strategy in PD, which is supported by observations that the DA toxin 6-OHDA upregulates mPGES-1 and triggers PGE2-dependent death of DA neurons (Ikeda-Matsuo et al., 2019). This therapeutic approach has been reviewed by Singh et al. (2021).
Aging is a physiological challenge that all organisms face throughout time, and it is also the leading risk factor for neurodegenerative illnesses. Therefore. effects of aging on the neuroinflammation within PD mice models should be considered. Zhao et al. showed that in aged mice, compared to young mice, behavioral performance decreased and DA neurons were depleted, which was followed by increased expression of pro-inflammatory factors (TLR2, p-NF-κB-p65, IL-1β, and TNF-α), as well as the pro-oxidative stress factor gp91phox. The inflammatory M1 microglia were increased by aging, and the equilibrium between oxidation and anti-oxidantswas disrupted. In LPS-treated, aged mice, poor behavioral performance and loss of DA neurons were observed, as well as upregulated TLR2, p-NF-κB-p65, IL-1β, TNF-α, iNOS, and gp91phox (Zhao et al., 2018). Also, Yao and Zhao demonstrated that in a MPTP-PD mouse model, aging enhanced M1 microglia activation while inhibiting M2 microglia activation in the substantia nigra, which was associated with an increase in proinflammatory cytokines TNF-α and IL-1β (Yao and Zhao, 2018).
We also reviewed cell models of PD used in the included studies. Rat/mice primary neuron/glia, BV2 murine microglial cells, and SH-SY5Y human neuroblastoma cell line were among the most common cell lines used for creating in vitro PD models. PD cell models have some advantages in comparison with animal models. First, PD-related genes can be efficiently overexpressed or knocked out in cultured cells. Second, in dopamine-producing cell lines, both MPP+ and 6-OHDA can be utilized to trigger cell death. Other benefits of these models include their unlimited proliferation which allows high-throughput experimentation with a wide range of experimental techniques and endpoints; homogeneity of cell populations which leads to high reproducibility; and the fact that some cell lines such as SH-SY5Y express important enzymes for dopamine metabolism and synapse formation (Han et al., 2003; Schildknecht et al., 2009; Lopes et al., 2010; Scholz et al., 2011; Thomas et al., 2013). Their main disadvantage is high proliferative capacity, which differs from neurons that do not divide. In comparison to primary neurons and organotypic cultures, immortalized cells are not only unable to replicate the appearance and physiology of a neuronal cell, but they also do not express many of the synaptic proteins. In addition, continuous proliferation induces a selection pressure that favors mutations that improve proliferation and survival, causing succeeding generations of cell lines to lose their DA phenotype in comparison to their parental lines. As a result, after repeated passaging, many cell lines become inappropriate for usage (Lopes et al., 2017a). Despite having important DA traits, SH-SY5Y cells lack neuronal characteristics. This cell line is in the early phases of neuronal development, with low numbers of neuronal markers. Furthermore, their oncogenic characteristics and persistent multiplication are incompatible with neurons (Gilany et al., 2008; Filograna et al., 2015; Lopes et al., 2017a). Both genetic and toxin-based techniques have been used to recreate PD pathology in this cell model, with 6-OHDA being the most widely used toxin. The majority of these investigations are concerned with achieving neuroprotection in cells exposed to 6-OHDA (Wei et al., 2015; Lin and Tsai, 2017). Undifferentiated SH-SY5Y cells have also been used to investigate the mechanisms of 6-OHDA toxicity (Soto-Otero et al., 2000; Izumi et al., 2005; Xicoy et al., 2017). 6-OHDA is taken up by DA neurons via DAT transporter and induces considerable oxidative stress. Undifferentiated cells do not mimic the 6-OHDA-induced cell death mechanisms that occur in vivo because they only express modest amounts of DAT (Lopes et al., 2017b).
In the current systematic review, we have collected data identifying the importance of several neuroinflammatory pathways and molecular mechanisms in the pathogenesis of PD. We conclude that neuroinflammation plays a role in both the initiation and progression of PD. We illustrate that neuroinflammatory reactions in PD models can be induced by various factors including MPTP, α-syn, 6-OHDA, and pKr-2, while other mechanisms, such as TLR2, NLRP3, IL-1β, TNF-α, NF-κB pathway, HMBG1, ROS production, CD36, Fyn, mast cells, ASK1, Lcn-2, TG2, CCL2, IL-33, TLR4, mPGES-1, and PGE2, contribute to the establishment and progression of the pathogenetic mechanisms in these models.
In addition, we identify several potential therapeutic approaches that may be effective in PD by alleviating neuroinflammation. They include miR-124 and miR-7 as well as inhibitors of NLRP3 inflammasomes, HMBG1, and TG2. We also acknowledge limitations to our work, which include very limited research on some of the mechanisms we review and a shortage of other comprehensive systematic reviews and meta-analyses that could be used to further validate our conclusions. Further research is needed to understand other neuroinflammatory mechanisms involved in the pathogenesis of PD and to develop new therapeutic approaches targeting them. By evaluating the relative impact of each factor and determining their collective contribution to neuroinflammation in PD, multitargeted therapeutic approaches could be developed that will hopefully solve the puzzle of PD, which currently lacks effective treatments.
Publicly available datasets were analyzed in this study. This data can be found at: https://pubmed.ncbi.nlm.nih.gov/.
MF, KV, and SY contributed to the conception and design of the study. MRT and FS contributed to the supervision of the manuscript. MD organized the database. AK edited the paper scientifically. All authors wrote the first draft of the manuscript, wrote sections of the manuscript, contributed to manuscript revision, read, and approved the submitted version.
This study was related to the Project (No. 1399/61320) from Student Research Committee, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We also appreciate the Student Research Committee and Research & Technology Chancellor in Shahid Beheshti University of Medical Sciences as well as the Jack Brown and Family Alzheimer's Disease Research Foundation for their financial support of this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.855776/full#supplementary-material
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W.-H., Castillo, P. E., et al. (2000). Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/S0896-6273(00)80886-7
Ahmed, I., Tamouza, R., Delord, M., Krishnamoorthy, R., Tzourio, C., Mulot, C., et al. (2012). Association between Parkinson's disease and the HLA-DRB1 locus. Mov. Disord. 27, 1104–1110. doi: 10.1002/mds.25035
Allen Reish, H. E., and Standaert, D. G. (2015). Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinsons Dis. 5, 1–19. doi: 10.3233/JPD-140491
Ambrosi, G., Kustrimovic, N., Siani, F., Rasini, E., Cerri, S., Ghezzi, C., et al. (2017). Complex changes in the innate and adaptive immunity accompany progressive degeneration of the nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine in the rat. Neurotox. Res. 32, 71–81. doi: 10.1007/s12640-017-9712-2
Ashany, D., Song, X., Lacy, E., Nikolic-Zugic, J., Friedman, S. M., and Elkon, K. B. (1995). Th1 CD4+ lymphocytes delete activated macrophages through the Fas/APO-1 antigen pathway. Proc. Natl. Acad. Sci. U.S.A. 92, 11225–11229. doi: 10.1073/pnas.92.24.11225
Bae, E., Lee, H., Jang, Y., Michael, S., Masliah, E., Min, D., et al. (2014). Phospholipase D1 regulates autophagic flux and clearance of α-synuclein aggregates. Cell Death Differ. 21, 1132–1141. doi: 10.1038/cdd.2014.30
Baeuerle, P. A., and Baltimore, D. (1996). NF-kappa B: ten years after. Cell 87, 13–20. doi: 10.1016/S0092-8674(00)81318-5
Baird, J. K., Bourdette, D., Meshul, C. K., and Quinn, J. F. (2019). The key role of T cells in Parkinson's disease pathogenesis and therapy. Parkinsonism Relat. Disord. 60, 25–31. doi: 10.1016/j.parkreldis.2018.10.029
Baltic, S., Perovic, M., Mladenovic, A., Raicevic, N., Ruzdijic, S., Rakic, L., et al. (2004). α-Synuclein is expressed in different tissues during human fetal development. J. Mol. Neurosci. 22, 199–203. doi: 10.1385/JMN:22:3:199
Bartel, D. P.. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. doi: 10.4049/jimmunol.0901363
Beck, K. E., De Girolamo, L. A., Griffin, M., and Billett, E. E. (2006). The role of tissue transglutaminase in 1-methyl-4-phenylpyridinium (MPP+)-induced toxicity in differentiated human SH-SY5Y neuroblastoma cells. Neurosci. Lett. 405, 46–51. doi: 10.1016/j.neulet.2006.06.061
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Benner, E. J., Banerjee, R., Reynolds, A. D., Sherman, S., Pisarev, V. M., Tsiperson, V., et al. (2008). Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 3, e1376. doi: 10.1371/journal.pone.0001376
Berry, C., La Vecchia, C., and Nicotera, P. (2010). Paraquat and Parkinson's disease. Cell Death Differ. 17, 1115–1125. doi: 10.1038/cdd.2009.217
Betarbet, R., Sherer, T. B., and Greenamyre, J. T. (2002). Animal models of Parkinson's disease. Bioessays 24, 308–318. doi: 10.1002/bies.10067
Bieganowska, K., Członkowska, A., Bidziński, A., Mierzewska, H., and Korlak, J. (1993). Immunological changes in the MPTP-induced Parkinson's disease mouse model. J. Neuroimmunol. 42, 33–37. doi: 10.1016/0165-5728(93)90209-H
Block, M. L., Zecca, L., and Hong, J.-S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Boireau, A., Bordier, F., Dubédat, P., and Doble, A. (1995). Methamphetamine and dopamine neurotoxicity: differential effects of agents interfering with glutamatergic transmission. Neurosci. Lett. 195, 9–12. doi: 10.1016/0304-3940(95)11765-O
Brochard, V., Combadière, B., Prigent, A., Laouar, Y., Perrin, A., Beray-Berthat, V., et al. (2009). Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 119, 182–192. doi: 10.1172/JCI36470
Burré, J., Sharma, M., and Südhof, T. C. (2014). α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U.S.A. 111, E4274–E4283. doi: 10.1073/pnas.1416598111
Burré, J., Sharma, M., and Südhof, T. C. (2015). Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. 35, 5221–5232. doi: 10.1523/JNEUROSCI.4650-14.2015
Burré, J., Sharma, M., and Südhof, T. C. (2018). Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 8, ea024091. doi: 10.1101/cshperspect.a024091
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Campolo, M., Paterniti, I., Siracusa, R., Filippone, A., Esposito, E., and Cuzzocrea, S. (2019). TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson's diseases in vivo model. Brain Behav. Immun. 76, 236–247. doi: 10.1016/j.bbi.2018.12.003
Cao, S., Standaert, D. G., and Harms, A. S. (2012). The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflammation 9, 259. doi: 10.1186/1742-2094-9-259
Caplan, I. F., and Maguire-Zeiss, K. A. (2018). Toll-like receptor 2 signaling and current approaches for therapeutic modulation in synucleinopathies. Front. Pharmacol. 9, 417. doi: 10.3389/fphar.2018.00417
Cenci, M. A., and Björklund, A. (2020). Animal models for preclinical Parkinson's research: an update and critical appraisal. Prog. Brain Res. 252, 27–59. doi: 10.1016/bs.pbr.2020.02.003
Chandra, G., Roy, A., Rangasamy, S. B., and Pahan, K. (2017). Induction of adaptive immunity leads to nigrostriatal disease progression in MPTP mouse model of Parkinson's disease. J. Immunol. 198, 4312–4326. doi: 10.4049/jimmunol.1700149
Chen, M. J., Yap, Y. W., Choy, M. S., Koh, C. H. V., Seet, S. J., Duan, W., et al. (2006). Early induction of calpains in rotenone-mediated neuronal apoptosis. Neurosci. Lett. 397, 69–73. doi: 10.1016/j.neulet.2005.12.011
Chen, P. E., Specht, C. G., Morris, R. G., and Schoepfer, R. (2002). Spatial learning is unimpaired in mice containing a deletion of the alpha-synuclein locus. Euro. J. Neurosci. 16, 154–158. doi: 10.1046/j.1460-9568.2002.02062.x
Chesselet, M.-F., and Richter, F. (2011). Modelling of Parkinson's disease in mice. Lancet Neurol. 10, 1108–1118. doi: 10.1016/S1474-4422(11)70227-7
Chesselet, M.-F., Richter, F., Zhu, C., Magen, I., Watson, M. B., and Subramaniam, S. R. (2012). A progressive mouse model of Parkinson's disease: the Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 9, 297–314. doi: 10.1007/s13311-012-0104-2
Chinta, S. J., Mallajosyula, J. K., Rane, A., and Andersen, J. K. (2010). Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 486, 235–239. doi: 10.1016/j.neulet.2010.09.061
Choi, C., and Benveniste, E. N. (2004). Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res. Rev. 44, 65–81. doi: 10.1016/j.brainresrev.2003.08.007
Choi, I., Zhang, Y., Seegobin, S. P., Pruvost, M., Wang, Q., Purtell, K., et al. (2016). Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 11, 1386. doi: 10.1038/s41467-020-15119-w
Choi, W.-S., Kim, H.-W., and Xia, Z. (2015). JNK inhibition of VMAT2 contributes to rotenone-induced oxidative stress and dopamine neuron death. Toxicology 328, 75–81. doi: 10.1016/j.tox.2014.12.005
Choubey, V., Safiulina, D., Vaarmann, A., Cagalinec, M., Wareski, P., Kuum, M., et al. (2011). Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 286, 10814–10824. doi: 10.1074/jbc.M110.132514
Cleeter, M., Cooper, J., and Schapira, A. (1992). Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: evidence for free radical involvement. J. Neurochem. 58, 786–789. doi: 10.1111/j.1471-4159.1992.tb09789.x
Conde, M. A., Alza, N. P., González, P. A. I., Bilbao, P. G. S., Campos, S. S., Uranga, R. M., et al. (2018). Phospholipase D1 downregulation by α-synuclein: Implications for neurodegeneration in Parkinson's disease. Biochim. Biophys. Acta 1863, 639–650. doi: 10.1016/j.bbalip.2018.03.006
Cunningham, C., Dunne, A., and Lopez-Rodriguez, A. B. (2019). Astrocytes: heterogeneous and dynamic phenotypes in neurodegeneration and innate immunity. Neuroscientist 25, 455–474. doi: 10.1177/1073858418809941
Członkowska, A., Kohutnicka, M., Kurkowska-Jastrzebska, I., and Członkowski, A. (1996). Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration. 5, 137–143. doi: 10.1006/neur.1996.0020
Dauer, W., Kholodilov, N., Vila, M., Trillat, A.-C., Goodchild, R., Larsen, K. E., et al. (2002). Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 99, 14524–14529. doi: 10.1073/pnas.172514599
Dekens, D. W., Eisel, U. L. M., Gouweleeuw, L., Schoemaker, R. G., and De Deyn, P. P. Naud,é P. J. W. (2021). Lipocalin 2 as a link between ageing, risk factor conditions and age-related brain diseases. Ageing Res. Rev. 70, 101414. doi: 10.1016/j.arr.2021.101414
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Deumens, R., Blokland, A., and Prickaerts, J. (2002). Modeling parkinson's disease in rats: an evaluation of 6-OHDA lesions of the nigrostriatal pathway. Exp. Neurol. 175, 303–317. doi: 10.1006/exnr.2002.7891
Earls, R. H., Menees, K. B., Chung, J., Barber, J., Gutekunst, C. A., Hazim, M. G., et al. (2019). Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflammation 16, 250. doi: 10.1186/s12974-019-1636-8
Emmanouilidou, E., and Vekrellis, K. (2016). Exocytosis and spreading of normal and aberrant α-synuclein. Brain Pathol. 26, 398–403. doi: 10.1111/bpa.12373
Engblom, D., Saha, S., Engström, L., Westman, M., Audoly, L. P., Jakobsson, P. J., et al. (2003). Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat. Neurosci. 6, 1137–1138. doi: 10.1038/nn1137
Falkenburger, B., and Schulz, J. (2006). Limitations of cellular models in Parkinson's disease research. Parkinsons Dis. Relat. Disord. 261–268. doi: 10.1007/978-3-211-45295-0_40
Faull, R., and Laverty, R. (1969). Changes in dopamine levels in the corpus striatum following lesions in the substantia nigra. Exp. Neurol. 23, 332–340. doi: 10.1016/0014-4886(69)90081-8
Fernagut, P.-O., and Chesselet, M.-F. (2004). Alpha-synuclein and transgenic mouse models. Neurobiol. Dis. 17, 123–130. doi: 10.1016/j.nbd.2004.07.001
Fernagut, P. O., Hutson, C. B., Fleming, S. M., Tetreaut, N. A., Salcedo, J., Masliah, E., et al. (2007). Behavioral and histopathological consequences of paraquat intoxication in mice: effects of α-synuclein over-expression. Synapse 61, 991–1001. doi: 10.1002/syn.20456
Ferrari, C. C., Godoy, M. C. P., Tarelli, R., Chertoff, M., Depino, A. M., and Pitossi, F. J. (2006). Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1β in the substantia nigra. Neurobiol. Dis. 24, 183–193. doi: 10.1016/j.nbd.2006.06.013
Ferrer, I., Blanco, R., Cutillas, B., and Ambrosio, S. (2000). Fas and Fas-L expression in Huntington's disease and Parkinson's disease. Neuropathol. Appl. Neurobiol. 26, 424–433. doi: 10.1046/j.1365-2990.2000.00267.x
Filograna, R., Civiero, L., Ferrari, V., Codolo, G., Greggio, E., Bubacco, L., et al. (2015). Analysis of the catecholaminergic phenotype in human SH-SY5Y and BE (2)-M17 neuroblastoma cell lines upon differentiation. PLoS ONE 10, e0136769. doi: 10.1371/journal.pone.0136769
Fleming, S. M., Tetreault, N. A., Mulligan, C. K., Hutson, C. B., Masliah, E., and Chesselet, M. F. (2008). Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur. J. Neurosci. 28, 247–256. doi: 10.1111/j.1460-9568.2008.06346.x
Freichel, C., Neumann, M., Ballard, T., Müller, V., Woolley, M., Ozmen, L., et al. (2007). Age-dependent cognitive decline and amygdala pathology in α-synuclein transgenic mice. Neurobiol. Aging 28, 1421–1435. doi: 10.1016/j.neurobiolaging.2006.06.013
Furukawa, K., Matsuzaki-Kobayashi, M., Hasegawa, T., Kikuchi, A., Sugeno, N., Itoyama, Y., et al. (2006). Plasma membrane ion permeability induced by mutant alpha-synuclein contributes to the degeneration of neural cells. J. Neurochem. 97, 1071–1077. doi: 10.1111/j.1471-4159.2006.03803.x
Gardai, S. J., Mao, W., Schüle, B., Babcock, M., Schoebel, S., Lorenzana, C., et al. (2013). Elevated alpha-synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson's disease. PLoS ONE. 8, e71634. doi: 10.1371/journal.pone.0071634
Gendelman, H. E., and Appel, S. H. (2011). Neuroprotective activities of regulatory T cells. Trends Mol. Med. 17, 687–688. doi: 10.1016/j.molmed.2011.08.005
Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2002). Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533. doi: 10.1016/S0896-6273(02)00682-7
Gilany, K., Van Elzen, R., Mous, K., Coen, E., Van Dongen, W., Vandamme, S., et al. (2008). The proteome of the human neuroblastoma cell line SH-SY5Y: an enlarged proteome. Biochim. Biophys. Acta 1784, 983–985. doi: 10.1016/j.bbapap.2008.03.003
Gispert, S., Turco, D. D., Garrett, L., Chen, A., Bernard, D. J., Hamm-Clement, J., et al. (2003). Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell. Neurosci. 24, 419–429. doi: 10.1016/S1044-7431(03)00198-2
Giuliani, F., Goodyer, C. G., Antel, J. P., and Yong, V. W. (2003). Vulnerability of human neurons to T cell-mediated cytotoxicity. J. Immunol. 171, 368–379. doi: 10.4049/jimmunol.171.1.368
Goedert, M.. (2001). Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2, 492–501. doi: 10.1038/35081564
Greenamyre, J. T., Betarbet, R., and Sherer, T. B. (2003). The rotenone model of Parkinson's disease: genes, environment and mitochondria. Parkinsonism Relat. Disord. 9, 59–64. doi: 10.1016/S1353-8020(03)00023-3
Gu, X. L., Long, C. X., Sun, L., Xie, C., Lin, X., and Cai, H. (2010). Astrocytic expression of Parkinson's disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 3, 12. doi: 10.1186/1756-6606-3-12
Guo, X., Namekata, K., Kimura, A., Harada, C., and Harada, T. (2017). ASK1 in neurodegeneration. Adv. Biol. Regul. 66, 63–71. doi: 10.1016/j.jbior.2017.08.003
Gustot, A., Gallea, J. I., Sarroukh, R., Celej, M. S., Ruysschaert, J.-M., and Raussens, V. (2015). Amyloid fibrils are the molecular trigger of inflammation in Parkinson's disease. Biochem. J. 471, 323–333. doi: 10.1042/BJ20150617
Hamza, T. H., Zabetian, C. P., Tenesa, A., Laederach, A., Montimurro, J., Yearout, D., et al. (2010). Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat. Genet. 42, 781–785. doi: 10.1038/ng.642
Han, B. S., Hong, H.-S., Choi, W.-S., Markelonis, G. J., Oh, T. H., and Oh, Y. J. (2003). Caspase-dependent and-independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J. Neurosci. 23, 5069–5078. doi: 10.1523/JNEUROSCI.23-12-05069.2003
Harms, A. S., Ferreira, S. A., and Romero-Ramos, M. (2021). Periphery and brain, innate and adaptive immunity in Parkinson's disease. Acta Neuropathol. 141, 527–545. doi: 10.1007/s00401-021-02268-5
Hasegawa, E., Takeshige, K., Oishi, T., Murai, Y., and Minakami, S. (1990). 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem. Biophys. Res. Commun. 170, 1049–1055. doi: 10.1016/0006-291X(90)90498-C
Hatami, A., and Chesselet, M. F. (2015). Transgenic rodent models to study alpha-synuclein pathogenesis, with a focus on cognitive deficits. Curr. Top. Behav. Neurosci. 22, 303–330. doi: 10.1007/7854_2014_355
Hawk, B. J., Khounlo, R., and Shin, Y.-K. (2019). Alpha-synuclein continues to enhance SNARE-dependent vesicle docking at exorbitant concentrations. Front. Neurosci. 13, 216. doi: 10.3389/fnins.2019.00216
Hayley, S., Crocker, S. J., Smith, P. D., Shree, T., Jackson-Lewis, V., Przedborski, S., et al. (2004). Regulation of dopaminergic loss by Fas in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J. Neurosci. 24, 2045–2053. doi: 10.1523/JNEUROSCI.4564-03.2004
Henn, A., Lund, S., Hedtjärn, M., Schrattenholz, A., Pörzgen, P., and Leist, M. (2009). The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26, 83–94. doi: 10.14573/altex.2009.2.83
Henry, C. J., Huang, Y., Wynne, A. M., and Godbout, J. P. (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23, 309–317. doi: 10.1016/j.bbi.2008.09.002
Hettiarachchi, N. T., Parker, A., Dallas, M. L., Pennington, K., Hung, C. C., Pearson, H. A., et al. (2009). alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J. Neurochem. 111, 1192–1201. doi: 10.1111/j.1471-4159.2009.06411.x
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. doi: 10.1016/S1474-4422(09)70062-6
Hirsch, E. C., Vyas, S., and Hunot, S. (2012). Neuroinflammation in Parkinson's disease. Parkinsonism Relat. Disord. 18, S210–S212. doi: 10.1016/S1353-8020(11)70065-7
Hong, G. U., Cho, J. W., Kim, S. Y., Shin, J. H., and Ro, J. Y. (2018). Inflammatory mediators resulting from transglutaminase 2 expressed in mast cells contribute to the development of Parkinson's disease in a mouse model. Toxicol. Appl. Pharmacol. 358, 10–22. doi: 10.1016/j.taap.2018.09.003
Hong-rong, X. I. E., Lin-sen, H. U., and Guo-yi, L. I. (2010). SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson's disease. Chin. Med. J. 123, 1086–1092. doi: 10.3760/cma.j.issn.0366-6999.2010.08.021
Hooijmans, C. R., Rovers, M. M., De Vries, R. B., Leenaars, M., Ritskes-Hoitinga, M., and Langendam, M. W. (2014). SYRCLE's risk of bias tool for animal studies. BMC Med. Res. Methodol. 14, 43. doi: 10.1186/1471-2288-14-43
Horvath, I., Iashchishyn, I. A., Moskalenko, R. A., Wang, C., Wärmländer, S., Wallin, C., et al. (2018). Co-aggregation of pro-inflammatory S100A9 with α-synuclein in Parkinson's disease: ex vivo and in vitro studies. J. Neuroinflammation 15, 172. doi: 10.1186/s12974-018-1210-9
Huang, J., Yang, J., Shen, Y., Jiang, H., Han, C., Zhang, G., et al. (2017). HMGB1 mediates autophagy dysfunction via perturbing beclin1-Vps34 complex in dopaminergic cell model. Front. Mol. Neurosci. 10, 13. doi: 10.3389/fnmol.2017.00013
Huang, M., Guo, M., Wang, K., Wu, K., Li, Y., Tian, T., et al. (2020). HMGB1 mediates paraquat-induced neuroinflammatory responses via activating RAGE signaling pathway. Neurotox. Res. 37, 913–925. doi: 10.1007/s12640-019-00148-1
Huang, M., Wang, B., Li, X., Fu, C., Wang, C., and Kang, X. (2019). α-synuclein: a multifunctional player in exocytosis, endocytosis, vesicle recycling. Front. Neurosci. 13, 28. doi: 10.3389/fnins.2019.00028
Hughes, C. D., Choi, M. L., Ryten, M., Hopkins, L., Drews, A., Botía, J. A., et al. (2019). Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson's disease pathogenesis. Acta Neuropathol. 137, 103–120. doi: 10.1007/s00401-018-1907-y
Iba, M., Kim, C., Sallin, M., Kwon, S., Verma, A., Overk, C., et al. (2020). Neuroinflammation is associated with infiltration of T cells in Lewy body disease and α-synuclein transgenic models. J. Neuroinflammation 17, 214–214. doi: 10.1186/s12974-020-01888-0
Ikeda-Matsuo, Y., Miyata, H., Mizoguchi, T., Ohama, E., Naito, Y., Uematsu, S., et al. (2019). Microsomal prostaglandin E synthase-1 is a critical factor in dopaminergic neurodegeneration in Parkinson's disease. Neurobiol. Dis. 124, 81–92. doi: 10.1016/j.nbd.2018.11.004
Ishola, I., Chaturvedi, J., Rai, S., Rajasekar, N., Adeyemi, O., Shukla, R., et al. (2013). Evaluation of amentoflavone isolated from Cnestis ferruginea Vahl ex DC (Connaraceae) on production of inflammatory mediators in LPS stimulated rat astrocytoma cell line (C6) and THP-1 cells. J. Ethnopharmacol. 146, 440–448. doi: 10.1016/j.jep.2012.12.015
Ishola, I. O., Akinyede, A., Adeluwa, T., and Micah, C. (2018). Novel action of vinpocetine in the prevention of paraquat-induced parkinsonism in mice: involvement of oxidative stress and neuroinflammation. Metab. Brain Dis. 33, 1493–1500. doi: 10.1007/s11011-018-0256-9
Izumi, Y., Sawada, H., Sakka, N., Yamamoto, N., Kume, T., Katsuki, H., et al. (2005). p-Quinone mediates 6-hydroxydopamine-induced dopaminergic neuronal death and ferrous iron accelerates the conversion of p-quinone into melanin extracellularly. J. Neurosci. Res. 79, 849–860. doi: 10.1002/jnr.20382
Jarrott, B., and Williams, S. J. (2016). Chronic brain inflammation: the neurochemical basis for drugs to reduce inflammation. Neurochem. Res. 41, 523–533. doi: 10.1007/s11064-015-1661-7
Javed, H., Thangavel, R., Selvakumar, G. P., Dubova, I., Schwartz, N., Ahmed, M. E., et al. (2020). NLRP3 inflammasome and glia maturation factor coordinately regulate neuroinflammation and neuronal loss in MPTP mouse model of Parkinson's disease. Int. Immunopharmacol. 83, 106441. doi: 10.1016/j.intimp.2020.106441
Jo, M. G., Ikram, M., Jo, M. H., Yoo, L., Chung, K. C., Nah, S. Y., et al. (2019). Gintonin mitigates MPTP-induced loss of nigrostriatal dopaminergic neurons and accumulation of α-synuclein via the Nrf2/HO-1 pathway. Mol. Neurobiol. 56, 39–55. doi: 10.1007/s12035-018-1020-1
Junn, E., Ronchetti, R. D., Quezado, M. M., Kim, S. Y., and Mouradian, M. M. (2003). Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for lewy body formation in Parkinson's disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 100, 2047–2052. doi: 10.1073/pnas.0438021100
Kahle, P. J.. (2008). α-Synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol. 115, 87–95. doi: 10.1007/s00401-007-0302-x
Kahle, P. J., Neumann, M., Ozmen, L., Müller, V., Odoy, S., Okamoto, N., et al. (2001). Selective insolubility of α-synuclein in human lewy body diseases is recapitulated in a transgenic mouse model. Am. J. Pathol. 159, 2215–2225. doi: 10.1016/S0002-9440(10)63072-6
Kamei, D., Yamakawa, K., Takegoshi, Y., Mikami-Nakanishi, M., Nakatani, Y., Oh-Ishi, S., et al. (2004). Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J. Biol. Chem. 279, 33684–33695. doi: 10.1074/jbc.M400199200
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. doi: 10.1016/j.cell.2007.11.024
Kannarkat, G. T., Boss, J. M., and Tansey, M. G. (2013). The role of innate and adaptive immunity in Parkinson's disease. J. Parkinsons. Dis. 3, 493–514. doi: 10.3233/JPD-130250
Karikari, A. A., McFleder, R. L., Ribechini, E., Blum, R., Bruttel, V., Knorr, S., et al. (2022). Neurodegeneration by α-synuclein-specific T cells in AAV-A53T-α-synuclein Parkinson's disease mice. Brain Behav. Immun. 101, 194–210. doi: 10.1016/j.bbi.2022.01.007
Kempuraj, D., Khan, M. M., Thangavel, R., Xiong, Z., Yang, E., and Zaheer, A. (2013). Glia maturation factor induces interleukin-33 release from astrocytes: implications for neurodegenerative diseases. J. Neuroimmune Pharmacol. 8, 643–650. doi: 10.1007/s11481-013-9439-7
Kempuraj, D., Thangavel, R., Fattal, R., Pattani, S., Yang, E., Zaheer, S., et al. (2016). Mast cells release chemokine CCL2 in response to parkinsonian toxin 1-methyl-4-phenyl-pyridinium (MPP(+)). Neurochem. Res. 41, 1042–1049. doi: 10.1007/s11064-015-1790-z
Kempuraj, D., Thangavel, R., Selvakumar, G. P., Ahmed, M. E., Zaheer, S., Raikwar, S. P., et al. (2019). Mast Cell proteases activate astrocytes and glia-neurons and release interleukin-33 by activating p38 and ERK1/2 MAPKs and NF-κB. Mol. Neurobiol. 56, 1681–1693. doi: 10.1007/s12035-018-1177-7
Kempuraj, D., Thangavel, R., Yang, E., Pattani, S., Zaheer, S., Santillan, D. A., et al. (2015). Dopaminergic toxin 1-methyl-4-phenylpyridinium, proteins α-synuclein and glia maturation factor activate mast cells and release inflammatory mediators. PLoS ONE 10, e0135776. doi: 10.1371/journal.pone.0135776
Kilpeläinen, T., Julku, U. H., Svarcbahs, R., and Myöhänen, T. T. (2019). Behavioural and dopaminergic changes in double mutated human A30P*A53T alpha-synuclein transgenic mouse model of Parkinson's disease. Sci. Rep. 9, 17382. doi: 10.1038/s41598-019-54034-z
Kim, B. W., Jeong, K. H., Kim, J. H., Jin, M., Kim, J. H., Lee, M. G., et al. (2016). Pathogenic upregulation of glial lipocalin-2 in the parkinsonian dopaminergic system. J. Neurosci. 36, 5608–5622. doi: 10.1523/JNEUROSCI.4261-15.2016
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562. doi: 10.1038/ncomms2534
Kim, J., Fiesel, F. C., Belmonte, K. C., Hudec, R., Wang, W.-X., Kim, C., et al. (2016). miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 11, 55. doi: 10.1186/s13024-016-0121-4
Klegeris, A.. (2021). Regulation of neuroimmune processes by damage- and resolution-associated molecular patterns. Neural Regen. Res. 16, 423–429. doi: 10.4103/1673-5374.293134
Klegeris, A., Giasson, B. I., Zhang, H., Maguire, J., Pelech, S., and McGeer, P. L. (2006). Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 20, 2000–2008. doi: 10.1096/fj.06-6183com
Kokhan, V., Afanasyeva, M., and Van'Kin, G. (2012). α-Synuclein knockout mice have cognitive impairments. Behav. Brain Res. 231, 226–230. doi: 10.1016/j.bbr.2012.03.026
Kumar, R., Agarwal, A. K., and Seth, P. K. (1995). Free radical-generated neurotoxicity of 6-hydroxydopamine. J. Neurochem. 64, 1703–1707. doi: 10.1046/j.1471-4159.1995.64041703.x
Kurkowska-Jastrzebska, I., Wrońska, A., Kohutnicka, M., Członkowski, A., and Członkowska, A. (1999). MHC class II positive microglia and lymphocytic infiltration are present in the substantia nigra and striatum in mouse model of Parkinson's disease. Acta Neurobiol. Exp. 59, 1–8.
La Vitola, P., Balducci, C., Baroni, M., Artioli, L., Santamaria, G., Castiglioni, M., et al. (2021). Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson's models. Neuropathol. Appl. Neurobiol. 47, 43–60. doi: 10.1111/nan.12644
Labzin, L. I., Heneka, M. T., and Latz, E. (2018). Innate immunity and neurodegeneration. Annu. Rev. Med. 69, 437–449. doi: 10.1146/annurev-med-050715-104343
Lai, T. T., Kim, Y. J., Nguyen, P. T., Koh, Y. H., Nguyen, T. T., Ma, H. I., et al. (2021). Temporal evolution of inflammation and neurodegeneration with alpha-synuclein propagation in Parkinson's disease mouse model. Front. Integr. Neurosci. 15, 715190. doi: 10.3389/fnint.2021.715190
Lawrence, T.. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651. doi: 10.1101/cshperspect.a001651
Lee, H. J., Bae, E. J., and Lee, S. J. (2014). Extracellular α-synuclein-a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 10, 92–98. doi: 10.1038/nrneurol.2013.275
Lee, K. W., Zhao, X., Im, J. Y., Grosso, H., Jang, W. H., Chan, T. W., et al. (2012). Apoptosis signal-regulating kinase 1 mediates MPTP toxicity and regulates glial activation. PLoS ONE 7, e29935. doi: 10.1371/journal.pone.0029935
Leitner, G. R., Wenzel, T. J., Marshall, N., Gates, E. J., and Klegeris, A. (2019). Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 23, 865–882. doi: 10.1080/14728222.2019.1676416
Li, H., Yang, J., Wang, Y., Liu, Q., Cheng, J., and Wang, F. (2019). Neuroprotective effects of increasing levels of HSP70 against neuroinflammation in Parkinson's disease model by inhibition of NF-κB and STAT3. Life Sci. 234, 116747. doi: 10.1016/j.lfs.2019.116747
Li, L., Nadanaciva, S., Berger, Z., Shen, W., Paumier, K., Schwartz, J., et al. (2013). Human A53T α-synuclein causes reversible deficits in mitochondrial function and dynamics in primary mouse cortical neurons. PLoS ONE 8, e85815. doi: 10.1371/journal.pone.0085815
Li, Y., Niu, M., Zhao, A., Kang, W., Chen, Z., Luo, N., et al. (2019). CXCL12 is involved in α-synuclein-triggered neuroinflammation of Parkinson's disease. J. Neuroinflammation 16, 263. doi: 10.1186/s12974-019-1646-6
Liberatore, G. T., Jackson-Lewis, V., Vukosavic, S., Mandir, A. S., Vila, M., McAuliffe, W. G., et al. (1999). Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 5, 1403–1409. doi: 10.1038/70978
Lim, R., Zaheer, A., Khosravi, H., Freeman, J. H. Jr, Halverson, H. E., Wemmie, J. A., and Yang, B. (2004). Impaired motor performance and learning in glia maturation factor-knockout mice. Brain Res. 1024, 225–232. doi: 10.1016/j.brainres.2004.08.003
Lin, C.-Y., and Tsai, C.-W. (2017). Carnosic acid attenuates 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y cells by inducing autophagy through an enhanced interaction of parkin and Beclin1. Mol. Neurobiol. 54, 2813–2822. doi: 10.1007/s12035-016-9873-7
Lopes, F. M., Bristot, I. J., da Motta, L. L., Parsons, R. B., and Klamt, F. (2017a). Mimicking Parkinson's disease in a dish: merits and pitfalls of the most commonly used dopaminergic in vitro models. Neuromol. Med. 19, 241–255. doi: 10.1007/s12017-017-8454-x
Lopes, F. M., da Motta, L. L., De Bastiani, M. A., Pfaffenseller, B., Aguiar, B. W., de Souza, L. F., et al. (2017b). RA differentiation enhances dopaminergic features, changes redox parameters, and increases dopamine transporter dependency in 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y cells. Neurotox. Res. 31, 545–559. doi: 10.1007/s12640-016-9699-0
Lopes, F. M., Schröder, R., da Frota Júnior, M. L. C., Zanotto-Filho, A., Müller, C. B., Pires, A. S., et al. (2010). Comparison between proliferative and neuron-like SH-SY5Y cells as an in vitro model for Parkinson disease studies. Brain Res. 1337, 85–94. doi: 10.1016/j.brainres.2010.03.102
Lunderius-Andersson, C., Enoksson, M., and Nilsson, G. (2012). Mast cells respond to cell injury through the recognition of IL-33. Front. Immunol. 3, 82. doi: 10.3389/fimmu.2012.00082
Lyman, M., Lloyd, D. G., Ji, X., Vizcaychipi, M. P., and Ma, D. (2014). Neuroinflammation: the role and consequences. Neurosci Res. 79, 1–12. doi: 10.1016/j.neures.2013.10.004
Magen, I., and Chesselet, M.-F. (2011). Mouse models of cognitive deficits due to alpha-synuclein pathology. J. Parkinsons Dis. 1, 217–227. doi: 10.3233/JPD-2011-11043
Main, B. S., Zhang, M., Brody, K. M., Ayton, S., Frugier, T., Steer, D., et al. (2016). Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson's disease. Glia. 64, 1590–1604. doi: 10.1002/glia.23028
Main, B. S., Zhang, M., Brody, K. M., Kirby, F. J., Crack, P. J., and Taylor, J. M. (2017). Type-I interferons mediate the neuroinflammatory response and neurotoxicity induced by rotenone. J. Neurochem. 141, 75–85. doi: 10.1111/jnc.13940
Mao, J., Gao, H., Bai, W., Zeng, H., Ren, Y., Liu, Y., et al. (2021). Lipoic acid alleviates LPS-evoked PC12 cell damage by targeting p53 and inactivating the NF-κB pathway. Acta Neurobiol. Exp. 81, 375–385. doi: 10.21307/ane-2021-037
Markowitz, J., and Carson, W. E. (2013). Review of S100A9 biology and its role in cancer. Biochim. Biophys. Acta. 1835, 100–109. doi: 10.1016/j.bbcan.2012.10.003
Marques, O., and Outeiro, T. F. (2012). Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 3, e350–e350. doi: 10.1038/cddis.2012.94
McGeer, E. G., and McGeer, P. L. (2007). The role of anti-inflammatory agents in Parkinson's disease. CNS Drugs 21, 789–797. doi: 10.2165/00023210-200721100-00001
Mendez-Gomez, H. R., Singh, J., Meyers, C., Chen, W., Gorbatyuk, O. S., and Muzyczka, N. (2018). The lipase activity of phospholipase D2 is responsible for nigral neurodegeneration in a rat model of Parkinson's disease. Neuroscience 377, 174–183. doi: 10.1016/j.neuroscience.2018.02.047
Miklossy, J., Doudet, D. D., Schwab, C., Yu, S., McGeer, E. G., and McGeer, P. L. (2006). Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 197, 275–283. doi: 10.1016/j.expneurol.2005.10.034
Min, B., and Chung, K. C. (2018). New insight into transglutaminase 2 and link to neurodegenerative diseases. BMB Rep. 51, 5–13. doi: 10.5483/BMBRep.2018.51.1.227
Min, B., Kwon, Y. C., Choe, K. M., and Chung, K. C. (2015). PINK1 phosphorylates transglutaminase 2 and blocks its proteasomal degradation. J. Neurosci. Res. 93, 722–735. doi: 10.1002/jnr.23535
Mitra, S., Chakrabarti, N., and Bhattacharyya, A. (2011). Differential regional expression patterns of α-synuclein, TNF-α, and IL-1β; and variable status of dopaminergic neurotoxicity in mouse brain after paraquat treatment. J. Neuroinflammation 8, 1–22. doi: 10.1186/1742-2094-8-163
Mohamadkhani, A.. (2018). Gut microbiota and fecal metabolome perturbation in children with autism spectrum disorder. Middle East J Dig Dis. 10, 205–212. doi: 10.15171/mejdd.2018.112
Mohamadkhani, A., Sotoudeh, M., Bowden, S., Poustchi, H., Jazii, F. R., Sayehmiri, K., et al. (2009). Downregulation of HLA class II molecules by G1896A pre-core mutation in chronic hepatitis B virus infection. Viral Immunol. 22, 295–300. doi: 10.1089/vim.2009.0031
Molteni, M., and Rossetti, C. (2017). Neurodegenerative diseases: the immunological perspective. J. Neuroimmunol. 313, 109–115. doi: 10.1016/j.jneuroim.2017.11.002
Molteni, R., Macchi, F., Zecchillo, C., Dell'Agli, M., Colombo, E., Calabrese, F., et al. (2013). Modulation of the inflammatory response in rats chronically treated with the antidepressant agomelatine. Euro. Neuropsychopharmacol. 23, 1645–1655. doi: 10.1016/j.euroneuro.2013.03.008
Morales-Garcia, J. A., Alonso-Gil, S., Gil, C., Martinez, A., Santos, A., and Perez-Castillo, A. (2015). Phosphodiesterase 7 inhibition induces dopaminergic neurogenesis in hemiparkinsonian rats. Stem Cells Transl. Med. 4, 564–575. doi: 10.5966/sctm.2014-0277
Morales-Garcia, J. A., Alonso-Gil, S., Santos, Á., and Perez-Castillo, A. (2020). Phosphodiesterase 7 regulation in cellular and rodent models of Parkinson's disease. Mol. Neurobiol. 57, 806–822. doi: 10.1007/s12035-019-01745-z
Morales-Garcia, J. A., Echeverry-Alzate, V., Alonso-Gil, S., Sanz-SanCristobal, M., Lopez-Moreno, J. A., Gil, C., et al. (2017). Phosphodiesterase7 inhibition activates adult neurogenesis in hippocampus and subventricular zone in vitro and in vivo. Stem Cells 35, 458–472. doi: 10.1002/stem.2480
Morales-Garcia, J. A., Redondo, M., Alonso-Gil, S., Gil, C., Perez, C., Martinez, A., et al. (2011). Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS ONE 6, e17240. doi: 10.1371/journal.pone.0017240
Mukai, K., Tsai, M., Saito, H., and Galli, S. J. (2018). Mast cells as sources of cytokines, chemokines growth factors. Immunol. Rev. 282, 121–150. doi: 10.1111/imr.12634
Murray, R. Z., Kay, J. G., Sangermani, D. G., and Stow, J. L. (2005). A role for the phagosome in cytokine secretion. Science 310, 1492–1495. doi: 10.1126/science.1120225
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/S1474-4422(19)30320-5
Neal, M. L., Boyle, A. M., Budge, K. M., Safadi, F. F., and Richardson, J. R. (2018). The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J. Neuroinflammation 15, 73. doi: 10.1186/s12974-018-1100-1
Nikodemova, M., and Watters, J. J. (2011). Outbred ICR/CD1 mice display more severe neuroinflammation mediated by microglial TLR4/CD14 activation than inbred C57Bl/6 mice. Neuroscience 190, 67–74. doi: 10.1016/j.neuroscience.2011.06.006
Nishibori, M., Mori, S., and Takahashi, H. K. (2019). Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J. Pharmacol. Sci. 140, 94–101. doi: 10.1016/j.jphs.2019.04.006
Noh, H., Jeon, J., and Seo, H. (2014). Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 69, 35–40. doi: 10.1016/j.neuint.2014.02.008
Olesen, M. N., Christiansen, J. R., Petersen, S. V., Jensen, P. H., Paslawski, W., Romero-Ramos, M., et al. (2018). CD4 T cells react to local increase of α-synuclein in a pathology-associated variant-dependent manner and modify brain microglia in absence of brain pathology. Heliyon 4, e00513. doi: 10.1016/j.heliyon.2018.e00513
Olson, K. E., and Gendelman, H. E. (2016). Immunomodulation as a neuroprotective and therapeutic strategy for Parkinson's disease. Curr. Opin. Pharmacol. 26, 87–95. doi: 10.1016/j.coph.2015.10.006
Oskvig, D. B., Elkahloun, A. G., Johnson, K. R., Phillips, T. M., and Herkenham, M. (2012). Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav. Immun. 26, 623–634. doi: 10.1016/j.bbi.2012.01.015
Panicker, N., Sarkar, S., Harischandra, D. S., Neal, M., Kam, T. I., Jin, H., et al. (2019). Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 216, 1411–1430. doi: 10.1084/jem.20182191
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2009). Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int. J. Biochem. Cell Biol. 41, 2015–2024. doi: 10.1016/j.biocel.2009.05.008
Park, D. R., Thomsen, A. R., Frevert, C. W., Pham, U., Skerrett, S. J., Kiener, P. A., et al. (2003). Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J. Immunol. 170, 6209–6216. doi: 10.4049/jimmunol.170.12.6209
Park, J., Lee, J. W., Cooper, S. C., Broxmeyer, H. E., Cannon, J. R., and Kim, C. H. (2017). Parkinson disease-associated LRRK2 G2019S transgene disrupts marrow myelopoiesis and peripheral Th17 response. J. Leukoc. Biol. 102, 1093–1102. doi: 10.1189/jlb.1A0417-147RR
Pathak, D., Sepp, K. J., and Hollenbeck, P. J. (2010). Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci. 30, 8984–8992. doi: 10.1523/JNEUROSCI.1621-10.2010
Paumier, K. L., Luk, K. C., Manfredsson, F. P., Kanaan, N. M., Lipton, J. W., Collier, T. J., et al. (2015). Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 82, 185–199. doi: 10.1016/j.nbd.2015.06.003
Perese, D., Ulman, J., Viola, J., Ewing, S., and Bankiewicz, K. (1989). A 6-hydroxydopamine-induced selective parkinsonian rat model. Brain Res. 494, 285–293. doi: 10.1016/0006-8993(89)90597-0
Perumal, A., Gopal, V., Tordzro, W., Cooper, T., and Cadet, J. (1992). Vitamin E attenuates the toxic effects of 6-hydroxydopamine on free radical scavenging systems in rat brain. Brain Res. Bull. 29, 699–701. doi: 10.1016/0361-9230(92)90142-K
Pourasgari, M., and Mohamadkhani, A. (2020). Heritability for stroke: essential for taking family history. Caspian J. Intern. Med. 11, 237–243. doi: 10.22088/cjim.11.3.237
Przedborski, S., Jackson-Lewis, V., Djaldetti, R., Liberatore, G., Vila, M., Vukosavic, S., et al. (2000). The parkinsonian toxin MPTP: action and mechanism. Restor. Neurol. Neurosci. 16, 135–142.
Przedborski, S., Tieu, K., Perier, C., and Vila, M. (2004). MPTP as a mitochondrial neurotoxic model of Parkinson's Disease. J. Bioenerg. Biomembr. 36, 375–379. doi: 10.1023/B:JOBB.0000041771.66775.d5
Przedbroski, S., Leviver, M., Jiang, H., Ferreira, M., Jackson-Lewis, V., Donaldson, D., et al. (1995). Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by instrastriatal injection of 6-hydroxydopamine. Neuroscience 67, 631–647. doi: 10.1016/0306-4522(95)00066-R
Purisai, M. G., McCormack, A. L., Cumine, S., Li, J., Isla, M. Z., and Di Monte, D. A. (2007). Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol. Dis. 25, 392–400. doi: 10.1016/j.nbd.2006.10.008
Qiao, Y., Wang, P., Qi, J., Zhang, L., and Gao, C. (2012). TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 586, 1022–1026. doi: 10.1016/j.febslet.2012.02.045
Qin, L., Wu, X., Block, M. L., Liu, Y., Breese, G. R., Hong, J. S., et al. (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462. doi: 10.1002/glia.20467
Qin, X. Y., Zhang, S. P., Cao, C., Loh, Y. P., and Cheng, Y. (2016). Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol. 73, 1316–1324. doi: 10.1001/jamaneurol.2016.2742
Ransohoff, R. M.. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 7777–83. doi: 10.1126/science.aag2590
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin. Invest. 122, 1164–1171. doi: 10.1172/JCI58644
Reynolds, A. D., Banerjee, R., Liu, J., Gendelman, H. E., and Mosley, R. L. (2007). Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J. Leukoc. Biol. 82, 1083–1094. doi: 10.1189/jlb.0507296
Rodrigues, M. C., Sanberg, P. R., Cruz, L. E., and Garbuzova-Davis, S. (2014). The innate and adaptive immunological aspects in neurodegenerative diseases. J. Neuroimmunol. 269, 1–8. doi: 10.1016/j.jneuroim.2013.09.020
Sachs, C., and Jonsson, G. (1975). Mechanisms of action of 6-hydroxydopamine. Biochem. Pharmacol. 24, 1–8. doi: 10.1016/0006-2952(75)90304-4
Sandhu, J. K., and Kulka, M. (2021). Decoding mast cell-microglia communication in neurodegenerative diseases. Int. J. Mol. Sci. 22, 1093. doi: 10.3390/ijms22031093
Sarkar, S., Dammer, E. B., Malovic, E., Olsen, A. L., Raza, S. A., Gao, T., et al. (2020a). Molecular signatures of neuroinflammation induced by αsynuclein aggregates in microglial cells. Front. Immunol. 11, 33. doi: 10.3389/fimmu.2020.00033
Sarkar, S., Nguyen, H. M., Malovic, E., Luo, J., Langley, M., Palanisamy, B. N., et al. (2020b). Kv1.3 modulates neuroinflammation and neurodegeneration in Parkinson's disease. J. Clin. Invest. 130, 4195–4212. doi: 10.1172/JCI136174
Schildknecht, S., Pöltl, D., Nagel, D. M., Matt, F., Scholz, D., Lotharius, J., et al. (2009). Requirement of a dopaminergic neuronal phenotype for toxicity of low concentrations of 1-methyl-4-phenylpyridinium to human cells. Toxicol. Appl. Pharmacol. 241, 23–35. doi: 10.1016/j.taap.2009.07.027
Scholz, D., Pöltl, D., Genewsky, A., Weng, M., Waldmann, T., Schildknecht, S., et al. (2011). Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem. 119, 957–971. doi: 10.1111/j.1471-4159.2011.07255.x
Schwarting, R., and Huston, J. (1996). Unilateral 6-hydroxydopamine lesions of meso-striatal dopamine neurons and their physiological sequelae. Prog. Neurobiol. 49, 215–266. doi: 10.1016/S0301-0082(96)00015-9
Senior, S. L., Ninkina, N., Deacon, R., Bannerman, D., Buchman, V. L., Cragg, S. J., et al. (2008). Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur J Neurosci. 27, 947–957. doi: 10.1111/j.1460-9568.2008.06055.x
Servier (2022). Smart News. Servier. Available online at: https://smart.servier.com/
Shao, Q.-H., Chen, Y., Li, F.-F, Wang, S., Zhang, X.-L., Yuan, Y.-H., et al. (2019). TLR4 deficiency has a protective effect in the MPTP/probenecid mouse model of Parkinson's disease. Acta Pharmacol. Sin. 40, 1503–1512. doi: 10.1038/s41401-019-0280-2
Shin, W. H., Jeon, M. T., Leem, E., Won, S. Y., Jeong, K. H., Park, S. J., et al. (2015). Induction of microglial toll-like receptor 4 by prothrombin kringle-2: a potential pathogenic mechanism in Parkinson's disease. Sci. Rep. 5, 14764. doi: 10.1038/srep14764
Simón-Sánchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312. doi: 10.1038/ng.487
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R., and Coyle, A. J. (2010). HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388. doi: 10.1146/annurev.immunol.021908.132603
Singh, A., Tripathi, P., and Singh, S. (2021). Neuroinflammatory responses in Parkinson's disease: relevance of ibuprofen in therapeutics. Inflammopharmacology 29, 5–14. doi: 10.1007/s10787-020-00764-w
Singh, S. S., Rai, S. N., Birla, H., Zahra, W., Rathore, A. S., and Singh, S. P. (2020). NF-κB-mediated neuroinflammation in Parkinson's disease and potential therapeutic effect of polyphenols. Neurotox. Res. 37, 491–507. doi: 10.1007/s12640-019-00147-2
Slanzi, A., Iannoto, G., Rossi, B., Zenaro, E., and Constantin, G. (2020). In vitro models of neurodegenerative diseases. Front. Cell Dev. Biol. 8, 328. doi: 10.3389/fcell.2020.00328
Soto-Otero, R., Méndez-Álvarez, E., Hermida-Ameijeiras, Á., Muñoz-Patiño, A. M., and Labandeira-Garcia, J. L. (2000). Autoxidation and neurotoxicity of 6-hydroxydopamine in the presence of some antioxidants: potential implication in relation to the pathogenesis of Parkinson's disease. J. Neurochem. 74, 1605–1612. doi: 10.1046/j.1471-4159.2000.0741605.x
Specht, C. G., and Schoepfer, R. (2004). Deletion of multimerin-1 in α-synuclein-deficient mice. Genomics 83, 1176–1178. doi: 10.1016/j.ygeno.2003.12.014
Srikrishna, G.. (2012). S100A8 and S100A9: new insights into their roles in malignancy. J. Innate Immun. 4, 31–40. doi: 10.1159/000330095
Steiner, J. A., Quansah, E., and Brundin, P. (2018). The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res. 373, 161–173. doi: 10.1007/s00441-018-2814-1
Stojkovska, I., Wagner, B. M., and Morrison, B. E. (2015). Parkinson's disease and enhanced inflammatory response. Exp. Biol. Med. 240, 1387–1395. doi: 10.1177/1535370215576313
Storelli, E., Cassina, N., Rasini, E., Marino, F., and Cosentino, M. (2019). Do Th17 lymphocytes and IL-17 contribute to Parkinson's disease? A systematic review of available evidence. Front. Neurol. 10, 13. doi: 10.3389/fneur.2019.00013
Subbarayan, M. S., Hudson, C., Moss, L. D., Nash, K. R., and Bickford, P. C. (2020). T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an α-synuclein rat model of Parkinson's disease. J. Neuroinflammation 17, 242. doi: 10.1186/s12974-020-01911-4
Sulzer, D., Alcalay, R. N., Garretti, F., Cote, L., Kanter, E., Agin-Liebes, J., et al. (2017). T cells from patients with Parkinson's disease recognize α-synuclein peptides. Nature 546, 656–661. doi: 10.1038/nature22815
Sun, Y., Wen, Y., Wang, L., Wen, L., You, W., Wei, S., et al. (2021). Therapeutic opportunities of interleukin-33 in the central nervous system. Front. Immunol. 12, 654626. doi: 10.3389/fimmu.2021.654626
Tansey, M. G., and Goldberg, M. S. (2010). Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518. doi: 10.1016/j.nbd.2009.11.004
Taracanova, A., Alevizos, M., Karagkouni, A., Weng, Z., Norwitz, E., Conti, P., et al. (2017). SP and IL-33 together markedly enhance TNF synthesis and secretion from human mast cells mediated by the interaction of their receptors. Proc. Natl. Acad. Sci. U.S.A. 114, E4002–e4009. doi: 10.1073/pnas.1524845114
Thakur, P., Breger, L. S., Lundblad, M., Wan, O. W., Mattsson, B., Luk, K. C., et al. (2017). Modeling Parkinson's disease pathology by combination of fibril seeds and α-synuclein overexpression in the rat brain. Proc. Natl. Acad. Sci. U.S.A. 114, E8284–e8293. doi: 10.1073/pnas.1710442114
Theodore, S., Cao, S., McLean, P. J., and Standaert, D. G. (2008). Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 67, 1149–1158. doi: 10.1097/NEN.0b013e31818e5e99
Thomas, M., Saldanha, M., Mistry, R., Dexter, D., Ramsden, D., and Parsons, R. (2013). Nicotinamide N-methyltransferase expression in SH-SY5Y neuroblastoma and N27 mesencephalic neurones induces changes in cell morphology via ephrin-B2 and Akt signalling. Cell Death Dis. 4, e669–e669. doi: 10.1038/cddis.2013.200
Titze-de-Almeida, R., and Titze-de-Almeida, S. S. (2018). miR-7 replacement therapy in Parkinson's Disease. Curr. Gene Ther. 18, 143–153. doi: 10.2174/1566523218666180430121323
Tobon-Velasco, C. J., Cuevas, E., and Torres-Ramos, M. A. (2014). Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targ. 13, 1615–1626. doi: 10.2174/1871527313666140806144831
Tore, F., and Tuncel, N. (2009). Mast cells: target and source of neuropeptides. Curr. Pharm. Des. 15, 3433–3445. doi: 10.2174/138161209789105036
Trudler, D., Nash, Y., and Frenkel, D. (2015). New insights on Parkinson's disease genes: the link between mitochondria impairment and neuroinflammation. J. Neural Transm. 122, 1409–1419. doi: 10.1007/s00702-015-1399-z
Trudler, D., Nazor, K. L., Eisele, Y. S., Grabauskas, T., Dolatabadi, N., Parker, J., et al. (2021). Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. U.S.A. 118, e2025847118. doi: 10.1073/pnas.2025847118
Tweedie, D., Sambamurti, K., and Greig, N. H. (2007). TNF-α inhibition as a treatment strategy for neurodegenerative disorders: new drug candidates and targets. Curr. Alzheimer Res. 4, 378–385. doi: 10.2174/156720507781788873
Uematsu, S., Matsumoto, M., Takeda, K., and Akira, S. (2002). Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 168, 5811–5816. doi: 10.4049/jimmunol.168.11.5811
Ungerstedt, U.. (1968). 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 5, 107–110. doi: 10.1016/0014-2999(68)90164-7
Verhaar, R., Drukarch, B., Bol, J. G., Jongenelen, C. A., Musters, R. J., and Wilhelmus, M. M. (2012). Increase in endoplasmic reticulum-associated tissue transglutaminase and enzymatic activation in a cellular model of Parkinson's disease. Neurobiol. Dis. 45, 839–850. doi: 10.1016/j.nbd.2011.10.012
Verhaar, R., Jongenelen, C. A., Gerard, M., Baekelandt, V., Van Dam, A. M., Wilhelmus, M. M., et al. (2011). Blockade of enzyme activity inhibits tissue transglutaminase-mediated transamidation of α-synuclein in a cellular model of Parkinson's disease. Neurochem. Int. 58, 785–793. doi: 10.1016/j.neuint.2011.03.004
von Euler Chelpin, M., and Vorup-Jensen, T. (2017). Targets and mechanisms in prevention of Parkinson's disease through immunomodulatory treatments. Scand. J. Immunol. 85, 321–330. doi: 10.1111/sji.12542
Wang, S., Yuan, Y.-H., Chen, N.-H., and Wang, H.-B. (2019). The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson's disease. Int. Immunopharmacol. 67, 458–464. doi: 10.1016/j.intimp.2018.12.019
Wang, X., and Schwarz, T. L. (2009). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. doi: 10.1016/j.cell.2008.11.046
Wang, Y., Li, L., Hou, C., Lai, Y., Long, J., Liu, J., et al. (2020). SNARE-mediated membrane fusion in autophagy. Semi. Cell Dev. Biol. 60, 97–104. doi: 10.1016/j.semcdb.2016.07.009
Wei, L., Ding, L., Mo, M.-,s, Lei, M., Zhang, L., Chen, K., and Xu, P. (2015). Wnt3a protects SH-SY5Y cells against 6-hydroxydopamine toxicity by restoration of mitochondria function. Transl. Neurodegener. 4, 1–8. doi: 10.1186/s40035-015-0033-1
Wenzel, T. J., Kwong, E., Bajwa, E., and Klegeris, A. (2020). Resolution-associated molecular patterns (RAMPs) as endogenous regulators of glia functions in neuroinflammatory disease. CNS Neurol. Disord. Drug Targets. 19, 483–494. doi: 10.2174/1871527319666200702143719
Wilhelmus, M. M., Verhaar, R., Andringa, G., Bol, J. G., Cras, P., Shan, L., et al. (2011). Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in Parkinson's disease brain. Brain Pathol. 21, 130–139. doi: 10.1111/j.1750-3639.2010.00429.x
Williams, G. P., Schonhoff, A. M., Jurkuvenaite, A., Gallups, N. J., Standaert, D. G., and Harms, A. S. (2021). CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson's disease. Brain 144, 2047–2059. doi: 10.1093/brain/awab103
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease—a double-edged sword. Neuron 35, 419–432. doi: 10.1016/S0896-6273(02)00794-8
Xiao, H. X., Song, B., Li, Q., Shao, Y. M., Zhang, Y. B., Chang, X. L., et al. (2022). Paraquat mediates BV-2 microglia activation by raising intracellular ROS and inhibiting Akt1 phosphorylation. Toxicol. Lett. 355, 116–126. doi: 10.1016/j.toxlet.2021.11.017
Xicoy, H., Wieringa, B., and Martens, G. J. M. (2017). The SH-SY5Y cell line in Parkinson's disease research: a systematic review. Mol. Neurodegener. 12, 10. doi: 10.1186/s13024-017-0149-0
Xie, W., and Chung, K. K. (2012). Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson's disease. J. Neurochem. 122, 404–414. doi: 10.1111/j.1471-4159.2012.07769.x
Yao, K., and Zhao, Y. F. (2018). Aging modulates microglia phenotypes in neuroinflammation of MPTP-PD mice. Exp. Gerontol. 111, 86–93. doi: 10.1016/j.exger.2018.07.010
Yao, L., Zhu, Z., Wu, J., Zhang, Y., Zhang, H., Sun, X., et al. (2019). MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson's disease. FASEB J. 33, 8648–8665. doi: 10.1096/fj.201900363R
Yavich, L., Tanila, H., Vepsäläinen, S., and Jäkälä, P. (2004). Role of α-synuclein in presynaptic dopamine recruitment. J. Neurosci. 24, 11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004
Ye, J., Jiang, Z., Chen, X., Liu, M., Li, J., and Liu, N. (2016). Electron transport chain inhibitors induce microglia activation through enhancing mitochondrial reactive oxygen species production. Exp. Cell Res. 340, 315–326. doi: 10.1016/j.yexcr.2015.10.026
Yi, M., Weaver, D., and Hajnóczky, G. (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167, 661–672. doi: 10.1083/jcb.200406038
Yu, Z., Jiang, N., Su, W., and Zhuo, Y. (2021). Necroptosis: a novel pathway in neuroinflammation. Front. Pharmacol. 12, 701564. doi: 10.3389/fphar.2021.701564
Zaheer, A., Knight, S., Zaheer, A., Ahrens, M., Sahu, S. K., and Yang, B. (2008a). Glia maturation factor overexpression in neuroblastoma cells activates glycogen synthase kinase-3beta and caspase-3. Brain Res. 1190, 206–214. doi: 10.1016/j.brainres.2007.11.011
Zaheer, A., Zaheer, S., Thangavel, R., Wu, Y., Sahu, S. K., and Yang, B. (2008b). Glia maturation factor modulates beta-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 1208, 192–203. doi: 10.1016/j.brainres.2008.02.093
Zhang, C., Sajith, A. M., Xu, X., Jiang, J., Phillip Bowen, J., Kulkarni, A., et al. (2022). Targeting NLRP3 signaling by a novel-designed sulfonylurea compound for inhibition of microglial inflammation. Bioorg. Med. Chem. 58, 116645. doi: 10.1016/j.bmc.2022.116645
Zhang, Y. N., Fan, J. K., Gu, L., Yang, H. M., Zhan, S. Q., and Zhang, H. (2021). Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson's disease. J. Neuroinflammation 18, 23. doi: 10.1186/s12974-021-02079-1
Zhao, J., Bi, W., Xiao, S., Lan, X., Cheng, X., Zhang, J., et al. (2019). Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 9, 5790. doi: 10.1038/s41598-019-42286-8
Zhao, Y. F., Qiong, Z., Zhang, J. F., Lou, Z. Y., Zu, H. B., Wang, Z. G., et al. (2018). The synergy of aging and LPS exposure in a mouse model of Parkinson's disease. Aging Dis. 9, 785–797. doi: 10.14336/AD.2017.1028
Zhou, Y., Lu, M., Du, R. H., Qiao, C., Jiang, C. Y., Zhang, K. Z., et al. (2016). MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Mol. Neurodegener. 11, 28. doi: 10.1186/s13024-016-0094-3
Zhu, J., Hu, Z., Han, X., Wang, D., Jiang, Q., Ding, J., et al. (2018). Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of β-arrestin2 and NLRP3. Cell Death Differ. 25, 2037–2049. doi: 10.1038/s41418-018-0127-2
Keywords: Parkinson's disease, nuclear factor kappa B (NF-κB), NLRP3 inflammasome, microglia, mast cells, neuroinflammation
Citation: Fathi M, Vakili K, Yaghoobpoor S, Qadirifard MS, Kosari M, Naghsh N, Asgari taei A, Klegeris A, Dehghani M, Bahrami A, Taheri H, Mohamadkhani A, Hajibeygi R, Rezaei Tavirani M and Sayehmiri F (2022) Pre-clinical Studies Identifying Molecular Pathways of Neuroinflammation in Parkinson's Disease: A Systematic Review. Front. Aging Neurosci. 14:855776. doi: 10.3389/fnagi.2022.855776
Received: 15 January 2022; Accepted: 23 May 2022;
Published: 04 July 2022.
Edited by:
Suman Dutta, University of California, Los Angeles, United StatesReviewed by:
Surya Pratap Singh, Banaras Hindu University, IndiaCopyright © 2022 Fathi, Vakili, Yaghoobpoor, Qadirifard, Kosari, Naghsh, Asgari taei, Klegeris, Dehghani, Bahrami, Taheri, Mohamadkhani, Hajibeygi, Rezaei Tavirani and Sayehmiri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mostafa Rezaei Tavirani, dGF2aXJhbnlAeWFob28uY29t; Fatemeh Sayehmiri, RnNheWVobWlyaUB5YWhvby5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.