 Roman Vozdek
Roman Vozdek Peter P. Pramstaller
Peter P. Pramstaller Andrew A. Hicks
Andrew A. Hicks
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Aging Neurosci. , 27 April 2022
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.806000
This article is part of the Research Topic Novel mechanisms involved in aging and neurodegeneration: Seeking potential therapeutic targets for neurodegenerative diseases. View all 13 articles
Idiopathic Parkinson’s disease (PD) is characterized by progressive loss of dopaminergic (DA) neurons during aging. The pathological hallmark of PD is the Lewy body detected in postmortem brain tissue, which is mainly composed of aggregated α-Synuclein (αSyn). However, it is estimated that 90% of PD cases have unknown pathogenetic triggers. Here, we generated a new transgenic Caenorhabditis elegans PD model eraIs1 expressing green fluorescent protein- (GFP-) based reporter of human αSyn in DA neurons, and exhibited a nice readout of the developed αSyn inclusions in DA neurons, leading to their degeneration during aging. Using these animals in a preliminary reverse genetic screening of >100-PD genome-wide association study- (GWAS-) based susceptibility genes, we identified 28 orthologs of C. elegans and their inactivation altered the phenotype of eraIs1; 10 knockdowns exhibited reduced penetrance of αSyn:Venus inclusions formed in the axons of cephalic (CEP) DA neurons, 18 knockdowns exhibited increased penetrance of disrupted CEP dendrite integrity among which nine knockdowns also exhibited disrupted neuronal morphology independent of the expressed αSyn reporter. Loss-of-function alleles of the five identified genes, such as sac-2, rig-6 or lfe-2, unc-43, and nsf-1, modulated the corresponding eraIs1 phenotype, respectively, and supported the RNA interference (RNAi) data. The Western blot analysis showed that the levels of insoluble αSyn:Venus were not correlated with the observed phenotypes in these mutants. However, RNAi of 12 identified modulators reduced the formation of pro-aggregating polyglutamine Q40:YFP foci in muscle cells, suggesting the possible role of these genes in cellular proteotoxicity. Therefore, modulators identified by their associated biological pathways, such as calcium signaling or vesicular trafficking, represent new potential therapeutic targets for neurodegenerative proteopathies and other diseases associated with aging.
Neurotoxicity of disordered α-synuclein (αSyn) is a pathogenetic hallmark of synucleinopathies, including Parkinson’s disease [PD; Spillantini et al. (1997)]. There are more than 20 reported genes associated with monogenic parkinsonism, including autosomal dominant SNCA encoding alpha-synuclein (αSyn) and LRRK2 encoding Leucine-rich repeat kinase 2, or autosomal recessive PRKN encoding the E3 ubiquitin ligase Parkin and PINK1 encoding Phosphatase and tensin homologue (PTEN)-induced kinase (Blauwendraat et al., 2020). However, familial PD with the identified genetic variants still accounts for only about 10% of diagnosed PD cases, while pathogenetic triggers in sporadic forms of PD are largely unknown (Reeve et al., 2014). Therefore, it is largely considered to be a complex disease with multifactorial etiology. In recent years, several genome-wide association studies (GWASs) have identified many tens of risk signals associated with sporadic PD surrounded by hundreds of potential susceptibility genes (Nalls et al., 2019). To date, there is little to no published functional validation of genes in these loci. In this study, we examined the role of approximately 100 of these genes in maintaining dopaminergic (DA) neurons upon exogenous expression of human αSyn in a newly constructed Caenorhabditis elegans genetic model designed to aid rapid initial functional screening.
The roundworm C. elegans, which does not possess the gene for αSyn, has been well-established as a PD model, which can help identify genes that protect against exogenous αSyn-induced degeneration of DA neurons or aggregation of αSyn in muscle cells (Cooper and Van Raamsdonk, 2018; Koopman et al., 2019). The C. elegans PD model, expressing αSyn tagged with green fluorescent protein (GFP) in the body wall muscle cells, was used to seek the effectors of αSyn misfolding through reverse genetic screens elicited by RNA interference (RNAi) (Hamamichi et al., 2008; van Ham et al., 2008; Jadiya et al., 2016). In addition, RNAi of 1,673 genes related to neuronal function revealed genes of the endocytic pathway in pan-neuronal αSyn-induced growth/motor abnormalities (Kuwahara et al., 2008). αSyn expressed in nematode DA neurons causes DA neurodegeneration characterized by neuronal loss or abnormal dendritic processing, and also dopamine-mediated locomotion deficits (Lakso et al., 2003; Kuwahara et al., 2006). In addition, in these PD worm models, the expression of human Torsin A and yeast Rab1, which play a role in vesicular trafficking, showed neuroprotective activity against α-synuclein-induced degeneration (Cao et al., 2005; Cooper et al., 2006). Several studies have also demonstrated that fluorescence-based reporters of αSyn expressed in neuronal tissues recapitulate DA neuronal deficits and show the spread of αSyn into the epithelium (Cooper et al., 2018; Sandhof et al., 2020). However, none of these previous nematode models have been used to identify modulators of αSyn aggregation in neurons. Here, we have generated a new nematode model, erals1, which allows monitoring of the expression of αSyn in DA neurons in vivo, and used these transgenic animals for functional screening of identified PD risk genes.
Unless otherwise stated, animals were maintained by standard procedures on nematode growth media (NGM) plates. Transgenic strains were generated by germline transformation using microinjection into Bristol strain N2. C. elegans constructs for the eraIs transgene were generated by direct PCR of the human SNCA gene cloned into the pDEST vector in front of, and in frame with, the Venus reporter gene with unc-54 3′UTR. The pDEST vectors carrying hSNCA:Venus or mCherry were subsequently recombined with a pENTRY vector carrying the dat-1 promoter sequence. Transgenic constructs were co-injected at 50 ng/μl, and stable extrachromosomal lines of mCherry- and Venus-positive animals were established. The extrachromosomal array was subsequently integrated by ultraviolet (UV) irradiation, and the lines carrying eraIs1 were subjected to 5× outcrossing. The strains used were as follows: eraIs1, otIs181, eraIs1;otIs181, eraIs1;pdr-1(gk448), eraIs1;pink-1(tm1779), eraIs1;wlzIs3, eraIs1;lfe-2(sy326), eraIs1;unc-43(n1186), eraIs1;nsf-1(ty10), eraIs1;sac-2(ok2743), eraIs1;unc-32(e189), eraIs1;rig-6(ok1589), and rmIs133 (unc-54p:Q40:YFP).
RNAi was fed to worms to knockdown the respective gene function. Gravid animals carrying eraIs1, otIs181, or rmsIs133 transgenes were placed on NGM media containing ampicillin 25 μg/ml and 1 mM IPTG and seeded with bacteria producing the desired double-stranded RNA (dsRNA). Progenies were subsequently grown at 23°C till the fourth larval stage (L4) stage in which the phenotype was scored via visual examination. eraIs1 animals were scored for the disruption of cephalic (CEP) integrity, which was defined by the presence of fluorescent inclusions in the area of the CEP axons (phenotype A) and CEP dendrite blebbing/loss (phenotype B). Both of these phenotypes were selected as being the most tractable by visual inspection due to the bright fluorescence of the αSyn reporter in CEP neurons, which allowed quantification of individual RNAi knockdowns in a relatively high-throughput way. Moreover, assessing animals at the L4 stage revealed modulators of phenotypes A and B. Each population of L4 knockdowns having 0–60% of individuals exhibiting phenotype A and 40–100% of individuals exhibiting phenotype B was classified as modulators. otIs181 and rmsIs133 animals were scored for the disruption of CEP integrity, defined by dendrite blebbing/loss and number of fluorescent foci, respectively. At least 20 eraIs1, 20 otIs181, or 3 rmsIs133 animals were visually examined for penetrance and fluorescent foci quantification, respectively. Visual examination was done using a fluorescent stereoscope (Nikon SMZ800N) by one researcher with coded plates to ensure blindness of the investigator. The results were recorded and subsequently decoded to reveal the names of RNAi targets. Bacterial clones were obtained from the C. elegans RNAi collection—Ahringer (Source: Bioscience).
Animals were grown at 23°C under non-starved conditions. One-day-old adult hermaphrodites were placed on NGM plates and recorded. For the crawling/swimming transition assay, animals were subjected to liquid exposure by dropping 30 μl of M9 buffer on the plate, which was dried within 7–8 min. For the mechanical stimuli assay, animals crawling on the NGM plate with seeded bacteria were stressed by five taps of the plate on the bench. For the foraging assay, well-fed animals were placed 1 cm away from the bacterial lawn. Animal movement was subsequently screened by quantifying body bends in the indicated time intervals of 30 s. During crawling, body bends were scored as head turns for moving forward. During swimming, body bends were scored as C-shaped movements. At least three biological replicates (five animals per assay) were used for statistical analysis. To compare the distribution of animals on and off the food area, 1-day old adults, which were starved for 1 h, were placed 1 cm away from the bacterial lawn on NGM plates, and their position on the plate was scored 30 min later. At least 50 animals for each group (N2 vs. eraIs1) with three independent biological replicates were used for statistical analysis.
Data are presented as mean ± standard deviation (SD) with p-values calculated by one-way analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons and the Mann–Whitney test for single comparison.
Worms were grown at 23°C as described above and collected as a mixed population of all larval and adult stages. Wet worm pellets were subsequently frozen at −20°C for 4 h to disrupt the nematode cuticle. Worm lysates were prepared by sonication of worm pellets resuspended in M9 buffer containing protease and phosphatase inhibitor cocktail. Crude extracts were immediately centrifuged for 1 h at 4°C and 20,000 g, and the supernatants (soluble fraction) and pellets (insoluble fraction) were used for the determination of αSyn:Venus levels in the indicated mutants by the Western blot analysis. Samples were boiled for 10 min in reducing lithium dodecyl sulfate (LDS) sample buffer and submitted to SDS-PAGE (4–12% precast gradient gel). Protein immunodetection was performed by the Western blot using a custom-made mouse monoclonal anti-αSyn antibody (Abnova, MAB5383 1:2,000). Ubiquitin, which was detected using a mouse monoclonal anti-ubiquitin Ab (CellSignal, P4D1 1:2,000), along with an unspecific signal, was used to demonstrate protein loading. Western blot signals were semi-quantified in ImageJ software (Fiji).
For confocal imaging, animals were mounted on a 2% agarose pad with 10 mM sodium azide and imaged on a Leica SP8-X confocal laser scanning microscope within 2–10 min. At least three images representing each C. elegans strain from three independent biological replicates were analyzed.
To study neuronal genes that mediate proteostasis upon the formation of αSyn inclusions, we have generated a new transgenic C. elegans carrying eraIs1(dat-1p:human SNCA:Venus; dat-1p:mCherry). The eraIs1 transgene uses the dat-1 promoter to drive the expression of a human hSNCA:Venus reporter along with an mCherry reporter specifically in DA neurons consisting of four CEP neurons, two anterior deirids (ADE) in the head, and two posterior deirids (PDE) in the tail. Bright GFP-based reporter Venus of human αSyn allowed us to monitor the spatial and temporal formation of its inclusions (fluorescent foci) in axons, dendrites, and cell bodies (Figure 1A). Notably, we observed that expressed mCherry, which is diffusely distributed in the cytosol under standard conditions throughout development and aging, forms inclusions in the presence of αSyn:Venus. Similar to the mCherry reporter, the expression of the untagged Venus reporter alone did not form fluorescent inclusions, indicating that the expression of exogenous human αSyn is crucial for the formation of inclusions (Figure 1A). Intriguingly, both αSyn:Venus and mCherry reporters in eraIs1 can also exhibit aggregated fluorescent signals away from intact neurons (Figure 1A). These isolated fluorescent foci of aggregated proteins presumably represent an extruded toxic material as an active neuronal self-maintaining mechanism against disrupted proteostasis rather than cell remnants of degenerated neurons (Melentijevic et al., 2017).
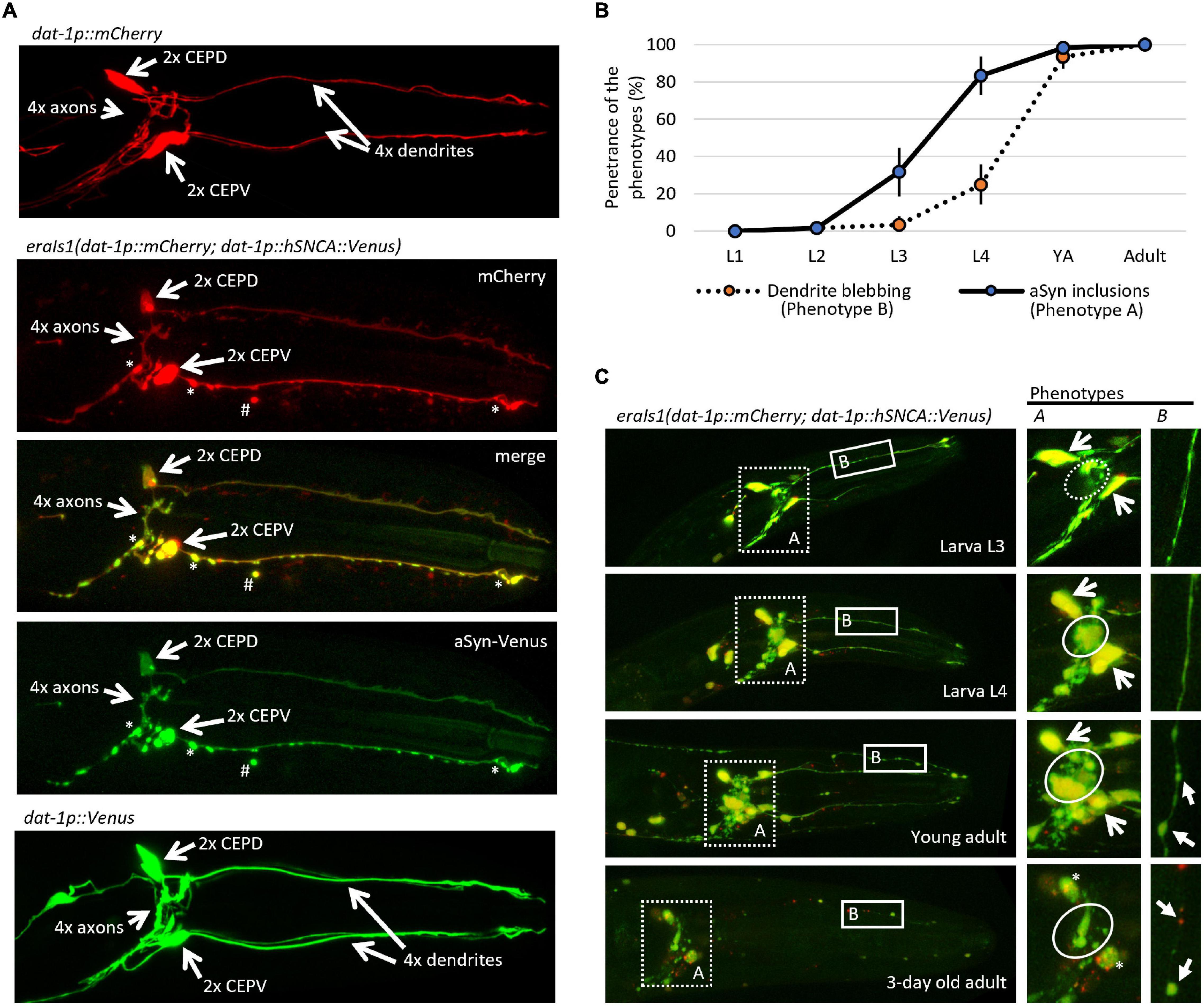
Figure 1. eraIs1 animals exhibit progressive formation of α-Synuclein (αSyn) inclusions and disruption of the dopaminergic (DA) neuron integrity during aging. (A) Representative fluorescent image of the head of third larval stage (L3) animals expressing the mCherry reporter alone, the mCherry reporter with the αSyn:Venus reporter, and the Venus reporter alone in DA neurons. Two head ventral CEPV and two dorsal CEPD neurons and their axons are indicated by open arrows. Stars indicate αSyn:Venus inclusions. Hashtag indicates extruded αSyn:Venus from DA neurons. (B) Distribution of phenotypes quantified at distinct developmental stages of eraIs1 animals. n ≥ 20 total animals for each group with three independent biological replicates. Mean ± standard deviation (SD) is shown. (C) Representative fluorescent images of the head of eraIs1 animals at different stages of development. Dotted square line highlights the area used for the quantification of phenotype A (αSyn:Venus inclusions) and the solid square line highlights the area used for the quantification of phenotype B [cephalic (CEP) dendrite blebbing]. On the right, there are enlarged images of the phenotypes A and B areas that show the phenotype identifiers for quantification. The circle line defines the area for the evaluation of phenotype A. The dashed circle indicates a normal phenotype, and the solid circle indicates phenotype A. Open arrows indicate CEP cell bodies. Three-day-old adult exhibits rounded CEP cell bodies indicated by the stars. Phenotype B is indicated by arrows.
During visual inspection of eraIs1 animals using a fluorescent stereoscope, we observed that eraIs1 nematodes exhibit the progressive formation of αSyn:Venus inclusions accompanied by morphological changes of CEP neurons during development, and ultimately the disruption of neuronal integrity in aged animals. The bright Venus fluorescence allowed us to determine which animals developed fluorescent inclusions in the axons of CEP neurons (phenotype A) and pathological morphology (blebbing) of CEP dendrites (phenotype B). We assessed the penetrance of these two phenotypes during animal development and determined that the penetrance of phenotype A increases from 32% at the third larval stage (L3) to 83% at L4, while the penetrance of phenotype B reaches 25% at L4 and 93% at the young adult stage. In 3-day-old adults, CEP neurons exhibited rounded cell bodies accompanied by dendritic disorganization, indicating the process of neurodegeneration associated with aging (Figures 1B,C; Chen et al., 2015).
Dopaminergic neurons regulate several animal behaviors, such as locomotory response to food availability (Sawin et al., 2000; Omura et al., 2012), foraging (Hills et al., 2004), or movement transitions between crawling and swimming (Vidal-Gadea et al., 2011). To determine whether eraIs1 animals exhibit respective behavioral phenotypes due to DA neuronal defects, we exposed 1-day-old adult eraIs1 and wild-type isolate N2 to various stimuli and compared their locomotor behavior by counting the body bends used for crawling or swimming. First, we exposed well-fed animals, crawling on the bacterial lawn on NGM plates, to M9 buffer. While N2 animals exposed to M9 liquid buffer responded immediately by swimming at the rate of 48 C-shaped body bends per 30 s, eraIs1 animals failed to respond, with an average rate of 3 C-shaped body bends per 30 s (Figure 2A). In addition, while crawling locomotion of well-fed eraIs1 animals was not altered before M9 buffer exposure, it was noticeably reduced after swimming compared to N2 animals (Figure 2A). The defective response of eraIs1 to M9 buffer is not caused by a locomotion defect as both N2and eraIs1 exhibited a swimming phenotype at the rate of 41 and 46 body bends per 30 s after 5 min in M9 buffer, respectively (Figure 2A). Moreover, mechanical stimuli elicited by plate tapping did not reveal significant behavioral changes between N2 and eraIs1 animals, indicating that the locomotion of eraIs1 animals is not impaired (Figure 2B). Next, we tested whether eraIs1 animals would exhibit foraging defects. We placed well-fed animals on an NGM plate outside the bacterial lawn and found that eraIs1 animals exhibit a reduced locomotor response to lack of food (Figure 2C). Moreover, when we used starved animals and examined the plates after 30 min, we observed that a greater number of eraIs1 animals remained outside, but in proximity to, the bacterial lawn, indicating a possible food sensing deficit (Figure 2D). Specifically, we observed that only 74% of eraIs1 animals were located inside the bacterial lawn compared to 95% of N2 animals (Figure 2E). Taken together, these behavioral data demonstrate that eraIs1 animals exhibit behavioral characteristics similar to that induced by dopamine deficiency.
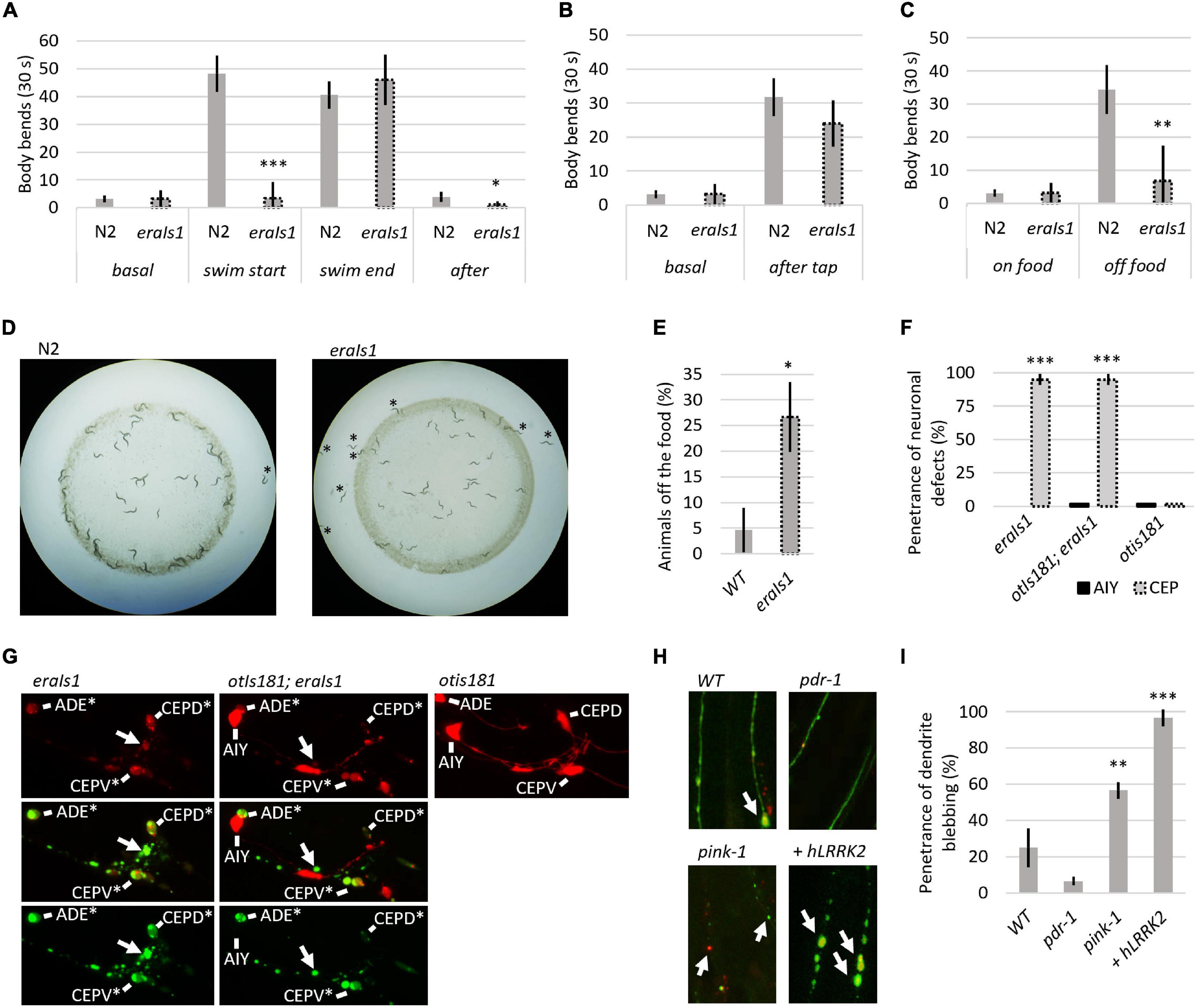
Figure 2. Evaluation of the eraIs1 model. (A) Locomotor phenotype of the indicated animals placed on a nematode growth media (NGM) plate seeded with OP 50 bacteria (basal), right after exposure to M9 buffer (swim start), 5 min after exposure to M9 (swim end), and right after swimming (after). n ≥ 10 young adults for each group with three independent biological replicates. During crawling, body bends were scored as head turns for moving forward. During swimming, body bends were scored as C-shaped movements. Mean ± SD is shown. *p < 0.05. ***p < 0.001. (B) Locomotory phenotype of the indicated animals localized in the bacterial lawn (basal) and right after being disturbed by tapping (after tap). n ≥ 10 young adults for each group with three independent biological replicates. Mean ± SD is shown. (C) Locomotory phenotype of indicated animals localized in bacterial lawn (on-food) and right after being placed off the bacteria (off-food). n ≥ 10 young adults for each group with three independent biological replicates. Mean ± SD is shown. **p < 0.01. (D) Distribution of the indicated animals on the NGM plate seeded with bacteria. Animals starved for 1 h are captured 30 min after being placed 1 cm away from the bacterial lawn. (E) Percentage of the indicated animal populations found outside the bacterial lawn. n ≥ 50 young adults for each group with three independent biological replicates. Mean ± SD is shown. *p < 0.05. (F) Percentage of young adult eraIs1, otIs181;eraIs1, and otIs181 animals exhibiting αSyn:Venus and mCherry inclusions in CEP and AIY neurons. n ≥ 20 animals for each group with three independent biological replicates. Mean ± SD is shown. ***p < 0.001. (G) Enlarged fluorescent images of the head of young adult eraIs1, eraIs1;otIs181, and otIs181 animals. Arrows indicate αSyn:Venus inclusions. CEP and AIY cell bodies are indicated. Stars indicate neuronal cell bodies with the αSyn:Venus and mCherry inclusions. (H) Representative fluorescent images of CEP dendrites in the indicated eraIs1 animals. eraIs1 wild type (WT), eraIs1;wlzIs3 carrying human LRRK2 p.G2019S, and pdr-1(gk448) and pink-1(tm1779) mutants are shown. Arrows indicate CEP dendrite blebbing. (I) The penetrance of dendrite blebbing in the indicated eraIs1 animals. n ≥ 20 fourth larval stage (L4) animals for each group with three independent biological replicates. Mean ± SD is shown. **p < 0.01. ***p < 0.001.
We verified the effect of αSyn on neuronal integrity with an independent neuronal mCherry reporter. We used the otIs181 transgene that uses dat-1:mCherry together with ttx-3:mCherry, which drives its expression in DA neurons and amphid interneurons (AIY), respectively, and crossed it with eraIs1. As expected, eraIs1 induced neuronal defects of the DA neurons but not the AIY neurons in young adults, suggesting cell-autonomous neurotoxicity of the expressed αSyn (Figures 2F,G). To further validate the PD model eraIs1, we tested whether known neurodegenerative triggers, such as loss-of-function of Parkin and PINK1 and gain of function of human LRRK2, which are associated with familial forms of PD, would induce neuronal defects upon αSyn expression (Yao et al., 2010; Blauwendraat et al., 2020). We used loss-of-function alleles of C. elegans prd-1/Parkin, pink-1/PINK1, and the transgene wlzIs3 expressing human LRRK2 p.G2019S (Saha et al., 2009) and crossed them with eraIs1. We assessed the toxicity of αSyn in these mutants at the L4 developmental stage, which allowed us to identify both suppressors of phenotype A and enhancers of phenotype B. In addition, the size of L4 animals reduces experimental bias resulting from impaired traceability of respective phenotypes in younger larval stages and phenotypic variability in adults. We found that dendrite blebbing was exacerbated in animals carrying pink-1 and wlzIs3 but not in pdr-1 mutants (Figures 2H,I). These data demonstrate that the phenotype modulated in eraIs1;pink-1 and eraIs1;wlzIs3 animals could be used as a readout to identify new modulators of αSyn-induced neurodegeneration.
We searched several current GWAS databases, such as iPDGC (Grenn et al., 2020), GWAS catalog (Buniello et al., 2019), and PDgene (Lill et al., 2012; Nalls et al., 2014) for genes associated with sporadic PD. Collectively, we identified 98 risk signals at different levels of statistical significance from 89 independent genomic loci. We selected 131 PD susceptibility genes based on their proximity and position relative to the PD risk signals and analyzed them in silico for evolutionary conservation in the C. elegans genome based on phylogenetic and structural data information. For 97 of the human-associated potential risk genes, we identified at least one C. elegans ortholog, collectively consisting of 127 different worm genes (Supplementary Table 1). We set out to perform a preliminary reverse genetic screen of these identified C. elegans orthologous genes using an RNA interference by “feeding approach” (genes for which an RNAi clone was not available were not included in the screen), or using previously isolated loss-of-function alleles, for modulated αSyn neurotoxicity in the eraIs1 strain. Overall, we have visually examined 98 knockdown/mutant animal populations and recorded the penetrance of phenotypes A (αSyn:Venus inclusions in CEP axons) and B (CEP dendrite blebbing/loss) at the L4 developmental stage (Figure 1C). The preliminary RNAi data obtained clustered the examined genes into two basic groups: (i) 10 genes whose inactivation reduced the penetrance of phenotype A (Figure 3A and Table 1) and (ii) 18 genes whose inactivation increased the penetrance of phenotype B (Figure 3B and Table 1).
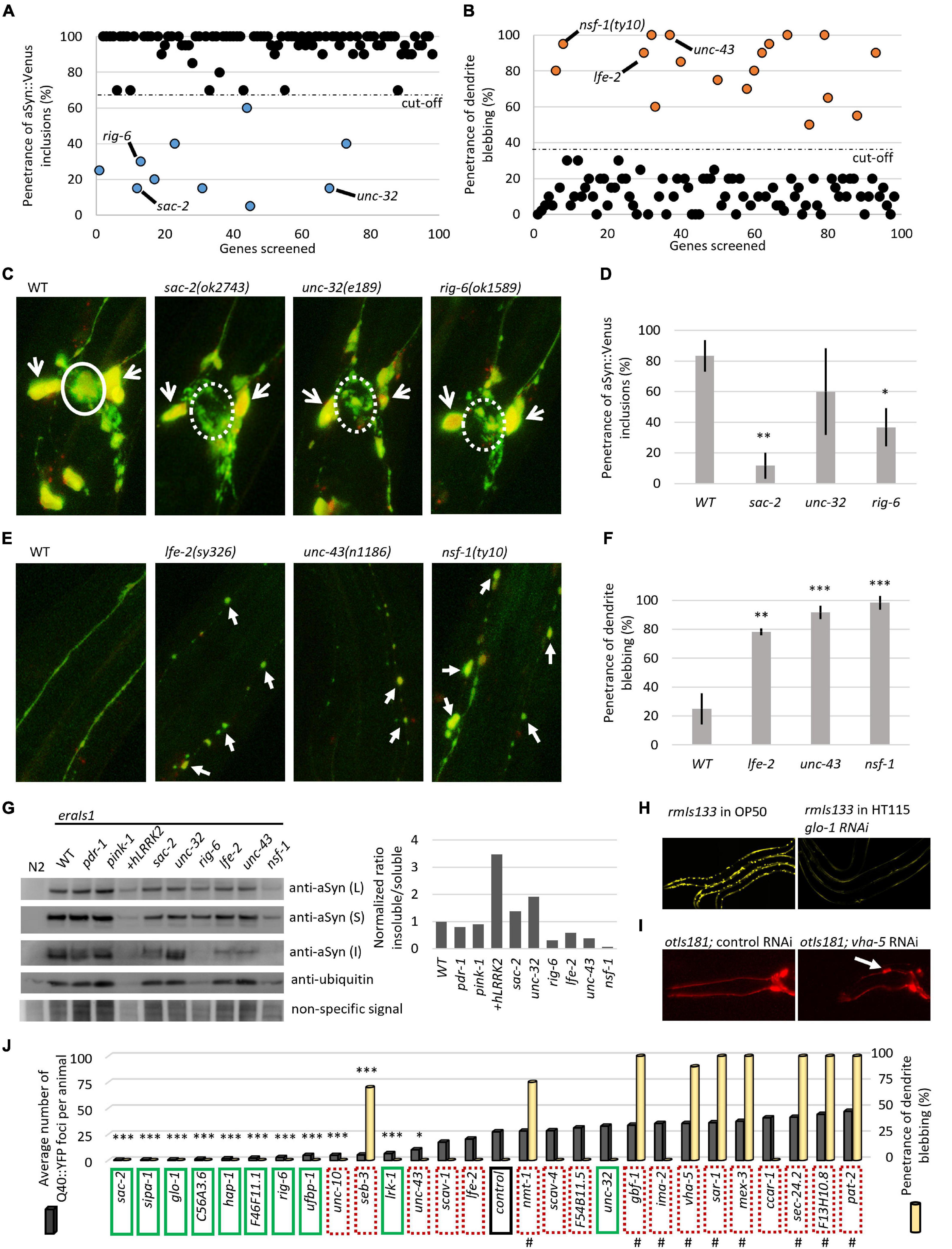
Figure 3. Reverse genetic screening for altered αSyn neurotoxicity in eraIs1 animals. (A) Distribution of the quantified phenotype of αSyn inclusions (phenotype A) in 98 different knockdowns. (B) Distribution of quantified phenotype B (CEP dendrite blebbing) in 98 different knockdowns. (C) Representative fluorescent images of CEP cell bodies and their axons of eraIs1 mutants at L4 stage. The solid circle line indicates phenotype A while the dotted circle line indicates a normal phenotype. Open arrows indicate CEP cell bodies. (D) Penetrance of phenotype A (αSyn:Venus inclusions) in the indicated mutants. n ≥ 20 L4 animals for each group with three independent biological replicates. Mean ± SD is shown. *p < 0.05. **p < 0.01. (E) Representative fluorescent images of CEP dendrites of eraIs1 mutants at the L4 stage. Arrows indicate CEP dendrite blebbing. (F) Penetrance of phenotype B (dendrite blebbing) in the indicated mutants. n ≥ 20 L4 animals for each group with three independent biological replicates. Mean ± SD is shown. **p < 0.01. ***p < 0.001. (G) αSyn:Venus protein levels determined by SDS-PAGE followed by western blot using anti-αSyn Ab, and anti-ubiquitin Ab and non-specific signal as a loading control, and the calculated ratio of insoluble/soluble αSyn levels normalized to WT are presented. Animals of mixed stages were collected from three biological replicates and 10 μl of either lysed Caenorhabditis elegans pellets of the same density (L) or soluble (S) and insoluble (I) fraction were loaded per lane. eraIs1 animals carry the following alleles: WT, pdr-1(gk448) and pink-1(tm1779), wlzIs3(snb-1p:hLRRK2 p.G2019S), lfe-2(sy326), unc-43(n1186), nsf-1(ty10), sac-2(ok2743), unc-32(e189), and rig-6(ok1589). (H) Representative fluorescent images of rmIs133 animals at the L4 stage expressing Q40:YFP in body wall muscles upon standard (feeding OP50 bacteria) and glo-1 RNA interference (RNAi). (I) Representative fluorescent images of the head of young adult otIs181 animals upon control and vha-5 RNAi. (J) Quantification of Q40:YFP fluorescent foci in rmIs133 (n ≥ 3 L4 animals) and penetrance of CEP dendrite blebbing in otIs181 animals (n ≥ 20 L4 animals) among RNAi knockdowns. Green boxes indicate identified modulators of phenotype A (αSyn:Venus inclusions in eraIs1), dotted red boxes indicate identified modulators of phenotype B (dendrite blebbing in eraIs1). Hashtags indicate knockdowns that fail to propagate (lethal or sterile phenotype). Stars indicate genes whose inactivation reduced Q40:YFP foci. *p < 0.05. ***p < 0.001.
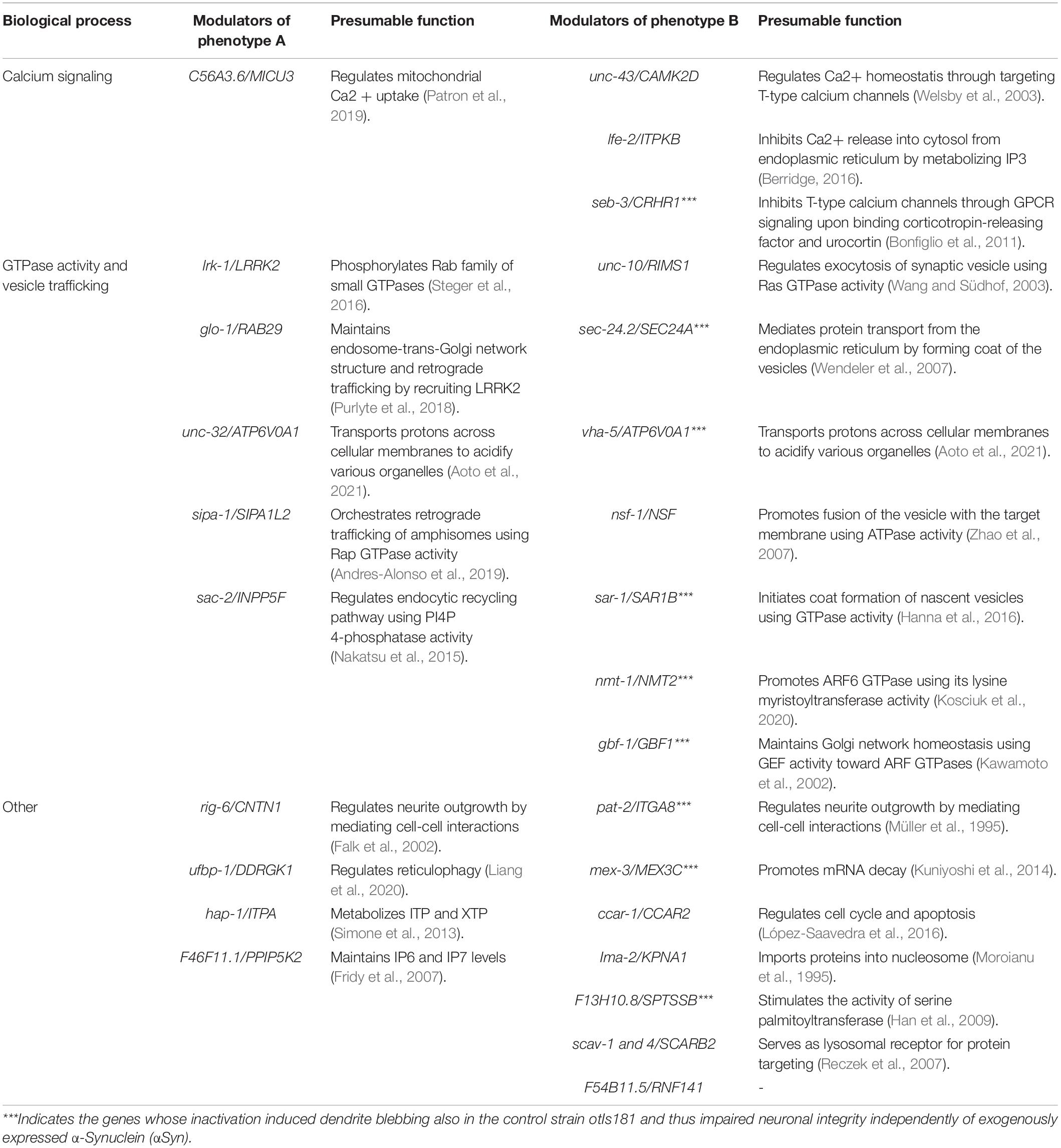
Table 1. Identified modulators.
We validated each RNAi phenotype with six previously characterized loss-of-function alleles, such as sac-2, rig-6, and unc-32, whose inactivation reduced the penetrance of phenotype A, and unc-43, lfe-2, and nsf-1, whose inactivation increased the penetrance of phenotype B. We confirmed reduced penetrance of αSyn:Venus inclusions (phenotype A) formed in sac-2 and rig-6 mutants even though confocal imaging of these mutants revealed that the formation of inclusions was not completely suppressed. However, the size of the formed inclusions cannot be recognized by visual inspection using a fluorescent stereoscope compared to the fluorescent inclusions accumulated in wild-type (WT) animals, which explains the observed reduction in penetrance in these mutants (Figures 3C,D). Next, we confirmed increased penetrance of dendrite blebbing (phenotype B) in lfe-2 and unc-43 that phenocopy nsf-1 mutants, and confocal imaging revealed the altered integrity of their CEP dendrites (Figures 3E,F). These data show that the preliminary results obtained from reverse genetic screening have been validated for five different genes confirming their modulatory role in αSyn neurotoxicity in C. elegans.
We investigated whether the altered penetrance of phenotypes A and B in the tested mutants was associated with altered levels, solubility, or processing of expressed αSyn:Venus. We used Western blot analysis to detect the levels of αSyn:Venus in crude, soluble and insoluble C. elegans extracts and the amount of αSyn:Venus its soluble and insoluble fractions. We used a detergent-free lysis buffer in which the insoluble αSyn should be composed of both aggregated/fibrillar form and monomeric αSyn bound to the cellular/organelle membrane. First, we did not detect any significant alterations of normalized αSyn levels in crude extracts of the tested mutants, indicating that modulated phenotypes are not mediated through altered expression of αSyn:Venus but rather to altered capability of neurons to cope with the stress induced by αSyn. However, we found various levels of insoluble αSyn among the tested mutants. We normalized the ratio of insoluble/soluble αSyn to WT and detected higher levels of insoluble αSyn in unc-32 mutants and in strain co-expressing human LRKK2 p.G2019S with increased penetrance of phenotype B. On the other hand, levels of insoluble αSyn were markedly decreased in rig-6 mutants, in which penetrance of phenotype A was reduced, and in nsf-1 mutants, in which the penetrance of phenotype B was increased (Figure 3G). These data suggest that both modulated phenotypes in eraIs1 mutants might have various pathogenetic basis that did not have to be necessarily associated with aggregation-related nor membrane-bound-related αSyn toxicity.
To better investigate the role of all identified modulators in cellular proteostasis, we next assessed the RNAi of the 28 identified genes for their capability to modulate the aggregation of the pro-aggregating poly-Glutamine (Q40):YFP protein expressed in muscle cells. We used rmIs133(unc-54p:Q40:YFP) animals, in which Q40:YFP progressively forms fluorescent foci during development (van Ham et al., 2008) and are easily tractable by visual inspection under a fluorescent stereoscope. We counted the fluorescent foci at the L4 stage among the tested RNAi conditions and found that nine modulators of phenotype A, such as RNAi of sac-2, sipa-1, glo-1, C56A3.6, hap-1, F46F11.1, rig-6, ufbp-1, and lrk-1, significantly reduced the number of formed Q40:YFP foci (Figures 3H,J). Interestingly, the RNAi against unc-10, seb-3, and unc-43, which were classified as modulators of phenotype B, also reduced the number of Q40:YFP foci (Figure 3J). These data suggest that 12 identified mediators of Q40:YFP aggregation may play a role in the maintenance of proteotoxicity, including neurotoxicity induced by the expression of αSyn in eraIs1 animals.
To evaluate the possibility that the identified modifiers could modulate neuronal integrity independent of αSyn expression, we retested all identified modulators in animals carrying otIs181(dat-1p:mCherry;ttx-3p:mCherry) (Flames and Hobert, 2009) expressing an mCherry reporter in DA neurons alone. We found that RNAi against nine genes that were identified as modulators of phenotype B, including seb-3, sec-24.2, vha-5, sar-1, nmt-1, gbf-1, pat-2, mex-3, and F13H10.8, induced CEP dendrite blebbing in otIs181 animals, indicating that inactivation of these genes disrupted neuronal integrity independent of neurotoxic αSyn expression (Figures 3I,J, and Table 1). Notably, RNAi of some of these genes caused sterility, larval arrest, or developmental delay of animals, indicating a widespread effect of gene inactivation on worm health. The complementary screens with eraIs1 and otIs181 animals identified 19 genes, whose inactivation modulated αSyn-mediated toxicity, and other nine genes, whose inactivation disrupted the integrity of DA neurons independent of αSyn-mediated toxicity.
Several studies over the years have revealed tens of genes that can modulate αSyn aggregation and associated neurodegeneration in various animal and cell model systems. However, a model that allows monitoring of the αSyn processing in neurons in vivo in a high-throughput way under various conditions is, until now, somewhat missing. Here, we generated a C. elegans model, in which αSyn inclusions and accompanying neuronal morphological processes can be monitored under a fluorescent stereoscope in vivo. We showed that the formation of αSyn inclusions was predominantly detected in the axons of CEP neurons at the L4 larval stage, which allowed the screening of inactivated genes that reduced the level of inclusion formation. We also showed that the expression of αSyn in eraIs1 induced the dendrite blebbing of CEP neurons in adults but was rare in larval stages, which allowed the screening of the genes, whose inactivation induced dendrite blebbing in the larval stages. Therefore, this study provides the first systematic functional screening of the genes identified in the PD GWAS data and reveals new genetic pathways that could mediate PD pathogenesis.
First, we identified 18 genes, whose inactivation exacerbated αSyn:Venus-induced neuronal defect characterized by CEP dendrite blebbing. Notably, RNAi of nine of these modulators induced dendrite blebbing independent of αSyn:Venus expression in these neurons and/or caused impaired propagation of animals, which indicates their crucial role in neuronal cell integrity and C. elegans biology. Therefore, we hypothesize that these genes, when misregulated, might induce neuronal defects in other organisms independent of the pathogenicity of αSyn. These data also raise speculation whether some PD GWAS genes could play a role in PD progression in an αSyn-independent manner. Second, we identified 10 genes whose inactivation by RNAi reduced the formation of αSyn:Venus inclusions in CEP axons.
RNAi screening identified a total of 19 genes, which, when inactivated, modulated αSyn-related toxicity; however, our data did not reveal the nature of such toxicity, which could be mediated by aggregation or non-aggregation. The Western blot analysis indicated that the observed neurodegenerative phenotype in eraIs1 animals does not have to be necessarily related to αSyn aggregation/fibrillation or membrane-bound αSyn as we detected contradicted insoluble αSyn levels among modifiers of phenotype A and among modifiers of phenotype B. In addition, RNAi of 12 identified modulators also reduced the formation of Q40:YFP inclusions in body wall muscle cells, of which three of these caused the dendrite blebbing in eraIs1 animals. Thus, we cannot exclude the possibility that some genetic conditions that modulated phenotypes of eraIs1 animals might have opposite consequences in other model systems. Notably, an increasing number of studies have shown that the formation of αSyn inclusions may be a beneficial mechanism for αSyn neurotoxicity (Wong and Krainc, 2017). We should also take into account that RNAi conditions utilizing the vector L4440 in HT115 bacteria might exert phenotype effects independent of the desired RNAi target and thus reduced YFP foci in some animals exposed to RNAi conditions could be an effect of transgene silencing (De-Souza et al., 2019); some RNAi knockdowns exhibited a decreased fluorescent signal of the soluble Q40:YFP reporter compared to animals exposed to standard OP50 bacteria (Figure 3I). Further investigations of these observed phenotypes and studying the aggregated vs. non-aggregated-related αSyn toxicity in the eraIs1 model in follow-up studies are needed. Lastly, it should be noted that other tested PD risk genes, which did not modulate eraIs1 phenotypes in this study, may play a role in αSyn neurotoxicity under different experimental designs or in other model systems or in humans.
Nonetheless, the physiological roles associated with the identified genes also reveal the biological processes that may constitute pathogenetic pathways in sporadic forms of PD. We found that human orthologs of the two identified modulators—unc-43/CAMK2D and lfe-2/ITPKs—regulate calcium release from the endoplasmic reticulum into the mitochondria while C56A3.6/MICU3 regulates mitochondrial calcium uptake (Table 1). These data are consistent with a recent observation revealing mammalian ITPKB as a protective gene against PD-like phenotypes triggered by mitochondrial calcium uptake (Apicco et al., 2021). We thus conclude that intracellular calcium signaling modulates αSyn neurotoxicity in C. elegans. In addition, we identified several specific GTPases and their regulators associated with vesicular trafficking, including synaptic, endosomal, ER/Golgi, and autophagosome/lysosome networks, which altered neuronal αSyn processing. The role of vesicular trafficking in PD is well-documented, and its impairment is one of the leading mechanisms of PD pathogenesis (Hunn et al., 2015). Several genes, such as sac-2/INPP5F and nsf-1/NSF have been shown to modulate PD-like phenotypes in mouse and fruit fly PD models, respectively (Babcock et al., 2015; Cao et al., 2020). Also, another identified modulator lrk-1/LRRK2 and its counterpart glo-1/RAB29 have been previously associated together in PD pathogenesis, but the role of RAB29 in αSyn pathology has not been validated (Kalogeropulou et al., 2020). These findings demonstrate that hypothesis-free identification of candidate PD genes through GWAS can be tracked functionally in a relatively high-throughput way with the eraIs1 model, to reveal new roles of evolutionary conserved genes in neuronal maintenance upon proteotoxic stress.
Together, reverse genetic screening using a new C. elegans PD model system identified 19 functionally interesting PD-risk genes involved in αSyn:Venus toxicity in nematode DA neurons and other nine genes involved in DA neuron maintenance in the αSyn-free system. The obtained data provide a strong foundation for follow-up studies aimed at further characterizing the role of these genes in PD, which represent new potential therapeutic targets for synucleinopathies and other neurodegenerative proteopathies associated with aging.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
RV designed, performed, and analyzed the experiments and wrote the manuscript. AH designed and analyzed the experiments. All authors acquired funding and reviewed the manuscript.
This project was funded by the European Union’s Horizon 2020 Research and Innovation Programme under Grant No. 844497. This research was partly funded by the Department of Educational Assistance, University and Research of the Autonomous Province of Bolzano, Italy, through the Institute for Biomedicine core funding initiative. The authors thank the Department of Innovation, Research University and Museums of the Autonomous Province of Bozen/Bolzano for covering the Open Access publication costs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Caenorhabditis Genetics Center for C. elegans strains and Dengke Ma lab for plasmids.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.806000/full#supplementary-material
Supplementary Table 1 | List of human Parkinson’s disease (PD) risk genes with respective Caenorhabditis elegans orthologs and identified RNA interference (RNAi) phenotype.
Andres-Alonso, M., Ammar, M. R., Butnaru, I., Gomes, G. M., Acuña Sanhueza, G., Raman, R., et al. (2019). SIPA1L2 controls trafficking and local signaling of TrkB-containing amphisomes at presynaptic terminals. Nat. Commun. 10:5448. doi: 10.1038/s41467-019-13224-z
Aoto, K., Kato, M., Akita, T., Nakashima, M., Mutoh, H., Akasaka, N., et al. (2021). ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H+-ATPases is essential for brain development in humans and mice. Nat. Commun. 12:2107. doi: 10.1038/s41467-021-22389-5
Apicco, D. J., Shlevkov, E., Nezich, C. L., Tran, D. T., Guilmette, E., Nicholatos, J. W., et al. (2021). The Parkinson’s disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release. Proc. Natl. Acad. Sci. U.S.A. 118:e2006476118. doi: 10.1073/pnas.2006476118
Babcock, D. T., Shen, W., and Ganetzky, B. (2015). A neuroprotective function of NSF1 sustains autophagy and lysosomal trafficking in Drosophila. Genetics 199, 511–522. doi: 10.1534/genetics.114.172403
Berridge, M. J. (2016). The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296. doi: 10.1152/physrev.00006.2016
Blauwendraat, C., Nalls, M. A., and Singleton, A. B. (2020). The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178.
Bonfiglio, J. J., Inda, C., Refojo, D., Holsboer, F., Arzt, E., and Silberstein, S. (2011). The corticotropin-releasing hormone network and the hypothalamic-pituitary-adrenal axis: molecular and cellular mechanisms involved. Neuroendocrinology 94, 12–20. doi: 10.1159/000328226
Buniello, A., MacArthur, J. A. L., Cerezo, M., Harris, L. W., Hayhurst, J., Malangone, C., et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012. doi: 10.1093/nar/gky1120
Cao, M., Park, D., Wu, Y., and Camilli, P. D. (2020). Absence of Sac2/INPP5F enhances the phenotype of a Parkinson’s disease mutation of synaptojanin 1. Proc. Natl. Acad. Sci. U.S.A. 117, 12428–12434. doi: 10.1073/pnas.2004335117
Cao, S., Gelwix, C. C., Caldwell, K. A., and Caldwell, G. A. (2005). Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J. Neurosci. 25, 3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005
Chen, X., Barclay, J. W., Burgoyne, R. D., and Morgan, A. (2015). Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Cent. J. 9:65. doi: 10.1186/s13065-015-0143-y
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). α-Synuclein blocks ER-Golgi traffic and rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328. doi: 10.1126/science.1129462
Cooper, J. F., and Van Raamsdonk, J. M. (2018). Modeling Parkinson’s disease in C. elegans. J. Parkinsons Dis. 8, 17–32.
Cooper, J. F., Spielbauer, K. K., Senchuk, M. M., Nadarajan, S., Colaiácovo, M. P., and Van Raamsdonk, J. M. (2018). α-synuclein expression from a single copy transgene increases sensitivity to stress and accelerates neuronal loss in genetic models of Parkinson’s disease. Exp. Neurol. 310, 58–69. doi: 10.1016/j.expneurol.2018.09.001
De-Souza, E. A., Camara, H., Salgueiro, W. G., Moro, R. P., Knittel, T. L., Tonon, G., et al. (2019). RNA interference may result in unexpected phenotypes in Caenorhabditis elegans. Nucleic Acids Res. 47, 3957–3969. doi: 10.1093/nar/gkz154
Falk, J., Bonnon, C., Girault, J.-A., and Faivre-Sarrailh, C. (2002). F3/contactin, a neuronal cell adhesion molecule implicated in axogenesis and myelination. Biol. Cell 94, 327–334. doi: 10.1016/s0248-4900(02)00006-0
Flames, N., and Hobert, O. (2009). Gene regulatory logic of dopamine neuron differentiation. Nature 458, 885–889. doi: 10.1038/nature07929
Fridy, P. C., Otto, J. C., Dollins, D. E., and York, J. D. (2007). Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J. Biol. Chem. 282, 30754–30762. doi: 10.1074/jbc.M704656200
Grenn, F. P., Kim, J. J., Makarious, M. B., Iwaki, H., Illarionova, A., Brolin, K., et al. (2020). The Parkinson’s disease genome-wide association study locus browser. Mov. Disord. Off. J. Mov. Disord. Soc. 35, 2056–2067. doi: 10.1002/mds.28197
Hamamichi, S., Rivas, R. N., Knight, A. L., Cao, S., Caldwell, K. A., and Caldwell, G. A. (2008). Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. U.S.A. 105, 728–733. doi: 10.1073/pnas.0711018105
Han, G., Gupta, S. D., Gable, K., Niranjanakumari, S., Moitra, P., Eichler, F., et al. (2009). Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc. Natl. Acad. Sci. U.S.A. 106, 8186–8191. doi: 10.1073/pnas.0811269106
Hanna, M. G., Mela, I., Wang, L., Henderson, R. M., Chapman, E. R., Edwardson, J. M., et al. (2016). Sar1 GTPase activity is regulated by membrane curvature. J. Biol. Chem. 291, 1014–1027. doi: 10.1074/jbc.M115.672287
Hills, T., Brockie, P. J., and Maricq, A. V. (2004). Dopamine and glutamate control area-restricted search behavior in Caenorhabditis elegans. J. Neurosci. 24, 1217–1225. doi: 10.1523/JNEUROSCI.1569-03.2004
Hunn, B. H. M., Cragg, S. J., Bolam, J. P., Spillantini, M.-G., and Wade-Martins, R. (2015). Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 38, 178–188. doi: 10.1016/j.tins.2014.12.009
Jadiya, P., Fatima, S., Baghel, T., Mir, S. S., and Nazir, A. (2016). A systematic RNAi Screen of neuroprotective genes identifies novel modulators of alpha-synuclein-associated effects in transgenic Caenorhabditis elegans. Mol. Neurobiol. 53, 6288–6300. doi: 10.1007/s12035-015-9517-3
Kalogeropulou, A. F., Freemantle, J. B., Lis, P., Vides, E. G., Polinski, N. K., and Alessi, D. R. (2020). Endogenous Rab29 does not impact basal or stimulated LRRK2 pathway activity. Biochem. J. 477, 4397–4423. doi: 10.1042/BCJ20200458
Kawamoto, K., Yoshida, Y., Tamaki, H., Torii, S., Shinotsuka, C., Yamashina, S., et al. (2002). GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic CPH Den. 3, 483–495. doi: 10.1034/j.1600-0854.2002.30705.x
Koopman, M., Seinstra, R. I., and Nollen, E. A. A. (2019). “C. elegans as a model for synucleinopathies and other neurodegenerative diseases: tools and techniques,” in Alpha-Synuclein: Methods and Protocols [Internet]. (Methods in Molecular Biology), ed. T. Bartels (New York, NY: Springer), 93–112. doi: 10.1007/978-1-4939-9124-2_9
Kosciuk, T., Price, I. R., Zhang, X., Zhu, C., Johnson, K. N., Zhang, S., et al. (2020). NMT1 and NMT2 are lysine myristoyltransferases regulating the ARF6 GTPase cycle. Nat. Commun. 11:1067. doi: 10.1038/s41467-020-14893-x
Kuniyoshi, K., Takeuchi, O., Pandey, S., Satoh, T., Iwasaki, H., Akira, S., et al. (2014). Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I–mediated antiviral innate immunity. Proc. Natl. Acad. Sci. U.S.A. 111, 5646–5651. doi: 10.1073/pnas.1401674111
Kuwahara, T., Koyama, A., Gengyo-Ando, K., Masuda, M., Kowa, H., Tsunoda, M., et al. (2006). Familial Parkinson mutant α-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans*. J. Biol. Chem. 281, 334–340. doi: 10.1074/jbc.M504860200
Kuwahara, T., Koyama, A., Koyama, S., Yoshina, S., Ren, C.-H., Kato, T., et al. (2008). A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in α-synuclein transgenic C. elegans. Hum. Mol. Genet. 17, 2997–3009. doi: 10.1093/hmg/ddn198
Lakso, M., Vartiainen, S., Moilanen, A.-M., Sirviö, J., Thomas, J. H., Nass, R., et al. (2003). Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human α-synuclein. J. Neurochem. 86, 165–172. doi: 10.1046/j.1471-4159.2003.01809.x
Liang, J. R., Lingeman, E., Luong, T., Ahmed, S., Muhar, M., Nguyen, T., et al. (2020). A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 180, 1160–1177.e20. doi: 10.1016/j.cell.2020.02.017
Lill, C. M., Roehr, J. T., McQueen, M. B., Kavvoura, F. K., Bagade, S., and Schjeide, B.-M. M., et al. (2012). Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet. 8:e1002548. doi: 10.1371/journal.pgen.1002548
López-Saavedra, A., Gómez-Cabello, D., Domínguez-Sánchez, M. S., Mejías-Navarro, F., Fernández-Ávila, M. J., Dinant, C., et al. (2016). A genome-wide screening uncovers the role of CCAR2 as an antagonist of DNA end resection. Nat. Commun. 7:12364. doi: 10.1038/ncomms12364
Melentijevic, I., Toth, M. L., Arnold, M. L., Guasp, R. J., Harinath, G., Nguyen, K. C., et al. (2017). C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 542, 367–371. doi: 10.1038/nature21362
Moroianu, J., Hijikata, M., Blobel, G., and Radu, A. (1995). Mammalian karyopherin alpha 1 beta and alpha 2 beta heterodimers: alpha 1 or alpha 2 subunit binds nuclear localization signal and beta subunit interacts with peptide repeat-containing nucleoporins. Proc. Natl. Acad. Sci. U.S.A. 92, 6532–6536. doi: 10.1073/pnas.92.14.6532
Müller, U., Bossy, B., Venstrom, K., and Reichardt, L. F. (1995). Integrin alpha 8 beta 1 promotes attachment, cell spreading, and neurite outgrowth on fibronectin. Mol. Biol. Cell 6, 433–448. doi: 10.1091/mbc.6.4.433
Nakatsu, F., Messa, M., Nández, R., Czapla, H., Zou, Y., Strittmatter, S. M., et al. (2015). Sac2/INPP5F is an inositol 4-phosphatase that functions in the endocytic pathway. J. Cell Biol. 209, 85–95. doi: 10.1083/jcb.201409064
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/S1474-4422(19)30320-5
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Omura, D. T., Clark, D. A., Samuel, A. D. T., and Horvitz, H. R. (2012). Dopamine signaling is essential for precise rates of locomotion by C. elegans. PLoS One 7:e38649. doi: 10.1371/journal.pone.0038649
Patron, M., Granatiero, V., Espino, J., Rizzuto, R., and De Stefani, D. (2019). MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 26, 179–195. doi: 10.1038/s41418-018-0113-8
Purlyte, E., Dhekne, H. S., Sarhan, A. R., Gomez, R., Lis, P., Wightman, M., et al. (2018). Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 37, 1–18. doi: 10.15252/embj.201798099
Reczek, D., Schwake, M., Schröder, J., Hughes, H., Blanz, J., Jin, X., et al. (2007). LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131, 770–783. doi: 10.1016/j.cell.2007.10.018
Reeve, A., Simcox, E., and Turnbull, D. (2014). Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res. Rev. 14, 19–30. doi: 10.1016/j.arr.2014.01.004
Saha, S., Guillily, M. D., Ferree, A., Lanceta, J., Chan, D., Ghosh, J., et al. (2009). LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci. 29, 9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009
Sandhof, C. A., Hoppe, S. O., Druffel-Augustin, S., Gallrein, C., Kirstein, J., Voisine, C., et al. (2020). Reducing INS-IGF1 signaling protects against non-cell autonomous vesicle rupture caused by SNCA spreading. Autophagy 16, 878–899. doi: 10.1080/15548627.2019.1643657
Sawin, E. R., Ranganathan, R., and Horvitz, H. R. (2000). C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619–631. doi: 10.1016/s0896-6273(00)81199-x
Simone, P. D., Pavlov, Y. I., and Borgstahl, G. E. O. (2013). ITPA (inosine triphosphate pyrophosphatase): from surveillance of nucleotide pools to human disease and pharmacogenetics. Mutat. Res. 753, 131–146. doi: 10.1016/j.mrrev.2013.08.001
Spillantini, M. G., Schmidt, M. L., Lee, V. M.-Y., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-Synuclein in Lewy bodies. Nature 388, 839–840.
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 5:e12813. doi: 10.7554/eLife.12813
van Ham, T. J., Thijssen, K. L., Breitling, R., Hofstra, R. M. W., Plasterk, R. H. A., and Nollen, E. A. A. (2008). C. elegans model identifies genetic modifiers of α-Synuclein inclusion formation during aging. PLoS Genet. 4:e1000027. doi: 10.1371/journal.pgen.1000027
Vidal-Gadea, A., Topper, S., Young, L., Crisp, A., Kressin, L., Elbel, E., et al. (2011). Caenorhabditis elegans selects distinct crawling and swimming gaits via dopamine and serotonin. Proc. Natl. Acad. Sci. U.S.A. 108, 17504–17509. doi: 10.1073/pnas.1108673108
Wang, Y., and Südhof, T. C. (2003). Genomic definition of RIM proteins: evolutionary amplification of a family of synaptic regulatory proteins. Genomics 81, 126–137. doi: 10.1016/s0888-7543(02)00024-1
Welsby, P. J., Wang, H., Wolfe, J. T., Colbran, R. J., Johnson, M. L., and Barrett, P. Q. (2003). A mechanism for the direct regulation of T-type calcium channels by Ca2+/Calmodulin-dependent kinase II. J. Neurosci. 23, 10116–10121. doi: 10.1523/JNEUROSCI.23-31-10116.2003
Wendeler, M. W., Paccaud, J.-P., and Hauri, H.-P. (2007). Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 8, 258–264. doi: 10.1038/sj.embor.7400893
Wong, Y. C., and Krainc, D. (2017). α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13. doi: 10.1038/nm.4269
Yao, C., El Khoury, R., Wang, W., Byrd, T. A., Pehek, E. A., Thacker, C., et al. (2010). LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 40, 73–81. doi: 10.1016/j.nbd.2010.04.002
Keywords: Parkinson’s disease, α-synuclein, GWAS, neurodegeneration, genetic screen, C. elegans
Citation: Vozdek R, Pramstaller PP and Hicks AA (2022) Functional Screening of Parkinson’s Disease Susceptibility Genes to Identify Novel Modulators of α-Synuclein Neurotoxicity in Caenorhabditis elegans. Front. Aging Neurosci. 14:806000. doi: 10.3389/fnagi.2022.806000
Received: 31 October 2021; Accepted: 18 March 2022;
Published: 27 April 2022.
Edited by:
Natalia Salvadores, Universidad Mayor, ChileReviewed by:
Leonidas Stefanis, National and Kapodistrian University of Athens Medical School, GreeceCopyright © 2022 Vozdek, Pramstaller and Hicks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roman Vozdek, cm9tYW4udm96ZGVrQGV1cmFjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.