
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci., 11 May 2022
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.849462
This article is part of the Research TopicGenetic and Molecular Diversity in Parkinson's DiseaseView all 9 articles
 Ping Hua1
Ping Hua1 Yuwen Zhao2,3
Yuwen Zhao2,3 Qian Zeng2
Qian Zeng2 Lanting Li1
Lanting Li1 Jingru Ren1
Jingru Ren1 Jifeng Guo2,3,4,5,6
Jifeng Guo2,3,4,5,6 Beisha Tang2,3,4,5,6*
Beisha Tang2,3,4,5,6* Weiguo Liu1*
Weiguo Liu1*
Background: Genetic factors play an important role in the pathogenesis of early-onset Parkinson’s disease (EOPD). To date, more than 20 pathogenic genes associated with Parkinson’s disease (PD) have been identified. This study aims to explore the mutation spectrum of EOPD and the clinical characteristics of mutation carriers in eastern China.
Methods: We recruited 155 unrelated EOPD patients, including 8 familial and 147 sporadic EOPD (age at onset ≤ 50 years). Overall, 24 known PD-associated genes were detected by whole exome sequencing and multiplex ligation-dependent probe amplification (MLPA) from patient samples. The genetic and clinical characteristics of pathogenic/likely pathogenic (P/LP) loci in this cohort were analyzed.
Results: Overall, 14 (9.03%) patients were detected with P/LP variants distributed in seven genes. The most frequent mutation occurred in PRKN (7/155, 4.52%), followed by LRRK2 (2/155, 1.29%), SNCA, CHCHD2, TMEM230, DNAJC13 and PLA2G6 (1/155, 0.64%, respectively). Exon rearrangement mutations accounted for 57.9% (11/19) of all mutations in PRKN. Four novel variants were detected: c.14T > C (p.M5T) in SNCA, c.297C > A (p.Y99X) in CHCHD2, c.2578C > T (p.R860C) in DNAJC13 and c.4C > T (p.Q2X) in TMEM230. We found the first case of LRRK2 c.6055G > A (p.G2019S) mutation in Chinese population. The median onset age of patients with P/LP mutations in autosomal recessive genes (PRKN and PLA2G6) was about 18.0 years earlier than patients without mutation. The proportion of patients with mutations were 63.64%, 27.03% and 9.68% when patients were stratified according to the age of onset at ≤ 30, ≤ 40 and ≤ 50 years, respectively.
Conclusion: Early-onset Parkinson’s disease patients from eastern China present a regional specific mutation spectrum. Analysis of larger patient cohorts is required to support these findings, and mechanistic studies of the four novel missense/non-sense mutations will clarify their role in the pathogenicity of EOPD.
Parkinson’s disease (PD) is a progressive neurodegenerative disease affecting 1% of people older than 60 years and 4% of people older than 85 years (Kalia and Lang, 2015). With aging of the population, prevalence is expected to exceed 5 million in China by 2030, accounting for almost half of the global number of patients with PD (Li et al., 2019). Parkinson’s disease involves complex pathogenic mechanisms, with advancing age as the greatest risk factor; both environmental and genetic factors also affect disease risk and progression. Approximately 5%–10% of patients develop PD early and display different clinical characteristics from PD in the elderly (Mehanna et al., 2014). Thus, PD is divided into early-onset Parkinson’s disease (EOPD) and late-onset Parkinson’s disease (LOPD) according to the age of onset. There is no unified international standard for defining the age of onset for EOPD. However, age boundaries have a significant impact on the statistical association of genetic mutations to PD. Taking the PRKN gene as an example, several European studies defining EOPD onset age at 45 found that more than 15% of sporadic EOPD patients carried pathogenic mutations in PRKN (Lücking et al., 2000; Periquet et al., 2003), while a Chinese study defining EOPD onset at 50 found only 2.66% of sporadic EOPD patients with such mutations (Zhao et al., 2020). At present, most studies set the age of onset for EOPD at ≤50 years old based on the genetic evidence from previous studies (Tanner et al., 1999). Genetic factors are closely related to EOPD. The earlier the onset age, the greater the possibility of detecting genetic variation (Kasten et al., 2018; Blauwendraat et al., 2020a,b; Zhao et al., 2020). Screening the pathogenic gene mutation spectrum in EOPD patients will help us to understand the pathogenesis of PD and explore targeted treatment strategies.
A growing number of pathogenic genes for PD have been identified and assigned PARK numbers according to their reported order (Lunati et al., 2018). Parkinson’s disease caused by pathogenic variants of these genes comply with Mendelian genetics, and are referred to as monogenic Parkinson’s disease. The rare pathogenic variants of these genes result in 20% of EOPD and no more than 3% of LOPD (Klein and Westenberger, 2012). The PRKN, PINK1 and DJ-1 genes are the three most common genes of autosomal-recessive EOPD, and account for approximately 13% of EOPD occurring before the age of 40 years (Puschmann, 2013). The SNCA, LRRK2 and VPS35 are autosomal-dominant genes with relatively clear pathogenic mechanisms (Lunati et al., 2018; Kluss et al., 2019; Cutillo et al., 2020). The significance of other genes requires further functional validation and replication in larger cohorts and different populations. These genes include PLOG, GBA, LRP10, RIC3, and RAB39b; suggesting a wide mutational spectrum for EOPD. Numerous studies around the world from Germany, Britain, Finland and South Korea, as well as southwest China, western China and Taiwan have revealed the genetic heterogeneity of different regions and races for EOPD (Siitonen et al., 2017; Lin et al., 2019; Tan et al., 2019; Trinh et al., 2019; Youn et al., 2019; Li et al., 2020; Zhao et al., 2020). At present, there is no genetic map of EOPD patients in eastern China.
To provide insights into the mutational profiles of eastern Chinese EOPD patients, this study screened mutations in 24 known PD-related genes through whole exome sequencing (WES) and multiplex ligation-dependent probe amplification (MLPA) in this cohort. In addition, the clinical characteristics of patients with pathogenic/likely pathogenic (P/LP) variants were analyzed to provide a correlation between genotype and phenotype.
A total of 155 patients with EOPD (age at onset ≤ 50 years) including 147 sporadic PD; 5 from families with autosomal-dominant (AD) PD and 3 from families with autosomal-recessive (AR) PD; were recruited from the Center for Parkinson and Movement Disorders at Nanjing Brain Hospital between September 2009 and June 2018. Among these patients, 136 cases came from the Jiangsu Province, while 19 cases came from the southern region of Anhui Province. Data from these patients were added into Parkinson’s Disease & Movement Disorders Multicenter Database and Collaborative Network in China (PD-MDCNC), established by Tang’s team. All the patients were diagnosed by two experienced neurologists, based on the United Kingdom PD Society Brain Bank diagnostic criteria for PD (Gibb and Lees, 1988) or the 2015 Movement Disorder Society clinical diagnostic criteria for PD (Postuma et al., 2015). Exclusion criteria included other neurodegenerative disorders, psychiatric disorders, and severe physical illnesses. Motor symptoms were evaluated by the Unified Parkinson’s disease Rating Scale (UPDRS) Parts I and II, and Hoehn-Yahr staging (H-Y). Non-motor symptoms were assessed by the 24-item Hamilton Rating Scale for Depression (HAMD), 14-item Hamilton Rating Scale for Anxiety (HAMA), Parkinson’s Disease Sleep Scale (PDSS), Mini-Mental State Examination (MMSE), Montreal Cognitive Assessment (MoCA), and Non-Motor Symptoms Screening Questionnaire (NMSQ). Medication was recorded and converted to the levodopa equivalent daily dose (LED) (Tomlinson et al., 2010).
Genomic DNA was isolated from citric acid anticoagulated peripheral blood leukocytes using the DNA Extraction Kit (Tiangen, Beijing, China). The exon and flanking regions of 24 PD-associated genes (Supplementary Table 1) were captured as previously described (Zhao et al., 2020). Whole-exome sequencing (WES) was performed on the Illumina HiSeq X10 platform with 150bp paired-end reads. The acquired reads were mapped onto the reference human genome (UCSC hg19) using the Burrow-Wheeler Aligner. Quality control and annotation were done as previously described (Zhao et al., 2020). Rare variants were filtered according to the minor allele frequency (1% for recessive genes; 0.1% for dominant genes) in East Asian population from public databases (GnomAD and ExAC).
Large exon deletions or duplications (copy-number variants, CNVs) of common PD-associated genes including SNCA, PRKN, PINK1, DJ-1, ATP13A2, and LRRK2 were detected using the Salsa MLPA kit P051-C1 (MRC-Holland, Amsterdam, The Netherlands).
The pathogenicity of missense mutations, non-sense variants, splicing site variants and Indels screened by WES was predicted by CADD,1 PolyPhen-2,2 SIFT,3 Mutation Taster4 and ReVe (Li et al., 2018). We also searched pathogenic variants in Pubmed,5 Gene4PD,6 and MDSGene,7 to determine whether they were reported before. According to the American College of Medical Genetics and Genomics (ACMG) recommendations (Richards et al., 2015), the candidate variants were further interpreted and classified into pathogenicity (P), likely pathogenicity (LP), uncertain significance (US), likely benign (LB) and benign (B). For pathogenic or likely pathogenic (P/LP) variants, patients carrying two or more allele variants and their first-degree relatives were sequenced by Sanger and/or MLPA for co-separation analysis.
Statistical analysis was performed using the SPSS software, version 18.0. Quantitative data with, or near, normal distribution were described by mean ± standard deviation, and t-test was used for comparison between the groups. Quantitative data with skewed distribution were described by median (minimum-maximum), and Mann-Whitney non-parametric test was used for comparison between the groups. The demographic and clinical data were compared between the P/LP variant carriers and non-carriers, and between carriers with AR/AD genetic variants and non-carriers. The frequency of gene mutations at different age-of-onset was compared. A two-tailed P < 0.05 was considered statistically significant.
The mean age at onset of the 155 patients was 43.39 ± 7.13 years, 58.7% were male; the mean age at assessment was 50.76 ± 9.28 years; and the mean duration of disease at assessment was 8.16 ± 7.40 years. The medium H-Y score was 2.0, ranging from 1.0 to 5.0, and 65% of patients were in the early stages of the disease. The motor and non-motor symptoms were relatively mild (Table 1).
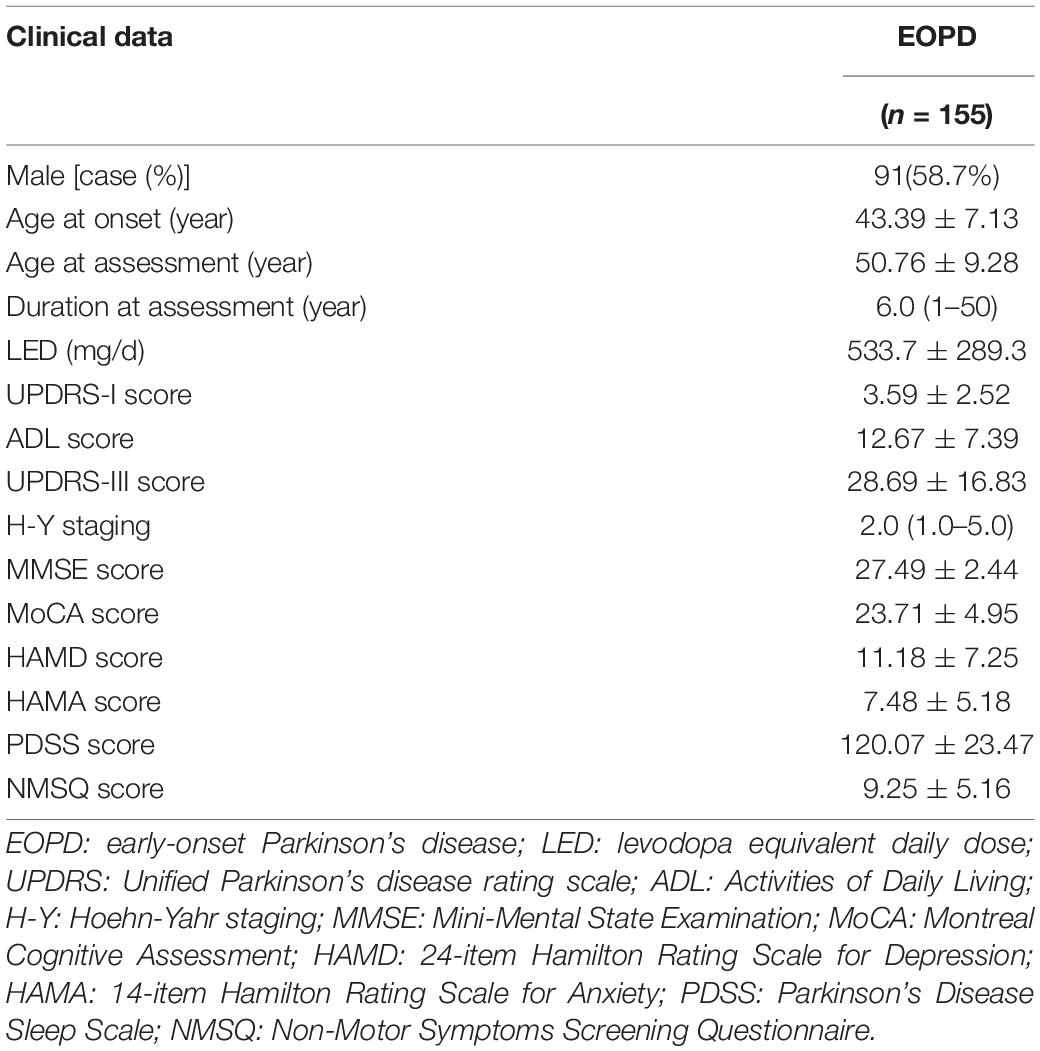
Table 1. Clinical data of 155 patients with EOPD.
P/LP mutations were detected in 14 (9.03%) patients, including one patient with family history of ADPD (1/5, 20%), one patient with family history of ARPD (1/3, 33.3%), and 12 sporadic patients (12/147, 8.8%) (Table 2). Among these 14 patients, immediate relatives of seven patients came for verification, and the genotypes of two patients’ parents could be inferred from their genotypes, so the pedigree chart of nine families is shown in Supplementary Figure 1.
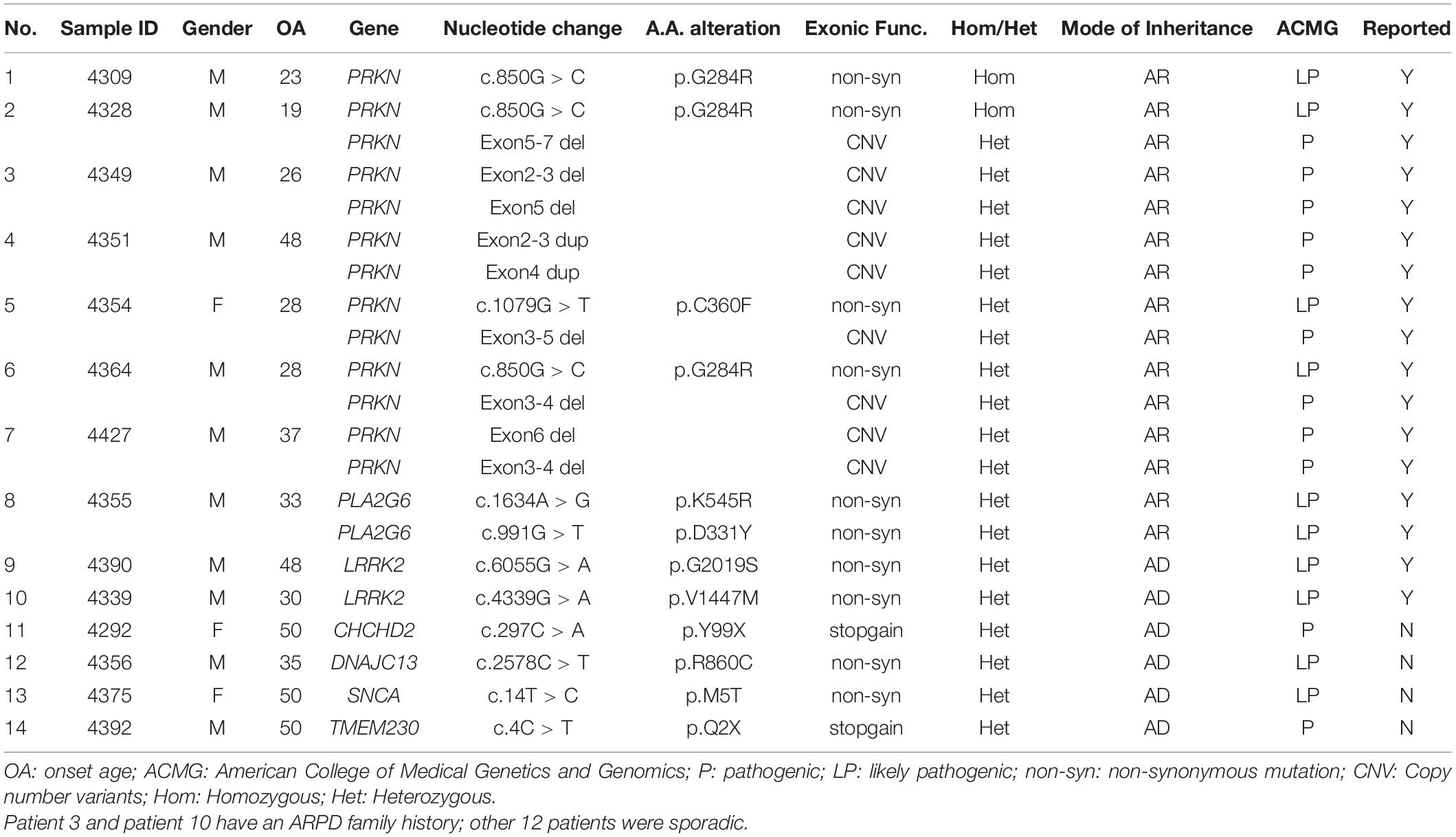
Table 2. List of 14 patients carrying rare P/LP variants in PD associated genes.
PRKN was the most prevalent causative gene in the 155 patients with EOPD (7/155, 4.52%). Among the seven P/LP-variants carriers, two had a homozygous variant (c.850G > C; one case also had a heterozygous deletion of exon 5–7), and five had compound heterozygous variants (Table 2). A compound heterozygous variant of PLA2G6 (c.1634A > G/c.991G > T) was also identified. All variants for both genes had been reported previously (Koh et al., 2019b), and included in the MDSGene and Gene4PD databases. Two LP variants of the LRRK2 were detected in this group, accounting for 1.3% of the total patients. One case was c.4339G > A (p.V1447M), which has been reported previously (Zhao et al., 2020). The other case detected was c.6055G > A (p.G2019S). This is the first case of LRRK2 c.6055G > A (p.G2019S) mutation detected in the Chinese population. In addition, four heterozygous missense/non-sense mutations were found in the ADPD-related genes CHCHD2, DNAJC13, SNCA and TMEM230, which have not been reported previously (Table 3).
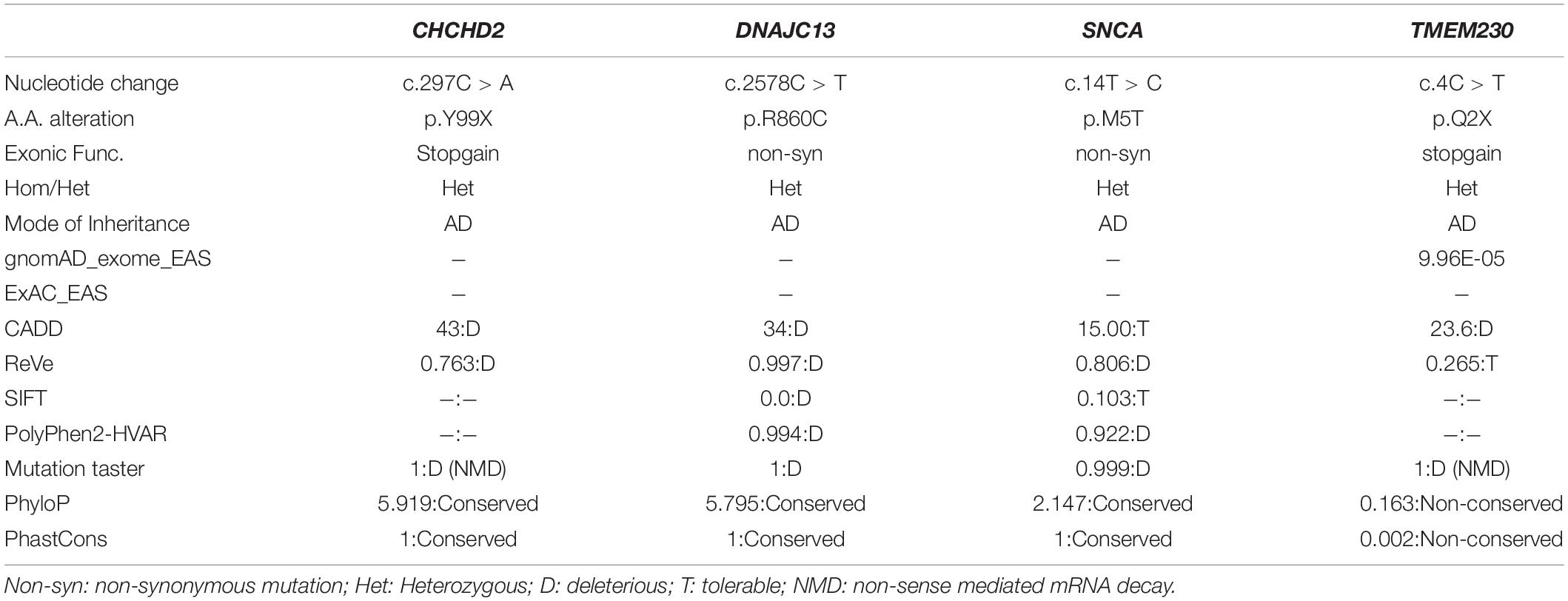
Table 3. Population frequency and pathogenicity prediction of four novel variants.
There were 22 patients that had non-pathogenic variants, including four variants of AD genes (Supplementary Table 2) that were classified as uncertain significance according to the ACMG recommendations (the population frequency and pathogenicity prediction of these four variants see in Supplementary Table 3), and 19 single heterozygous variants of AR genes (one patient carried two simple heterozygous variants; see details in Supplementary Table 4). Although a single heterozygous variant of the AR gene is not genetically pathogenic, six variants (c.1285 + 2T > C, c.850G > C, deletion of exon 2–3, deletion of exon 8-9 of PRKN, p.R3296Gfs*2 frameshift mutation of VPS13c, and p.N801Rfs*11 frameshift mutation of SYNJ1) among these 19 single heterozygous variants need to be paid attention to as these six variants may cause coding abnormalities of single chromosomes and become pathogenic, if they exist in homozygous form. Among them, the patient with c.1285 + 2T > C of PRKN has a history of ARPD. We failed to get the families of these patients to do co-separation analysis, otherwise it may help us explore whether these heterozygous variants increase the risk of PD.
Clinical phenotypes were compared between carriers of P/LP variants and non-carriers, and between carriers of US variants and non-carriers (Table 4). The results showed that the median age at onset of patients with P/LP variants (mean 36.07 years; median 34 years) was 12 years earlier than that of patients without these gene variants (mean 44.52 years; median 46 years). However, there was no significant difference in the severity of motor and non-motor symptoms between these two groups. Comparing the US variant carriers and non-carriers, there was no significant difference in age of onset of clinical manifestation.
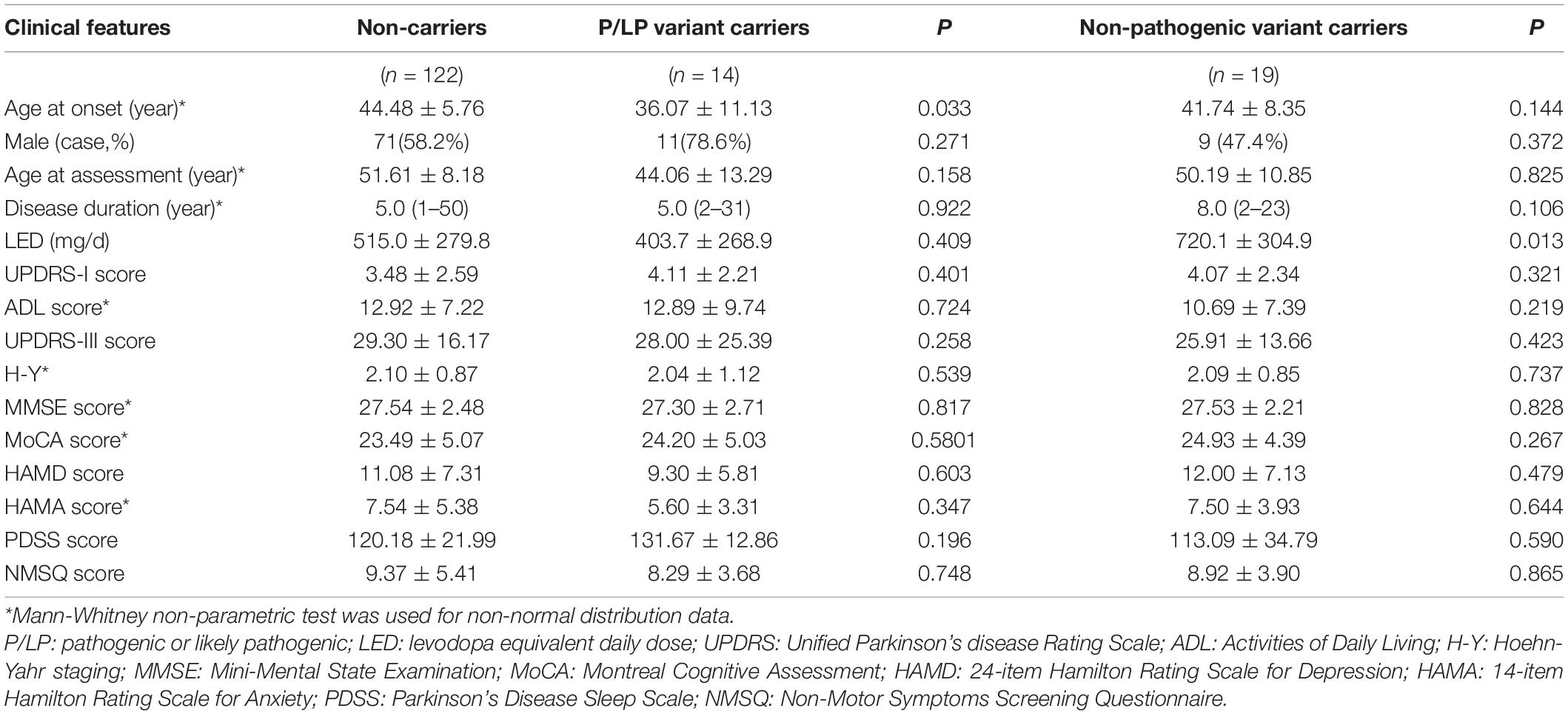
Table 4. Comparison of clinical phenotypes between P/LP variant carriers and non-carriers, and between with non- pathogenic variant carriers and non-carriers.
The age at onset and NMSQ score of patients with PRKN variants were significantly lower than those without mutation in this gene, and the course of disease at the time of evaluation was significantly longer than those without gene mutation. However, there was no significant difference in the severity of motor and non-motor symptoms between the two groups, indicating that disease progression in PRKN variant patients was relatively slower in the tested Chinese population (Table 5).
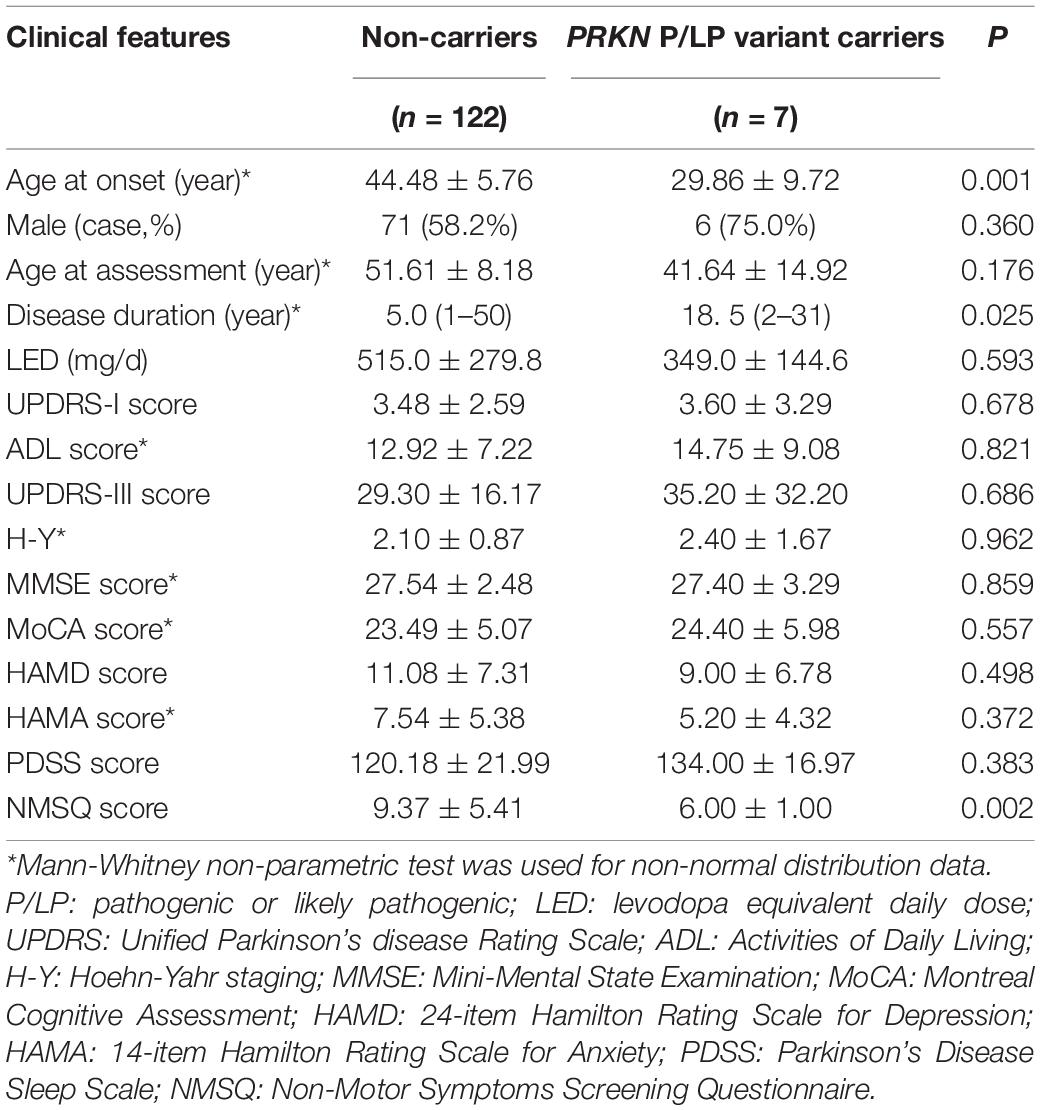
Table 5. Comparation of clinical phenotypes between carriers with P/LP variants in PRKN and non-carriers.
In this cohort, the onset age of carriers with AD gene variants (mean 43.83 years, median 49 years) was older than carriers with AR gene variants (mean 30.25 years, median 28 years) (Supplementary Table 5). Three patients with variants in CHCHD2, SNCA and TMEM230 were 50 years old, and two patients with variants in LRRK2 were 30 and 48 years old, respectively. The median onset age of patients with the PRKN mutation was only 27 years, while the onset age of patients with PLA2G6 mutation was 33 years. The molecular diagnosis rate of 155 patients with EOPD in this cohort was inversely proportional to the age at onset. In this sample, the rate of P/LP variants occurring in patients with onset age below 30 years was as high as 54.55% (6/11), reducing to 24.32% (9/37) in patients with onset age below 40, and the rate in patients with onset age below 50 was only 9.03% (14/155).
This study investigated the genetic causes of EOPD patients in eastern China using WES integrated with MLPA. The results identified 14 patients (9.03%) with P/LP variants in PRKN, PLA2G6, LRRK2, SNCA, CHCHD2, TMEM230 and GIGYF2; the first case of LRRK2 c.6055G > A (p.G2019S) mutation in a Chinese population, and four novel P/LP mutation sites. This revealed some geographical heterogeneities.
The proportion of familial PD in this study (5.2%) was lower than in the study conducted in southwest (19.6%) of China (Li et al., 2020). However, the proportion of patients carrying P/LP variants in this study was a bit higher than the study in southwest (7.5%). Additionally, the proportion of patients carrying the P/LP variants in the sporadic EOPD cohort in southcentral of China was only 4.59% (Zhao et al., 2020). The most common mutated gene across all three studies was PRKN, as it accounted for nearly 50.0% of all patients with identifiable mutations. The most common form of pathogenic mutation in PRKN was exon rearrangement with compound heterozygous mutation. We also found that the mean age of onset for patients with PRKN mutations was 15 years earlier than that of EOPD patients without mutations, and this is consistent with previous reports (Kasten et al., 2018; Zhao et al., 2020). In addition to PRKN, an LP variant in PLA2G6 was also detected. The age of onset for this patient was 35 years, which was consistent with a previous report that patients with ARPD-related gene mutations had an early age of onset (Kasten et al., 2018). Previous studies have reported that patients with PRKN, PLA2G6, PINK1 and ATP13A2 mutations showed significantly earlier onset than patients with VPS13C and RIC3 variants, while those with LRRK2, SNCA and GIGYF2 mutations generally showed later onset (Zhao et al., 2020). Therefore, genetic testing can be helpful in diagnosing EOPD when symptoms occur before 40 years of age, especially for those with a family history of ARPD. In this regard, the MLPA technology is particularly useful in detecting exon rearrangement variations in EOPD patients, and should be considered for routine use in clinics.
It has been reported that the mutation frequency of PINK1 is secondary to PRKN, and occurs in 3.7% of AR-EOPD patients (Hernandez et al., 2016). However, pathogenic variants of PINK1 were not detected in our cohort, and one patient carrying the PINK1 variant was detected in the study of southwest and southcentral regions, respectively. A compound heterozygous variant of PLA2G6 (c.1634A > G, c.991G > T) was detected, which is the only mutation in a recessive gene detected in our study, aside from PRKN. The 33-year-old patient suffered from dystonia of the left foot, and developed dyskinesia after two years despite responding well to levodopa in early treatment, which was consistent with the clinical manifestation of dystonia-Parkinson’s syndrome caused by PLA2G6 (Paisan-Ruiz et al., 2009). The study in southwest China also detected one compound heterozygous variant of PLA2G6 (Li et al., 2020). The southcentral study detected three compound heterozygous variants of PLA2G6 in sporadic EOPD, and was found to be the second most frequent mutation in the ARPD cohort (Zhao et al., 2020). It seems that the mutation frequency of PLA2G6 in the Chinese population is higher than PINK1 and DJ-1, and most of them exist in the compound heterozygous state. Moreover, several single heterozygous mutations of PLA2G6 detected in Chinese PD patients were associated with impaired PLA2G6 phospholipase activity (Gui et al., 2013). Of particular interest is mutations in PLA2G6 have been linked to several neurological diseases including infantile neuroaxonal dystrophy (INAD) (Khateeb et al., 2006), neurodegeneration with brain iron accumulation (NBIA) (Morgan et al., 2006), hereditary spastic paraplegia (HSP) (Koh et al., 2019a) and PD, but the underlying mechanism is still unknown. Thus, further research on the complexity of gene-gene and gene-environment interactions will be required to unveil the role of PLA2G6 in the Chinese population.
Four novel mutations were detected in this cohort, all of which were SNVs of ADPD related genes. Two non-sense mutations, c.297C > A (p.Y99X) of CHCHD2 and c.4C > T (p.Q2X) of TMEM230, resulted in premature termination codons, which may produce truncated proteins and caused the disease through haploinsufficiency effect or non-sense mediated mRNA decay (NMD). TMEM230 is a recently discovered PD-associated gene and encodes membrane transporter 230. The physiological function of this protein is unclear; but it can be found in vesicle structures in dopaminergic neurons of the substantia nigra and Purkinje neurons of the cerebellum; and may participate in the transport and circulation of synaptic vesicles, autophagy, neurotoxicity, and Golgi secretion. Several PD-linked TMEM230 mutations have been reported in North American (Deng et al., 2016), Caucasian (Giri et al., 2017), and Chinese populations (Fan et al., 2017; Yang et al., 2017). However, functional studies are still required to determine the role of these variants. The CHCHD2 protein is located in the mitochondrial intermembrane space and is highly expressed in dopaminergic neurons of the substantia nigra. This protein is involved in the mitochondrial-mediated apoptotic pathway and regulates the function of complex IV in the mitochondrial respiratory chain (Imai et al., 2019). Abl2 kinase phosphorylation of CHCHD2 at Tyr99 is involved in the regulation of complex IV (Aras et al., 2017); the c.297C > A (p.Y99X) mutation identified in our study may be pathogenic by affecting CHCHD2 phosphorylation. There is a high linkage disequilibrium between the two mutations of DNAJC13, c.2578C > T (p.R860C) and c.2564A > G (p.N855S), which were reported to be the pathogenic variants of a large four-generation ADPD family (Vilariño-Güell et al., 2014). There may be synergism between these two mutations, affecting the endosomal transport and sorting function of the DNAJC13 protein, and participating in the deposition of α-synuclein aggregates and Lewy bodies in specific brain regions (Vilariño-Güell et al., 2014). Point mutations in SNCA may upregulate α-synuclein expression (Cronin et al., 2009), alter its solubility (Porcari et al., 2015), promote early oligomerization and accelerate α-synuclein fibrillation (Mohite et al., 2018). The c.14T > C (p.M5T) mutation in SNCA detected in our study may result in two possible pathogenic mechanisms. The first may be a gain-of-function (GoF) effect, resulting in increased oligomerization of α-synuclein, while the second may be a dominant negative (DN) effect, with compensatory over-expression of wild-type single-stranded DNA of the heterozygous variant. However, functional studies will be required to validate the pathogenic role of SNCA c.14T > C (p.M5T). The clinical manifestation of this patient is consistent with the phenotypic characteristics of missense mutation in SNCA: the age of onset is late (50 years old), the progress is rapid, with the development of obvious cognitive impairment (MMSE 24 points, MoCA 17 points) and mental symptoms (hallucinations, spousal infidelity delusions) after 2 years.
Finally, we report here the first case of LRRK2 c.6055G > A (p.G2019S) mutation in a Chinese population. This is a 48-year-old man with no family history of PD. His symptoms are characterized by rigidity and bradykinesia of both lower limbs with no obvious tremor; increased axial muscular tension; Pisa syndrome; few non-motor symptoms; slow disease progression and good response to piribedil. These are consistent with the clinical phenotype of Ashkenazi Jews with LRRK2 c.6055G > A (p.G2019S) mutations where carriers had longer disease duration; lower extremity onset; postural instability and gait difficulty; persistent response to levodopa for more than 5 years (Alcalay et al., 2013). The mutation frequency of this locus is high in North African Arabs and German Jews, but the positive rate in Eurasian populations is only 0.1%–4% (Lesage et al., 2006; Ozelius et al., 2006). Our detection of the first Chinese case with this mutation appears to be in line with these reported statistics; reinforcing the need for a deeper understanding of these population differences and their implications for diagnosis and treatment strategies.
There are several limitations in this study: first, the first identified case of LRRK2 c.6055G > A (p.G2019S) mutation in the Chinese population can benefit from more in-depth genetic analysis. Second, the four novel P/LP variants require functional experiments to clarify their pathogenic mechanism. Finally, we only detected rare mutations in the coding region and splicing site of known pathogenic genes; evaluation of non-coding regions may reveal more pathogenic variants. With whole genome sequencing combined with MLPA technology, this may become less challenging, and comprehensive mapping of the mutation spectrum of PD will be possible.
The mutation spectrum of EOPD patients in eastern China displayed different characteristics compared to previously reported populations. Larger scale epidemiological investigations across centers and regions, as well as in-depth mechanistic studies, will further improve our understanding of the etiology of EOPD. The EOPD subtyping system with a combination of clinical markers such as clinical phenotype, imaging and abnormal protein expression will facilitate early diagnosis, guide individualized treatment, and be an important means to realize clinical translation of genetic research.
According to national legislation/guidelines, specifically the Administrative Regulations of the People’s Republic of China on Human Genetic Resources (http://www.gov.cn/zhengce/content/2019-06/10/content_5398829.htm, http://english.www.gov.cn/policies/latest_releases/2019/06/10/content_281476708945462.htm), no additional raw data are available at this time. Data of this project can be accessed after an approved application to the China National Genebank (CNGB, https://db.cngb.org/cnsa/). Please refer to https://db.cngb.org/, or email: Q05HQmRiQGNuZ2Iub3Jn for detailed application guidance. The accession code CNP0002762 should be included in the application.
The studies involving human participants were reviewed and approved by the Ethics Committee of Nanjing Medical University. The patients/participants provided their written informed consent to participate in this study.
PH and JG: conceptualization. YZ, QZ, and PH: data curation and formal analysis. PH, LL, and JR: investigation. PH: writing – original draft. JG, BT, and WL: supervision and writing – review and editing. WL: funding acquisition. BT and WL: resources. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key Research and Development Program of China (2017YFC1310300), the National Natural Science Foundation of China (NSFC) (No. 81571348), the Science and Technology Program of Jiangsu Province (Nos. BE2019611 and BE2018608), the Jiangsu Provincial Natural Science Foundation of China (BK20151077), Key Project supported by Medical Science and Technology Development Foundation, Nanjing Department of Health (No. JQX18005).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are indebted to the participation of patients and their family members that participated in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.849462/full#supplementary-material
Alcalay, R. N., Mirelman, A., Saunders-Pullman, R., Tang, M. X., Mejia Santana, H., Raymond, D., et al. (2013). Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov. Disord. 28, 1966–1971. doi: 10.1002/mds.25647
Aras, S., Arrabi, H., Purandare, N., Hüttemann, M., Kamholz, J., Züchner, S., et al. (2017). Abl2 kinase phosphorylates Bi-organellar regulator MNRR1 in mitochondria, stimulating respiration. Biochim. Biophys. Acta Mol. Cell Res. 1864, 440–448. doi: 10.1016/j.bbamcr.2016.11.029
Blauwendraat, C., Nalls, M. A., and Singleton, A. B. (2020a). The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178.
Blauwendraat, C., Reed, X., Krohn, L., Heilbron, K., Bandres-Ciga, S., Tan, M., et al. (2020b). Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143, 234–248. doi: 10.1093/brain/awz350
Cronin, K. D., Ge, D., Manninger, P., Linnertz, C., Rossoshek, A., Orrison, B. M., et al. (2009). Expansion of the Parkinson disease-associated SNCA-Rep1 allele upregulates human alpha-synuclein in transgenic mouse brain. Hum. Mol. Genet. 18, 3274–3285. doi: 10.1093/hmg/ddp265
Cutillo, G., Simon, D. K., and Eleuteri, S. (2020). VPS35 and the mitochondria: connecting the dots in Parkinson’s disease pathophysiology. Neurobiol. Dis. 145:105056. doi: 10.1016/j.nbd.2020.105056
Deng, H. X., Shi, Y., Yang, Y., Ahmeti, K. B., Miller, N., Huang, C., et al. (2016). Identification of TMEM230 mutations in familial Parkinson’s disease. Nat. Genet. 48, 733–739. doi: 10.1038/ng.3589
Fan, T. S., Lin, C. H., Lin, H. I., Chen, M. L., and Wu, R. M. (2017). Lack of TMEM230 mutations in patients with familial and sporadic Parkinson’s disease in a Taiwanese population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 751–756. doi: 10.1002/ajmg.b.32576
Gibb, W. R., and Lees, A. J. (1988). The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 51, 745–752. doi: 10.1136/jnnp.51.6.745
Giri, A., Mok, K. Y., Jansen, I., Sharma, M., Tesson, C., Mangone, G., et al. (2017). Lack of evidence for a role of genetic variation in TMEM230 in the risk for Parkinson’s disease in the Caucasian population. Neurobiol. Aging 50, .e111–.e167. doi: 10.1016/j.neurobiolaging.2016.10.004
Gui, Y. X., Xu, Z. P., Lv, Wen, Liu, H. M., Zhao, J. J., and Hu, X. Y. (2013). Four novel rare mutations of PLA2G6 in Chinese population with Parkinson’s disease. Parkinsonism Relat. Disord. 19, 21–26. doi: 10.1016/j.parkreldis.2012.07.016
Hernandez, D. G., Reed, X., and Singleton, A. B. (2016). Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 139(Suppl. 1) 59–74. doi: 10.1111/jnc.13593
Imai, Y., Meng, H., Shiba-Fukushima, K., and Hattori, N. (2019). Twin CHCH proteins, CHCHD2, and CHCHD10: key molecules of Parkinson’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Int. J. Mol. Sci. 20:908. doi: 10.3390/ijms20040908
Kasten, M., Hartmann, C., Hampf, J., Schaake, S., Westenberger, A., Vollstedt, E. J., et al. (2018). Genotype-phenotype relations for the Parkinson’s disease genes parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741. doi: 10.1002/mds.27352
Khateeb, S., Flusser, H., Ofir, R., Shelef, I., Narkis, G., Vardi, G., et al. (2006). PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am. J. Hum. Genet. 79, 942–948. doi: 10.1086/508572
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888.
Kluss, J. H., Mamais, A., and Cookson, M. R. (2019). LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 47, 651–661. doi: 10.1042/BST20180462
Koh, K., Ichinose, Y., Ishiura, H., Nan, H., Mitsui, J., Takahashi, J., et al. (2019a). Correction: PLA2G6-associated neurodegeneration presenting as a complicated form of hereditary spastic paraplegia. J. Hum. Genet. 64, 61–63. doi: 10.1038/s10038-018-0533-9
Koh, K., Ichinose, Y., Ishiura, H., Nan, H., Mitsui, J., Takahashi, J., et al. (2019b). PLA2G6-associated neurodegeneration presenting as a complicated form of hereditary spastic paraplegia. J. Hum. Genet. 64, 55–59. doi: 10.1038/s10038-018-0519-7
Lesage, S., Dürr, A., Tazir, M., Lohmann, E., Leutenegger, A. L., Janin, S., et al. (2006). LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N. Engl. J. Med. 354, 422–423. doi: 10.1056/NEJMc055540
Li, G., Ma, J., Cui, S., He, Y., Xiao, Q., Liu, J., et al. (2019). Parkinson’s disease in China: a forty-year growing track of bedside work. Transl. Neurodegener. 8:22. doi: 10.1186/s40035-019-0162-z
Li, J., Zhao, T., Zhang, Y., Zhang, K., Shi, L., Chen, Y., et al. (2018). Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 46, 7793–7804. doi: 10.1093/nar/gky678
Li, N., Wang, L., Zhang, J., Tan, E. K., Li, J., Peng, J., et al. (2020). Whole-exome sequencing in early-onset Parkinson’s disease among ethnic Chinese. Neurobiol. Aging 90, .e5–.e150. doi: 10.1016/j.neurobiolaging.2019.12.023
Lin, C. H., Chen, P. L., Tai, C. H., Lin, H. I., Chen, C. S., Chen, M. L., et al. (2019). A clinical and genetic study of early-onset and familial parkinsonism in taiwan: an integrated approach combining gene dosage analysis and next-generation sequencing. Mov. Disord. 34, 506–515. doi: 10.1002/mds.27633
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Lunati, A., Lesage, S., and Brice, A. (2018). The genetic landscape of Parkinson’s disease. Rev. Neurol. (Paris) 174, 628–643.
Mehanna, R., Moore, S., Hou, J. G., Sarwar, A. I., and Lai, E. C. (2014). Comparing clinical features of young onset, middle onset and late onset Parkinson’s disease. Parkinsonism Relat. Disord. 20, 530–534. doi: 10.1016/j.parkreldis.2014.02.013
Mohite, G. M., Kumar, R., Panigrahi, R., Navalkar, A., Singh, N., Datta, D., et al. (2018). Comparison of kinetics, toxicity, oligomer formation, and membrane binding capacity of α-synuclein familial mutations at the A53 site, including the newly discovered A53V mutation. Biochemistry 57, 5183–5187. doi: 10.1021/acs.biochem.8b00314
Morgan, N. V., Westaway, S. K., Morton, J. E., Gregory, A., Gissen, P., Sonek, S., et al. (2006). PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 38, 752–754. doi: 10.1038/ng1826
Ozelius, L. J., Senthil, G., Saunders-Pullman, R., Ohmann, E., Deligtisch, A., Tagliati, M., et al. (2006). LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 354, 424–425. doi: 10.1056/nejmc055509
Paisan-Ruiz, C., Bhatia, K. P., Li, A., Hernandez, D., Davis, M., Wood, N. W., et al. (2009). Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 65, 19–23. doi: 10.1002/ana.21415
Periquet, M., Latouche, M., Lohmann, E., Rawal, N., De Michele, G., Ricard, S., et al. (2003). Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 126, 1271–1278. doi: 10.1093/brain/awg136
Porcari, R., Proukakis, C., Waudby, C. A., Bolognesi, B., Mangione, P. P., Paton, J. F., et al. (2015). The H50Q mutation induces a 10-fold decrease in the solubility of α-synuclein. J. Biol. Chem. 290, 2395–2404. doi: 10.1074/jbc.M114.610527
Postuma, R. B., Berg, D., Stern, M., Poewe, W., Olanow, C. W., Oertel, W., et al. (2015). MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601. doi: 10.1002/mds.26424
Puschmann, A. (2013). Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat. Disord. 19, 407–415. doi: 10.1016/j.parkreldis.2013.01.020
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Siitonen, A., Nalls, M. A., Hernández, D., Gibbs, J. R., Ding, J., Ylikotila, P., et al. (2017). Genetics of early-onset Parkinson’s disease in finland: exome sequencing and genome-wide association study. Neurobiol. Aging 53, .e7–.e195. doi: 10.1016/j.neurobiolaging.2017.01.019
Tan, M. M. X., Malek, N., Lawton, M. A., Hubbard, L., Pittman, A. M., Joseph, T., et al. (2019). Genetic analysis of Mendelian mutations in a large UK population-based Parkinson’s disease study. Brain 142, 2828–2844. doi: 10.1093/brain/awz191
Tanner, C. M., Ottman, R., Goldman, S. M., Ellenberg, J., Chan, P., Mayeux, R., et al. (1999). Parkinson disease in twins: an etiologic study. JAMA 281, 341–346. doi: 10.1001/jama.281.4.341
Tomlinson, C. L., Stowe, R., Patel, S., Rick, C., Gray, R., and Clarke, C. E. (2010). Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov. Disord. 25, 2649–2653. doi: 10.1002/mds.23429
Trinh, J., Lohmann, K., Baumann, H., Balck, A., Borsche, M., Brüggemann, N., et al. (2019). Utility and implications of exome sequencing in early-onset Parkinson’s disease. Mov. Disord. 34, 133–137. doi: 10.1002/mds.27559
Vilariño-Güell, C., Rajput, A., Milnerwood, A. J., Shah, B., Szu-Tu, C., Trinh, J., et al. (2014). DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 23, 1794–1801.
Yang, X., An, R., Xi, J., Zheng, J., Chen, Y., Huang, H., et al. (2017). Sequencing TMEM230 in Chinese patients with sporadic or familial Parkinson’s disease. Mov. Disord. 32, 800–802. doi: 10.1002/mds.26996
Youn, J., Lee, C., Oh, E., Park, J., Kim, J. S., Kim, H. T., et al. (2019). Genetic variants of PARK genes in Korean patients with early-onset Parkinson’s disease. Neurobiol. Aging 75, .e9–.e224. doi: 10.1016/j.neurobiolaging.2018.10.030
Keywords: Parkinson’s disease, early onset, whole exome sequencing, genetics, Chinese population
Citation: Hua P, Zhao Y, Zeng Q, Li L, Ren J, Guo J, Tang B and Liu W (2022) Genetic Analysis of Patients With Early-Onset Parkinson’s Disease in Eastern China. Front. Aging Neurosci. 14:849462. doi: 10.3389/fnagi.2022.849462
Received: 06 January 2022; Accepted: 04 April 2022;
Published: 11 May 2022.
Edited by:
Kin Ying Mok, University College London, United KingdomReviewed by:
Vinita Ganesh Chittoor, University of California, San Francisco, United StatesCopyright © 2022 Hua, Zhao, Zeng, Li, Ren, Guo, Tang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiguo Liu, bGl1d2VpZ3VvMTExMUBzaW5hLmNvbQ==; Beisha Tang, YnN0YW5nNzM5OEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.