 Jarosław Dulski
Jarosław Dulski Ryan J. Uitti1
Ryan J. Uitti1 Owen A. Ross
Owen A. Ross Zbigniew K. Wszolek
Zbigniew K. Wszolek- 1Department of Neurology, Mayo Clinic, Jacksonville, FL, United States
- 2Division of Neurological and Psychiatric Nursing, Faculty of Health Sciences, Medical University of Gdańsk, Gdańsk, Poland
- 3Department of Neurology, St. Adalbert Hospital, Copernicus PL Ltd., Gdańsk, Poland
- 4Department of Neuroscience, Mayo Clinic, Jacksonville, FL, United States
The heterogeneity of Parkinson’s disease (PD) has been recognized since its description by James Parkinson over 200 years ago. The complexity of motor and non-motor PD manifestations has led to many attempts of PD subtyping with different prognostic outcomes; however, the pathophysiological foundations of PD heterogeneity remain elusive. Genetic contributions to PD may be informative in understanding the underpinnings of PD subtypes. As such, recognizing genotype-phenotype associations may be crucial for successful gene therapy. We review the state of knowledge on the genetic architecture underlying PD subtypes, discussing the monogenic forms, as well as oligo- and polygenic risk factors associated with various PD subtypes. Based on our review, we argue for the unification of PD subtyping classifications, the dichotomy of studies on genetic factors and genetic modifiers of PD, and replication of results from previous studies.
Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder after Alzheimer’s disease. The prevalence in the general population is low (0.3%); however, it increases rapidly with age and is estimated at 1 and 3% after 60 and 80 years, respectively (Balestrino and Schapira, 2020). Furthermore, among neurological disorders, PD displayed the most prominent growth in prevalence and disability in the last three decades (Ou et al., 2021). Most of the growing trend in PD incidence has been attributed to the aging of the population; however, other factors, including lifestyle choices, environmental pollution, occupational exposure, and genetic composition, and evolving treatments, are considered to play a role (Ou et al., 2021).
Traditionally, PD is characterized by four cardinal motor symptoms: bradykinesia, tremor, rigidity, and postural instability. However, patients differ in the severity of these symptoms, and distinct combinations of symptoms may predominate in the clinical picture. The heterogeneity of clinical presentations led to many attempts of PD subtyping. The research on PD subtypes evolved from studies on arbitrarily pre-defined groups into data-driven clustering of PD patients. The former (older) approach most often divides patients into tremor-dominant (TD)/indeterminate/postural instability and gait difficulty (PIGD) (Zetusky et al., 1985; Jankovic et al., 1990), TD/mixed/akinetic-rigid (AR) (Schiess et al., 2000; Korchounov et al., 2004; Kang et al., 2005) or TD/AR/gait difficulty (GD)/mixed (Konno et al., 2018) subtypes. In contrast, the latter (novel) approach is more advantageous in the research setting, as it allows hypothesis-free analysis of multiple domains; but it is rarely translated into an algorithm applicable in clinical practice (Fereshtehnejad et al., 2017; Lawton et al., 2018; Zhang et al., 2019; Mestre et al., 2021). To further confound the picture, non-motor symptoms have been recognized, including behavioral and mood disturbances, cognitive impairment, sensory symptoms, autonomic dysfunction, and sleep and wakefulness disorders (Dulski et al., 2015, 2018, 2019, 2022, forthcoming; Balestrino and Schapira, 2020; Siuda, 2021; Tipton, 2021). Table 1 presents the various PD subtypes investigated in the previous studies.
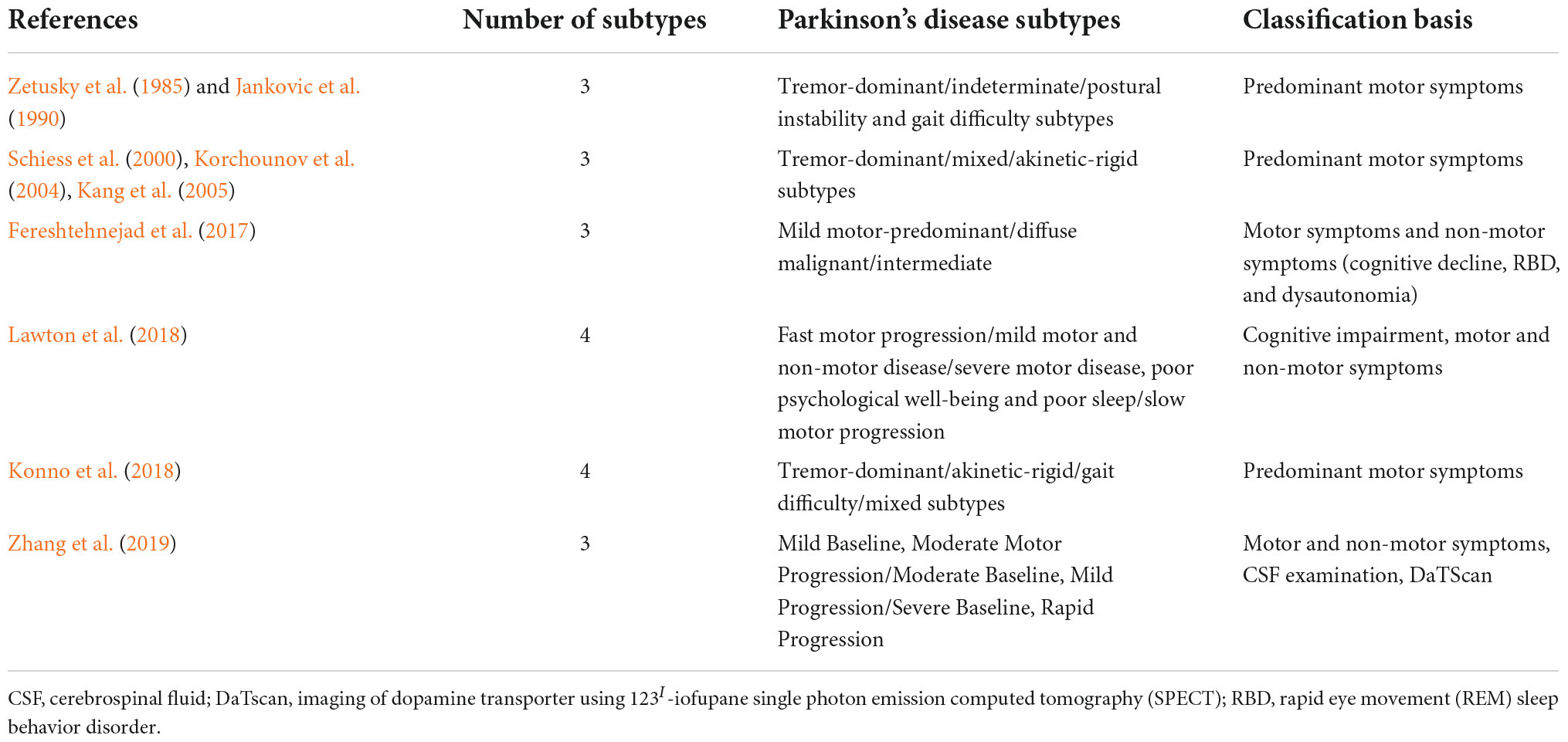
Table 1. Parkinson’s disease subtypes.
Having a first-degree relative with PD increases the risk by two-three fold; however, only 15% of patients have a positive family history of PD, and less than 10% are diagnosed with monogenic forms (Balestrino and Schapira, 2020; Hall et al., 2020). Furthermore, as of June 2022, over 100 genetic loci were associated with PD and other forms of parkinsonism; genetic contributions have been legion in the last decade (Dulski et al., forthcoming). Genetic studies are increasingly sophisticated and less costly, leading to more widespread application in research and clinical settings. In particular, with genome-wide association studies (GWAS), new variants or combinations of them (oligo- and polygenic inheritance) are being discovered.
In this manuscript, we review the state of knowledge on the genetic architecture underlying PD heterogeneity and subtypes. We summarize the phenotypes associated with mutations according to classification from our study on PD (TD/AR/GD/mixed subtypes) that we find most helpful in clinical settings (Konno et al., 2018). Finally, we provide implications of various genetic profiles for clinical management and suggest future research directions.
Monogenic forms of Parkinson’s disease
Monogenic forms of PD may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner, and account for approximately 30% of familial PD forms (Milanowski et al., 2021). Of note, single-gene forms of PD are not always familial, as a patient may have a recessive variant, incomplete penetrance, or de novo mutation; they are likely responsible for 3–5% of sporadic PD forms (Milanowski et al., 2021). Monogenic PD may present at different ages as juvenile-onset PD (JOPD) (below 20 years), early-onset PD (EOPD) (between 20 and 50 years), and late-onset PD (LOPD) (after 50 years) (Dulski et al., forthcoming). Table 2 presents characteristics of the monogenic forms of PD and their practical implications.
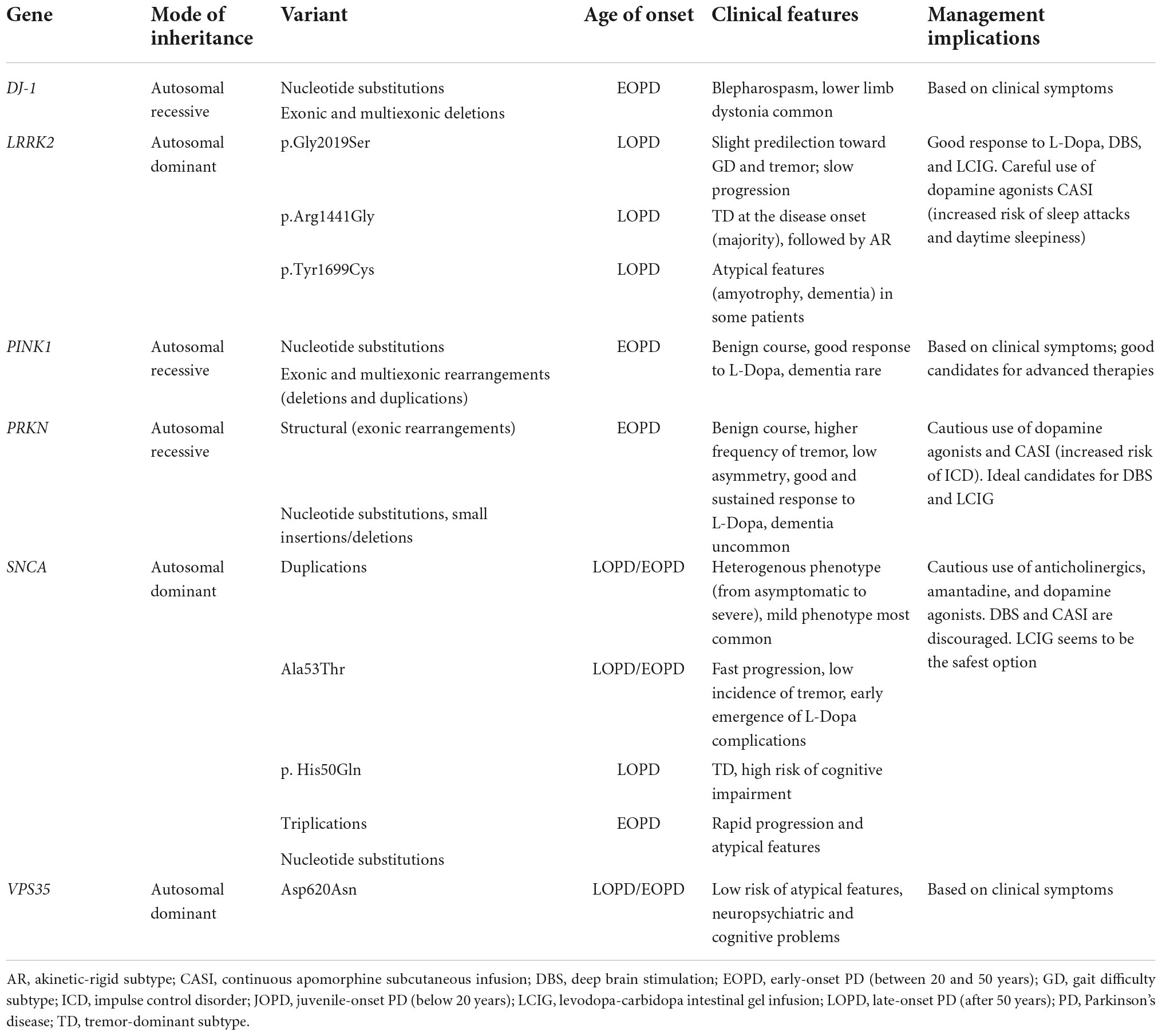
Table 2. Characteristics of the monogenic forms of Parkinson’s disease (PD) and their practical implications.
Monogenic forms of Parkinson’s disease predominantly presenting with tremor-dominant subtype
LRRK2
Mutations in the Leucine-Rich Repeat Kinase 2 (LRRK2) gene are the most common cause of genetic PD and are responsible for 1% of sporadic and 5% of familial cases worldwide (Guadagnolo et al., 2021; Dulski et al., forthcoming). They are inherited in an autosomal dominant fashion. Most cases are of Caucasian descent (63%), followed by Hispanics (8%), and Asians (11%) (Trinh et al., 2018). The LRRK2 gene encodes a mainly cytoplasmic protein involved in diverse biological activities, including neuronal vesicular trafficking, mitochondrial functions, and autophagy (Madureira et al., 2020; Guadagnolo et al., 2021; Lee et al., 2022). The phenotype resembles a classic LOPD, and at the clinical and neuropathological level, it cannot be distinguished from idiopathic PD (Trinh et al., 2018; Guadagnolo et al., 2021; Dulski et al., forthcoming). Numerous variants in the LRRK2 have been reported; however, only some were associated with PD. At least seven variants (p.Arg1441Gly/Cys/His, p.Asn1437His, p.Gly2019Ser, p.Ile2020Thr, and p.Tyr1699Cys) are considered pathogenic, and a few others (p.Arg1628Pro, p.Gly2385Arg, and p.Ser1761Arg) are reported risk factors for PD (Guadagnolo et al., 2021; Simpson et al., 2022; Dulski et al., forthcoming). The most prevalent variant is p.Gly2019Ser, which alone may account for up 10–40% of the familial PD cases in the North African, Ashkenazi Jewish, and Mediterranean populations (Mirelman et al., 2013; Marras et al., 2016; Trinh et al., 2018; Guadagnolo et al., 2021). The p.Gly2019Ser variant and other studied LRRK2 mutations display incomplete penetrance, increasing with age and impacted by other genetic variants and ethnical ancestry, with Northern Europeans developing symptoms later than Northern Africans (Mirelman et al., 2013; Hentati et al., 2014; Marras et al., 2016; Trinh et al., 2018; Guadagnolo et al., 2021). The p.Gly2019Ser variant has a heterogeneous presentation, but there seems to be slight predilection toward the postural instability, tremor, and generally a milder progression of motor and non-motor manifestations (including cognitive deterioration) (Mirelman et al., 2013; Marras et al., 2016; Trinh et al., 2018; Guadagnolo et al., 2021). The p.Arg1441Gly variant is manifested at the disease onset as a TD PD subtype in most patients, followed by an AR subtype (a quarter of patients), progression is slow, and dementia risk is low (Vinagre-Aragón et al., 2021). The p.Gly2385Arg variant predisposes to the PIGD subtype in the first few years of PD, faster motor progression, and cognitive decline (Marras et al., 2016). The p.Tyr1699Cys variant has various presentations, with atypical features (amyotrophy, dementia) in some patients (Kim et al., 2012). The LRRK2 mutation carriers tend to respond well to dopaminergic therapy. In the advanced stages of the disease, they are good candidates for deep brain stimulation (DBS), and levodopa-carbidopa intestinal gel infusion (LCIG) (Trinh et al., 2018; Guadagnolo et al., 2021; Salles et al., 2021; Lee et al., 2022). However, the dopamine agonists and continuous apomorphine subcutaneous infusion (CASI) should be considered with caution, as LRRK2 mutation carriers are at higher risk of sleep attacks and daytime sleepiness (Salles et al., 2021). And while LRRK2 neuropathology typically reflects “usual” PD pathology, with prominent synuclein inclusions and nigral cell loss, there may be significant variability in neuropathological findings leading some patients to be reclassified atypical PD despite having usual clinical presentations (Uitti et al., 2004). In summary, the LRRK2 mutations are most often associated with TD and GD subtypes (Kim et al., 2012; Mirelman et al., 2013; Marras et al., 2016; Trinh et al., 2018; Madureira et al., 2020; Guadagnolo et al., 2021; Vinagre-Aragón et al., 2021; Lee et al., 2022; Simpson et al., 2022; Dulski et al., forthcoming).
Monogenic forms of Parkinson’s disease predominantly presenting with akinetic-rigid subtype
PRKN
Mutations in the PRKN (Parkin) are the most common cause of EOPD, responsible for up to 15% of all PD cases with onset below 50 years old (Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). They are inherited in an autosomal recessive manner. Most patients were Asians (39%) and Caucasians (31%), followed by Hispanics (10%) (Kasten et al., 2018). The PRKN gene encodes a protein essential for mitochondrial homeostasis and mitophagy. Numerous pathogenic variants were reported, the most common being structural variants (exonic rearrangements), followed by missense and frameshift mutations (Dulski et al., forthcoming). The median age of onset is 31 years of age (Kasten et al., 2018). The phenotype resembles classic PD; however, it tends to have a more benign disease course, better responsiveness to levodopa, high frequency of bradykinesia, rigidity, tremor, and less asymmetry (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). Given that dementia, dysautonomia, and atypical features are uncommon and patients frequently develop fluctuations and dyskinesia, PRKN-PD patients are ideal candidates for DBS and infusion therapies (Kasten et al., 2018; Guadagnolo et al., 2021; Salles et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). Of note, at least one study demonstrated that PRKN-PD patients might be more prone to impulse control disorder (ICD); therefore, caution should be used when administering dopamine agonists and CASI (Morgante et al., 2016). Approximately 2% of the population may carry one pathogenic variant in PRKN, and they seem to be at the same risk for developing PD as the non-carriers (Zhu et al., 2022; Dulski et al., forthcoming). In summary, carriers of PRKN mutations tend to manifest with an AR subtype.
PINK1
Pathogenic variants in the PINK1 account for up to 5% of EOPD worldwide (Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). They are inherited in an autosomal recessive fashion. Most patients were Caucasians (33%), followed by mixed ethnicity (31%), and Asians (18%) (Kasten et al., 2018). The gene encodes a protein involved in mitochondrial functions. Numerous variants have been reported, with missense mutation responsible for nearly half of the cases (Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). The median age of onset is 32 years (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). The phenotype is similar to that of PRKN, but with a higher frequency of dystonia, dysautonomia, and a lower frequency of tremor (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). Cognitive impairment may be present but generally is rare (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022). The patients respond well to dopaminergic therapy and are good candidates for advanced treatment options (Kasten et al., 2018; Guadagnolo et al., 2021; Salles et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). The studies on the carriers of one pathogenic variant did not show increased susceptibility for PD (Krohn et al., 2020; Dulski et al., forthcoming). In summary, PINK1 mutations are most often linked with an AR subtype.
DJ-1
The mutations in the DJ1 gene are explain up to 1% of EOPD cases (Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). They are inherited in an autosomal recessive manner. Most patients were of mixed ethnicity (38%), followed by Asiana (25%), Hispanics (13%), Ashkenazi Jews (13%), and Caucasians (13%) (Kasten et al., 2018). Like PRKN and PINK1, DJ-1 encodes a protein crucial for the proper functioning of the mitochondria (Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). At least 27 pathogenic variants were reported, with missense mutations being the most common (Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). The median age of onset is 27 years (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). The phenotype is similar to that of PRKN and PINK1, with more frequent psychiatric symptoms (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). Blepharospasm and lower limb dystonia are also common (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). Most patients respond well to dopaminergic therapy; however, the data on this subject are limited (Kasten et al., 2018; Guadagnolo et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). Therefore, every patient should be considered individually, with clinical symptoms determining the choice of oral and advanced therapies (Kasten et al., 2018; Guadagnolo et al., 2021; Salles et al., 2021; Jia et al., 2022; Dulski et al., forthcoming). In summary, DJ-1 mutations are most often linked with an AR subtype.
Monogenic forms of Parkinson’s disease predominantly presenting with mixed subtype
SNCA
Mutations in the SNCA (alpha-synuclein) gene are the second most prevalent cause of autosomal dominant PD and are responsible for 1–2% of autosomal dominant familial PD cases worldwide (Lesage et al., 2020). Most patients were Caucasians (70%), followed by Asians (16%) and Hispanics (14%) (Trinh et al., 2018). Pathogenic variants disrupt the lysosomal degradation of alpha-synuclein and lead to its aggregation (Guadagnolo et al., 2021). The most common are whole gene duplications, followed by a few missense mutations (most often p.Ala53Thr, but also p.Ala30Pro, p.Glu46Lys, p.Glu51Asp, p. Ala53Glu, and p. His50Gln) and triplications (Trinh et al., 2018; Lesage et al., 2020; Guadagnolo et al., 2021; Dulski et al., forthcoming). The SNCA duplications have a heterogeneous phenotype, including asymptomatic status at the one end of the spectrum, benign phenotype with slow progression in the middle (most common), and rapidly progressive parkinsonism on the opposite end of the spectrum (rare) (Guadagnolo et al., 2021). The p.Ala53Thr variant manifests with classic PD, albeit the TD PD subtype is infrequent and is associated with faster disease progression and early emergence of levodopa complications (Guadagnolo et al., 2021). The p. His50Gln often manifests as a TD PD subtype, and cognitive impairment is common (Appel-Cresswell et al., 2013; Guadagnolo et al., 2021). The other mutations are very rare and there is insufficient data to draw genotype-phenotype conclusions. The SNCA triplications present with EOPD with a rapid progression and atypical features. Generally, SNCA-related PD is associated with a high rate of psychotic symptoms and depression, early onset of cognitive decline, and autonomic dysfunction (Guadagnolo et al., 2021). Therefore, anticholinergics, amantadine, and dopamine agonists should be limited in these patients. In addition, these patients are at high risk of adverse effects related to DBS and CASI, and LCIG seems to be the safest option (Salles et al., 2021). In summary, SNCA mutations are most often associated with a mixed subtype (Trinh et al., 2018; Lesage et al., 2020; Guadagnolo et al., 2021; Salles et al., 2021; Dulski et al., forthcoming).
Monogenic forms of Parkinson’s disease without clear predilection toward a specific subtype
Autosomal dominant forms of monogenic Parkinson’s disease
Until now, mutations in several other genes have been in autosomal dominant familial PD. This gene list includes, but is not limited to, VPS35, EIF4G1, TMEM230, CHCHD2, LRP10, RIC3, ARSA, and PSAP (Guadagnolo et al., 2021; Dulski et al., forthcoming).
Pathogenic variants in the VPS35 gene are responsible for less than 1% of familial PD cases worldwide (Trinh et al., 2018; Guadagnolo et al., 2021; Dulski et al., forthcoming). They are inherited in an autosomal dominant manner. Most patients were Caucasians (45%), followed by Asians (35%) and Ashkenazi Jews (20%) (Trinh et al., 2018). The VPS35 gene encodes the protein involved in the endosome-trans-Golgi network trafficking (Trinh et al., 2018; Guadagnolo et al., 2021; Dulski et al., forthcoming). At least 11 variants were reported, but until now, pathogenicity was confirmed only for the p.Asp620Asn variant (Trinh et al., 2018; Guadagnolo et al., 2021; Dulski et al., forthcoming). The phenotype resembles classic PD with a mean age of onset at 50; however, a milder course, with a low risk of atypical features, neuropsychiatric and cognitive problems, was noted (Trinh et al., 2018; Guadagnolo et al., 2021; Dulski et al., forthcoming). The reports on the prevalence of tremor, postural instability, dysautonomia, and daytime sleepiness provided inconsistent results and seemed similar to sporadic PD (Trinh et al., 2018; Liu and Le, 2020; Guadagnolo et al., 2021; Salles et al., 2021; Dulski et al., forthcoming). Most studies reported a good response to levodopa. Therefore, the patient’s management should be tailored to clinical findings. Based on previous studies, no conclusion can be drawn regarding the most often associated phenotype (Trinh et al., 2018; Liu and Le, 2020; Guadagnolo et al., 2021; Salles et al., 2021; Dulski et al., forthcoming).
Mutations in other genes were reported only in a few families with PD, and the causative role of some of them in PD is debated. In particular, recent studies suggest that EIF4G1 may not play a role in PD (Saini et al., 2021). Pathogenic variants in these genes may present as LOPD (EIF4G1, TMEM230, CHCHD2, LRP10, RIC3, ARSA, PSAP), EOPD (CHCHD2, RIC3, ARSA, and PSAP), and JOPD (ARSA). LRP-10-associated phenotype is often accompanied by dementia, which may be the first presentation (Guadagnolo et al., 2021; Dulski et al., forthcoming). Patients with PD due to the ARSA mutations often present with a tremor in their youth and develop TD PD in their 5th–6th decade (Dulski et al., forthcoming). Mutations in these genes may manifest as classic PD with a good and long-lasting response to dopaminergic therapy. Due to the scarcity of information on the genotype-phenotype associations, the therapy should be tailored based on clinical symptomatology. The advanced treatment options should also be considered (Guadagnolo et al., 2021; Saini et al., 2021; Dulski et al., forthcoming).
Autosomal recessive forms of monogenic Parkinson’s disease
At least seven other known genetic loci are associated with autosomal recessive parkinsonism, including ATP13A2 (Kufor-Rakeb syndrome), PLA2G6 (neurodegeneration with brain iron accumulation 2A), FBXO7, DNAJC6, SYNJ1, VPS13C, and WASL (Kumar et al., 2021; Dulski et al., forthcoming). Mutations in these genes present as JOPD (ATP13A2, PLA2G6, FBXO7, DNAJC6, and SYNJ1) or EOPD (VPS13C and WASL) (Kumar et al., 2021; Dulski et al., forthcoming). The WASL-associated phenotype is that of slowly progressive PD with a good response to levodopa (Kumar et al., 2021; Dulski et al., forthcoming). However, the pathogenic variants in the other genes usually lead to PD accompanied by atypical features, the onset of dementia early in the disease course, fast progression, and variable response to levodopa (Kumar et al., 2021; Wittke et al., 2021; Dulski et al., forthcoming). Hence, except for WASL-related PD, the treatment should mainly consist of oral pharmacological therapy, preferentially with levodopa. In WASL-associated PD, DBS and other advanced therapies may be considered.
X-linked forms of Parkinson’s disease
Mutations in at least eight genetic loci (TAF1, FMR1, RAB39B, WDR45, GLA, MeCP2, PGK1, and ATP6AP) on the X chromosome were associated with the development of PD (Di Lazzaro et al., 2021; Dulski et al., forthcoming). Contrary to the autosomal monogenic forms, pathogenic variants on the X chromosome usually manifest at a young age (JOPD or EOPD), with parkinsonism accompanied by many (atypical) features including intellectual disability or early cognitive decline, psychiatric features, spasticity, seizures (Di Lazzaro et al., 2021; Dulski et al., forthcoming), myoclonus, dystonia, and usually display a poor response to dopaminergic treatment. Most of the affected individuals are males; however, rarely, female carriers may present with a mild phenotype of pure PD with a good response to levodopa and no atypical features (Di Lazzaro et al., 2021; Dulski et al., forthcoming). Based on previous studies, no conclusion regarding the most often associated PD subtypes can be drawn.
Genetic risk factors associated with Parkinson’s disease symptomatology
Table 3 summarizes the genetic factors associated with PD symptomatology.
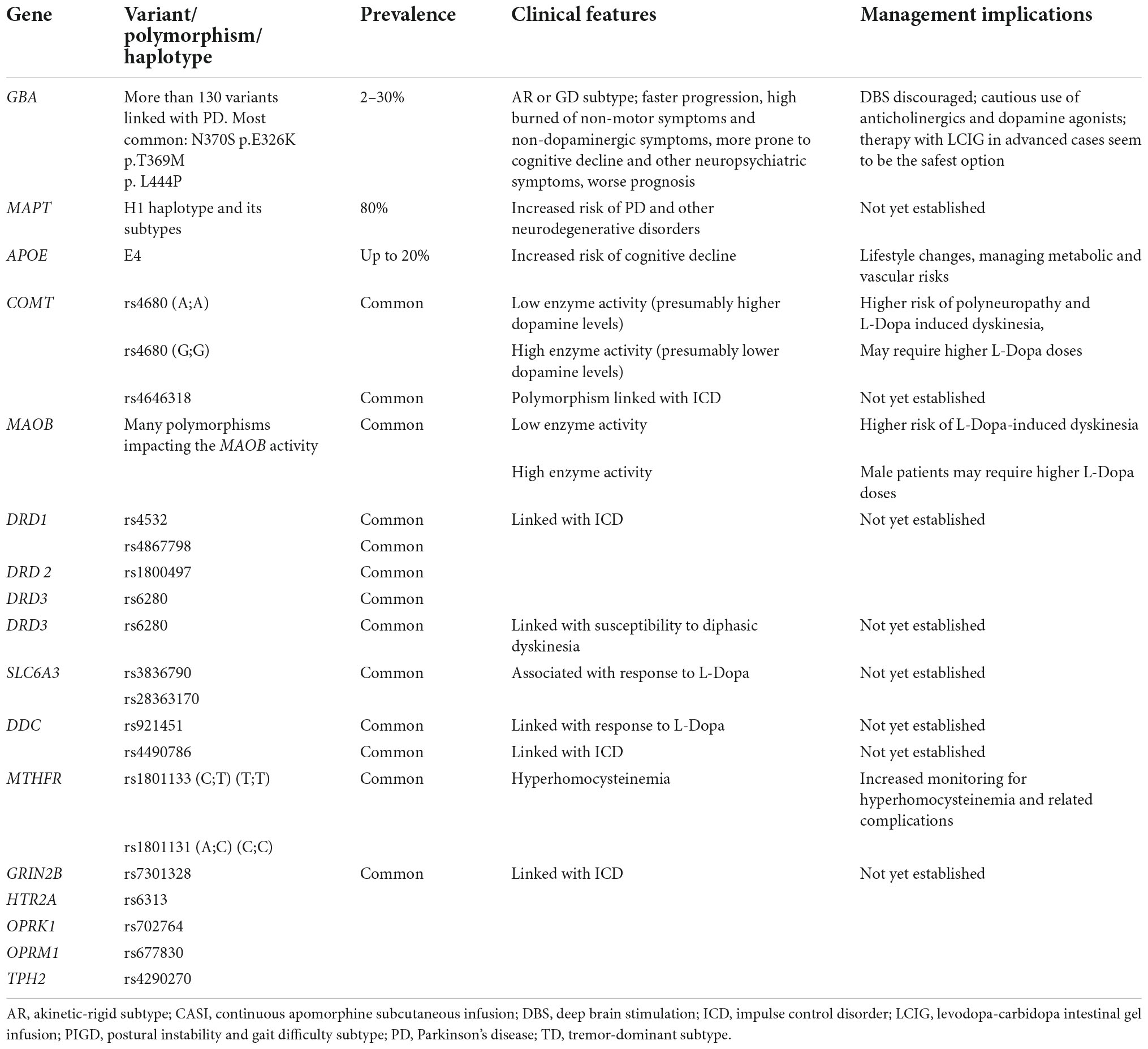
Table 3. The genetic risk factors associated with Parkinson’s disease (PD) symptomatology.
Genetic risk factors for akinetic-rigid/gait difficulty subtypes
GBA
Biallelic pathogenic variants in the GBA gene were classically linked with Gaucher’s disease, an autosomal recessive multisystem disorder with varied neurological manifestations. In recent years, heterozygous variants in GBA were found to be the most common genetic risk factors for PD, with an estimated prevalence of 2–30% worldwide (Jesús et al., 2016; Iwaki et al., 2019b; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Dulski et al., forthcoming). The penetrance is low, and over the lifespan is estimated at 9.1%; however, it increases with age up to 30% at 80 years (Jesús et al., 2016; Iwaki et al., 2019b; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Dulski et al., forthcoming). As penetrance is higher in carriers with a positive family history of PD than without such history, other genetic factors may be at play (Jesús et al., 2016; Iwaki et al., 2019b; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Dulski et al., forthcoming).
More than 130 genetic variants associated with PD have been described (Jesús et al., 2016; Iwaki et al., 2019b; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Dulski et al., forthcoming). Generally, heterozygote status increases the risk for PD by five times (Jesús et al., 2016; Iwaki et al., 2019b; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Dulski et al., forthcoming). In addition, the GBA mutations worsen the prognosis of PD (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). Carriers of GBA mutations develop PD a few years earlier, more often they present with the AR or PIGD subtype, tend to progress faster, and have a higher burden of non-non-motor symptoms (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). In particular, cognitive decline and other neuropsychiatric symptoms are common and occur earlier than in non-carriers (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). Motor non-dopaminergic symptoms, like dysphagia, dysarthria and freezing of gait were also reported to be more common (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). However, individual variants harbor the different risks of developing PD and have a diverse impact on the disease course (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). The four missense variants (p.N370S, p.E326K, p.T369M, and p. L444P) account for more than 80% of GBA mutations associated with PD (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). The p.L444P is considered the most deleterious, showing the strongest link with PD and dementia, the highest penetrance, and the mean age of disease onset of 51 years (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). The p.N370S is the most common variant representing more than 70% of GBA mutations in certain populations (i.e., Ashkenazi Jewish) (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). The p.N370S is also linked with earlier age of onset (mean of 54 years), higher rate of dementia, and intermediate penetrance (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). The p.T369M is the most benign, with the lowest penetrance (2.15%), low risk of dementia, and the normal age of PD onset (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). The previous studies provided inconsistent data on the effects of p.E326K; however, recently, it was shown to confer a higher risk of PIGD subtype, faster motor progression, and a higher rate of dementia than expected (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming). Other GBA variants (including p.E365K, p.T408M) were also linked with faster motor and cognitive progression (Jesús et al., 2016; Lythe et al., 2017; Iwaki et al., 2019b; Mangone et al., 2020; Petrucci et al., 2020; Straniero et al., 2020; Menozzi and Schapira, 2021; Pal et al., 2022; Dulski et al., forthcoming).
Given the preponderance of neuropsychiatric features, anticholinergics and dopamine agonists should be used with caution in the GBA carriers. Even more, caution should be exercised when considering therapy with DBS. A few studies have demonstrated that GBA mutation carriers are at higher risk of cognitive decline following DBS and thus a worse outcome (Lythe et al., 2017; Mangone et al., 2020; Pal et al., 2022). In light of these findings, therapy with LCIG in advanced PD with levodopa complications seems to be the safest option.
In summary, GBA mutations are most often associated with AR and GD subtypes.
MAPT
Pathogenic variants in MAPT, encoding microtube-associated protein tau, were described in various forms of inherited neurodegenerative disorders (Paul et al., 2016; Deutschlander et al., 2020; Dulski et al., forthcoming). Most of them manifested with autosomal dominant atypical parkinsonisms and dementia; however, in rare cases, they presented as classic PD (Paul et al., 2016; Deutschlander et al., 2020; Dulski et al., forthcoming). Recently, the MAPT locus was divided into two main haplotypes, H1 and H2 (Paul et al., 2016; Deutschlander et al., 2020; Dulski et al., forthcoming). The H1 haplotype is the most common, occurring in more than 80% of the general population, and associated with an increased risk of PD, atypical parkinsonisms, Alzheimer’s disease, and multiple other neurodegenerative diseases (Paul et al., 2016; Deutschlander et al., 2020; Dulski et al., forthcoming). On the contrary, the rare haplotype H2 seems to have a protective effect over neurodegeneration (Paul et al., 2016; Deutschlander et al., 2020; Dulski et al., forthcoming). In line with this, patients with TD-PD tend to have a higher prevalence of H2 haplotype than other PD subtypes (non-tremor dominant), which, by contrast, are more likely to carry H1 haplotype (Pascale et al., 2016). The haplotype H1 was further divided into 16 subtypes. Patients with PD and H1j subhaplotype had a lower risk of tremor and were more prone to rapid eye movement (REM) sleep behavior disorder (RBD), whereas H1r conferred a lower risk of bradykinesia, and H1b increased the risk for orthostatic hypotension (Deutschlander et al., 2020).
Genetic risk factors for complications of levodopa therapy
The COMT and MAOB genes encode two major enzymes involved in catecholamine metabolism. Most studies focused on the impact of COMT polymorphisms resulting in low, medium, and high COMT activity (Bialecka et al., 2008; Andréasson et al., 2017; Erga et al., 2018; Sampaio et al., 2018; Falla et al., 2021; Soraya et al., 2022). Some studies showed that PD patients with high COMT activity require higher levodopa doses and are at and lower risk of levodopa-induced dyskinesia, compared to the PD patients with low COMT activity. However, other studies provided contradictory results concerning the dyskinesia risk and COMT activity (Bialecka et al., 2008; Andréasson et al., 2017; Erga et al., 2018; Sampaio et al., 2018; Falla et al., 2021; Soraya et al., 2022). One COMT polymorphism (rs4646318) was found linked with impulse control disorder in PD (Erga et al., 2018). Of note, PD patients with low COMT activity may be at increased risk of developing polyneuropathy compared to those with high COMT activity (Andréasson et al., 2017). Only a few studies investigated the impact of MAO-B polymorphisms and PD symptomatology. Patients with MAOB polymorphism associated with a lower enzyme activity are more susceptible to levodopa-induced dyskinesia (Sampaio et al., 2018). In addition, male patients with polymorphisms resulting in high MAO-B activity tend to be treated with higher levodopa doses (Sampaio et al., 2018).
Dopamine D receptors 1–5 are encoded by the DRD 1–5 genes. Most of the previous studies focused on the DRD 1–3 genes (Lee et al., 2011; Zainal Abidin et al., 2015; Miller et al., 2018; Li et al., 2022). The DRD3 rs6280 (T > C) polymorphism was linked with higher susceptibility for diphasic dyskinesia (Lee et al., 2011). Another polymorphism within the DRD2, rs1076560 (G > T) was associated with gait dysfunction and better gait improvement in dopaminergic treatment compared to homozygous G allele carriers (Miller et al., 2018).
The SLC6A3 gene encodes for the dopamine transporter (DAT). The SLC6A3 polymorphisms (rs3836790 and rs28363170) were associated with motor response to levodopa treatment. However, some of the other studies did not confirm this observation (Moreau et al., 2015; Li et al., 2020, 2022; Soraya et al., 2022).
The polymorphisms in the DDC gene, coding for L-amino acid decarboxylase (AAAD), were investigated only in a few studies concerning PD (Moreau et al., 2015; Erga et al., 2018; Li et al., 2020; Soraya et al., 2022). In one recent study, the DDC polymorphism (rs921451) was found to modulate the motor response to levodopa; however, it was not confirmed in other studies (Moreau et al., 2015; Li et al., 2020).
The mutations in the MTHFR gene (rs1801133, C > T; rs1801131, A > C) are a known cause of hyperhomocysteinemia in the general population (Szadejko et al., 2016; Liu et al., 2018). Furthermore, hyperhomocysteinemia was associated with an increased risk of polyneuropathy and cardiovascular disease (Białecka et al., 2012; Szadejko et al., 2016; Liu et al., 2018). Since levodopa may increase homocysteine levels, PD patients have a particularly high risk of hyperhomocysteinemia. In addition, rs1801131 and rs1801133 MTHFR polymorphisms were linked with an increased risk of developing PD (Szadejko et al., 2016; Liu et al., 2018). However, recent metanalysis could not replicate the proposed association between rs1801131 and rs1801133 MTHFR polymorphisms and higher risk of developing PD (Diao et al., 2019). Screening PD patients treated with LCIG or high doses of levodopa for MTHFR mutations may select a subpopulation of patients with the highest risk of hyperhomocysteinemia-related complications, enabling monitoring this subpopulation and choosing optimal pharmacological therapy.
Genetic risk factors for cognitive decline
GBA variants seem to be the most prevalent genetic risk factors modifying the susceptibility to developing cognitive decline in PD patients. However, variants in APOE and MTHFR were also linked with a higher propensity for cognitive decline. The APOE gene codes for Apolipoprotein E and occurs in three alleles, E2, E3, and E4. The E3 is the most prevalent and has a frequency of approximately 80%. The E4 may be found in up to 20% of the population and was associated with a higher risk of Alzheimer’s dementia (AD), whereas the rarest E2 was linked with a lower risk of cognitive decline (Paul et al., 2016; Iwaki et al., 2019b; Dilliott et al., 2021; Tipton et al., 2021; Counsell et al., 2022; Li et al., 2022). Previous studies have demonstrated that patients with PD and at least one E4 allele are at increased risk of cognitive deterioration (Paul et al., 2016; Iwaki et al., 2019b; Dilliott et al., 2021; Tipton et al., 2021; Counsell et al., 2022; Li et al., 2022). However, contrary to AD, the protective effect of E2 on cognition was not confirmed in PD, and some showed that it might even increase the risk of PD and atypical parkinsonisms (Dilliott et al., 2021; Li et al., 2022). The MTHFR gene and its implications and in PD were discussed in-depth above. Nonetheless, as hyperhomocysteinemia was linked with an increased risk of cognitive decline, PD patients carrying an MTHFR mutation (rs1801133, C > T; rs1801131, A > C) are more prone to developing dementia (Białecka et al., 2012; Liu et al., 2018).
Genetic risk factors for impulse control disorder
Several polymorphisms of the DRD 1 (rs4532, C > T; rs4867798, T > C), DRD 2 (rs1800497, C > T), DRD 3 rs6280 (T > C) were associated with an increased risk of impulsive behaviors in PD patients (Zainal Abidin et al., 2015).
Other studies demonstrated that DDC (rs4490786), GRIN2B (rs7301328), HTR2A (rs6313), OPRK1 (rs702764), OPRM1 (rs677830), and TPH2 (rs4290270) polymorphisms were also associated with ICD in PD patients (Lee et al., 2011, 2012; Zainal Abidin et al., 2015; Erga et al., 2018; Miller et al., 2018; Li et al., 2022).
Oligo- and polygenic genetic factors behind Parkinson’s disease and its heterogeneity
Table 4 summarizes the previous genetic studies on PD heterogeneity.
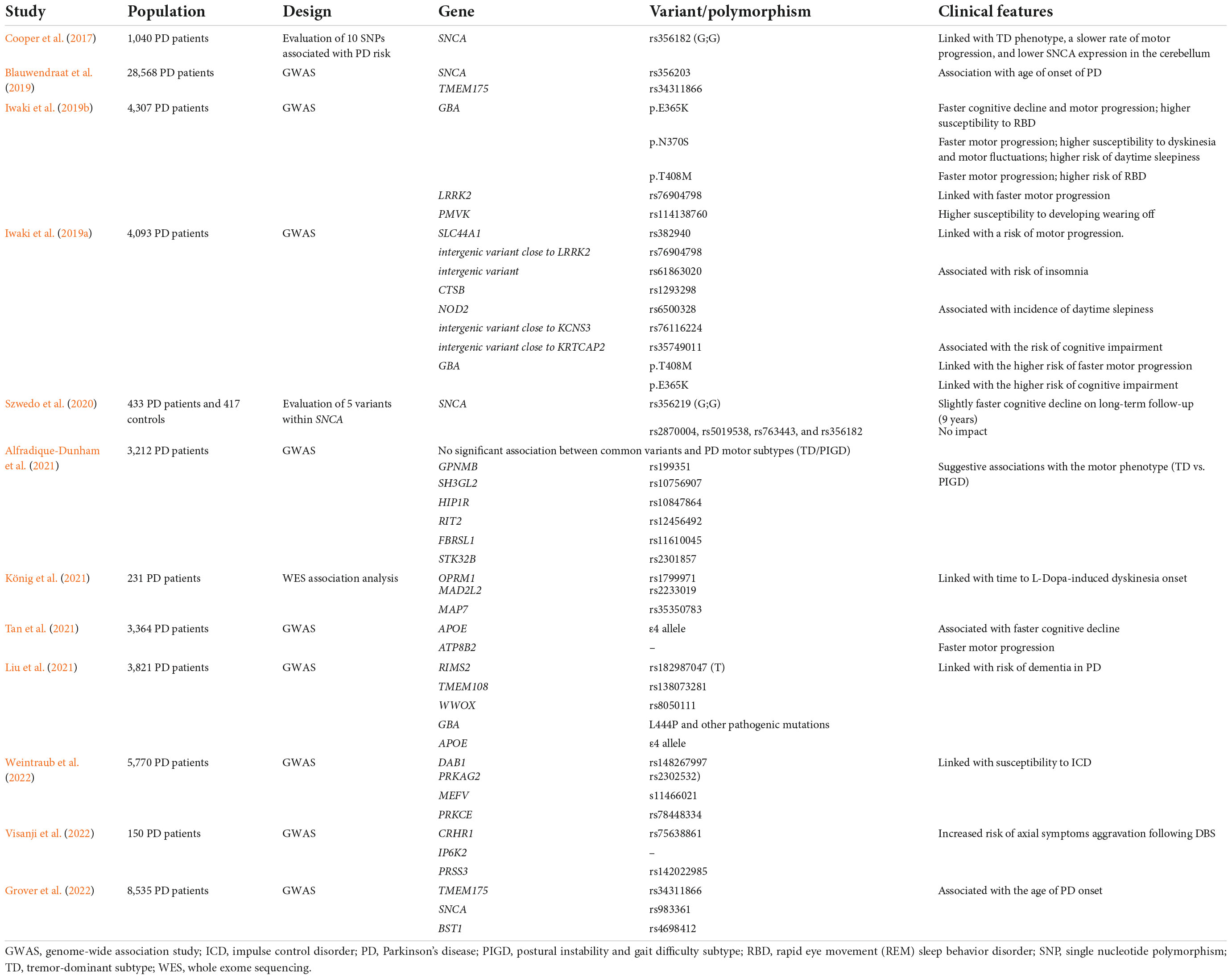
Table 4. Previous genetic studies on Parkinson’s disease (PD) heterogeneity.
In 2019, the largest GWAS in PD, involving almost 38 thousand patients, found 90 common independent variants, which together accounted for 16–36% of the observed PD heritability (Nalls et al., 2019). Recently, a large meta-analysis on PD GWAS on 8,535 patients revealed that polymorphisms of the TMEM175 (rs34311866), SNCA (rs983361), and BST1 (rs4698412) correlated with the age of the disease onset (Grover et al., 2022).
The rs356182 polymorphism near the SNCA gene was linked with the TD/PIGD phenotype. The GG genotype correlated with TD subtype, showed a slower rate of motor progression, and had lower SNCA expression in the cerebellum (Cooper et al., 2017). However, in another study, SNCA polymorphisms (rs356219, rs2870004, rs5019538, rs763443, and rs356182) that were previously linked with PD risk, were found not to contribute significantly to PD heterogeneity (Szwedo et al., 2020). A large GWAS study on 28,568 PD patients found that two genetic loci (SNCA, TMEM175) were modulating the age of the disease onset (Blauwendraat et al., 2019). In 2019, a GWAS study on 4,307 PD patients found that GBA variants (p.E365K, p.N370S, and p.T408M) correlated with rate of cognitive and motor progression, risk of developing dyskinesia, motor fluctuations, RBD, and daytime sleepiness (Iwaki et al., 2019b).
Another GWAS study in 2019, on 4,039 PD patients confirmed the association of GBA variants with higher risk of cognitive impairment (p.E365K) and faster motor progression (p.T408M), as well as, found a few genetic loci linked with the risk of sleep disturbances (rs61863020, rs1293298, rs6500328, and rs76116224), worsening of motor symptoms (rs382940, rs382940) and cognitive decline (rs35749011) (Iwaki et al., 2019a). However, only two polymorphisms (rs382940 in SLC44A1, and rs61863020 – intergenic variant) reached the genomewide significance (Iwaki et al., 2019a). The same study found that the T allele of rs76904798 polymorphism within LRRK2 increased susceptibility to motor progression, and rs114138760 within PMVK was linked with the risk of developing wearing off (Iwaki et al., 2019b). Two large studies from 2021, confirmed that PD patients with APOE E4 allele or GBA variants are at increased risk of faster cognitive deterioration and identified novel polymorphism linked with faster cognitive (RIMS2, TMEM108, and WWOX) and motor (ATP8B2) progression (Liu et al., 2021; Tan et al., 2021). Another GWAS study in 2021, on 3,212 PD cases, did not find any significant association between common variants and PD motor subtypes (TD/PIGD) (Alfradique-Dunham et al., 2021). However, the authors reported multiple suggestive associations, including the polymorphisms of GPNMB (rs199351), SH3GL2 (rs10756907), HIP1R (rs10847864), RIT2 (rs12456492), FBRSL1 (rs11610045), and STK32B (rs2301857) genes (Alfradique-Dunham et al., 2021). Interestingly, a variant in the STK32B was previously reported in other GWAS as a risk factor for essential tremor (Müller et al., 2016). Whole-exome sequencing (WES) association analysis of 231 PD patients found the polymorphisms in the OPRM1 (rs1799971), MAD2L2 (rs2233019), and MAP7 (rs35350783) genes to be linked with time to levodopa-induced dyskinesia onset (König et al., 2021). In 2022, GWAS on 5,770 patients with PD detected polymorphisms of DAB1 (rs148267997), PRKAG2 (rs2302532), MEFV (rs11466021), and PRKCE (rs78448334) genes associated with impulse control disorders (Weintraub et al., 2022). In 2022, a study on 150 PD patients treated with DBS found associations between the deterioration of axial symptoms following surgery and polymorphisms of CRHR1 (rs75638861), IP6K2 (SNP not listed), and PRSS3 (rs142022985) genes (Visanji et al., 2022).
Discussion
The majority of studies on PD subtypes used the data-driven (hypothesis-free) approach to uncover phenotypic clusters. The most frequently assessed phenotypic domains were the motor features, cognitive functions, and other neuropsychiatric symptoms. Nevertheless, despite being superseded by the data-driven clustering in the research, the PD subtyping, based mainly on studies on pre-defined (hypothesis-driven) motor phenotypes, is still the one most commonly used in everyday clinical practice. Neurologists thoroughly consider the patient’s motor predominant symptomatology, which significantly influences management strategy. Levodopa is the most effective medication for PD and is considered the gold standard against which other drugs and therapies are compared. Levodopa is the mainstay of PD therapy in most patients; however, in patients with certain phenotypes, other treatments should also be considered.
In patients with the GD subtype, an improvement on levodopa alone is often not satisfactory, and additional medications are given to improve gait and balance. In clinical settings, amantadine and rivastigmine are most often used (Cui and Lewis, 2021). Patients with advanced PD, motor fluctuations, and levodopa-induced dyskinesia are good candidates for DBS and infusional therapies. In such cases, the phenotype (mainly motor symptoms, cognitive status, and autonomic functions) guides the choice of therapies. Additionally, the predominant symptomatology determines the type of physical therapy, selection of assistive devices, and others (Dijk et al., 2020). Nevertheless, the clinical implications of PD subtypes go beyond the choice of therapy. Previous studies consistently demonstrated that different motor subtypes vary in disease progression and the burden of non-dopaminergic (e.g., cognitive decline, dysautonomia, RBD) symptoms. The TD subtype is considered the phenotype with a lower risk of cognitive decline, slower progression, and lesser impact of non-dopaminergic symptoms compared to GD and AR subtypes (Marras et al., 2020; Ren et al., 2020, 2021; Kohat et al., 2021; Myers et al., 2021; Lee et al., 2022). Lastly, the PD subtyping is relevant for clinical trials, as some protocols include the presence of specific phenotypes in the inclusion or exclusion criteria. Notably, that may be relevant to gene therapy, which raised high hopes among PD patients and physicians (Fiandaca et al., 2020). Taken together, determining the subtype of individual PD patients is of great importance for the time being and long-term prognosis.
Nonetheless, some studies demonstrated that patients might change their phenotypes over time, whereas most did not provide longitudinal data (Kohat et al., 2021; Mestre et al., 2021). Thus, the stability of PD subtypes over time is largely unknown, and there is a need to find markers that predict the susceptibility of a patient to develop certain symptoms. Therefore, in the PD subtyping research, high expectations were placed on studies investigating the biomarkers of the disease progression. However, the biological underpinnings of PD are complex and not completely understood. The neurodegenerative process is characterized by progressive loss of dopaminergic neurons in the pars compacta of substantia nigra, and misfolding and aggregation of α-synuclein in the cytoplasm of the neurons (Lewy bodies) (Balestrino and Schapira, 2020). Most studies on PD subtypes evaluated biomarkers in the cerebrospinal fluid (CSF) and blood, and the results held promise to define the pathophysiological bases of the different PD subtypes in the future. The CSF levels of α-synuclein, total tau, phosphorylated tau, and beta-amyloid 42 differed in PD patients and healthy controls; however, the attempts to correlate them with clinical symptoms provided inconsistent results. Blood studies showed that higher levels of pro-inflammatory markers were linked with a more severe motor phenotype (IFN-γ, TNF-α, IL-2, and IL-10), faster motor progression (CRP and IL-6), cognitive impairment (IFN-γ, TNF-α, IL-2, and IL-10), and worse sleep quality (CRP). In contrast, higher levels of anti-inflammatory markers (IL-4 and IL-13) correlated with milder motor progression (Williams-Gray et al., 2016; Lawton et al., 2020; Lee et al., 2022). Yet, these studies have potential drawbacks making their application in clinical practice challenging. The concomitant inflammatory process or medications may change the levels of inflammatory markers. Additionally, a routine application of CSF assays to monitor PD patients is challenging (Figura and Friedman, 2020; Mestre et al., 2021; Lee et al., 2022).
In light of this, the genetic markers seem to be the most promising to be used in clinical practice in the future. However, we recognize several issues to be addressed in future research. First, the previous studies used different PD subtyping classifications, which was one of the most significant factors behind the incompatibility of the studies. Therefore, we think there is a strong need to unify the PD subtyping. Of note, the classification should consider the needs of clinicians and should be easy to use in everyday clinical practice. We propose incorporating the subtyping from Konno et al. (2018). Contrary to the two other commonly used classification systems (Zetusky et al., 1985; Jankovic et al., 1990; Schiess et al., 2000; Korchounov et al., 2004; Kang et al., 2005), it is the only one considering AR and GD separately, besides the TD and mixed subtypes.
Secondly, based on our review, previous studies found little overlap between genetic risk factors for PD and genetic modifiers of PD subtypes and progression, and it seems that different genetic loci underlie the susceptibility for PD and its heterogeneity. As lumping them together may impede the research yield, we believe they should be investigated separately. Lastly, the advances in genetic research moved the spotlight from rare and highly penetrant monogenic variants to common variants (polymorphisms) that have low penetrance but a higher impact on the general population. However, despite using more advanced methods and large cohorts, most studies provided results that were not replicated. The failure to implement the findings into clinical practice may be partially caused by a number of studies with often discrepant methods and different results. Therefore, we call for more consistency in the studies by first assessing the known variants’ associations and then investigating the new variants and their links.
As the treatment landscape continues to evolve, precision medicine combining PD subtyping, genetic profile, and biomarkers offers the opportunity to tailor disease treatment to an individual patient. For example, patients with LRRK2 mutations resulting in a gain of LRRK2 kinase activity are potential candidates for drugs inhibiting its activity or antisense oligonucleotides silencing the altered LRRK2 gene (Prasuhn and Brüggemann, 2021; Lee et al., 2022). Patients with mitochondrial dysfunction, as with PRKN or PINK1 mutations, are being tested for the potential benefits of coenzyme Q10 or vitamin K2 (Prasuhn and Brüggemann, 2021; Lee et al., 2022). Reduced activity of glucocerebrosidase (GCase) in GBA mutation carriers prompted studies aiming at enhancing its activity, its external augmentation, or reduction of its substance (glucosylceramide) (Prasuhn and Brüggemann, 2021; Lee et al., 2022). As LRRK2 activity is elevated in idiopathic PD and GCase can decrease α-synuclein accumulation, these therapies may also be helpful in idiopathic PD patients (Prasuhn and Brüggemann, 2021; Lee et al., 2022).
In summary, the findings from the genetic studies on PD heterogeneity hold promise for utility in clinical practice. However, it is necessary to unify PD subtyping classification and facilitate the link between research and clinical practice. As there is growing evidence that different genetic factors underlie the development of PD and its heterogeneous presentation, we believe the dichotomy is needed to address the unmet needs of research and clinical practice aspects of PD.
Author contributions
JD took the lead in writing the manuscript. RU and OR revised the manuscript. ZW supervised the preparation of the manuscript and revised it. All authors contributed to the article and approved the submitted version.
Funding
The Haworth Family Professorship in Neurodegenerative Diseases fund sponsored the fellowship of JD at Mayo Clinic, RU and ZW have a financial interest in technologies entitled, “Identification of Mutations in PARK8, a Locus for Familial Parkinson’s Disease” and “Identification of a Novel LRRK2 Mutation, 6055G > A (G2019S), Linked to Autosomal Dominant Parkinsonism in Families from Several European Populations”. Those technologies have been licensed to a commercial entity, and to date, RU and ZW have received royalties <$1.500 through Mayo Clinic in accordance with its royalty sharing policies. OR Grants: NIH/NINDS U54-NS100693, UG3-NS104095, U54- NS110435, DOD (W81XWH-17-1-0249), The Michael J. Fox Foundation and American Parkinson Disease Association Center for Advanced Research. ZW Grants: NIH/NIA and NIH/NINDS (1U19AG063911, FAIN: U19AG063911), Mayo Clinic Center for Regenerative Medicine, Mayo Clinic in Florida Focused Research Team Program, the gifts from The Sol Goldman Charitable Trust, and the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, and The Albertson Parkinson’s Research Foundation. He serves as PI or Co-PI on Biohaven Pharmaceuticals, Inc. (BHV4157-206 and BHV3241-301), Neuraly, Inc. (NLY01-PD-1), and Vigil Neuroscience, Inc. (VGL101-01.001) grants.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alfradique-Dunham, I., Al-Ouran, R., von Coelln, R., Blauwendraat, C., Hill, E., Luo, L., et al. (2021). Genome-wide association study meta-analysis for Parkinson disease motor subtypes. Neurol. Genet. 7:e557. doi: 10.1212/nxg.0000000000000557
Andréasson, M., Brodin, L., Laffita-Mesa, J. M., and Svenningsson, P. (2017). Correlations between methionine cycle metabolism, COMT genotype, and polyneuropathy in L-Dopa Treated Parkinson’s disease: A preliminary cross-sectional study. J. Parkinsons Dis. 7, 619–628. doi: 10.3233/JPD-171127
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Bialecka, M., Kurzawski, M., Klodowska-Duda, G., Opala, G., Tan, E. K., and Drozdzik, M. (2008). The association of functional catechol-O-methyltransferase haplotypes with risk of Parkinson’s disease, levodopa treatment response, and complications. Pharmacogenet. Genomics 18, 815–821. doi: 10.1097/FPC.0b013e328306c2f2
Białecka, M., Kurzawski, M., Roszmann, A., Robowski, P., Sitek, E. J., Honczarenko, K., et al. (2012). Association of COMT, MTHFR, and SLC19A1(RFC-1) polymorphisms with homocysteine blood levels and cognitive impairment in Parkinson’s disease. Pharmacogenet. Genomics 22, 716–724. doi: 10.1097/FPC.0b013e32835693f7
Blauwendraat, C., Heilbron, K., Vallerga, C. L., Bandres-Ciga, S., von Coelln, R., Pihlstrøm, L., et al. (2019). Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 34, 866–875. doi: 10.1002/mds.27659
Cooper, C. A., Jain, N., Gallagher, M. D., Weintraub, D., Xie, S. X., Berlyand, Y., et al. (2017). Common variant rs356182 near SNCA defines a Parkinson’s disease endophenotype. Ann. Clin. Transl. Neurol. 4, 15–25. doi: 10.1002/acn3.371
Counsell, C., Giuntoli, C., Khan, Q. I., Maple-Grødem, J., and Macleod, A. D. (2022). The incidence, baseline predictors, and outcomes of dementia in an incident cohort of Parkinson’s disease and controls. J. Neurol. 269, 4288–4298. doi: 10.1007/s00415-022-11058-2
Cui, C. K., and Lewis, S. J. G. (2021). Future therapeutic strategies for freezing of gait in Parkinson’s Disease. Front. Hum. Neurosci. 15:741918. doi: 10.3389/fnhum.2021.741918
Deutschlander, A. B., Konno, T., Soto-Beasley, A. I., Walton, R. L., van Gerpen, J. A., Uitti, R. J., et al. (2020). Association of MAPT subhaplotypes with clinical and demographic features in Parkinson’s disease. Ann. Clin. Transl. Neurol. 7, 1557–1563. doi: 10.1002/acn3.51139
Di Lazzaro, G., Magrinelli, F., Estevez-Fraga, C., Valente, E. M., Pisani, A., and Bhatia, K. P. (2021). X-linked Parkinsonism: Phenotypic and genetic heterogeneity. Mov. Disord. 36, 1511–1525.
Diao, H. M., Song, Z. F., and Xu, H. D. (2019). Association between MTHFR genetic polymorphism and Parkinson’s disease susceptibility: A Meta-analysis. Open Med. 14, 613–624. doi: 10.1515/med-2019-0069
Dijk, J. M., Espay, A. J., Katzenschlager, R., and de Bie, R. M. A. (2020). The choice between advanced therapies for Parkinson’s disease patients: Why, What, and When? J. Parkinsons Dis. 10, S65–S73. doi: 10.3233/jpd-202104
Dilliott, A. A., Sunderland, K. M., McLaughlin, P. M., Roberts, A. C., Evans, E. C., Abrahao, A., et al. (2021). Association of apolipoprotein E variation with cognitive impairment across multiple neurodegenerative diagnoses. Neurobiol. Aging 105, 378.e1–378.e9. doi: 10.1016/j.neurobiolaging.2021.04.011
Dulski, J., Fujioka, S., and Wszolek, Z. K. (forthcoming). “Genetic parkinsonisms,” in IAPRD textbook of movement disorders, eds E. Wolters, C. Baumann, and D. Truong (Cambridge: Cambridge University Press).
Dulski, J., Schinwelski, M., Konkel, A., Grabowski, K., Libionka, W., Wa̧ż, P., et al. (2018). The impact of subthalamic deep brain stimulation on polysomnographic sleep pattern in patients with Parkinson’s disease - Preliminary report. Neurol. Neurochir. Pol. 52, 514–518. doi: 10.1016/j.pjnns.2018.05.006
Dulski, J., Schinwelski, M., Konkel, A., Grabowski, K., Libionka, W., Wa̧ż, P., et al. (2019). The impact of subthalamic deep brain stimulation on sleep and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat. Disord. 64, 138–144.
Dulski, J., Schinwelski, M., Konkel, A., and Sławek, J. (2015). Sleep disorders in Parkinson’s disease. Postępy Psychiatr. Neurol. 24, 147–155.
Dulski, J., Wa̧ż, P., Konkel, A., Grabowski, K., Libionka, W., Schinwelski, M., et al. (2022). The Impact of subthalamic deep brain stimulation on restless legs syndrome in Parkinson’s disease. Neuromodulation 25, 904–910. doi: 10.1111/ner.13462
Erga, A. H., Dalen, I., Ushakova, A., Chung, J., Tzoulis, C., Tysnes, O. B., et al. (2018). Dopaminergic and opioid pathways associated with impulse control disorders in Parkinson’s disease. Front. Neurol. 9:109. doi: 10.3389/fneur.2018.00109
Falla, M., Di Fonzo, A., Hicks, A. A., Pramstaller, P. P., and Fabbrini, G. (2021). Genetic variants in levodopa-induced dyskinesia (LID): A systematic review and meta-analysis. Parkinsonism Relat. Disord. 84, 52–60. doi: 10.1016/j.parkreldis.2021.01.020
Fereshtehnejad, S. M., Zeighami, Y., Dagher, A., and Postuma, R. B. (2017). Clinical criteria for subtyping Parkinson’s disease: Biomarkers and longitudinal progression. Brain 140, 1959–1976. doi: 10.1093/brain/awx118
Fiandaca, M. S., Lonser, R. R., Elder, J. B., Za̧bek, M., and Bankiewicz, K. S. (2020). Advancing gene therapies, methods, and technologies for Parkinson’s Disease and other neurological disorders. Neurol. Neurochir. Pol. 54, 220–231.
Figura, M., and Friedman, A. (2020). In search of Parkinson’s disease biomarkers - is the answer in our mouths? A systematic review of the literature on salivary biomarkers of Parkinson’s disease. Neurol. Neurochir. Pol. 54, 14–20. doi: 10.5603/PJNNS.a2020.0011
Grover, S., Ashwin, A. K. S., Pihlstrom, L., Domenighetti, C., Schulte, C., Sugier, P. E., et al. (2022). Genome-wide association and meta-analysis of age-at-onset in Parkinson disease: Evidence from COURAGE-PD Consortium. Neurology 99, e698–e710. doi: 10.1212/wnl.0000000000200699
Guadagnolo, D., Piane, M., Torrisi, M. R., Pizzuti, A., and Petrucci, S. (2021). Genotype-phenotype correlations in monogenic Parkinson disease: A review on clinical and molecular findings. Front. Neurol. 12:648588. doi: 10.3389/fneur.2021.648588
Hall, A., Bandres-Ciga, S., Diez-Fairen, M., Quinn, J. P., and Billingsley, K. J. (2020). Genetic risk profiling in Parkinson’s disease and utilizing genetics to gain insight into disease-Related Biological Pathways. Int. J. Mol. Sci. 21:7332. doi: 10.3390/ijms21197332
Hentati, F., Trinh, J., Thompson, C., Nosova, E., Farrer, M. J., and Aasly, J. O. (2014). LRRK2 parkinsonism in Tunisia and Norway: A comparative analysis of disease penetrance. Neurology 83, 568–569. doi: 10.1212/wnl.0000000000000675
Iwaki, H., Blauwendraat, C., Leonard, H. L., Liu, G., Maple-Grødem, J., Corvol, J. C., et al. (2019b). Genetic risk of Parkinson disease and progression: An analysis of 13 longitudinal cohorts. Neurol. Genet. 5:e348. doi: 10.1212/nxg.0000000000000348
Iwaki, H., Blauwendraat, C., Leonard, H. L., Kim, J. J., Liu, G., Maple-Grødem, J., et al. (2019a). Genomewide association study of Parkinson’s disease clinical biomarkers in 12 longitudinal patients’ cohorts. Mov. Disord. 34, 1839–1850. doi: 10.1002/mds.27845
Jankovic, J., McDermott, M., Carter, J., Gauthier, S., Goetz, C., Golbe, L., et al. (1990). Variable expression of Parkinson’s disease. A base-line analysis of the DAT ATOP cohort The Parkinson Study Group. Neurology 40, 1529–1529. doi: 10.1212/wnl.40.10.1529
Jesús, S., Huertas, I., Bernal-Bernal, I., Bonilla-Toribio, M., Cáceres-Redondo, M. T., Vargas-González, L., et al. (2016). GBA variants influence motor and non-motor features of Parkinson’s disease. PLoS One 11:e0167749. doi: 10.1371/journal.pone.0167749
Jia, F., Fellner, A., and Kumar, K. R. (2022). Monogenic Parkinson’s Disease: Genotype, phenotype, pathophysiology, and genetic testing. Genes 13:471.
Kang, G. A., Bronstein, J. M., Masterman, D. L., Redelings, M., Crum, J. A., and Ritz, B. (2005). Clinical characteristics in early Parkinson’s disease in a central California population-based study. Mov. Disord. 20, 1133–1142. doi: 10.1002/mds.20513
Kasten, M., Hartmann, C., Hampf, J., Schaake, S., Westenberger, A., Vollstedt, E. J., et al. (2018). Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 33, 730–741. doi: 10.1002/mds.27352
Kim, J. S., Cho, J. W., Shin, H., Lee, W. Y., Ki, C. S., Cho, A. R., et al. (2012). A Korean Parkinson’s disease family with the LRRK2 p.Tyr1699Cys mutation showing clinical heterogeneity. Mov. Disord. 27, 320–324. doi: 10.1002/mds.24033
Kohat, A. K., Ng, S. Y. E., Wong, A. S. Y., Chia, N. S. Y., Choi, X., Heng, D. L., et al. (2021). Stability of MDS-UPDRS motor subtypes over three years in early Parkinson’s disease. Front. Neurol. 12:704906. doi: 10.3389/fneur.2021.704906
König, E., Nicoletti, A., Pattaro, C., Annesi, G., Melotti, R., Gialluisi, A., et al. (2021). Exome-wide association study of levodopa-induced dyskinesia in Parkinson’s disease. Sci. Rep. 11:19582. doi: 10.1038/s41598-021-99393-8
Konno, T., Deutschländer, A., Heckman, M. G., Ossi, M., Vargas, E. R., Strongosky, A. J., et al. (2018). Comparison of clinical features among Parkinson’s disease subtypes: A large retrospective study in a single center. J. Neurol. Sci. 386, 39–45. doi: 10.1016/j.jns.2018.01.013
Korchounov, A., Schipper, H. I., Preobrazhenskaya, I. S., Kessler, K. R., and Yakhno, N. N. (2004). Differences in age at onset and familial aggregation between clinical types of idiopathic Parkinson’s disease. Mov. Disord. 19, 1059–1064. doi: 10.1002/mds.20061
Krohn, L., Grenn, F. P., Makarious, M. B., Kim, J. J., Bandres-Ciga, S., Roosen, D. A., et al. (2020). Comprehensive assessment of PINK1 variants in Parkinson’s disease. Neurobiol. Aging 91, 168.e1–168.e5. doi: 10.1016/j.neurobiolaging.2020.03.003
Kumar, S., Abbas, M. M., Govindappa, S. T., Muthane, U. B., Behari, M., Pandey, S., et al. (2021). Compound heterozygous variants in Wiskott-Aldrich syndrome like (WASL) gene segregating in a family with early onset Parkinson’s disease. Parkinsonism Relat. Disord. 84, 61–67. doi: 10.1016/j.parkreldis.2021.02.001
Lawton, M., Baig, F., Toulson, G., Morovat, A., Evetts, S. G., Ben-Shlomo, Y., et al. (2020). Blood biomarkers with Parkinson’s disease clusters and prognosis: The oxford discovery cohort. Mov. Disord. 35, 279–287. doi: 10.1002/mds.27888
Lawton, M., Ben-Shlomo, Y., May, M. T., Baig, F., Barber, T. R., Klein, J. C., et al. (2018). Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J. Neurol. Neurosurg. Psychiatry 89, 1279–1287. doi: 10.1136/jnnp-2018-318337
Lee, J. Y., Cho, J., Lee, E. K., Park, S. S., and Jeon, B. S. (2011). Differential genetic susceptibility in diphasic and peak-dose dyskinesias in Parkinson’s disease. Mov. Disord. 26, 73–79. doi: 10.1002/mds.23400
Lee, J.-Y., Jeon, B. S., Kim, H.-J., and Park, S.-S. (2012). Genetic variant of HTR2A associates with risk of impulse control and repetitive behaviors in Parkinson’s disease. Parkinsonism Relat. Disord. 18, 76–78. doi: 10.1016/j.parkreldis.2011.08.009
Lee, S. H., Park, S. M., Yeo, S. S., Kwon, O., Lee, M. K., Yoo, H., et al. (2022). Parkinson’s disease subtyping using clinical features and biomarkers: Literature review and preliminary study of subtype clustering. Diagnostics 12:112. doi: 10.3390/diagnostics12010112
Lesage, S., Houot, M., Mangone, G., Tesson, C., Bertrand, H., Forlani, S., et al. (2020). Genetic and phenotypic basis of autosomal dominant Parkinson’s disease in a large multi-center cohort. Front. Neurol. 11:682. doi: 10.3389/fneur.2020.00682
Li, J., Yi, M., Li, B., Yin, S., Zhang, Y., Huang, Z., et al. (2022). Polymorphism of neurodegeneration-related genes associated with Parkinson’s disease risk. Neurol. Sci. 43, 5301–5312. doi: 10.1007/s10072-022-06192-8
Li, L., Lin, H., Hua, P., Yan, L., Dong, H., Li, T., et al. (2020). Polymorphism of the Dopa-decarboxylase gene modifies the motor response to levodopa in chinese patients with Parkinson’s disease. Front. Neurol. 11:520934. doi: 10.3389/fneur.2020.520934
Liu, G., Peng, J., Liao, Z., Locascio, J. J., Corvol, J. C., Zhu, F., et al. (2021). Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nat. Genet. 53, 787–793. doi: 10.1038/s41588-021-00847-6
Liu, L., Zhang, L., Guo, L., Yu, Q., Li, H., Teng, J., et al. (2018). MTHFR C677T and A1298C polymorphisms may contribute to the risk of Parkinson’s disease: A meta-analysis of 19 studies. Neurosci. Lett. 662, 339–345. doi: 10.1016/j.neulet.2017.10.060
Liu, X., and Le, W. (2020). Profiling non-motor symptoms in monogenic Parkinson’s disease. Front. Aging Neurosci. 12:591183. doi: 10.3389/fnagi.2020.591183
Lythe, V., Athauda, D., Foley, J., Mencacci, N. E., Jahanshahi, M., Cipolotti, L., et al. (2017). GBA-associated Parkinson’s disease: Progression in a deep brain stimulation cohort. J. Parkinsons Dis. 7, 635–644. doi: 10.3233/JPD-171172
Madureira, M., Connor-Robson, N., and Wade-Martins, R. (2020). LRRK2: Autophagy and lysosomal activity. Front. Neurosci. 14:498. doi: 10.3389/fnins.2020.00498
Mangone, G., Bekadar, S., Cormier-Dequaire, F., Tahiri, K., Welaratne, A., Czernecki, V., et al. (2020). Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat. Disord. 76, 56–62. doi: 10.1016/j.parkreldis.2020.04.002
Marras, C., Alcalay, R. N., Caspell-Garcia, C., Coffey, C., Chan, P., Duda, J. E., et al. (2016). Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov. Disord. 31, 1192–1202. doi: 10.1002/mds.26614
Marras, C., Chaudhuri, K. R., Titova, N., and Mestre, T. A. (2020). Therapy of Parkinson’s disease subtypes. Neurotherapeutics 17, 1366–1377. doi: 10.1007/s13311-020-00894-7
Menozzi, E., and Schapira, A. H. V. (2021). Exploring the genotype–phenotype correlation in GBA-Parkinson disease: Clinical aspects, biomarkers, and potential Modifiers. Front. Neurol. 12:694764. doi: 10.3389/fneur.2021.694764
Mestre, T. A., Fereshtehnejad, S. M., Berg, D., Bohnen, N. I., Dujardin, K., Erro, R., et al. (2021). Parkinson’s disease subtypes: Critical appraisal and recommendations. J. Parkinsons Dis. 11, 395–404. doi: 10.3233/JPD-202472
Milanowski, Ł. M., Ross, O. A., Friedman, A., Hoffman-Zacharska, D., Gorka-Skoczylas, P., Jurek, M., et al. (2021). Genetics of Parkinson’s disease in the Polish population. Neurol. Neurochir. Pol. 55, 241–252.
Miller, N. S., Chou, K. L., Bohnen, N. I., Müller, M. L. T. M., and Seidler, R. D. (2018). Dopaminergic polymorphisms associated with medication responsiveness of gait in Parkinson’s disease. Parkinsonism Relat. Disord. 48, 54–60. doi: 10.1016/j.parkreldis.2017.12.010
Mirelman, A., Heman, T., Yasinovsky, K., Thaler, A., Gurevich, T., Marder, K., et al. (2013). Fall risk and gait in Parkinson’s disease: The role of the LRRK2 G2019S mutation. Mov. Disord. 28, 1683–1690. doi: 10.1002/mds.25587
Moreau, C., Meguig, S., Corvol, J. C., Labreuche, J., Vasseur, F., Duhamel, A., et al. (2015). Polymorphism of the dopamine transporter type 1 gene modifies the treatment response in Parkinson’s disease. Brain 138(Pt 5), 1271–1283. doi: 10.1093/brain/awv063
Morgante, F., Fasano, A., Ginevrino, M., Petrucci, S., Ricciardi, L., Bove, F., et al. (2016). Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology 87, 1436–1441. doi: 10.1212/wnl.0000000000003177
Müller, S. H., Girard, S. L., Hopfner, F., Merner, N. D., Bourassa, C. V., Lorenz, D., et al. (2016). Genome-wide association study in essential tremor identifies three new loci. Brain 139(Pt 12), 3163–3169. doi: 10.1093/brain/aww242
Myers, P. S., Jackson, J. J., Clover, A. K., Lessov-Schlaggar, C. N., Foster, E. R., Maiti, B., et al. (2021). Distinct progression patterns across Parkinson disease clinical subtypes. Ann. Clin. Transl. Neurol. 8, 1695–1708. doi: 10.1002/acn3.51436
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/s1474-4422(19)30320-5
Ou, Z., Pan, J., Tang, S., Duan, D., Yu, D., Nong, H., et al. (2021). Global Trends in the Incidence, Prevalence, and Years Lived With Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front. Public Health 9:776847. doi: 10.3389/fpubh.2021.776847
Pal, G., Mangone, G., Hill, E. J., Ouyang, B., Liu, Y., Lythe, V., et al. (2022). Parkinson disease and subthalamic nucleus deep brain stimulation: Cognitive effects in GBA mutation carriers. Ann. Neurol. 91, 424–435. doi: 10.1002/ana.26302
Pascale, E., Di Battista, M. E., Rubino, A., Purcaro, C., Valente, M., Fattapposta, F., et al. (2016). Genetic architecture of MAPT gene region in Parkinson disease subtypes. Front. Cell. Neurosci. 10:96. doi: 10.3389/fncel.2016.00096
Paul, K. C., Rausch, R., Creek, M. M., Sinsheimer, J. S., Bronstein, J. M., Bordelon, Y., et al. (2016). APOE, MAPT, and COMT and Parkinson’s disease susceptibility and cognitive symptom progression. J. Parkinsons Dis. 6, 349–359. doi: 10.3233/JPD-150762
Petrucci, S., Ginevrino, M., Trezzi, I., Monfrini, E., Ricciardi, L., Albanese, A., et al. (2020). GBA-related Parkinson’s disease: Dissection of genotype-phenotype correlates in a large italian cohort. Mov. Disord. 35, 2106–2111. doi: 10.1002/mds.28195
Prasuhn, J., and Brüggemann, N. (2021). Genotype-driven therapeutic developments in Parkinson’s disease. Mol. Med. 27:42. doi: 10.1186/s10020-021-00281-8
Ren, J., Hua, P., Li, Y., Pan, C., Yan, L., Yu, C., et al. (2020). Comparison of three motor subtype classifications in de novo Parkinson’s disease patients. Front. Neurol. 11:601225. doi: 10.3389/fneur.2020.601225
Ren, J., Pan, C., Li, Y., Li, L., Hua, P., Xu, L., et al. (2021). Consistency and stability of motor subtype classifications in patients with de novo Parkinson’s disease. Front. Neurosci. 15:637896. doi: 10.3389/fnins.2021.637896
Saini, P., Rudakou, U., Yu, E., Ruskey, J. A., Asayesh, F., Laurent, S. B., et al. (2021). Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson’s disease. Neurobiol. Aging 100, 119e7–119.e13. doi: 10.1016/j.neurobiolaging.2020.10.019
Salles, P. A., Mata, I. F., and Fernandez, H. H. (2021). Should we start integrating genetic data in decision-making on device-aided therapies in Parkinson disease? A point of view. Parkinsonism Relat. Disord. 88, 51–57. doi: 10.1016/j.parkreldis.2021.05.013
Sampaio, T. F., dos Santos, E. U. D., de Lima, G. D. C., dos Anjos, R. S. G., da Silva, R. C., Asano, A. G. C., et al. (2018). MAO-B and COMT genetic variations associated with levodopa treatment response in patients with Parkinson’s disease. J. Clin. Pharmacol. 58, 920–926. doi: 10.1002/jcph.1096
Schiess, M. C., Zheng, H., Soukup, V. M., Bonnen, J. G., and Nauta, H. J. (2000). Parkinson’s disease subtypes: Clinical classification and ventricular cerebrospinal fluid analysis. Parkinsonism Relat. Disord. 6, 69–76. doi: 10.1016/s1353-8020(99)00051-6
Simpson, C., Vinikoor-Imler, L., Nassan, F. L., Shirvan, J., Lally, C., Dam, T., et al. (2022). Prevalence of ten LRRK2 variants in Parkinson’s disease: A comprehensive review. Parkinsonism Relat. Disord. 98, 103–113. doi: 10.1016/j.parkreldis.2022.05.012
Siuda, J. (2021). Importance of non-motor symptoms in PD and atypical parkinsonism. Neurol. Neurochir. Pol. 55, 503–507.
Soraya, G. V., Ulhaq, Z. S., Shodry, S., A’Raaf Sirojan Kusuma, M., Herawangsa, S., Sativa, M. O., et al. (2022). Polymorphisms of the dopamine metabolic and signaling pathways are associated with susceptibility to motor levodopa-induced complications (MLIC) in Parkinson’s disease: A systematic review and meta-analysis. Neurol. Sci. 43, 3649–3670. doi: 10.1007/s10072-021-05829-4
Straniero, L., Asselta, R., Bonvegna, S., Rimoldi, V., Melistaccio, G., Soldà, G., et al. (2020). The SPID-GBA study: Sex distribution, penetrance, incidence, and dementia in GBA-PD. Neurol. Genet. 6:e523. doi: 10.1212/nxg.0000000000000523
Szadejko, K., Dziewiatowski, K., Szabat, K., Robowski, P., Schinwelski, M., Sitek, E., et al. (2016). Polyneuropathy in levodopa-treated Parkinson’s patients. J. Neurol. Sci. 371, 36–41. doi: 10.1016/j.jns.2016.09.061
Szwedo, A. A., Pedersen, C. C., Ushakova, A., Forsgren, L., Tysnes, O. B., Counsell, C. E., et al. (2020). Association of SNCA Parkinson’s disease risk polymorphisms with disease progression in newly diagnosed patients. Front. Neurol. 11:620585. doi: 10.3389/fneur.2020.620585
Tan, M. M. X., Lawton, M. A., Jabbari, E., Reynolds, R. H., Iwaki, H., Blauwendraat, C., et al. (2021). Genome-Wide association studies of cognitive and motor progression in Parkinson’s disease. Mov. Disord. 36, 424–433. doi: 10.1002/mds.28342
Tipton, P. W. (2021). Dissecting Parkinsonism: Cognitive and gait disturbances. Neurol. Neurochir. Pol. 55, 513–524. doi: 10.5603/PJNNS.a2021.0084
Tipton, P. W., Bülbül, N., Crook, J., Quicksall, Z., Ross, O. A., Uitti, R. J., et al. (2021). Effects of sex and APOE on Parkinson’s Disease-related cognitive decline. Neurol. Neurochir. Pol. 55, 559–566. doi: 10.5603/PJNNS.a2021.0071
Trinh, J., Zeldenrust, F. M. J., Huang, J., Kasten, M., Schaake, S., Petkovic, S., et al. (2018). Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. 33, 1857–1870.
Uitti, R. J., Calne, D. B., Dickson, D. W., and Wszolek, Z. K. (2004). Is the neuropathological ‘gold standard’ diagnosis dead? Implications of clinicopathological findings in an autosomal dominant neurodegenerative disorder. Parkinsonism Relat. Disord. 10, 461–463. doi: 10.1016/j.parkreldis.2004.10.001
Vinagre-Aragón, A., Campo-Caballero, D., Mondragón-Rezola, E., Pardina-Vilella, L., Hernandez Eguiazu, H., Gorostidi, A., et al. (2021). A More homogeneous phenotype in Parkinson’s disease related to R1441G Mutation in the LRRK2 Gene. Front. Neurol. 12:635396. doi: 10.3389/fneur.2021.635396
Visanji, N. P., Ghani, M., Yu, E., Kakhki, E. G., Sato, C., Moreno, D., et al. (2022). Axial impairment following deep brain stimulation in Parkinson’s disease: A surgicogenomic approach. J. Parkinsons Dis. 12, 117–128. doi: 10.3233/jpd-212730
Weintraub, D., Posavi, M., Fontanillas, P., Tropea, T. F., Mamikonyan, E., Suh, E., et al. (2022). Genetic prediction of impulse control disorders in Parkinson’s disease. Ann. Clin. Transl. Neurol. 9, 936–949. doi: 10.1002/acn3.51569
Williams-Gray, C. H., Wijeyekoon, R., Yarnall, A. J., Lawson, R. A., Breen, D. P., Evans, J. R., et al. (2016). Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 31, 995–1003. doi: 10.1002/mds.26563
Wittke, C., Petkovic, S., Dobricic, V., Schaake, S., Respondek, G., Weissbach, A., et al. (2021). Genotype-phenotype relations for the atypical Parkinsonism genes: MDSGene systematic review. Mov. Disord. 36, 1499–1510. doi: 10.1002/mds.28517
Zainal Abidin, S., Tan, E. L., Chan, S. C., Jaafar, A., Lee, A. X., Abd Hamid, M. H., et al. (2015). DRD and GRIN2B polymorphisms and their association with the development of impulse control behaviour among Malaysian Parkinson’s disease patients. BMC Neurol. 15:59. doi: 10.1186/s12883-015-0316-2
Zetusky, W. J., Jankovic, J., and Pirozzolo, F. J. (1985). The heterogeneity of Parkinson’s disease: Clinical and prognostic implications. Neurology 35, 522–526. doi: 10.1212/wnl.35.4.522
Zhang, X., Chou, J., Liang, J., Xiao, C., Zhao, Y., Sarva, H., et al. (2019). Data-Driven Subtyping of Parkinson’s disease using longitudinal clinical records: A cohort study. Sci. Rep. 9:797. doi: 10.1038/s41598-018-37545-z
Keywords: genotype, phenotype, monogenic, oligogenic and polygenic inheritance, heterogeneity, genome-wide association study (GWAS), PD
Citation: Dulski J, Uitti RJ, Ross OA and Wszolek ZK (2022) Genetic architecture of Parkinson’s disease subtypes – Review of the literature. Front. Aging Neurosci. 14:1023574. doi: 10.3389/fnagi.2022.1023574
Received: 19 August 2022; Accepted: 03 October 2022;
Published: 20 October 2022.
Edited by:
R. M. Damian Holsinger, The University of Sydney, AustraliaReviewed by:
Simona Petrucci, Sapienza University of Rome, ItalyQian Xu, Xiangya Hospital, Central South University, China
Copyright © 2022 Dulski, Uitti, Ross and Wszolek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zbigniew K. Wszolek, V3N6b2xlay5aYmlnbmlld0BtYXlvLmVkdQ==