 Bei Huang
Bei Huang Xiaoling Lang
Xiaoling Lang Xihong Li1,2*
Xihong Li1,2*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 09 November 2022
Sec. Neuroinflammation and Neuropathy
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.1023161
TP53-induced glycolysis and apoptosis regulator (TIGAR) mainly regulates pentose phosphate pathway by inhibiting glycolysis, so as to synthesize ribose required by DNA, promote DNA damage repair and cell proliferation, maintain cell homeostasis and avoid body injury. Its physiological functions include anti-oxidative stress, reducing inflammation, maintaining mitochondrial function, inhibiting apoptosis, reducing autophagy etc. This paper reviews the research of TIGAR in neurological diseases, including stroke, Parkinson’s disease (PD), Alzheimer’s disease (AD), seizures and brain tumors, aiming to provide reference for the development of new therapeutic targets.
The TP53-induced glycolysis and apoptosis regulator (TIGAR), first reported in 2006 (Bensaad et al., 2006; Green and Chipuk, 2006), is a downstream target gene of p53 and an important factor involved in glycolysis and apoptosis (Hu et al., 2019b). It is indispensable in metabolism and is involved in metabolic syndrome, including hyperglycemia, insulin resistance, alcoholic fatty liver and tissue ischemia (Bensaad et al., 2006; Geng et al., 2018).
TIGAR is highly expressed in many cancer cells to promote cell survival (Bensaad et al., 2006, 2009; Xie et al., 2014; Yu et al., 2015). This may be related to its role in reducing reactive oxygen species (ROS), reducing DNA damage and improving DNA repair in these cells (Tang et al., 2021). TIGAR can aggravate myocardial ischemia injury and heart failure in animal and cell models (Kimata et al., 2010; Hoshino et al., 2012; Tang et al., 2021). In lung epithelial cells, PM2.5 exposure induces LKB1/p53/TIGAR dependent autophagy. TIGAR triggers autophagy, upregulates vascular endothelial growth factor (VEGF) transcription and protein synthesis, thereby aggravating chronic lung inflammation (Xu et al., 2019). TIGAR inhibits excessive autophagy and the degradation of tight junction proteins by promoting the flux of pentose phosphate pathway (PPP) and the production of nicotinamide adenine dinucleotide phosphate (NADPH), and then protects the structural integrity of tight junctions in cerebral microvascular endothelial cells (Wang et al., 2017a). Fructose-2,6-diphosphate (Fru-2,6-P2) can promote glycolysis and inhibit gluconeogenesis, normalizing the blood glucose level of diabetes animals (Monge et al., 1986; Rodríguez-Gil et al., 1991), while TIGAR can reduce Fru-2,6-P2 level and inhibit glycolysis, which may lead to elevated blood glucose and insulin resistance (Derdak et al., 2011). In 2015, the protective effect of TIGAR on cerebral ischemia–reperfusion injury and brain preconditioning was first reported (Zhou et al., 2016).
At present, the research on TIGAR in nervous system diseases is relatively rare, but targeting it may be a valuable strategy. TIGAR can increase the survival rate of stroke animals, improve motor function, and promote the recovery of cognitive function (Li et al., 2014). Because the oxidative damage of dopaminergic nigrostriatal neuronsis important in Parkinson’s disease (PD) pathogenesis, and TIGAR is significantly associated with neuronal glucose metabolism, PPP activity and oxidative stress, it may participate in the pathogenesis of PD (Tang et al., 2021). Decreased levels of TIGAR protein are found in different stages of Alzheimer’s disease (AD) dementia (Katsel et al., 2013). Oxidative stress and oxidative stress-induced neuronal apoptosis may be related to the occurrence and development of epilepsy, and TIGAR has antioxidant effect (Chen et al., 2019). TIGAR can also improve the survival rate of glioma cells under hypoxia (Wanka et al., 2012). This paper reviews researches on TIGAR in nervous system diseases, including stroke, PD, AD, seizures and brain tumors, and summarizes the potential role and mechanism of TIGAR, aiming to provide reference for the development of new therapeutic targets.
TIGAR gene is located on chromosome 12p13-3 and contains 6 exons and two p53 binding sites (Bensaad et al., 2006). One site is upstream of the first exon and the other is in the first intron (Bensaad et al., 2006). Transcription factors such as cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) (Zou et al., 2013), SP1 (Sun et al., 2015), p53 (Wang et al., 2016), hypoxia-inducible factor-1α (HIF-1α) (Geng et al., 2018), hormones (Sun et al., 2015) and MicroRNA (Chen et al., 2015; Xu et al., 2017) can regulate the expression of TIGAR. (Table 1).

Table 1. Factors regulating the expression of TIGAR.
TIGAR protein is mainly found in the endoplasmic reticulum, cytoplasm, matrix and mitochondrial membrane of brain tissues (Geng et al., 2018). The comparison of the predicted amino acid sequences of TIGAR protein shows that it is highly conservative in vertebrates (Bensaad et al., 2006). TIGAR is functionally similar to the diphosphatase domain of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2) in regulating glycolysis, ROS level and cell apoptosis (Bensaad et al., 2006). TIGAR is a double phosphatase with two conserved pockets that bind phosphate molecules on the substrate by dephosphorylation (Geng et al., 2018). One pocket is located near the core histidine residues composed of His11, His183, Arg10, Arg61 and Glu89, and the other is located at the C-terminal near Arg10 and Asn217, according to the analysis of zebrafish TIGAR crystal structure (Geng et al., 2018). The two active sites of TIGAR are open, positively charged and can bind to other small molecules (Li and Jogl, 2009). TIGAR protein is abundant in brain (Li et al., 2014), heart (Okawa et al., 2019), kidney (Kim et al., 2015a), skeletal muscle (Geng et al., 2019) and a variety of cancer cells (Xie et al., 2014; Ma et al., 2017; Shen et al., 2018; Chu et al., 2020; Wang et al., 2020). Its expression level is high in olfactory bulb, cerebellum and cortex (Li et al., 2014).
Previous studies have found that TIGAR can alleviate ischemic brain injury and play a neuroprotective role through reducing oxidative stress, inflammation, mitochondrial dysfunction, apoptosis and autophagy. (Table 2).

Table 2. The physiological function of TIGAR.
Oxidative stress is characterized by excessive production of ROS and reactive nitrogen species (RNS) under harmful stimulation, leading to tissue damage and neuronal death (Kleinschnitz et al., 2010; Jiang et al., 2018). After ischemia/reperfusion (I/R), the inhibition of ROS has neuroprotective effect (Crack and Taylor, 2005). ROS is mainly generated from mitochondria (Niizuma et al., 2009) and NADPH oxidase (NOX) family-mediated reactions (Bedard and Krause, 2007),applying oxidative stimulation to proteins, DNA, lipids and organelles in cells (Rahman, 2007; Kalogeris et al., 2012). NOX is the only known enzyme family acting exclusively for ROS production (Opitz et al., 2007). It transfers electrons from NADPH to molecular oxygen and then produces superoxide (Li et al., 2014). Four rodent genes catalyzing the subunit NOX have been identified, namely Nox1, Nox2, Nox3 and Nox4 (Kleinschnitz et al., 2010). Among them, NOX1, NOX2 and NOX4 are the main subtypes of NOX in the central system (Liu et al., 2020). NOX4-mediated oxidative stress led to neuronal damage through the leakage of blood–brain barrier and neuronal apoptosis (Kleinschnitz et al., 2010). The upregulation of NOX4 expression in striatum, especially mitochondria, increased the risk of oxidative stress (Liu et al., 2021). The induction of TIGAR was earlier than that of NOX4, and the application of NADPH could increase the content of glutathione (GSH) rather than ROS (Li et al., 2014). This indicated that the increase of endogenous NADPH caused by TIGAR upregulation would not produce more ROS through NOX4 under I/R conditions (Li et al., 2014).
The antioxidant activity of TIGAR is related to its regulation of glucose metabolism, maintenance of NADPH level, and regeneration of antioxidant GSH (Cheung et al., 2016). NADPH supplementation inhibited ROS and autophagy/lysosomal pathway (Liu et al., 2020). TIGAR can be activated by p53. It activates hexokinase and intracellular glucose metabolism shifts from glycolysis to PPP, leading to reduced ROS production and autophagy (Dodson et al., 2013). Under oxidative stress, neurons can use glucose to maintain their antioxidant state through PPP (Herrero-Mendez et al., 2009). PPP provides NADPH for antioxidant stress and energy supply (Li et al., 2014). Glucose-6-phosphate dehydrogenase (G6PD) is the rate limiting enzyme in PPP (Jain et al., 2004), and therefore, the level of G6PD would also determine the flux of PPP and the generation rate of NADPH (Li et al., 2014). After the overexpression of lentivirus-mediated TIGAR gene, G6PD was upregulated and PPP flux was increased, leading to a rise in NADPH production, and vice versa (Li et al., 2014). Overexpression of TIGAR gene can reduce intracellular ROS significantly. Thus, the PPP metabolic pathway regulated by TIGAR gene may be an effective mechanism against oxidative stress. Besides, neither G6PD silencing nor TIGAR mutation without enzymatic activity abolishes the antioxidative effects of TIGAR (Liu et al., 2022). After 6 h of OGD, both exogenous and mutant TIGAR failed to rescue NADPH loss in cells, reproducing the PPP deficiency observed in ischemic mouse brains, but both wild-type and mutant TIGAR significantly reversed ROS burst in cells (Liu et al., 2022). G6PD silencing did not eliminate the antioxidant and neuroprotective effects of TIGAR in cells with 6 h of OGD (Liu et al., 2022). These indicate that TIGAR has PPP-independent antioxidant activity in prolonged cerebral ischemia.
Oxygen glucose deprivation/reoxygenation (OGD/R) or I/R induced the upregulation of inducible nitric oxide synthase (iNOS), cyclooxygenases 2 (COX2) and the releasing of proinflammatory cytokine interleukin 1 beta (IL-1β) and tumor necrosis factor-α (TNF-α) (Chen et al., 2018). TIGAR overexpression could inhibit the increase of these inflammatory factors effectively and TIGAR knockout had the opposite effect. This suggests that TIGAR may play an anti-inflammatory role by inhibiting the release of inflammatory factors and the activation of transcription factors.
Transcription factor nuclear factor kappaB (NF-κB) is the main regulator of ischemic injury severity. Reducing NF-κB activation can decrease inflammation after cerebral ischemia, and then alleviate brain injury (Dvoriantchikova et al., 2009). NF-κB promotes inflammation by regulating the expression of a series of inflammation-related genes, while TIGAR can inhibit NF-κB directly and inhibit NF-κB-mediated signals and inflammatory cytokines (Tang et al., 2018), preventing neuroinflammation (Li et al., 2021). NF-κB inhibitor protein (IκBα) is a protein inhibiting NF-κB. The level of total IκBα protein was decreased and NF-κB was increased significantly in cultured primary astrocytes during OGD/R prophase, accompanied by phosphorylation of IκBα and nuclear transmutation of NF-κB. TIGAR overexpression inhibited these changes, suggesting that TIGAR can alleviate inflammation by inhibiting the phosphorylation and degradation of IκBα and NF-κB expression (Chen et al., 2018). In addition, exogenous NADPH plays an anti-inflammatory role by inhibiting IκB kinase (IKK)/IκBα/NF-κB signal transduction (Qin et al., 2017), suggesting that TIGAR may inhibit NF-κB signal transduction by promoting NADPH production through PPP (Li et al., 2021).
The mechanism of TIGAR inhibiting neuroinflammation may be related to the clearance of ROS, because ROS is a powerful promoter of neuroinflammation (Giffard and Swanson, 2005). ROS stimulates the overexpression of proinflammatory genes such as TNF-α and IL-1β in the cerebral microvascular system, causing inflammatory responses (del Zoppo and Mabuchi, 2003; Tsai and Albers, 2015). ROS induces the activation of NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammatory bodies (Minutoli et al., 2016). ROS can activate Toll-like receptor (TLRs) and peroxisome proliferator-activated receptors-γ (PPAR-γ), and microglia are sensitive to signals from TLRs and PPAR-γ (Kim et al., 2015b), triggering a series of inflammatory responses in turn. Studies have shown that TIGAR can increase the levels of NADPH and GSH, promoting the clearance of ROS (Quaegebeur et al., 2016).
As the motility of cells, mitochondria are critical in cellular energy homeostasis and also involved in regulating the different cell death mechanisms (He et al., 2020). Mitochondrial dysfunction is one of the markers of I/R damage, leading to neuronal death (Vosler et al., 2009). Fusion and division of membrane allow the mitochondrial contents to mix to maintain integrity, and the function of damaged mitochondria can be restored via fusion with adjacent intact mitochondria (Detmer and Chan, 2007).
TIGAR affected the integrity and degradation of mitochondria in 5-8F cells (Feng et al., 2018). Reduction of mitochondrial proteins and genes involved in mitochondrial biogenesis, replication, citric acid cycle, and fatty acid oxidation was observed in TIGAR-knockout mice (Geng et al., 2019). Mitochondrial TIGAR overexpression enhanced the generation of Adenosine triphosphate (ATP), maintained mitochondrial membrane potential and reduced mitochondrial oxidative stress under hypoxia condition (Geng et al., 2019). TIGAR increases exercise endurance, promotes remodeling of skeletal muscle, and regulates mitochondrial content and function through sirtuin 1 (SIRT1)-peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) pathways; in addition, during the exhaustive exercise in mice, TIGAR was translocated to mitochondria and combined with ATP synthase F1 subunit α (ATP5A1) to protect mitochondrial function (Geng et al., 2019).
TIGAR relocates to mitochondria under hypoxia, forms a complex with hexokinase 2 (HK2) on the outer membrane of mitochondria, and regulates the activity of HK2, helping to control the ROS of mitochondria (Cheung et al., 2012). Li et al. (Li et al., 2014) confirmed the localization of TIGAR in mitochondria and observed that the mitochondrial localization of TIGAR increased under the conditions of I/R and OGD/R, thereby believing that TIGAR could protect mitochondrial function. Their results showed that the knockout of TIGAR gene further reduced mitochondrial membrane potential, increased mitochondrial ROS and the activation of caspase-3, and triggered mitochondria-mediated apoptosis of damaged neurons (Li et al., 2014). Exogenous NADPH can reverse OGD-induced mitochondrial changes, caspase-3 activation and neuronal death resulting from TIGAR gene knockout. On the contrary, the overexpression of TIGAR had a beneficial effect on mitochondria (Li et al., 2014). Therefore, TIGAR protects neurons from I/R injury by clearing intracellular ROS and maintaining mitochondrial function.
Apoptosis is a form of programmed death, which is crucial for the development and homeostasis of organisms (Birkinshaw and Czabotar, 2017). ROS is a key mediator of cellular oxidative stress and neuronal apoptosis during cerebral ischemia (Broughton et al., 2009). Its interaction with other tissue components produces a variety of other free radicals. Particularly, the interaction between O2-and NO produces highly toxic molecule peroxynitrite. This oxidant causes tissue damage and is one of the important trigger molecules of apoptosis (Wang et al., 2018). B cell lymphoma-2 (Bcl-2) family proteins are involved in the regulation of mitochondria-dependent apoptosis pathway (Gross et al., 1999). Anti-apoptotic protein Bcl-2 inhibits apoptosis via preventing mitochondrial membrane depolarization and inhibiting caspase-3 activation, while pro-apoptotic protein Bcl-2 associated protein X (Bax) promotes apoptosis via inducing mitochondrial membrane depolarization (Broughton et al., 2009). Bcl-2 participates in ischemic tolerance induced by preconditioning (Shimizu et al., 2001; Gwak et al., 2011). Isoflurane preconditioning (ISO) increases the expression of Bcl-2 to prevent the release of cytochrome c from mitochondria and inhibit neuronal apoptosis (Li et al., 2008). Bcl-2/B-cell lymphoma-extra large (Bcl-xL) may prevent the production of ROS in the process of apoptosis (Gottlieb et al., 2000). Bcl-2-deficient mice showed greater brain oxidative stress and sensitivity to oxidants than wild-type mice (Hochman et al., 1998). ROS may induce cell apoptosis through regulating Bcl-2/Bax-dependent mitochondrial apoptosis pathway (Zhou et al., 2016). ROS can also regulate Bcl-2 phosphorylation, ubiquitination, or cystine oxidation, resulting in decreased Bcl-2 expression and increased Bax expression (Li et al., 2004; Luanpitpong et al., 2013). The expression of TIGAR reduced ROS levels and thereby protected ROS-sensitive apoptotic responses (Bensaad et al., 2006). TIGAR cut down ROS-dependent apoptosis through PPP flux (Zhou et al., 2016). In TIGAR-deficient cells, ROS-dependent apoptosis were increased (Bensaad et al., 2009).
In addition, the activation of caspase is the most recognized biochemical feature in the early and late stage of apoptosis, and detection of active caspase-3 in cells and tissues is an important method to induce apoptosis by various apoptotic signals (Choudhary et al., 2015). Pretreatment of Zhou et al. (Zhou et al., 2016) inhibited the activation of caspase-3 in neurons induced by OGD/reperfusion, while TIGAR knockout increased the division of caspase-3, indicating that knockout of TIGAR gene can cancel the anti-apoptotic effect induced by pretreatment.
Autophagy is a cellular catabolic pathway that degrades and recycles proteins, damaged organelles, and misfolded proteins to maintain cell dynamic balance and normal cellular function (Wang et al., 2018). Moderate autophagy is neuroprotective, while excessive autophagy may lead to neuron cell damage in the central nervous system (CNS) (Shi et al., 2012). Autophagy impairment participates in the pathogenesis and progression of neurodegenerative diseases including PD (Zhu et al., 2022). Selective autophagy is an active mechanism in AD pathology, and regulating selective autophagy will be an effective strategy to control this pathogenesis (Guan et al., 2022). Proper PPP function and GSH levels are essential for normal autophagy (Dodson et al., 2013). The maintenance of GSH reduced form requires the participation of NADPH produced by PPP (Russell et al., 1999). TIGAR may raise GSH/oxidized glutathione (GSSG) ratio by increasing PPP flux while reducing glycolysis (Bensaad et al., 2006).
TIGAR play a neuroprotective role by inhibiting autophagy. Excessive ROS accumulation would disrupt the stable state of cells, not only causing oxidative stress, but also promoting the activation of autophagy (Bensaad et al., 2009). TIGAR affects autophagy by regulating ROS levels, and the lack of TIGAR can increase autophagy activity significantly (Bensaad et al., 2006). Severely damaged mitochondria are selectively removed by autophagy, called mitochondrial phagocytosis, to prevent apoptosis (Matsuda et al., 2010). BCL-2/adenovirus E1B 19KD interacting protein 3 (BNIP3) protein is an effective inducer of cell death and autophagy, causing a protective stress response to remove damaged mitochondria (Hamacher-Brady et al., 2007). Upregulation of TIGAR reduces BNIP3 activation by inhibiting ROS signal (Hoshino et al., 2012).
Mammalian target of rapamycin (mTOR) is the primary regulator of autophagy (Ghiglieri et al., 2010), and inhibiting mTOR-S6KP70 signaling pathway is one of the main signals to activate autophagy. Apart from reducing ROS, TIGAR also may limit autophagy activation through the mTOR-S6KP70 signaling pathway (Zhang et al., 2019). These suggest that autophagy inhibition is at least partially the basis of TIGAR’s neuroprotective mechanism in brain I/R-induced neuron injury (Zhang et al., 2019). How TIGAR regulates mTOR-S6KP70 signaling pathway remains to be further studied.
Beclin-1 protein is essential for autophagy (Xu and Qin, 2019). Microtubule-associated protein light chain 3 (LC3) is widely used to monitor autophagy, and the number of LC3-II is significantly related to that of autophagosomes (Mizushima and Yoshimori, 2007). After brain I/R in mice, autophagosomes were produced in large quantities, the expression of Beclin-1 and LC3II protein increased significantly, and p62 protein expression was significantly decreased, indicating that autophagy was activated after brain I/R (Zhang et al., 2019). After TIGAR overexpression, the expression levels of Beclin-1 and LC3II decreased significantly, and P62 expression increased, while the inhibition of TIGAR gene expression in mice had the opposite effect. Supplementation of autophagy inhibitor 3-methyladenine (3-MA) could reverse the damage caused by inhibiting TIGAR gene, indicating that the decrease of autophagy activation is related to the neuroprotective effect of TIGAR.
In the lung tissue of patients with idiopathic pulmonary fibrosis, the level of TIGAR increased, which was associated with decreased LC3, p62, autophagosomes and overall autophagy flux (Patel et al., 2012). TIGAR was activated during nutrient starvation of HeLa cells, reducing reactive species and inhibiting the induction of autophagy (Bensaad et al., 2009). Thus, regulation of TIGAR has been proposed as a target for anticancer therapy, since blocking TIGAR activity can upregulate autophagy and improves the survival of cancer cells (Dewaele et al., 2010; Figure 1).
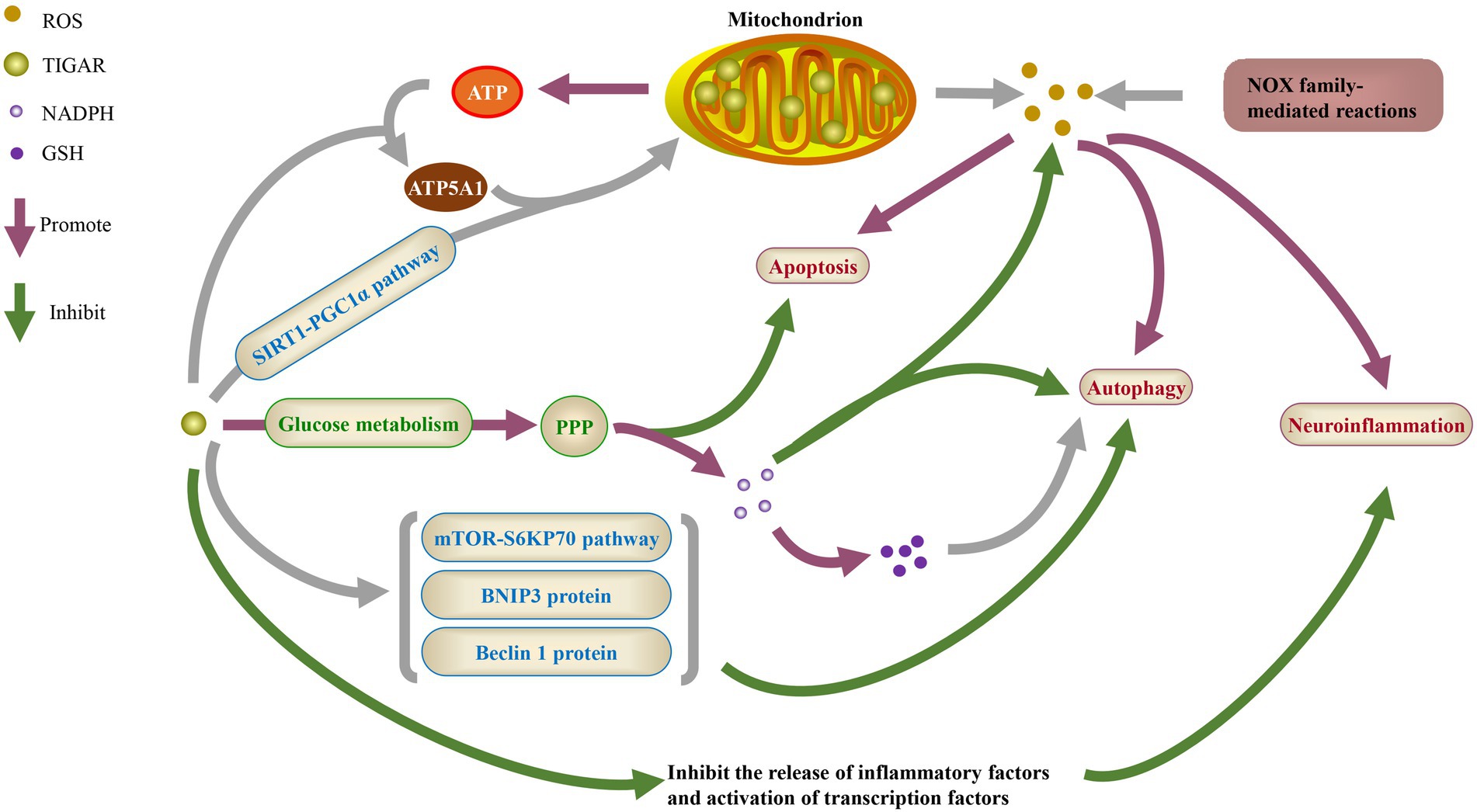
Figure 1. The physiological function of TIGAR. ROS, reactive oxygen species; NADPH, nicotinamide adenine dinucleotide phosphate; GSH, glutathione; ATP, adenosine triphosphate; ATP5A1, ATP synthase F1 subunit a; SIRT1, sirtuin 1; PGC1α, peroxisome proliferator-activated receptor γ coactivator 1α; PPP, pentose phosphate pathway; mTOR, mammalian target of rapamycin; BNIP3, BCL-2/adenovirus E1B 19KD interacting protein 3; NOX, NADPH oxidases.
TIGAR plays an important role in nervous system diseases due to its multiple physiological functions. Its mechanisms in the occurrence and development of stroke, PD, AD, seizures and brain tumors provide references for treatment.
Stroke is caused by sudden interruption of cerebral blood flow (Chan, 1996), consisting of bleeding due to ruptured blood vessels and ischemia resulting from blocked vessels (Jiang et al., 2018). About 87% of strokes are ischemic (Benjamin et al., 2019).
Neuroinflammation is generally believed to cause brain damage or repair after strokes (Famakin, 2014). Astrocytes and microglia play crucial regulatory roles in the inflammatory response to ischemic injury. In ischemic conditions, astrocytes are activated and produce pro-inflammatory mediators and cytokines, including TNF-α, IL-1β and ROS (Giffard and Swanson, 2005; Iadecola and Anrather, 2011). In some cases, regulated astrocyte response may help repair nerve injury (Rossi et al., 2007). However, dysregulated astrocyte response or high levels of pro-inflammatory cytokines can be harmful to ischemic recovery by increasing the level of toxic nitric oxide and/or inducing neuronal apoptosis directly (Giffard and Swanson, 2005; Gelderblom et al., 2009). TIGAR can play an anti-inflammatory role by inhibiting the release of inflammatory factors and the activation of transcription factors (Chen et al., 2018).
NADPH, a metabolite of PPP, protects neurons from I/R-induced damage in rodent and primate stroke models (Li et al., 2016). GSH is the main source of sulfhydryl groups in most living cells and is of vital importance in maintaining the redox state of appropriate protein thiols, while NADPH can maintain the level of reduced GSH (Bensaad et al., 2006; Gu et al., 2015). ROS is a key mediator of cellular oxidative stress and neuronal apoptosis during cerebral ischemia (Broughton et al., 2009). I/R increased the production of ROS, applying oxidative stress to proteins, DNA, lipids and organelles in cells (Rahman, 2007; Kalogeris et al., 2012). TIGAR is relatively abundant in brain neurons and responds rapidly to I/R injury. The overexpression of TIGAR reduced OGD/R-induced astrocyte damage significantly, while TIGAR gene knockout aggravated such damage (Chen et al., 2018). The overexpression of adenovirus-mediated TIGAR not only reduced infarct volume, but also inhibited the decline of glial fibrillary acidic protein (GFAP) 24 h after reperfusion, reduced long-term mortality significantly and improved the recovery of neurological function in vivo, while TIGAR knockout had the opposite effect (Chen et al., 2018). TIGAR inhibit glycolysis and improve PPP as compensation, resulting in increased NADPH production, which helps to limit ROS (Bensaad et al., 2009; Cheung et al., 2012; Yin et al., 2014).
In animal and cellular stroke models, I/R increased mitochondrial localization of TIGAR (Li et al., 2014). TIGAR overexpression or NADPH supplementation saved OGD/R-induced ROS increase, GSH decrease, mitochondrial dysfunction and caspase-3 activation, which were exacerbated by TIGAR knockout (Li et al., 2014). The expression change and expression level of TIGAR during development may be related to the susceptibility of neurons to ischemic injury (Cao et al., 2015). Tripartite motif (TRIM) 31 is a new E3 ubiquitin ligase of TIGAR. TRIM31 deficiency alleviates mitochondrial damage induced by cerebral ischemia by upregulating TIGAR protein level (Zeng et al., 2021). TIGAR protects against ischemic brain injury by enhancing PPP flux and maintaining mitochondrial function, so it may be a valuable therapeutic target for ischemic brain injury (Li et al., 2014). Besides, TIGAR participates in ischemic preconditioning by clearing ROS and inhibiting neuronal apoptosis (Zhou et al., 2016). The combination of TIGAR and cerebral ischemic preconditioning also may be a new target for stroke prevention.
A key pathological feature of PD is the loss of dopaminergic (DA) neurons in substantia nigra (López et al., 2019). The lack of TIGAR protein in the pathological inclusion bodies of motor neurone disease (MND) or multiple system atrophy (MSA) indicates the specificity of disease, further enhancing the possibility that TIGAR may participate in the pathogenesis of PD (López et al., 2019). Apart from that, overexpression of mitochondrial HK2 conferred neuronal protection and survival in animal and cell models of PD and neurodegeneration (Gimenez-Cassina et al., 2009; Corona et al., 2010). Further studies are needed to elucidate TIGAR’s role in the pathogenesis of PD and figure out whether it is associated with hexokinase expression.
In the brain samples of PD patients, the progressive loss of DA neurons in the substantia nigra pars compacta (SNpc) is partly attributed to the defects in autophagy flux (Zatloukal et al., 2002; Tanji et al., 2011). Lack of DA neurons is associated with lysosomal dysfunction in PD (Wan et al., 2017). Reduced fusion between autophagosome and lysosome or reduced proteolysis in lysosomes leads to autophagy damage (Ge et al., 2021). Lysosome loss may cause PD, so restoring the function of lysosome seems to be a viable neuroprotective strategy for PD (Ge et al., 2021). Studies have shown that oxidative neurotoxin aroused lysosomal membrane permeabilization (LMP) in PD rodent model (Dehay et al., 2010; Wu et al., 2015), and selective autophagy is one of the protective processes concerning the clearance of damaged lysosomes (Papadopoulos et al., 2017). SP1-induced TIGAR could save the neurotoxin 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced lysosome dysfunction, avoiding the damage of DA neurons in PD (Ge et al., 2021). This identified that TIGAR-mediated lysosome repair may be a potential intervention for neurodegenerative diseases such as PD.
MPTP contributes to PD (Langston, 2017). Exogenous NADPH can prevent the toxicity of MPTP to dopaminergic neurons by increasing GSH, reducing ROS and inhibiting the neuroinflammation of substantia nigra compacta in mice (Zhou et al., 2018). Overexpression of G6PD in mouse dopaminergic neurons shows a protective effect against MPTP toxicity (Mejías et al., 2006). TIGAR may be a potential therapeutic target for PD by increasing the production of NADPH and G6PD.
AD is characterized by amyloid-β (Aβ) plaques and hyperphosphorylated Tau-associated neurofibrillary tangles in the brain (Durairajan et al., 2022; Iyaswamy et al., 2022). Oxidative stress, caused by the imbalance between ROS production and accumulation, is the main cause of Aβ-induced neurotoxicity in AD (Wang et al., 2022).
Studies have assessed TIGAR mRNA (via microarray) and protein levels in tissue homogenates of the superior temporal gyrus in patients with AD and control group after death (Katsel et al., 2013). Microarray data and protein levels assessed by westernblotting showed a decrease in TIGAR in AD patients compared with the control group.
Ataxia-telangiectasia mutated (ATM) can act as a functional component and effector of cell redox sensing (Cosentino et al., 2011). ATM and its downstream effector p53 are involved in tumor inhibition and the regulation of neural cell cycle (CCL), oxidative phosphorylation and glycolysis rate (Katsel et al., 2013). TIGAR protein levels decreased at different stages of AD dementia severity, indicating a diminished role of ATM-p53 signaling in countering glycolysis/oxygen-phosphorus-induced cell death (Katsel et al., 2013). TP53 activates TIGAR at low pressure (Madan et al., 2012). However, after long-term exposure to stress environment and induced TP53-mediated apoptosis, the expression of TIGAR decreased, suggesting that the induced apoptosis might reflect the loss of protection of survival signal induced by TP53 (Bensaad et al., 2006). Therefore, TIGAR may play a key role in the transformation of TP53-induced stress response, and its decreased expression may negatively affect cell survival during the progression of dementia (Katsel et al., 2013).
Hippocampal neurons are very important for the occurrence and development of AD (Hu et al., 2019a). AD is highly associated with neuroinflammation and oxidative stress in the brain leading to neuronal loss. The pro-inflammatory and redox-sensitive transcription factor NF-κB acts essentially in AD (Ju Hwang et al., 2019). MiR-146a-5pis deregulates in the peripheral blood of patients with AD and may serve as a biomarker for the diagnosis or treatment of AD (Gupta et al., 2017; Fransquet and Ryan, 2018). It is also involved in the progression from mild cognitive impairment to AD (Ansari et al., 2019). In AD patients, NF-κBis positively correlated with the expression level of miR-146a-5p, but there is a negative correlation between NF-κB mRNA and TIGAR mRNA, and between miR-146a-5p and TIGAR mRNA (Lei et al., 2021). NF-κB-induced upregulation of miR-146a-5p promoted oxidative stress and pyroptosis in hippocampal neuronal models of AD via TIGAR (Lei et al., 2021), which may be instructive to explore new therapeutic targets.
Oxidative stress in the brain can lead to neuronal apoptosis, leading to neurological diseases such as epilepsy thereby (Méndez-Armenta et al., 2014). Oxidative stress itself is the result of the first seizure and can trigger subsequent seizures (Patel, 2004; Puttachary et al., 2015). A growing body of evidence shows that overproduction of ROS in neurons disrupts intracellular Ca2+ homeostasis and leads to overload of calcium, causing neuronal hyperexcitation and the release of pro-apoptotic factors (Aguiar et al., 2012; Puttachary et al., 2015). The imbalance between ROS production and degradation would trigger oxidative stress, resulting in high ROS levels, which in turn damage macromolecules and cell membranes (Davies, 2000; Chrissobolis and Faraci, 2008).
In kainic acid (KA)-treated rats, TIGAR upregulation in cortex and hippocampus was accompanied by increased ROS levels, decreased antioxidant indices and increased pro-apoptotic factors. Increased TIGAR expression may be a feedback mechanism that promotes NADPH production and inhibits oxidative stress after seizures (Chen et al., 2019). TIGAR inhibits KA-induced seizures via inhibiting oxidative stress and neuronal apoptosis (Chen et al., 2019). Lentivirus-mediated TIGAR overexpression reduced KA-induced oxidative stress and neuronal apoptosis significantly, resulting in prolonged seizure latency, suggesting that TIGAR has anti-epileptic, antioxidant and anti-apoptotic effects, and therefore is a promising therapeutic target for seizures (Chen et al., 2019).
Glioblastoma multiforme (GBM), the most aggressive malignant brain tumor, is characterized by an imbalance between the growth of glioblastoma cells and glucose metabolism (Wang et al., 2017b). At present, it is highly resistant to treatment, including radiotherapy (Sinha et al., 2013). NFκB can be activated by ionizing radiation (IR) (Brach et al., 1991), while radiation-induced NFκB mediates radiation resistance in glioma cells by defending against oxidative stress (Iwanaga et al., 1998). The resistance to tumor necrosis factor-alpha (TNFα)-induced apoptosis of some tumors (Tsujimoto et al., 1985) is attributed to TNFα-mediated activation of NFκB (Beg and Baltimore, 1996; Wang et al., 1996). The radiation-induced cross effect of TNFα-NFκB promotes the survival of neuroblastoma cells (Veeraraghavan et al., 2011). NFκB enhances metabolic adaptation in cancer (Mauro et al., 2011), and P53 blocks activation of NFκB by inhibiting glycolysis (Kawauchi et al., 2008). P53 induction in these cells is regulated by TIGAR (Sinha et al., 2013). ROS sensitize glioma cells to chemotherapeutic agents (Sharma et al., 2007; Dixit et al., 2009), while TIGAR protects cells from ROS-related apoptosis (Sinha et al., 2013).
Gliomas, mainly composed of astrocytomas and oligodendrogliomas, are one of the most deadly cancers in the CNS (Giese et al., 2003). Haapasalo et al. (Haapasalo et al., 2003) analyzed 433 cases of astrocytoma and showed that the expression of thioredoxin reductase-1 (TrxR1) was upregulated in more than 66% of cases, which was significantly associated with relatively high proliferative activity and poor prognosis. Another study showed that immune reactivity of TrxR1 was observed in more than 50% of recurrent oligodendrogliomas, 1.5 times that of primary oligodendrogliomas, indicating that TrxR1 acts positively in the malignant progression of oligodendrogliomas (Järvelä et al., 2006). TrxR1 overexpresses in more than half of glioma patients (Haapasalo et al., 2003; Järvelä et al., 2006), so there is an urgent need for effective treatment of gliomas with TrxR1 overexpression. Overexpression of TrxR1 reduces the radiosensitivity of glioma cells significantly. Knockout of TIGAR gene can radio-sensitize TrxR1-overexpressed gliomas through inhibiting the nuclear transport of IR-induced thioredoxin-1 (Trx1) (Zhang et al., 2017). This suggests that elimination of TIGAR may be a strategy to radio-sensitize TrxR1-overexpressed gliomas.
TIGAR overexpressed in glioblastoma, and ectopic expression of TIGAR reduced cell death induced by glucose and oxygen restriction (Wanka et al., 2012). Metabolic analysis showed that TIGAR inhibited glycolysis and promoted respiration. Glioma cells benefit from TIGAR expression through raising energy production of glucose and enhancing defense mechanism against ROS via increasing respiration. This indicates that TIGAR is a major metabolic regulator, which can improve the survival rate of glioma cells under hypoxia (Wanka et al., 2012). Therefore, targeting metabolic regulators such as TIGAR may be a valuable strategy (Table 3).

Table 3. The role of TIGAR in nervous system diseases.
TIGAR plays an important role in antioxidant stress, reducing inflammation, maintaining mitochondrial function, inhibiting apoptosis, and reducing autophagy. Existing studies have shown that TIGAR may be a valuable therapeutic strategy in nervous system diseases. The combination of TIGAR and cerebral ischemic preconditioning probably become a new target for stroke prevention and treatment. TIGAR-mediated lysosomal repair also may be a potential intervention method for neurodegenerative diseases such as PD. The decrease of TIGAR expression may have a negative impact on cell survival during the progression of dementia. The increase of TIGAR expression is possibly a feedback mechanism to promote NADPH production and inhibit oxidative stress after epilepsy. TIGAR improve the survival rate of glioma cells under hypoxia.
At present, the research on TIGAR in nervous system diseases is relatively rare. There still remain many problems that need to be further clarified. The distribution of TIGAR in organelles, the regulatory mechanism of its expression, and the proteins interacting with TIGAR still need to be further studied. The regulatory mechanism of TIGAR repositioning in response to stress in the endoplasmic reticulum and nucleus is still unclear, which is worthy of further study. TIGAR’s ability to regulate ROS levels also plays a crucial role in autophagy, but the specific mechanism needs to be further explored. Overexpression of TIGAR inhibited autophagy activation and reduced ischemic brain damage in the one-hour/two-hour occlusion model. Further studies are needed to investigate the dynamic changes of autophagy and its role in ischemic stroke. Overexpression of HK2 can protect neurons in PD model. Further researches are expected to clarify the role of TIGAR in the pathogenesis of PD and figure out whether it is related to the expression of hexokinase. The molecular mechanism of oxidative stress and apoptosis in AD hippocampal neurons may have certain guiding significance for exploring new therapeutic targets, such as the overexpression of TIGAR, inhibition of oxidative stress and the knockdown of NF-κB or miR-146a-5p. The protective effect of TIGAR on seizures is not fully understood. Clinical application of lentivirus-mediated promotion of TIGAR overexpression still needs to be explored. Further studies are needed to elucidate the complex role and mechanism of TIGAR in the occurrence, development and treatment resistance of brain tumors. Although the treatment of nervous system diseases targeting TIGAR has great application potential, it is unknown that whether promoting its overexpression would affect other tissues and organs or not, because TIGAR is expressed in many tissues and organs throughout the body. The complex role of TIGAR in nervous system diseases and its neuroprotective effects through different mechanisms need to be further revealed, including potential agonists, inhibitors and related pathways, so as to provide references for the research of TIGAR-targeted drugs.
BH drafted manuscript and prepared tables. XLi and XLa edited and revised manuscript. BH, XLa, and XLi approved final version of manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by The National Natural Science Foundation of China (82071353 to XLi); The National Key Research and Development Program of China (2017YFA 0104201 to XLi) and Key Research and Development Projects of Sichuan Province in China (2021YFS0029 to XLi).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aguiar, C. C., Almeida, A. B., Araújo, P. V., De Abreu, R. N., Chaves, E. M., Do Vale, O. C., et al. (2012). Oxidative stress and epilepsy: literature review. Oxidative Med. Cell. Longev. 2012:795259. doi: 10.1155/2012/795259
Ansari, A., Maffioletti, E., Milanesi, E., Marizzoni, M., Frisoni, G. B., Blin, O., et al. (2019). Mir-146a and Mir-181a are involved in the progression of mild cognitive impairment to Alzheimer's disease. Neurobiol. Aging 82, 102–109. doi: 10.1016/j.neurobiolaging.2019.06.005
Bedard, K., and Krause, K. H. (2007). The Nox Family of Ros-generating Nadph oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Beg, A. A., and Baltimore, D. (1996). An essential role for Nf-Kappab in preventing Tnf-alpha-induced cell death. Science 274, 782–784. doi: 10.1126/science.274.5288.782
Benjamin, E. J., Muntner, P., Alonso, A., Bittencourt, M. S., Callaway, C. W., Carson, A. P., et al. (2019). Heart disease and stroke Statistics-2019 update: a report from the American Heart Association. Circulation 139, E56–E528. doi: 10.1161/CIR.0000000000000659
Bensaad, K., Cheung, E. C., and Vousden, K. H. (2009). Modulation of intracellular Ros levels by Tigar controls autophagy. EMBO J. 28, 3015–3026. doi: 10.1038/emboj.2009.242
Bensaad, K., Tsuruta, A., Selak, M. A., Vidal, M. N., Nakano, K., Bartrons, R., et al. (2006). Tigar, a P53-inducible regulator of glycolysis and apoptosis. Cells 126, 107–120. doi: 10.1016/j.cell.2006.05.036
Birkinshaw, R. W., and Czabotar, P. E. (2017). The Bcl-2 family of proteins and mitochondrial outer membrane Permeabilisation. Semin. Cell Dev. Biol. 72, 152–162. doi: 10.1016/j.semcdb.2017.04.001
Brach, M. A., Hass, R., Sherman, M. L., Gunji, H., Weichselbaum, R., and Kufe, D. (1991). Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J. Clin. Invest. 88, 691–695. doi: 10.1172/JCI115354
Broughton, B. R., Reutens, D. C., and Sobey, C. G. (2009). Apoptotic mechanisms after cerebral ischemia. Stroke 40, E331–E339. doi: 10.1161/STROKEAHA.108.531632
Cao, L., Chen, J., Li, M., Qin, Y. Y., Sun, M., Sheng, R., et al. (2015). Endogenous level of Tigar in brain is associated with vulnerability of neurons to ischemic injury. Neurosci. Bull. 31, 527–540. doi: 10.1007/s12264-015-1538-4
Chan, P. H. (1996). Role of oxidants in ischemic brain damage. Stroke 27, 1124–1129. doi: 10.1161/01.STR.27.6.1124
Chen, S., Li, P., Li, J., Wang, Y., Du, Y., Chen, X., et al. (2015). Mir-144 inhibits proliferation and induces apoptosis and autophagy in lung cancer cells by targeting Tigar. Cell. Physiol. Biochem. 35, 997–1007. doi: 10.1159/000369755
Chen, C., Mei, Q., Wang, L., Feng, X., Tao, X., Qiu, C., et al. (2019). Tigar suppresses seizures induced by Kainic acid through inhibiting oxidative stress and neuronal apoptosis. Biochem. Biophys. Res. Commun. 515, 436–441. doi: 10.1016/j.bbrc.2019.05.156
Chen, J., Zhang, D. M., Feng, X., Wang, J., Qin, Y. Y., Zhang, T., et al. (2018). Tigar inhibits ischemia/reperfusion-induced inflammatory response of astrocytes. Neuropharmacology 131, 377–388. doi: 10.1016/j.neuropharm.2018.01.012
Cheung, E. C., Lee, P., Ceteci, F., Nixon, C., Blyth, K., Sansom, O. J., et al. (2016). Opposing effects of Tigar-and Rac1-derived Ros on Wnt-driven proliferation in the mouse intestine. Genes Dev. 30, 52–63. doi: 10.1101/gad.271130.115
Cheung, E. C., Ludwig, R. L., and Vousden, K. H. (2012). Mitochondrial localization of Tigar under hypoxia stimulates Hk2 and lowers Ros and cell death. Proc. Natl. Acad. Sci. U. S. A. 109, 20491–20496. doi: 10.1073/pnas.1206530109
Choudhary, G. S., Al-Harbi, S., and Almasan, A. (2015). Caspase-3 activation is a critical determinant of Genotoxic stress-induced apoptosis. Methods Mol. Biol. 1219, 1–9. doi: 10.1007/978-1-4939-1661-0_1
Chrissobolis, S., and Faraci, F. M. (2008). The role of oxidative stress and Nadph oxidase in cerebrovascular disease. Trends Mol. Med. 14, 495–502. doi: 10.1016/j.molmed.2008.09.003
Chu, J., Niu, X., Chang, J., Shao, M., Peng, L., Xi, Y., et al. (2020). Metabolic remodeling by Tigar overexpression is a therapeutic target in esophageal squamous-cell carcinoma. Theranostics 10, 3488–3502. doi: 10.7150/thno.41427
Corona, J. C., Gimenez-Cassina, A., Lim, F., and Díaz-Nido, J. (2010). Hexokinase ii gene transfer protects against Neurodegeneration in the rotenone and Mptp mouse models of Parkinson's disease. J. Neurosci. Res. 88, 1943–1950. doi: 10.1002/jnr.22357
Cosentino, C., Grieco, D., and Costanzo, V. (2011). Atm activates the pentose phosphate pathway promoting anti-oxidant Defence and DNA repair. EMBO J. 30, 546–555. doi: 10.1038/emboj.2010.330
Crack, P. J., and Taylor, J. M. (2005). Reactive oxygen species and the modulation of stroke. Free Radic. Biol. Med. 38, 1433–1444. doi: 10.1016/j.freeradbiomed.2005.01.019
Davies, K. J. (2000). Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 50, 279–289. doi: 10.1080/15216540051081010
Dehay, B., Bové, J., Rodríguez-Muela, N., Perier, C., Recasens, A., Boya, P., et al. (2010). Pathogenic Lysosomal depletion in Parkinson's disease. J. Neurosci. 30, 12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010
Del Zoppo, G. J., and Mabuchi, T. (2003). Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab. 23, 879–894. doi: 10.1097/01.WCB.0000078322.96027.78
Derdak, Z., Lang, C. H., Villegas, K. A., Tong, M., Mark, N. M., De La Monte, S. M., et al. (2011). Activation of P53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J. Hepatol. 54, 164–172. doi: 10.1016/j.jhep.2010.08.007
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879. doi: 10.1038/nrm2275
Dewaele, M., Maes, H., and Agostinis, P. (2010). Ros-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 6, 838–854. doi: 10.4161/auto.6.7.12113
Dixit, D., Sharma, V., Ghosh, S., Koul, N., Mishra, P. K., and Sen, E. (2009). Manumycin inhibits Stat3, telomerase activity, and growth of Glioma cells by elevating intracellular reactive oxygen species generation. Free Radic. Biol. Med. 47, 364–374. doi: 10.1016/j.freeradbiomed.2009.04.031
Dodson, M., Darley-Usmar, V., and Zhang, J. (2013). Cellular metabolic and Autophagic pathways: traffic control by redox signaling. Free Radic. Biol. Med. 63, 207–221. doi: 10.1016/j.freeradbiomed.2013.05.014
Durairajan, S. S. K., Selvarasu, K., Bera, M. R., Rajaram, K., Iyaswamy, A., and Li, M. (2022). Alzheimer's disease and other Tauopathies: exploring efficacy of medicinal plant-derived compounds in alleviating tau-mediated Neurodegeneration. Curr. Mol. Pharmacol. 15, 361–379. doi: 10.2174/1874467214666210906125318
Dvoriantchikova, G., Barakat, D., Brambilla, R., Agudelo, C., Hernandez, E., Bethea, J. R., et al. (2009). Inactivation of Astroglial Nf-kappa B promotes survival of retinal neurons following ischemic injury. Eur. J. Neurosci. 30, 175–185. doi: 10.1111/j.1460-9568.2009.06814.x
Famakin, B. M. (2014). The immune response to acute focal cerebral ischemia and associated post-stroke Immunodepression: a focused review. Aging Dis. 5, 307–326. doi: 10.14336/AD.2014.0500307
Feng, J., Luo, L., Liu, Y., Fu, S., Chen, J., Duan, X., et al. (2018). Tp53-induced glycolysis and apoptosis regulator is indispensable for mitochondria quality control and degradation following damage. Oncol. Lett. 15, 155–160. doi: 10.3892/ol.2017.7303
Fransquet, P. D., and Ryan, J. (2018). Micro Rna as a potential blood-based epigenetic biomarker for Alzheimer's disease. Clin. Biochem. 58, 5–14. doi: 10.1016/j.clinbiochem.2018.05.020
Ge, J., Lin, H., Yang, J., Li, Q., Zhou, J., Qin, Z., et al. (2021). Tp53-induced glycolysis and apoptosis regulator (Tigar) ameliorates Lysosomal damage in the 1-Methyl-4-Phenyl-1, 2, 3, 6-Tetrahydropyridine-mediated mouse model of Parkinson's disease. Toxicol. Lett. 339, 60–69. doi: 10.1016/j.toxlet.2020.12.011
Gelderblom, M., Leypoldt, F., Steinbach, K., Behrens, D., Choe, C. U., Siler, D. A., et al. (2009). Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40, 1849–1857. doi: 10.1161/STROKEAHA.108.534503
Geng, J., Wei, M., Yuan, X., Liu, Z., Wang, X., Zhang, D., et al. (2019). Tigar regulates mitochondrial functions through Sirt1-Pgc1α pathway and translocation of Tigar into mitochondria in skeletal muscle. FASEB J. 33, 6082–6098. doi: 10.1096/fj.201802209R
Geng, J., Yuan, X., Wei, M., Wu, J., and Qin, Z. H. (2018). The diverse role of Tigar in cellular homeostasis and cancer. Free Radic. Res. 52, 1240–1249. doi: 10.1080/10715762.2018.1489133
Ghiglieri, V., Pendolino, V., Bagetta, V., Sgobio, C., Calabresi, P., and Picconi, B. (2010). Mtor inhibitor Rapamycin suppresses striatal post-ischemic Ltp. Exp. Neurol. 226, 328–331. doi: 10.1016/j.expneurol.2010.09.012
Giese, A., Bjerkvig, R., Berens, M. E., and Westphal, M. (2003). Cost of migration: invasion of malignant Gliomas and implications for treatment. J. Clin. Oncol. 21, 1624–1636. doi: 10.1200/JCO.2003.05.063
Giffard, R. G., and Swanson, R. A. (2005). Ischemia-induced programmed cell death in astrocytes. Glia 50, 299–306. doi: 10.1002/glia.20167
Gimenez-Cassina, A., Lim, F., Cerrato, T., Palomo, G. M., and Diaz-Nido, J. (2009). Mitochondrial hexokinase ii promotes neuronal survival and acts downstream of glycogen synthase Kinase-3. J. Biol. Chem. 284, 3001–3011. doi: 10.1074/jbc.M808698200
Gottlieb, E., Vander Heiden, M. G., and Thompson, C. B. (2000). Bcl-X(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol. Cell. Biol. 20, 5680–5689. doi: 10.1128/MCB.20.15.5680-5689.2000
Green, D. R., and Chipuk, J. E. (2006). P53 and metabolism: inside the Tigar. Cells 126, 30–32. doi: 10.1016/j.cell.2006.06.032
Gross, A., Mcdonnell, J. M., and Korsmeyer, S. J. (1999). Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911. doi: 10.1101/gad.13.15.1899
Gu, F., Chauhan, V., and Chauhan, A. (2015). Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 18, 89–95. doi: 10.1097/MCO.0000000000000134
Guan, X., Iyaswamy, A., Sreenivasmurthy, S. G., Su, C., Zhu, Z., Liu, J., et al. (2022). Mechanistic insights into selective autophagy subtypes in Alzheimer's disease. Int. J. Mol. Sci. 23: 25–3609. doi: 10.3390/ijms23073609
Gupta, P., Bhattacharjee, S., Sharma, A. R., Sharma, G., Lee, S. S., and Chakraborty, C. (2017). Mirnas in Alzheimer disease: a therapeutic perspective. Curr. Alzheimer Res. 14, 1198–1206. doi: 10.2174/1567205014666170829101016
Gwak, M. S., Cao, L., Li, L., and Zuo, Z. (2011). Isoflurane preconditioning reduces oxygen-glucose deprivation-induced neuronal injury via B-cell lymphoma 2 protein. Environ. Toxicol. Pharmacol. 31, 262–265. doi: 10.1016/j.etap.2010.10.009
Haapasalo, H., Kyläniemi, M., Paunul, N., Kinnula, V. L., and Soini, Y. (2003). Expression of antioxidant enzymes in Astrocytic brain tumors. Brain Pathol. 13, 155–164. doi: 10.1111/j.1750-3639.2003.tb00015.x
Hamacher-Brady, A., Brady, N. R., Logue, S. E., Sayen, M. R., Jinno, M., Kirshenbaum, L. A., et al. (2007). Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 14, 146–157. doi: 10.1038/sj.cdd.4401936
He, Z., Ning, N., Zhou, Q., Khoshnam, S. E., and Farzaneh, M. (2020). Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med. 146, 45–58. doi: 10.1016/j.freeradbiomed.2019.11.005
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S., and Bolaños, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by Apc/C-Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Hochman, A., Sternin, H., Gorodin, S., Korsmeyer, S., Ziv, I., Melamed, E., et al. (1998). Enhanced oxidative stress and altered antioxidants in brains of Bcl-2-deficient mice. J. Neurochem. 71, 741–748.
Hoshino, A., Matoba, S., Iwai-Kanai, E., Nakamura, H., Kimata, M., Nakaoka, M., et al. (2012). P53-Tigar Axis attenuates Mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell. Cardiol. 52, 175–184. doi: 10.1016/j.yjmcc.2011.10.008
Hu, W., Chen, S., Thorne, R. F., and Wu, M. (2019b). Tp53, Tp53 target genes (dram, Tigar), and autophagy. Adv. Exp. Med. Biol. 1206, 127–149. doi: 10.1007/978-981-15-0602-4_6
Hu, K., Li, Y., Yu, H., and Hu, Y. (2019a). Ctbp1 confers protection for hippocampal and cortical neurons in rat models of Alzheimer's disease. Neuroimmunomodulation 26, 139–152. doi: 10.1159/000500942
Iadecola, C., and Anrather, J. (2011). Stroke research at a crossroad: asking the brain for directions. Nat. Neurosci. 14, 1363–1368. doi: 10.1038/nn.2953
Iwanaga, M., Mori, K., Iida, T., Urata, Y., Matsuo, T., Yasunaga, A., et al. (1998). Nuclear factor kappa B dependent induction of gamma Glutamylcysteine Synthetase by ionizing radiation in T98g human Glioblastoma cells. Free Radic. Biol. Med. 24, 1256–1268. doi: 10.1016/S0891-5849(97)00443-7
Iyaswamy, A., Wang, X., Krishnamoorthi, S., Kaliamoorthy, V., Sreenivasmurthy, S. G., Kumar Durairajan, S. S., et al. (2022). Theranostic F-Sloh mitigates Alzheimer’s disease pathology involving Tfeb and ameliorates cognitive functions in Alzheimer's disease models. Redox Biol. 51:102280. doi: 10.1016/j.redox.2022.102280
Jain, M., Cui, L., Brenner, D. A., Wang, B., Handy, D. E., Leopold, J. A., et al. (2004). Increased myocardial dysfunction after ischemia-reperfusion in mice lacking Glucose-6-phosphate dehydrogenase. Circulation 109, 898–903. doi: 10.1161/01.CIR.0000112605.43318.CA
Järvelä, S., Bragge, H., Paunu, N., Järvelä, T., Paljärvi, L., Kalimo, H., et al. (2006). Antioxidant enzymes in Oligodendroglial brain tumors: association with proliferation, apoptotic activity and survival. J. Neuro-Oncol. 77, 131–140. doi: 10.1007/s11060-006-9118-0
Jiang, S., Li, T., Ji, T., Yi, W., Yang, Z., Wang, S., et al. (2018). Ampk: potential therapeutic target for ischemic stroke. Theranostics 8, 4535–4551. doi: 10.7150/thno.25674
Ju Hwang, C., Choi, D. Y., Park, M. H., and Hong, J. T. (2019). Nf-Κb as a key mediator of brain inflammation in Alzheimer's disease. CNS Neurol. Disord. Drug Targets 18, 3–10. doi: 10.2174/1871527316666170807130011
Kalogeris, T., Baines, C. P., Krenz, M., and Korthuis, R. J. (2012). Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 298, 229–317. doi: 10.1016/B978-0-12-394309-5.00006-7
Katsel, P., Tan, W., Fam, P., Purohit, D. P., and Haroutunian, V. (2013). Cell cycle checkpoint abnormalities during dementia: a plausible association with the loss of protection against oxidative stress in Alzheimer's disease [corrected]. PLoS One 8:E68361. doi: 10.1371/journal.pone.0068361
Kawauchi, K., Araki, K., Tobiume, K., and Tanaka, N. (2008). P53 regulates glucose metabolism through An Ikk-Nf-Kappab pathway and inhibits cell transformation. Nat. Cell Biol. 10, 611–618. doi: 10.1038/ncb1724
Kim, J., Devalaraja-Narashimha, K., and Padanilam, B. J. (2015a). Tigar regulates glycolysis in ischemic kidney proximal tubules. Am. J. Physiol. Renal Physiol. 308, F298–F308. doi: 10.1152/ajprenal.00459.2014
Kim, J. Y., Kim, N., and Yenari, M. A. (2015b). Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci. Ther. 21, 309–319. doi: 10.1111/cns.12360
Kimata, M., Matoba, S., Iwai-Kanai, E., Nakamura, H., Hoshino, A., Nakaoka, M., et al. (2010). P53 and Tigar regulate cardiac Myocyte energy homeostasis under hypoxic stress. Am. J. Physiol. Heart Circ. Physiol. 299, H1908–H1916. doi: 10.1152/ajpheart.00250.2010
Kleinschnitz, C., Grund, H., Wingler, K., Armitage, M. E., Jones, E., Mittal, M., et al. (2010). Post-stroke inhibition of induced Nadph oxidase type 4 prevents oxidative stress and Neurodegeneration. PLoS Biol. 8:e1000479. doi: 10.1371/journal.pbio.1000479
Langston, J. W. (2017). The MPTP story. The Mptp Story. J Parkinsons Dis 7, S11–S19. doi: 10.3233/JPD-179006
Lei, B., Liu, J., Yao, Z., Xiao, Y., Zhang, X., Zhang, Y., et al. (2021). Nf-Κb-induced Upregulation of Mir-146a-5p promoted hippocampal neuronal oxidative stress and Pyroptosis via Tigar in a model of Alzheimer's disease. Front. Cell. Neurosci. 15:653881. doi: 10.3389/fncel.2021.653881
Li, H., and Jogl, G. (2009). Structural and biochemical studies of Tigar (Tp53-induced glycolysis and apoptosis regulator). J. Biol. Chem. 284, 1748–1754. doi: 10.1074/jbc.M807821200
Li, Q. Q., Li, J. Y., Zhou, M., Qin, Z. H., and Sheng, R. (2021). Targeting Neuroinflammation to treat cerebral ischemia-the role of Tigar/Nadph Axis. Neurochem. Int. 148:105081. doi: 10.1016/j.neuint.2021.105081
Li, L., Peng, L., and Zuo, Z. (2008). Isoflurane preconditioning increases B-cell Lymphoma-2 expression and reduces cytochrome C release from the mitochondria in the ischemic penumbra of rat brain. Eur. J. Pharmacol. 586, 106–113. doi: 10.1016/j.ejphar.2008.02.073
Li, M., Sun, M., Cao, L., Gu, J. H., Ge, J., Chen, J., et al. (2014). A Tigar-regulated metabolic pathway is critical for protection of brain ischemia. J. Neurosci. 34, 7458–7471. doi: 10.1523/JNEUROSCI.4655-13.2014
Li, D., Ueta, E., Kimura, T., Yamamoto, T., and Osaki, T. (2004). Reactive oxygen species (Ros) control the expression of Bcl-2 family proteins by regulating their phosphorylation and Ubiquitination. Cancer Sci. 95, 644–650. doi: 10.1111/j.1349-7006.2004.tb03323.x
Li, M., Zhou, Z. P., Sun, M., Cao, L., Chen, J., Qin, Y. Y., et al. (2016). Reduced Nicotinamide adenine dinucleotide phosphate, a pentose phosphate pathway product, might be a novel drug candidate for ischemic stroke. Stroke 47, 187–195. doi: 10.1161/STROKEAHA.115.009687
Liu, N., Lin, M. M., Huang, S. S., Liu, Z. Q., Wu, J. C., Liang, Z. Q., et al. (2021). Nadph and Mito-Apocynin treatment protects against Ka-induced Excitotoxic injury through autophagy pathway. Front. Cell Dev. Biol. 9:612554. doi: 10.3389/fcell.2021.612554
Liu, Z. Q., Liu, N., Huang, S. S., Lin, M. M., Qin, S., Wu, J. C., et al. (2020). Nadph protects against Kainic acid-induced Excitotoxicity via autophagy-lysosome pathway in rat striatum and primary cortical neurons. Toxicology 435:152408. doi: 10.1016/j.tox.2020.152408
Liu, M., Zhou, X., Li, Y., Ma, S., Pan, L., Zhang, X., et al. (2022). Tigar alleviates oxidative stress in brain with extended ischemia via a pentose phosphate pathway-independent manner. Redox Biol. 53:102323. doi: 10.1016/j.redox.2022.102323
López, K. L. R., Simpson, J. E., Watson, L. C., Mortiboys, H., Hautbergue, G. M., Bandmann, O., et al. (2019). Tigar inclusion pathology is specific for Lewy body diseases. Brain Res. 1706, 218–223. doi: 10.1016/j.brainres.2018.09.032
Luanpitpong, S., Chanvorachote, P., Stehlik, C., Tse, W., Callery, P. S., Wang, L., et al. (2013). Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells. Mol. Biol. Cell 24, 858–869. doi: 10.1091/mbc.e12-10-0747
Ma, T., Zhang, Y., Zhang, C., Luo, J. G., and Kong, L. Y. (2017). Downregulation of Tigar sensitizes the antitumor effect of Physapubenolide through increasing intracellular Ros levels to trigger apoptosis and Autophagosome formation in human breast carcinoma cells. Biochem. Pharmacol. 143, 90–106. doi: 10.1016/j.bcp.2017.07.018
Madan, E., Gogna, R., Kuppusamy, P., Bhatt, M., Pati, U., and Mahdi, A. A. (2012). Tigar induces P53-mediated cell-cycle arrest by regulation of Rb-E2f1 complex. Br. J. Cancer 107, 516–526. doi: 10.1038/bjc.2012.260
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). Pink1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for Mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Mauro, C., Leow, S. C., Anso, E., Rocha, S., Thotakura, A. K., Tornatore, L., et al. (2011). Nf-Κb controls energy homeostasis and metabolic adaptation by Upregulating mitochondrial respiration. Nat. Cell Biol. 13, 1272–1279. doi: 10.1038/ncb2324
Mejías, R., Villadiego, J., Pintado, C. O., Vime, P. J., Gao, L., Toledo-Aral, J. J., et al. (2006). Neuroprotection by transgenic expression of Glucose-6-phosphate dehydrogenase in dopaminergic Nigrostriatal neurons of mice. J. Neurosci. 26, 4500–4508. doi: 10.1523/JNEUROSCI.0122-06.2006
Méndez-Armenta, M., Nava-Ruíz, C., Juárez-Rebollar, D., Rodríguez-Martínez, E., and Gómez, P. Y. (2014). Oxidative stress associated with neuronal apoptosis in experimental models of epilepsy. Oxidative Med. Cell. Longev. 2014:293689. doi: 10.1111/j.1750-3639.2003.tb00015.x
Minutoli, L., Puzzolo, D., Rinaldi, M., Irrera, N., Marini, H., Arcoraci, V., et al. (2016). Ros-mediated Nlrp3 Inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxidative Med. Cell. Longev. 2016:2183026. doi: 10.1111/j.1750-3639.2003.tb00015.x
Mizushima, N., and Yoshimori, T. (2007). How to interpret Lc3 Immunoblotting. Autophagy 3, 542–545. doi: 10.4161/auto.4600
Monge, L., Mojena, M., Ortega, J. L., Samper, B., Cabello, M. A., and Feliu, J. E. (1986). Chlorpropamide raises Fructose-2,6-Bisphosphate concentration and inhibits gluconeogenesis in isolated rat hepatocytes. Diabetes 35, 89–96. doi: 10.2337/diab.35.1.89
Niizuma, K., Endo, H., and Chan, P. H. (2009). Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 109, 133–138. doi: 10.1111/j.1471-4159.2009.05897.x
Okawa, Y., Hoshino, A., Ariyoshi, M., Kaimoto, S., Tateishi, S., Ono, K., et al. (2019). Ablation of cardiac Tigar preserves myocardial energetics and cardiac function in the pressure overload heart failure model. Am. J. Physiol. Heart Circ. Physiol. 316, H1366–H1377. doi: 10.1152/ajpheart.00395.2018
Opitz, N., Drummond, G. R., Selemidis, S., Meurer, S., and Schmidt, H. H. (2007). The 'A's and 'O's of Nadph oxidase regulation: a commentary on “subcellular localization and function of alternatively spliced Noxo1 isoforms”. Free Radic. Biol. Med. 42, 175–179. doi: 10.1016/j.freeradbiomed.2006.11.003
Papadopoulos, C., Kirchner, P., Bug, M., Grum, D., Koerver, L., Schulze, N., et al. (2017). Vcp/P97 cooperates with Yod1, Ubxd1 and Plaa to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36, 135–150. doi: 10.15252/embj.201695148
Patel, M. (2004). Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic. Biol. Med. 37, 1951–1962. doi: 10.1016/j.freeradbiomed.2004.08.021
Patel, A. S., Lin, L., Geyer, A., Haspel, J. A., An, C. H., Cao, J., et al. (2012). Autophagy in idiopathic pulmonary fibrosis. PLoS One 7:E41394:e41394. doi: 10.1371/journal.pone.0041394
Puttachary, S., Sharma, S., Stark, S., and Thippeswamy, T. (2015). Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed. Res. Int. 2015:745613. doi: 10.1155/2015/745613
Qin, Y. Y., Li, M., Feng, X., Wang, J., Cao, L., Shen, X. K., et al. (2017). Combined Nadph and the Nox inhibitor Apocynin provides greater anti-inflammatory and Neuroprotective effects in a mouse model of stroke. Free Radic. Biol. Med. 104, 333–345. doi: 10.1016/j.freeradbiomed.2017.01.034
Quaegebeur, A., Segura, I., Schmieder, R., Verdegem, D., Decimo, I., Bifari, F., et al. (2016). Deletion or inhibition of the oxygen sensor Phd1 protects against ischemic stroke via reprogramming of neuronal metabolism. Cell Metab. 23, 280–291. doi: 10.1016/j.cmet.2015.12.007
Rahman, K. (2007). Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2, 219–236.
Rodríguez-Gil, J. E., Gómez-Foix, A. M., Fillat, C., Bosch, F., and Guinovart, J. J. (1991). Activation by vanadate of glycolysis in hepatocytes from diabetic rats. Diabetes 40, 1355–1359. doi: 10.2337/diab.40.10.1355
Rossi, D. J., Brady, J. D., and Mohr, C. (2007). Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 10, 1377–1386. doi: 10.1038/nn2004
Russell, R. L., Siedlak, S. L., Raina, A. K., Bautista, J. M., Smith, M. A., and Perry, G. (1999). Increased neuronal Glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch. Biochem. Biophys. 370, 236–239. doi: 10.1006/abbi.1999.1404
Sharma, V., Joseph, C., Ghosh, S., Agarwal, A., Mishra, M. K., and Sen, E. (2007). Kaempferol induces apoptosis in Glioblastoma cells through oxidative stress. Mol. Cancer Ther. 6, 2544–2553. doi: 10.1158/1535-7163.MCT-06-0788
Shen, M., Zhao, X., Zhao, L., Shi, L., An, S., Huang, G., et al. (2018). Met is involved in Tigar-regulated metastasis of non-small-cell lung cancer. Mol. Cancer 17:88. doi: 10.1186/s12943-018-0839-4
Shi, R., Weng, J., Zhao, L., Li, X. M., Gao, T. M., and Kong, J. (2012). Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci. Ther. 18, 250–260. doi: 10.1111/j.1755-5949.2012.00295.x
Shimizu, S., Nagayama, T., Jin, K. L., Zhu, L., Loeffert, J. E., Watkins, S. C., et al. (2001). Bcl-2 antisense treatment prevents induction of tolerance to focal ischemia in the rat brain. J. Cereb. Blood Flow Metab. 21, 233–243. doi: 10.1097/00004647-200103000-00007
Sinha, S., Ghildiyal, R., Mehta, V. S., and Sen, E. (2013). Atm-Nfκb Axis-driven Tigar regulates sensitivity of Glioma cells to Radiomimetics in the presence of Tnfα. Cell Death Dis. 4:E615. doi: 10.1038/cddis.2013.128
Sun, M., Li, M., Huang, Q., Han, F., Gu, J. H., Xie, J., et al. (2015). Ischemia/reperfusion-induced Upregulation of Tigar in brain is mediated by Sp1 and modulated by Ros and hormones involved in glucose metabolism. Neurochem. Int. 80, 99–109. doi: 10.1016/j.neuint.2014.09.006
Tang, J., Chen, L., Qin, Z. H., and Sheng, R. (2021). Structure, regulation, and biological functions of Tigar and its role in diseases. Acta Pharmacol. Sin. 42, 1547–1555. doi: 10.1038/s41401-020-00588-y
Tang, Y., Kwon, H., Neel, B. A., Kasher-Meron, M., Pessin, J. B., Yamada, E., et al. (2018). The Fructose-2,6-Bisphosphatase Tigar suppresses Nf-Κb signaling by directly inhibiting the linear ubiquitin assembly complex Lubac. J. Biol. Chem. 293, 7578–7591. doi: 10.1074/jbc.RA118.002727
Tanji, K., Mori, F., Kakita, A., Takahashi, H., and Wakabayashi, K. (2011). Alteration of Autophagosomal proteins (Lc3, Gabarap and Gate-16) in Lewy body disease. Neurobiol. Dis. 43, 690–697. doi: 10.1016/j.nbd.2011.05.022
Tsai, J. P., and Albers, G. W. (2015). Reperfusion versus recanalization: the winner is…. Stroke 46, 1433–1434. doi: 10.1161/STROKEAHA.115.009268
Tsujimoto, M., Yip, Y. K., and Vilcek, J. (1985). Tumor necrosis factor: specific binding and internalization in sensitive and resistant cells. Proc. Natl. Acad. Sci. U. S. A. 82, 7626–7630. doi: 10.1073/pnas.82.22.7626
Veeraraghavan, J., Natarajan, M., Aravindan, S., Herman, T. S., and Aravindan, N. (2011). Radiation-triggered tumor necrosis factor (Tnf) alpha-Nfkappab cross-signaling favors survival advantage in human Neuroblastoma cells. J. Biol. Chem. 286, 21588–21600. doi: 10.1074/jbc.M110.193755
Vosler, P. S., Graham, S. H., Wechsler, L. R., and Chen, J. (2009). Mitochondrial targets for stroke: focusing basic science research toward development of clinically translatable therapeutics. Stroke 40, 3149–3155. doi: 10.1161/STROKEAHA.108.543769
Wan, W., Jin, L., Wang, Z., Wang, L., Fei, G., Ye, F., et al. (2017). Iron deposition leads to neuronal Α-Synuclein pathology by inducing autophagy dysfunction. Front. Neurol. 8:1. doi: 10.3389/fneur.2017.00001
Wang, C. K., Ahmed, M. M., Jiang, Q., Lu, N. N., Tan, C., and Gao, Y. P. (2017a). Melatonin ameliorates hypoglycemic stress-induced brain endothelial tight junction injury by inhibiting protein nitration of Tp53-induced glycolysis and apoptosis regulator. J. Pineal Res. 63:e12440. doi: 10.1111/jpi.12440
Wang, G., Fu, X. L., Wang, J. J., Guan, R., and Tang, X. J. (2017b). Novel strategies to discover effective drug targets in metabolic and immune therapy for Glioblastoma. Curr. Cancer Drug Targets 17, 17–39. doi: 10.3389/fneur.2017.00001
Wang, X., Iyaswamy, A., Xu, D., Krishnamoorthi, S., Sreenivasmurthy, S. G., Yang, Y., et al. (2022). Real-time detection and visualization of amyloid-Β aggregates induced by hydrogen peroxide in cell and mouse models of Alzheimer's disease. ACS Appl. Mater. Interfaces. doi: 10.1021/acsami.2c07859
Wang, X., Li, R., Chen, R., Huang, G., Zhou, X., and Liu, J. (2020). Prognostic values of Tigar expression and (18)F-Fdg pet/Ct in clear cell renal cell carcinoma. J. Cancer 11, 1–8. doi: 10.7150/jca.33442
Wang, S. J., Li, D., Ou, Y., Jiang, L., Chen, Y., Zhao, Y., et al. (2016). Acetylation is crucial for P53-mediated Ferroptosis and tumor suppression. Cell Rep. 17, 366–373. doi: 10.1016/j.celrep.2016.09.022
Wang, C. Y., Mayo, M. W., and Baldwin, A. S. (1996). Tnf-and cancer therapy-induced apoptosis: potentiation by inhibition of Nf-Kappab. Science 274, 784–787. doi: 10.1126/science.274.5288.784
Wang, P., Shao, B. Z., Deng, Z., Chen, S., Yue, Z., and Miao, C. Y. (2018). Autophagy in ischemic stroke. Prog. Neurobiol. 163-164, 98–117. doi: 10.1016/j.pneurobio.2018.01.001
Wanka, C., Steinbach, J. P., and Rieger, J. (2012). Tp53-induced glycolysis and apoptosis regulator (Tigar) protects Glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J. Biol. Chem. 287, 33436–33446. doi: 10.1074/jbc.M112.384578
Wu, F., Xu, H. D., Guan, J. J., Hou, Y. S., Gu, J. H., Zhen, X. C., et al. (2015). Rotenone impairs Autophagic flux and Lysosomal functions in Parkinson's disease. Neuroscience 284, 900–911. doi: 10.1016/j.neuroscience.2014.11.004
Xie, J. M., Li, B., Yu, H. P., Gao, Q. G., Li, W., Wu, H. R., et al. (2014). Tigar has a dual role in cancer cell survival through regulating apoptosis and autophagy. Cancer Res. 74, 5127–5138. doi: 10.1158/0008-5472.CAN-13-3517
Xu, X., Liu, C., and Bao, J. (2017). Hypoxia-induced Hsa-Mir-101 promotes glycolysis by targeting Tigar Mrna in clear cell renal cell carcinoma. Mol. Med. Rep. 15, 1373–1378. doi: 10.3892/mmr.2017.6139
Xu, H. D., and Qin, Z. H. (2019). Beclin 1, Bcl-2 and autophagy. Adv. Exp. Med. Biol. 1206, 109–126. doi: 10.1007/978-981-15-0602-4_5
Xu, H., Xu, X., Wang, H., Qimuge, A., Liu, S., Chen, Y., et al. (2019). Lkb1/P53/Tigar/autophagy-dependent Vegf expression contributes to Pm2.5-induced pulmonary inflammatory responses. Sci. Rep. 9:16600. doi: 10.1038/s41598-019-53247-6
Yin, L., Kufe, T., Avigan, D., and Kufe, D. (2014). Targeting Muc1-C is synergistic with Bortezomib in Downregulating Tigar and inducing Ros-mediated myeloma cell death. Blood 123, 2997–3006. doi: 10.1182/blood-2013-11-539395
Yu, H. P., Xie, J. M., Li, B., Sun, Y. H., Gao, Q. G., Ding, Z. H., et al. (2015). Tigar regulates Dna damage and repair through Pentosephosphate pathway and Cdk5-Atm pathway. Sci. Rep. 5:9853. doi: 10.1038/srep09853
Zatloukal, K., Stumptner, C., Fuchsbichler, A., Heid, H., Schnoelzer, M., et al. (2002). P62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 160, 255–263. doi: 10.1016/S0002-9440(10)64369-6
Zeng, S., Zhao, Z., Zheng, S., Wu, M., Song, X., Li, Y., et al. (2021). The E3 ubiquitin ligase Trim31 is involved in cerebral ischemic injury by promoting degradation of Tigar. Redox Biol. 45:102058. doi: 10.1016/j.redox.2021.102058
Zhang, Y., Chen, F., Tai, G., Wang, J., Shang, J., Zhang, B., et al. (2017). Tigar knockdown Radiosensitizes Trxr1-overexpressing Glioma in vitro and in vivo via inhibiting Trx1 nuclear transport. Sci. Rep. 7:42928. doi: 10.1038/srep42928
Zhang, D. M., Zhang, T., Wang, M. M., Wang, X. X., Qin, Y. Y., Wu, J., et al. (2019). Tigar alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radic. Biol. Med. 137, 13–23. doi: 10.1016/j.freeradbiomed.2019.04.002
Zhou, Y., Wu, J., Sheng, R., Li, M., Wang, Y., Han, R., et al. (2018). Reduced Nicotinamide adenine dinucleotide phosphate inhibits Mptp-induced Neuroinflammation and neurotoxicity. Neuroscience 391, 140–153. doi: 10.1016/j.neuroscience.2018.08.032
Zhou, J. H., Zhang, T. T., Song, D. D., Xia, Y. F., Qin, Z. H., and Sheng, R. (2016). Tigar contributes to ischemic tolerance induced by cerebral preconditioning through scavenging of reactive oxygen species and inhibition of apoptosis. Sci. Rep. 6:27096. doi: 10.1038/srep27096
Zhu, Z., Liu, L. F., Su, C. F., Liu, J., Tong, B. C., Iyaswamy, A., et al. (2022). Corynoxine B derivative Cb6 prevents Parkinsonian toxicity in mice by inducing Pik3c3 complex-dependent autophagy. Acta Pharmacol. Sin. doi: 10.3389/fneur.2017.00001
Keywords: TIGAR, stroke, PD, AD, seizures, brain tumors, nervous system diseases
Citation: Huang B, Lang X and Li X (2022) The role of TIGAR in nervous system diseases. Front. Aging Neurosci. 14:1023161. doi: 10.3389/fnagi.2022.1023161
Edited by:
Manoj K. Mishra, University of Calgary, CanadaReviewed by:
Ashok Iyaswamy, Hong Kong Baptist University, Hong Kong SAR, ChinaCopyright © 2022 Huang, Lang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoling Lang, NDkxMzc1MTVAcXEuY29t; Xihong Li, bGl4aWhvbmdoeGV5QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.